
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pharmacol. , 25 September 2020
Sec. Renal Pharmacology
Volume 11 - 2020 | https://doi.org/10.3389/fphar.2020.585633
This article is part of the Research Topic Combating Diabetes and Diabetic Kidney Disease View all 13 articles
 Keiichiro Matoba1*
Keiichiro Matoba1* Yusuke Takeda1Yosuke Nagai1Kensuke Sekiguchi1Tamotsu Yokota1Kazunori Utsunomiya2Rimei Nishimura1
Yusuke Takeda1Yosuke Nagai1Kensuke Sekiguchi1Tamotsu Yokota1Kazunori Utsunomiya2Rimei Nishimura1Rho-associated coiled-coil-containing protein kinase (ROCK) is a serine/threonine kinase that was originally identified as RhoA interacting protein. A diverse array of cellular functions, including migration, proliferation, and phenotypic modulation, are orchestrated by ROCK through a mechanism involving cytoskeletal rearrangement. Mammalian cells express two ROCK isoforms: ROCK1 (Rho-kinase β/ROKβ) and ROCK2 (Rho-kinase α/ROKα). While both isoforms have structural similarities and are widely expressed across multiple tissues, investigations in gene knockout animals and cell-based studies have revealed distinct functions of ROCK1 and ROCK2. With respect to the kidney, inhibiting ROCK activity has proven effective for the preventing diabetic kidney disease (DKD) in both type 1 and type 2 diabetic rodent models. However, despite significant progress in the understanding of the renal ROCK biology over the past decade, the pathogenic roles of the ROCK isoforms is only beginning to be elucidated. Recent studies have demonstrated the involvement of renal ROCK1 in mitochondrial dynamics and cellular transdifferentiation, whereas ROCK2 activation leads to inflammation, fibrosis, and cell death in the diabetic kidney. This review provides a conceptual framework for dissecting the molecular underpinnings of ROCK-driven renal injury, focusing on the differences between ROCK1 and ROCK2.
The World Health Organization estimates that, each year, around 1.2 million people worldwide die from end-stage renal disease (ESRD). Artificial kidneys and miniaturized dialysis save millions of lives, however dialysis requires cost up to US$91,000 per patient per year in the United States (End chronic kidney disease neglect, 2020), and fewer than half of those on dialysis survive for more than 5 years from the onset of ESRD. Diabetic kidney disease (DKD) in particular has had a devastating impact on the increasing frequency of ESRD.
One major breakthrough in the management of DKD came in the past two decades, when inhibitors of the renin-angiotensin system (RAS) were proven to attenuate the progressive impairment of the renal function. While cardiovascular outcome trials with sodium glucose co-transporter 2 (SGLT2) inhibitors demonstrated these agents’ renoprotective actions (Zinman et al., 2015; Kosiborod et al., 2017; Neal et al., 2017), the details are undoubtedly much more complex, with key concerns that current standards of care do not elicit complete remission. Given the limited drugs available to suppress DKD progression, there has been an ongoing effort to identify factors inducing renal injury and to develop effective therapeutic strategies.
Rho-associated protein kinase (ROCK) belongs to the family of serine/threonine kinases and is a major downstream effector of the small GTP-binding protein RhoA. ROCK signaling is involved in the regulation of a plethora of cellular functions. Due to its centrality in most cellular events, robust temporospatial and context-dependent regulation of ROCK is needed for cell homeostasis. In the kidney, over-activation of the ROCK pathway is clearly harmful; it promotes glomerular fibrosis and podocyte loss in the setting of a variety of diseases including but not limited to diabetes (Matoba et al., 2010; Meyer-Schwesinger et al., 2012; Matoba et al., 2013; Matoba et al., 2017). In addition, elevated ROCK activity results in the increase of oxidative stress, sodium retention, and vascular tone (Bussemaker et al., 2009; Calo et al., 2016; Calo et al., 2017). The beneficial effects of ROCK inhibition have been described in rodent models of DKD (Gojo et al., 2007; Kolavennu et al., 2008).
Two mammalian ROCK isoforms, ROCK1 (also known as Rho-kinase β/ROKβ) and ROCK2 (also referred to as Rho-kinase α/ROKα), have been identified (Nakagawa et al., 1996). The ROCK1 gene is located on chromosome 18 and consists of 1354 amino acids, while the ROCK2 gene is located on chromosome 2 and consists of 1388 amino acids. While these isoforms share 65% overall identity in amino acid sequence, ROCK1 and ROCK2 are differentially regulated, with distinct functions.
This review focuses on the pathophysiological functions of ROCK1 and ROCK2, and discusses the therapeutic effects of ROCK isoform inhibition in DKD.
Among protein kinase neighbors, ROCKs are closely associated with myotonic dystrophy kinase-related Cdc42-binding kinase (MRCK) and citron kinase. These kinases have the same domain structure, which consists of an N-terminal kinase domain, a central coiled-coil region, and various functional motifs at their respective C-terminal (Figure 1). In ROCKs, these functional motifs contain Rho-binding domain (RBD) and pleckstrin homology domain (PHD) that is split into two by an internal cysteine-rich C1 domain (CRD). Under natural conditions, PHD blunts ROCK activity by sequestering kinase interface (Wen et al., 2008). Supporting of this is the fact that deletion of the C-terminal region including the PHD results in constitutive activation in vitro (Wen et al., 2008). However, when the RBD binds to GTP-bound active RhoA, RhoB, or RhoC, or PHD is removed, ROCK is constitutively activated. Despite the high sequence homology in their kinase domains, different machinery is involved in the activation process, with ROCK1 activated through the cleavage of the C-terminal PHD by caspase-3 and ROCK2 activation mediated by granzyme B-regulated cleavage. In addition, the inactivation process differs between these two isoforms: ROCK1 is negatively controlled by Rad GTP-binding protein, whereas ROCK2 is inhibited by Gem GTP-binding protein (Ward et al., 2002).
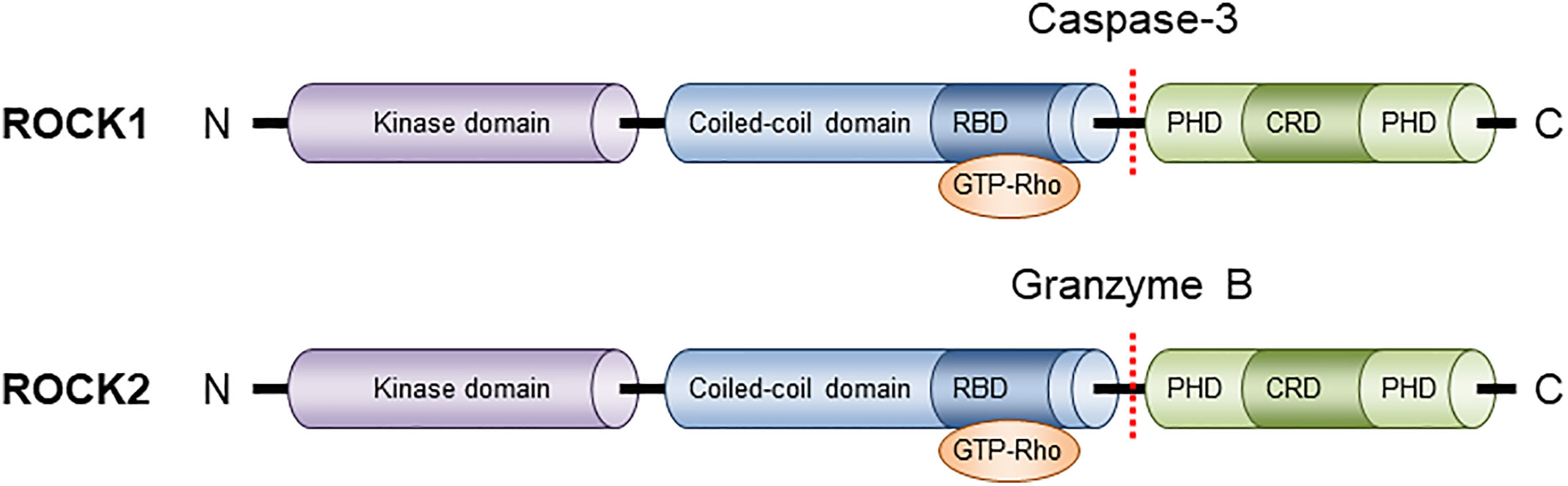
Figure 1 Structure of ROCK isoforms. ROCK1 and ROCK2 are known as ROKβ and ROKα respectively. Both isoforms consist of three major domains: a kinase domain in the N-terminal domain, a coiled-coil domain that contains a Rho-binding domain (RBD), and a putative pleckstrin homology domain (PHD) at its C-terminal end.
While ROCK1 is predominantly distributed in non-neural tissues including the gastrointestinal tract and lung, ROCK2 is found in the brain, kidney, and bladder (Nakagawa et al., 1996; Iizuka et al., 2012), indicating distinct actions of each isoform in these tissues. At the cellular level, ROCK1 has been detected in the cell membrane (Glyn et al., 2003), actin filaments, and lysosomes (Iizuka et al., 2012); however, the subcellular distribution of ROCK1 has not been fully clarified. ROCK2 activates p300 acetyltransferase to mediate gene transcription in vitro, which might explain why ROCK2 is predominantly localized to the nuclei (Tanaka et al., 2006). Consistently, ROCK2 is detected in euchromatin, where transcriptional events take place. ROCK1 and ROCK2 thus have different tissue and cellular distributions, which may affect their functions.
Findings obtained from global knockout of ROCK1 or ROCK2 have expanded our understanding regarding the function of each isoform. Mice harboring systemic ROCK1 deletion display impaired eye closure and an abnormal umbilical ring (Shimizu et al., 2005), whereas ROCK2 deficiency leads to intrauterine growth retardation (Thumkeo et al., 2003). While these data, coupled with other findings, suggest divergent physiological and pathological functions of ROCK isoforms, the specificity of those substrates has not been fully characterized (Hartmann et al., 2015).
Renal ROCK signaling is activated in rodent models of diabetes, regardless of the diabetes type (Gojo et al., 2007; Matoba et al., 2013). The ROCK-mediated molecular basis of DKD progression has been shaped by researchers using pharmacological inhibitors of ROCK (Y27632 and fasudil). Both of these agents ameliorate ROCK activity by competitively combining the ATP sites of the ROCK catalytic domain. While these studies have expanded ROCK research in the field of renal biology, these compounds inhibit both ROCK1 and ROCK2 with equal potency and have non-specific targets, such as protein kinase C, A, and mitogen-activated protein kinases at higher doses (Liao et al., 2007). Some of these disadvantages have been overcome by gene silencing approach, such as with small interfering RNA (siRNA) and systemic or conditional knockout. The distinct actions of each ROCK isoform in DKD are summarized in Figure 2.
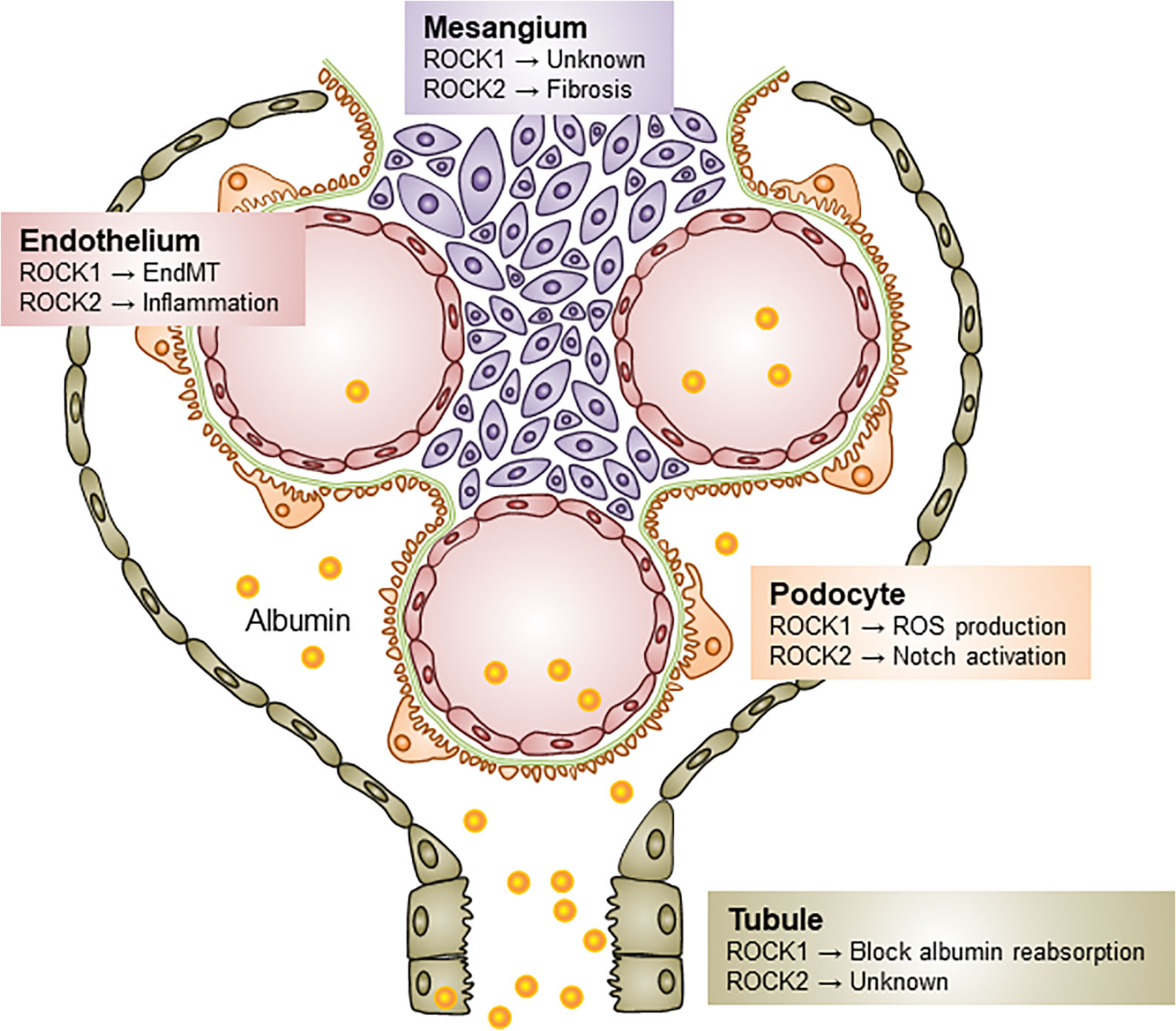
Figure 2 Distinct roles of ROCK isoforms in diabetic kidney disease. Both ROCK1 and ROCK2 contribute to the pathogenesis of DKD via different mechanisms. ROCK1 activation induces podocyte ROS production, EndMT, and blocks albumin endocytosis in tubular epithelial cells. Little is known about the role of ROCK1 on the mesangial biology, but ROCK2 elevation induces as the progression of mesangial expansion, Notch activation in podocytes, and endothelial inflammation. ROS, Reactive oxygen species; EndMT, Endothelial-to-mesenchymal transition.
The upregulation of the ROCK1 isoform is detected in the glomerular endothelium and mesangium of db/db mice (Peng et al., 2016) as well as in the distal tubules of streptozotocin (STZ)-induced diabetic rats (Wu et al., 2013). In cell-based experiments, tubular ROCK1 is activated by the CXC chemokine ligand 16 (Liang et al., 2018), a cytokine produced by diabetic kidney (Ye et al., 2017), to drive production of pro-inflammatory cytokines including tumor necrosis factor α (TNF-α), interleukin 1β, and caspase-3 activation and apoptosis.
From a transcriptional standpoint, we previously showed that siRNA-mediated gene ablation of ROCK1 was sufficient to induce a reduction in hypoxia-inducible factor 1α (HIF-1α) under diabetic conditions (Matoba et al., 2013). In that study, the HIF-1α expression was also suppressed by ROCK2 inhibition, suggesting that both ROCK1 and ROCK2 are requisite for glomerular HIF-1α generation and downstream fibrotic reactions in mesangial cells. The specific action of mesangial ROCK1 has not yet been clarified.
A series of elegant and comprehensive investigations from the Danesh laboratory identified ROCK1-mediated molecular events in DKD using gain- and loss-of-function studies in mice (Wang W. et al., 2012). Intriguingly, ROCK1-deficient mice showed attenuation of albuminuria and histological abnormalities in these models. Conversely, podocyte-specific ROCK1 knockin confers a phenotype that has many of the features of DKD. Mechanistically, they described an unexpected direct action of ROCK1 for regulating mitochondrial fission through phosphorylation and the recruitment of dynamin-related protein-1 (Drp1). The results of that study implicate ROCK1 as a critical regulator of the mitochondrial dynamics in diabetes and suggest that ROCK1 may be a relevant therapeutic target for the generation of oxidative stress in podocytes.
The permselectivity of the glomerular filtration barrier limits the passage of albumin into the Bowman’s capsule, resulting in the loss of transport selectivity and culminating in albuminuria, as is common among individuals in DKD. Glomerular endothelium, a key component of the filtration barrier, is converted into the mesenchymal phenotype in cases of diabetes, a process termed endothelial-to-mesenchymal transition (EndMT). Peng et al. investigated the contribution of ROCK1 to EndMT using ROCK1-overexpressing glomerular endothelial cells (Peng et al., 2016). The authors performed quantitative polymerase chain reaction (qPCR) and Western blotting and observed the increased expression of mesenchymal markers (e.g. α-SMA and Snail), together with the loss of endothelial junctional molecules, particularly VE-cadherin. Collectively, they reported that the activation of ROCK1 triggers EndMT, resulting in the loss of cellular attachment to each other and vascular hyper-permeability. These data provide critical insights into the heretofore unclear functions of ROCK1 in the signaling pathway that mediates the damage to glomerular tight junctions and albuminuria in DKD.
Zhou et al. investigated the function of ROCK1 in STZ-induced DKD models (Zhou et al., 2011). To determine the pathological contribution of tubular ROCK1, the authors analyzed the phenotype of diabetic ROCK1-deficient mice. They found that genetic ablation of ROCK1 prevented the development of albuminuria, and this effect was associated with protection against the loss of megalin and cubulin, members of the low-density lipoprotein receptor family that mediate albumin endocytosis in proximal tubular epithelial cells (Zhai et al., 2000). That study provided novel insights into the role of ROCK1 in albumin reabsorption in tubules. Interestingly, benidipine, a calcium channel blocker, has been suggested to inhibit proteinuria by suppressing ROCK1 and the transdifferentiation of renal tubular epithelium without affecting the glucose metabolism or blood pressure (Wu et al., 2013).
The inhibition of both ROCK isoforms by Y27632 or fasudil is effective for preventing tubulointestinal fibrosis in unilateral ureteral obstruction (UUO) models (Nagatoya et al., 2002; Baba et al., 2015); however, the systemic deletion of ROCK1 did not protect against the obstructive kidney damage (Fu et al., 2006). There was no recovery of transforming growth factor β (TGF-β)/SMAD signaling or structural derangement in the kidney of ROCK1-deficient mice. As such, we may reasonably suggest that targeting ROCK1 alone may not be adequate for attenuating tubular fibrosis, at least in UUO models, and the pathological contribution of ROCK1 to tubules may differ between DKD and other renal disease.
Whether or not ROCK1 exerts other functions in DKD is not completely understood. Genome-wide screening approaches will be required to define ROCK1 targets and the precise mechanisms of action. Such analyses will also provide promising opportunities for the development of ROCK1 inhibitors and their translation into clinical medicine.
Initial insights linking ROCK2 to diseases were gleaned from studies implicating ROCK2 as a regulator of, among others, immunity, inflammation, and fibrosis (Yang et al., 2018; Stam et al., 2019; Ricker et al., 2020). With regard to the kidney, we provided the first evidence indicating ROCK2 to be a core component of signaling circuitry that governs DKD progression. Nagai et al. demonstrated the upregulation of ROCK2 in the renal cortex of type 2 diabetic db/db mice (Nagai et al., 2019). In that study, ROCK2 inhibitors were evaluated for their efficacy against glomerular expansion and albuminuria in vivo. As a result, the preventive effects of these histological and functional abnormalities were confirmed. The authors also performed a loss of function analysis and revealed that gene deletion of ROCK2, but not ROCK1, decreased the fibrogenic response, concomitant with the suppression of phosphorylation of JNK and Erk, which in turn blocks the nuclear translocation of nuclear factor κB (NF-κB). Hence, ROCK2 inhibition appears to be a promising pharmacological intervention against DKD.
The podocyte slit diaphragm proteins nephrin and podocin are critical component forming the filtration barrier. In the context of diabetes, these components are damaged, mainly by the activation of Notch signaling pathways (Mathieson, 2011; Loeffler and Wolf, 2014). After the binding of Notch receptors to Notch ligands, such as Jagged-like and Delta-like, the C-terminal Notch intracellular domain (NICD) is cleaved from the cell membrane by γ-secretase and translocates into the nucleus, where the formation of recombination signal binding protein for immunoglobulin κJ region (Rbpj) and mastermind-like (MAML) proteins occurs in order to induce the expression of gene sets important for the development of the kidney (Malashicheva et al., 2020). The Notch pathway is reactivated in renal tissue obtained from diabetic mice to regulate the expression of Notch ligands (Niranjan et al., 2008). High-glucose conditions, TGF-β, or vascular endothelial growth factor (VEGF) are postulated to be the molecular basis for the upregulation of Notch signaling (Bonegio and Susztak, 2012). Of note, ROCK2-deficient podocytes are characterized by a significant reduction in TGF-β-induced Notch ligand expression (Matoba et al., 2017). In contrast, the induction of Notch ligand was not inhibited by ROCK1 gene deletion. These findings indicate the isoform-specific role of ROCK2 in podocytes and provide critical insights into potential strategies against albuminuria seen in DKD. Studies aimed at revealing the interdependency between ROCK2 and Notch modules through the generation of conditional knockout models are thus expected to be beneficial.
There is growing appreciation for the influence of vascular inflammation on regulating the progression of diabetic renal damage (Matoba et al., 2019). In addition to its effect in mesangial cells and podocytes, ROCK2 also plays important roles in endothelial cells. Takeda et al. conducted a series of studies to unravel the mechanisms by which ROCK2 activates vascular inflammation (Takeda et al., 2019). The qPCR array analysis of the mRNA expression profiles in ROCK2-null endothelium revealed differentially expressed genes related to vascular inflammation. Since chemokines and E-selectin production were downregulated in the endothelium, the authors examined monocyte migration and cell to cell adhesion, and found that these activities were abolished compared with those in endothelium with normal levels of ROCK2. These observations will need to be considered when establishing the contribution of ROCK2 to DKD, and when administering ROCK2 inhibitors to patients.
The impressive journey of ROCK2 inhibitors started with the development of KD-25 (formally SLx-2119), which is an orally available and selective inhibitor with a half maximal inhibitory concentration (IC50) and an inhibitory constant (Ki) of 60 nM and 41 nM, respectively (Boerma et al., 2008). Since this drug is used in clinical trials for patients with graft versus host disease (GVHD) and psoriasis (Yiu and Warren, 2016; MacDonald et al., 2017) (Table 1), ROCK2 inhibitors may could be used to treat DKD. The success of ROCK2 inhibitor clinical trials will hopefully inspire researchers to redouble their efforts to determine the molecular profiles responsible for ROCK2-regulated events in DKD.
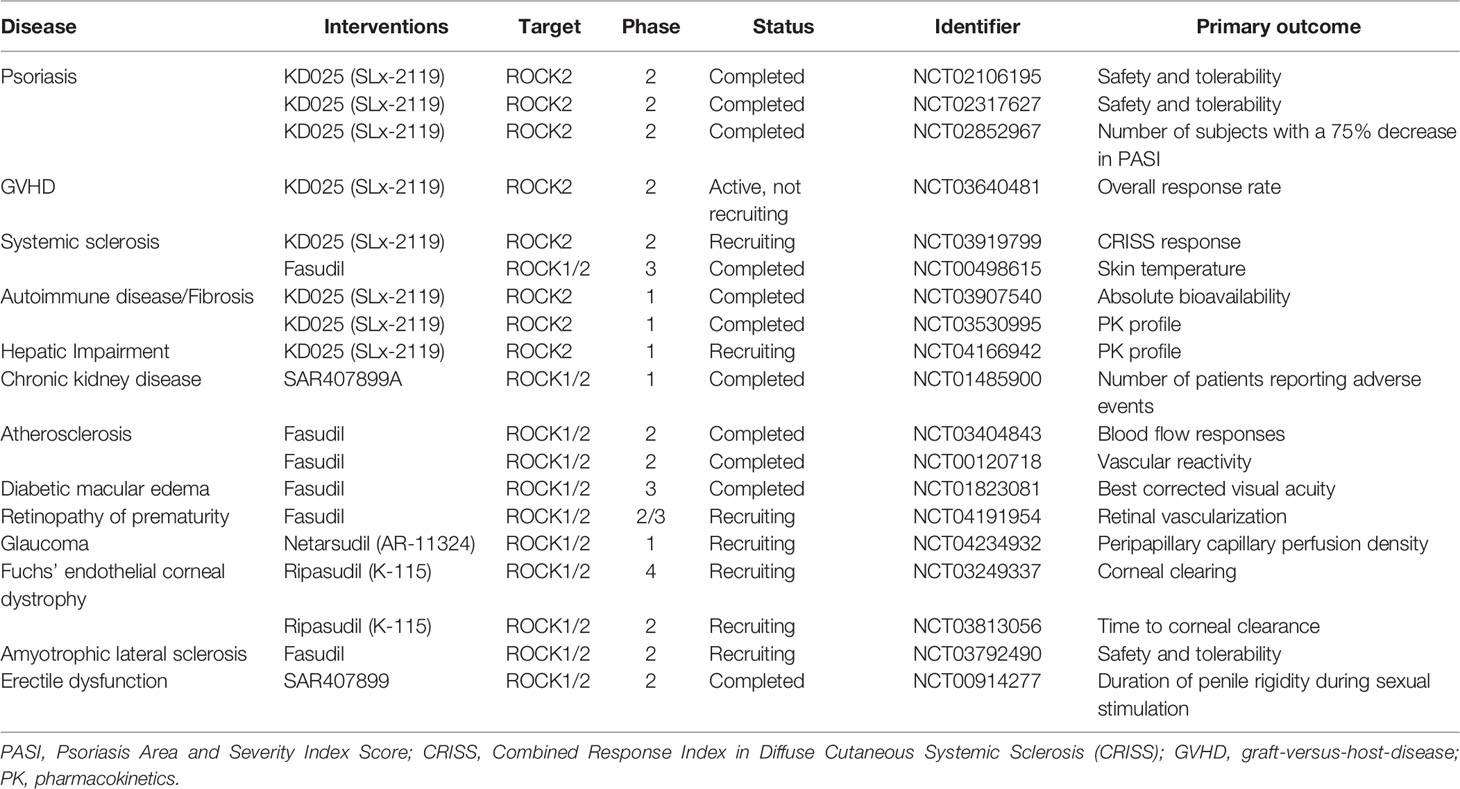
Table 1 Clinical trials of ROCK inhibitors.
Cardiovascular events are pertinent to morbidity and mortality in patients with DKD. Therefore, elucidation of molecular circuitry that governs atherogenic changes remains a major area of research. Recently, critical roles of ROCK isoforms in vascular disease have been evaluated by researchers. James Liao from Chicago and Hiroaki Shimokawa from Sendai are leaders in this field. Liao et al. identified macrophage ROCK1 as an essential element in the development of atherosclerosis through the modulation of foam cell formation and macrophage chemotaxis (Wang et al., 2008). ROCK2 also influences foam cell formation by inhibiting peroxisome proliferator-activated receptor-γ-mediated reverse cholesterol transport in inflammatory cells (Zhou et al., 2012). In vascular smooth muscle cells, ROCK2 controls migration and proliferation activities (Shimizu et al., 2013). In addition, Shimizu et al. focused on the pathologic role of ROCK2 in heart disease and showed that ROCK2 regulates hypertrophy of cardiomyocyte and cell death through interaction with serum response factor and ERK (Shimizu and Liao, 2016). These important findings coupled with the work of others have led to an increasing appreciation for ROCK2 as a critical molecule for not only renal disease but also cardiovascular disease.
As discussed above, published data have added to a burgeoning body of evidence that ROCKs are critical therapeutic targets against DKD and its related cardiovascular events. However, some caveats must be considered before this concept is accepted. First, the development of ROCK1-specific inhibitors and prospective intervention studies using ROCK1 or ROCK2 inhibitors are required in order to justify targeting ROCK isoforms to treat DKD. Second, whether an isoform-specific approach or pan ROCK inhibition would provide a better therapeutic outcome has yet to be clarified. The comparison of circulating and tissue levels of ROCK1 and ROCK2 between DKD patients and healthy subjects would facilitate our understanding the contribution of each isoform to the pathogenesis of DKD. These studies will also help identify useful targets of DKD therapy, which may vary by clinical stage, and allow for the earlier recognition of patients with diabetes who are at risk of DKD. Third, an open and thorough discussion of the risks while balancing potential clinical benefits of ROCK isoform inhibition is warranted. RhoA activation as well as RhoA inhibition results in podocyte damage (Wang L. et al., 2012), indicating that there is likely a narrow therapeutic window for ROCK isoform activity. This information will provide important insights to consider before commencing with ROCK isoform-selective inhibition in patients. In addition, given the impairment of insulin signaling in skeletal muscle observed in ROCK1 knockout mice (Lee et al., 2009), drugs with limited access to the kidney may be beneficial for patients with diabetes. However, it should be noted that the feasibility of ROCK inhibition has already been established with fasudil, a pan ROCK inhibitor, in patients with stroke (Shibuya et al., 2005). Moreover, clinical data of statins, which inhibit both ROCK1 and ROCK2 through the regulation of RhoA prenylation, demonstrate this medication to be well tolerated and safe during long-term treatment (Ford et al., 2016). Considering these findings alongside cogent evidence that ROCK is critical in versatile pathological aspects of diabetes, targeting ROCK1 and/or ROCK2 is expected to have therapeutic value for not only DKD but also other microvascular complications (i.e. retinopathy, neuropathy) (Yokota et al., 2007; Kanazawa et al., 2013). A deeper understanding of both the divergent and redundant roles of each isoform is therefore considered to be important for the development of effective therapeutic strategies, and for improving the prognosis of patients with diabetes.
KM wrote the manuscript. YT, YN, KS, TY, KU, and RN helped edit and revised the manuscript for important intellectual content. All authors contributed to the article and approved the submitted version.
This work was supported by JSPS KAKENHI Grant Number 20K08645 and 18K15985 (to KM), the Yokoyama Foundation for Clinical Pharmacology (to KM), the MSD Life Science Foundation (to KM), the Takeda Science Foundation (to KM), the Suzuken Memorial Foundation (to KM), the Ichiro Kanehara Foundation (to KM) and the Japan Diabetes Foundation (to RN).
KM has received research support from Sanofi KK, Tanabe Pharma, and Takeda Pharmaceutical. RN has received speaker honoraria from Astellas Pharma, Nippon Boehringer Ingelheim, Eli Lilly Japan KK, Kissei Pharmaceutical, Medtronic Japan, MSD, Novartis Pharma KK, Novo Nordisk Pharma, Sanofi KK, and Takeda Pharmaceutical.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Some of the contents of this review were generated based on the works of authors (Fu et al., 2006; Matoba et al., 2010; Meyer-Schwesinger et al., 2012; Matoba et al., 2013; Shimizu et al., 2013; Loeffler and Wolf, 2014; Shimizu and Liao, 2016; Matoba et al., 2017; Malashicheva et al., 2020).
Baba, I., Egi, Y., Utsumi, H., Kakimoto, T., Suzuki, K. (2015). Inhibitory effects of fasudil on renal interstitial fibrosis induced by unilateral ureteral obstruction. Mol. Med. Rep. 12, 8010–8020. doi: 10.3892/mmr.2015.4467
Boerma, M., Fu, Q., Wang, J., Loose, D. S., Bartolozzi, A., Ellis, J. L., et al. (2008). Comparative gene expression profiling in three primary human cell lines after treatment with a novel inhibitor of Rho kinase or atorvastatin. Blood Coagul. Fibrinol. 19, 709–718. doi: 10.1097/MBC.0b013e32830b2891
Bonegio, R., Susztak, K. (2012). Notch signaling in diabetic nephropathy. Exp. Cell Res. 318, 986–992. doi: 10.1016/j.yexcr.2012.02.036
Bussemaker, E., Herbrig, K., Pistrosch, F., Palm, C., Passauer, J. (2009). Role of rho-kinase in the regulation of vascular tone in hypertensive renal transplant recipients. Atherosclerosis 207, 567–572. doi: 10.1016/j.atherosclerosis.2009.05.025
Calo, L. A., Vertolli, U., Pagnin, E., Ravarotto, V., Davis, P. A., Lupia, M., et al. (2016). Increased rho kinase activity in mononuclear cells of dialysis and stage 3-4 chronic kidney disease patients with left ventricular hypertrophy: Cardiovascular risk implications. Life Sci. 148, 80–85. doi: 10.1016/j.lfs.2016.02.019
Calo, L. A., Ravarotto, V., Simioni, F., Naso, E., Marchini, F., Bonfante, L., et al. (2017). Pathophysiology of Post Transplant Hypertension in Kidney Transplant: Focus on Calcineurin Inhibitors Induced Oxidative Stress and Renal Sodium Retention and Implications with RhoA/Rho Kinase Pathway. Kidney Blood Press Res. 42, 676–685. doi: 10.1159/000483023
Ford, I., Murray, H., McCowan, C., Packard, C. J. (2016). Long-Term Safety and Efficacy of Lowering Low-Density Lipoprotein Cholesterol With Statin Therapy: 20-Year Follow-Up of West of Scotland Coronary Prevention Study. Circulation 133, 1073–1080. doi: 10.1161/CIRCULATIONAHA.115.019014
Fu, P., Liu, F., Su, S., Wang, W., Huang, X. R., Entman, M. L., et al. (2006). Signaling mechanism of renal fibrosis in unilateral ureteral obstructive kidney disease in ROCK1 knockout mice. J. Am. Soc. Nephrol. 17, 3105–3114. doi: 10.1681/ASN.2005121366
Glyn, M. C., Lawrenson, J. G., Ward, B. J. A. (2003). Rho-associated kinase mitigates reperfusion-induced change in the shape of cardiac capillary endothelial cells in situ. Cardiovasc. Res. 57, 195–206. doi: 10.1016/s0008-6363(02)00616-8
Gojo, A., Utsunomiya, K., Taniguchi, K., Yokota, T., Ishizawa, S., Kanazawa, Y., et al. (2007). The Rho-kinase inhibitor, fasudil, attenuates diabetic nephropathy in streptozotocin-induced diabetic rats. Eur. J. Pharmacol. 568, 242–247. doi: 10.1016/j.ejphar.2007.04.011
Hartmann, S., Ridley, A. J., Lutz, S. (2015). The Function of Rho-Associated Kinases ROCK1 and ROCK2 in the Pathogenesis of Cardiovascular Disease. Front. Pharmacol. 6, 276. doi: 10.3389/fphar.2015.00276
Iizuka, M., Kimura, K., Wang, S., Kato, K., Amano, M., Kaibuchi, K., et al. (2012). Distinct distribution and localization of Rho-kinase in mouse epithelial, muscle and neural tissues. Cell Struct. Funct. 37, 155–175. doi: 10.1247/csf.12018
Kanazawa, Y., Takahashi-Fujigasaki, J., Ishizawa, S., Takabayashi, N., Ishibashi, K., Matoba, K., et al. (2013). The Rho-kinase inhibitor fasudil restores normal motor nerve conduction velocity in diabetic rats by assuring the proper localization of adhesion-related molecules in myelinating Schwann cells. Exp. Neurol. 247, 438–446. doi: 10.1016/j.expneurol.2013.01.012
Kolavennu, V., Zeng, L., Peng, H., Wang, Y., Danesh, F. R. (2008). Targeting of RhoA/ROCK signaling ameliorates progression of diabetic nephropathy independent of glucose control. Diabetes 57, 714–723. doi: 10.2337/db07-1241
Kosiborod, M., Cavender, M. A., Fu, A. Z., Wilding, J. P., Khunti, K., Holl, R. W., et al. (2017). Lower Risk of Heart Failure and Death in Patients Initiated on Sodium-Glucose Cotransporter-2 Inhibitors Versus Other Glucose-Lowering Drugs: The CVD-REAL Study (Comparative Effectiveness of Cardiovascular Outcomes in New Users of Sodium-Glucose Cotransporter-2 Inhibitors). Circulation 136, 249–259. doi: 10.1161/CIRCULATIONAHA.117.029190
Lee, D. H., Shi, J., Jeoung, N. H., Kim, M. S., Zabolotny, J. M., Lee, S. W., et al. (2009). Targeted disruption of ROCK1 causes insulin resistance in vivo. J. Biol. Chem. 284, 11776–11780. doi: 10.1074/jbc.C900014200
Liang, H., Liao, M., Zhao, W., Zheng, X., Xu, F., Wang, H., et al. (2018). CXCL16/ROCK1 signaling pathway exacerbates acute kidney injury induced by ischemia-reperfusion. BioMed. Pharmacother. 98, 347–356. doi: 10.1016/j.biopha.2017.12.063
Liao, J. K., Seto, M., Noma, K. (2007). Rho kinase (ROCK) inhibitors. J. Cardiovasc. Pharmacol. 50, 17–24. doi: 10.1097/FJC.0b013e318070d1bd
Loeffler, I., Wolf, G. (2014). Transforming growth factor-beta and the progression of renal disease. Nephrol. Dial. Transplant. 29 Suppl 1, i37–i45. doi: 10.1093/ndt/gft267
MacDonald, K. P., Blazar, B. R., Hill, G. R. (2017). Cytokine mediators of chronic graft-versus-host disease. J. Clin. Invest. 127, 2452–2463. doi: 10.1172/JCI90593
Malashicheva, A., Kostina, A., Kostareva, A., Irtyuga, O., Gordeev, M., Uspensky, V., et al. (2020). Notch signaling in the pathogenesis of thoracic aortic aneurysms: A bridge between embryonic and adult states. Biochim. Biophys. Acta Mol. Basis Dis. 1866, 165631. doi: 10.1016/j.bbadis.2019.165631
Mathieson, P. W. (2011). The podocyte as a target for therapies–new and old. Nat. Rev. Nephrol. 8, 52–56. doi: 10.1038/nrneph.2011.171
Matoba, K., Kawanami, D., Ishizawa, S., Kanazawa, Y., Yokota, T., Utsunomiya, K., et al. (2010). Rho-kinase mediates TNF-alpha-induced MCP-1 expression via p38 MAPK signaling pathway in mesangial cells. Biochem. Biophys. Res. Commun. 402, 725–730. doi: 10.1016/j.bbrc.2010.10.093
Matoba, K., Kawanami, D., Okada, R., Tsukamoto, M., Kinoshita, J., Ito, T., et al. (2013). Rho-kinase inhibition prevents the progression of diabetic nephropathy by downregulating hypoxia-inducible factor 1alpha. Kidney Int. 84, 545–554. doi: 10.1038/ki.2013.130
Matoba, K., Kawanami, D., Nagai, Y., Takeda, Y., Akamine, T., Ishizawa, S., et al. (2017). Rho-Kinase Blockade Attenuates Podocyte Apoptosis by Inhibiting the Notch Signaling Pathway in Diabetic Nephropathy. Int. J. Mol. Sci. 18. doi: 10.3390/ijms18081795
Matoba, K., Takeda, Y., Nagai, Y., Kawanami, D., Utsunomiya, K., Nishimura, R., et al. (2019). Unraveling the Role of Inflammation in the Pathogenesis of Diabetic Kidney Disease. Int. J. Mol. Sci. 20. doi: 10.3390/ijms20143393
Meyer-Schwesinger, C., Dehde, S., Sachs, M., Mathey, S., Arefi, K., Gatzemeier, S., et al. (2012). Rho-kinase inhibition prevents proteinuria in immune-complex-mediated antipodocyte nephritis. Am. J. Physiol. Renal Physiol. 303, F1015–F1025. doi: 10.1152/ajprenal.00380.2011
Nagai, Y., Matoba, K., Kawanami, D., Takeda, Y., Akamine, T., Ishizawa, S., et al. (2019). ROCK2 regulates TGF-beta-induced expression of CTGF and profibrotic genes via NF-kappaB and cytoskeleton dynamics in mesangial cells. Am. J. Physiol. Renal Physiol. 317, F839–F851. doi: 10.1152/ajprenal.00596.2018
Nagatoya, K., Moriyama, T., Kawada, N., Takeji, M., Oseto, S., Murozono, T., et al. (2002). Y-27632 prevents tubulointerstitial fibrosis in mouse kidneys with unilateral ureteral obstruction. Kidney Int. 61, 1684–1695. doi: 10.1046/j.1523-1755.2002.00328.x
Nakagawa, O., Fujisawa, K., Ishizaki, T., Saito, Y., Nakao, K., Narumiya, S., et al. (1996). ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett. 392, 189–193. doi: 10.1016/0014-5793(96)00811-3
Neal, B., Perkovic, V., Mahaffey, K. W., de Zeeuw, D., Fulcher, G., Erondu, N., et al. (2017). Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N Engl. J. Med. 377, 644–657. doi: 10.1056/NEJMoa1611925
Niranjan, T., Bielesz, B ., Gruenwald, A., Ponda, M. P., Kopp, J. B., Thomas, D. B., et al. (2008). The Notch pathway in podocytes plays a role in the development of glomerular disease. Nat. Med. 14, 290–298. doi: 10.1038/nm1731
Peng, H., Li, Y., Wang, C., Zhang, J., Chen, Y., Chen, W., et al. (2016). ROCK1 Induces Endothelial-to-Mesenchymal Transition in Glomeruli to Aggravate Albuminuria in Diabetic Nephropathy. Sci. Rep. 6, 20304. doi: 10.1038/srep20304
Ricker, E., Chinenov, Y., Pannellini, T., Flores-Castro, D., Ye, C., Gupta, S., et al. (2020). Serine-threonine kinase ROCK2 regulates germinal center B cell positioning and cholesterol biosynthesis. J. Clin. Invest. 3654–3670 doi: 10.1172/JCI132414
Shibuya, M., Hirai, S., Seto, M., Satoh, S., Ohtomo, E. (2005). Effects of fasudil in acute ischemic stroke: results of a prospective placebo-controlled double-blind trial. J. Neurol. Sci. 238, 31–39. doi: 10.1016/j.jns.2005.06.003
Shimizu, T., Liao, J. K. (2016). Rho Kinases and Cardiac Remodeling. Circ. J. 80, 1491–1498. doi: 10.1253/circj.CJ-16-0433
Shimizu, Y., Thumkeo, D., Keel, J., Ishizaki, T., Oshima, H., Oshima, M., et al. (2005). ROCK-I regulates closure of the eyelids and ventral body wall by inducing assembly of actomyosin bundles. J. Cell Biol. 168, 941–953. doi: 10.1083/jcb.200411179
Shimizu, T., Fukumoto, Y., Tanaka, S., Satoh, K., Ikeda, S., Shimokawa, H., et al. (2013). Crucial role of ROCK2 in vascular smooth muscle cells for hypoxia-induced pulmonary hypertension in mice. Arterioscler. Thromb. Vasc. Biol. 33, 2780–2791. doi: 10.1161/ATVBAHA.113.301357
Stam, K., Cai, Z., van der Velde, N., van Duin, R., Lam, E., van der Velden, J., et al. (2019). Cardiac remodelling in a swine model of chronic thromboembolic pulmonary hypertension: comparison of right vs. left ventricle. J. Physiol. 597, 4465–4480. doi: 10.1113/JP277896
Takeda, Y., Matoba, K., Kawanami, D., Nagai, Y., Akamine, T., Ishizawa, S., et al. (2019). ROCK2 Regulates Monocyte Migration and Cell to Cell Adhesion in Vascular Endothelial Cells. Int. J. Mol. Sci. 20. doi: 10.3390/ijms20061331
Tanaka, T., Nishimura, D., Wu, R. C., Amano, M., Iso, T., Kedes, L., et al. (2006). Nuclear Rho kinase, ROCK2, targets p300 acetyltransferase. J. Biol. Chem. 281, 15320–15329. doi: 10.1074/jbc.M510954200
Thumkeo, D., Keel, J., Ishizaki, T., Hirose, M., Nonomura, K., Oshima, H., et al. (2003). Targeted disruption of the mouse rho-associated kinase 2 gene results in intrauterine growth retardation and fetal death. Mol. Cell Biol. 23, 5043–5055. doi: 10.1128/MCB.23.14.5043-5055.2003
Wang, H. W., Liu, P. Y., Oyama, N., Rikitake, Y., Kitamoto, S., Gitlin, J., et al. (2008). Deficiency of ROCK1 in bone marrow-derived cells protects against atherosclerosis in LDLR-/- mice. FASEB J. 22, 3561–3570. doi: 10.1096/fj.08-108829
Wang, W., Wang, Y., Long, J., Wang, J., Haudek, S. B., Overbeek, P., et al. (2012). Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 15, 186–200. doi: 10.1016/j.cmet.2012.01.009
Wang, L., Ellis, M. J., Gomez, J. A., Eisner, W., Fennell, W., Howell, D. N., et al. (2012). Mechanisms of the proteinuria induced by Rho GTPases. Kidney Int. 81, 1075–1085. doi: 10.1038/ki.2011.472
Ward, Y., Yap, S. F., Ravichandran, V., Matsumura, F., Ito, M., Spinelli, B., et al. (2002). The GTP binding proteins Gem and Rad are negative regulators of the Rho-Rho kinase pathway. J. Cell Biol. 157, 291–302. doi: 10.1083/jcb.200111026
Wen, W., Liu, W., Yan, J., Zhang, M. (2008). Structure basis and unconventional lipid membrane binding properties of the PH-C1 tandem of rho kinases. J. Biol. Chem. 283, 26263–26273. doi: 10.1074/jbc.M803417200
Wu, G., Xu, M., Xu, K., Hu, Y. (2013). Benidipine protects kidney through inhibiting ROCK1 activity and reducing the epithelium-mesenchymal transdifferentiation in type 1 diabetic rats. J. Diabetes Res. 2013, 174526. doi: 10.1155/2013/174526
Yang, W., Zhou, G., Yu, T., Chen, L., Yu, L., Guo, Y., et al. (2018). Critical role of ROCK2 activity in facilitating mucosal CD4(+) T cell activation in inflammatory bowel disease. J. Autoimmun. 89, 125–138. doi: 10.1016/j.jaut.2017.12.009
Ye, Y., Chen, Q., Li, J., Jin, L., Zheng, J., Li, X., et al. (2017). CXCL16 deficiency attenuates diabetic nephropathy through decreasing oxidative stress and inflammation. Biochem. Biophys. Res. Commun. 491, 848–854. doi: 10.1016/j.bbrc.2017.05.013
Yiu, Z. Z., Warren, R. B. (2016). Novel Oral Therapies for Psoriasis and Psoriatic Arthritis. Am. J. Clin. Dermatol. 17, 191–200. doi: 10.1007/s40257-016-0179-3
Yokota, T., Utsunomiya, K., Taniguchi, K., Gojo, A., Kurata, H., Tajima, N., et al. (2007). Involvement of the Rho/Rho kinase signaling pathway in platelet-derived growth factor BB-induced vascular endothelial growth factor expression in diabetic rat retina. Jpn. J. Ophthalmol. 51, 424–430. doi: 10.1007/s10384-007-0471-0
Zhai, X. Y., Nielsen, R., Birn, H., Drumm, K., Mildenberger, S., Freudinger, R., et al. (2000). Cubilin- and megalin-mediated uptake of albumin in cultured proximal tubule cells of opossum kidney. Kidney Int. 58, 1523–1533. doi: 10.1046/j.1523-1755.2000.00314.x
Zhou, L., Liu, F., Huang, X. R., Liu, F., Chen, H., Chung, A. C., et al. (2011). Amelioration of albuminuria in ROCK1 knockout mice with streptozotocin-induced diabetic kidney disease. Am. J. Nephrol. 34, 468–475. doi: 10.1159/000332040
Zhou, Q., Mei, Y., Shoji, T., Han, X., Kaminski, K., Oh, G. T., et al. (2012). Rho-associated coiled-coil-containing kinase 2 deficiency in bone marrow-derived cells leads to increased cholesterol efflux and decreased atherosclerosis. Circulation 126, 2236–2247. doi: 10.1161/CIRCULATIONAHA.111.086041
Keywords: notch, hypoxia, inflammation, Rho (Rho GTPase), ROCK1/ROCK2, diabetic kidney disease (DKD)
Citation: Matoba K, Takeda Y, Nagai Y, Sekiguchi K, Yokota T, Utsunomiya K and Nishimura R (2020) The Physiology, Pathology, and Therapeutic Interventions for ROCK Isoforms in Diabetic Kidney Disease. Front. Pharmacol. 11:585633. doi: 10.3389/fphar.2020.585633
Received: 21 July 2020; Accepted: 07 September 2020;
Published: 25 September 2020.
Edited by:
Keizo Kanasaki, Shimane University, JapanReviewed by:
Onkar Prakash Kulkarni, Birla Institute of Technology and Science, IndiaCopyright © 2020 Matoba, Takeda, Nagai, Sekiguchi, Yokota, Utsunomiya and Nishimura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Keiichiro Matoba, bWF0b2JhQGppa2VpLmFjLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.