 Lucy Perrem1,2,3*
Lucy Perrem1,2,3* Felix Ratjen1,2,3
Felix Ratjen1,2,3- 1Division of Respiratory Medicine, The Hospital for Sick Children, Toronto, ON, Canada
- 2Department of Paediatrics, University of Toronto, Toronto, ON, Canada
- 3Translational Medicine Program, SickKids Research Institute, Toronto, ON, Canada
The inflammatory response in the CF airway begins early in the disease process and becomes persistent through life in most patients. Inflammation, which is predominantly neutrophilic, worsens airway obstruction and plays a critical role in the development of structural lung damage. While cystic fibrosis transmembrane regulator modulators will likely have a dramatic impact on the trajectory of CF lung disease over the coming years, addressing other important aspects of lung disease such as inflammation will nevertheless remain a priority. Considering the central role of neutrophils and their products in the inflammatory response, potential therapies should ultimately affect neutrophils and their products. The ideal anti-inflammatory therapy would exert a dual effect on the pro-inflammatory and pro-resolution arms of the inflammatory cascade, both of which contribute to dysregulated inflammation in CF. This review outlines the key factors to be considered in the design of clinical trials evaluating anti-inflammatory therapies in CF. Important lessons have been learned from previous clinical trials in this area and choosing the right efficacy endpoints is key to the success of any anti-inflammatory drug development program. Identifying and validating non-invasive biomarkers, novel imaging techniques and sensitive lung function tests capable of monitoring disease activity and therapeutic response are important areas of research and will be useful for the design of future anti-inflammatory drug trials.
Introduction
Airway inflammation plays a critical role in the development of bronchiectasis in cystic fibrosis (CF) and contributes to the progressive decline in lung function (Pillarisetti et al., 2011; Sagel et al., 2012; Sly et al., 2013; Nichols and Chmiel, 2015). Inflammation typically beings early in the disease process and becomes persistent in most patients (Pillarisetti et al., 2011; Sagel et al., 2012; Sly et al., 2013; Nichols and Chmiel, 2015).
The inflammatory process in CF is predominantly driven by neutrophils (Cohen-Cymberknoh et al., 2013). Neutrophils are recruited into the airway in response to infection and then kill bacteria by releasing their contents including peroxidases and proteases (Watt et al., 2005). A key feature of inflammation in CF is that the response is excessive relative to the burden of infection due to a combination of increased influx and decreased clearance of pathogens and inflammatory cells (Nichols and Chmiel, 2015). Normally, once the intracellular contents are released, neutrophils undergo apoptosis by alveolar macrophages. However, in the CF airway, neutrophils undergo necrosis rather than apoptosis and this results in the release of damaging intracellular contents and chemoattractants and fuels further neutrophil influx (Watt et al., 2005; Cohen-Cymberknoh et al., 2016). The release of massive quantities of neutrophil elastase overwhelms the endogenous anti-proteases, such as anti-1 antitrypsin, needed to neutralize proteolytic activity, and this ultimately leads to the destruction of lung tissue (Birrer et al., 1994). The actions of neutrophil elastase also work to make the neutrophils in CF airways less effective at killing bacteria, partly due to cleavage of immunoglobulins and complement (McElvaney, 2016). This becomes a vicious cycle of neutrophilic inflammation, protease release, and oxidative stress which leads to tissue destruction and fibrosis of the lungs (Cohen-Cymberknoh et al., 2013).
Cystic fibrosis transmembrane regulator (CFTR) modulators are a new class of therapy that targets the basic genetic defect and could dramatically change the landscape of CF care. These drugs are potentially efficacious for approximately 90% of the CF population (Bell et al., 2020). However, despite the impressive improvements in lung function observed with ivacaftor in CF patients with gating mutations (Whiting et al., 2014), there is limited evidence that CFTR modulators impact inflammation. In a multicenter prospective cohort study in the post-approval setting, Rowe and co-workers found that patients on ivacaftor experienced significant improvements in forced expiratory volume in 1 second (FEV1) and reductions in sweat chloride, but there were no significant changes in any sputum inflammatory markers (Rowe et al., 2014). In contrast, Hisert and co-workers evaluated the impact of ivacaftor on airway inflammation over a longer period of time and reported a significant reduction in sputum inflammatory markers including neutrophil elastase, IL-8 and IL-1b (Rowe et al., 2014). However, even in this study, most patients had ongoing chronic infection and inflammation, albeit at a lower level. A large observational study designed to evaluate the effects of the triple combination CFTR modulator (elexacaftor/tezacaftor/ivacaftor) may be able to more definitively establish how this class of drug impacts airway inflammation (Block et al., 2006).
Considering that the clinical response to CFTR modulators is variable, they are not universally available and, if bronchiectasis is already established, chronic infection and inflammation persist and lung function continues to decline (Sawicki et al., 2015), developing new therapies that target other aspects of CF lung disease remains a priority (Perrem and Ratjen, 2019). The purpose of this review is to outline key aspects to be considered in the design of clinical trials evaluating the efficacy of anti-inflammatory therapies.
What Is the Ideal Inflammatory Pathway to Target?
The complexity of the inflammatory process in CF provides multiple potential targets for intervention. Due to the central role of neutrophils in the inflammatory process, effective anti-inflammatory therapies must target neutrophils or their products (Torphy et al., 2015).
The CF airway contains a broad spectrum of pro-inflammatory mediators, such as TNF-alpha, IL-1beta, IL-6, IL-8, IL-17, IL-33. GM-CSF, G-CSF, and HMGB-1 (Nichols and Chmiel, 2015). The downregulation of inflammation is also defective, with many studies demonstrating a deficiency in counter-regulatory molecules such as IL-10 and lipoxin-A4 (LXA4). The multiplicity of inflammatory pathways and inherent redundancy in the process makes it challenging to target specific components, both the pro-inflammatory and pro-resolving pathways offer potential targets for therapeutic interventions.
Longitudinal data from the Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST CF) surveillance program demonstrated that free neutrophil elastase activity in bronchoalveolar lavage (BAL) fluid at 3 months of age was associated with bronchiectasis on computed tomography (CT) scan at 1 and 3 years of age (Sly et al., 2013). While it is unclear whether targeting a specific mediator of inflammation will be successful, given the intense burden of neutrophil elastase in CF and the predictive value of this biomarker, neutrophil elastase is a key target for anti-inflammatory therapy. Studies investigating alpha-1 antitrypsin therapy as a way to restore the protease-antiprotease balance in the CF airway have shown an improvement in neutrophil function, a reduction in inflammation, and augmentation on bacterial clearance (McElvaney et al., 1991; Birrer et al., 1994; Griese et al., 2007; McElvaney, 2016). This provides proof of concept that this approach could be successful in future clinical trials.
Lessons Learned From Previous Trials
Previous studies investigating anti-inflammatory therapies have demonstrated the importance of choosing the right target and the right dose of active drug.
The major concern in undertaking trials with anti-inflammatory agents in CF is the potential that suppressing the inflammatory response in a chronically infected airway will impair host defense and exacerbate infections (Torphy et al., 2015). We learned these lessons from a large phase II clinical trial of an LTB4 receptor antagonist (BIIL 284 BS). The CF inflammatory response is mediated in part by LTB4, a key modulator of inflammation that attracts and activates neutrophils in the airway. Preclinical studies suggested that LTB4-receptor inhibition could have positive clinical effects (Konstan et al., 1993) but the phase II BIL 284BS trial was terminated early because of increased pulmonary exacerbations in the adults treated with the study drug, combined with evidence of decreased pulmonary function and increased circulating neutrophils (Konstan et al., 2014). To understand the mechanisms underlying these negative results, subsequent studies in mice found that lower doses of the drug attenuated the inflammatory response without increasing infection, but high doses, like the dose used in the clinical trial, overly suppressed the inflammatory response and were associated with increased bacterial colony counts (Döring et al., 2014). Therefore, future trials evaluating anti-inflammatory drugs in the clinical trial setting should first have generated sufficient information about the possible harms of the drug. Preclinical studies or data available from studies investigating the same drug in other patient populations are essential to ensure a drug is safe for clinical trials in CF. Table 1 describes how the study design and outcome measure will vary with the development phase when investigating anti-inflammatory therapies in cystic fibrosis.
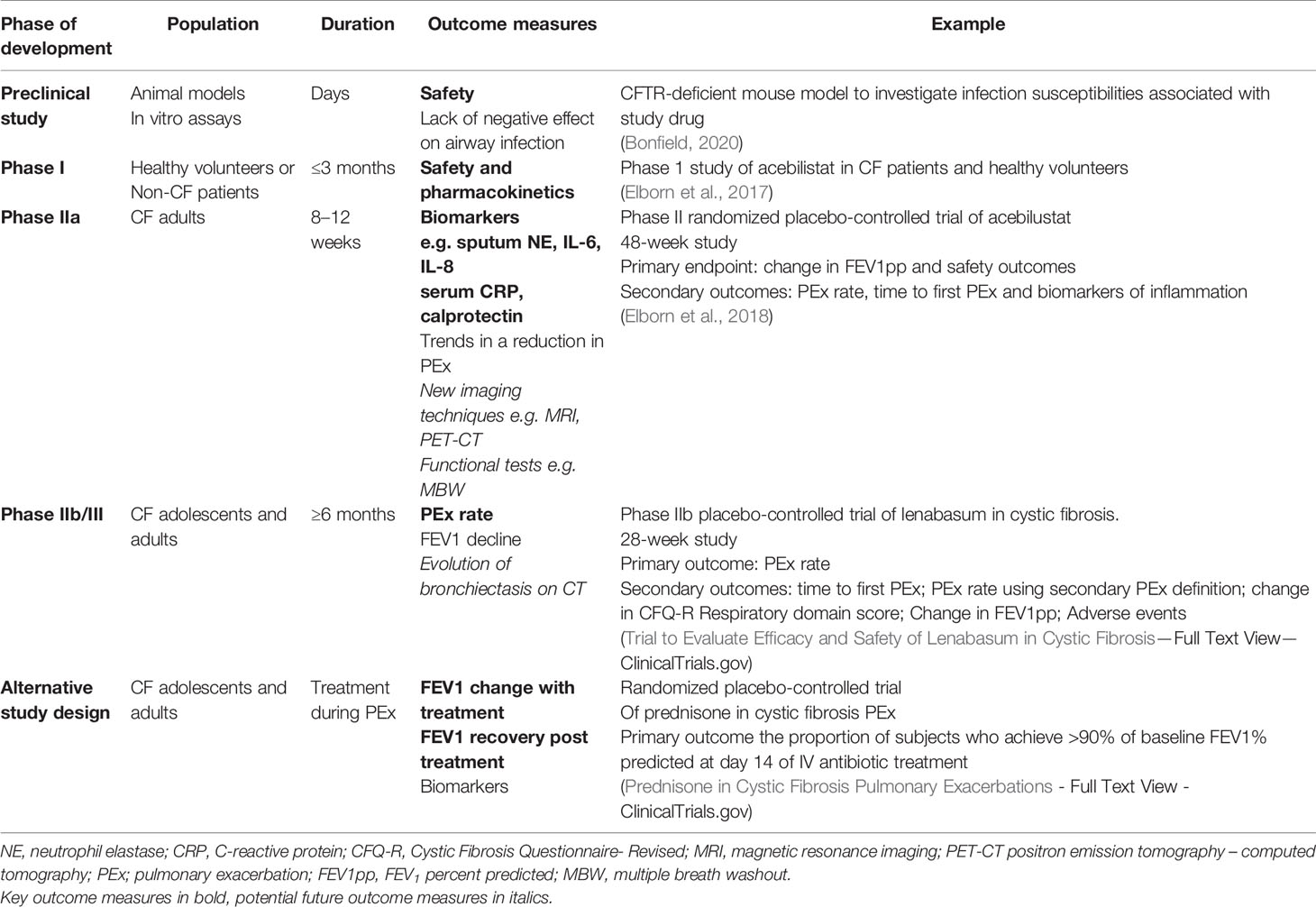
Table 1 The study design and outcome measure will vary with the development phase of an anti-inflammatory therapy.
Role of Biomarkers
Given the important role of inflammation in CF lung disease, identifying biomarkers capable of monitoring disease activity or therapeutic response would be very useful (Tiddens et al., 2015). In clinical drug development, biomarkers can be used in early phase studies to demonstrate the biological safety and efficacy of new therapies, confirm the mechanism of action and inform dose selection (Muhlebach et al., 2016). Biomarkers can also be useful to compare results from preclinical and clinical studies (Torphy et al., 2015; Muhlebach et al., 2016). The relatively poor success rate in developing new anti-inflammatory therapies can, in part, be attributed to a lack of accurate, reproducible, noninvasive biomarkers that reflect the anti-inflammatory process (Martinez et al., 2011).
Lung-Derived Inflammatory Biomarkers
Bronchoalveolar lavage (BAL) is considered the gold standard for quantifying airway inflammation in the CF (Tiddens et al., 2015). However, the use of BAL longitudinally is limited by the invasiveness of the procedure. Sputum is an alternative way of obtaining material directly from the site of inflammation. For younger subjects or those with mild lung disease who cannot expectorate, sputum induction improves sample acquisition and biomarker measurements and are reasonably comparable with expectorated sputum (Sagel et al., 2001; Zemanick et al., 2015). However, both spontaneously expectorated and induced sputum are variable, making it difficult to track patients over time (Sagel et al., 2007; Chmiel et al., 2015).
The utility of sputum biomarkers of inflammation in CF clinical trials has been previously reviewed in detail, with findings from multiple studies supporting the association between pro-inflammatory cytokines and disease status in CF subjects (Sagel et al., 2007; Tiddens et al., 2015; Muhlebach et al., 2016). A secondary analysis of data from four randomized controlled trials, which included a diverse CF population, demonstrated that free neutrophil elastase and IL-8 were negatively correlated with FEV1. Neutrophil elastase, a key mediator of lung damage, was the inflammatory marker with the strongest relationship with FEV1 (Mayer-Hamblett et al., 2007), correlating with FEV1 both cross-sectionally and longitudinally. As neutrophil elastase correlates with bronchiectasis (DeBoer et al., 2014), tracks with and is predictive of future lung function decline (Mayer-Hamblett et al., 2007; Sagel et al., 2012), relates to treatment response and predicts time to next exacerbation (Ordoñez et al., 2003; Waters et al., 2015), it is currently considered the most informative sputum biomarker to monitor CF lung disease.
Another sputum biomarker with supportive longitudinal data is high-mobility group box-1 protein (HMGB-1); this inflammatory marker predicted subsequent pulmonary exacerbations and survival during 7 years of follow-up (Liou et al., 2001).
Despite these strong observational data, it is still not clear what magnitude of biomarker change that could be considered clinically meaningful in the interventional setting. Furthermore, for biomarkers to convincingly reflect the treatment effect of an investigational drug, multiple biomarkers should improve rather than just a single one. Chmiel and colleagues designed a placebo-controlled randomized trial to investigate whether sputum biomarkers could be used to screen candidate anti-inflammatory therapies over a short period of time (Chmiel et al., 2015). Results from a screening study such as this could provide a go-no-go decision on whether to proceed with a phase 2 trial. Given than high-dose ibuprofen is the only anti-inflammatory drug recommended for use in CF (Mogayzel et al., 2013), it was chosen to test this hypothesis. Ibuprofen is a non-steroidal anti-inflammatory drug that affects the cyclo-oxygenase pathway and results in the inhibition of prostaglandin synthesis and, at high doses, is associated with a reduction in neutrophil migration into the lung (Konstan et al., 2003). However, in this proof of concept study, there was no significant change in key inflammatory markers, including neutrophil elastase, over a 28-day trial period. It is conceivable that a longer trial is required to see a decrease in inflammatory markers in a chronically infected airway; alternatively, ibuprofen may not have been the ideal drug to test the hypothesis.
Blood-Based Inflammatory Markers
Blood-based markers are relatively non-invasive, are easily standardized and can be obtained from subjects of any age and disease severity. Although the data linking blood-based inflammatory markers to clinical outcomes is less extensive than for sputum and BAL markers, systemic inflammatory biomarkers correlate with important clinical events including pulmonary exacerbations and lung function decline (Proesmans et al., 2011; Shoki et al., 2013; Reid et al., 2015; Quon et al., 2016).
A clinical trial of school-age children with CF uninfected with Pseudomonas aeruginosa investigated the responsiveness of a panel of systemic inflammatory markers to treatment with azithromycin (Ratjen et al., 2012). Azithromycin, a macrolide antibiotic, is presumed to exert its proinflammatory effect in the CF airway by reducing proinflammatory cytokine production by cells such as neutrophils, monocytes, and bronchial epithelial cells; although its precise mechanism of action remains unclear (Parnham et al., 2005). The trial by Ratjen and co-workers demonstrated that circulating neutrophil counts, C-reactive protein, serum amyloid A, and calprotectin all significantly reduced within 28-days of treatment. Furthermore, reductions in these inflammatory markers were correlated with improvements in lung function and weight gain, providing indirect evidence that these changes were associated with clinically meaningful outcomes. A secondary analysis of this study data showed that early changes in serum calprotectin levels after the first 28 days of azithromycin treatment were predictive of pulmonary exacerbation risk by day 168 (Dong et al., 2019). This demonstrates that early changes in biomarkers have the potential to predict meaningful longer-term outcomes and could be useful outcome measures in interventional trials.
Capturing the Effect on Lung Disease
Spirometry
Reduced FEV1, derived from spirometry, is strongly linked with increased morbidity and mortality and is, therefore, a key outcome measure in CF clinical studies (Kerem et al., 1992). Unlike drugs targeting other aspects of CF lung disease (e.g., CFTR modulators and mucoactive drugs) which have shown improvements in FEV1 within 14 to 28 days (Fuchs et al., 1994; Ramsey et al., 2011), trials evaluating anti-inflammatory drugs have not reported immediate effects on lung function (Perrem and Ratjen, 2019). However, failure to show short-term improvements in expiratory flows airway resistance does not necessarily predict long-term benefits in lung function decline. This was demonstrated by trials investigating ibuprofen, a non-steroidal anti-inflammatory drug that reduces neutrophil influx into the lung a at higher dose (Mogayzel et al., 2013). High dose ibuprofen does not improve FEV1, but data from two prospective clinical trials that included 226 participants, showed that the use of ibuprofen twice daily slows the decline of FEV1 (Konstan et al., 2007; Lands et al., 2007; Lands and Stanojevic, 2019). A recent observational study also demonstrated that this beneficial effect of high-dose ibuprofen translates to improved survival (Konstan et al., 2018). Ibuprofen, therefore, provides proof of concept that targeting inflammation might improve outcomes for patients with CF, but that short-term benefits in lung function may not be seen. However, this does not exclude the possibility that a more potent anti-inflammatory compound could potentially achieve this. Interventional studies using lung function decline as an endpoint would need to be conducted over multiple years with large sample sizes size and few drug development programs would be willing to take this route. This highlights the need to identify sensitive biomarkers that can more rapidly screen candidate drugs and be used as surrogate endpoints in phase II trials reducing the failure of drugs in phase III.
A post hoc analysis of data from the ibuprofen trial from Konstan et al. showed a slower rate of annual decline in lung function in the ibuprofen group in younger children than in those 13 years and older (Konstan et al., 2018). The findings were consistent for all lung function outcomes (FEV1, FVC, and FEF 25%–75%) and suggest that ibuprofen is more efficacious when used in individuals with mild CF lung disease. It is also possible that it is the trajectory of lung function and not just baseline FEV1 that influences response to therapy. Future trials should consider stratifying patients not only based on the severity of lung disease but also on the trajectory of lung function over time.
Multiple Breath Washout Test
With more CF patients categorized as early lung disease with FEV1 in the normal range, there is an urgent need for more sensitive outcome measures (Tiddens et al., 2015). The lung clearance index (LCI), derived from the multiple breath washout (MBW) test, reflects ventilation inhomogeneity with higher values indicating more severe lung disease. LCI is a reliable, valid, and responsive functional test and is now an established outcome measure in interventional trials (Kent et al., 2014). LCI also correlates with markers of systemic inflammation(Horsley et al., 2013; O’Neill et al., 2018), including CRP and calprotectin, and with the extent of airway inflammation (Ramsey et al., 2017). The LCI has been shown to detect treatment effects to medications such as hypertonic saline in trials involving both preschool (Subbarao et al., 2013; Ratjen et al., 2019) and school-age CF subjects with preserved spirometry (Amin et al., 2010; Amin et al., 2011; Davies et al., 2013; Ratjen et al., 2017), where a change in FEV1 with treatment was not detected. The published treatment effects for LCI range from less than 1 units (0.6 for hypertonic saline, 0.9 units for dornase alfa) (Amin et al., 2011; Stanojevic and Ratjen, 2016) up to 2.2 units for ivacaftor in patients with CFTR gating mutations (Davies et al., 2013). LCI has not yet been incorporated into the design of a clinical trial for an anti-inflammatory therapy but the enhanced sensitivity to detect treatment effects compared to FEV1 may potentially facilitate anti-inflammatory trials in the future.
Pulmonary Exacerbations
Pulmonary exacerbations are important clinical events in the disease process and directly contribute to the progression of lung disease (Sanders et al., 2010; Heltshe et al., 2016; Stanojevic et al., 2017; van Horck et al., 2018). Therefore, pulmonary exacerbations also serve as meaningful clinical efficacy endpoints in interventional trials. Multiple clinical trials investigating different classes of CF medications, such as CFTR modulators (Davies et al., 2013; Ratjen et al., 2018) dornase alfa (Fuchs et al., 1994), hypertonic saline (Elkins and Dentice, 2020), and tobramycin (Ramsey et al., 1999) have shown a reduction in pulmonary exacerbations compared with placebo. Given the role of inflammation in pulmonary exacerbations, targeting the inflammatory process should logically result in a reduction in pulmonary exacerbations but this has not yet been proven in phase III trials. Furthermore, given the improved overall state of health in CF, powering a study to pulmonary exacerbation endpoints, particularly in patients with mild lung disease, require large numbers of subjects, and longer follow-up times to demonstrate a treatment effect. With respect to pulmonary exacerbations, it is unclear as to what is the best definition and surrogate outcome measure — risk, frequency, or time to the next pulmonary exacerbation event. Studies using pulmonary exacerbations as an endpoint could reduce the number of patients required by limiting recruitment to individuals with a recent history of exacerbations as this has predictive value for future events (Block et al., 2006). This concept has been implemented in the design of a phase 2 study investigating lenabasum (Chmiel and Elborn); a drug that acts as a selective agonist of the cannabinoid receptor on type 2 immune cells and exerts anti-inflammatory and pro-resolution effects without suppressing the immune system (Motwani et al., 2018).
The ideal timing to initiate anti-inflammatory therapies is unclear, whether they would be most efficacious if initiated when a patient is clinically stable or during a pulmonary exacerbation when the inflammatory process is at its peak. A pilot randomized controlled trial investigating the short-term use of oral prednisone in CF patients presenting with pulmonary exacerbations demonstrated a modest improvement in lung function (Dovey et al., 2007). The PIPE study is an ongoing multisite randomized placebo-controlled trial that is investigating the efficacy of oral prednisone as an adjunctive therapy during pulmonary exacerbations. If successful, this study design could provide a model for evaluating other anti-inflammatory therapies in the future (Prednisone in Cystic Fibrosis Pulmonary Exacerbations - Full Text View - ClinicalTrials.gov).
Other Outcome Measures
Various types of imaging techniques are now available to determine the presence and extent of lung disease in patients with CF, including CT and chest magnetic resonance imaging (MRI) (Tiddens et al., 2015). Studies have demonstrated that infection, inflammation, and abnormal chest CT findings are already present in a significant proportion of asymptomatic infants with CF at 3-months of age (Sly et al., 2009) and that these early structural changes are progressive (Mott et al., 2012). On the other end of the spectrum, in CF patients screened for lung transplantation, those with a higher volume of infection/inflammation-like changes were shown to have a higher risk of dying on the waiting list (Loeve et al., 2009). Furthermore, individuals with more extensive structural lung disease on CT experience more pulmonary exacerbations (Brody et al., 2005; Loeve et al., 2011).
While chest CT scans are sensitive at detecting structural changes, the evolution of these changes over time is slow, with bronchiectasis deteriorating at about 1.5% per year on serial CT scans in CF patients(De Jong et al., 2006). Therefore, trials using the development of bronchiectasis on CT as the primary endpoint would take multiple years and/or large numbers of subjects (De Jong et al., 2006; Owens et al., 2011).
Pulmonary MRI can now provide high‐resolution images that are sensitive to early disease and specific to inflammation in cystic fibrosis (CF) lung disease (Amin and Ratjen, 2008; Tiddens et al., 2015; Ciet et al., 2017; Woods et al., 2019). Unlike CT, MRI does not use ionizing radiation, and this is particularly advantageous in children and when scans need to be repeated within a relatively short time period (Amin and Ratjen, 2008; Tiddens et al., 2015). MRI techniques can track changes in lung function longitudinally and quantify treatment response (Rayment et al., 2018; Santyr et al., 2019; Woods et al., 2019). Proton density and T1/T2 contrast images can be obtained within a single breath‐hold, providing a depiction of structural abnormalities and active inflammation. Hyperpolarized‐gas MRI, increasingly using 129Xe, is now becoming more widespread and has been demonstrated to have high sensitivity to early airway obstruction in CF and could have utility as an endpoint in future clinical trials, particularly in the acute setting of pulmonary exacerbations.
Another promising non-invasive imaging technique that provides information about the level of inflammation is positron emission tomography (PET) with [18]fluorodeoxyglucose ([18]FDG; FDG-PET). 18F-FDG is taken up by activated neutrophils, macrophages, and lymphocytes so in contrast to other imaging techniques, FDG-PET scans assess CF airway inflammation directly. Several observational studies have shown that 18F-FDG uptake can determine the location and intensity of pulmonary inflammation and, combined with CT, PET can be used to assess anatomy and structures. (Labiris et al., 2003; Chen et al., 2006; Klein et al., 2009; Amin et al., 2012). A study by Amin and co-workers, demonstrated that FDG PET/CT depicts changes in inflammation in the lung after intravenous antibiotics for a pulmonary exacerbation, with the PET signal correlating with the burden of sputum neutrophils (Amin et al., 2012). Data from this study support the utility of FDG PET/CT as an outcome measure in treatment studies, although it is not yet a validated outcome for use in interventional trials.
CFTR Modulators and Anti-Inflammatory Therapies
Time will tell how increasing CFTR modulator access will affect future anti-inflammatory therapeutic development. The positive effects of CFTR modulators on the progression of CF lung disease are undoubtedly positive but this will make the issues of endpoints for anti-inflammatory trials event more challenging.
Acebilustat is an inhibitor of leukotriene A4 hydrolase (LTA4H), an enzyme that catalyzes the rate-limiting step in the formation of leukotriene B4 (LTB4), a potent chemoattractant and activator of inflammatory immune cells including neutrophils (Rao et al., 2010). This anti-inflammatory drug is also proposed to work by shunting substrates down the metabolic pathway to produce the pro-resolving mediator LXA4 (Tobin et al., 2010). In the Acebilustat phase IIb trial, the observed reduction in pulmonary exacerbations during the 48-week study period was also observed in the subgroup of patients taking CFTR modulators (Griese et al., 2007). This suggests that patients taking modulators may receive additional benefit from anti-inflammatory drugs. It would be important for future trials to take this into account and stratify groups based on whether subjects are taking CFTR modulators.
Conclusion
Designing trials of anti-inflammatory therapies in CF faces specific challenges different from other drug development programs. Demonstrating safety data from preclinical studies, choosing appropriate and realistic efficacy endpoints and integrating sensitive imaging and lung function outcomes into future trials are important considerations. These measures will increase the likelihood that potentially efficacious therapies are not abandoned prematurely and that efficacy can be conclusively demonstrated in studies that can be completed in reasonable time frames.
Author Contributions
Both authors conceptualized the content of the review. LP wrote the first draft and FR reviewed and edited the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
CF, cystic fibrosis; CFTR, cystic fibrosis transmembrane regulator; NE, neutrophil elastase; MRI, magnetic resonance imaging; CT, computed tomography; PET, positron emission tomography; MBW, multiple breath washout; FEV1, forced expiratory volume in 1 s.
References
Amin, R., Ratjen, F. (2008). MRI: the next frontier for cystic fibrosis clinicians? Expert Rev. Respir. Med. 2, 179–182. doi: 10.1586/17476348.2.2.179
Amin, R., Subbarao, P., Jabar, A., Balkovec, S., Jensen, R., Kerrigan, S., et al. (2010). Hypertonic saline improves the LCI in paediatric patients with CF with normal lung function. Thorax 65, 379–383. doi: 10.1136/thx.2009.125831
Amin, R., Subbarao, P., Lou, W., Jabar, A., Balkovec, S., Jensen, R., et al. (2011). The effect of dornase alfa on ventilation inhomogeneity in patients with cystic fibrosis. Eur. Respir. J. 37, 806–812. doi: 10.1183/09031936.00072510
Amin, R., Charron, M., Grinblat, L., Shammas, A., Grasemann, H., Graniel, K., et al. (2012). Cystic fibrosis: Detecting changes in airway inflammation with FDG PET/CT. Radiology 264, 868–875. doi: 10.1148/radiol.12111873
Bell, S. C., Mall, M. A., Gutierrez, H., Macek, M., Madge, S., Davies, J. C., et al. (2020). The future of cystic fibrosis care: a global perspective. Lancet Respir. Med. 8, 65–124. doi: 10.1016/S2213-2600(19)30337-6
Birrer, P., McElvaney, N. G., Rüdeberg, A., Sommer, C. W., Liechti-Gallati, S., Kraemer, R., et al. (1994). Protease-antiprotease imbalance in the lungs of children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 150, 207–213. doi: 10.1164/ajrccm.150.1.7912987
Block, J. K., Vandemheen, K. L., Tullis, E., Fergusson, D., Doucette, S., Haase, D., et al. (2006). Predictors of pulmonary exacerbations in patients with cystic fibrosis infected with multi-resistant bacteria. Thorax 61, 969–974. doi: 10.1136/thx.2006.061366
Bonfield, T. L. (2020). Preclinical modeling for therapeutic development in cystic fibrosis. Am. J. Respir. Crit. Care Med. 201, 267–268. doi: 10.1164/rccm.201910-2056ED
Brody, A. S., Sucharew, H., Campbell, J. D., Millard, S. P., Molina, P. L., Klein, J. S., et al. (2005). Computed tomography correlates with pulmonary exacerbations in children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 172, 1128–1132. doi: 10.1164/rccm.200407-989OC
Chen, D. L., Ferkol, T. W., Mintun, M. A., Pittman, J. E., Rosenbluth, D. B., Schuster, D. P. (2006). Quantifying pulmonary inflammation in cystic fibrosis with positron emission tomography. Am. J. Respir. Crit. Care Med. 173, 1363–1369. doi: 10.1164/rccm.200506-934OC
Chmiel, J., Elborn, S. A Multicenter, Randomized, Double-Blind, Placebo-Controlled Phase 2 Trial to Evaluate Efficacy and Safety of Lenabasum in Cystic Fibrosis. Available at: https://clinicaltrials.gov/ct2/show/NCT03451045 (Accessed March 26, 2019).
Chmiel, J. F., Konstan, M. W., Accurso, F. J., Lymp, J., Mayer-Hamblett, N., VanDevanter, D. R., et al. (2015). Use of ibuprofen to assess inflammatory biomarkers in induced sputum: Implications for clinical trials in cystic fibrosis. J. Cyst. Fibros. 14, 720–726. doi: 10.1016/j.jcf.2015.03.007
Ciet, P., Bertolo, S., Ros, M., Andrinopoulou, E. R., Tavano, V., Lucca, F., et al. (2017). Detection and monitoring of lung inflammation in cystic fibrosis during respiratory tract exacerbation using diffusion-weighted magnetic resonance imaging. Eur. Respir. J. 50, 1601437. doi: 10.1183/13993003.01437-2016
Cohen-Cymberknoh, M., Kerem, E., Ferkol, T., Elizur, A. (2013). Airway inflammation in cystic fibrosis: molecular mechanisms and clinical implications. Thorax 68, 1157–1162. doi: 10.1136/thoraxjnl-2013-203204
Cohen-Cymberknoh, M., Gilead, N., Gartner, S., Rovira, S., Blau, H., Mussaffi, H., et al. (2016). Eradication failure of newly acquired Pseudomonas aeruginosa isolates in cystic fibrosis. J. Cyst. Fibros. 15, 776–782. doi: 10.1016/j.jcf.2016.04.006
Davies, J., Sheridan, H., Bell, N., Cunningham, S., Davis, S. D., Elborn, J. S., et al. (2013). Assessment of clinical response to ivacaftor with lung clearance index in cystic fibrosis patients with a G551D-CFTR mutation and preserved spirometry: A randomised controlled trial. Lancet Respir. Med. 1, 630–638. doi: 10.1016/S2213-2600(13)70182-6
De Jong, P. A., Lindblad, A., Rubin, L., Hop, W. C. J., De Jongste, J. C., Brink, M., et al. (2006). Progression of lung disease on computed tomography and pulmonary function tests in children and adults with cystic fibrosis. Thorax 61, 80–85. doi: 10.1136/thx.2005.045146
DeBoer, E. M., Swiercz, W., Heltshe, S. L., Anthony, M. M., Szefler, P., Klein, R., et al. (2014). Automated ct scan scores of bronchiectasis and air trapping in cystic fibrosis. Chest 145, 593–603. doi: 10.1378/chest.13-0588
Dong, K., Singh, A., Ng, R. T., Sin, D. D., Tebbutt, S. J., Ratjen, F., et al. (2019). “Proteomic Profiling to Identify Blood Biomarkers Predictive of Response to Azithromycin in Children and Adolescents With Cystic Fibrosis,” in Chest (Elsevier Inc), 667–673. doi: 10.1016/j.chest.2019.05.017
Döring, G., Bragonzi, A., Paroni, M., Aktürk, F.-F., Cigana, C., Schmidt, A., et al. (2014). BIIL 284 reduces neutrophils numbers but increases P. aeruginosa bacteraemia and inflammation in mouse lungs. J. Cyst. Fibros. 13, 156. doi: 10.1016/J.JCF.2013.10.007
Dovey, M., Aitken, M. L., Emerson, J., McNamara, S., Waltz, D. A., Gibson, R. L. (2007). Oral corticosteroid therapy in cystic fibrosis patients hospitalized for pulmonary exacerbation: A pilot study. Chest 132, 1212–1218. doi: 10.1378/chest.07-0843
Elborn, J. S., Bhatt, L., Grosswald, R., Ahuja, S., Springman, E. B. (2017). Phase I Studies of Acebilustat: Pharmacokinetics, Pharmacodynamics, Food Effect, and CYP3A Induction. Clin. Transl. Sci. 10, 20–27. doi: 10.1111/cts.12426
Elborn, J. S., Ahuja, S., Springman, E., Mershon, J., Grosswald, R., Rowe, S. M. (2018). EMPIRE-CF: A phase II randomized placebo-controlled trial of once-daily, oral acebilustat in adult patients with cystic fibrosis – Study design and patient demographics. Contemp. Clin. Trials 72, 86–94. doi: 10.1016/j.cct.2018.07.014
Elkins, M., Dentice, R. (2020). Timing of hypertonic saline inhalation for cystic fibrosis. Cochrane Database Syst. Rev. 2020. doi: 10.1002/14651858.CD008816.pub4
Fuchs, H. J., Borowitz, D. S., Christiansen, D. H., Morris, E. M., Nash, M. L., Ramsey, B. W., et al. (1994). Effect of Aerosolized Recombinant Human DNase on Exacerbations of Respiratory Symptoms and on Pulmonary Function in Patients with Cystic Fibrosis. N. Engl. J. Med. 331, 637–642. doi: 10.1056/NEJM199409083311003
Griese, M., Latzin, P., Kappler, M., Weckerle, K., Heinzimaier, T., Bernhardt, T., et al. (2007). α1-Antitrypsin inhalation reduces airway inflammation in cystic fibrosis patients. Eur. Respir. J. 29, 240–250. doi: 10.1183/09031936.00047306
Heltshe, S. L., Goss, C. H., Thompson, V., Sagel, S. D., Sanders, D. B., Marshall, B. C., et al. (2016). Short-term and long-term response to pulmonary exacerbation treatment in cystic fibrosis. Thorax 71, 223–229. doi: 10.1136/thoraxjnl-2014-206750
Horsley, A. A. R., Davies, J. C. J., Gray, R. R. D., Macleod, K. A., Donovan, J., Aziz, Z. A., et al. (2013). Changes in physiological, functional and structural markers of cystic fibrosis lung disease with treatment of a pulmonary exacerbation. Thorax 68, 532–539. doi: 10.1136/thoraxjnl-2012-202538
Kent, L., Reix, P., Innes, J. A., Zielen, S., Le Bourgeois, M., Braggion, C., et al. (2014). Lung clearance index: evidence for use in clinical trials in cystic fibrosis. J. Cyst. Fibros. 13, 123–138. doi: 10.1016/j.jcf.2013.09.005
Kerem, E., Reisman, J., Corey, M., Canny, G. J., Levison, H. (1992). Prediction of mortality in patients with cystic fibrosis. N. Engl. J. Med. 326, 1187–1191. doi: 10.1056/NEJM199204303261804
Klein, M., Cohen-Cymberknoh, M., Armoni, S., Shoseyov, D., Chisin, R., Orevi, M., et al. (2009). 18F-fluorodeoxyglucose-PET/CT imaging of lungs in patients with cystic fibrosis. Chest 136, 1220–1228. doi: 10.1378/chest.09-0610
Konstan, M. W., Walenga, R. W., Hilliard, K. A., Hilliard, J. B. (1993). Leukotriene B4 markedly elevated in the epithelial lining fluid of patients with cystic fibrosis. Am. Rev. Respir. Dis. 148, 896–901. doi: 10.1164/ajrccm/148.4_Pt_1.896
Konstan, M. W., Krenicky, J. E., Finney, M. R., Kirchner, H. L., Hilliard, K. A., Hilliard, J. B., et al. (2003). Effect of ibuprofen on neutrophil migration in vivo in cystic fibrosis and healthy subjects. J. Pharmacol. Exp. Ther. 306, 1086–1091. doi: 10.1124/jpet.103.052449
Konstan, M. W., Schluchter, M. D., Xue, W., Davis, P. B. (2007). Clinical use of Ibuprofen is associated with slower FEV1 decline in children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 176, 1084–1089. doi: 10.1164/rccm.200702-181OC
Konstan, M. W., Döring, G., Heltshe, S. L., Lands, L. C., Hilliard, K. A., Koker, P., et al. (2014). A randomized double blind, placebo controlled phase 2 trial of BIIL 284 BS (an LTB4 receptor antagonist) for the treatment of lung disease in children and adults with cystic fibrosis. J. Cyst. Fibros. 13, 148–155. doi: 10.1016/j.jcf.2013.12.009
Konstan, M. W., VanDevanter, D. R., Sawicki, G. S., Pasta, D. J., Foreman, A. J., Neiman, E. A., et al. (2018). Association of High-Dose Ibuprofen Use, Lung Function Decline, and Long-Term Survival in Children with Cystic Fibrosis. Ann. Am. Thorac. Soc 15, 485–493. doi: 10.1513/AnnalsATS.201706-486OC
Labiris, N. R., Nahmias, C., Freitag, A. P., Thompson, M. L., Dolovich, M. B. (2003). Uptake of 18fluorodeoxyglucose in the cystic fibrosis lung: A measure of lung inflammation? Eur. Respir. J. 21, 848–854. doi: 10.1183/09031936.03.00065102
Lands, L. C., Stanojevic, S. (2019). Oral non-steroidal anti-inflammatory drug therapy for lung disease in cystic fibrosis. Cochrane Database Syst. Rev. 2019. doi: 10.1002/14651858.CD001505.pub5
Lands, L. C., Milner, R., Cantin, A. M., Manson, D., Corey, M. (2007). High-dose ibuprofen in cystic fibrosis: Canadian safety and effectiveness trial. J. Pediatr. 151, 249–254. doi: 10.1016/j.jpeds.2007.04.009
Liou, T. G., Adler, F. R., Fitzsimmons, S. C., Cahill, B. C., Hibbs, J. R., Marshall, B. C. (2001). Predictive 5-year survivorship model of cystic fibrosis. Am. J. Epidemiol. 153, 345–352. doi: 10.1093/aje/153.4.345
Loeve, M., van Hal, P. T. W., Robinson, P., de Jong, P. A., Lequin, M. H., Hop, W. C., et al. (2009). The spectrum of structural abnormalities on CT scans from patients with CF with severe advanced lung disease. Thorax 64, 876–882. doi: 10.1136/thx.2008.110908
Loeve, M., Gerbrands, K., Hop, W. C., Rosenfeld, M., Hartmann, I. C., Tiddens, H. A. (2011). Bronchiectasis and pulmonary exacerbations in children and young adults with cystic fibrosis. Chest 140, 178–185. doi: 10.1378/chest.10-1152
Martinez, F. J., Donohue, J. F., Rennard, S., II (2011). The future of chronic obstructive pulmonary disease treatment - Difficulties of and barriers to drug development. Lancet 378, 1027–1037. doi: 10.1016/S0140-6736(11)61047-7
Mayer-Hamblett, N., Aitken, M. L., Accurso, F. J., Kronmal, R. A., Konstan, M. W., Burns, J. L., et al. (2007). Association between pulmonary function and sputum biomarkers in cystic fibrosis. Am. J. Respir. Crit. Care Med. 175, 822–828. doi: 10.1164/rccm.200609-1354OC
McElvaney, N. G., Hubbard, R. C., Birrer, P., Crystal, R. G., Chernick, M. S., Frank, M. M., et al. (1991). Aerosol α1 -antitrypsin treatment for cystic fibrosis. Lancet 337, 392–394. doi: 10.1016/0140-6736(91)91167-S
McElvaney, N. G. (2016). “Alpha-1 antitrypsin therapy in cystic fibrosis and the lung disease associated with alpha-1 antitrypsin deficiency,” in Annals of the American Thoracic Society (American Thoracic Society), S191–S196. doi: 10.1513/AnnalsATS.201504-245KV
Mogayzel, P. J., Naureckas, E. T., Robinson, K. A., Mueller, G., Hadjiliadis, D., Hoag, J. B., et al. (2013). Cystic fibrosis pulmonary guidelines: Chronic medications for maintenance of lung health. Am. J. Respir. Crit. Care Med. 187, 680–689. doi: 10.1164/rccm.201207-1160OE
Mott, L. S., Park, J., Murray, C. P., Gangell, C. L., De Klerk, N. H., Robinson, P. J., et al. (2012). Progression of early structural lung disease in young children with cystic fibrosis assessed using CT. Thorax 67, 509–516. doi: 10.1136/thoraxjnl-2011-200912
Motwani, M. P., Bennett, F., Norris, P. C., Maini, A. A., George, M. J., Newson, J., et al. (2018). Potent Anti-Inflammatory and Pro-Resolving Effects of Anabasum in a Human Model of Self-Resolving Acute Inflammation. Clin. Pharmacol. Ther. 104, 675–686. doi: 10.1002/cpt.980
Muhlebach, M. S., Clancy, J. P., Heltshe, S. L., Ziady, A., Kelley, T., Accurso, F., et al. (2016). Biomarkers for cystic fibrosis drug development. J. Cyst. Fibros. 15, 714–723. doi: 10.1016/j.jcf.2016.10.009
Nichols, D. P., Chmiel, J. F. (2015). Inflammation and its genesis in cystic fibrosis. Pediatr. Pulmonol. 50, S39–S56. doi: 10.1002/ppul.23242
Ordoñez, C. L., Henig, N. R., Mayer-Hamblett, N., Accurso, F. J., Burns, J. L., Chmiel, J. F., et al. (2003). Inflammatory and Microbiologic Markers in Induced Sputum after Intravenous Antibiotics in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 168, 1471–1475. doi: 10.1164/rccm.200306-731OC
O’Neill, K., Bradley, J. M., Reid, A., Downey, D. G., Rendall, J., McCaughan, J., et al. (2018). Airway infection, systemic inflammation and lung clearance index in children and adults with cystic fibrosis. Eur. Respir. J. 51, 1701704. doi: 10.1183/13993003.01704-2017
Owens, C. M., Aurora, P., Stanojevic, S., Bush, A., Wade, A., Oliver, C., et al. (2011). Lung clearance index and HRCT are complementary markers of lung abnormalities in young children with CF. Thorax 66, 481–488. doi: 10.1136/thx.2010.150375
Parnham, M. J., Čulić, O., Eraković, V., Munić, V., Popović-Grle, S., Barišić, K., et al. (2005). Modulation of neutrophil and inflammation markers in chronic obstructive pulmonary disease by short-term azithromycin treatment. Eur. J. Pharmacol. 517, 132–143. doi: 10.1016/j.ejphar.2005.05.023
Perrem, L., Ratjen, F. (2019). Anti-inflammatories and mucociliary clearance therapies in the age of CFTR modulators. Pediatr. Pulmonol. 54 Suppl 3, S46–S55. doi: 10.1002/ppul.24364
Pillarisetti, N., Williamson, E., Linnane, B., Skoric, B., Robertson, C. F., Robinson, P., et al. (2011). Infection, Inflammation, and Lung Function Decline in Infants with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 184, 75–81. doi: 10.1164/rccm.201011-1892OC
Prednisone in Cystic Fibrosis Pulmonary Exacerbations Full Text View - ClinicalTrials.gov. Available at: https://clinicaltrials.gov/ct2/show/NCT03070522 (Accessed August 10, 2020).
Proesmans, M., Els, C., Vermeulen, F., De Boeck, K. (2011). Change in IgG and evolution of lung function in children with cystic fibrosis. J. Cyst. Fibros. 10, 128–131. doi: 10.1016/j.jcf.2010.12.004
Quon, B. S., Dai, D. L. Y., Hollander, Z., Ng, R. T., Tebbutt, S. J., Man, S. F. P., et al. (2016). Discovery of novel plasma protein biomarkers to predict imminent cystic fibrosis pulmonary exacerbations using multiple reaction monitoring mass spectrometry. Thorax 71, 216–222. doi: 10.1136/thoraxjnl-2014-206710
Ramsey, B. W., Pepe, M. S., Quan, J. M., Otto, K. L., Montgomery, A. B., Williams-Warren, J., et al. (1999). Intermittent Administration of Inhaled Tobramycin in Patients with Cystic Fibrosis. N. Engl. J. Med. 340, 23–30. doi: 10.1056/NEJM199901073400104
Ramsey, B. W., Davies, J., McElvaney, N. G., Tullis, E., Bell, S. C., Dřevínek, P., et al. (2011). A CFTR Potentiator in Patients with Cystic Fibrosis and the G551D Mutation. N. Engl. J. Med. 365, 1663–1672. doi: 10.1056/NEJMoa1105185
Ramsey, K., Foong, R., Grdosic, J. (2017). Multiple breath washout outcomes are sensitive to inflammation and infection in children with cystic fibrosis. Ann. Am. Thorac. Soc. 14, 1436–1442. doi: 10.1513/AnnalsATS.201611-935OC
Rao, N. L., Riley, J. P., Banie, H., Xue, X., Sun, B., Crawford, S., et al. (2010). Leukotriene A4 Hydrolase Inhibition Attenuates Allergic Airway Inflammation and Hyperresponsiveness. Am. J. Respir. Crit. Care Med. 181, 899–907. doi: 10.1164/rccm.200807-1158OC
Ratjen, F., Saiman, L., Mayer-Hamblett, N., Lands, L. C., Kloster, M., Thompson, V., et al. (2012). Effect of Azithromycin on Systemic Markers of Inflammation in Patients With Cystic Fibrosis Uninfected With Pseudomonas aeruginosa. Chest 142, 1259–1266. doi: 10.1378/chest.12-0628
Ratjen, F., Hug, C., Marigowda, G., Tian, S., Huang, X., Stanojevic, S., et al. (2017). Efficacy and safety of lumacaftor and ivacaftor in patients aged 6–11 years with cystic fibrosis homozygous for F508del-CFTR: a randomised, placebo-controlled phase 3 trial. Lancet Respir. Med. 5, 557–567. doi: 10.1016/S2213-2600(17)30215-1
Ratjen, F., Davis, S. D., Stanojevic, S., Kronmal, R. A., Hinckley Stukovsky, K. D., Jorgensen, N., et al. (2019). Inhaled hypertonic saline in preschool children with cystic fibrosis (SHIP): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Respir. Med. 9, 802–809. doi: 10.1016/S2213-2600(19)30187-0
Rayment, J. H., Stanojevic, S., Davis, S. D., Retsch-Bogart, G., Ratjen, F. (2018). Lung clearance index to monitor treatment response in pulmonary exacerbations in preschool children with cystic fibrosis. Thorax 73, 451–458. doi: 10.1136/thoraxjnl-2017-210979
Reid, P. A., McAllister, D. A., Boyd, A. C., Innes, J. A., Porteous, D., Greening, A. P., et al. (2015). Measurement of serum calprotectin in stable patients predicts exacerbation and lung function decline in cystic fibrosis. Am. J. Respir. Crit. Care Med. 191, 233–236. doi: 10.1164/rccm.201407-1365LE
Rowe, S. M., Heltshe, S. L., Gonska, T., Donaldson, S. H., Borowitz, D., Gelfond, D., et al. (2014). Clinical Mechanism of the Cystic Fibrosis Transmembrane Conductance Regulator Potentiator Ivacaftor in G551D-mediated Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 190, 175–184. doi: 10.1164/rccm.201404-0703OC
Sagel, S. D., Kapsner, R., Osberg, I., Sontag, M. K., Accurso, F. J. (2001). Airway inflammation in children with cystic fibrosis and healthy children assessed by sputum induction. Am. J. Respir. Crit. Care Med. 164, 1425–1431. doi: 10.1164/ajrccm.164.8.2104075
Sagel, S. D., Chmiel, J. F., Konstan, M. W. (2007). “Sputum biomarkers of inflammation in cystic fibrosis lung disease,” in Proceedings of the American Thoracic Society (Proc Am Thorac Soc), 406–417. doi: 10.1513/pats.200703-044BR
Sagel, S. D., Wagner, B. D., Anthony, M. M., Emmett, P., Zemanick, E. T. (2012). Sputum Biomarkers of Inflammation and Lung Function Decline in Children with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 186, 857–865. doi: 10.1164/rccm.201203-0507OC
Sanders, D. B., Bittner, R. C. L., Rosenfeld, M., Hoffman, L. R., Redding, G. J., Goss, C. H. (2010). Failure to recover to baseline pulmonary function after cystic fibrosis pulmonary exacerbation. Am. J. Respir. Crit. Care Med. 182, 627–632. doi: 10.1164/rccm.200909-1421OC
Santyr, G., Kanhere, N., Morgado, F., Rayment, J. H., Ratjen, F., Couch, M. J. (2019). Hyperpolarized Gas Magnetic Resonance Imaging of Pediatric Cystic Fibrosis Lung Disease. Acad. Radiol. 26, 344–354. doi: 10.1016/j.acra.2018.04.024
Sawicki, G. S., McKone, E. F., Pasta, D. J., Millar, S. J., Wagener, J. S., Johnson, C. A., et al. (2015). Sustained Benefit from Ivacaftor Demonstrated by Combining Clinical Trial and Cystic Fibrosis Patient Registry Data. Am. J. Respir. Crit. Care Med. 192, 836–842. doi: 10.1164/rccm.201503-0578OC
Shoki, A. H., Mayer-Hamblett, N., Wilcox, P. G., Sin, D. D., Quon, B. S. (2013). Systematic review of blood biomarkers in cystic fibrosis pulmonary exacerbations. Chest 144, 1659–1670. doi: 10.1378/chest.13-0693
Sly, P. D., Brennan, S., Gangell, C., de Klerk, N., Murray, C., Mott, L., et al. (2009). Lung Disease at Diagnosis in Infants with Cystic Fibrosis Detected by Newborn Screening. Am. J. Respir. Crit. Care Med. 180, 146–152. doi: 10.1164/rccm.200901-0069OC
Sly, P. D., Gangell, C. L., Chen, L., Ware, R. S., Ranganathan, S., Mott, L. S., et al. (2013). Risk Factors for Bronchiectasis in Children with Cystic Fibrosis. NEJM. Org. N Engl. J. Med. 21, 1963–1970. doi: 10.1056/NEJMoa1301725
Stanojevic, S., Ratjen, F. (2016). Physiologic endpoints for clinical studies for cystic fibrosis. J. Cyst. Fibros. 15, 416–423. doi: 10.1016/j.jcf.2016.05.014
Stanojevic, S., McDonald, A., Waters, V., MacDonald, S., Horton, E., Tullis, E., et al. (2017). Effect of pulmonary exacerbations treated with oral antibiotics on clinical outcomes in cystic fibrosis. Thorax 72, 327–332. doi: 10.1136/thoraxjnl-2016-208450
Subbarao, P., Stanojevic, S., Brown, M., Jensen, R., Rosenfeld, M., Davis, S., et al. (2013). Lung Clearance Index as an Outcome Measure for Clinical Trials in Young Children with Cystic Fibrosis A Pilot Study Using Inhaled Hypertonic Saline. Am. J. Respir. Crit. Care Med. 188, 456–460. doi: 10.1164/rccm.201302-0219OC
Tiddens, H. A. W. M., Puderbach, M., Venegas, J. G., Ratjen, F., Donaldson, S. H., Davis, S. D., et al. (2015). Novel outcome measures for clinical trials in cystic fibrosis. Pediatr. Pulmonol. 50, 302–315. doi: 10.1002/ppul.23146
Tobin, D. M., Vary, J. C., Ray, J. P., Walsh, G. S., Dunstan, S. J., Bang, N. D., et al. (2010). The lta4h Locus Modulates Susceptibility to Mycobacterial Infection in Zebrafish and Humans. Cell 140, 717–730. doi: 10.1016/J.CELL.2010.02.013
Torphy, T. J., Allen, J., Cantin, A. M., Konstan, M. W., Accurso, F. J., Joseloff, E., et al. (2015). “Considerations for the conduct of clinical trials with antiinflammatory agents in cystic fibrosis: A cystic fibrosis foundation workshop report,” in Annals of the American Thoracic Society (American Thoracic Society), 1398–1406. doi: 10.1513/AnnalsATS.201506-361OT
Trial to Evaluate Efficacy and Safety of Lenabasum in Cystic Fibrosis Full Text View - ClinicalTrials.gov. Available at: https://clinicaltrials.gov/ct2/show/NCT03451045 (Accessed August 13, 2020).
van Horck, M., van de Kant, K., Winkens, B., Wesseling, G., Gulmans, V., Hendriks, H., et al. (2018). Risk factors for lung disease progression in children with cystic fibrosis. Eur. Respir. J. 51, 1702509. doi: 10.1183/13993003.02509-2017
Waters, V. J., Stanojevic, S., Sonneveld, N., Klingel, M., Grasemann, H., Yau, Y. C. W., et al. (2015). Factors associated with response to treatment of pulmonary exacerbations in cystic fibrosis patients. J. Cyst. Fibros. 14, 755–762. doi: 10.1016/j.jcf.2015.01.007
Watt, A. P., Courtney, J., Moore, J., Ennis, M., Elborn, J. S. (2005). Neutrophil cell death, activation and bacterial infection in cystic fibrosis. Thorax 60, 659–664. doi: 10.1136/thx.2004.038240
Whiting, P., Maiwenn, Al, Burgers, L., Westwood, M., Ryder, S., Hoogendoorn, M., et al. (2014). Ivacaftor for the treatment of patients with cystic fibrosis and the G551D mutation: A systematic review and cost-effectiveness analysis. Health Technol. Assess. (Rockv). 18, 1–106. doi: 10.3310/hta18180
Woods, J. C., Wild, J. M., Wielpütz, M. O., Clancy, J. P., Hatabu, H., Kauczor, H., et al. (2019). Current state of the art MRI for the longitudinal assessment of cystic fibrosis. J. Magn. Reson. Imaging 10.1002, jmri.27030. doi: 10.1002/jmri.27030
Keywords: anti-inflammatories, cystic fibrosis, clinical trials, lung, airway inflammation
Citation: Perrem L and Ratjen F (2020) Designing Clinical Trials for Anti-Inflammatory Therapies in Cystic Fibrosis. Front. Pharmacol. 11:576293. doi: 10.3389/fphar.2020.576293
Received: 25 June 2020; Accepted: 24 August 2020;
Published: 11 September 2020.
Edited by:
Noel Gerard McElvaney, Royal College of Surgeons in Ireland, IrelandReviewed by:
Luigi Brunetti, University of Studies G. d’Annunzio Chieti and Pescara, ItalyGiustino Orlando, University of Studies G. d’Annunzio Chieti and Pescara, Italy
Copyright © 2020 Perrem and Ratjen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lucy Perrem, bHVjeS5wZXJyZW1Ac2lja2tpZHMuY2E=