
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 11 December 2020
Sec. Cardiovascular and Smooth Muscle Pharmacology
Volume 11 - 2020 | https://doi.org/10.3389/fphar.2020.569251
 Tiantian Wang1†
Tiantian Wang1† Chunxia Liu1†
Chunxia Liu1† Ling-hui Pan1Zhen Liu1Chang-long Li1
Ling-hui Pan1Zhen Liu1Chang-long Li1 Jin-yuan Lin1
Jin-yuan Lin1 Yi He1Jing-yuan Xiao1Siyi Wu1Yi Qin1
Yi He1Jing-yuan Xiao1Siyi Wu1Yi Qin1 Zhao Li2*
Zhao Li2* Fei Lin1*
Fei Lin1*Background: Lung ischemia reperfusion injury (LIRI) is a complex pathophysiological process activated by lung transplantation and acute lung injury. The p38 mitogen-activated protein kinase (MAPK) is involved in breakdown of the endothelial barrier during LIRI, but the mechanism is still unclear. Therefore, we investigated the function of p38 MAPK in LIRI in vivo and in vitro.
Methods: Sprague–Dawley rats were subjected to ischemia reperfusion with or without pretreatment with a p38 MAPK inhibitor. Lung injury was assessed using hematoxylin and eosin staining, and pulmonary blood–air barrier permeability was evaluated using Evans blue staining. A rat pulmonary microvascular endothelial cell line was infected with lentiviral expressing short hairpin (sh)RNA targeting p38 MAPK and then cells were subjected to oxygen/glucose deprivation and reoxygenation (OGD/R). Markers of endothelial destruction were measured by western blot and immunofluorescence.
Results:In vivo LIRI models showed structural changes indicative of lung injury and hyperpermeability of the blood–air barrier. Inhibiting p38 MAPK mitigated these effects. Oxygen/glucose deprivation and reoxygenation promoted hyperpermeability of the endothelial barrier in vitro, but knockdown of p38 MAPK attenuated cell injury; maintained endothelial barrier integrity; and partially reversed injury-induced downregulation of permeability protein AQP1, endothelial protective protein eNOS, and junction proteins ZO-1 and VE-cadherin while downregulating ICAM-1, a protein involved in destroying the endothelial barrier, and ET-1, a protein involved in endothelial dysfunction.
Conclusion: Inhibition of p38 MAPK alleviates LIRI by decreasing blood–air hyperpermeability. Blocking p38 MAPK may be an effective treatment against acute lung injury.
Lung ischemia reperfusion injury (LIRI) is a complex pathophysiological process (Lau et al., 2009). LIRI is characterized by increased pulmonary vascular resistance, increased microvascular permeability, pulmonary edema, pulmonary hypertension, and impaired oxygenation (de Perrot et al., 2003). Many clinical conditions, including lung transplantation, acute lung injury, and acute respiratory distress syndrome, can induce LIRI, which is a risk factor for late graft failure in lung transplantation (Lee et al., 2010; Sun et al., 2017).
LIRI disrupts the blood–air barrier at the interface of an alveolar epithelial cell layer and endothelial cell layer, promoting lung inflammation and acute lung injury (Zhang et al., 2015; Bärnthaler et al., 2017; Kling et al., 2017). Current methods to prevent breakdown of the blood–air barrier are ineffective; thus, it is crucial to clarify the mechanisms of blood–air barrier dysfunction in LIRI.
Microvascular endothelial cells are targeted for blood–air barrier destruction during LIRI (Zhang et al., 2015), and several proteins may be dysregulated during disruption of the blood–air barrier. AQP1, a water-selective channel protein, is upregulated when lung endothelial cells are more permeable to water (Ma et al., 2004); mice lacking AQP1 show reduced movement of endothelial fluid, and their microvascular endothelium is less permeable to water (Towne et al., 2000; Hua et al., 2019). eNOS/NO signaling, an endothelial homeostasis protective pathway, improved endothelial barrier during ischemia/reperfusion through suppressing inflammation (Qi et al., 2016; Zhang et al., 2019). Conversely, overexpression of ET-1, an endothelial dysfunction maker protein, promoted endothelial permeability during short-term ischemia with long-term reperfusion (Zhang et al., 2013). ICAM-1 plays an important role in destroying the endothelial barrier (Wang et al., 1999). It increases permeability of human dermal microvascular endothelial cells by changing their cell junctions or cytoskeleton organization (Clark et al., 2007). Endothelial cells form tight barriers through tight junctions in a process regulated by the protein ZO-1 (Tornavaca et al., 2015), and VE-cadherin is an adhesion protein specifically located at tight junctions (Li et al., 2019). F-actin is the main cytoskeletal protein involved in cellular contraction, which is important for maintaining normal morphology and function (Deng et al., 2018). LIRI induces degradation of tight junctions by downregulating ZO-1 and VE-cadherin while stress fibers accumulate, compromising the endothelial barrier (Kim et al., 2020).
Inflammation during LIRI has been linked to activation of p38 mitogen-activated protein kinase (MAPK), which can influence cell differentiation, proliferation, and survival and production of proinflammatory mediators (Boyle et al., 2006; Wada et al., 2017; Xiao et al., 2017). Although p38 MAPK is known to help damage the endothelial barrier during inflammation-induced acute lung injury (Tan et al., 2013; Li et al., 2015; Song et al., 2017), whether and how it contributes to blood–air barrier hyperpermeability is unclear.
We hypothesized that p38 MAPK participates in the process of LIRI by altering expression of proteins that regulate permeability of the blood–air barrier. Specifically, we speculated that inhibiting p38 MAPK may upregulate AQP1 while downregulating ICAM-1, thereby reversing LIRI-induced changes in expression of ZO-1, VE-cadherin, and F-actin. We tested this possibility in vivo and in vitro.
Animal protocols were approved by the Institutional Animal Care and Use Committee of Guangxi Medical University (Nanning, China). Adult male Sprague–Dawley rats (220–250 g, 7–8 weeks) were acquired from the animal center of Guangxi Medical University. Animals were maintained in a 12 h light–dark cycle in humidity-controlled rooms with freely accessible food and water.
Rats were randomly divided into four groups (n = 15 per group): DMSO, SB203580 (p38 MAPK inhibitor, dissolved in DMSO), I/R (ischemia reperfusion), and I/R + SB203580. The rats in the control and the SB203580 groups received DMSO as a vehicle control or SB203580 (10 mg/kg) by intraperitoneal injection 30 min before the sham surgery (Liu et al., 2008; Xiong et al., 2016; Zhang et al., 2020a). LIRI was induced in the I/R and I/R + SB203580 groups. Rats were sedated and mechanically ventilated using an ALC-V9A–type small animal ventilator (ALCOTT BIOTECH of Shanghai, China). Inspiratory-to-expiratory ratio was set to 1:1 and respiration rate to 60 breaths/min. The tidal volume was 8–10 ml/kg with the required fraction of inspired oxygen of 100%. In the I/R and I/R + SB203580 groups, rats were subjected to thoracotomy, and the left hilus pulmonis was occluded for 1 h using a vascular clamp. Then, the clamp was removed, and reperfusion was allowed for 4 h before sacrifice by bloodletting through the carotid artery (Fei et al., 2017). The rats in the I/R and I/R + SB203580 groups received DMSO as vehicle control or SB203580 by intraperitoneal injection 30 min before surgery.
Lung tissue specimens were fixed in 4% paraformaldehyde and then embedded in paraffin. Samples were stained with hematoxylin and eosin and photographed under a light microscope (EVOS FL AutoLife Technologies). Lung injury was scored as previously described (Fei et al., 2017). Briefly, scoring was based on the infiltration of inflammatory cells, the extent of pulmonary edema, and the interstitial congestion and hemorrhage. For each mouse, six fields were assessed at 400X magnification. The score of lung injury is based on three criteria: aggregation or infiltration of inflammatory cells in vessel walls or air spaces [1 point = only wall, 2 = rare cells in air space, 3 = intermediate, and 4 = severe (air space congested)], hyaline membrane formation and interstitial congestion in the lung [1 point = normal lung, 2 = moderate (>25% of lung section), 3 = intermediate (25%–50% of lung section), and 4 = severe (>50% of lung section)], and presence (1) or absence (0) of hemorrhage. The scores for each criterion were summed to obtain the score for each animal. Six sections were independently assessed by two pathologists blinded to animal grouping.
Lung tissue samples were minced into pieces approximately 1 mm and fixed in 3% glutaraldehyde for at least 2 h, followed by 1% osmic acid for 1–2 h. Tissue was dehydrated in different concentrations of acetone before embedding in resin. Samples were cut into ultrathin sections with an ultramicrotome and observed with an H-7560 transmission electron microscope (H-7560, Tokyo, Japan).
After sacrifice, wet left lung tissue was weighed, incubated at 60°C for 96 h, and then weighed again. These values were used to calculate wet-to-dry ratios (Dai et al., 2015).
After the right lung hilum was clipped, the left lung was flushed three times with 5 ml of cold phosphate-buffered saline (PBS) using a tracheal catheter to collect BALF (Fei et al., 2017). Total protein in the BALF was detected by a bicinchoninic acid (BCA) protein assay kit following the manufacturer’s instructions (Beyotime Biotechnology, China).
Next, 0.5% Evans blue (4 mg/kg) was injected via the jugular vein 1 h before sacrifice. Then, the left lung tissue was weighed, homogenized, treated with formamide (100 mg/ml; Sigma-Aldrich, Shanghai, China), incubated at 37°C for 24 h, and centrifuged for 20 min at 10,000 g. Absorbance of the supernatant was measured at 620 nm, and the concentration of Evans blue was calculated from the absorbance according to a standard curve.
After sacrifice, lung tissue was homogenized. The concentrations of IL-6 and IL-1β in lung tissues or in BALF (acquired as previously described in the total protein in bronchoalveolar lavage fluid section) were quantified using ELISA kits (Elabscience Biotechnology, Wuhan, China) following the manufacturer’s instructions.
Lung tissue specimens were embedded in paraffin and sliced 2–4 μm thick. The sections were dewaxed, rehydrated, immersed in 0.01 M citrate buffer (pH 6.0), and boiled in a pressure cooker for 2 min. The sections were blocked with 3% hydrogen peroxide for 10 min and then incubated overnight at 4°C with rabbit monoclonal antibody against ZO-1 (1:400; catalog no. ab221547, Abcam, UK) or rabbit polyclonal antibody against VE-cadherin (1:100; catalog no. ab231227, Abcam). The sections were exposed to bio-goat antirabbit IgG as a secondary antibody from an SP (mouse/rabbit IgG)-POD kit (Catalog no. SP0041, Solarbio Life Sciences) following the manufacturer's instructions. Then, the sections were stained with DAB from the SP (mouse/rabbit IgG)-POD kit. Normal rabbit IgG (1:100, catalog no. AB-105-C, R&D) in PBS was used instead of a primary antibody (ZO-1 or VE-cadherin) in the immunohistochemistry procedure as a negative control. Spleen and kidney tissues instead of lung tissue were used for ZO-1 and VE-cadherin IHC as a positive control.
Expression of ZO-1 and VE-cadherin was visualized by a light microscope (EVOS FL AutoLife Technologies) and quantified from six randomly selected fields of view using Image-Pro-Plus 6. The integrated optical density (IOD) was measured from areas of protein staining, and the ratio of IOD area was used in statistical analysis.
Rat pulmonary microvascular endothelial cells (rPMVECs) were purchased from the BeNa Culture Collection (catalog no. BNCC-338210; Beijing, China) and cultured in DMEM (Gibco, China) supplemented with 10% fetal bovine serum (FBS; Gibco, Australia), 100 μg/ml penicillin/streptomycin (Gibco) and 1% endothelial cell growth supplement (ScienCell) at 37°C in an atmosphere of 5% CO2 and 95% air. rPMVECs were infected with lentivirus expressing short hairpin (sh)RNA against p38 MAPK (5’-GACCGTTTCAGTCCATCAT-3’) or a control shRNA (5’-TTCTCCGAACGTGTCACGT-3’) in the presence of polybrene (5 μg/ml). Cells were cultured for 72 h after infection. The lentiviruses were synthesized by Genechem (Shanghai, China).
To mimic ischemic-like conditions in vitro, we subjected rPMVECs to OGD/R as described (Shen et al., 2004). Briefly, rPMVECs were washed three times with PBS, fed serum- and glucose-free medium (Gibco), and then placed for 1 h at 37°C in a Whitley H35 Hypoxystation (Don Whitley Scientific) in an atmosphere of 1% O2, 5% CO2, and 94% N2. Then, cells were cultured for 4 h at 37°C in glucose-containing medium in an atmosphere of 5% CO2 and 95% O2.
Cell viability of rPMVECs infected with control or p38 MAPK shRNA before OGD/R was assayed using the CCK-8 kit (JDOJINDO, Japan) following the manufacturer’s instructions. Apoptosis was assessed using a caspase-3 spectrophotometric detection kit (Wanleibio, China) following the manufacturer’s instructions. The level of NO in cell supernatant was assessed using an NO detect kit (Beyotime, China) following the manufacturer's instructions.
Cells (1 × 105) were seeded into the upper chamber of polyester membranes in a 24-well Transwell (pore size 0.4 μm; diameter 6.5 mm; Costar, NY) and cultured in 0.5 ml of DMEM. Medium (1.5 ml) was added to the lower chamber, and Transwells were incubated for 2 days until the cells reached 100% confluence. The cells were subjected to the OGD/R procedure described above, and at 1 h before the end of the reoxygenation period, FITC-dextran solution was added to the upper chamber of each Transwell. Sample readings were converted to FITC-dextran concentrations based on a standard curve. The permeability coefficient (Pc) of FITC-dextran was described as Pc = [A]/t × 1/A × V/[L], where [A] denotes the concentration of FITC-dextran in the lower chamber; [L], the concentration in the upper chamber; A, the area of the membrane (cm2); t, time (s); and V, the volume of the lower chamber (Zhang et al., 2015).
Cells were fixed in 4% paraformaldehyde for at least 10 min, washed three times with PBS, permeabilized with 0.5% Triton X-100, and blocked for 1 h with 5% bovine serum albumin. Then cells were incubated overnight at 4°C on a shaking table with goat polyclonal anti-ZO-1 antibody (1:100; catalog no. ab190085; Abcam) or rabbit polyclonal anti-VE-cadherin antibody (1:300; catalog no. ab33168, Abcam). PBS instead of primary antibody was used as a negative control in immunofluorescence staining. Then, the cells were exposed to an appropriate secondary antibody, donkey antigoat IgG (H + L) cross-adsorbed secondary antibody, Alexa Fluor 568 (1:1000, catalog no. A-11057, invitrogen) for ZO-1 and goat antirabbit IgG (H + L) highly cross-adsorbed secondary antibody, Alexa Fluor 568 (1:1000, catalog no. A-11036, invitrogen) for VE-cadherin, for 1 h at room temperature. Finally, cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Solarbio, China), and cells were viewed under a fluorescence microscope (Olympus BX51). Cytoskeletal changes were examined using rhodamine-conjugated phalloidin (Solarbio, China) to target F-actin.
Total RNA was extracted from cells using TRIzol (catalog no. 15596-026, Invitrogen) following the manufacturer’s instructions. Total RNA (1 μg) was used as a template in a 20-μl reverse transcription reaction using the Prime Script™ RT reagent kit (Takara, Japan) following the manufacturer’s instructions. The cDNA was amplified using the following primers: p38 MAPK forward, 5'-GAACAACATCGTGAAGTGTCAGAAGC-3'; p38 MAPK reverse, 5'-CCTGTGGATTATGTCAGCCGAGTG-3'; HIF-1α forward, 5'-TCTGGGTTGAAACTCAAGCAACTA-3'; HIF-1α reverse, 5'-CAACCGGTTTAAGGACACATTCTG-3'; GAPDH forward, 5'-AGAAGGCTGGGGCTCATTTG-3'; and GAPDH reverse, 5'-AGGGGCCATCCACAGTCTTC-3'.
Relative gene expression was quantified using the 2−∆∆ct method relative to the expression of GAPDH.
Left lung tissues were removed and digested with 0.25% EDTA in centrifuge tubes. Digested tissues or harvested rPMVECs were homogenized in RIPA buffer (Solarbio) containing protease inhibitor cocktail (Solarbio) and phosphatase inhibitor cocktail (Cell Signaling Technology, USA) and then centrifuged at 12,000 g for 15 min at 4°C. The supernatant protein concentration was assayed using a Bicinchoninic acid kit (Beyotime Biotechnology), and equal amounts of proteins were separated on 10% SDS-PAGE and transferred to 0.22 μm PVDF membranes (Millipore) at 4°C. The membranes were incubated overnight at 4°C, shaking with the following primary antibodies: anti-GAPDH (1:1000, Santa Cruz), rabbit polyclonal anti-phospho-p38 MAPK (1:1000; clone D3F9, Cell Signaling Technology), rabbit polyclonal anti-p38 MAPK (1:1000; catalog no. 9212, Cell Signaling Technology), rabbit monoclonal anti-AQP1 (1:5000; catalog no. ab168387, Abcam), mouse monoclonal anti-ICAM-1 (1:2000; catalog no. ab171123, Abcam), goat polyclonal anti-ZO-1 (1:1000; catalog no. ab190085, Abcam), rabbit polyclonal anti-VE-cadherin (1:500; catalog no. ab33168, Abcam), mouse monoclonal anti-HIF-1α (5 μg/ml; catalog no. ab1, Abcam), Endothelin 1 monoclonal antibody (1:10000, catalog no. MA3-005, Invitrogen), and eNOS polyclonal antibody (1:1000, catalog no. PA1-037, Invitrogen). After being washed three times by TBST, the membranes were incubated with the appropriate secondary antibody, goat antimouse H&L IRDye@800 CW (1:10000, catalog no. ab216722), goat antirabbit H&L IRDye@800 CW (1:10000, catalog no. ab216723), or donkey antigoat H&L IRDye@800 CW (1:10000, catalog no. ab216725) for 1 h, and band density was quantified based on enhanced chemiluminescence using the Alpha Innotech system (BioRad).
Data were expressed as mean ± SD and analyzed using SPSS 17.0. Data were plotted using Graphpad Prism 5.0. Comparisons among multiple groups were analyzed by one-way analysis of variance, followed by the Tukey test for intergroup comparisons. Differences associated with p < 0.05 were considered statistically significant.
To investigate the function of p38 MAPK in an in vivo model of LIRI, we blocked its function using SB203580. The morphological changes in lung tissues were assessed using hematoxylin and eosin staining (Figure 1A). In the control groups, the pulmonary alveoli and interstitium were intact, and the alveolar septum showed uniform thickness without signs of alveolar wall thickening or inflammatory cell infiltration. However, the I/R group showed severe congestion and hemorrhage in the lung tissues as well as infiltration by inflammatory and red blood cells. Blocking p38 MAPK remarkably attenuated these changes (Figure 1B).
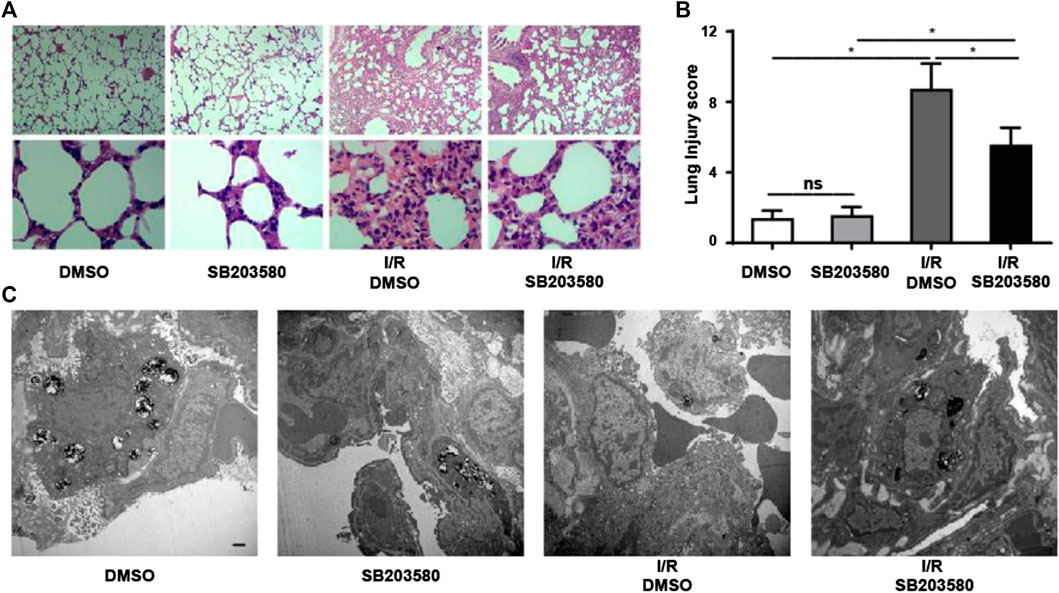
FIGURE 1. Inhibiting p38 Mitogen-activated protein kinase (MAPK) mitigates Lung ischemia reperfusion injury (LIRI) in vivo. (A) Lung sections were stained by hematoxylin and eosin and morphological changes assessed. Magnification, 40X and 400X. (B) Lung injury was scored based on images in panel A (n = 6 sections per group). ns, not significant, *p < 0.05. (C) Ultrastructural changes were assessed using transmission electron microscopy. Scale bar, 1 μm. Blocking P38 MAPK in vivo attenuates I/R-induced leakage at the pulmonary blood–air barrier
Transmission electron microscopy confirmed these results at the ultrastructural level (Figure 1C). In groups without lung injury, pulmonary endothelial cells were tightly connected with integral type II alveolar epithelial cells and an intact basement membrane, and microvilli and lamellar bodies appeared normal. In the I/R group, pulmonary endothelial cells were swollen, type II epithelial microvilli were reduced in number, and lamellar bodies were absent. These changes were partially reversed when p38 MAPK was blocked. Taken together, our results indicate that blocking p38 MAPK using SB203580 attenuates lung injury in an in vivo LIRI model.
Pulmonary blood–air barrier leakage plays a crucial role in I/R-induced lung injury (de Perrot et al., 2003). We assessed lung edema by measuring pulmonary wet-to-dry ratios. I/R injury increased the ratio that blocking p38 MAPK reduced (Figure 2A). Furthermore, I/R increased the level of total proteins in BALF, and antagonizing p38 MAPK partially reversed this (Figure 2B). Consistently, Evans blue dye binding to albumin, which can be used to evaluate the permeability of pulmonary microvasculature (Zhang et al., 2020b), was significantly higher in the I/R group than in all other groups, indicating increased pulmonary microvascular leakage (Figures 2C,D). SB203580 significantly reduced the amount of Evans blue binding, indicating less leakage, which was presumably due to less injury.
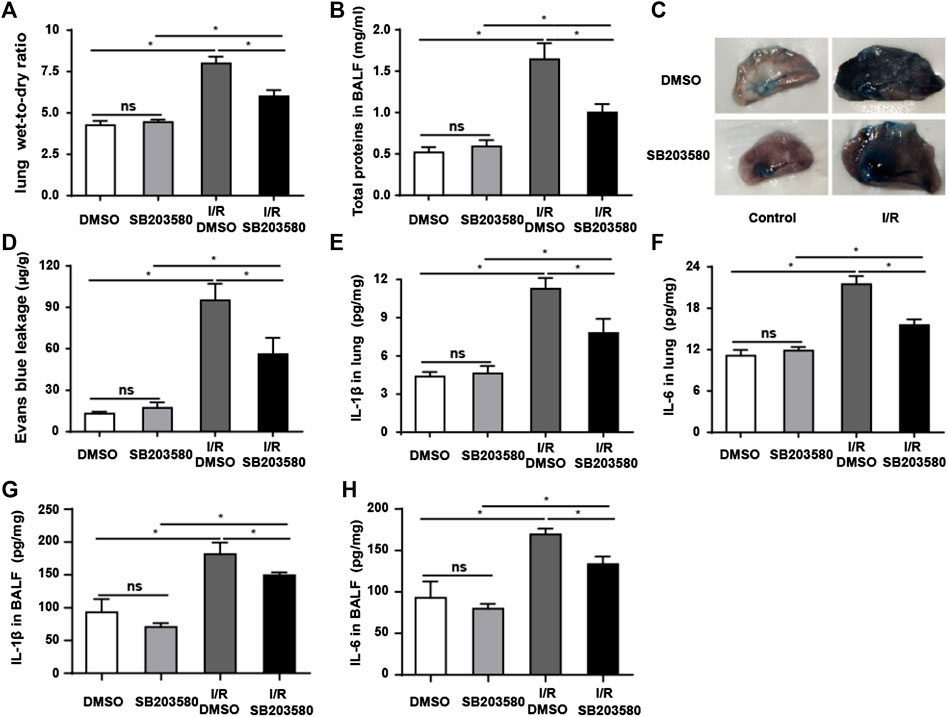
FIGURE 2. p38 MAPK inhibition attenuates I/R-induced leakage of the pulmonary blood–air barrier in vivo. (A) Wet-to-dry ratios of lung tissues as an index of lung edema. (B) Quantification of total protein in bronchoalveolar lavage fluid. (C,D) Macroscopic images of lungs and quantification of Evans blue leakage. (E,F) Quantification of IL-1β and IL-6 abundance in lung tissues by Enzyme-Linked Immunosorbent Assay (ELISA). (G,H) Quantification of IL-1β and IL-6 abundance in Bronchoalveolar Lavage Fluid (BALF) by ELISA. Data shown are mean ± SD from n = 7 per group. ns, not significant, *p < 0.05.
The levels of IL-6 and IL-1β in lung tissues and BALF were used to evaluate the inflammatory response (Sivaraman et al., 2015; Li et al., 2020). Both were significantly higher in the I/R group in lung tissue (Figures 2E,F) and in BALF (Figures 2G,H) than in the control group, which was partially reversed by SB203580. Our findings support the idea that blocking p38 MAPK protects against the onset of pulmonary blood–air barrier leakage.
Previous studies show that the AQP1 level varies inversely with the degree of pulmonary edema (Su et al., 2004). We found that I/R injury sharply downregulated AQP1, which was reversed by SB203580 (Figures 3A–Cfig3). Disruption of endothelial cell tight junctions and adhesion junctions contributes to vascular barrier dysfunction (Yamazaki et al., 2019), and high levels of ICAM-1 can destroy endothelial junctions (Clark et al., 2007). Thus, we examined the expression of ZO-1, a marker of endothelial tight junctions; VE-cadherin, a marker of endothelial adhesion junction (Li et al., 2020); and ICAM-1 to assess the extent of endothelial barrier destruction. I/R significantly upregulated ICAM-1 and downregulated ZO-1 and VE-cadherin, which SB203580 partially reversed (Figures 3A,D–F).
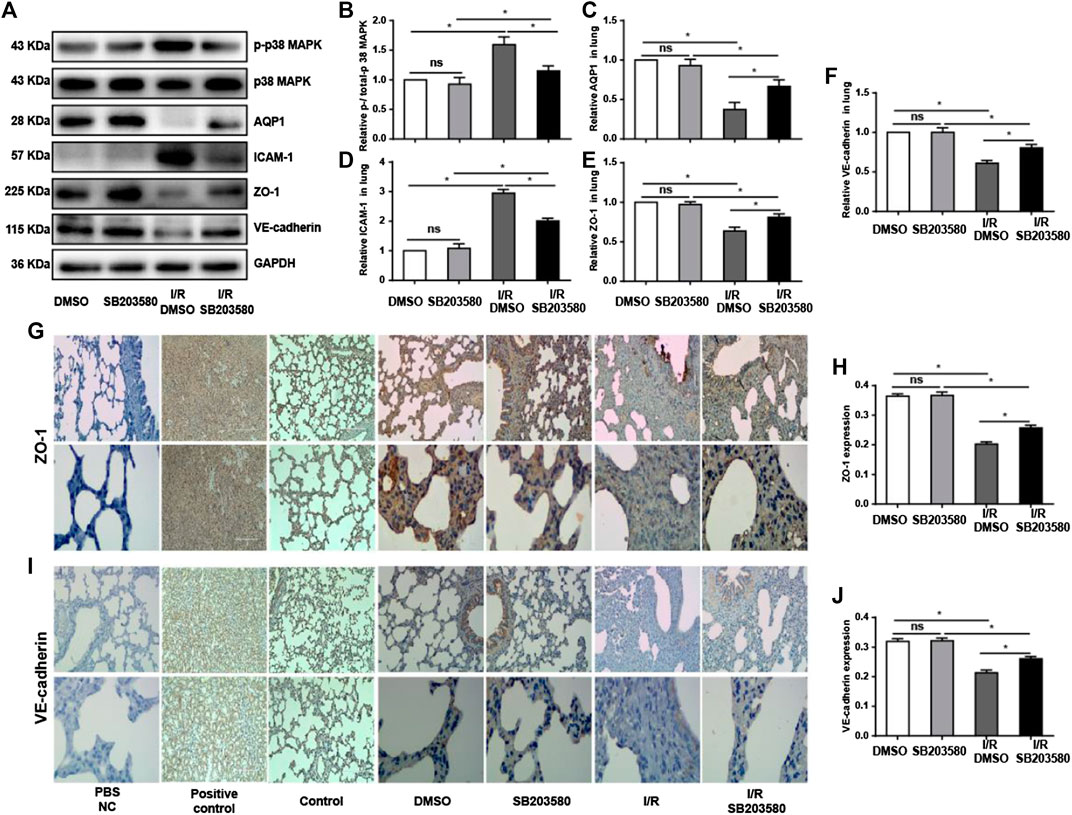
FIGURE 3. p38 MAPK inhibition attenuates I/R injury-induced damage to endothelial integrity. (A) Western blot of phospho-p38 MAPK, p38 MAPK, AQP1, ICAM-1, ZO-1, and VE-cadherin in lung tissues. (B–F) Densitometry of western blots from panel A. Levels were normalized to the control, which was defined as 1.0. (G,I) Immunohistochemistry of ZO-1 and VE-cadherin in lung tissues. Normal rabbit IgG in Phosphate-buffered saline (PBS) was used in the immunohistochemistry experiment instead of the primary antibody as a negative control. Rat spleen and kidney tissue instead of lung tissue were used for ZO-1 and VE-cadherin immunohistochemistry, respectively, as a positive control. Control group mice were treated without vehicle, drug, or surgery. Scale bar: 200 μm. (H,J) Quantification of ZO-1 and VE-cadherin immunohistochemical staining. n = 6 tissue sections per group. ns, not significant, *p < 0.05.
The results with western blotting were confirmed using immunofluorescence (Figures 3G,I), which also showed that ZO-1 and VE-cadherin protein levels were markedly decreased after I/R injury compared with control groups although pretreatment with SB203580 increased their expression. These findings suggest that inhibition of p38 MAPK can significantly alleviate I/R-induced destruction of the endothelial barrier.
To gain mechanistic insights into our results with the in vivo model, we moved into experiments in rPMVECs. First, we confirmed that we could knock down p38 MAPK in this cell line. Cells were transfected with a lentiviral vector expressing shRNA targeting p38 MAPK. Green fluorescence indicated a transfection efficiency greater than 80% (Figures 4A). The shRNA targeting p38 MAPK led to significantly lower levels of p38 MAPK mRNA and phospho-p38 MAPK than the control shRNA (Figures 4B,C). Next, we confirmed that we could induce OGD/R in the cells as a disease model as described (Shen et al., 2004). HIF-1α is a crucial transcriptional factor induced by hypoxia conditions (Ryou et al., 2015). As expected, OGD/R upregulated HIF-1α. This upregulation was partially reversed by p38 MAPK knockdown (Figures 4D,E).

FIGURE 4. Inhibiting p38 MAPK protects rPMVECs from OGD/R injury. (A) Fluorescent and bright field micrographs of rPMVECs transfected with lentivirus-expressing control or p38 MAPK-targeting shRNA. Levels of (B) p38 MAPK mRNA, (C) p-p38 MAPK protein, (D) HIF-1α mRNA, and (E) HIF-1α protein were measured. (F) Viability of rPMVECs was measured using the CCK-8 assay. (G) Activity of caspase 3 in rPMVECs was measured as an index of apoptosis. (H) The permeability coefficient in rPMVECs was measured as an index of the pulmonary blood–air barrier based on flux of the fluorescent marker FITC-dextran. (G) The level of NO in cultured rPMVEC supernatant in different groups was detected. ns: no statistical significance, *p < 0.05, each experiment was performed at least three times independently. Blocking p38 MAPK mitigates OGD/R-induced endothelial dysfunction.
In addition, OGD/R reduced cell viability by 50% (Figure 4F) and upregulated pro-apoptotic caspase-3 activity (West et al., 2006) (Figure 4G), both of which were reversed by p38 MAPK knockdown. OGD/R markedly increased FITC-dextran flux (Figure 4H), indicating hyperpermeability of the pulmonary blood–air barrier (Hoffmann et al., 2011), which p38 MAPK knockdown significantly decreased. Regarding endothelial dysfunction, OGD/R decreased the NO level in the cell supernatant, and p38 MAPK knockdown attenuated the decrease (Figure 4G). Together, these results indicate that p38 MAPK knockdown can protect against OGD/R injury.
To confirm our in vivo findings, we measured levels of phospho-p38 MAPK, AQP1, ICAM-1, VE-cadherin, and ZO-1 expression in rPMVECs. OGD/R upregulated phospho-p38 MAPK and ICAM-1, which p38 MAPK knockdown reversed (Figures 5A,B,D). Conversely, OGD/R downregulated AQP1, ZO-1, and VE-cadherin, which p38 MAPK knockdown reversed (Figures 5A,C,E,F). Also, we found that OGD/R promoted endothelial inflammation and dysfunction indicated by the decrease of eNOS and increase of ET-1, which p38 MAPK knockdown reversed (Figures 5G–I). These results support our in vivo findings that OGR/D induces endothelial inflammatory damage and compromises the permeability barrier, which inhibition of p38 MAPK can mitigate.
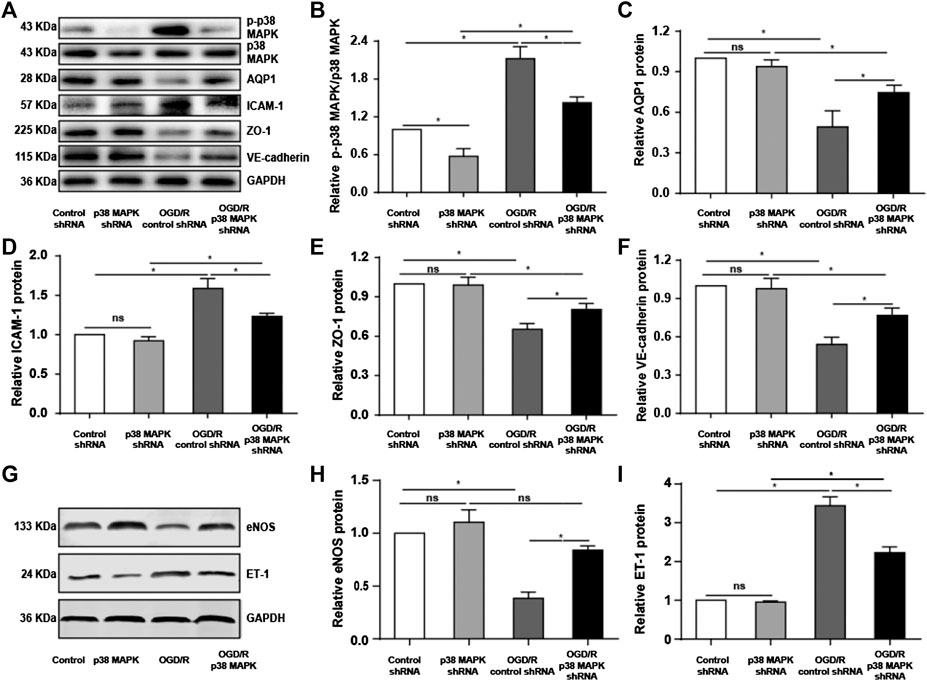
FIGURE 5. Knocking down p38 MAPK protects against endothelial destruction induced by OGD/R. (A) Western blot of phospho-p38 MAPK, p38 MAPK, AQP1, ICAM-1, ZO-1, and VE-cadherin in rPMVECs. (B–F) Quantification of proteins from western blots in panel A. (G) Western blot of eNOS and ET-1 in rPMVECs. (H,I) Quantification of proteins from western blots in panel G. Each experiment was performed independently at least three times. ns, not significant, *p < 0.05.
To further confirm the role of p38 in mediating endothelial destruction induced by OGD/R, we performed immunofluorescence staining, which showed that OGD/R downregulated ZO-1 and VE-cadherin, while p38 MAPK knockdown reversed this (Figures 6A,B). In control rPMVECs, F-actin localized mainly on the cell membrane such that cell borders were obvious. OGD/R led to accumulation of F-actin stress fibers and blurred cell boundaries, which p38 MAPK knockdown mitigated. These results suggest that inhibition of p38 MAPK preserves expression of proteins that maintain the endothelial barrier.
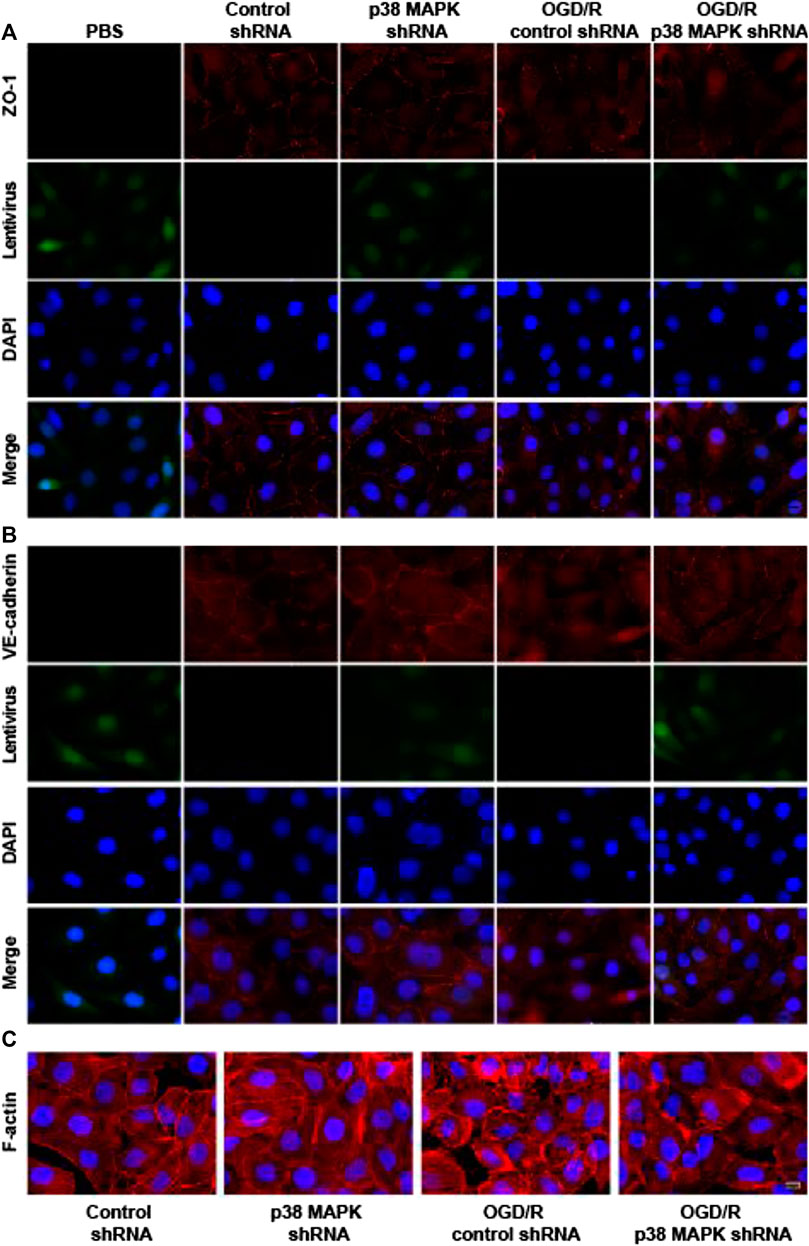
FIGURE 6. p38 MAPK knockdown mitigates OGD/R-induced changes in expression of ZO-1 and VE-cadherin and in F-actin distribution. (A–C) Immunofluorescence micrographs showing ZO-1, VE-cadherin, and F-actin in rPMVECs. PBS was used instead of the primary antibody as a negative control in the immunofluorescence experiment. Green indicates lentivirus. Nuclei were stained with DAPI (blue). Scale bar, 10 μm.
Inhibition of p38 MAPK has been shown to therapeutically treat acute lung injury induced by intestinal I/R in rats (Xiong et al., 2016) or by swine influenza virus or lipopolysaccharide in mice (Gao et al., 2013; Wei et al., 2014). Therefore, in the present study, we explored the potential therapeutic effect of inhibiting p38 MAPK against LIRI in rats and OGD/R injury in rPMVEC cultures. We found that the rats subjected to LIRI exhibited severe pathological changes in lung morphology and ultrastructure, and rPMVECs subjected to OGD/R strongly upregulated HIF-1α (Ryou et al., 2015), indicating that we established both disease models. In both models, inhibition or knockdown of p38 MAPK mitigated injury, suggesting that p38 MAPK contributes to tissue damage during LIRI and may be a good therapeutic target.
The blood–air barrier contributes to pulmonary gas exchange (Bärnthaler et al., 2017), which is compromised during LIRI when the microvascular endothelial layer is damaged (Zhang et al., 2015). Subsequent inflammatory cell infiltration and edema are important features of acute lung injury (Herold et al., 2013) as well as inflammation driven by IL-1β and IL-6 (Bosmann and Ward, 2013). Consistently, I/R in vivo and OGD/R in vitro both increased the high permeability of the endothelial barrier, which may be driven by the increased level of IL-1β and IL-6 and contribute to inflammatory cell infiltration and edema. Inhibition of p38 MAPK attenuated the increase of IL-1β and IL-6 in the lung and BALF, providing a potential target for inflammation and blood–air barrier damage during LIRI.
Edema resulting from lung injury is inversely related to levels of AQP1, which can reduce lung edema (Han et al., 2015; Xu et al., 2016; Li et al., 2018). Consistently, we found that the level of AQP1 expression decreased due to injury in vivo and in vitro but was partially restored when p38 MAPK was inhibited. Conversely, ICAM-1 contributes to injury by altering cell junctions or the cytoskeleton to increase microvascular endothelial cell permeability (Clark et al., 2007). In rats exposed to lung I/R, knockdown of p38 MAPK downregulated ICAM-1 and mitigated the resulting injury (Lv et al., 2012). In our study, we detected increased levels of ICAM-1 at sites of structural destruction, and we found that p38 MAPK inhibition or knockdown attenuated I/R-induced upregulation of ICAM-1 in vivo and in vitro, which was associated with less severe damage to cellular morphology and tight junctions. Collectively, our results illustrate that knockdown of p38 MAPK can affect positive and negative regulators of endothelial architecture.
Likewise, we found that p38 MAPK can regulate specific mediators of tight junctions that maintain the endothelial cell barrier. ZO-1 forms part of the endothelial tight junctions and helps maintain the integrity of the endothelial barrier (Tornavaca et al., 2015). VE-cadherin is an adhesion protein located specifically at the tight junctions of endothelial cells (Wu et al., 2016), and F-actin is the main cytoskeletal contraction protein, which is important for maintaining the morphology and normal function of cells (Deng et al., 2018). I/R induced the degradation of tight junctions and the accumulation of F-actin stress fibers, which results in the loss of endothelial barrier function (Deng et al., 2018). We found that ZO-1 and VE-cadherin were dramatically downregulated by LIRI or OGD/R and that p38 MAPK inhibition reversed these effects. Our results are consistent with previous work linking p38 MAPK to ZO-1. Inhibition of p38 MAPK has been shown to increase the expression of ZO-1, thereby alleviating dysfunction of the human endothelial barrier induced by high mobility group tbox1 protein (Luan et al., 2018). Clostridium tyrobutyricum inhibits the p38 MAPK signaling pathway to upregulate ZO-1 and thereby protect against dysfunction of the intestinal barrier induced by lipopolysaccharide (Xiao et al., 2018). Our results and previous studies suggest that inhibition of p38 MAPK can protect tight junctions and the cytoskeleton to maintain the integrity of the endothelial barrier in the face of I/R injury.
Our study with in vivo and in vitro injury models suggests that inhibiting p38 MAPK mitigates LIRI and I/R-induced inflammatory responses at least in part by restoring the integrity of the pulmonary blood–air barrier. Such inhibition may work by upregulating AQP1 and downregulating ICAM-1. Our findings support antagonizing p38 MAPK as a new therapeutic approach to limit LIRI and, ultimately, acute lung injury.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
The animal study was reviewed and approved by Guangxi Medical University Institutional Animal Care and Use Committee.
TW curated data and wrote the original draft of the manuscript. CL handled the animal model, IHC, WB, and QPCR experiments and wrote part of the manuscript. L-hP analyzed data. ZL collected microscopy images. C-lL performed a literature search, and J-yL performed statistical analysis. YH collected data, and J-yX handled the animal model. SW and YQ did QPCR and WB. ZL and FL wrote and edited the manuscript. FL conceptualized and managed the project.
This study was supported by the National Natural Science Foundation of China (81560018, 8196022, and 81900234), the 2017 Young Anesthesiologist Research Fund of the Anesthesiology Branch of the Chinese Medical Doctor Association, the Guangxi Thousands of Young and Middle-aged Backbone Teacher Training Program and Guangxi Medical High-level Talents Program (G201903011), the Postdoctoral Innovative Talent Support Program (BX20180393), and the China Postdoctoral Science Foundation (2018M640859).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Bärnthaler, T., Maric, J., Platzer, W., Konya, V., Theiler, A., Hasenöhrl, C., et al. (2017). The role of PGE2 in alveolar epithelial and lung microvascular endothelial crosstalk. Sci. Rep. 7, 7923. doi:10.1038/s41598-017-08228-y
Bosmann, M., and Ward, P. A. (2013). The inflammatory response in sepsis. Trends Immunol. 34, 129–136. doi:10.1016/j.it.2012.09.004
Boyle, D. L., Jones, T. L., Hammaker, D., Svensson, C. I., Rosengren, S., Albani, S., et al. (2006). Regulation of peripheral inflammation by spinal p38 MAP kinase in rats. PLoS Medicine 3, e338. doi:10.1371/journal.pmed.0030338
Clark, P. R., Manes, T. D., Pober, J. S., and Kluger, M. S. (2007). Increased ICAM-1 expression causes endothelial cell leakiness, cytoskeletal reorganization and junctional alterations. J. Invest. Dermatol. 127, 762–774. doi:10.1038/sj.jid.5700670
Dai, H., Pan, L., Lin, F., Ge, W., Li, W., and He, S. (2015). Mechanical ventilation modulates Toll-like receptors 2, 4, and 9 on alveolar macrophages in a ventilator-induced lung injury model. J. Thorac. Dis. 7, 616–624. doi:10.3978/j.issn.2072-1439.2015.02.10
de Perrot, M., Liu, M., Waddell, T. K., and Keshavjee, S. (2003). Ischemia-reperfusion-induced lung injury. Am. J. Respir. Crit. Care Med. 167, 490–511. doi:10.1164/rccm.200207-670SO
Deng, R., Li, F., Wu, H., Wang, W. Y., Dai, L., Zhang, Z. R., et al. (2018). Anti-inflammatory mechanism of geniposide: inhibiting the hyperpermeability of fibroblast-like synoviocytes via the RhoA/p38MAPK/NF-κB/F-actin signal pathway. Front. Pharmacol. 9, 105. doi:10.3389/fphar.2018.00105
Fei, L., Jifeng, F., Tiantian, W., Yi, H., and Linghui, P. (2017). Glycyrrhizin ameliorate ischemia reperfusion lung injury through downregulate TLR2 signaling cascade in alveolar macrophages. Front. Pharmacol. 8, 389. doi:10.3389/fphar.2017.00389
Gao, M., Chen, L., Yu, H., Sun, Q., Kou, J., and Yu, B. (2013). Diosgenin down-regulates NF-κB p65/p50 and p38MAPK pathways and attenuates acute lung injury induced by lipopolysaccharide in mice. Int. Immunopharm. 15, 240–245. doi:10.1016/j.intimp.2012.11.019
Han, G., Ma, L., Guo, Y., Li, L., Li, D., and Liu, H. (2015). Hyperbaric oxygen therapy palliates lipopolysaccharide-induced acute lung injury in rats by upregulating AQP1 and AQP5 expression. Exp. Lung Res. 41, 444–449. doi:10.3109/01902148.2015.1064189
Herold, S., Gabrielli, N. M., and Vadász, I. (2013). Novel concepts of acute lung injury and alveolar-capillary barrier dysfunction. Am. J. Physiol. Lung Cell Mol. Physiol. 305, L665–L681. doi:10.1152/ajplung.00232.2013
Hoffmann, A., Bredno, J., Wendland, M., Derugin, N., Ohara, P., and Wintermark, M. (2011). High and low molecular weight fluorescein isothiocyanate (FITC)-Dextrans to assess blood-brain barrier disruption: technical considerations. Transl. Stroke Res. 2, 106–111. doi:10.1007/s12975-010-0049-x
Hua, Y., Ying, X., Qian, Y., Liu, H., Lan, Y., Xie, A., et al. (2019). Physiological and pathological impact of AQP1 knockout in mice. Biosci. Rep. 39, BSR20182303. doi:10.1042/BSR20182303
Kim, D. Y., Zhang, H., Park, S., Kim, Y., Bae, C. R., Kim, Y. M., et al. (2020). CU06-1004 (endothelial dysfunction blocker) ameliorates astrocyte end-feet swelling by stabilizing endothelial cell junctions in cerebral ischemia/reperfusion injury. J. Mol. Med. (Berl). 98, 875–886. doi:10.1007/s00109-020-01920-z
Kling, K. M., Lopez-Rodriguez, E., Pfarrer, C., Mühlfeld, C., and Brandenberger, C. (2017). Aging exacerbates acute lung injury-induced changes of the air-blood barrier, lung function, and inflammation in the mouse. Am. J. Physiol. Lung Cell Mol. Physiol. 312, L1–L12. doi:10.1152/ajplung.00347.2016
Lau, C. L., Zhao, Y., Kim, J., Kron, I. L., Sharma, A., Yang, Z., et al. (2009). Enhanced fibrinolysis protects against lung ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 137, 1241–1248. doi:10.1016/j.jtcvs.2008.12.029
Lee, J. C., Christie, J. D., and Keshavjee, S. (2010). Primary graft dysfunction: definition, risk factors, short- and long-term outcomes. Semin. Respir. Crit. Care Med. 31, 161–171. doi:10.1055/s-0030-1249111
Li, H., Shi, H., Gao, M., Ma, N., and Sun, R. (2018). Long non-coding RNA CASC2 improved acute lung injury by regulating miR-144-3p/AQP1 axis to reduce lung epithelial cell apoptosis. Cell Biosci. 8, 15. doi:10.1186/s13578-018-0205-7
Li, L., Hu, J., He, T., Zhang, Q., Yang, X., Lan, X., et al. (2015). P38/MAPK contributes to endothelial barrier dysfunction via MAP4 phosphorylation-dependent microtubule disassembly in inflammation-induced acute lung injury. Sci. Rep. 5, 8895. doi:10.1038/srep08895
Li, X. X., Ling, S. K., Hu, M. Y., Ma, Y., Li, Y., and Huang, P. L. (2019). Protective effects of acarbose against vascular endothelial dysfunction through inhibiting Nox4/NLRP3 inflammasome pathway in diabetic rats. Free Radic. Biol. Med. 145, 175–186. doi:10.1016/j.freeradbiomed.2019.09.015
Li, Z., Yin, M., Zhang, H., Ni, W., Pierce, R., Zhou, H. J., et al. (2020). BMX represses thrombin-PAR1-mediated endothelial permeability and vascular leakage during early sepsis. Circ. Res. 126, 471–485. doi:10.1161/CIRCRESAHA.119.315769
Liu, S., Feng, G., Wang, G. L., and Liu, G. J. (2008). p38MAPK inhibition attenuates LPS-induced acute lung injury involvement of NF-κB pathway. Eur. J. Pharmacol. 584, 159–165. doi:10.1016/j.ejphar.2008.02.009
Luan, Z., Hu, B., Wu, L., Jin, S., Ma, X., Zhang, J., et al. (2018). Unfractionated heparin alleviates human lung endothelial barrier dysfunction induced by high mobility group box 1 through regulation of P38-GSK3β-snail signaling pathway. Cell. Physiol. Biochem. 46, 1907–1918. doi:10.1159/000489375
Lv, X., Tan, J., Liu, D., Wu, P., and Cui, X. (2012). Intratracheal administration of p38α short-hairpin RNA plasmid ameliorates lung ischemia-reperfusion injury in rats. J. Heart Lung Transplant. 31, 655–662. doi:10.1016/j.healun.2012.03.001
Ma, B., Xiang, Y., Li, T., Yu, H. M., and Li, X. J. (2004). Inhibitory effect of topiramate on Lewis lung carcinoma metastasis and its relation with AQP1 water channel. Acta Pharmacol. Sin. 25, 54–60.
Qi, D., Tang, X., He, J., Wang, D., Zhao, Y., Deng, W., et al. (2016). Omentin protects against LPS-induced ARDS through suppressing pulmonary inflammation and promoting endothelial barrier via an Akt/eNOS-dependent mechanism. Cell Death Dis. 7, e2360. doi:10.1038/cddis.2016.265
Ryou, M. G., Choudhury, G. R., Li, W., Winters, A., Yuan, F., Liu, R., et al. (2015). Methylene blue-induced neuronal protective mechanism against hypoxia-reoxygenation stress. Neuroscience 301, 193–203. doi:10.1016/j.neuroscience.2015.05.064
Shen, C. C., Huang, H. M., Ou, H. C., Chen, H. L., Chen, W. C., and Jeng, K. C. (2004). Protective effect of nicotinamide on neuronal cells under oxygen and glucose deprivation and hypoxia/reoxygenation. J. Biomed. Sci. 11, 472–481. doi:10.1159/000077897
Sivaraman, V., Pechous, R. D., Stasulli, N. M., Eichelberger, K. R., Miao, E. A., and Goldman, W. E. (2015). Yersinia pestis activates both IL-1β and IL-1 receptor antagonist to modulate lung inflammation during pneumonic plague. PLoS Pathog. 11, e1004688. doi:10.1371/journal.ppat.1004688
Song, L., Li, D., Wang, J., Meng, C., and Cui, X. (2017). Effects of p38 mitogen-activated protein kinase on lung ischemia-reperfusion injury in diabetic rats. J. Surg. Res. 216, 9–17. doi:10.1016/j.jss.2017.03.024
Su, X., Song, Y., Jiang, J., and Bai, C. (2004). The role of aquaporin-1 (AQP1) expression in a murine model of lipopolysaccharide-induced acute lung injury. Respir. Physiol. Neurobiol. 142, 1–11. doi:10.1016/j.resp.2004.05.001
Sun, Q., Wu, Y., Zhao, F., and Wang, J. (2017). Maresin 1 ameliorates lung ischemia/reperfusion injury by suppressing oxidative stress via activation of the Nrf-2-mediated HO-1 signaling pathway. Oxid. Med. Cell. Longev. 2017, 9634803. doi:10.1155/2017/9634803
Tan, J., Liu, D., Lv, X., Wang, L., Zhao, C., Che, Y., et al. (2013). MAPK mediates inflammatory response and cell death in rat pulmonary microvascular endothelial cells in an ischemia-reperfusion model of lung transplantation. J. Heart Lung Transplant. 32, 823–831. doi:10.1016/j.healun.2013.05.005
Tornavaca, O., Chia, M., Dufton, N., Almagro, L. O., Conway, D. E., Randi, A. M., et al. (2015). ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation. J. Cell Biol. 208, 821–838. doi:10.1083/jcb.201404140
Towne, J. E., Harrod, K. S., Krane, C. M., and Menon, A. G. (2000). Decreased expression of aquaporin (AQP)1 and AQP5 in mouse lung after acute viral infection. Am. J. Respir. Cell Mol. Biol. 22, 34–44. doi:10.1165/ajrcmb.22.1.3818
Wada, M., Canals, D., Adada, M., Coant, N., Salama, M. F., Helke, K. L., et al. (2017). P38 delta MAPK promotes breast cancer progression and lung metastasis by enhancing cell proliferation and cell detachment. Oncogene 36, 6649–6657. doi:10.1038/onc.2017.274
Wang, X., Sun, Z., Börjesson, A., and Andersson, R. (1999). Inhibition of platelet-activating factor, intercellular adhesion molecule 1 and platelet endothelial cell adhesion molecule 1 reduces experimental pancreatitis-associated gut endothelial barrier dysfunction. Br. J. Surg. 86, 411–416. doi:10.1046/j.1365-2168.1999.01028.x
Wei, D., Huang, Z. H., Zhang, R. H., Wang, C. L., Xu, M. J., Liu, B. J., et al. (2014). Roles of p38 MAPK in the regulation of the inflammatory response to swine influenza virus-induced acute lung injury in mice. Acta Virol. 58, 374–379. doi:10.4149/av_2014_04_374
West, T., Atzeva, M., and Holtzman, D. M. (2006). Caspase-3 deficiency during development increases vulnerability to hypoxic-ischemic injury through caspase-3-independent pathways. Neurobiol. Dis. 22, 523–537. doi:10.1016/j.nbd.2005.12.017
Wu, Z., Wang, Z., Dai, F., Liu, H., Ren, W., Chang, J., et al. (2016). Dephosphorylation of Y685-VE-cadherin involved in pulmonary microvascular endothelial barrier injury induced by angiotensin II. Mediat. Inflamm. 2016, 8696481. doi:10.1155/2016/8696481
Xiao, Y. T., Yan, W. H., Cao, Y., Yan, J. K., and Cai, W. (2017). P38 MAPK pharmacological inhibitor SB203580 alleviates total parenteral Nutrition-induced loss of intestinal barrier function but promotes hepatocyte lipoapoptosis. Cell. Physiol. Biochem. 41, 623–634. doi:10.1159/000457933
Xiao, Z., Liu, L., Tao, W., Pei, X., Wang, G., and Wang, M. (2018). Clostridium tyrobutyricum protect intestinal barrier function from LPS-induced apoptosis via P38/JNK signaling pathway in IPEC-J2 cells. Cell. Physiol. Biochem. 46, 1779–1792. doi:10.1159/000489364
Xiong, L. L., Tan, Y., Ma, H. Y., Dai, P., Qin, Y. X., Yang, R. A., et al. (2016). Administration of SB239063, a potent p38 MAPK inhibitor, alleviates acute lung injury induced by intestinal ischemia reperfusion in rats associated with AQP4 downregulation. Int. Immunopharm. 38, 54–60. doi:10.1016/j.intimp.2016.03.036
Xu, J., Huang, B., Wang, Y., Tong, C., Xie, P., Fan, R., et al. (2016). Emodin ameliorates acute lung injury induced by severe acute pancreatitis through the up-regulated expressions of AQP1 and AQP5 in lung. Clin. Exp. Pharmacol. Physiol. 43, 1071–1079. doi:10.1111/1440-1681.12627
Yamazaki, Y., Shinohara, M., Shinohara, M., Yamazaki, A., Murray, M. E., Liesinger, A. M., et al. (2019). Selective loss of cortical endothelial tight junction proteins during Alzheimer's disease progression. Brain : a journal of neurology 142, 1077–1092. doi:10.1093/brain/awz011
Zhang, D., Li, C., Zhou, J., Song, Y., Fang, X., Ou, J., et al. (2015). Autophagy protects against ischemia/reperfusion-induced lung injury through alleviating blood-air barrier damage. J. Heart Lung Transplant. 34, 746–755. doi:10.1016/j.healun.2014.12.008
Zhang, H. W., Ding, J. D., Zhang, Z. S., Zhao, S. S., Duan, K. Y., Zhu, B. Q., et al. (2020a). Critical role of p38 in spinal cord injury by regulating inflammation and apoptosis in a rat model. Spine 45, E355–E363. doi:10.1097/BRS.0000000000003282
Zhang, J., He, Y., Yan, X., Chen, S., He, M., Lei, Y., et al. (2020b). MicroRNA-483 amelioration of experimental pulmonary hypertension. EMBO Molecular Medicine 12, e11303. doi:10.15252/emmm.201911303
Zhang, S., Hu, X., Guo, S., Shi, L., He, Q., Zhang, P., et al. (2019). Myricetin ameliorated ischemia/reperfusion-induced brain endothelial permeability by improvement of eNOS uncoupling and activation eNOS/NO. J. Pharmacol. Sci. 140, 62–72. doi:10.1016/j.jphs.2019.04.009
Zhang, X., Yeung, P. K., McAlonan, G. M., Chung, S. S., and Chung, S. K. (2013). Transgenic mice over-expressing endothelial endothelin-1 show cognitive deficit with blood-brain barrier breakdown after transient ischemia with long-term reperfusion. Neurobiol. Learn. Mem. 101, 46–54. doi:10.1016/j.nlm.2013.01.002
Keywords: p38 mitogen-activated protein kinase, lung ischemia reperfusion injury, blood-air barrier hyperpermeability, zonulae occludente 1, vascular endothelial cadherin
Citation: Wang T, Liu C, Pan L-h, Liu Z, Li C-l, Lin J-y, He Y, Xiao J-y, Wu S, Qin Y, Li Z and Lin F (2020) Inhibition of p38 MAPK Mitigates Lung Ischemia Reperfusion Injury by Reducing Blood–Air Barrier Hyperpermeability. Front. Pharmacol. 11:569251. doi: 10.3389/fphar.2020.569251
Received: 03 June 2020; Accepted: 29 October 2020;
Published: 11 December 2020.
Edited by:
Chrishan S. Samuel, Monash University, AustraliaReviewed by:
Simon Royce, Monash College, AustraliaCopyright © 2020 Wang, Pan, Liu, Li, Lin, He, Xiao, Li and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhao Li, NDM1MDM2NDc3QHFxLmNvbQ==; Fei Lin, Zmx5bGluMDIwOEAxNjMuY29t
† These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.