 Meng Geng†
Meng Geng† Andrew Lin
Andrew Lin Thao P. Nguyen
Thao P. Nguyen- Division of Cardiology, Department of Medicine, The Cardiovascular Research Laboratory, David Geffen School of Medicine at UCLA, Los Angeles, CA, United States
Since the clinical use of digitalis as the first pharmacological therapy for atrial fibrillation (AF) 235 years ago in 1785, antiarrhythmic drug therapy has advanced considerably and become a cornerstone of AF clinical management. Yet, a preventive or curative panacea for sustained AF does not exist despite the rise of AF global prevalence to epidemiological proportions. While multiple elevated risk factors for AF have been established, the natural history and etiology of AF remain incompletely understood. In the present article, the first section selectively highlights some disappointing shortcomings and current efforts in antiarrhythmic drug therapy to uncover reasons why AF is such a clinical challenge. The second section discusses some modern takes on the natural history of AF as a relentless, progressive fibro-inflammatory “atriomyopathy.” The final section emphasizes the need to redefine therapeutic strategies on par with new insights of AF pathophysiology.
Introduction
Atrial fibrillation (AF) is the most common sustained arrhythmia with rising global prevalence and a 37% lifetime risk. In AF, the chaotic rapid atrial excitation renders atrial contractions ineffective and ventricular contractions irregularly irregular. The resulting hemodynamic disturbances in AF lead to deleterious clinical consequences. Blood stagnation and flow turbulence in the fibrillatory atrium increase the risk of stroke by five-fold (Wolf et al., 1991), heart failure by three-fold (Stewart et al., 2002), dementia by two-fold (Ott et al., 1997), and death by two-fold (Stewart et al., 2002). The rapid ventricular response in AF often causes symptoms and if left untreated, prolonged tachycardia can induce cardiomyopathy over time (Nerheim et al., 2004). Even when the ventricular response is normal or slow, AF can still cause symptoms that limit exercise tolerance and reduce quality of life. While there was just under half a million of AF-related hospitalizations in the United States in 2010 (Patel et al., 2014), this number has been increasing with each subsequent year (Nisar et al., 2016). AF treatment costs $26 billion annually in the United States alone (Kim et al., 2011; Rozen et al., 2018) and this financial burden will continue to rise as AF soars to epidemic proportions.
Since the first recognition of AF in animals 392 years ago by William Harvey in 1,628 (McMichael, 1982) and since the first ECG depiction of AF in humans 113 years ago by Willem Einthoven in 1906 (Einthoven, 1906; Silverman, 1994), several pathogenic mechanisms have been proposed (Burstein and Nattel, 2008; Greiser et al., 2011). Elevated risk factors for AF were established (January et al., 2014), with advancing age as the most prominent of all (Benjamin et al., 1994; Schnabel et al., 2015). In the United States, extended longevity from medical advances has led to the projection of up to 15 million patients diagnosed with AF by 2050 (Miyasaka et al., 2006; Colilla et al., 2013). Modifiable risk factors for developing AF within 10 years (Schnabel et al., 2009) include hypertension, diabetes mellitus (Patel et al., 2014), coronary artery disease (Peigh et al., 2020), heart failure, obesity (Thacker et al., 2013), and obstructive sleep apnea (Youssef et al., 2018). Non-modifiable AF risk factors include valvular heart disease (Banerjee et al., 2019), hypertrophic cardiomyopathy (Olivotto et al., 2001), and congenital heart disease (Hu and Lin, 2019). The lifetime AF risk of 20% for a patient without risk factors rises to 38% in the presence of just one elevated risk factor (Staerk et al., 2018).
In the interim period between the first pharmacological therapy for heart failure-associated AF using the digitalis leaf 234 years ago by William Withering in 1785 (Withering, 1785; Wray et al., 1985) and the explosive growth of catheter ablation therapy for AF in the last decade, several additional antiarrhythmic drugs have been developed. Yet, none has translated into significant gains in AF clinical management. With each new antiarrhythmic drug discovery, hopes for an effective and safe AF panacea rose, but soon ebbed away. However, despite all its limitations during more than two centuries to prevent or permanently cure AF, antiarrhythmic drug therapy has been and will remain a cornerstone of AF clinical management.
Review Scope
In this primarily pharmacological review, our goal is to examine the benefits (Table 1) and limitations (Table 2) of early, current, and emerging antiarrhythmic drug therapy for AF control. We will also examine the translation gap between the latest mechanistic insights and the presently outdated therapeutic paradigms. We will not acute management of the first AF occurrence or anticoagulant therapy for AF-related stroke prevention. Nor will we discuss antiarrhythmic therapy for lone AF, postoperative AF, AF precipitated by acute precipitating illnesses, or atrial flutter. Instead, we will review some prominent antiarrhythmic drug therapies for recurrent AF. We will focus particularly on the antiarrhythmic drugs featured in selected landmark AF clinical trials that shifted therapeutic paradigms and shaped management guidelines. We will emphasize why we must understand AF natural history to redefine strategies for antiarrhythmic drug development.

TABLE 1. Old and current Atrial Fibrillation antiarrhythmic drugs: therapeutic mechanisms and benefits.
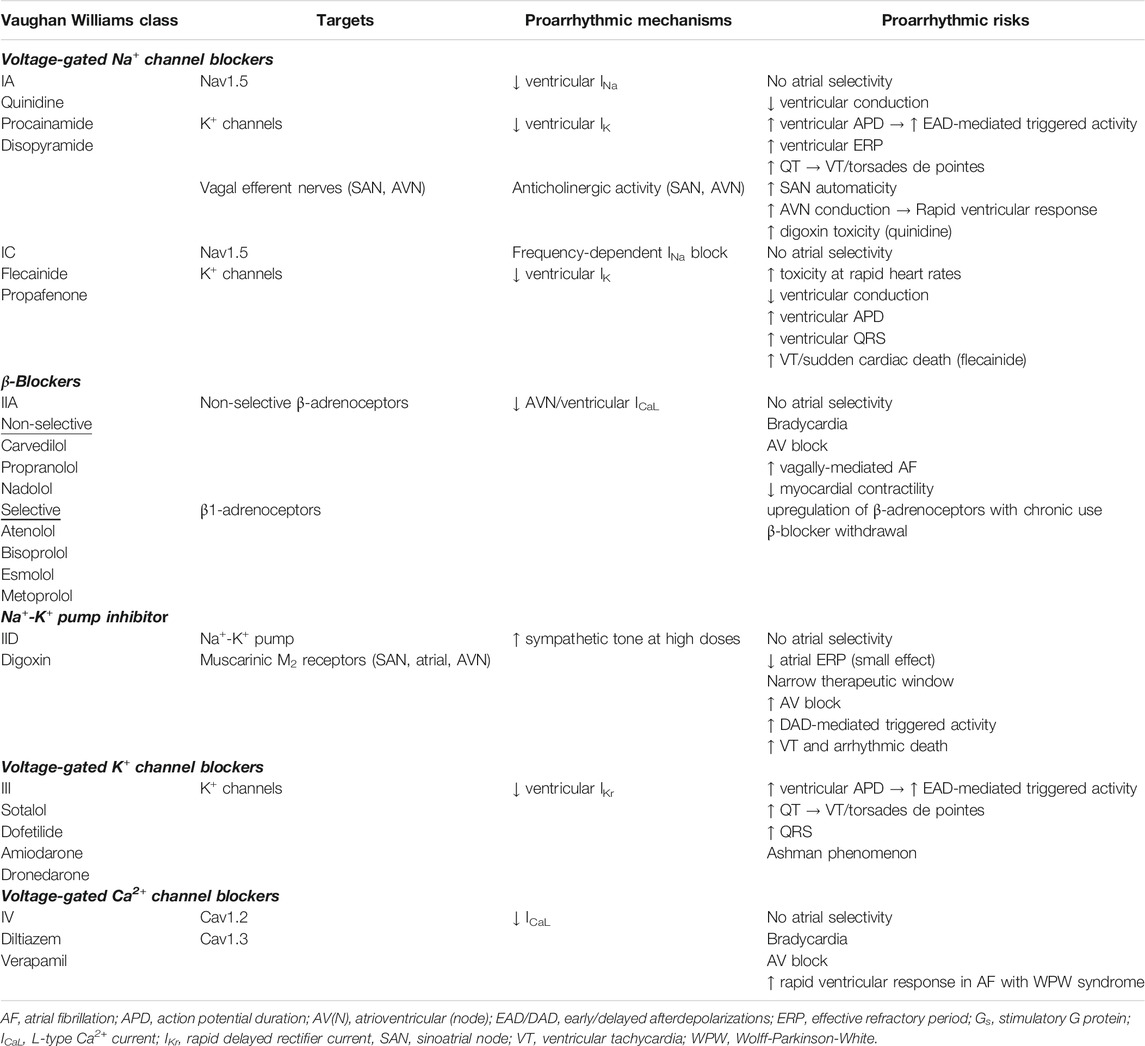
TABLE 2. Old and current Atrial Fibrillation antiarrhythmic drugs: therapeutic mechanisms and benefits.
Early Antiarrhythmic Drugs for Rhythm Control
Historically, the therapeutic approach was a “one-fits-all” paradigm that focused on the antiarrhythmic drugs rather than on the patients’ AF. The classical concept of an ideal pharmacotherapy for AF is an antiarrhythmic drug that can restore and maintain sinus rhythm with minimal or acceptable systemic side effects. In the 20th century, while both rhythm control and rate control were generally acceptable strategies for long-term AF management, the rhythm control strategy, which relies on cardioversion and antiarrhythmic drug therapy, was by far the preferred initial therapy. Classical AF antiarrhythmic drugs most commonly target ion channels for current inhibition rather than current augmentation (Table 1). Unfortunately, because these ion channel blockers commonly lack exquisite target specificity, they block both atrial and ventricular ion channels. As a result, adverse effects, largely from ventricular ion channel blockade (Table 2), often blunt atrial therapeutic benefits (Table 1).
Class IA Drugs (Quinidine, Procainamide, Disopyramide)
Quinidine, one of the oldest antiarrhythmic drugs, was used extensively in the 20th century for a variety of arrhythmias, including AF (Askey, 1946; McMillan and Welfare, 1947; Wegria, 1947). The combination of quinidine and verapamil has similar efficacy in preventing AF recurrences as sotalol but with better safety profile (Fetsch et al., 2004; Patten et al., 2004).
Procainamide and disopyramide came next on the market as promising new agents for AF in the mid-1900s (McCord and Taguchi, 1951; Miller et al., 1952; Hartel et al., 1974). Classified as Vaughan Williams class IA, quinidine, procainamide, and disopyramide block not only Na+ channels, but also K+ channels, thereby prolonging effective refractory period (ERP) (Vaughan Williams, 1984). ERP prolongation is beneficial in reducing susceptibility to reentry and therefore AF recurrences.
However, the lack of atrial selectivity of these antiarrhythmics contributes to QRS and QT prolongation, thereby increasing the risk of torsades de pointes, a most serious adverse effect that can lead to sudden cardiac arrest. There is also evidence that class IA drugs, especially disopyramide, decrease vagal tone by inhibiting cholinergic activity, which may require the concomitant use of β-blockers or Ca2+ channel blockers (Warrington and Hamer, 1980; Nakajima et al., 1989). For example, thanks to its negative inotropy, disopyramide received class IIa recommendation with level of evidence C for maintenance of sinus rhythm in AF patients with underlying hypertrophic cardiomyopathy, but must be used in conjunction with a β-blocker or nondihydropyridine Ca2+ channel blocker (January et al., 2014).
Thus, despite moderate efficacy in preventing AF recurrences, clinical adverse events, especially ventricular proarrhythmia and resultant increased mortality (quinidine and disopyramide), mar the safety profile of these class IA rhythm-control drugs (Lafuente-Lafuente et al., 2015).
Evolving Therapeutic Strategies
The 21st century marks critical milestones that shifted existing therapeutic paradigms and shaped management guidelines. The first milestone came in 2001, when the ACC, the AHA, and the European Society of Cardiology (ESC) jointly established a unified classification scheme for AF (Fuster et al., 2001; Calkins et al., 2007). Nonsustained recurrent AF is designated “paroxysmal” if AF does not sustain past 7 days. Past 7 days, AF is considered sustained. Sustained AF is designated “persistent” if not lasting longer than 1 year, “longstanding persistent” if lasting longer than 1 year (Calkins et al., 2007; Kirchhof et al., 2016), and “permanent” if rhythm control is unsuccessful or not attempted. Permanent AF is reclassified as longstanding persistent if a rhythm control strategy is adopted after 1 year of continuous AF. By separating AF presentations based on their temporal heterogeneities, this classification scheme marked an important shift in the therapeutic paradigm. The classical “one-fits-all,” drug-centered approach was replaced by the modern arrhythmia-centered realization that no single therapeutic strategy best serves all AF classes. Different AF classes are associated with different prognoses and require different therapeutic strategies, such as rhythm vs. rate control, pharmacological vs. non-pharmacological intervention (Table 3).
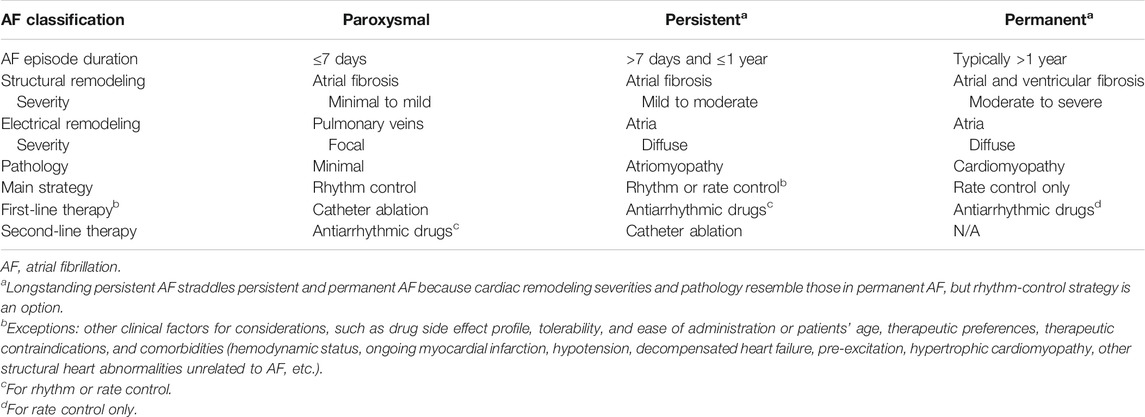
TABLE 3. Management strategy for recurrent Atrial Fibrillation based on episode duration.
The next milestone came from the revolutionary insights of the Atrial Fibrillation Follow-up Investigation of Rhythm Management (AFFIRM) (Wyse et al., 2002; Corley et al., 2004), the largest randomized controlled clinical trial at the time (n = 4,060 AF patients; 4-years follow up). The goal of the AFFIRM trial was to address the controversy of anticoagulated rhythm control vs. anticoagulated rate control as the main long-term therapeutic strategy with superior survival benefits. The AFFIRM trial revealed that while rhythm control improved survival, the existing antiarrhythmic drugs themselves did not improve survival because adverse effects of the drugs offset any survival benefits by the drugs in maintaining sinus rhythm (Corley et al., 2004). Therefore, antiarrhythmic drug therapies achieved similar outcomes with either the rhythm-control or the rate-control strategy. Nonetheless, this trial contributed in shifting the therapeutic paradigm from prior rhythm control to rate control as the main strategy.
However, improved survival is only one of the two primary clinical outcome endpoints in AF clinical management. The other primary therapeutic outcome is improved quality of life. Following the AFFIRM trial, multiple other randomized controlled trials addressed the question whether rhythm control or rate control was the strategy to superior improvement in quality of life. For example, the Controlled Study of Rate vs. Rhythm Control in Patients with Chronic AF and Heart Failure (CAFÉ-II) found that the rhythm-control strategy, even when achieved by an antiarrhythmic drug such as amiodarone, was associated with greater symptomatic relief and improved quality of life as compared to the rate-control strategy (Shelton et al., 2009).
Although an in-depth discussion of non-pharmacological interventions to control AF is beyond the scope of this pharmacological review, a brief comparison of the different merits and shortcomings of pharmacological and non-pharmacological approach, most notably catheter ablation, is warranted.
Overall, non-pharmacological interventions for AF has evolved a long way from electrical cardioversion and surgical ablation to device-based therapy with Holter or pacemaker telemonitoring and catheter ablation. The first non-pharmacological intervention for AF was electrical cardioversion to defibrillate the atria for rhythm control of non-persistent AF or symptomatic management, but its recent use as a diagnostic rather than therapeutic procedure has emerged. To the same end, implantable atrial defibrillator (IAD) for symptomatic, drug-refractory AF was evaluated in a multicenter study (n = 136 patients, 40-months median follow-up) but long-term outcome was not encouraging (Geller et al., 2003). Next, instead of atrial defibrillation, permanent atrial pacing to prevent and/or terminate AF has been explored, but there is insufficient evidence to support recommendation for AF therapy unless otherwise indicated for symptomatic bradycardia or sick sinus syndrome (Ellenbogen, 2007).
In the interim, ablation of AF has evolved from the first classical labyrinth Cox-Maze open-heart surgery with cardiopulmonary bypass support in 1987 to less invasive surgical and particularly catheter procedures. Various energy sources (such as cryothermia or ultra-low temperature cryoablation, unipolar or bipolar radiofrequency, microwave, laser, ultrasound, or electroporation) have been experimented to effectively and safely cause atrial myocyte death in a contiguous, transmural, permanent, and linear fashion. The goal of AF ablation is to electrically isolate common sites of triggers that initiate and/or maintain AF. The most common targets for ablation are the pulmonary veins (Haissaguerre et al., 1998) and left atrial appendage (Di Biase et al., 2010; Hocini et al., 2011); less common targets include the left atrial roof, mitral valve isthmus, and atrioventricular junction. Various refinements beyond pulmonary vein isolation, including complex fractionated atrial electrograms (CFAEs), focal impulse and rotor modulation (FIRM), electrocardiographic imaging (ECGI), have been explored but met with variable, inconsistent success (Weiss et al., 2016). For left atrial appendage occlusion, the AtriClip device has gained increasing popularity over the last decade as a safe and efficacious alternative to surgical closure and can be easily deployed during concomitant open chest surgery or stand-alone minimally invasive procedure (Ellis et al., 2017; Caliskan et al., 2018; Toale et al., 2019).
Taken together, while antiarrhythmic drug therapy repeatedly fell short of efficacy and safety expectations, the last 2 decades and particularly the last 5 years mark a revolutionizing milestone in non-pharmacological interventions for rhythm control with tremendous advances in AF catheter ablation (Kuck et al., 2016). For example, the Catheter Ablation vs. Standard Conventional Therapy in Patients with Left Ventricular Dysfunction and Atrial Fibrillation randomized clinical trial (CASTLE-AF; n = 363 patients, 37.8-months median follow-up) (Marrouche et al., 2018) showed that AF catheter ablation led to reduced overall mortality and heart failure-related hospitalizations. Despite all the controversies that it generated, CASTLE-AF contributed to the most recent clinical guideline revision. In early July 2019, the AHA/ACC/HRS published a focused update containing only one revision for rhythm control maintenance: a new, weak class IIb recommendation with level of evidence B-R for AF catheter ablation in heart failure (January et al., 2019).
However, concerning the two primary endpoints of improved survival and improved quality of life, catheter ablation has not yet proven superior to antiarrhythmic drug therapy as demonstrated by the Catheter Ablation vs. Antiarrhythmic Drug Therapy for AF randomized clinical trial (CABANA; n = 2,204 patients; 5-years follow up) (Moreno and Zamorano, 2014; Packer et al., 2018). In addition, AF catheter ablation faces its own host of considerable limitations, such as higher costs, limited availabilities of skilled electrophysiologists and experienced centers, procedural risks, and patients’ ablation candidacy, to name just a few. Thus, aside from patients’ own values and preferences for a pharmacological vs. non-pharmacological approach, expert consensus in both the ESC and AHA/ACC//HRS guidelines indicates a stronger class of recommendation for catheter ablation to maintain sinus rhythm in the presence of three critical variables: symptomatic recurrences, AF class (≤80% success rate for paroxysmal vs. ≤50% for persistent and longstanding persistent) (Kirchhof et al., 2016; Calkins et al., 2018), and heart failure (class of recommendation IIb) (January et al., 2019).
Current Antiarrhythmic Drugs for Rate Control
Recommendations for antiarrhythmic drug therapy for the ventricular rate-control strategy have not changed (January et al., 2019) since the 2014 AHA/ACC/HRS guideline and are summarized in Table 4 (January et al., 2014). For ventricular rate-control of AF patients with preserved left ventricular ejection fraction (LVEF), β-blockers and nondihydropyridine Ca2+-channel blockers (CCBs) remain first-line therapies, but differences between their mechanisms of action and pharmacokinetics lead to differences in therapeutic applications. For AF patients with reduced LVEF, β-blockers are the only first-line rate-control drugs whereas CCBs are contraindicated.
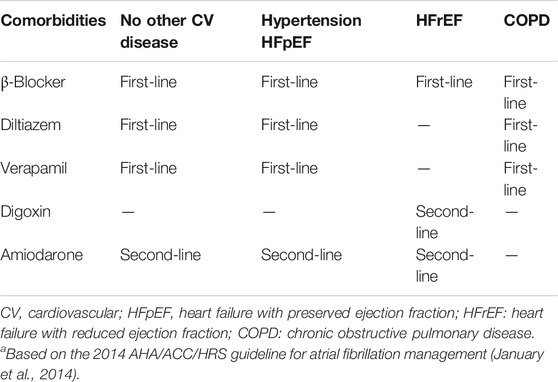
TABLE 4. Antiarrhythmic drug recommendations for ventricular rate control since 2014.a
Class IIA Drugs (β-Blockers)
The β-adrenergic system is an important regulator of the L-type Ca2+ current (ICaL). Cardiac β-adrenoceptors (80% β1, 20% β2) belong to the G protein-coupled receptor superfamily. β-Blockers slows ventricular response in AF by indirect inhibition of ICaL via competitive inhibition with β-agonists for binding to β-adrenoceptors. Binding of β-agonists to β-adrenoceptors activates stimulatory G-proteins (Gs) that activate adenylyl cyclase to generate cyclic adenosine monophosphate (cAMP), which in turn activates protein kinase A to phosphorylate phospholamban, ryanodine receptors, myofilament proteins, and L-type Ca2+ channels, thereby increasing ICaL. Binding of β-blockers to β-adrenoceptors prevents β-agonists from binding and activating Gs-dependent signaling. Because the β-blocker dose-response curve for rate control is relatively flat over the clinically-relevant dose range, once the high dose-range is reached, further dose uptitration produces only minimal gains in additional heart rate reduction but can substantially increase the likelihood of adverse events (hypotension). In that case, another drug class should be added.
Class IID Drugs (Digoxin)
Digoxin has a direct action on the myocardium and an indirect action on the autonomic nervous system. Digoxin directly blocks the myocardial Na + -K+ pump, leading to increased intracellular Ca2+ and slightly improved cardiac contractility. However, more prominent and relevant to AF rate control, digoxin indirectly modulates the electrophysiology of the atria, sinoatrial node, and particularly atrioventricular node by modulating the cardiac autonomic nervous system. In AF patients without heart failure, the indirect neural action of digoxin predominates because it is more robust and occurs at lower therapeutic doses (Watanabe, 1985). This explains why digoxin toxicity in the whole animal results primarily from the indirect neural effect but in the isolated heart from the direct myocardial effect (Levitt et al., 1976b). Of note, in heart failure, because baseline vagal tone is reduced and sympathetic tone is increased, the direct myocardial action of digoxin may predominate (Goodman et al., 1975; Watanabe, 1985).
Interestingly, the mechanisms underlying the pharmacodynamics effects of digoxin on the atrioventricular node caused quite a controversy, but they highlight both the influence of digoxin on the autonomic nervous system and the complex contributions of the autonomic nervous system to AF pathogenesis. Digoxin minimally modulates the electrophysiology of the atria (by reducing atrial refractoriness) and the sinoatrial node (by depressing its activity). In contrast, digoxin exerts profound vagomimetic and small sympatholytic effects on the atrioventricular node, leading to marked increase of atrioventricular nodal refractoriness. This results in reduced impulse conduction to the ventricles and slowing of ventricular response (Goodman et al., 1975; Watanabe, 1985). An important clinical implication is that because the therapeutic mechanism of digoxin depends on intact rich autonomic innervation of the atrioventricular node; its rate-control effect will be attenuated during exercise (that causes withdrawal of vagal tone), blunted in failing hearts, and ineffective in denervated transplanted hearts (Goodman et al., 1975).
Another important clinical implication is that the synergistic or antagonistic interactions of digoxin with other drugs that also act on the autonomic nervous system may potentially lead to disastrous clinical consequences. For example, the potent anticholinergic activity of Class IA antiarrhythmic drugs may antagonize the vagal stimulation by digitalis, potentially leading to deleterious increases in ventricular response. On the other hand, the antiadrenergic activity of β-blockers and other antihypertensive drugs may potentiate the sympathetic inhibition by digoxin, leading to excessively slow ventricular response or even atrioventricular block with resultant fatigue, syncope, or even sudden death. Likewise, the impairment of atrioventricular conduction by Ca2+-channel blockers may potentiate that by digoxin, leading to dangerously slow ventricular response (Watanabe, 1985).
A most important digoxin cardiotoxicity is cardiac arrhythmias, typically in the form of atrioventricular block with or without enhanced automaticity. Additionally, although at therapeutic doses the predominant action of digoxin is vagal stimulation, at high doses digoxin action may paradoxically switch from sympathetic inhibition to sympathetic stimulation, potentially causing lethal ventricular tachyarrhythmias (Watanabe, 1985).
Class IV Drugs (Nondihydropyridine Ca2+-Channel Blockers)
ICaL contributes to the upstroke of atrioventricular-node action potentials. Unlike β-blockers, CCBs inhibit ICaL directly by binding to L-type Ca2+ channels. Therefore, while both β-blockers and CCBs block ICaL, the difference in their mechanisms of action allows synergism. In contrast to the flat β-blocker dose-response curve, the CCB dose-response curve is relatively linear over the clinically relevant dose range. Therefore, CCB dose uptitration produces appreciable gains in additional ventricular rate reduction throughout the full therapeutic range. In a head-to-head comparative clinical trial of emergency patients presenting with AF and rapid ventricular response, diltiazem achieves rate-control targets faster without any increase in adverse events compared to metoprolol (Fromm et al., 2015).
Current Antiarrhythmic Drugs for Rhythm Control
Recommendations for antiarrhythmic drug therapy for the rhythm control strategy have not changed (January et al., 2019) since the 2014 AHA/ACC/HRS guideline (Table 5) (January et al., 2014).

TABLE 5. Antiarrhythmic drug recommendations for rhythm control since 2014.a
Class IC Drugs (Flecainide, Propafenone)
Both flecainide and propafenone are recommended first-line therapy for AF patients with no structural heart disease to restore sinus rhythm acutely or to maintain sinus rhythm long-term (Anderson et al., 1989; Meinertz et al., 2002). These two potent Na+ channel blockers slow atrial conduction, thereby reducing atrial automaticity and excitability. Due to their frequency dependence, their potency and, unfortunately, their proarrhythmic toxicity increase with rapid heart rates. Because these drugs lack atrial selectivity, they also act on the ventricular tissue to prolong both action potential and QRS. A QRS prolongation of 25% on therapy over baseline would require dose reduction or drug discontinuation due to the risk of lethal ventricular tachyarrhythmias and hemodynamic collapse. In fact, the contraindication of these drugs in structural heart disease stems from the demonstration of increased mortality associated with flecainide in the Cardiac Arrhythmia Suppression Trial (CAST) (Echt et al., 1991; Anderson et al., 1994). Due to similar concern for increased risk of sudden cardiac death, another important contraindication is the congenital loss-of-function Na+ channel mutations, such as in Brugada syndrome.
Class III Drugs (Sotalol, Dofetilide, Amiodarone)
Together with flecainide and propafenone, sotalol is also indicated first-line therapy in the maintenance of sinus rhythm for AF patients with no structural heart disease (Camm et al., 2010; Fuster et al., 2011). The antiarrhythmic benefit for AF and proarrhythmic toxicity of the class III drugs come from their potency in prolonging the action potential and refractoriness of the atria and ventricles, respectively.
Sotalol is particularly potent and proarrhythmic at slower heart rates due to its reverse frequency dependence. Thus, unlike the frequency-dependent class IC antiarrhythmics (flecainide) that are effective in terminating AF, the reverse-frequency dependent class III antiarrhythmics like sotalol and dofetilide are ineffective in terminating AF, but effective in preventing AF. One randomized placebo-controlled clinical trial (n = 300 patients with recurrent symptomatic AF; 1-year follow up) found that sotalol and propafenone are equally effective in maintaining sinus rhythm (73 and 63%, respectively), both being superior to placebo (35%) (Bellandi et al., 2001). However, only sotalol caused malignant proarrhythmia (4%) within 72 h of therapy initiation (Bellandi et al., 2001). Due to the lack of atrial selectivity, the class III proarrhythmic toxicity of sotalol relates to its potency in prolonging the QT interval, resulting in its propensity to cause torsades de pointes. The higher the dose and the slower the heart rate, the higher the risk of severe QT prolongation and consequently the higher the risk of torsades de pointes and mortality (Lafuente-Lafuente et al., 2015).
Whereas sotalol has mixed non-cardioselective β-blocking property, dofetilide is a “pure” class III antiarrhythmic, blocking only the voltage-gated K+ channels. Dofetilide was approved by United States Food and Drug Administration (FDA) for use in the United States in 1999 for AF patients without structural heart disease and without severely diminished creatinine clearance. Due to reverse-frequency dependence, dofetilide is more effective in maintaining sinus rhythm than in cardioverting AF as demonstrated by the Symptomatic AF Investigative Research on Dofetilide (SAFIRE-D) study (Singh et al., 2000). However, as with sotalol, because dofetilide causes dose-dependent QT prolongation, its major proarrhythmic toxicity is torsades de pointes. With incidence of dofetilide-associated torsades de pointes reportedly as high as 4.7%, drug initiation requires inpatient monitoring and certified physicians.
Amiodarone, an iodinated benzofuran derivative, is the most widely used off-label antiarrhythmic for AF due to its superior efficacy in both cardioversion (Khan et al., 2003) and particularly sinus rhythm maintenance (Roy et al., 2000; Wyse et al., 2002; Singh et al., 2005; Freemantle et al., 2011). Amiodarone displays properties of all four Vaughan Williams classes, but is classified as class III due to its major potency in blocking K+ channels to prolong cardiac action potential and refractoriness. Therefore, amiodarone also causes QT prolongation, but not torsades de pointes, probably because it also inhibits multiple other ion channels. Although a major cardiac adverse effect of amiodarone is sinus bradycardia, the use of amiodarone is unfortunately limited by a plethora of non-cardiac toxicities, attributed to the progressive accumulation of its damaging metabolite desethylamiodarone in multiple organs including the lungs, thyroid, liver, as well as the central nervous system. Amiodarone also exhibits multiple drug interactions (e.g., with warfarin) due to its inhibition of the cytochrome P450. Therefore, current clinical practice reserves amiodarone for AF with severe left heart failure or for refractory AF when options for other antiarrhythmic drugs have been exhausted.
Emerging Antiarrhythmic Drugs for Rhythm Control
There have been renewed efforts in the 21st century to discover either new antiarrhythmic agents or new synergistic combinations of existing and new agents to improve both efficacy and safety for AF antiarrhythmic drug therapy (Table 6). To this end, the modern mechanistic prototype for the ideal antiarrhythmic drug to control AF should include two critical specificities: first, specificity for the atrial tissue (to avoid ventricular proarrhythmia) and second, specificity for AF pathology.
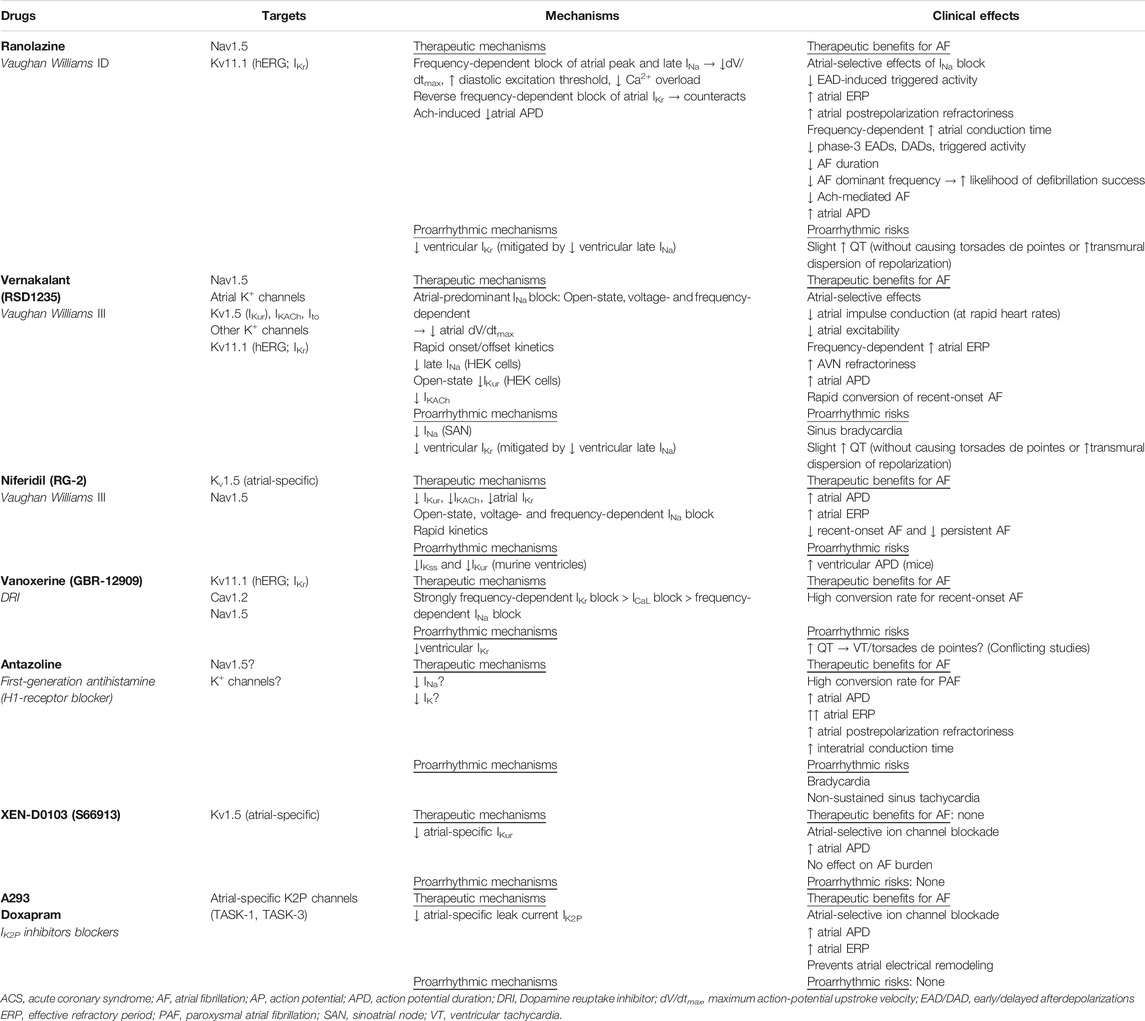
TABLE 6. Emergent antiarrhythmic drugs for AF rhythm control: mechanisms and clinical effects.
The atrial specificity of an antiarrhythmic drug can be enhanced by targeting atrial-predominant currents for blockade, particularly those affected by AF electrical remodeling, such as the ultrarapid delayed rectifier K+ current (IKur), the acetylcholine-activated inward-rectifier K+ current (IKACh), the two-pore-domain K+ current (IK2P), and the small-conductance Ca2+-activated K+ current (ISK). The AF-pathology specificity of an antiarrhythmic drug can be enhanced by exploiting the voltage- and frequency dependence of its channel blockade. Because atrial tissue with active AF is typically depolarized and has high frequency of depolarization, the more the drug blocking potency depends on depolarized voltage and high frequency of depolarization, the greater its specificity for AF pathology.
New Class I Drug (Ranolazine)
Ranolazine was initially designed as an anti-ischemic, antianginal drug because it blocks both atrial and ventricular late Na+ current (late INa), which increases pathologically during myocardial ischemia. Increased late INa is proarrhythmic because the resulting intracellular Na+ overload activates Na+-Ca2+ exchanger (NCX) reverse-mode, causing intracellular Ca2+ overload that leads to sarcoplasmic reticulum Ca2+ leak, diastolic dysfunction, and increased oxygen demand (Hale et al., 2006; Hale et al., 2008). However, ranolazine anti-AF efficacy in AF may also be due to its reduction of peak INa and especially its atrial-selective inhibition of the rapid delayed rectifier K+ current (IKr) carried by Kv11.1, the cardiac human ether-a-go-go-related gene (hERG) K+ channel. Together, these three mechanisms allow ranolazine to increase atrial postrepolarization refractoriness and slow interatrial conduction without affecting atrial action potential duration (Burashnikov et al., 2007; Kumar et al., 2009; Antzelevitch et al., 2011).
Ranolazine plus amiodarone is one particular combination of antiarrhythmic drug therapy that has shown promising results for synergistic efficacy for conversion of post-cardiac surgery and other recent-onset AF (Koskinas et al., 2014; Simopoulos et al., 2018). For new-onset AF, the combination of ranolazine and amiodarone had higher efficacy in terminating AF than amiodarone alone (Koskinas et al., 2014).Used alone, ranolazine decreased AF recurrence after electrical cardioversion (White and Nguyen, 2017) and reduced the frequency of new onset AF as well recurrences following coronary artery bypass grafting surgery (De Vecchis et al., 2018).
Likewise, the combination of ranolazine and low-dose dronedarone reduced the AF burden (mean duration of AF episodes) by 59% as compared to placebo in the HARMONY clinical trial (Reiffel et al., 2015). The synergistic activity between ranolazine and dronedarone can potentially reduce the dose needed and reduce the adverse effects associated with dronedarone monotherapy.
New Class III Drugs (Dronedarone, Vernakalant, Niferidil)
Dronedarone
Dronedarone is a newer, non-iodinated benzofuran derivative of amiodarone, designed to reduce amiodarone-related non-cardiac toxicities. Iodine moieties were removed to reduce thyroid toxicity. A methylsulfonamide group was added to reduce lipophilicity and consequent accumulation in lipid-rich organs (Zareba, 2006). Disappointingly, comparing with amiodarone, dronedarone efficacy is inferior (Le Heuzey et al., 2010; Freemantle et al., 2011) and its safety profile is different, but not necessarily superior (Hohnloser et al., 2009; Connolly et al., 2011). The most concerning dronedarone non-cardiac toxicity, which received FDA warning, is severe liver injury with rare risk of acute failure requiring transplant.
Vernakalant (RSD1235)
Another notable contender is vernakalant (RSD1235), a new class III antiarrhythmic drug with frequency- and voltage-dependent blockade. Originally developed for Kv1.5 (IKur) block to treat AF, vernakalant was shown experimentally to block multiple ion channels, including those mediating IKur, IKACh, the transient outward K+ current (Ito), IKr, peak and late INa. Interestingly, vernakalant acts as an atrial-selective antiarrhythmic drug but is not an atrial-selective ion channel blocker. At least two reasons account for its atrial selectivity. First, because the resting membrane potential is more depolarized in the atria than ventricles, plus this voltage difference is further amplified during AF, the dependence of its potency on depolarized membrane voltage confers atrial selectivity. Second, because Kv1.5 channels are detected only in human atria, but not in human ventricles, the blockade of human Kv1.5 by vernakalant is atrial-specific. Aside from its atrial selectivity, its rapid onset and offset kinetics confers a low proarrhythmic risk. Of note, while the Kv1.5 block by vernakalant was demonstrated in Kv1.5-expressing HEK293 cells (Eldstrom et al., 2007) and a rat model of myocardial infarction (Fedida et al., 2005), there has been no evidence yet for Kv1.5 block by vernakalant in human atrial myocytes (Wettwer et al., 2013). Likewise, to date the block of INaL by vernakalant was demonstrated only in Nav1.5-expressing HEK293 cells and rabbit Purkinje fibers (Fedida et al., 2006; Orth et al., 2006), but not in human atrial tissue (Wettwer et al., 2013).
Instead, the clinical efficacy of vernakalant in acute rhythm control of recent-onset AF is largely attributed to its frequency- and voltage-dependent open-state blockade of Na+ channels. Nav1.5 block by vernakalant reduces maximum action-potential upstroke velocity (dV/dtmax), thereby impairing atrial impulse conduction (Roy et al., 2008; Kowey et al., 2009; Camm et al., 2011). Additionally, its rapid unbinding kinetics of Nav1.5 block by vernakalant contributes to augmenting atrial selectivity and lowering ventricular proarrhythmic risks (Burashnikov and Antzelevitch, 2009).
Multiple placebo-controlled phase 3 studies have demonstrated the superior efficacy of vernakalant in rapid AF cardioversion (8–14 min after the first dose) (Roy et al., 2008; Kowey et al., 2009; Pratt et al., 2010; Stiell et al., 2010; Beatch et al., 2017), particularly in comparison to other antiarrhythmic drugs, such as amiodarone (Camm et al., 2011), propafenone (Conde et al., 2013a), flecainide (Conde et al., 2013b), and ibutilide (Simon et al., 2017).
In Europe, per the 2012 ESC Guidelines on AF management, vernakalant received class I recommendation with level of evidence A for cardioversion of AF with structurally normal heart or minimal heart disease and class IIb recommendation with level of evidence B for cardioversion of patients with moderate structural heart disease (Camm et al., 2012). Unfortunately, despite its high conversion rate for recent-onset AF that would allow rapid and efficacious treatment in the emergency department to reduce cost of care, vernakalant has not been approved by the United States FDA due to lingering safety concerns, including hypotension, bradycardia (a common adverse event), and the increased risk of serious adverse events (≤2%) with usage in heart failure or post-acute coronary syndrome (Lindsay, 2011). The risk of vernakalant inducing sustained ventricular arrhythmias or torsades de pointes is low but may increase with concomitant use of Class I/III antiarrhythmic drugs (Beatch and Mangal, 2016; Akel and Lafferty, 2018). The opposing conclusions regarding vernakalant usage by the regulatory bodies from the two sides of the Atlantic is puzzling considering their examination of the same clinical evidence. Because of the United States’ dominant position in the global marketplace, the FDA decision to withhold approval for vernakalant led sponsors of vernakalant to abandon further refinement pursuits for the drug.
Niferidil (RG-2)
Niferidil (RG-2) is a new class III antiarrhythmic drug, first introduced for human use in early 2010s. In patients, niferidil is highly effective for both persistent AF (85% conversion rate within 24 h) and recent-onset AF of less than 3-months duration (92% conversion rate) (Maykov et al., 2014). Because the study lacked randomization and placebo controls, more robust clinical trials are needed to demonstrate niferidil promising efficacy and safety profile in cardioversion of persistent AF. The ionic mechanisms responsible for its unique antiarrhythmic efficacy in human persistent AF have not been investigated. However, in guinea pigs, niferidil prolongs atrial action potential duration via IKr inhibition (Abramochkin et al., 2017). However, as demonstrated in mice, niferidil has off-target effects in the ventricular myocardium, where it also prolongs ventricular action potential duration in mice by inhibiting two other delayed rectifier K+ current, IKur and the steady-state current (Iss) (Abramochkin et al., 2015). It is unclear how probable off-target inhibition of human ventricular K+ currents by niferidil affects its efficacy and safety profile as an antiarrhythmic drug for AF. Therefore, minimizing off-target effects to improve efficacy and safety is the motivation to improve atrial selectivity of new antiarrhythmic drugs for AF control.
New Atrial-Selective Atrial Fibrillation Antiarrhythmic Drugs (Vanoxerine, Antazoline, XEN-D0103, IK2P Blockers)
The need for atrial selectivity galvanized efforts to repurpose existent drugs (vanoxerine, antazoline) and to develop novel antiarrhythmic drugs (XEN-D0103, IK2P blockers) (Table 6).
Vanoxerine (GBR-12909)
Vanoxerine is a 1,4-dialkylpiperazine derivative, a potent and selective dopamine reuptake inhibitor originally designed to treat Parkinson’s disease and depression due to its potent dopamine reuptake inhibition. Vanorexine is a potent blocker of IKr, ICaL, and INa in decreased order of potency. ICaL and INa blocks are strongly frequency-dependent and can offset IKr block, such that vanorexine does not prolong ventricular action potential duration or the QT interval, nor increase of the transmural dispersion of ventricular repolarization (Lacerda et al., 2010). These mechanisms likely explain why, unlike other class III antiarrhythmic drugs, vanorexine does not cause ventricular arrhythmias in animals (Matsumoto et al., 2010). Therefore, vanorexine resembles amiodarone in therapeutic benefits with its multichannel block and repolarization uniformity, but thankfully not in amiodarone toxicity (Lacerda et al., 2010).
Subsequently, a randomized, double-blinded study confirmed vanoxerine superior efficacy to placebo (70% vs. 20%). However, the study was terminated early due to the occurrence of ventricular arrhythmias and torsades de pointes in the treatment group (Piccini et al., 2016). This concerning finding regarding vanorexine safety profile was in contrast to an earlier phase 2B clinical trial, the Safety and Efficacy of Vanoxerine for Conversion of Atrial Fibrillation or Flutter to Normal Sinus Rhythm (COR-ART) trial, which found vanoxerine to have high conversion rate (84%), to be well tolerated, and to cause no ventricular arrhythmias (Dittrich et al., 2015). Clearly, larger studies are required to study the safety and efficacy of vanoxerine before definitive conclusions can be drawn. This may be a worthy endeavor if vanoxerine becomes a viable option for a pill-in-the-pocket approach for paroxysmal AF in structural heart disease where standard antiarrhythmic drugs are contraindicated.
Antazoline
Antazoline is a first-generation antihistamine with known antiarrhythmic properties and quinidine-like activities, first reported back in 1960s. Early data of the use of intravenous antazoline in AF cardioversion showed disappointing minimal efficacy (Gehring and Kehler, 1970). Since then, studies on antazoline have been sparse until recent renewed interest. Antazoline has been shown experimentally to reduce AF inducibility by increasing atrial APD, atrial ERP, atrial postrepolarization refractoriness, and interatrial conduction time. Inhibition of several ion currents, including both Na+ and K+ currents, presumably accounts for its antiarrhythmic effects (Frommeyer et al., 2017).
In the Efficacy and Safety of Antazoline in the Rapid Cardioversion of Paroxysmal Atrial Fibrillation (AnPAF) randomized clinical trial (n = 74 patients with paroxysmal AF) that antazoline could rapidly restore sinus rhythm (72.2% efficacy in the treatment group vs. 10.5% in the placebo group) and its only serious proarrhythmic effect was non-sustained sinus tachycardia in 5.6% of the treatment group (Maciag et al., 2017). A subsequent retrospective review of medical records from 450 Polish patients with short-duration AF, the Cardioversion with Antazoline Mesylate (CANT) study, demonstrated that antazoline represents an efficacious and safe method of pharmacological cardioversion with superior success rate compared to amiodarone and propafenone (Wybraniec et al., 2018). Thus, antazoline has emerged as a promising antiarrhythmic drug with cardioversion efficacy potentially surpassing amiodarone. However, additional randomized clinical trials are necessary to confirm this finding and to investigate its safety profile.
XEN-D0103/S66913
In the pipeline is XEN-D0103 (also known as S66913), a novel atrial-selective inhibitor of IKur carried by the Kv1.5 channel, which expresses only in atria (Ford et al., 2016). A phase one study demonstrated that XEN-D-0103/S66913 may have promising therapeutic benefit for reducing AF burden because it prolonged the atrial action potential duration specifically without affecting the ventricles (Ford et al., 2016). However, a recent prospective single-center phase two double-blind, randomized, placebo-controlled, cross-over study (n = 21 patients with paroxysmal AF and implanted dual-chamber permanent pacemaker for conduction defects) found that although XEN-D0103 was well-tolerated, it did not reduce AF burden when used alone (Shunmugam et al., 2018).
A most recent randomized, double-blind, placebo-controlled trial, the Double-blind, International study AssessinG efficacy of S 066913 in paRoxysmal Atrial Fibrillation-IKur inhibitor (DIAGRAF-IKUR; n = 58 patients with paroxysmal AF being considered for ablation), confirmed prior findings that S66913 was safe but ineffective in reducing AF burden (Camm et al., 2019). However, due to the small sample sizes of these studies, further trials in larger patient samples are still needed to assess its efficacy in cardioversion and maintenance.
IK2P Blockers (A293, Doxapram)
Another attractive target in the pipeline are atrial-selective inhibitors of IK2P, a K+ leak current carried by the most recently discovered K2P channels (Goldstein et al., 2001). Like other leak currents, IK2P contributes to the resting membrane potential and controls myocyte excitability by shaping action potential amplitude, duration, and frequency. Several classic antiarrhythmic drugs, such as amiodarone, dronedarone, carvedilol, are non-selective IK2P blockers; hence, the pressing need to develop selective IK2P blockers for similar efficacy but improved safety.
The Two-pore-domain Weak Inward rectifying K+ channel (TWIK)-related Acid-Sensitive K+ channel-1 (TASK-1; K2P3.1) is a member of the K2P channel family (Enyedi and Czirják, 2010). Because TASK-1 channels are abundant in human atria but not in ventricles (Heijman et al., 2017; Schmidt et al., 2017), TASK-1 channels regulate atrial action potential duration (Limberg et al., 2011). In chronic AF, but not in paroxysmal AF, atrial TASK-1-mediated K2P3.1 current expression is upregulated and causes action potential shortening (Schmidt et al., 2015). In a preclinical trial with an AF pig model, inhibition of atrial-specific K2P3.1 current using the TASK-1 channel blocker A293 reverses pathologic AF-related atrial action potential shortening and significantly prolongs atrial ERP without causing ventricular arrhythmias (Wiedmann et al., 2020). In another more recent pig preclinical trial, K2P3.1 current inhibition by genetic ablation of TASK-1 channels using small interfering RNA (siRNA) prevents atrial electrical remodeling and suppresses AF (Schmidt et al., 2019). However, an important caveat is that in chronic-AF patients with heart failure due to left ventricular dysfunction, atrial K2P3.1 expression is downregulated instead of upregulated, making it unlikely for IK2P blockers to be an efficient anti-AF approach in patients with structurally remodeled atria (Schmidt et al., 2017).
These encouraging preclinical findings highlight the need to further assess in clinical trials the potential of atrial-selective inhibition of IK2P as a novel, promising mechanism-based strategy to treat AF. In fact, an ongoing clinical trial in Germany, the DOxapram Conversion TO Sinus rhythm study (DOCTOS Trial, EudraCT no. 2018-002979-17, n = 40 AF patients aged 18–64 years and n = 40 AF patients aged ≥65 years), is evaluating the efficacy and safety of doxapram, a potent atrial-selective IK2P blocker, in cardioverting paroxysmal and persistent non-valvular AF to sinus rhythm within 6 h of intravenous administration. Doxapram, a known ventilator stimulant, blocks both TASK-1 and TASK-3 (K2P9.1) channels. TASK-3, the closest relative of TASK-1, expresses prominently in human right auricles. Interestingly, human atrial TASK-1 and TASK-3 can form heterodimers (Rinné et al., 2015).
Redefining Natural History and Pathophysiological Mechanisms
Paradigm Shift: From Three Distinct Atrial Fibrillation Temporal Patterns to One Progressive Fibro-Inflammatory “Atriomyopathy”
Electrical Remodeling
An early breakthrough in AF pathophysiology came with the recognition of the pathogenic electrical remodeling process. In electrical remodeling, “AF begets AF”—that is AF leads to electrical remodeling of the atrial electrophysiology to facilitate further AF induction and maintenance (Wijffels et al., 1995)—such that by the time AF progresses to the permanent stage, attempts to restore sinus rhythm are doomed to fail and therefore should not be attempted. The main perturbations in AF electrical remodeling that lead to shortening of the atrial action potential duration and resultant shortening of atrial refractoriness include downregulation of ICaL (Yue et al., 1997; Van Wagoner et al., 1999), upregulation of the inward rectifier current (IK1) (Biliczki et al., 2019), upregulation of ISK (Qi et al., 2014), and constitutive activation of IKACh (Dobrev et al., 2001; Dobrev et al., 2005; Heijman et al., 2018). The main perturbations in AF electrical remodeling leading to aberrant Ca2+ handling and resultant increased likelihood of reentry (Liu et al., 2020) and delayed afterdepolarization (a well-known trigger of AF) include persistent diastolic ryanodine receptor (RyR2) leak due to both sustained sarcoplasmic reticulum Ca2+ overloading, Ca2+/calmodulin kinase type II (CaMKII) activation, and upregulation of NCX (Nattel and Dobrev, 2012).
Structural Remodeling
The next breakthrough in AF pathophysiology came with the recognition of the pathogenic structural remodeling process. Fibrosis and inflammation were recognized as the hallmark of atrial structural remodeling in AF (Nattel, 2016; Yao et al., 2018). The knowledge that fibrosis and inflammation in AF correspond with disease severity and have prognostic value came with the technological advent of catheter ablation interventions for AF and late-gadolinium enhancement magnetic resonance imaging (LGE-MRI) to detect atrial fibrosis in vivo. LGE-MRI revealed that atrial fibrosis severity determines AF ablation outcome, likelihood of AF recurrence, stroke risk, and significant sinus node dysfunction; therefore, assessment of atrial fibrosis can guide patient selection and ablation procedures (Akoum and Marrouche, 2014; McGann et al., 2014). Extensive atrial structural remodeling by fibrosis (≥30% LA wall enhancement) predicts poor response to catheter ablation therapy for AF (Marrouche et al., 2014; McGann et al., 2014). Importantly, even when ablation succeeds in restoring sinus rhythm, ablation causes additional inflammation and fibrosis that perpetuate the original electrical and structural remodeling caused by AF (Teh et al., 2012). After AF ablation, the atrial conduction slowing, the decrease in left atrial ejection fraction with or without atrial dilation, and the development of new ablation-induced inflammation and fibrosis (Okumura et al., 2011) indicate worsening atrial function and predict increased risk of AF recurrence (Marrouche et al., 2014; Kim et al., 2018; Kim et al., 2019). Marked atrial fibrosis and elevated levels of inflammatory markers in AF recurrences are evident both in vivo (Qu et al., 2009; Sonmez et al., 2014) and postmortem (Ito et al., 2013; Corradi et al., 2014). Collagen volume, activity of nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB, a crucial inflammatory signaling transcription factor) (Qu et al., 2009) and serum levels of fibro-inflammatory biomarkers are all elevated in persistent-AF (Sonmez et al., 2014) and post-ablation AF recurrence (Okumura et al., 2011). Importantly, this elevated fibro-inflammatory state positively correlates with left atrial dilatation, an index of atrial structural (Sonmez et al., 2014).
Natural History
A breakthrough in AF natural history came with the realization of the prognostic value of the AF burden (duration and frequency of AF episodes) on disease progression. The knowledge of this longitudinal evolution of AF prompted the American College of Cardiology (ACC), American Heart Association (AHA), and ESC to classify AF based on temporal rhythm patterns into paroxysmal, persistent, and permanent and to recommend indications for therapeutic interventions based on AF class (Fuster et al., 2006; Jahangir et al., 2007; Ogawa et al., 2018).
Progressive “Atriomyopathy”
A most significant breakthrough in AF natural history and pathophysiology came with the appreciation that atrial electrical remodeling is intertwined with atrial structural remodeling (Li et al., 1999; Nattel et al., 2008) in a regenerative positive feedback loop that amplifies the pathophysiology of the initial AF episodes. We coined the term “atriomyopathy” to describe the ultimate consequence of the synergy of these two relentless AF pathogenic remodeling processes for the atrial myocardium. The progressive nature of AF atriomyopathy implies a critical need for early rhythm-control therapy in AF because timing matters in preventing or delaying the progression of AF atriomyopathy. This paradigm-shifting therapeutic strategy is most recently validated by the Early Treatment of Atrial Fibrillation for Stroke Prevention Trial (EAST-AFNET 4), an international, randomized, open, blinded-outcome-assessment clinical trial (Kirchhof et al., 2020). In fact, the most recently published 2020 AF guidelines mark a revolutionizing paradigm shift in management strategies because the pressing need for primary prevention to thwart AF atriomyopathy onset is emphasized for the first time (Hindricks et al., 2020).
Redefining Therapeutic Goals
Paradigm Shift: The Need for a Mechanism-Based Multipronged Strategy
To date, AF has been treated for rate control by antiarrhythmic drugs and for rhythm control by either antiarrhythmic drugs or invasive ablation. While the invasive Cox-maze and catheter ablation have evolved over the last three-and-a-half and 2 decades, respectively, antiarrhythmic drugs for AF have evolved over three-and-a-half centuries. Yet, our expectations for an efficacious and safe AF management strategy have not been met. Over that same time, thanks to basic and clinical research, our understanding of AF pathophysiology has also evolved from the assumption of AF as a static disease to the appreciation of AF as a progressive fibro-inflammatory atriomyopathy driven by a relentless positive feedback loop between electrical and structural remodeling. Therefore, our AF management focus also needs to evolve from a pure curative intent to incorporate preventive intent. Importantly, our AF management strategy also needs to evolve from the historical “one-fits-all” approach to a modern mechanism-based multipronged strategy to target both pathogenic electrical and structural remodeling processes (Figure 1).
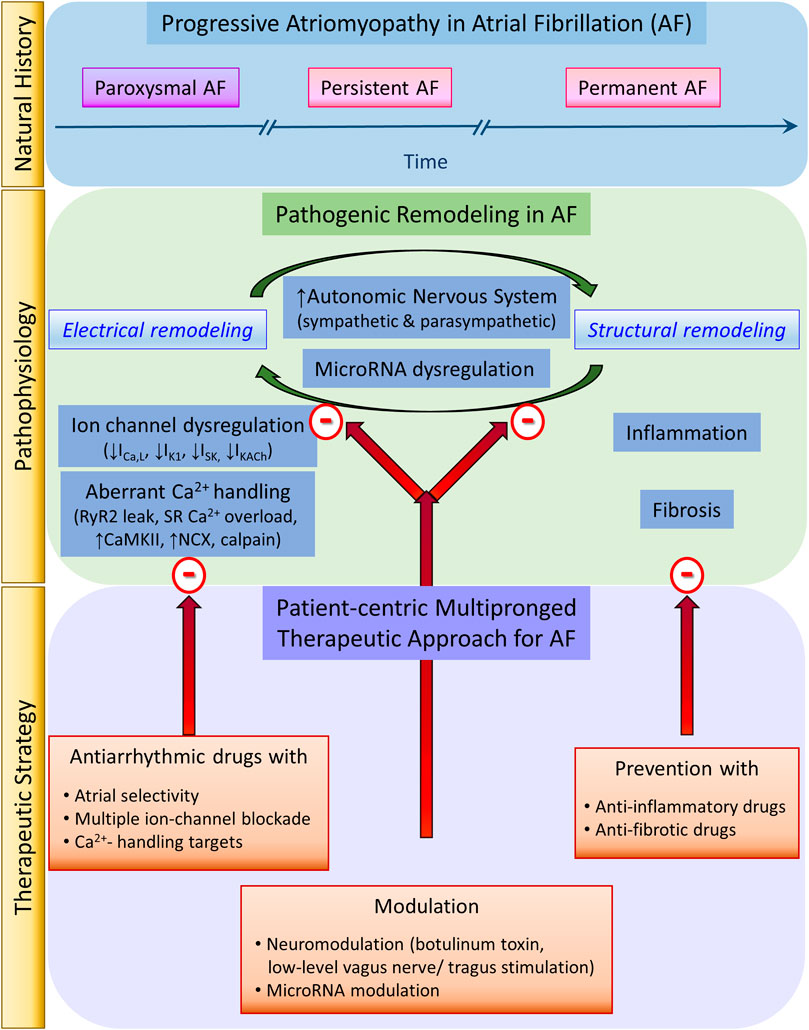
FIGURE 1. Atrial fibrillation natural history, pathophysiology, and therapeutic strategy.
Targeting Electrical Remodeling
To prevent or forestall pathogenic electrical remodeling with efficacy and safety, basic research has revealed at least three novel aspects to focus the development of antiarrhythmic drugs: atrial selectivity, multiple-ion-channel blockade, and Ca2+-mishandling targets. Novel potential ionic current targets include the currents carried by K+ channels with atrial-predominant expression, such as IKur, IK2P, ISK, and IKACh. Ca2+ mishandling also offers promising targets for stabilizers of RyR2, modulators of the sarcoplasmic reticulum Ca2+-ATPase 2a (SERCA2a), and inhibitors of NCX, CaMKII, and Ca2+-dependent protease calpain.
Targeting Structural Remodeling
Unlike electrical remodeling, which offers opportunities for reversibility, to date structural remodeling has not been meaningfully reversed once established. Therefore, prevention is the best approach to target pathogenic structural remodeling (Benjamin et al., 2009; Savelieva et al., 2011a; Savelieva et al., 2011b). Clinical targeting of modifiable AF risk factors such as diabetes, hypertension, obesity, and obstructive sleep apnea should be early and aggressive. Statins, and anti-inflammatory drugs targeting cytokines (IL-1β, IL-6, and TNF-α) and the renin-angiotensin-aldosterone system (RAAS) have been tried as potential therapies to forestall the progression of inflammation and fibrosis (Savelieva et al., 2011a; Savelieva et al., 2011b; Chaugai et al., 2016). Unfortunately, clinical success is still lacking, consistent with the irreversibility of structural remodeling.
Targeting Both Electrical and Structural Remodeling
Other promising areas of active research in modulation of microRNA (miRNA) expression and of the autonomic nervous system focus on mechanisms that target both electrical and structural remodeling simultaneously. Basic and clinical science have recently discovered the central signaling role of miRNAs in AF pathogenesis (Luo et al., 2015; Shen et al., 2020). Multiple dysregulated miRNAs, including miR-1, miR-26, miR-106b-25, miR-208a, miR-328, and miR-499, contribute to AF electrical remodeling (Lu et al., 2010; Ling et al., 2013; Luo et al., 2013; Soeki et al., 2016). These miRNAs target and dysregulate the expression of genes encoding critical atrial K+ and Ca2+ ion channels to cause shortening atrial action potential duration and effective refractory period to favor reentry for AF maintenance. Other multiple dysregulated miRNAs, including miR-21, miR-26, miR-29, miR-30, miR-133, and miR-590, contribute to AF pro-fibrotic structural remodeling (Dawson et al., 2013; Cheng et al., 2019; Sieweke et al., 2020). Therefore, modulating miRNAs has the potential of an innovative AF management strategy of important therapeutic, diagnostic, and prognostic values.
Neural modulation promises another frontier in AF rate and rhythm control given the prominent role of neural remodeling in AF initiation and maintenance (Chen et al., 2014). Autonomic nervous system activation of either limb or both limbs simultaneously can promote AF (Coumel, 1993). Neural remodeling perpetuates AF by engaging a positive feedback loop with adverse atrial electrical and structural remodeling. Sympathetic stimulation induces adrenergic AF (Lu et al., 2008) and facilitates cholinergic AF induction (Hashimoto et al., 1968; Sharifov et al., 2004). Sympathetic stimulation induces adverse atrial neural remodeling by heterogeneous sympathetic hyperinnervation and extensive nerve sprouting (Jayachandran et al., 2000; Chang et al., 2001). Sympathetic stimulation induces adverse atrial electrical remodeling by causing cellular Ca2+ overload and mishandling, activating NCX forward-mode and CaMKII, thereby promoting the emergence of AF triggers and shifting of pacemaker activity to ultrarapid-firing ectopic atrial sites (Geesbreght and Randall, 1971). Sympathetic stimulation induces adverse structural remodeling by causing myocyte apoptosis and myocardial fibrosis. Hence, to prevent or reverse AF electrical and structural remodeling, suppression of sympathetic outflow has been investigated using antiadrenergic drugs, renal sympathetic denervation (Wang et al., 2013), or ablation of atrial ganglionated plexi. Sympathetic suppression may reduce AF burden and inducibility and improve the success of pulmonary vein isolation (Katritsis et al., 2011), but success has proved inconsistent.
More curious are the paradoxical contributions of vagal stimulation to both AF pathogenesis and AF therapy (Chen et al., 2014). Vagal stimulation induces cholinergic AF (Hashimoto et al., 1968; Levitt et al., 1976a), also by causing adverse electrical and structural remodeling. Vagal stimulation induces adverse atrial electrical remodeling by heterogeneous shortening of atrial ERP (Liu and Nattel, 1997) and increasing dispersion of atrial refractoriness (Alessi et al., 1958), heterogeneous shortening of atrial action potential duration and increasing dispersion of atrial repolarization as well as increasing interatrial dyssynchrony (Ninomiya, 1966). Vagal stimulation induces profibrotic cardiac structural remodeling. For example, following T5 spinal cord transection, parasympathetic hyperinnervation causes dilation of left ventricular chamber, thinning of myocardial wall, and increased collagen content (Lujan et al., 2014). Therefore, it comes as no surprise that suppression of vagal stimulation by anticholinergic drugs or vagal denervation may improve the efficacy of pulmonary vein isolation to prevent AF recurrence (Chen and Tan, 2007) and reduces AF inducibility (Elvan et al., 1995). Likewise, suppression of vagal stimulation by ablation of pulmonary vein ganglionated plexi may increase the success of pulmonary vein isolation by mitigating vagally-induced shortening of atrial ERP (Lemola et al., 2008). Interestingly, botulinum toxin, known to inhibit vesicular release of intracellular acetylcholine from presynaptic neurons, presents a novel neuromodulation approach in AF therapeutics by simultaneous sympathovagal suppression. Recently, two prospective, double-blind study of patients with paroxysmal AF undergoing coronary artery bypass grafting, found that botulinum toxin injected into the epicardial fat pad suppressed AF both early post-operatively and, surprisingly, long-term during 1-year (Pokushalov et al., 2015) and 3-years follow up (Romanov et al., 2019). The unexpected salutary long-term antiarrhythmic effects of botulinum toxin were attributed to its suppression of the ganglionated plexi activity (Pokushalov et al., 2015).
Thus, it may seem shockingly heretical at first to suggest vagal stimulation as a promising candidate for the new frontier in AF therapeutics. Yet, it has long been recognized that digoxin controls ventricular response during AF by vagal stimulation of the atrioventricular node. Likewise, direct electrical stimulation of the right inferior ganglionated plexus may slow ventricular response in postoperative AF by slowing atrioventricular node conduction. However, the emerging paradigm of AF neuromodulation by vagal stimulation relies on another mechanism to suppress AF, namely by preventing or reversing atrial electrical and structural remodeling. Not all vagal stimulation is arrhythmogenic; the proarrhythmic vs. antiarrhythmic effects are dictated by the strength and duration of vagal stimulation and the complex but poorly understood interactions between the extrinsic and intrinsic cardiac autonomic nervous system. For example, in a dog model of tachypacing-induced AF, strong vagal nerve stimulation that slows the sinus rate by at least 60% facilitates AF inducibility, but moderate vagal nerve stimulation that slows the sinus rate by only 10–30% does not shorten atrial ERP or affect AF inducibility (Takei et al., 2001; Zhang et al., 2009a). Low-level stimulation of the extrinsic cardiac autonomic nervous system can suppress AF by inhibiting the autonomic outflow of intrinsic cardiac autonomic nervous system (Zhang et al., 2009c; Shen et al., 2011; Yu et al., 2011). In fact, low-level vagus nerve stimulation (LLVS) can suppress tachypacing-induced adrenergic AF as well was cholinergic-induced AF by mitigating neural remodeling by the intrinsic cardiac autonomic nervous system, thereby preventing and reversing adverse atrial electrical and structural remodeling (Sheng et al., 2011; Kusunose et al., 2014; Beaumont et al., 2015). LLVS suppresses post-operative AF and attenuates inflammation (decreased serum TNF-α and IL-6) (Zhang et al., 2009b; Rossi et al., 2012; Stavrakis et al., 2017). Likewise, in dogs and humans, transcutaneous low-level tragus electrical stimulation (LLTS) suppresses AF and attendant generation of inflammatory cytokines in patients with paroxysmal AF (Stavrakis et al., 2015). The Transcutaneous Electrical Vagus Nerve Stimulation to Suppress Atrial Fibrillation (TREAT-AF), a sham-controlled, double-blind, randomized clinical trial just completed in April 2020, found that intermittent LLTS lowers the AF burden in patients with paroxysmal AF compared to sham control stimulation (Stavrakis et al., 2020).
From these exciting experimental and clinical findings regarding neuromodulation as AF treatment, we learn at least two important lessons. First, to improve efficacy and safety, future antiarrhythmic drugs should have not only atrial selectivity but also dual adrenergic and cholinergic activities (like amiodarone) instead of only a single activity. Second, discrepancies in findings and inconsistencies in success surrounding the use of neuromodulation for AF control highlight the complexity of the dynamic influences of the cardiac autonomic nervous system on AF pathogenesis and the need for future research to guide appropriate identification of patients who would most benefit from neuromodulation for AF prevention or AF control based on individual variability in autonomic influences on existing cardiac structural and electrical remodeling.
Paradigm Shift: From Drug-Centricity to Patient-Centricity
In summary, as the progressive nature of AF atriomyopathy and the synergy of the two pathogenic remodeling processes in AF becomes clearer, identifying novel therapeutic targets and strategies will progress. We may also become better at considering individual variabilities in genes, environment, and lifestyle to bring precision medicine to AF therapeutics. The hope is to shift the overarching AF therapeutic paradigm from drug-centricity to patient-centricity.
Author Contributions
MG and AL performed literature search and assisted with manuscript preparation. TN conceived the review, performed literature search, and prepared the manuscript. All authors contributed to manuscript revision, approved the submitted version, and agreed to be accountable for all aspects of the work.
Funding
This work was supported by the National Institutes of Health R01 HL141452 to TN.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Clinical Trial Acronyms
AFFIRM, Atrial Fibrillation Follow-up Investigation of Rhythm Management; AnPAF, Efficacy and Safety of Antazoline in the Rapid Cardioversion of Paroxysmal Atrial Fibrillation; CABANA, Catheter Ablation vs. Antiarrhythmic Drug Therapy for AF; CAFÉ-II, Controlled Study of Rate vs. Rhythm Control in Patients with Chronic AF and Heart Failure; CANT, Cardioversion with Antazoline Mesylate; CAST, Cardiac Arrhythmia Suppression Trial; CASTLE-AF, Catheter Ablation vs. Standard Conventional Therapy in Patients with Left Ventricular Dysfunction and Atrial Fibrillation; COR-AT, Safety and Efficacy of Vanoxerine for Conversion of Atrial Fibrillation or Flutter to Normal Sinus Rhythm; DIAGRAF-IKUR, Double-blind, International study AssessinG efficacy of S 066913 in paRoxysmal Atrial Fibrillation-IKur inhibitor; DOCTOS, DOxapram Conversion TO Sinus rhythm study; EAST-AFNET 4, Early Treatment of Atrial Fibrillation for Stroke Prevention Trial; SAFIRE-D, Symptomatic AF Investigative Research on Dofetilide; TREAT-AF, Transcutaneous Electrical Vagus Nerve Stimulation to Suppress Atrial Fibrillation.
References
Abramochkin, D. V., Kuzmin, V. S., and Rosenshtraukh, L. V. (2015). Effects of new class III antiarrhythmic drug niferidil on electrical activity in murine ventricular myocardium and their ionic mechanisms. Naunyn-Schmiedeberg’s Arch. Pharmacol. 388, 1105–1112. doi:10.1007/s00210-015-1146-x
Abramochkin, D. V., Kuzmin, V. S., and Rosenshtraukh, L. V. (2017). A new class III antiarrhythmic drug niferidil prolongs action potentials in Guinea pig atrial myocardium via inhibition of rapid delayed rectifier. Cardiovasc. Drugs Ther. 31, 525–533. doi:10.1007/s10557-017-6762-x
Akel, T., and Lafferty, J. (2018). Efficacy and safety of intravenous vernakalant for the rapid conversion of recent-onset atrial fibrillation: a meta-analysis. Ann. Noninvasive Electrocardiol. 23, e12508. doi:10.1111/anec.12505
Akoum, N., and Marrouche, N. (2014). Assessment and impact of cardiac fibrosis on atrial fibrillation. Curr. Cardiol. Rep. 16, 518. doi:10.1007/s11886-014-0518-z
Alessi, R., Nusynowitz, M., Abildskov, J. A., and Moe, G. K. (1958). Nonuniform distribution of vagal effects on the atrial refractory period. Am. J. Physiol. Legacy Content 194, 406–410. doi:10.1152/ajplegacy.1958.194.2.406
Anderson, J. L., Gilbert, E. M., Alpert, B. L., Henthorn, R. W., Waldo, A. L., Bhandari, A. K., et al. (1989). Prevention of symptomatic recurrences of paroxysmal atrial fibrillation in patients initially tolerating antiarrhythmic therapy. A multicenter, double-blind, crossover study of flecainide and placebo with transtelephonic monitoring. Flecainide Supraventricular Tachycardia Study Group. Circulation 80, 1557–1570. doi:10.1161/01.cir.80.6.1557
Anderson, J. L., Platia, E. V., Hallstrom, A., Henthorn, R. W., Buckingham, T. A., Carlson, M. D., et al. (1994). Interaction of baseline characteristics with the hazard of encainide, flecainide, and moricizine therapy in patients with myocardial infarction. A possible explanation for increased mortality in the Cardiac Arrhythmia Suppression Trial (CAST). Circulation 90, 2843–2852. doi:10.1161/01.cir.90.6.2843
Antzelevitch, C., Burashnikov, A., Sicouri, S., and Belardinelli, L. (2011). Electrophysiologic basis for the antiarrhythmic actions of ranolazine. Heart Rhythm 8, 1281–1290. doi:10.1016/j.hrthm.2011.03.045
Askey, J. M. (1946). Quinidine in the treatment of auricular fibrillation in association with congestive failure. Ann. Intern. Med. 24, 371–384. doi:10.7326/0003-4819-24-3-371
Banerjee, A., Allan, V., Denaxas, S., Shah, A., Kotecha, D., Lambiase, P. D., et al. (2019). Subtypes of atrial fibrillation with concomitant valvular heart disease derived from electronic health records: phenotypes, population prevalence, trends and prognosis. Europace 21 (12), 1776–1784. doi:10.1093/europace/euz323
Beatch, G. N., Bhirangi, K., Juul-Moller, S., and Rustige, J. (2017). Efficacy and safety of vernakalant for cardioversion of recent-onset atrial fibrillation in the asia-pacific region. J. Cardiovasc. Pharmacol. 69, 86–92. doi:10.1097/fjc.0000000000000445
Beatch, G. N., and Mangal, B. (2016). Safety and efficacy of vernakalant for the conversion of atrial fibrillation to sinus rhythm; a phase 3b randomized controlled trial. BMC Cardiovasc. Disord. 16, 113. doi:10.1186/s12872-016-0289-0
Beaumont, E., Southerland, E. M., Hardwick, J. C., Wright, G. L., Ryan, S., Li, Y., et al. (2015). Vagus nerve stimulation mitigates intrinsic cardiac neuronal and adverse myocyte remodeling postmyocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 309, H1198–H1206. doi:10.1152/ajpheart.00393.2015
Bellandi, F., Simonetti, I., Leoncini, M., Frascarelli, F., Giovannini, T., Maioli, M., et al. (2001). Long-term efficacy and safety of propafenone and sotalol for the maintenance of sinus rhythm after conversion of recurrent symptomatic atrial fibrillation. Am. J. Cardiol. 88, 640–645. doi:10.1016/s0002-9149(01)01806-9
Benjamin, E. J., Chen, P.-S., Bild, D. E., Mascette, A. M., Albert, C. M., Alonso, A., et al. (2009). Prevention of atrial fibrillation: report from a national heart, lung, and blood institute workshop. Circulation 119, 606–618. doi:10.1161/circulationaha.108.825380
Benjamin, E. J., Levy, D., Vaziri, S. M., D’agostino, R. B., Belanger, A. J., and Wolf, P. A. (1994). Independent risk factors for atrial fibrillation in a population-based cohort. The Framingham Heart Study. JAMA 271, 840–844. doi:10.1001/jama.271.11.840
Biliczki, P., Boon, R. A., Girmatsion, Z., Bukowska, A., Ördög, B., Kaess, B. M., et al. (2019). Age-related regulation and region-specific distribution of ion channel subunits promoting atrial fibrillation in human left and right atria. Europace 21, 1261–1269. doi:10.1093/europace/euz135
Burashnikov, A., and Antzelevitch, C. (2009). Atrial-selective sodium channel block for the treatment of atrial fibrillation. Expet Opin. Emerg. Drugs 14, 233–249. doi:10.1517/14728210902997939
Burashnikov, A., Di Diego, J. M., Zygmunt, A. C., Belardinelli, L., and Antzelevitch, C. (2007). Atrium-selective sodium channel block as a strategy for suppression of atrial fibrillation: differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation 116, 1449–1457. doi:10.1161/circulationaha.107.704890
Burstein, B., and Nattel, S. (2008). Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J. Am. Coll. Cardiol. 51, 802–809. doi:10.1016/j.jacc.2007.09.064CrossRef Full Text
Caliskan, E., Sahin, A., Yilmaz, M., Seifert, B., Hinzpeter, R., Alkadhi, H., et al. (2018). Epicardial left atrial appendage AtriClip occlusion reduces the incidence of stroke in patients with atrial fibrillation undergoing cardiac surgery. Europace 20, e105-e114. doi:10.1093/europace/eux211
Calkins, H., Brugada, J., Packer, D. L., Cappato, R., Chen, S.-A., Crijns, H. J. G., et al. (2007). HRS/EHRA/ECAS expert consensus statement on catheter and surgical ablation of atrial fibrillation: recommendations for personnel, policy, procedures and follow-up. A report of the Heart Rhythm Society (HRS) Task Force on catheter and surgical ablation of atrial fibrillation. Heart Rhythm 4, 816–861. doi:10.1016/j.hrthm.2007.04.005
Calkins, H., Hindricks, G., Cappato, R., Kim, Y.-H., Saad, E. B., Aguinaga, L., et al. (2018). 2017 HRS/EHRA/ECAS/APHRS/SOLAECE expert consensus statement on catheter and surgical ablation of atrial fibrillation: executive summary. Europace 20, 157–208. doi:10.1093/europace/eux275
Camm, A. J., Capucci, A., Hohnloser, S. H., Torp-Pedersen, C., Van Gelder, I. C., Mangal, B., et al. (2011). A randomized active-controlled study comparing the efficacy and safety of vernakalant to amiodarone in recent-onset atrial fibrillation. J. Am. Coll. Cardiol. 57, 313–321. doi:10.1016/j.jacc.2010.07.046
Camm, A. J., Dorian, P., Hohnloser, S. H., Kowey, P. R., Tyl, B., Ni, Y., et al. (2019). A randomized, double-blind, placebo-controlled trial assessing the efficacy of S66913 in patients with paroxysmal atrial fibrillation. Eur. Heart J. Cardiovasc. Pharmacother. 5, 21–28. doi:10.1093/ehjcvp/pvy022CrossRef Full Text
Camm, A. J., Kirchhof, P., Camm, A. J., Kirchhof, P., Lip, G. Y., Schotten, U., et al. (2010). Guidelines for the management of atrial fibrillation: the task force for the management of atrial fibrillation of the European Society of Cardiology (ESC). Eur. Heart J. 31, 2369–2429. doi:10.1093/eurheartj/ehq278
Camm, A. J., Lip, G. Y., De Caterina, R., Savelieva, I., Atar, D., Hohnloser, S. H., et al. (2012). 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation--developed with the special contribution of the European Heart Rhythm Association. Europace 14, 1385–1413. doi:10.1093/europace/eus305
Chang, C.-M., Wu, T.-J., Zhou, S., Doshi, R. N., Lee, M.-H., Ohara, T., et al. (2001). Nerve sprouting and sympathetic hyperinnervation in a canine model of atrial fibrillation produced by prolonged right atrial pacing. Circulation 103, 22–25. doi:10.1161/01.cir.103.1.22
Chaugai, S., Meng, W. Y., and Ali Sepehry, A. (2016). Effects of RAAS blockers on atrial fibrillation prophylaxis. J. Cardiovasc. Pharmacol. Therapeut. 21, 388–404. doi:10.1177/1074248415619490
Chen, P.-S., Chen, L. S., Fishbein, M. C., Lin, S.-F., and Nattel, S. (2014). Role of the autonomic nervous system in atrial fibrillation. Circ. Res. 114, 1500–1515. doi:10.1161/circresaha.114.303772
Chen, P.-S., and Tan, A. Y. (2007). Autonomic nerve activity and atrial fibrillation. Heart Rhythm 4, S61–S64. doi:10.1016/j.hrthm.2006.12.006
Cheng, W. L., Kao, Y. H., Chao, T. F., Lin, Y. K., Chen, S. A., and Chen, Y. J. (2019). MicroRNA‐133 suppresses ZFHX3‐dependent atrial remodelling and arrhythmia. Acta Physiol. 227, e13322. doi:10.1111/apha.13322.
Colilla, S., Crow, A., Petkun, W., Singer, D. E., Simon, T., and Liu, X. (2013). Estimates of current and future incidence and prevalence of atrial fibrillation in the U.S. adult population. Am. J. Cardiol. 112, 1142–1147. doi:10.1016/j.amjcard.2013.05.063
Conde, D., Costabel, J. P., Aragon, M., Lambardi, F., Klein, A., Corrales Barbosa, A., et al. (2013a). Propafenone versus vernakalant for conversion of recent-onset atrial fibrillation. Cardiovasc Ther 31, 377–380. doi:10.1111/1755-5922.12036.
Conde, D., Costabel, J. P., Caro, M., Ferro, A., Lambardi, F., Corrales Barboza, A., et al. (2013b). Flecainide versus vernakalant for conversion of recent-onset atrial fibrillation. Int. J. Cardiol. 168, 2423–2425. doi:10.1016/j.ijcard.2013.02.006
Connolly, S. J., Camm, A. J., Halperin, J. L., Joyner, C., Alings, M., Amerena, J., et al. (2011). Dronedarone in high-risk permanent atrial fibrillation. N. Engl. J. Med. 365, 2268–2276. doi:10.1056/nejmoa1109867
Corley, S. D., Epstein, A. E., Dimarco, J. P., Domanski, M. J., Geller, N., Greene, H. L., et al. (2004). Relationships between sinus rhythm, treatment, and survival in the atrial fibrillation follow-up investigation of rhythm management (AFFIRM) study. Circulation 109, 1509–1513. doi:10.1161/01.CIR.0000121736.16643.11
Corradi, D., Callegari, S., Manotti, L., Ferrara, D., Goldoni, M., Alinovi, R., et al. (2014). Persistent lone atrial fibrillation: clinicopathologic study of 19 cases. Heart Rhythm 11, 1250–1258. doi:10.1016/j.hrthm.2014.02.008
Coumel, P. (1993). Cardiac arrhythmias and the autonomic nervous system. J. Cardiovasc. Electrophysiol. 4, 338–355. doi:10.1111/j.1540-8167.1993.tb01235.x
Dawson, K., Wakili, R., Ördög, B., Clauss, S., Chen, Y., Iwasaki, Y., et al. (2013). MicroRNA29: a mechanistic contributor and potential biomarker in atrial fibrillation. Circulation 127, 1466–1475. doi:10.1161/circulationaha.112.001207
De Vecchis, R., Ariano, C., Giasi, A., and Cioppa, C. (2018). Antiarrhythmic effects of ranolazine used both alone for prevention of atrial fibrillation and as an add-on to intravenous amiodarone for its pharmacological cardioversion: a meta-analysis. Minerva Cardioangiol. 66, 349–359. doi:10.23736/s0026-4725.17.04382-1
Di Biase, L., Burkhardt, J. D., Mohanty, P., Sanchez, J., Mohanty, S., Horton, R., et al. (2010). Left atrial appendage: an underrecognized trigger site of atrial fibrillation. Circulation 122, 109–118. doi:10.1161/circulationaha.109.928903
Dittrich, H. C., Feld, G. K., Bahnson, T. D., Camm, A. J., Golitsyn, S., Katz, A., et al. (2015). COR-ART: a multicenter, randomized, double-blind, placebo-controlled dose-ranging study to evaluate single oral doses of vanoxerine for conversion of recent-onset atrial fibrillation or flutter to normal sinus rhythm. Heart Rhythm 12, 1105–1112. doi:10.1016/j.hrthm.2015.02.014
Dobrev, D., Friedrich, A., Voigt, N., Jost, N., Wettwer, E., Christ, T., et al. (2005). The G protein-gated potassium current I(K,ACh) is constitutively active in patients with chronic atrial fibrillation. Circulation 112, 3697–3706. doi:10.1161/circulationaha.105.575332
Dobrev, D., Graf, E., Wettwer, E., Himmel, H. M., Hála, O., Doerfel, C., et al. (2001). Molecular basis of downregulation of G-protein-coupled inward rectifying K(+) current (I(K,ACh) in chronic human atrial fibrillation: decrease in GIRK4 mRNA correlates with reduced I(K,ACh) and muscarinic receptor-mediated shortening of action potentials. Circulation 104, 2551–2557. doi:10.1161/hc4601.099466
Echt, D. S., Liebson, P. R., Mitchell, L. B., Peters, R. W., Obias-Manno, D., Barker, A. H., et al. (1991). Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N. Engl. J. Med. 324, 781–788. doi:10.1056/nejm199103213241201
Eldstrom, J., Wang, Z., Xu, H., Pourrier, M., Ezrin, A., Gibson, K., et al. (2007). The molecular basis of high-affinity binding of the antiarrhythmic compound vernakalant (RSD1235) to Kv1.5 channels. Mol. Pharmacol. 72, 1522–1534. doi:10.1124/mol.107.039388
Ellenbogen, K. A. (2007). Pacing therapy for prevention of atrial fibrillation. Heart Rhythm 4, S84–S87. doi:10.1016/j.hrthm.2006.12.005
Ellis, C. R., Aznaurov, S. G., Patel, N. J., Williams, J. R., Sandler, K. L., Hoff, S. J., et al. (2017). Angiographic efficacy of the atriclip left atrial appendage exclusion device placed by minimally invasive thoracoscopic approach. JACC Clinical Electrophysiol. 3, 1356–1365. doi:10.1016/j.jacep.2017.03.008
Elvan, A., Pride, H. P., Eble, J. N., and Zipes, D. P. (1995). Radiofrequency catheter ablation of the atria reduces inducibility and duration of atrial fibrillation in dogs. Circulation 91, 2235–2244. doi:10.1161/01.cir.91.8.2235
Enyedi, P., and Czirják, G. (2010). Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol. Rev. 90, 559–605. doi:10.1152/physrev.00029.2009
Fedida, D., Orth, P. M. R., Chen, J. Y. C., Lin, S., Plouvier, B., Jung, G., et al. (2005). The mechanism of atrial antiarrhythmic action of RSD1235. J. Cardiovasc. Electrophysiol. 16, 1227–1238. doi:10.1111/j.1540-8167.2005.50028.x
Fedida, D., Orth, P. M. R., Hesketh, J. C., and Ezrin, A. M. (2006). The role of late INa and antiarrhythmic drugs in EAD formation and termination in Purkinje fibers. J. Cardiovasc. Electrophysiol. 17 (Suppl. 1), S71-s78. doi:10.1111/j.1540-8167.2006.00386.x
Fetsch, T., Bauer, P., Engberding, R., Koch, H. P., Lukl, J., Meinertz, T., et al. (2004). Prevention of atrial fibrillation after cardioversion: results of the PAFAC trial. Eur. Heart J. 25, 1385–1394. doi:10.1016/j.ehj.2004.04.015
Ford, J., Milnes, J., El Haou, S., Wettwer, E., Loose, S., Matschke, K., et al. (2016). The positive frequency-dependent electrophysiological effects of the IKur inhibitor XEN-D0103 are desirable for the treatment of atrial fibrillation. Heart Rhythm 13, 555–564. doi:10.1016/j.hrthm.2015.10.003
Freemantle, N., Lafuente-Lafuente, C., Mitchell, S., Eckert, L., and Reynolds, M. (2011). Mixed treatment comparison of dronedarone, amiodarone, sotalol, flecainide, and propafenone, for the management of atrial fibrillation. Europace 13, 329–345. doi:10.1093/europace/euq450
Fromm, C., Suau, S. J., Cohen, V., Likourezos, A., Jellinek-Cohen, S., Rose, J., et al. (2015). Diltiazem vs. Metoprolol in the management of atrial fibrillation or flutter with rapid ventricular rate in the emergency department. J. Emerg. Med. 49, 175–182. doi:10.1016/j.jemermed.2015.01.014
Frommeyer, G., Sterneberg, M., Dechering, D. G., Kaese, S., Bogeholz, N., Pott, C., et al. (2017). Effective suppression of atrial fibrillation by the antihistaminic agent antazoline: first experimental insights into a novel antiarrhythmic agent. Cardiovasc. Ther. 35 (2), e12244. doi:10.1111/1755-5922.12244
Fuster, V., Rydén, L. E., Asinger, R. W., Cannom, D. S., Crijns, H. J., Frye, R. L., et al. (2001). ACC/AHA/ESC guidelines for the management of patients with atrial fibrillation: executive summary. A report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the European Society of Cardiology committee for practice guidelines and policy conferences (committee to develop guidelines for the management of patients with atrial fibrillation). J. Am. Coll. Cardiol. 38, 1231–1265. doi:10.1016/s0735-1097(01)01587-x
Fuster, V., Rydén, L. E., Cannom, D. S., Crijns, H. J., Curtis, A. B., Ellenbogen, K. A., et al. (2006). ACC/AHA/ESC 2006 guidelines for the management of patients with atrial fibrillation--executive summary: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the European Society of Cardiology committee for practice guidelines (writing committee to revise the 2001 guidelines for the management of patients with atrial fibrillation). J. Am. Coll. Cardiol. 48, 854–906. doi:10.1016/j.jacc.2006.07.009
Fuster, V., Rydén, L. E., Cannom, D. S., Crijns, H. J., Curtis, A. B., Ellenbogen, K. A., et al. (2011). 2011 ACCF/AHA/HRS focused updates incorporated into the ACC/AHA/ESC 2006 guidelines for the management of patients with atrial fibrillation: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 123, e269-367. doi:10.1161/cir.0b013e318214876d
Geesbreght, J., and Randall, W. (1971). Area localization of shifting cardiac pacemakers during sympathetic stimulation. American J. Physiol. Legacy Content 220, 1522–1527. doi:10.1152/ajplegacy.1971.220.5.1522
Gehring, D. A., and Kehler, J. G. (1970). Conversion of atrial fibrillation with antazoline hydrochloride (Arithmin). Angiology 21, 11–17. doi:10.1177/000331977002100104
Geller, J., Reek, S., Timmermans, C., Kayser, T., Tse, H. F., Wolpert, C., et al. (2003). Treatment of atrial fibrillation with an implantable atrial defibrillator - long term results. Eur. Heart J. 24, 2083–2089. doi:10.1016/j.ehj.2003.09.033
Goldstein, S. A. N., Bockenhauer, D., O’kelly, I., and Zilberberg, N. (2001). Potassium leak channels and the KCNK family of two-P-domain subunits. Nat. Rev. Neurosci. 2, 175–184. doi:10.1038/35058574
Goodman, D. J., Rossen, R. M., Cannom, D. S., Rider, A. K., and Harrison, D. C. (1975). Effect of digoxin on atioventricular conduction. Studies in patients with and without cardiac autonomic innervation. Circulation 51, 251–256. doi:10.1161/01.cir.51.2.251
Greiser, M., Lederer, W. J., and Schotten, U. (2011). Alterations of atrial Ca2+ handling as cause and consequence of atrial fibrillation. Cardiovasc. Res. 89, 722–733. doi:10.1093/cvr/cvq389
Haïssaguerre, M., Jaïs, P., Shah, D. C., Takahashi, A., Hocini, M., Quiniou, G., et al. (1998). Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N. Engl. J. Med. 339, 659–666. doi:10.1056/nejm199809033391003
Hale, S. L., Leeka, J. A., and Kloner, R. A. (2006). Improved left ventricular function and reduced necrosis after myocardial ischemia/reperfusion in rabbits treated with ranolazine, an inhibitor of the late sodium channel. J. Pharmacol. Exp. Therapeut. 318, 418–423. doi:10.1124/jpet.106.103242
Hale, S. L., Shryock, J. C., Belardinelli, L., Sweeney, M., and Kloner, R. A. (2008). Late sodium current inhibition as a new cardioprotective approach. J. Mol. Cell. Cardiol. 44, 954–967. doi:10.1016/j.yjmcc.2008.03.019
Härtel, G., Louhija, A., and Konttinen, A. (1974). Disopyramide in the prevention of recurrence of atrial fibrillation after electroconversion. Clin. Pharmacol. Ther. 15, 551–555. doi:10.1002/cpt1974156551
Hashimoto, K., Chiba, S., Tanaka, S., Hirata, M., and Suzuki, Y. (1968). Adrenergic mechanism participating in induction of atrial fibrillation by ACh. American J. Physiol. Legacy Content 215, 1183–1191. doi:10.1152/ajplegacy.1968.215.5.1183
Heijman, J., Ghezelbash, S., and Dobrev, D. (2017). Investigational antiarrhythmic agents: promising drugs in early clinical development. Expet Opin. Invest. Drugs 26, 897–907. doi:10.1080/13543784.2017.1353601
Heijman, J., Kirchner, D., Kunze, F., Chrétien, E. M., Michel-Reher, M. B., Voigt, N., et al. (2018). Muscarinic type-1 receptors contribute to I K,ACh in human atrial cardiomyocytes and are upregulated in patients with chronic atrial fibrillation. Int. J. Cardiol. 255, 61–68. doi:10.1016/j.ijcard.2017.12.050
Hindricks, G., Potpara, T., Dagres, N., Arbelo, E., Bax, J. J., Blomström-Lundqvist, C., et al. (2020). 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association of Cardio-Thoracic Surgery (EACTS). Eur. Heart J. [Epub ahead of print]. doi:10.1093/eurheartj/ehaa612CrossRef Full Text
Hocini, M., Shah, A. J., Nault, I., Sanders, P., Wright, M., Narayan, S. M., et al. (2011). Localized reentry within the left atrial appendage: arrhythmogenic role in patients undergoing ablation of persistent atrial fibrillation. Heart Rhythm 8, 1853–1861. doi:10.1016/j.hrthm.2011.07.013
Hohnloser, S. H., Crijns, H. J. G. M., Van Eickels, M., Gaudin, C., Page, R. L., Torp-Pedersen, C., et al. (2009). Effect of dronedarone on cardiovascular events in atrial fibrillation. N. Engl. J. Med. 360, 668–678. doi:10.1056/nejmoa0803778
Hu, W.-S., and Lin, C.-L. (2019). Risk of atrial fibrillation in patients with congenital heart disease: results of a propensity score-matched, nationwide cohort study. J. Atheroscler. Thromb. 26, 670–677. doi:10.5551/jat.48835
Ito, K., Date, T., Ikegami, M., Hongo, K., Fujisaki, M., Katoh, D., et al. (2013). An immunohistochemical analysis of tissue thrombin expression in the human atria. PLoS One 8, e65817. doi:10.1371/journal.pone.0065817
Jahangir, A., Lee, V., Friedman, P. A., Trusty, J. M., Hodge, D. O., Kopecky, S. L., et al. (2007). Long-term progression and outcomes with aging in patients with lone atrial fibrillation: a 30-year follow-up study. Circulation 115, 3050–3056. doi:10.1161/circulationaha.106.644484
January, C. T., Wann, L. S., Alpert, J. S., Calkins, H., Cigarroa, J. E., Cleveland, J. C., et al. (2014). 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the Heart Rhythm Society. J. Am. Coll. Cardiol. 64, e1–e76. doi:10.1016/j.jacc.2014.03.022CrossRef Full Text
January, C. T., Wann, L. S., Calkins, H., Chen, L. Y., Cigarroa, J. E., Cleveland, J. C., et al. (2019). 2019 AHA/ACC/HRS focused update of the 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines and the Heart Rhythm Society. J. Am. Coll. Cardiol. 74, 104–132. doi:10.1016/j.jacc.2019.01.011
Jayachandran, J. V., Sih, H. J., Winkle, W., Zipes, D. P., Hutchins, G. D., and Olgin, J. E. (2000). Atrial fibrillation produced by prolonged rapid atrial pacing is associated with heterogeneous changes in atrial sympathetic innervation. Circulation 101, 1185–1191. doi:10.1161/01.cir.101.10.1185
Katritsis, D. G., Giazitzoglou, E., Zografos, T., Pokushalov, E., Po, S. S., and Camm, A. J. (2011). Rapid pulmonary vein isolation combined with autonomic ganglia modification: a randomized study. Heart Rhythm 8, 672–678. doi:10.1016/j.hrthm.2010.12.047
Khan, I. A., Mehta, N. J., and Gowda, R. M. (2003). Amiodarone for pharmacological cardioversion of recent-onset atrial fibrillation. Int. J. Cardiol. 89, 239–248. doi:10.1016/s0167-5273(02)00477-1
Kim, M. H., Johnston, S. S., Chu, B.-C., Dalal, M. R., and Schulman, K. L. (2011). Estimation of total incremental health care costs in patients with atrial fibrillation in the United States. Circ. Cardiovasc. Qual. Outcomes 4, 313–320. doi:10.1161/circoutcomes.110.958165
Kim, Y. G., Choi, J. I., Boo, K. Y., Kim, D. Y., Oh, S. K., Park, H. S., et al. (2019). Clinical and echocardiographic risk factors predict late recurrence after radiofrequency catheter ablation of atrial fibrillation. Sci. Rep. 9, 6890. doi:10.1038/s41598-019-43283-7
Kim, Y. G., Shim, J., Oh, S. K., Park, H. S., Lee, K. N., Hwang, S. H., et al. (2018). Different responses of left atrium and left atrial appendage to radiofrequency catheter ablation of atrial fibrillation: a follow up MRI study. Sci. Rep. 8, 7871. doi:10.1038/s41598-018-26212-y
Kirchhof, P., Benussi, S., Kotecha, D., Ahlsson, A., Atar, D., Casadei, B., et al. (2016). 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Kardiologia Polska 18, 1609–1678. doi:10.1093/europace/euw295
Kirchhof, P., Camm, A. J., Goette, A., Brandes, A., Eckardt, L., Elvan, A., et al. (2020). Early rhythm-control therapy in patients with atrial fibrillation. N. Engl. J. Med. 383 (14), 1305–1316. doi:10.1056/NEJMoa2019422
Koskinas, K. C., Fragakis, N., Katritsis, D., Skeberis, V., and Vassilikos, V. (2014). Ranolazine enhances the efficacy of amiodarone for conversion of recent-onset atrial fibrillation. Europace 16, 973–979. doi:10.1093/europace/eut407
Kowey, P. R., Dorian, P., Mitchell, L. B., Pratt, C. M., Roy, D., Schwartz, P. J., et al. (2009). Vernakalant hydrochloride for the rapid conversion of atrial fibrillation after cardiac surgery: a randomized, double-blind, placebo-controlled trial. Circ. Arrhythm. Electrophysiol. 2, 652–659. doi:10.1161/circep.109.870204CrossRef Full Text
Kuck, K.-H., Brugada, J., Fürnkranz, A., Metzner, A., Ouyang, F., Chun, K. R. J., et al. (2016). Cryoballoon or radiofrequency ablation for paroxysmal atrial fibrillation. N. Engl. J. Med. 374, 2235–2245. doi:10.1056/nejmoa1602014
Kumar, K., Nearing, B. D., Carvas, M., Nascimento, B. C. G., Acar, M., Belardinelli, L., et al. (2009). Ranolazine exerts potent effects on atrial electrical properties and abbreviates atrial fibrillation duration in the intact porcine heart. J. Cardiovasc. Electrophysiol. 20, 796–802. doi:10.1111/j.1540-8167.2009.01437.x
Kusunose, K., Zhang, Y., Mazgalev, T. N., Van Wagoner, D. R., Thomas, J. D., and Popović, Z. B. (2014). Impact of vagal nerve stimulation on left atrial structure and function in a canine high-rate pacing model. Circ. Heart Fail. 7, 320–326. doi:10.1161/circheartfailure.113.000937
Lacerda, A. E., Kuryshev, Y. A., Yan, G.-X., Waldo, A. L., and Brown, A. M. (2010). Vanoxerine: cellular mechanism of a new antiarrhythmic. J. Cardiovasc. Electrophysiol. 21, 301–310. doi:10.1111/j.1540-8167.2009.01623.x
Lafuente-Lafuente, C., Valembois, L., Bergmann, J. F., and Belmin, J. (2015). Antiarrhythmics for maintaining sinus rhythm after cardioversion of atrial fibrillation. Cochrane Database Syst. Rev., CD005049. doi:10.1002/14651858.CD005049.pub4
Le Heuzey, J.-Y., De Ferrari, G. M., Radzik, D., Santini, M., Zhu, J., and Davy, J.-M. (2010). A short-term, randomized, double-blind, parallel-group study to evaluate the efficacy and safety of dronedarone versus amiodarone in patients with persistent atrial fibrillation: the DIONYSOS study. J. Cardiovasc. Electrophysiol. 21, 597–605. doi:10.1111/j.1540-8167.2010.01764.x
Lemola, K., Chartier, D., Yeh, Y.-H., Dubuc, M., Cartier, R., Armour, A., et al. (2008). Pulmonary vein region ablation in experimental vagal atrial fibrillation: role of pulmonary veins versus autonomic ganglia. Circulation 117, 470–477. doi:10.1161/circulationaha.107.737023
Levitt, B., Cagin, N. A., Kleid, J., Somberg, J., and Gillis, R. (1976a). Role of the nervous system in the genesis of cardiac rhythm disorders. Am. J. Cardiol. 37, 1111–1113. doi:10.1016/0002-9149(76)90435-5
Levitt, B., Cagin, N. A., Somberg, J. C., and Kleid, J. J. (1976b). Neural basis for the genesis and control of digitalis arrhythmias. Cardiology 61, 50–60. doi:10.1159/000169747
Li, D., Fareh, S., Leung, T. K., and Nattel, S. (1999). Promotion of atrial fibrillation by heart failure in dogs: atrial remodeling of a different sort. Circulation 100, 87–95. doi:10.1161/01.cir.100.1.87
Limberg, S. H., Netter, M. F., Rolfes, C., Rinné, S., Schlichthörl, G., Zuzarte, M., et al. (2011). TASK-1 channels may modulate action potential duration of human atrial cardiomyocytes. Cell. Physiol. Biochem. 28, 613–624. doi:10.1159/000335757
Lindsay, B. D. (2011). Vernakalant: additional evidence for safety and efficacy for new onset atrial fibrillation. J. Am. Coll. Cardiol. 57, 322–323. doi:10.1016/j.jacc.2010.08.633
Ling, T.-Y., Wang, X.-L., Chai, Q., Lau, T.-W., Koestler, C. M., Park, S. J., et al. (2013). Regulation of the SK3 channel by microRNA-499--potential role in atrial fibrillation. Heart Rhythm 10, 1001–1009. doi:10.1016/j.hrthm.2013.03.005
Liu, L., and Nattel, S. (1997). Differing sympathetic and vagal effects on atrial fibrillation in dogs: role of refractoriness heterogeneity. Am. J. Physiol. Heart Circ. Physiol. 273, H805–H816. doi:10.1152/ajpheart.1997.273.2.h805
Liu, T., Xiong, F., Qi, X. Y., Xiao, J., Villeneuve, L., Abu-Taha, I., et al. (2020). Altered calcium handling produces reentry-promoting action potential alternans in atrial fibrillation-remodeled hearts. JCI Insight 5, e133754. doi:10.1172/jci.insight.133754CrossRef Full Text
Lu, Y., Zhang, Y., Wang, N., Pan, Z., Gao, X., Zhang, F., et al. (2010). MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation 122, 2378–2387. doi:10.1161/circulationaha.110.958967
Lu, Z., Scherlag, B. J., Lin, J., Niu, G., Fung, K.-M., Zhao, L., et al. (2008). Atrial fibrillation begets atrial fibrillation: autonomic mechanism for atrial electrical remodeling induced by short-term rapid atrial pacing. Circ. Arrhythm. Electrophysiol. 1, 184–192. doi:10.1161/circep.108.784272
Lujan, H. L., Janbaih, H., and Dicarlo, S. E. (2014). Structural remodeling of the heart and its premotor cardioinhibitory vagal neurons following T(5) spinal cord transection. J. Appl. Physiol. 116, 1148–1155. doi:10.1152/japplphysiol.01285.2013
Luo, X., Pan, Z., Shan, H., Xiao, J., Sun, X., Wang, N., et al. (2013). MicroRNA-26 governs profibrillatory inward-rectifier potassium current changes in atrial fibrillation. J. Clin. Invest. 123, 1939–1951. doi:10.1172/jci62185
Luo, X., Yang, B., and Nattel, S. (2015). MicroRNAs and atrial fibrillation: mechanisms and translational potential. Nat. Rev. Cardiol. 12, 80–90. doi:10.1038/nrcardio.2014.178
Maciag, A., Farkowski, M. M., Chwyczko, T., Beckowski, M., Syska, P., Kowalik, I., et al. (2017). Efficacy and safety of antazoline in the rapid cardioversion of paroxysmal atrial fibrillation (the AnPAF Study). Europace 19, 1637–1642. doi:10.1093/europace/euw384CrossRef Full Text
Marrouche, N. F., Brachmann, J., Andresen, D., Siebels, J., Boersma, L., Jordaens, L., et al. (2018). Catheter ablation for atrial fibrillation with heart failure. N. Engl. J. Med. 378, 417–427. doi:10.1056/nejmoa1707855
Marrouche, N. F., Wilber, D., Hindricks, G., Jais, P., Akoum, N., Marchlinski, F., et al. (2014). Association of atrial tissue fibrosis identified by delayed enhancement MRI and atrial fibrillation catheter ablation: the DECAAF study. JAMA 311, 498–506. doi:10.1001/jama.2014.3
Matsumoto, N., Khrestian, C. M., Ryu, K., Lacerda, A. E., Brown, A. M., and Waldo, A. L. (2010). Vanoxerine, a new drug for terminating atrial fibrillation and flutter. J. Cardiovasc. Electrophysiol. 21, 311–319. doi:10.1111/j.1540-8167.2009.01622.x
Maykov, E. B., Yuricheva, Y. A., Mironov, N. Y., Sokolov, S. F., Golitsyn, S. P., Rosenshtraukh, L. V., et al. (2014). Efficacy of a new class III drug niferidil in cardioversion of persistent atrial fibrillation and flutter. J. Cardiovasc. Pharmacol. 64, 247–255. doi:10.1097/fjc.0000000000000112
Mccord, M. C., and Taguchi, J. T. (1951). A study of the effect of procaine amide hydrochloride in supraventricular arrhythmias. Circulation 4, 387–393. doi:10.1161/01.cir.4.3.387
Mcgann, C., Akoum, N., Patel, A., Kholmovski, E., Revelo, P., Damal, K., et al. (2014). Atrial fibrillation ablation outcome is predicted by left atrial remodeling on MRI. Circ. Arrhythm. Electrophysiol. 7, 23–30. doi:10.1161/circep.113.000689
Mcmichael, J. (1982). History of atrial fibrillation 1628-1819 Harvey - de Senac - laennec. Heart 48, 193–197. doi:10.1136/hrt.48.3.193
Mcmillan, R. L., and Welfare, C. R. (1947). Chronic auricular fibrillation; its treatment with quinidine sulfate. J. Am. Med. Assoc. 135, 1132–1136. doi:10.1001/jama.1947.02890170010003
Meinertz, T., Lip, G. Y. H., Lombardi, F., Sadowski, Z. P., Kalsch, B., Camez, A., et al. (2002). Efficacy and safety of propafenone sustained release in the prophylaxis of symptomatic paroxysmal atrial fibrillation (The European Rythmol/Rytmonorm Atrial Fibrillation Trial [ERAFT] Study). Am. J. Cardiol. 90, 1300–1306. doi:10.1016/s0002-9149(02)02867-9
Miller, G., Weinberg, S. L., and Pick, A. (1952). The effect of procaine amide (pronestyl) in clinical auricular fibrillation and flutter. Circulation 6, 41–50. doi:10.1161/01.cir.6.1.41
Miyasaka, Y., Barnes, M. E., Gersh, B. J., Cha, S. S., Bailey, K. R., Abhayaratna, W. P., et al. (2006). Secular trends in incidence of atrial fibrillation in Olmsted County, Minnesota, 1980 to 2000, and implications on the projections for future prevalence. Circulation 114, 119–125. doi:10.1161/circulationaha.105.595140
Moreno, J., and Zamorano, J. L. (2014). The CABANA trial. Eur. Heart J. 35, 1908–1909Pubmed
Nakajima, T., Kurachi, Y., Ito, H., Takikawa, R., and Sugimoto, T. (1989). Anti-cholinergic effects of quinidine, disopyramide, and procainamide in isolated atrial myocytes: mediation by different molecular mechanisms. Circ. Res. 64, 297–303. doi:10.1161/01.res.64.2.297
Nattel, S. (2016). How does fibrosis promote atrial fibrillation persistence: in silico findings, clinical observations, and experimental data. Cardiovasc. Res. 110, 295–297. doi:10.1093/cvr/cvw092
Nattel, S., Burstein, B., and Dobrev, D. (2008). Atrial remodeling and atrial fibrillation: mechanisms and implications. Circ. Arrhythm. Electrophysiol. 1, 62–73. doi:10.1161/circep.107.754564
Nattel, S., and Dobrev, D. (2012). The multidimensional role of calcium in atrial fibrillation pathophysiology: mechanistic insights and therapeutic opportunities. Eur. Heart J. 33, 1870–1877. doi:10.1093/eurheartj/ehs079
Nerheim, P., Birger-Botkin, S., Piracha, L., and Olshansky, B. (2004). Heart failure and sudden death in patients with tachycardia-induced cardiomyopathy and recurrent tachycardia. Circulation 110, 247–252. doi:10.1161/01.cir.0000135472.28234.cc
Ninomiya, I. (1966). Direct evidence of nonuniform distribution of vagal effects on dog atria. Circ. Res. 19, 576–583. doi:10.1161/01.res.19.3.576CrossRef Full Text
Nisar, M. U., Sharbaugh, M. S., Thoma, F. W., Munir, M. B., Kamran, A. S., Althouse, A. D., et al. (2016). Trends in atrial fibrillation hospitalizations from 1996 to 2010 using data from national hospital discharge survey. Circulation 134
Ogawa, H., An, Y., Ikeda, S., Aono, Y., Doi, K., Ishii, M., et al. (2018). Progression from paroxysmal to sustained atrial fibrillation is associated with increased adverse events. Stroke 49, 2301–2308. doi:10.1161/strokeaha.118.021396
Okumura, Y., Watanabe, I., Nakai, T., Ohkubo, K., Kofune, T., Kofune, M., et al. (2011). Impact of biomarkers of inflammation and extracellular matrix turnover on the outcome of atrial fibrillation ablation: importance of matrix metalloproteinase-2 as a predictor of atrial fibrillation recurrence. J. Cardiovasc. Electrophysiol. 22, 987–993. doi:10.1111/j.1540-8167.2011.02059.x
Olivotto, I., Cecchi, F., Casey, S. A., Dolara, A., Traverse, J. H., and Maron, B. J. (2001). Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 104, 2517–2524. doi:10.1161/hc4601.097997
Orth, P., Hesketh, J., Mak, C., Yang, Y., Lin, S., Beatch, G., et al. (2006). RSD1235 blocks late INa and suppresses early afterdepolarizations and torsades de pointes induced by class III agents. Cardiovasc. Res. 70, 486–496. doi:10.1016/j.cardiores.2006.01.026
Ott, A., Breteler, M. M. B., De Bruyne, M. C., Van Harskamp, F., Grobbee, D. E., and Hofman, A. (1997). Atrial fibrillation and dementia in a population-based study. The Rotterdam Study. Stroke 28, 316–321. doi:10.1161/01.str.28.2.316
Packer, D. L., Mark, D. B., Robb, R. A., Monahan, K. H., Bahnson, T. D., Moretz, K., et al. (2018). Catheter ablation versus antiarrhythmic drug therapy for atrial fibrillation (CABANA) trial: study rationale and design. Am. Heart J. 199, 192–199. doi:10.1016/j.ahj.2018.02.015
Patel, N. J., Deshmukh, A., Pant, S., Singh, V., Patel, N., Arora, S., et al. (2014). Contemporary trends of hospitalization for atrial fibrillation in the United States, 2000 through 2010: implications for healthcare planning. Circulation 129, 2371–2379. doi:10.1161/circulationaha.114.008201
Patten, M., Maas, R., Bauer, P., Luderitz, B., Sonntag, F., Dluzniewski, M., et al. (2004). Suppression of paroxysmal atrial tachyarrhythmias--results of the SOPAT trial. Eur. Heart J. 25, 1395–1404. doi:10.1016/j.ehj.2004.06.014
Peigh, G., Kaplan, R. M., Bavishi, A., Diaz, C. L., Baman, J. R., Matiasz, R., et al. (2020). A novel risk model for very late return of atrial fibrillation beyond 1 year after cryoballoon ablation: the SCALE-CryoAF score. J. Intervent. Card. Electrophysiol. 58 (2), 209–217. doi:10.1007/s10840-019-00588-x
Piccini, J. P., Pritchett, E. L. C., Davison, B. A., Cotter, G., Wiener, L. E., Koch, G., et al. (2016). Randomized, double-blind, placebo-controlled study to evaluate the safety and efficacy of a single oral dose of vanoxerine for the conversion of subjects with recent onset atrial fibrillation or flutter to normal sinus rhythm: restore SR. Heart Rhythm 13, 1777–1783. doi:10.1016/j.hrthm.2016.04.012
Pokushalov, E., Kozlov, B., Romanov, A., Strelnikov, A., Bayramova, S., Sergeevichev, D., et al. (2015). Long-term suppression of atrial fibrillation by botulinum toxin injection into epicardial fat pads in patients undergoing cardiac surgery: one-year follow-up of a randomized pilot study. Circ. Arrhythm. Electrophysiol. 8, 1334–1341. doi:10.1161/circep.115.003199
Pratt, C. M., Roy, D., Torp-Pedersen, C., Wyse, D. G., Toft, E., Juul-Moller, S., et al. (2010). Usefulness of vernakalant hydrochloride injection for rapid conversion of atrial fibrillation. Am. J. Cardiol. 106, 1277–1283. doi:10.1016/j.amjcard.2010.06.054CrossRef Full Text
Qi, X.-Y., Diness, J. G., Brundel, B. J. J. M., Zhou, X.-B., Naud, P., Wu, C.-T., et al. (2014). Role of small-conductance calcium-activated potassium channels in atrial electrophysiology and fibrillation in the dog. Circulation 129, 430–440. doi:10.1161/circulationaha.113.003019
Qu, Y.-C., Du, Y.-M., Wu, S.-L., Chen, Q.-X., Wu, H.-L., and Zhou, S.-F. (2009). Activated nuclear factor-kappaB and increased tumor necrosis factor-alpha in atrial tissue of atrial fibrillation. Scand. Cardiovasc. J. 43, 292–297. doi:10.1080/14017430802651803
Reiffel, J. A., Camm, A. J., Belardinelli, L., Zeng, D., Karwatowska-Prokopczuk, E., Olmsted, A., et al. (2015). The HARMONY trial: combined ranolazine and dronedarone in the management of paroxysmal atrial fibrillation: mechanistic and therapeutic synergism. Circ. Arrhythm. Electrophysiol. 8, 1048–1056. doi:10.1161/circep.115.002856
Rinné, S., Kiper, A. K., Schlichthörl, G., Dittmann, S., Netter, M. F., Limberg, S. H., et al. (2015). TASK-1 and TASK-3 may form heterodimers in human atrial cardiomyocytes. J. Mol. Cell. Cardiol. 81, 71–80. doi:10.1016/j.yjmcc.2015.01.017CrossRef Full Text
Romanov, A., Pokushalov, E., Ponomarev, D., Bayramova, S., Shabanov, V., Losik, D., et al. (2019). Long-term suppression of atrial fibrillation by botulinum toxin injection into epicardial fat pads in patients undergoing cardiac surgery: three-year follow-up of a randomized study. Heart Rhythm 16, 172–177. doi:10.1016/j.hrthm.2018.08.019
Rossi, P., Ricci, A., De Paulis, R., Papi, E., Pavaci, H., Porcelli, D., et al. (2012). Epicardial ganglionated plexus stimulation decreases postoperative inflammatory response in humans. Heart Rhythm 9, 943–950. doi:10.1016/j.hrthm.2012.01.025
Roy, D., Pratt, C. M., Torp-Pedersen, C., Wyse, D. G., Toft, E., Juul-Moller, S., et al. (2008). Vernakalant hydrochloride for rapid conversion of atrial fibrillation: a phase 3, randomized, placebo-controlled trial. Circulation 117, 1518–1525. doi:10.1161/circulationaha.107.723866
Roy, D., Talajic, M., Dorian, P., Connolly, S., Eisenberg, M. J., Green, M., et al. (2000). Amiodarone to prevent recurrence of atrial fibrillation. Canadian trial of atrial fibrillation investigators. N. Engl. J. Med. 342, 913–920. doi:10.1056/nejm200003303421302
Rozen, G., Hosseini, S. M., Kaadan, M. I., Biton, Y., Heist, E. K., Vangel, M., et al. (2018). Emergency department visits for atrial fibrillation in the United States: trends in admission rates and economic burden from 2007 to 2014. J. Am. Heart Assoc. 7, e009024. doi:10.1161/jaha.118.009024
Savelieva, I., Kakouros, N., Kourliouros, A., and Camm, A. J. (2011a). Upstream therapies for management of atrial fibrillation: review of clinical evidence and implications for European Society of Cardiology guidelines. Part I: primary prevention. Europace 13, 308–328. doi:10.1093/europace/eur002
Savelieva, I., Kakouros, N., Kourliouros, A., and Camm, A. J. (2011b). Upstream therapies for management of atrial fibrillation: review of clinical evidence and implications for European Society of Cardiology guidelines. Part II: secondary prevention. Europace 13, 610–625. doi:10.1093/europace/eur023
Schmidt, C., Wiedmann, F., Beyersdorf, C., Zhao, Z., El-Battrawy, I., Lan, H., et al. (2019). Genetic ablation of TASK-1 (tandem of P domains in a weak inward rectifying K(+) channel-related acid-sensitive K(+) channel-1) (K(2P)3.1) K(+) channels suppresses atrial fibrillation and prevents electrical remodeling. Circ. Arrhythmia Electrophysiol. 12, e007465. doi:10.1161/circep.119.007465CrossRef Full Text
Schmidt, C., Wiedmann, F., Voigt, N., Zhou, X.-B., Heijman, J., Lang, S., et al. (2015). Upregulation of K(2P)3.1 K+ current causes action potential shortening in patients with chronic atrial fibrillation. Circulation 132, 82–92. doi:10.1161/circulationaha.114.012657
Schmidt, C., Wiedmann, F., Zhou, X. B., Heijman, J., Voigt, N., Ratte, A., et al. (2017). Inverse remodelling of K2P3.1 K+ channel expression and action potential duration in left ventricular dysfunction and atrial fibrillation: implications for patient-specific antiarrhythmic drug therapy. Eur. Heart J. 38, 1764–1774. doi:10.1093/eurheartj/ehx493.p5835
Schnabel, R. B., Sullivan, L. M., Levy, D., Pencina, M. J., Massaro, J. M., D’agostino, R. B., et al. (2009). Development of a risk score for atrial fibrillation (Framingham Heart Study): a community-based cohort study. Lancet 373, 739–745. doi:10.1016/s0140-6736(09)60443-8
Schnabel, R. B., Yin, X., Gona, P., Larson, M. G., Beiser, A. S., Mcmanus, D. D., et al. (2015). 50 year trends in atrial fibrillation prevalence, incidence, risk factors, and mortality in the Framingham Heart Study: a cohort study. Lancet 386, 154–162. doi:10.1016/s0140-6736(14)61774-8
Sharifov, O. F., Fedorov, V. V., Beloshapko, G. G., Glukhov, A. V., Yushmanova, A. V., and Rosenshtraukh, L. V. (2004). Roles of adrenergic and cholinergic stimulation in spontaneous atrial fibrillation in dogs. J. Am. Coll. Cardiol. 43, 483–490. doi:10.1016/j.jacc.2003.09.030
Shelton, R. J., Clark, A. L., Goode, K., Rigby, A. S., Houghton, T., Kaye, G. C., et al. (2009). A randomised, controlled study of rate versus rhythm control in patients with chronic atrial fibrillation and heart failure: (CAFE-II Study). Heart 95, 924–930. doi:10.1136/hrt.2008.158931
Shen, M. J., Shinohara, T., Park, H.-W., Frick, K., Ice, D. S., Choi, E.-K., et al. (2011). Continuous low-level vagus nerve stimulation reduces stellate ganglion nerve activity and paroxysmal atrial tachyarrhythmias in ambulatory canines. Circulation 123, 2204–2212. doi:10.1161/circulationaha.111.018028
Shen, N.-N., Zhang, C., Li, Z., Kong, L.-C., Wang, X.-H., Gu, Z.-C., et al. (2020). MicroRNA expression signatures of atrial fibrillation: the critical systematic review and bioinformatics analysis. Exp. Biol. Med. 245, 42–53. doi:10.1177/1535370219890303
Sheng, X., Scherlag, B. J., Yu, L., Li, S., Ali, R., Zhang, Y., et al. (2011). Prevention and reversal of atrial fibrillation inducibility and autonomic remodeling by low-level vagosympathetic nerve stimulation. J. Am. Coll. Cardiol. 57, 563–571. doi:10.1016/j.jacc.2010.09.034
Shunmugam, S. R., Sugihara, C., Freemantle, N., Round, P., Furniss, S., and Sulke, N. (2018). A double-blind, randomised, placebo-controlled, cross-over study assessing the use of XEN-D0103 in patients with paroxysmal atrial fibrillation and implanted pacemakers allowing continuous beat-to-beat monitoring of drug efficacy. J. Intervent. Card Electrophysiol. 51, 191–197. doi:10.1007/s10840-018-0318-2
Sieweke, J. T., Pfeffer, T. J., Biber, S., Chatterjee, S., Weissenborn, K., Grosse, G. M., et al. (2020). miR-21 and NT-proBNP correlate with echocardiographic parameters of atrial dysfunction and predict atrial fibrillation. J. Clin. Med. 9, 1118. doi:10.3390/jcm9041118
Silverman, M. E. (1994). From rebellious palpitations to the discovery of auricular fibrillation: contributions of Mackenzie, Lewis and Einthoven. Am. J. Cardiol. 73, 384–389. doi:10.1016/0002-9149(94)90013-2
Simon, A., Niederdoeckl, J., Skyllouriotis, E., Schuetz, N., Herkner, H., Weiser, C., et al. (2017). Vernakalant is superior to ibutilide for achieving sinus rhythm in patients with recent-onset atrial fibrillation: a randomized controlled trial at the emergency department. Europace 19, 233–240. doi:10.1093/europace/euw052
Simopoulos, V., Hevas, A., Hatziefthimiou, A., Dipla, K., Skoularigis, I., Tsilimingas, N., et al. (2018). Amiodarone plus ranolazine for conversion of post-cardiac surgery atrial fibrillation: enhanced effectiveness in reduced versus preserved ejection fraction patients. Cardiovasc. Drugs Ther. 32, 559–565. doi:10.1007/s10557-018-6832-8
Singh, B. N., Singh, S. N., Reda, D. J., Tang, X. C., Lopez, B., Harris, C. L., et al. (2005). Amiodarone versus sotalol for atrial fibrillation. N. Engl. J. Med. 352, 1861–1872. doi:10.1056/nejmoa041705
Singh, S., Zoble, R. G., Yellen, L., Brodsky, M. A., Feld, G. K., Berk, M., et al. (2000). Efficacy and safety of oral dofetilide in converting to and maintaining sinus rhythm in patients with chronic atrial fibrillation or atrial flutter: the symptomatic atrial fibrillation investigative research on dofetilide (SAFIRE-D) study. Circulation 102, 2385–2390. doi:10.1161/01.cir.102.19.2385
Soeki, T., Matsuura, T., Bando, S., Tobiume, T., Uematsu, E., Ise, T., et al. (2016). Relationship between local production of microRNA-328 and atrial substrate remodeling in atrial fibrillation. J. Cardiol. 68, 472–477. doi:10.1016/j.jjcc.2015.12.007
Sonmez, O., Ertem, F. U., Vatankulu, M. A., Erdogan, E., Tasal, A., Kucukbuzcu, S., et al. (2014). Novel fibro-inflammation markers in assessing left atrial remodeling in non-valvular atrial fibrillation. Med. Sci. Monit. 20, 463–470. doi:10.12659/MSM.890635
Staerk, L., Wang, B., Preis, S. R., Larson, M. G., Lubitz, S. A., Ellinor, P. T., et al. (2018). Lifetime risk of atrial fibrillation according to optimal, borderline, or elevated levels of risk factors: cohort study based on longitudinal data from the Framingham Heart Study. BMJ 361, k1453. doi:10.1136/bmj.k1453
Stavrakis, S., Humphrey, M. B., Scherlag, B. J., Hu, Y., Jackman, W. M., Nakagawa, H., et al. (2015). Low-level transcutaneous electrical vagus nerve stimulation suppresses atrial fibrillation. J. Am. Coll. Cardiol. 65, 867–875. doi:10.1016/j.jacc.2014.12.026
Stavrakis, S., Humphrey, M. B., Scherlag, B. J., Iftikhar, O., Parwani, P., Abbas, M., et al. (2017). Low-level vagus nerve stimulation suppresses post-operative atrial fibrillation and inflammation: a randomized study. JACC Clinical Electrophysiol. 3, 929–938. doi:10.1016/j.jacep.2017.02.019
Stavrakis, S., Stoner, J. A., Humphrey, M. B., Morris, L., Filiberti, A., Reynolds, J. C., et al. (2020). TREAT AF (transcutaneous electrical vagus nerve stimulation to suppress atrial fibrillation): a randomized clinical trial. JACC Clinical Electrophysiol. 6, 282–291. doi:10.1016/j.jacep.2019.11.008
Stewart, S., Hart, C. L., Hole, D. J., and Mcmurray, J. J. V. (2002). A population-based study of the long-term risks associated with atrial fibrillation: 20-year follow-up of the Renfrew/Paisley study. Am. J. Med. 113, 359–364. doi:10.1016/s0002-9343(02)01236-6
Stiell, I. G., Roos, J. S., Kavanagh, K. M., and Dickinson, G. (2010). A multicenter, open-label study of vernakalant for the conversion of atrial fibrillation to sinus rhythm. Am. Heart J. 159, 1095–1101. doi:10.1016/j.ahj.2010.02.035
Takei, M., Tsuboi, M., Usui, T., Hanaoka, T., Kurogouchi, F., Aruga, M., et al. (2001). Vagal stimulation prior to atrial rapid pacing protects the atrium from electrical remodeling in anesthetized dogs. Jpn. Circ. J. 65, 1077–1081. doi:10.1253/jcj.65.1077
Teh, A. W., Kistler, P. M., Lee, G., Medi, C., Heck, P. M., Spence, S. J., et al. (2012). Electroanatomic remodeling of the left atrium in paroxysmal and persistent atrial fibrillation patients without structural heart disease. J. Cardiovasc. Electrophysiol. 23, 232–238. doi:10.1111/j.1540-8167.2011.02178.x
Thacker, E. L., Mcknight, B., Psaty, B. M., Longstreth, W. T., Dublin, S., Jensen, P. N., et al. (2013). Association of body mass index, diabetes, hypertension, and blood pressure levels with risk of permanent atrial fibrillation. J. Gen. Intern. Med. 28, 247–253. doi:10.1007/s11606-012-2220-4
Toale, C., Fitzmaurice, G. J., Eaton, D., Lyne, J., and Redmond, K. C. (2019). Outcomes of left atrial appendage occlusion using the AtriClip device: a systematic review. Interact. Cardiovasc. Thorac. Surg. 29, 655–662. doi:10.1093/icvts/ivz156
Van Wagoner, D. R., Pond, A. L., Lamorgese, M., Rossie, S. S., Mccarthy, P. M., and Nerbonne, J. M. (1999). Atrial L-type Ca2+ currents and human atrial fibrillation. Circ. Res. 85, 428–436. doi:10.1161/01.res.85.5.428
Vaughan Williams, E. M. (1984). A classification of antiarrhythmic actions reassessed after a decade of new drugs. J. Clin. Pharmacol. 24, 129–147. doi:10.1002/j.1552-4604.1984.tb01822.x
Wang, X., Zhao, Q., Huang, H., Tang, Y., Xiao, J., Dai, Z., et al. (2013). Effect of renal sympathetic denervation on atrial substrate remodeling in ambulatory canines with prolonged atrial pacing. PLoS One 8, e64611. doi:10.1371/journal.pone.0064611
Warrington, S. J., and Hamer, J. (1980). Some cardiovascular problems with disopyramide. Postgrad. Med. 56, 229–233. doi:10.1136/pgmj.56.654.229
Watanabe, A. M. (1985). Digitalis and the autonomic nervous system. J. Am. Coll. Cardiol. 5, 35a–42a. doi:10.1016/s0735-1097(85)80461-7
Wegria, R. (1947). Correlation between the effect of quinidine sulfate on the rate of the circus movement in auricular fibrillation and its concentration in the blood plasma. J. Clin. Invest. 26, 1200Pubmed |
Weiss, J. N., Qu, Z., and Shivkumar, K. (2016). Ablating atrial fibrillation: a translational science perspective for clinicians. Heart Rhythm 13, 1868–1877. doi:10.1016/j.hrthm.2016.05.026
Wettwer, E., Christ, T., Endig, S., Rozmaritsa, N., Matschke, K., Lynch, J. J., et al. (2013). The new antiarrhythmic drug vernakalant: ex vivo study of human atrial tissue from sinus rhythm and chronic atrial fibrillation. Cardiovasc. Res. 98, 145–154. doi:10.1093/cvr/cvt006
White, C. M., and Nguyen, E. (2017). Novel use of ranolazine as an antiarrhythmic agent in atrial fibrillation. Ann. Pharmacother. 51, 245–252. doi:10.1177/1060028016673073
Wiedmann, F., Beyersdorf, C., Zhou, X., Büscher, A., Kraft, M., Nietfeld, J., et al. (2020). Pharmacologic TWIK-related acid-sensitive K+ channel (TASK-1) potassium channel inhibitor A293 facilitates acute cardioversion of paroxysmal atrial fibrillation in a porcine large animal model. J. Am. Heart Assoc. 9, e015751. doi:10.1161/jaha.119.015751
Wijffels, M. C. E. F., Kirchhof, C. J. H. J., Dorland, R., and Allessie, M. A. (1995). Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 92, 1954–1968. doi:10.1161/01.cir.92.7.1954
Withering, W. (1785). An account of the foxglove and some of its medical uses: with practical remarks on dropsy and other disesases. Birmingham.
Wolf, P. A., Abbott, R. D., and Kannel, W. B. (1991). Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke 22, 983–988. doi:10.1161/01.str.22.8.983
Wray, S., Eisner, D. A., and Allen, D. G. (1985). Two hundred years of the foxglove. Med. Hist. 29, 132–150. doi:10.1017/s0025727300070551
Wybraniec, M. T., Wróbel, W., Wilkosz, K., Wrona, K., Bula, K., and Mizia‐Stec, K. (2018). Pharmacological cardioversion with antazoline in atrial fibrillation: the results of the CANT study. J. Am. Heart Assoc. 7, e010153. doi:10.1161/jaha.118.010153
Wyse, D. G., Waldo, A. L., Dimarco, J. P., Domanski, M. J., Rosenberg, Y., Schron, E. B., et al. (2002). A comparison of rate control and rhythm control in patients with atrial fibrillation. N. Engl. J. Med. 347, 1825–1833. doi:10.1056/nejmoa021328
Yao, C., Veleva, T., Scott, L., Cao, S., Li, L., Chen, G., et al. (2018). Enhanced cardiomyocyte NLRP3 inflammasome signaling promotes atrial fibrillation. Circulation 138, 2227–2242. doi:10.1161/circulationaha.118.035202
Youssef, I., Kamran, H., Yacoub, M., Patel, N., Goulbourne, C., Kumar, S., et al. (2018). Obstructive sleep apnea as a risk factor for atrial fibrillation: a meta-analysis. J. Sleep Disord. Ther. 7, 282. doi:10.4172/2167-0277.1000282
Yu, L., Scherlag, B. J., Li, S., Sheng, X., Lu, Z., Nakagawa, H., et al. (2011). Low-level vagosympathetic nerve stimulation inhibits atrial fibrillation inducibility: direct evidence by neural recordings from intrinsic cardiac ganglia. J. Cardiovasc. Electrophysiol. 22, 455–463. doi:10.1111/j.1540-8167.2010.01908.xCrossRef Full Text
Yue, L., Feng, J., Gaspo, R., Li, G.-R., Wang, Z., and Nattel, S. (1997). Ionic remodeling underlying action potential changes in a canine model of atrial fibrillation. Circ. Res. 81, 512–525. doi:10.1161/01.res.81.4.512
Zareba, K. M. (2006). Dronedarone: a new antiarrhythmic agent. Drugs Today 42, 75–86. doi:10.1358/dot.2006.42.2.925346
Zhang, Y., Ilsar, I., Sabbah, H. N., Ben David, T., and Mazgalev, T. N. (2009a). Relationship between right cervical vagus nerve stimulation and atrial fibrillation inducibility: therapeutic intensities do not increase arrhythmogenesis. Heart Rhythm 6, 244–250. doi:10.1016/j.hrthm.2008.10.043
Zhang, Y., Popović, Z. B., Bibevski, S., Fakhry, I., Sica, D. A., Van Wagoner, D. R., et al. (2009b). Chronic vagus nerve stimulation improves autonomic control and attenuates systemic inflammation and heart failure progression in a canine high-rate pacing model. Circ. Heart Fail. 2, 692–699. doi:10.1161/circheartfailure.109.873968
Zhang, Y., Scherlag, B. J., Lu, Z., Niu, G.-D., Yamanashi, W. S., Hogan, C., et al. (2009c). Comparison of atrial fibrillation inducibility by electrical stimulation of either the extrinsic or the intrinsic autonomic nervous systems. J. Intervent. Card Electrophysiol. 24, 5–10. doi:10.1007/s10840-008-9297-z
Keywords: atrial fibrillation, a-fib, afib, antiarrhythmic, antiarrhythmic drug therapy, paroxysmal AF, persistent AF, permanent AF
Citation: Geng M, Lin A and Nguyen TP (2020) Revisiting Antiarrhythmic Drug Therapy for Atrial Fibrillation: Reviewing Lessons Learned and Redefining Therapeutic Paradigms. Front. Pharmacol. 11:581837. doi: 10.3389/fphar.2020.581837
Received: 09 July 2020; Accepted: 17 September 2020;
Published: 09 November 2020.
Edited by:
Chrishan S. Samuel, Monash University, AustraliaReviewed by:
Tamer M. A. Mohamed, University of Louisville, United StatesUrsula Ravens, Technische Universität Dresden, Germany
Copyright © 2020 Geng, Lin and Nguyen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thao P. Nguyen, dHBuZ3V5ZW5AbWVkbmV0LnVjbGEuZWR1
†Present address: Saint Mary’s Hospital, Waterbury, CT, United States
Abbreviations: ACC, American College of Cardiology; ADT, antiarrhythmic drug therapy; AF, atrial fibrillation; AHA, American Heart Association; CaMKII, Ca2+/calmodulin kinase type II; CCB, Ca2+ channel blocker; CFAEs, complex fractionated atrial electrograms; dV/dtmax, maximum upstroke velocity; ECGI, electrocardiographic imaging; ERP, effective refractory period; ESC, European Society of Cardiology; FDA, Food and Drug Administration; FIRM, focal impulse and rotor modulation; Gs, stimulatory G-protein; hERG, human ether-a-go-go-related gene; HFpEF (HFrEF), heart failure with preserved (reduced) ejection fraction; ICaL, L-type Ca2+ current; If, funny current; IK1, inward rectifier current; IK2P, two-pore-domain K+ current; IKACh, acetylcholine-activated inward-rectifier K+ current; IKr/IKur, rapid/ultrarapid delayed rectifier K+ current; (late) INa, (late) Na+ current; ISK, small-conductance Ca2+-activated K+ current; Iss, steady-state current; LVEF, left ventricular ejection fraction (LVEF)LGE-MRI, late-gadolinium enhancement magnetic resonance imaging; LLTS, low-level tragus electrical stimulation; LLVS, low-level vagus nerve stimulation; miRNA, microRNA; Nav1.5, voltage-gated sodium channel; NCX, Na+-Ca2+ exchanger; NF-κB, nuclear factor κ-light-chain-enhancer of activated B cells; RAAS, renin-angiotensin-aldosterone system; RyR2, ryanodine receptor; SERCA2a, sarcoplasmic reticulum Ca2+-ATPase 2a; siRNA, small interfering RNA; SK, small-conductance Ca2+-activated K+ channel; TASK-1/-3, two-pore-domain Weak Inward rectifying K+ channel (TWIK)-related Acid-Sensitive K+ channel-1/-3.