 Zhen Li
Zhen Li Wei Zou2
Wei Zou2 Yunjiao Zhang
Yunjiao Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 16 November 2020
Sec. Pharmacology of Anti-Cancer Drugs
Volume 11 - 2020 | https://doi.org/10.3389/fphar.2020.580251
This article is part of the Research Topic Molecular Mechanisms of Drug Resistance And Strategies of Sensitization in Breast Cancer View all 16 articles
As a new-generation CDK inhibitor, a CDK4/6 inhibitor combined with endocrine therapy has been successful in the treatment of advanced estrogen receptor–positive (ER+) breast cancer. Although there has been overall progress in the treatment of cancer, drug resistance is an emerging cause for breast cancer–related death. Overcoming CDK4/6 resistance is an urgent problem. Overactivation of the cyclin-CDK-Rb axis related to uncontrolled cell proliferation is the main cause of CDK4/6 inhibitor resistance; however, the underlying mechanisms need to be clarified further. We review various resistance mechanisms of CDK4/6 inhibitors in luminal breast cancer. The cell signaling pathways involved in therapy resistance are divided into two groups: upstream response mechanisms and downstream bypass mechanisms. Finally, we discuss possible strategies to overcome CDK4/6 inhibitor resistance and identify novel resistance targets for future clinical application.
Breast cancer (BC) is a common women-related malignant tumor disease in developed countries. Estrogen receptor–positive (ER-positive) breast cancer represents approximately 70% of all BC (Goldhirsch et al., 2011; Malvezzi et al., 2013). ER-positive breast cancer can be further stratified into pathological subtypes, such as ductal or mixed ductal and lobular, mucinous, and tubular carcinomas, which are referred to as luminal breast cancer (Ignatiadis and Sotiriou, 2013). Luminal breast tumors are highly heterogeneous in terms of histology and response to treatment. Luminal A and B are two main ER-positive breast cancer subtypes, based on different gene expression profiles, prognosis, and clinical therapy responses (Sotiriou and Pusztai, 2009).
The difference between luminal A and B is mainly related to the expression of hormone receptors. Luminal B tumors have lower levels of ER expression, lower or no levels of progesterone receptor (PR) expression, but higher tumor grade and higher Ki-67–positive staining than luminal A tumors (Goldhirsch et al., 2011; Creighton, 2012). Endocrine therapy, such as ER downregulators, selective ER modulators, and aromatase inhibitors, is considered to be the primary treatment for luminal A and luminal B. However, in the clinic, the main therapy for luminal B is chemotherapy, due to the lower sensitivity of these patients to endocrine treatment or drug resistance (Rouzier et al., 2005; Ignatiadis et al., 2012). In fact, endocrine resistance is an unavoidable problem in clinical therapy of luminal tumors. Development of new therapy methods to avert endocrine resistance is an urgent challenge in clinical medicine (Anurag et al., 2020).
It is well known that the cell cycle is driven by cyclin-dependent kinases (CDKs), such as CDK4 and CDK6, which are also closely associated with tumor initiation and progression Yu et al., 2006; Choi et al., 2012). The activity of cyclin D and CDK4/6 complexes is considered to play the major role in tumor cell proliferation driven by estrogen, especially in breast cancer (Filmus et al., 1994). In recent years, it has been established that targeting the cell cycle for anticancer treatment is a rational option that could be combined with endocrine therapy.
CDK inhibitors, which target overactive CDK activities in tumor cells, have been widely used in preclinical or clinical trials. In the clinic, three CDK4/6 inhibitors, namely, palbociclib (Fry et al., 2004), ribociclib (Infante et al., 2016), and abemaciclib (Patnaik et al., 2016), have been successfully used in combination with other endocrine therapy drugs for ER-positive and human epidermal growth factor receptor-2 (HER2)–negative advanced breast cancer treatment (Ribnikar et al., 2019); in addition, significant overall survival (OS) benefits have been confirmed at ESMO2019 conference.
Despite the fact that the new guidelines for the therapy of advanced breast cancer includes a CDK4/6 inhibitor combined with endocrine treatment as the first- or second-line drug in most countries, most patients eventually develop acquired drug resistance to CDK4/6 inhibitors (Konecny et al., 2011). Several factors affect the effectiveness of CDK4/6 inhibitors, such as continuous expression of G1-S-phase cyclins and gene mutations in key cell signaling pathways (Herrera-Abreu et al., 2016). Research on the molecular mechanisms or clinical strategies to overcome CDK4/6 inhibitor resistance is ongoing (Pandey et al., 2019; Portman et al., 2019). Therefore, the major emerging consideration in treatment of advanced luminal breast cancer is now CDK4/6 inhibitor resistance.
In this review, we discuss three CDK4/6 inhibitors with different clinical trial results and various resistance mechanisms, aiming to help identify novel clinical therapeutic targets to improve endocrine therapy resistance and provide possible strategies to overcome resistance to CDK4/6 inhibitors in advanced luminal breast cancer.
In malignant cells, overactive CDK activities are targeted by CDK inhibitors. The major barrier limiting CDK inhibitors from further development is the lack of selectivity, due to similar structures among CDKs (Shapiro, 2006; Michaud et al., 2010). In the meantime, some biocomputing technologies, such as computer-aided (Kalra et al., 2017) and pharmacological (Tadesse et al., 2018; Yin et al., 2018) approaches, have been employed to develop a new-generation CDK inhibitor with higher selectivity. Recently, there has been great progress in CDK inhibitor design, especially the design of CDK4/6 inhibitors, which have been successfully used in clinical trials.
ATP-binding domains are the main drug targets of CDK4/6 inhibitors to block cell cycle G1-S transition (Asghar et al., 2015). Three third-generation CDK inhibitors, palbociclib, ribociclib, and abemaciclib, have higher specificity to CDK4/6 than other members of the CDK family and have been translated into clinical use against advanced luminal breast cancer. The phase III MONALEESA-3 trial used a combination of ribociclib and fulvestrant in advanced ER+/HER2 breast cancer demonstrated an increased PFS (progression-free survival) (Slamon et al., 2018) and an improved OS compared with fulvestrant alone (Slamon et al., 2019). The phase III MONARCH-plus trial with abemaciclib and nonsteroidal aromatase inhibitor (NSAI) or fulvestrant treatment showed improved PFS in predominantly Chinese postmenopausal women with ER+/HER2 breast cancer (Jiang et al., 2019a). Moreover, in the phase III MONARCH HER trial, triple treatment with abemaciclib, trastuzumab (Herceptin), and fulvestrant showed better therapy outcomes than trastuzumab plus chemotherapy in ER+/HER2+ patients. In addition, phase II/III trials of the three CDK4/6 inhibitors in combination with letrozole, tamoxifen, fulvestrant, and herceptin in the first-/second-line setting have already been completed (Table 1).
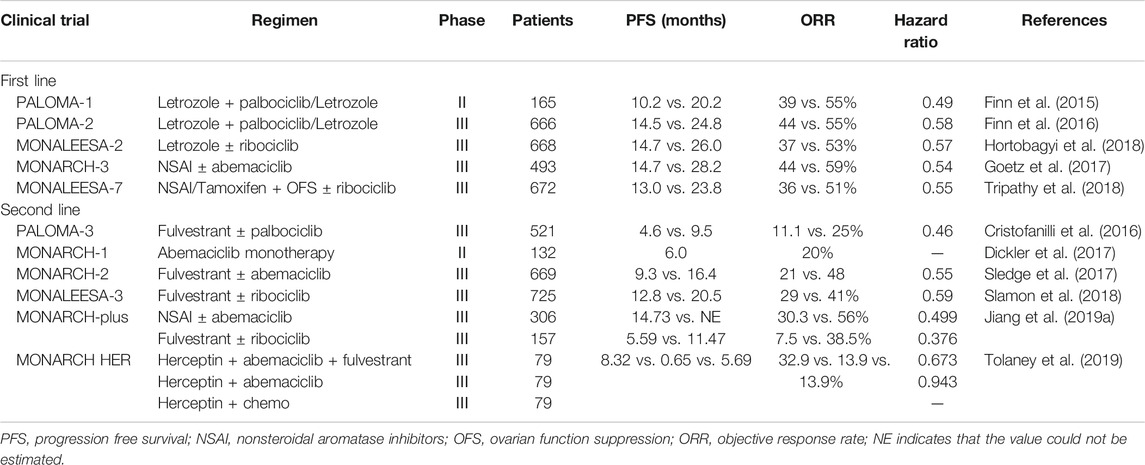
TABLE 1. CDK4/6 inhibitors for the treatment of advanced luminal breast cancer in phase II/III trials.
CDK4/6 inhibitors are not a panacea due to the therapy resistance. It has been reported in the PALOMA-2 trial that more than 30% patients experienced recurrence of their cancer within 2 years of CDK4/6 inhibitor treatment (Finn et al., 2016), indicating that palbociclib combined with endocrine therapy may affect CDK inhibitor sensitivity and allow tumor cells to return to a proliferative phenotype. However, whether the mechanism of endocrine therapy resistance is associated with the inhibition of cell cycle or activation of other “bypass” signaling pathways is not fully understood. We summarized the molecular mechanisms of CDK4/6 inhibitor resistance below (Figure 1).
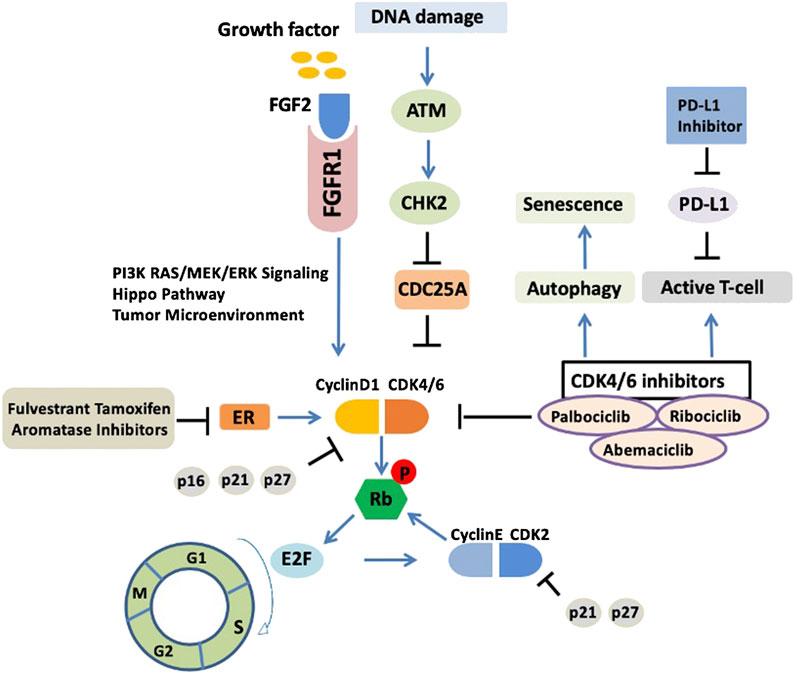
FIGURE 1. Upstream response and downstream bypass signaling mechanisms of CDK4/6 inhibitor resistance. Current molecular mechanisms of CDK4/6 inhibitor resistance are highlighted. ER, estrogen receptor; CDK, cyclin dependent kinases; ATM, ataxia telangiectasia mutated; CHK2, checkpoint kinase 2; Rb, retinoblastoma protein.
Upregulation of cyclin–CDK activity promotes the cell cycle and proliferation (Hanahan and Weinberg, 2011). The retinoblastoma (Rb) protein acts as a gatekeeper to prevent the cell cycle from progressing from G1 phase into S phase. CDK4/6 forms heterodimers with D-type cyclins (particularly D1) to phosphorylate Rb. As a result, Rb protein phosphorylation leads to the release of transcription factor E2Fs, which activates the DNA transcriptional program for cell cycle G1/S.
In luminal breast cancer, the development of resistance to endocrine therapy is associated with the function and integrity of Rb (Musgrove et al., 2011). Fortunately, the low incidence of Rb gene deletion/mutation (3.9%) in luminal-like breast tumors offers the possibility for CDK4/6 inhibition (Ciriello et al., 2015). This viewpoint has been solidified by a study that showed that the effects of clinical therapy in luminal breast cancer were not sensitive to palbociclib when Rb expression is absent (Dean et al., 2012). In addition, the function of Rb can also be regulated by E2F transcriptional factors. Malorni et al. indicated that the expression of both E2F1 and E2F2 could cause loss of Rb and predict the sensitivity of cell lines to palbociclib in luminal breast cancer (Malorni et al., 2016).
Overexpression of CDK4 or CDK6 is the main mechanism of resistance to CDK4/6 inhibitors. Studies have shown that increased expression of CDK6 reduced the response of CDK4/6 inhibitors in luminal cell line models. At the same time, knockdown of CDK6 rescued the therapy sensitivity, which indicated that CDK6-mediated drug resistance may be independent of CDK4 expression (Yang et al., 2017). In addition, either high or low expression of CDK4 has been detected in CDK4/6 inhibitor–resistant breast cancer cells (Bollard et al., 2017). Therefore, whether the expression of CDK4 is associated with CDK4/6 inhibitor resistance requires further investigation.
As a member of the INK4 family, p16 is a natural inhibitor of CDK4 and plays a vital role in the regulation of the cell cycle (Serrano et al., 1993). In general, p16 severs as a tumor suppressor and targets the CDK4/6 complex in dysregulatory cells depending on the function of Rb (Medema et al., 1995). For example, Dean JL et al. reported that the resistance to CDK4/6 inhibitors was caused by the absence of Rb, regardless of p16 expression (Dean et al., 2012). On the other hand, the expression level of p16 affected the effectiveness of CDK4/6 inhibition. Overexpression of p16-mediated resistance to CDK4/6 inhibitors in the absence of Rb (Witkiewicz et al., 2011) and low expression of p16 did not rescue the clinical benefit in Rb-positive luminal breast cancer patients in the phase II palbociclib monotherapy trial (DeMichele et al., 2015). The potential mechanism is that p16 overexpression suppresses the activity of CDK4 and expression of cyclin D1 (Witkiewicz et al., 2011), which are the main targets of CDK4/6 inhibitors, thus leading to reduced or no effects of CDK4/6 inhibition (Elvin et al., 2017). Whether p16 amplification and loss of Rb work together in CDK4/6 inhibitor resistance is not clearly understood. Further studies revealing the mechanistic association between p16 and Rb might be beneficial to avert acquired resistance to CDK4/6 inhibitors.
Deficiency of mismatch repair may lead to the endocrine therapy resistance in luminal breast cancer through the abrogation of CHK2-mediated inhibition of CDK4. A recent study showed that defects in single-strand break repair in luminal breast cancer can drive endocrine therapy resistance and is closely associated with the ATM-CHK2-CDC25A pathway (Anurag et al., 2018). ATM, as a DNA damage sensor, activates CHK2, which in turn phosphorylates CDC25A at S123 for degradation. Importantly, as a phosphatase, CDC25A could inhibit the phosphorylation of CDK4/6. The CDK4/6 complex activity could be reactivated with the “on state” of CDC25A. Therefore, the cross talk between the CDK4/6–Rb and ATM–CHK2–CDC25A axes is very important. Moreover, recently, Haricharan et al. demonstrated that for the efficacy of endocrine agents in luminal tumors, both ATM and CHK2 are required; inactivation of either of these negative cell cycle regulators prevents cell cycle arrest upon ER inhibition (Haricharan et al., 2017).
In luminal breast cancer, activation of ER is the major driver of CDK4/6. Selective ER-related endocrine therapy, such as ER downregulators (fulvestrant), ER modulators (tamoxifen), and aromatase inhibitors (AIs), have been combined with CDK4/6 inhibitors and broadly used in the treatment of advanced ER-positive breast cancer. The expression level of cyclin D1 could be upregulated by ER (Du et al., 2014). Resistance to CDK4/6 inhibitors may be related to the decrease in cyclin D1 due to the loss of ER (Gong et al., 2017b). For instance, resistance to the CDK4/6 inhibitor abemaciclib occurred in preclinical trials and was associated with the loss of cyclin D1 and ER/PR expression. In addition, studies showed that CDK6 overexpression diminished the responsiveness to ER antagonism and mediated the resistance to CDK4/6 inhibitors by decreasing the expression of ER and PR (Yang et al., 2017). Moreover, tumor biopsy specimens from patients associated with changes in ER/PR levels showed resistance to CDK4/6 inhibitors mediated by low ER/PR expression. Moreover, it also has been indicated that luminal tumors are resistant to endocrine therapy when they have an activating ESR1 mutation; however, CDK4/6 inhibitors take effect regardless of ESR1 mutation status (Fribbens et al., 2016).
The PI3K–AKT–mTOR signaling pathway is involved in tumor cell growth, survival, and metastasis. In luminal breast cancer, ER transcriptional activity could be enforced by the activation of PI3K–AKT–mTOR signaling, which drives endocrine therapy resistance (Miller et al., 2011). Furthermore, activation of the PI3K–AKT–mTOR pathway can also promote the stability of the CDK4/6 complex, thus reversing the effects of CDK4/6 inhibition (Miller et al., 2011). A recent study indicated that loss of PTEN expression could mediate CDK4/6 inhibitor resistance by increasing AKT activation and decreasing the expression of p27, which leads to the excessive activation of CDK4 and CDK2 (Costa et al., 2020). CDK4 in lysosome activates mTORC1 and is also associated with cancer progression (Martinez-Carreres et al., 2019). Moreover, it has been reported that CDK4/6 inhibitors preferred to activate PI3K–AKT–mTOR pathway than ER signaling (Takeshita et al., 2018), with the reactivation of E2F (Jansen et al., 2017). Therefore, the therapeutic trial, endocrine therapy backbone combined with PI3K and mTOR inhibition, and CDK4/6 inhibitors can be combined.
Strategies that inhibit PI3K and mTOR activities have been shown to restore sensitivity to endocrine therapy. Everolimus, a mTOR inhibitor, was the first drug developed to overcome endocrine therapy resistance in combination with AI (Pronzato, 2017). Some other mTORC1/2 inhibitors also restored the sensitivity of CDK4/6 inhibitors in resistant cells by suppressing Rb phosphorylation (Michaloglou et al., 2018). PI3K inhibitors, such as alpelisib, combined with fulvestrant prolonged PFS among patients with mutated PIK3CA in advanced luminal breast cancer who had previously received endocrine therapy (Andre et al., 2019). In addition, PI3K inhibitors have been implicated in the prevention of early CDK4/6 inhibitor adaptions by decreasing the expression of cyclin D1 (Herrera-Abreu et al., 2016). In the future, a combination of a PI3K–AKT–mTOR pathway inhibitor and a CDK4/6 inhibitor may be a valuable therapeutic strategy.
The fibroblast growth factor receptor (FGFR) pathway is involved the proliferation and survival in luminal breast cancer (Sahores et al., 2018). Like other mitogenic pathways, FGFR is relevant in the crosslinking of cyclin D and CDK4/6. Of the five FGFRs, FGFR1 is associated with CDK4/6 inhibitor resistance. FGFR1 activates the PI3K–AKT–mTOR and RAS–MEK–ERK signaling pathways (Turner et al., 2010). In the clinic, FGFR1 overexpression mediated resistance to palbociclib or ribociclib when combined with endocrine therapy (fulvestrant) (Formisano et al., 2017). This could be reversed by the FGFR tyrosine kinase inhibitor (TKI) lucitanib (Formisano et al., 2019). FGFR2 amplification has also been reported in metastatic luminal breast cancer and the response to an mTOR inhibitor (Wein et al., 2017). In addition, FGF2 could also activate FGFR signaling and mediate endocrine therapy resistance in preclinical research (Turner et al., 2010). A previous study showed that the FGFR2 inhibitor formononetin had a strong inhibitory effect on angiogenesis and tumor growth (Wu et al., 2015). Therefore, targeting FGFR1/2 in luminal breast cancer may be a viable option combined with the inhibition of CDK4/6 to overcome CDK4/6 inhibitor resistance.
The Hippo pathway is closely related to the development and progression of breast cancer and has emerged as a linchpin in breast cancer therapy resistance (Gujral and Kirschner, 2017) (Shi et al., 2015). Hippo pathway effectors, such as YAP, TAZ, and TEAD, have been employed as drug targets to hit other signaling pathways (Dey et al., 2020). In ovarian cancer, YAP expression is associated with PI3K inhibitor resistance (Muranen et al., 2016). TEADs have also been shown to be a mediator of CDK6 induction (Xie et al., 2013). Importantly, alterations in the Hippo pathway are related to CDK4/6 inhibitor resistance. In the latter clinical case, loss of FAT1 is associated with CDK4/6 inhibitor resistance caused by YAP/TAZ nuclear localization and CDK6 overexpression in ER-positive breast cancer (Li et al., 2018). Therefore, targeting the Hippo pathway offers a new therapeutic strategy against CDK4/6 inhibitor resistance.
The molecular mechanisms responsible for resistance to CDK4/6 inhibitors are diverse and complicated, and the current knowledge is far from complete. Recently, several new “bypass” signaling pathway mechanisms on CDK4/6 inhibitor adaption have been discovered.
Cyclin E–CDK2 complexes can also inactivate Rb by phosphorylating Rb and releasing transcriptional factor E2F to initiate the cell cycle. However, as the “second wave” that phosphorylates Rb, the efficiency of this process is subsequent to CDK4/6 complexes. Excessive activation of the CDK2 pathway mediates resistance to CDK4/6 inhibitors because released E2F reverse targets cyclin E2, stabilizing the cyclin E2–CDK2 complexes and reducing CDK4/6 inhibition (Caldon et al., 2009). The abnormal expression of cyclin E1/2-CDK2 and persistent activation of E2F are associated with resistance to CDK4/6 inhibitors (Taylor-Harding et al., 2015). For instance, CCNE1 gene amplification also induces resistance in the CDK4/6 single agent model; CCNE2 gene amplification has been found in patients in whom palbociclib treatment failed (Hortobagyi et al., 2016). Moreover, in the clinic, lower CCNE1 messenger RNA expression is often associated with improved palbociclib efficacy in ER-positive metastatic breast cancer (Turner et al., 2019). Activity of cyclin E1–CDK2 complexes could be suppressed by p21Waf1/Cip1 and p27Kip1 (Martin et al., 2017); therefore, the development of CDK2 inhibitors have the potency and advantage as bypass signals to reduce CDK4/6 inhibitor resistance by the inhibition of cyclin E1/2–CDK2 (Caldon et al., 2012).
Autophagy is generally thought of as a cell survival mechanism. The activation of autophagy induces cell cycle arrest and cell senescence (Glick et al., 2010). Targeting autophagy is an available strategy for novel drug development and tumor treatment. Autophagy inhibition is relevant to the efficacy of anti–breast cancer drugs (Chittaranjan et al., 2014). An accumulation of evidence suggests that autophagy activation is involved in resistance to CDK4/6 inhibitors. Studies have shown that breast cancer cells activate autophagy in response to palbociclib, possibly through the inhibition of cyclin D1 expression, and the combination of autophagy and CDK4/6 inhibitors induces irreversible growth inhibition and senescence in vitro (Vijayaraghavan et al., 2017b). More work is being done to increase the efficacy of CDK4/6 inhibitors by inhibiting autophagy, which may help avert CDK4/6 inhibitor resistance.
The adaptive immune response plays a role in CDK4/6 inhibitor efficacy. CDK4/6 inhibitors promote tumor immunogenicity, and the effects of CDK4/6 inhibitors targeting both tumor T cells and regulatory T cells are associated with reduced activity of E2F transcription factors and DNA methyltransferase (Goel et al., 2017). In addition, CDK4/6 inhibitors enhance antitumor immunity by increasing T-cell activation and promoting T cells to kill tumor cells (Deng et al., 2018). Moreover, immunotherapeutic approaches combined with CDK4/6 inhibitors could achieve better therapeutic effects. CDK4/6 inhibitors increase the expression of PD-L1 (programmed cell death ligand 1), thus inducing the inflammatory microenvironment and improving tumor immunogenicity (Minton, 2017; Schaer et al., 2018). Therefore, CDK4/6 inhibitors combined with a PD-L1 immune checkpoint inhibitor can improve the effect of tumor immunotherapy. Currently, there are several ongoing clinical trials of immune checkpoint antibodies, including pembrolizumab and avelumab (Anurag et al., 2020). However, immune evasion or alterations in the immune microenvironment eventually leads to CDK4/6 inhibitor resistance (Goel et al., 2017; Teh and Aplin, 2019). In terms of mechanism, immune evasion may be associated with the abnormal expression of immune-related regulators, such as IFN-α and IFN-β, and change in tumor microenvironment of CDK4/6 inhibitor–resistant breast tumors (Vijayaraghavan et al., 2017a). Future investigations using tumor-infiltrating lymphocyte analyses are needed to better understand CDK4/6 inhibitor resistance mechanisms of immune evasion.
Histone deacetylases (HDACs) can increase CDK4/6 inhibition efficacy and mediate cell cycle arrest by upregulating p21 expression in CDK4/6 inhibitor resistant tumors (Lee et al., 2018). Even though the mechanism is not very clear, HDAC inhibition works synergistically with CDK4/6 inhibitors in luminal breast cancer. Cornell et al. demonstrated that miR-432-5p–mediated suppression of the TGF-β signaling pathway via SMAD4 knockdown and increased CDK6 expression, thus conferring transmissible and reversible CDK4/6 inhibitor adaptation (Cornell et al., 2019). In addition, a recent study showed that LncRNA TROJAN could mediate resistance to CDK4/6 inhibitors by increasing CDK2 activation in ER+ breast cancer (Jin et al., 2020). Analysis of patient plasma exosomes may identify emerging resistance mechanisms.
In the clinic, treatment effectiveness is based on the improved survival of patients. Currently, endocrine targeted therapy and chemotherapy are common options for the treatment of luminal breast cancer. CDK4/6 inhibitors have been used in advanced ER-positive breast cancer patients with antimitosis, but they eventually develop resistance to the CDK4/6 inhibitors (Franco et al., 2014; Yoshida et al., 2016). In the past 5 years, endocrine therapy combined with PI3K and mTOR inhibitors and CDK4/6 inhibitors has gradually become a new therapeutic strategy. Several studies have confirmed that CDK4/6 inhibitors combined with PI3K inhibitors (Vora et al., 2014) or mTORC1/2 inhibitors could reverse resistance (Michaloglou et al., 2018). Furthermore, studies have shown that CDK4/6 inhibitors may increase tumor immunogenicity, which provides a rationale for combination regimens composed of CDK4/6 inhibitors and immunotherapies. Therefore, CDK4/6 inhibitors combined with other clinical therapies might be a cautious approach to overcome therapy resistance. We summarized possible strategies to overcome resistance to CDK4/6 inhibitors in Table 2.

TABLE 2. Possible strategies to overcome resistance to CDK4/6 inhibitors in ER-positive BC.
Whether CDK4/6 inhibition is truly suitable for patients with advanced ER-positive breast cancer and whether resistance develops are being studied in a number of preclinical studies and models. Rb may be a biomarker. It has been demonstrated that fully functional Rb is required for the effective use of CDK4/6 inhibitors in the clinic (Karakas et al., 2016). However, not all Rb+/ER+ patients would benefit from CDK4/6 inhibitor therapy, even though the mutation of Rb is very rare (3.9%) in ER-positive breast cancer. The utility of Rb as biomarker combined with low-molecular-weight cyclin E1 (LMWE) is associated with CDK4/6 inhibitor sensitivity (Hunt et al., 2017). A cohort of 109 patients with Rb-/LMWE+ had shorter PFS when treated with palbociclib plus endocrine therapy (Vijayaraghavan et al., 2017b). Although cyclin D1 plays a vital role in CDK4/6 inhibition, unfortunately, CCND1 amplification as single biomarker for CDK4/6 inhibitor sensitivity needs to be refined further. In the PALOMA-1 study, patients treated with palbociclib plus letrozole had no beneficial outcomes regardless of CCND1 status (Finn et al., 2015). Moreover, CDK4 phosphorylation status shows the potential as a biomarker to predict the sensitivity to palbociclib but needs further clinical observation (Raspe et al., 2017).
The development of CDK4/6 inhibitors has been a significant advancement in luminal breast cancer therapy. In other breast cancer subtypes, such as triple negative breast cancer, clinical trials of CDK4/6 inhibitors in combination with anti-androgen inhibitors are still ongoing. However, resistance to CDK4/6 inhibitors in clinical treatment is an unavoidable problem. Although CDK4/6 inhibitor resistance has been well investigated and different mechanisms have been revealed, systematic and comprehensive clinical trials are required to develop new strategies to overcome CDK4/6 inhibitor resistance. Therefore, further efforts to investigate much more precise resistance mechanisms to CDK4/6 inhibitors or to develop more successful CDK inhibitors are needed in order to explore new therapeutic approaches to avert or overcome resistance.
ZL: conceptual design and article writing; WZ: work drawing; JZ: conceptual design; YZ: make keyword tables; QX: work drawing; SL: conceptual design and article writing; CC: conceptual design and article writing
This study was supported in part by grants from the National Key R&D Program of China [2018YFC2000400], the National Nature Science Foundation of China [81830087, U1602221, 31771516], Science and Technology Innovation Team of Yunnan Province [2018HC002, 2019HC005], Basic Research Program of Yunnan Province [2019FE001(-309)], and Research Institutions attached to the Health and Family Planning Commission of Yunnan Province [2017NS180].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Andre, F., Ciruelos, E., Rubovszky, G., Campone, M., Loibl, S., and Rugo, H. S. (2019). Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N. Engl. J. Med. 380(20), 1929–1940. doi:10.1056/NEJMoa1813904
Anurag, M., Punturi, N., Hoog, J., Bainbridge, M. N., Ellis, M. J., and Haricharan, S. (2018). Comprehensive profiling of DNA repair defects in breast cancer identifies a novel class of endocrine therapy resistance drivers. Clin. Canc. Res. 24 (19), 4887–4899. doi:10.1158/1078-0432.CCR-17-3702
Anurag, M., Haricharan, S., and Ellis, M. J. (2020). CDK4/6 inhibitor biomarker research: are we barking up the wrong tree? Clin. Canc. Res. 26 (1), 3–5. doi:10.1158/1078-0432.CCR-19-3119
Asghar, U., Witkiewicz, A. K., Turner, N. C., and Knudsen, E. S. (2015). The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 14 (2), 130–146. doi:10.1038/nrd4504
Bian, X., Liang, Z., Feng, A., Salgado, E., and Shim, H. (2018). HDAC inhibitor suppresses proliferation and invasion of breast cancer cells through regulation of miR-200c targeting CRKL. Biochem. Pharmacol. 147, 30–37. doi:10.1016/j.bcp.2017.11.008
Bollard, J., Miguela, V., Ruiz de Galarreta, M., Venkatesh, A., Bian, C. B., and Roberto, M. P. (2017). Palbociclib (PD-0332991), a selective CDK4/6 inhibitor, restricts tumour growth in preclinical models of hepatocellular carcinoma. Gut 66 (7), 1286–1296. doi:10.1136/gutjnl-2016-312268
Caldon, C. E., Sergio, C. M., Schutte, J., Boersma, M. N., Sutherland, R. L., and Carroll, J. S. (2009). Estrogen regulation of cyclin E2 requires cyclin D1 but not c-Myc. Mol. Cell Biol. 29 (17), 4623–4639. doi:10.1128/MCB.00269-09
Caldon, C. E., Sergio, C. M., Kang, J., Muthukaruppan, A., Boersma, M. N., and Stone, A. (2012). Cyclin E2 overexpression is associated with endocrine resistance but not insensitivity to CDK2 inhibition in human breast cancer cells. Mol. Canc. Therapeut. 11 (7), 1488–1499. doi:10.1158/1535-7163.MCT-11-0963
Chittaranjan, S., Bortnik, S., Dragowska, W. H., Xu, J., Abeysundara, N., and Leung, A. (2014). Autophagy inhibition augments the anticancer effects of epirubicin treatment in anthracycline-sensitive and -resistant triple-negative breast cancer. Clin. Canc. Res. 20 (12), 3159–3173. doi:10.1158/1078-0432.CCR-13-2060
Choi, Y. J., Li, X., Hydbring, P., Sanda, T., Stefano, J., and Christie, A. L. (2012). The requirement for cyclin D function in tumor maintenance. Canc. Cell 22 (4), 438–451. doi:10.1016/j.ccr.2012.09.015
Ciriello, G., Gatza, M. L., Beck, A. H., Wilkerson, M. D., Rhie, S. K., and Pastore, A. (2015). Comprehensive molecular portraits of invasive lobular breast cancer. Cell 163 (2), 506–519. doi:10.1016/j.cell.2015.09.033
Cornell, L., Wander, S. A., Visal, T., Wagle, N., and Shapiro, G. I. (2019). MicroRNA-mediated suppression of the TGF-beta pathway confers transmissible and reversible CDK4/6 inhibitor resistance. Cell Rep. 26 (10), 2667–2680.e7. doi:10.1016/j.celrep.2019.02.023
Costa, C., Wang, Y., Ly, A., Hosono, Y., Murchie, E., and Walmsley, C. S. (2020). PTEN loss mediates clinical cross-resistance to CDK4/6 and PI3Kalpha inhibitors in breast cancer. Canc. Discov. 10 (1), 72–85. doi:10.1158/2159-8290.CD-18-0830
Creighton, C. J. (2012). The molecular profile of luminal B breast cancer. Biologics 6, 289–297. doi:10.2147/BTT.S29923
Cristofanilli, M., Turner, N. C., Bondarenko, I., Ro, J., Im, S. A., and Masuda, N. (2016). Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 17 (4), 425–439. doi:10.1016/S1470-2045(15)00613-0
Cui, Z., Zhang, Y., Xia, K., Yan, Q., Kong, H., and Zhang, J. (2018). Nanodiamond autophagy inhibitor allosterically improves the arsenical-based therapy of solid tumors. Nat. Commun. 9 (1), 4347. doi:10.1038/s41467-018-06749-2
Dean, J. L., McClendon, A. K., Hickey, T. E., Butler, L. M., Tilley, W. D., and Witkiewicz, A. K. (2012). Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell Cycle 11 (14), 2756–2761. doi:10.4161/cc.21195
DeMichele, A., Clark, A. S., Tan, K. S., Heitjan, D. F., Gramlich, K., and Gallagher, M. (2015). CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: phase II activity, safety, and predictive biomarker assessment. Clin. Canc. Res. 21 (5), 995–1001. doi:10.1158/1078-0432.CCR-14-2258
Deng, J., Wang, E. S., Jenkins, R. W., Li, S., Dries, R., and Yates, K. (2018). CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Canc. Discov. 8 (2), 216–233. doi:10.1158/2159-8290.Cd-17-0915
Dey, A., Varelas, X., and Guan, K. L. (2020). Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 19 (7), 480–494. doi:10.1038/s41573-020-0070-z
Dickler, M. N., Tolaney, S. M., Rugo, H. S., Cortes, J., Dieras, V., and Patt, D. (2017). MONARCH 1, A phase II study of abemaciclib, a CDK4 and CDK6 inhibitor, as a single agent, in patients with refractory HR(+)/HER2(−) metastatic breast cancer. Clin. Canc. Res. 23 (17), 5218–5224. doi:10.1158/1078-0432.CCR-17-0754
Du, C., Li, Z., Wang, S., Zhou, Z., Wang, J., and Dong, J. (2014). Tongshu capsule down-regulates the expression of estrogen receptor alpha and suppresses human breast cancer cell proliferation. PLoS One 9 (8), e104261. doi:10.1371/journal.pone.0104261
Elvin, J. A., Gay, L. M., Ort, R., Shuluk, J., Long, J., and Shelley, L. (2017). Clinical benefit in response to palbociclib treatment in refractory uterine leiomyosarcomas with a common CDKN2A alteration. Oncologist 22 (4), 416–421. doi:10.1634/theoncologist.2016-0310
Filmus, J., Robles, A. I., Shi, W., Wong, M. J., Colombo, L. L., and Conti, C. J. (1994). Induction of cyclin D1 overexpression by activated ras. Oncogene 9 (12), 3627–3633.
Finn, R. S., Crown, J. P., Lang, I., Boer, K., Bondarenko, I. M., and Kulyk, S. O. (2015). The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 16 (1), 25–35. doi:10.1016/S1470-2045(14)71159-3
Finn, R. S., Martin, M., Rugo, H. S., Jones, S., Im, S. A., and Gelmon, K. (2016). Palbociclib and letrozole in advanced breast cancer. N. Engl. J. Med. 375 (20), 1925–1936. doi:10.1056/NEJMoa1607303
Formisano, L., Stauffer, K. M., Young, C. D., Bhola, N. E., Guerrero-Zotano, A. L., and Jansen, V. M. (2017). Association of FGFR1 with ERalpha maintains ligand-independent ER transcription and mediates resistance to estrogen deprivation in ER(+) breast cancer. Clin. Canc. Res. 23 (20), 6138–6150. doi:10.1158/1078-0432.CCR-17-1232
Formisano, L., Lu, Y., Servetto, A., Hanker, A. B., Jansen, V. M., and Bauer, J. A. (2019). Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat. Commun. 10 (1), 1373. doi:10.1038/s41467-019-09068-2
Franco, J., Witkiewicz, A. K., and Knudsen, E. S. (2014). CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget 5 (15), 6512–6525. doi:10.18632/oncotarget.2270
Fribbens, C., O’Leary, B., Kilburn, L., Hrebien, S., Garcia-Murillas, I., and Beaney, M. (2016). Plasma ESR1 mutations and the treatment of estrogen receptor-positive advanced breast cancer. J. Clin. Oncol. 34 (25), 2961–2968. doi:10.1200/JCO.2016.67.3061
Fry, D. W., Harvey, P. J., Keller, P. R., Elliott, W. L., Meade, M., and Trachet, E. (2004). Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Canc. Therapeut. 3 (11), 1427–1438.
Glick, D., Barth, S., and Macleod, K. F. (2010). Autophagy: cellular and molecular mechanisms. J. Pathol. 221 (1), 3–12. doi:10.1002/path.2697
Goel, S., DeCristo, M. J., Watt, A. C., BrinJones, H., Sceneay, J., and Li, B. B. (2017). CDK4/6 inhibition triggers anti-tumour immunity. Nature 548 (7668), 471–475. doi:10.1038/nature23465
Goetz, M. P., Toi, M., Campone, M., Sohn, J., Paluch-Shimon, S., and Huober, J. (2017). Monarch 3: abemaciclib as initial therapy for advanced breast cancer. J. Clin. Oncol. 35 (32), 3638–3646. doi:10.1200/JCO.2017.75.6155
Goldhirsch, A., Wood, W. C., Coates, A. S., Gelber, R. D., Thurlimann, B., and Senn, H. J. (2011). Strategies for subtypes–Dealing with the diversity of breast cancer: highlights of the St. Gallen international expert consensus on the primary therapy of early breast cancer 2011. Ann. Oncol. 22 (8), 1736–1747. doi:10.1093/annonc/mdr304
Gong, C., Hu, C., Gu, F., Xia, Q., Yao, C., and Zhang, L. (2017a). Co-delivery of autophagy inhibitor ATG7 siRNA and docetaxel for breast cancer treatment. J. Contr. Release 266, 272–286. doi:10.1016/j.jconrel.2017.09.042
Gong, X., Litchfield, L. M., Webster, Y., Chio, L. C., Wong, S. S., and Stewart, T. R. (2017b). Genomic aberrations that activate D-type cyclins are associated with enhanced sensitivity to the CDK4 and CDK6 inhibitor abemaciclib. Canc. Cell 32 (6), 761–776.e6. doi:10.1016/j.ccell.2017.11.006
Gujral, T. S., and Kirschner, M. W. (2017). Hippo pathway mediates resistance to cytotoxic drugs. Proc. Natl. Acad. Sci. U. S. A. 114 (18), E3729–E3738. doi:10.1073/pnas.1703096114
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144 (5), 646–674. doi:10.1016/j.cell.2011.02.013
Haricharan, S., Punturi, N., Singh, P., Holloway, K. R., Anurag, M., and Schmelz, J. (2017). Loss of MutL disrupts CHK2-dependent cell-cycle control through CDK4/6 to promote intrinsic endocrine therapy resistance in primary breast cancer. Canc. Discov. 7 (10), 1168–1183. doi:10.1158/2159-8290.CD-16-1179
Herrera-Abreu, M. T., Palafox, M., Asghar, U., Rivas, M. A., Cutts, R. J., and Garcia-Murillas, I. (2016). Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Canc. Res. 76 (8), 2301–2313. doi:10.1158/0008-5472.CAN-15-0728
Hortobagyi, G. N., Stemmer, S. M., Burris, H. A., Yap, Y. S., Sonke, G. S., and Paluch-Shimon, S. (2016). Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N. Engl. J. Med. 375 (18), 1738–1748. doi:10.1056/NEJMoa1609709
Hortobagyi, G. N., Stemmer, S. M., Burris, H. A., Yap, Y. S., Sonke, G. S., and Paluch-Shimon, S. (2018). Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2-negative advanced breast cancer. Ann. Oncol. 29 (7), 1541–1547. doi:10.1093/annonc/mdy155
Hunt, K. K., Karakas, C., Ha, M. J., Biernacka, A., Yi, M., and Sahin, A. A. (2017). Cytoplasmic cyclin E predicts recurrence in patients with breast cancer. Clin. Canc. Res. 23 (12), 2991–3002. doi:10.1158/1078-0432.CCR-16-2217
Ignatiadis, M., Singhal, S. K., Desmedt, C., Haibe-Kains, B., Criscitiello, C., and Andre, F. (2012). Gene modules and response to neoadjuvant chemotherapy in breast cancer subtypes: a pooled analysis. J. Clin. Oncol. 30 (16), 1996–2004. doi:10.1200/JCO.2011.39.5624
Ignatiadis, M., and Sotiriou, C. (2013). Luminal breast cancer: from biology to treatment. Nat. Rev. Clin. Oncol. 10 (9), 494–506. doi:10.1038/nrclinonc.2013.124
Infante, J. R., Cassier, P. A., Gerecitano, J. F., Witteveen, P. O., Chugh, R., and Ribrag, V. (2016). A phase I study of the cyclin-dependent kinase 4/6 inhibitor ribociclib (LEE011) in patients with advanced solid tumors and lymphomas. Clin. Canc. Res. 22 (23), 5696–5705. doi:10.1158/1078-0432.CCR-16-1248
Jansen, V. M., Bhola, N. E., Bauer, J. A., Formisano, L., Lee, K. M., and Hutchinson, K. E. (2017). Kinome-wide RNA interference screen reveals a role for PDK1 in acquired resistance to CDK4/6 inhibition in ER-positive breast cancer. Canc. Res. 77 (9), 2488–2499. doi:10.1158/0008-5472.CAN-16-2653
Jiang, Z., Hu, X., Zhang, Q., Sun, T., Yin, Y., and Li, H. (2019a). MONARCHplus: a phase III trial of abemaciclib plus nonsteroidal aromatase inhibitor (NSAI) or fulvestrant (F) for women with HR+/HER2− advanced breast cancer (ABC). Ann. Oncol. 30, v863. doi:10.1093/annonc/mdz394.014
Jiang, Z., Li, W., Hu, X., Zhang, Q., Sun, T., and Cui, S. (2019b). Tucidinostat plus exemestane for postmenopausal patients with advanced, hormone receptor-positive breast cancer (ACE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 20 (6), 806–815. doi:10.1016/S1470-2045(19)30164-0.
Jin, X., Ge, L. P., Li, D. Q., Shao, Z. M., Di, G. H., and Xu, X. E. (2020). LncRNA TROJAN promotes proliferation and resistance to CDK4/6 inhibitor via CDK2 transcriptional activation in ER+ breast cancer. Mol. Canc. 19 (1), 87. doi:10.1186/s12943-020-01210-9
Kalra, S., Joshi, G., Munshi, A., and Kumar, R. (2017). Structural insights of cyclin dependent kinases: implications in design of selective inhibitors. Eur. J. Med. Chem. 142, 424–458. doi:10.1016/j.ejmech.2017.08.071
Karakas, C., Biernacka, A., Bui, T., Sahin, A. A., Yi, M., and Akli, S. (2016). Cytoplasmic cyclin E and phospho-cyclin-dependent kinase 2 are biomarkers of aggressive breast cancer. Am. J. Pathol. 186 (7), 1900–1912. doi:10.1016/j.ajpath.2016.02.024
Kok, M., Voorwerk, L., Horlings, H., Sikorska, K., Vijver, K. v. d., and Slagter, M. (2018). Adaptive phase II randomized trial of nivolumab after induction treatment in triple negative breast cancer (TONIC trial): final response data stage I and first translational data. J. Clin. Oncol. 36 (Suppl. 15), 1012. doi:10.1200/JCO.2018.36.15_suppl.1012
Konecny, G. E., Winterhoff, B., Kolarova, T., Qi, J., Manivong, K., and Dering, J. (2011). Expression of p16 and retinoblastoma determines response to CDK4/6 inhibition in ovarian cancer. Clin. Canc. Res. 17 (6), 1591–1602. doi:10.1158/1078-0432.CCR-10-2307
Lang, L., Hou, Y., Chen, Y., Tu, G., Tao, J., and Yang, D. (2018). ATM-mediated phosphorylation of cortactin involved in actin polymerization promotes breast cancer cells migration and invasion. Cell. Physiol. Biochem. 51 (6), 2972–2988. doi:10.1159/000496048
Lee, J., Lim, B., Pearson, T., Tripathy, D., Ordentlich, P., and Ueno, N. (2018). Abstract P5-21-15: the synergistic antitumor activity of entinostat (MS-275) in combination with palbociclib (PD 0332991) in estrogen receptor-positive and triple-negative breast cancer. Canc. Res. 78 (Suppl. 4), 893–905.e8. doi:10.1158/1538-7445.Sabcs17-p5-21-15
Li, Z., Razavi, P., Li, Q., Toy, W., Liu, B., and Ping, C. (2018). Loss of the FAT1 tumor suppressor promotes resistance to CDK4/6 inhibitors via the Hippo pathway. Canc. Cell 34 (6), 893–905.e8. doi:10.1016/j.ccell.2018.11.006
Liang, D. H., Choi, D. S., Ensor, J. E., Kaipparettu, B. A., Bass, B. L., and Chang, J. C. (2016). The autophagy inhibitor chloroquine targets cancer stem cells in triple negative breast cancer by inducing mitochondrial damage and impairing DNA break repair. Canc. Lett. 376 (2), 249–258. doi:10.1016/j.canlet.2016.04.002
Liu-Chittenden, Y., Huang, B., Shim, J. S., Chen, Q., Lee, S. J., and Anders, R. A. (2012). Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 26 (12), 1300–1305. doi:10.1101/gad.192856.112
Malorni, L., Piazza, S., Ciani, Y., Guarducci, C., Bonechi, M., and Biagioni, C. (2016). A gene expression signature of retinoblastoma loss-of-function is a predictive biomarker of resistance to palbociclib in breast cancer cell lines and is prognostic in patients with ER positive early breast cancer. Oncotarget 7 (42), 68012–68022. doi:10.18632/oncotarget.12010
Malvezzi, M., Bertuccio, P., Levi, F., La Vecchia, C., and Negri, E. (2013). European cancer mortality predictions for the year 2013. Ann. Oncol. 24 (3), 792–800. doi:10.1093/annonc/mdt010
Martin, L.-A., Pancholi, S., Ribas, R., Gao, Q., Simigdala, N., and Nikitorowicz-Buniak, J. (2017). Abstract P3-03-09: resistance to palbociclib depends on multiple targetable mechanisms highlighting the potential of drug holidays and drug switching to improve therapeutic outcome. Canc. Res. 77 (Suppl. 4), P3-03-09. doi:10.1158/1538-7445.Sabcs16-p3-03-09
Martinez-Carreres, L., Puyal, J., Leal-Esteban, L. C., Orpinell, M., Castillo-Armengol, J., and Giralt, A. (2019). CDK4 regulates lysosomal function and mTORC1 activation to promote cancer cell survival. Canc. Res. 79 (20), 5245–5259. doi:10.1158/0008-5472.CAN-19-0708
Medema, R. H., Herrera, R. E., Lam, F., and Weinberg, R. A. (1995). Growth suppression by p16ink4 requires functional retinoblastoma protein. Proc. Natl. Acad. Sci. U.S.A. 92 (14), 6289–6293. doi:10.1073/pnas.92.14.6289
Michaloglou, C., Crafter, C., Siersbaek, R., Delpuech, O., Curwen, J. O., and Carnevalli, L. S. (2018). Combined inhibition of mTOR and CDK4/6 is required for optimal blockade of E2F function and long-term growth inhibition in estrogen receptor-positive breast cancer. Mol. Canc. Therapeut. 17 (5), 908–920. doi:10.1158/1535-7163.MCT-17-0537
Michaud, K., Solomon, D. A., Oermann, E., Kim, J. S., Zhong, W. Z., and Prados, M. D. (2010). Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 70 (8), 3228–3238. doi:10.1158/0008-5472.CAN-09-4559
Miller, T. W., Rexer, B. N., Garrett, J. T., and Arteaga, C. L. (2011). Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 13 (6), 224. doi:10.1186/bcr3039
Minton, K. (2017). Tumour immunology: cell cycle inhibitors boost tumour immunogenicity. Nat. Rev. Immunol. 17 (9), 529. doi:10.1038/nri.2017.104
Muranen, T., Selfors, L. M., Hwang, J., Gallegos, L. L., Coloff, J. L., and Thoreen, C. C. (2016). ERK and p38 MAPK activities determine sensitivity to PI3K/mTOR inhibition via regulation of MYC and YAP. Canc. Res. 76 (24), 7168–7180. doi:10.1158/0008-5472.CAN-16-0155
Musgrove, E. A., Caldon, C. E., Barraclough, J., Stone, A., and Sutherland, R. L. (2011). Cyclin D as a therapeutic target in cancer. Nat. Rev. Canc. 11 (8), 558–572. doi:10.1038/nrc3090
Pandey, K., An, H. J., Kim, S. K., Lee, S. A., Kim, S., and Lim, S. M. (2019). Molecular mechanisms of resistance to CDK4/6 inhibitors in breast cancer: a review. Int. J. Canc. 145 (5), 1179–1188. doi:10.1002/ijc.32020
Parry, D., Guzi, T., Shanahan, F., Davis, N., Prabhavalkar, D., and Wiswell, D. (2010). Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol. Canc. Therapeut. 9 (8), 2344–2353. doi:10.1158/1535-7163.MCT-10-0324
Patnaik, A., Rosen, L. S., Tolaney, S. M., Tolcher, A. W., Goldman, J. W., and Gandhi, L. (2016). Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non-small cell lung cancer, and other solid tumors. Canc. Discov. 6 (7), 740–753. doi:10.1158/2159-8290.CD-16-0095
Portman, N., Alexandrou, S., Carson, E., Wang, S., Lim, E., and Caldon, C. E. (2019). Overcoming CDK4/6 inhibitor resistance in ER-positive breast cancer. Endocr. Relat. Canc. 26 (1), R15–R30. doi:10.1530/ERC-18-0317.
Pronzato, P. (2017). Role of everolimus in the treatment of metastatic HER2-negative/HR-positive breast cancer. Future Oncol. 13 (16), 1371–1384. doi:10.2217/fon-2017-0098
Raspe, E., Coulonval, K., Pita, J. M., Paternot, S., Rothe, F., and Twyffels, L. (2017). CDK4 phosphorylation status and a linked gene expression profile predict sensitivity to palbociclib. EMBO Mol. Med. 9 (8), 1052–1066. doi:10.15252/emmm.201607084
Ribnikar, D., Volovat, S. R., and Cardoso, F. (2019). Targeting CDK4/6 pathways and beyond in breast cancer. Breast 43, 8–17. doi:10.1016/j.breast.2018.10.001
Robertson, F. M., Chu, K., Boley, K. M., Ye, Z., Liu, H., and Wright, M. C. (2013). The class I HDAC inhibitor Romidepsin targets inflammatory breast cancer tumor emboli and synergizes with paclitaxel to inhibit metastasis. J. Exp. Therapeut. Oncol. 10 (3), 219–233.
Rouzier, R., Perou, C. M., Symmans, W. F., Ibrahim, N., Cristofanilli, M., and Anderson, K. (2005). Breast cancer molecular subtypes respond differently to preoperative chemotherapy. Clin. Canc. Res. 11 (16), 5678–5685. doi:10.1158/1078-0432.CCR-04-2421
Sahores, A., May, M., Sequeira, G. R., Fuentes, C., Jacobsen, B., and Lanari, C. (2018). Targeting FGFR with BGJ398 in breast cancer: effect on tumor growth and metastasis. Curr. Cancer Drug Targets 18 (10), 979–987. doi:10.2174/1568009618666171214114706
Schaer, D. A., Beckmann, R. P., Dempsey, J. A., Huber, L., Forest, A., and Amaladas, N. (2018). The CDK4/6 inhibitor abemaciclib induces a T cell inflamed tumor microenvironment and enhances the efficacy of PD-L1 checkpoint blockade. Cell Rep. 22 (11), 2978–2994. doi:10.1016/j.celrep.2018.02.053
Shapiro, G. I. (2006). Cyclin-dependent kinase pathways as targets for cancer treatment. J. Clin. Oncol. 24 (11), 1770–1783. doi:10.1200/JCO.2005.03.7689
Schmid, P., Cortés, J., Dent, R., Pusztai, L., McArthur, H. L., and Kuemmel, S. (2019). LBA8_PR–KEYNOTE-522: phase III study of pembrolizumab (pembro) + chemotherapy (chemo) vs placebo (pbo) + chemo as neoadjuvant treatment, followed by pembro vs pbo as adjuvant treatment for early triple-negative breast cancer (TNBC). Ann. Oncol. 30, v851–v934. /doi:10.1093/annonc/mdz394.003
Schneeweiss, A., Rugo, H. S., Winer, E. P., Barrios, C. H., Iwata, H., and Dieras, V. (2019). IMpassion130: expanded safety analysis from a P3 study of atezolizumab (A) + nab-paclitaxel (nP) in patients (pts) with treatment (tx)-naïve, locally advanced or metastatic triple-negative breast cancer (mTNBC). J. Clin. Oncol. 37 (Suppl. 15), 1068. doi:10.1200/JCO.2019.37.15_suppl.1068
Serrano, M., Hannon, G. J., and Beach, D. (1993). A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 366 (6456), 704–707. doi:10.1038/366704a0
Shi, P., Feng, J., and Chen, C. (2015). Hippo pathway in mammary gland development and breast cancer. Acta Biochim. Biophys. Sin. 47 (1), 53–59. doi:10.1093/abbs/gmu114
Slamon, D. J., Neven, P., Chia, S., Fasching, P. A., De Laurentiis, M., and Im, S. A. (2018). Phase III randomized study of ribociclib and fulvestrant in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: MONALEESA-3. J. Clin. Oncol. 36 (24), 2465–2472. doi:10.1200/JCO.2018.78.9909
Slamon, D. J., Neven, P., Chia, S., Fasching, P. A., De Laurentiis, M., and Im, S.-A. (2019). Overall survival with ribociclib plus fulvestrant in advanced breast cancer. N. Engl. J. Med. 382 (6), 514–524. doi:10.1056/NEJMoa1911149
Sledge, G. W., Toi, M., Neven, P., Sohn, J., Inoue, K., and Pivot, X. (2017). MONARCH 2: abemaciclib in combination with fulvestrant in women with HR+/HER2− advanced breast cancer who had progressed while receiving endocrine therapy. J. Clin. Oncol. 35 (25), 2875–2884. doi:10.1200/JCO.2017.73.7585
Smith, S. A., Sessions, R. B., Shoemark, D. K., Williams, C., Ebrahimighaei, R., and McNeill, M. C. (2019). Antiproliferative and antimigratory effects of a novel YAP–TEAD interaction inhibitor identified using in silico molecular docking. J. Med. Chem. 62 (3), 1291–1305. doi:10.1021/acs.jmedchem.8b01402
Song, S., Xie, M., Scott, A. W., Jin, J., Ma, L., and Dong, X. (2018). A novel YAP1 inhibitor targets CSC-enriched radiation-resistant cells and exerts strong antitumor activity in esophageal adenocarcinoma. Mol. Canc. Therapeut. 17 (2), 443–454. doi:10.1158/1535-7163.MCT-17-0560
Sotiriou, C., and Pusztai, L. (2009). Gene-expression signatures in breast cancer. N. Engl. J. Med. 360 (8), 790–800. doi:10.1056/NEJMra0801289
Squires, M. S., Feltell, R. E., Wallis, N. G., Lewis, E. J., Smith, D. M., and Cross, D. M. (2009). Biological characterization of AT7519, a small-molecule inhibitor of cyclin-dependent kinases, in human tumor cell lines. Mol. Canc. Therapeut. 8 (2), 324–332. doi:10.1158/1535-7163.MCT-08-0890
Tadesse, S., Bantie, L., Tomusange, K., Yu, M., Islam, S., and Bykovska, N. (2018). Discovery and pharmacological characterization of a novel series of highly selective inhibitors of cyclin-dependent kinases 4 and 6 as anticancer agents. Br. J. Pharmacol. 175 (12), 2399–2413. doi:10.1111/bph.13974
Takeshita, T., Yamamoto, Y., Yamamoto-Ibusuki, M., Tomiguchi, M., Sueta, A., and Murakami, K. (2018). Clinical significance of plasma cell-free DNA mutations in PIK3CA, AKT1, and ESR1 gene according to treatment lines in ER-positive breast cancer. Mol. Canc. 17 (1), 67. doi:10.1186/s12943-018-0808-y
Tan, A. R., Yang, X., Berman, A., Zhai, S., Sparreboom, A., and Parr, A. L. (2004). Phase I trial of the cyclin-dependent kinase inhibitor flavopiridol in combination with docetaxel in patients with metastatic breast cancer. Clin. Canc. Res. 10 (15), 5038–5047. doi:10.1158/1078-0432.CCR-04-0025
Taylor-Harding, B., Aspuria, P. J., Agadjanian, H., Cheon, D. J., Mizuno, T., and Greenberg, D. (2015). Cyclin E1 and RTK/RAS signaling drive CDK inhibitor resistance via activation of E2F and ETS. Oncotarget 6 (2), 696–714. doi:10.18632/oncotarget.2673
Teh, J. L. F., and Aplin, A. E. (2019). Arrested developments: CDK4/6 inhibitor resistance and alterations in the tumor immune microenvironment. Clin. Canc. Res. 25 (3), 921–927. doi:10.1158/1078-0432.CCR-18-1967
Tolaney, S. M., Wardley, A. M., Zambelli, S., Hilton, J., Troso-Sandoval, T., and Ricci, F. (2019). MonarcHER: a randomized phase II study of abemaciclib plus trastuzumab with or without fulvestrant versus trastuzumab plus standard-of-care chemotherapy in women with HR+, HER2+ advanced breast cancer (ABC). Ann. Oncol. 30, v861–v862. doi:10.1093/annonc/mdz394.012
Tripathy, D., Im, S. A., Colleoni, M., Franke, F., Bardia, A., and Harbeck, N. (2018). Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): a randomised phase 3 trial. Lancet Oncol. 19 (7), 904–915. doi:10.1016/S1470-2045(18)30292-4
Turner, N., Pearson, A., Sharpe, R., Lambros, M., Geyer, F., and Lopez-Garcia, M. A. (2010). FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Canc. Res. 70 (5), 2085–2094. doi:10.1158/0008-5472.CAN-09-3746
Turner, N. C., Neven, P., Loibl, S., and Andre, F. (2017). Advances in the treatment of advanced oestrogen-receptor-positive breast cancer. Lancet 389 (10087), 2403–2414. doi:10.1016/S0140-6736(16)32419-9
Turner, N. C., Liu, Y., Zhu, Z., Loi, S., Colleoni, M., and Loibl, S. (2019). Cyclin E1 expression and palbociclib efficacy in previously treated hormone receptor-positive metastatic breast cancer. J. Clin. Oncol. 37 (14), 1169–1178. doi:10.1200/JCO.18.00925
Vijayaraghavan, S., Doostan, I., Carey, J. P. W., and Keyomarsi, K. (2017a). Abstract 2060: characterizing acquired resistance to palbociclib in breast cancer. Canc. Res. 77 (Suppl. 13), 2060. doi:10.1158/1538-7445.Am2017-2060
Vijayaraghavan, S., Karakas, C., Doostan, I., Chen, X., Bui, T., and Yi, M. (2017b). CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat. Commun. 8, 15916. doi:10.1038/ncomms15916
Vora, S. R., Juric, D., Kim, N., Mino-Kenudson, M., Huynh, T., and Costa, C. (2014). CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Canc. Cell 26 (1), 136–149. doi:10.1016/j.ccr.2014.05.020
Wein, L., Savas, P., Van Geelen, C., Caramia, F., Moodie, K., and Joshi, S. (2017). FGFR2 amplification in metastatic hormone-positive breast cancer and response to an mTOR inhibitor. Ann. Oncol. 28 (8), 2025–2027. doi:10.1093/annonc/mdx194
Witkiewicz, A. K., Knudsen, K. E., Dicker, A. P., and Knudsen, E. S. (2011). The meaning of p16(ink4a) expression in tumors: functional significance, clinical associations and future developments. Cell Cycle 10 (15), 2497–2503. doi:10.4161/cc.10.15.16776
Wu, X. Y., Xu, H., Wu, Z. F., Chen, C., Liu, J. Y., and Wu, G. N. (2015). Formononetin, a novel FGFR2 inhibitor, potently inhibits angiogenesis and tumor growth in preclinical models. Oncotarget 6 (42), 44563–44578. doi:10.18632/oncotarget.6310
Xie, Q., Chen, J., Feng, H., Peng, S., Adams, U., and Bai, Y. (2013). YAP/TEAD-mediated transcription controls cellular senescence. Canc. Res. 73 (12), 3615–3624. doi:10.1158/0008-5472.CAN-12-3793
Yang, C., Li, Z., Bhatt, T., Dickler, M., Giri, D., and Scaltriti, M. (2017). Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 36 (16), 2255–2264. doi:10.1038/onc.2016.379
Yin, L., Li, H., Liu, W., Yao, Z., Cheng, Z., and Zhang, H. (2018). A highly potent CDK4/6 inhibitor was rationally designed to overcome blood brain barrier in gliobastoma therapy. Eur. J. Med. Chem. 144, 1–28. doi:10.1016/j.ejmech.2017.12.003
Yoshida, A., Lee, E. K., and Diehl, J. A. (2016). Induction of therapeutic senescence in vemurafenib-resistant melanoma by extended inhibition of CDK4/6. Cancer Res. 76 (10), 2990–3002. doi:10.1158/0008-5472.CAN-15-2931
Keywords: luminal breast cancer, endocrine resistance, upstream response signaling, downstream bypass signaling, CDK4/6 inhibitor
Citation: Li Z, Zou W, Zhang J, Zhang Y, Xu Q, Li S and Chen C (2020) Mechanisms of CDK4/6 Inhibitor Resistance in Luminal Breast Cancer. Front. Pharmacol. 11:580251. doi: 10.3389/fphar.2020.580251
Received: 05 July 2020; Accepted: 30 September 2020;
Published: 16 November 2020.
Edited by:
Daotai Nie, Southern Illinois University Carbondale, United StatesReviewed by:
Lianjun Zhang, Chinese Academy of Medical Sciences, ChinaCopyright © 2020 Li, Zou, Zhang, Zhang, Xu, Li and Chen . This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ceshi Chen Y2hlbmNAbWFpbC5raXouYWMuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.