 Julie Schnipper
Julie Schnipper Isabelle Dhennin-Duthille1
Isabelle Dhennin-Duthille1 Halima Ouadid-Ahidouch
Halima Ouadid-Ahidouch- 1Laboratory of Cellular and Molecular Physiology, UR-4667, University of Picardie Jules Verne, Amiens, France
- 2Department of Biology, Faculty of Sciences, Ibn Zohr University, Agadir, Morocco
Pancreatic ductal adenocarcinoma (PDAC) is the fourth most common cause of cancer-related deaths in United States and Europe. It is predicted that PDAC will become the second leading cause of cancer-related deaths during the next decades. The development of PDAC is not well understood, however, studies have shown that dysregulated exocrine pancreatic fluid secretion can contribute to pathologies of exocrine pancreas, including PDAC. The major roles of healthy exocrine pancreatic tissue are secretion of enzymes and bicarbonate rich fluid, where ion channels participate to fine-tune these biological processes. It is well known that ion channels located in the plasma membrane regulate multiple cellular functions and are involved in the communication between extracellular events and intracellular signaling pathways and can function as signal transducers themselves. Hereby, they contribute to maintain resting membrane potential, electrical signaling in excitable cells, and ion homeostasis. Despite their contribution to basic cellular processes, ion channels are also involved in the malignant transformation from a normal to a malignant phenotype. Aberrant expression and activity of ion channels have an impact on essentially all hallmarks of cancer defined as; uncontrolled proliferation, evasion of apoptosis, sustained angiogenesis and promotion of invasion and migration. Research indicates that certain ion channels are involved in the aberrant tumor growth and metastatic processes of PDAC. The purpose of this review is to summarize the important expression, localization, and function of ion channels in normal exocrine pancreatic tissue and how they are involved in PDAC progression and development. As ion channels are suggested to be potential targets of treatment they are furthermore suggested to be biomarkers of different cancers. Therefore, we describe the importance of ion channels in PDAC as markers of diagnosis and clinical factors.
Introduction
Ion channels are plasma membrane spanning proteins found in all human tissues, allowing rapid transport of ions and fluids between the extracellular and intracellular milieu (Niemeyer et al., 2001; Gouaux and Mackinnon, 2005). Opening of ion channels can result in redistribution of different ions, which changes the electrical and chemical properties of the cell leading to several cellular processes (Roux, 2017). These include multiple signal transduction and downstream signaling events, including regulation of gene expression, secretion of enzymes and hormones, and intracellular communication between compartments (Chen et al., 1994; Tolon et al., 1996; Stock and Schwab, 2015). A stable regulation of these processes maintains normal tissue homeostasis, such as cell cycle progression, migration, and apoptosis (Kunzelmann, 2005; Kunzelmann, 2016; Prevarskaya et al., 2018; Anderson et al., 2019). Accordingly, dysregulated expression as well as altered function of ion channels are related to a great number of diseases (Kim, 2014), and can drive the transformation from normal to malignant cell behavior (Litan and Langhans, 2015). Over the past decades, aberrant and even cancer-specific expression of numerous ion channels have been demonstrated in various types of cancers (Pedersen and Stock, 2013; Djamgoz et al., 2014). Together, the abnormal expression and activity of ion channels can be categorized as “hallmarks of cancer” (Hanahan and Weinberg, 2011).
The pancreas is a complex organ, which has two main functions exerted by an exocrine and endocrine compartment (Pandiri, 2014). Dysregulation of exocrine pancreatic fluid secretion can contribute to pathologies such as pancreatitis and neoplasms such as pancreatic ductal adenocarcinoma (PDAC), whereas a well-known disorder related to dysfunction of the endocrine pancreas is diabetes mellitus (Pallagi et al., 2015; Kirkegard et al., 2017). The exocrine pancreas ensures enzymatic secretion for digesting fats and proteins in the intestines and, in parallel, the secretion of abundant fluid rich in bicarbonate ions, which serves to neutralize the acidic chime in the duodenum (Ishiguro et al., 2012; Lee et al., 2012; Pallagi et al., 2015). The bicarbonate secretion involves a tightly coordinated network of ion channels and transporters (Novak et al., 2013). The ductal epithelial cells comprising the exocrine pancreas are, as other types of epithelia, well-organized and exhibit epithelial features such as a polarized morphology and specialized cell-to-cell contact with tight junctions (Rodriguez-Boulan and Nelson, 1989). The ductal cells are equipped with a highly polarized set of ion channels and transporters, enabling the net bicarbonate excretion at the apical membrane, balanced by the net efflux of acid via the basolateral membrane to maintain their intracellular pH (Steward et al., 2005). Therefore, a correct distribution of ion channels and transporters is important to maintain the secreting function of exocrine pancreas (Lee et al., 2012). Moreover, expression, function, and localization of ion channels in the plasma membrane are involved in the development and progression of PDAC (Pedersen et al., 2017). PDAC can arise from ductal cells (Schneider et al., 2005) or from acinar cells transforming to ductal cells by acinar–to-ductal-metaplasia, resulting in these cells possessing a ductal phenotype (Aichler et al., 2012). The transformation-associated loss of cell polarity and cell-cell adhesions of the epithelial cell layer will result in an altered localization of ion channels (Coradini et al., 2011; Pedersen and Stock, 2013).
Several reports and reviews about the role of transporters in bicarbonate, pancreatic fluid secretion and PDAC have been published (Novak, 2000; Lee et al., 2001; Novak et al., 2011; Ishiguro et al., 2012; Lee et al., 2012; Kong et al., 2014; Lemstrova et al., 2014; Pedersen et al., 2017; Yamaguchi et al., 2017). However, the role of ion channels in exocrine pancreas and in PDAC is not well understood. In this review, we aim to make a synthesis of the important role of ion channels and their localization and function in fluid secretion in healthy exocrine pancreatic tissue (see Table 1 and Figure 1). Next, we summarize the sparse knowledge of the involvement of ion channels in PDAC progression and development via effects on proliferation, apoptosis, invasion and migration (see Table 2 and Figure 2). Finally, we describe how ion channels are important novel biomarkers in PDAC (see Table 2 and Figure 3).
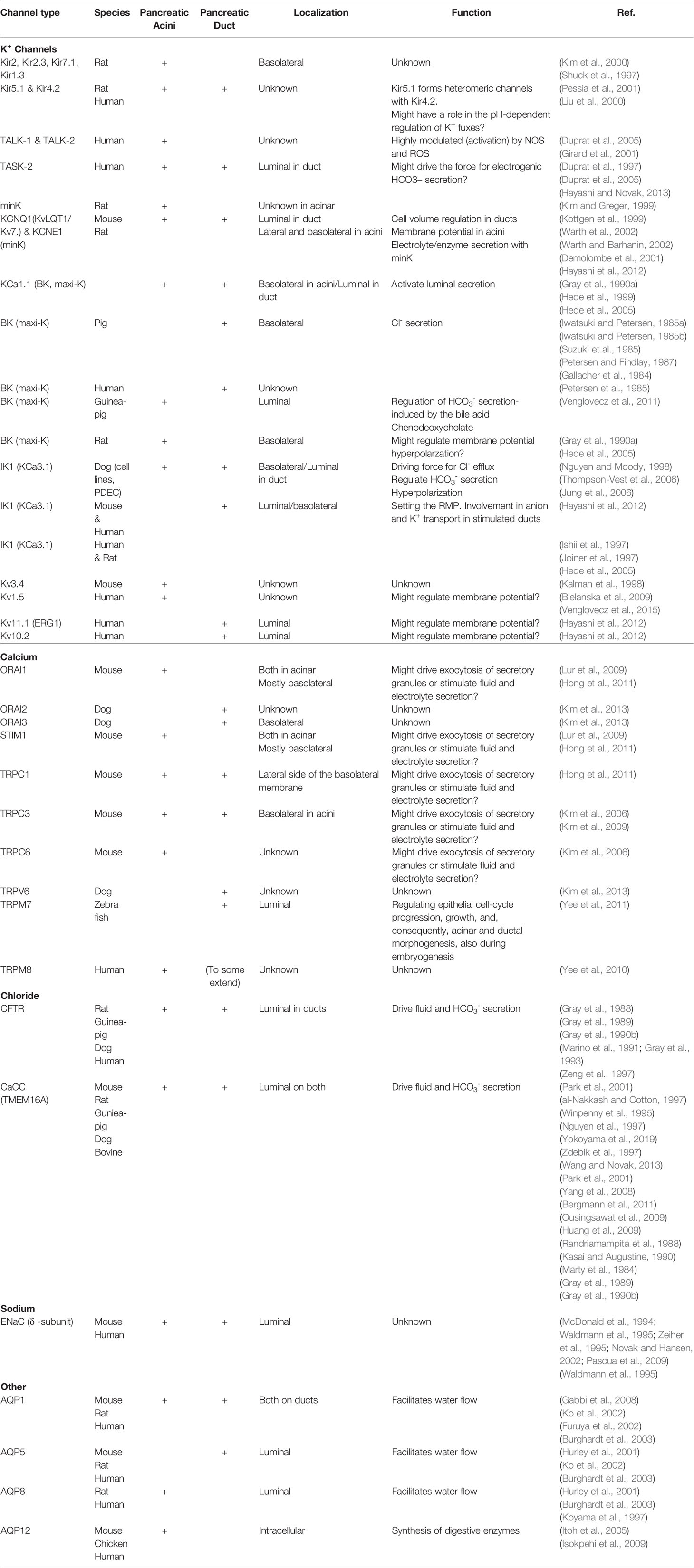
Table 1 Expression, localization, and the potential role of ion channels in exocrine pancreas.
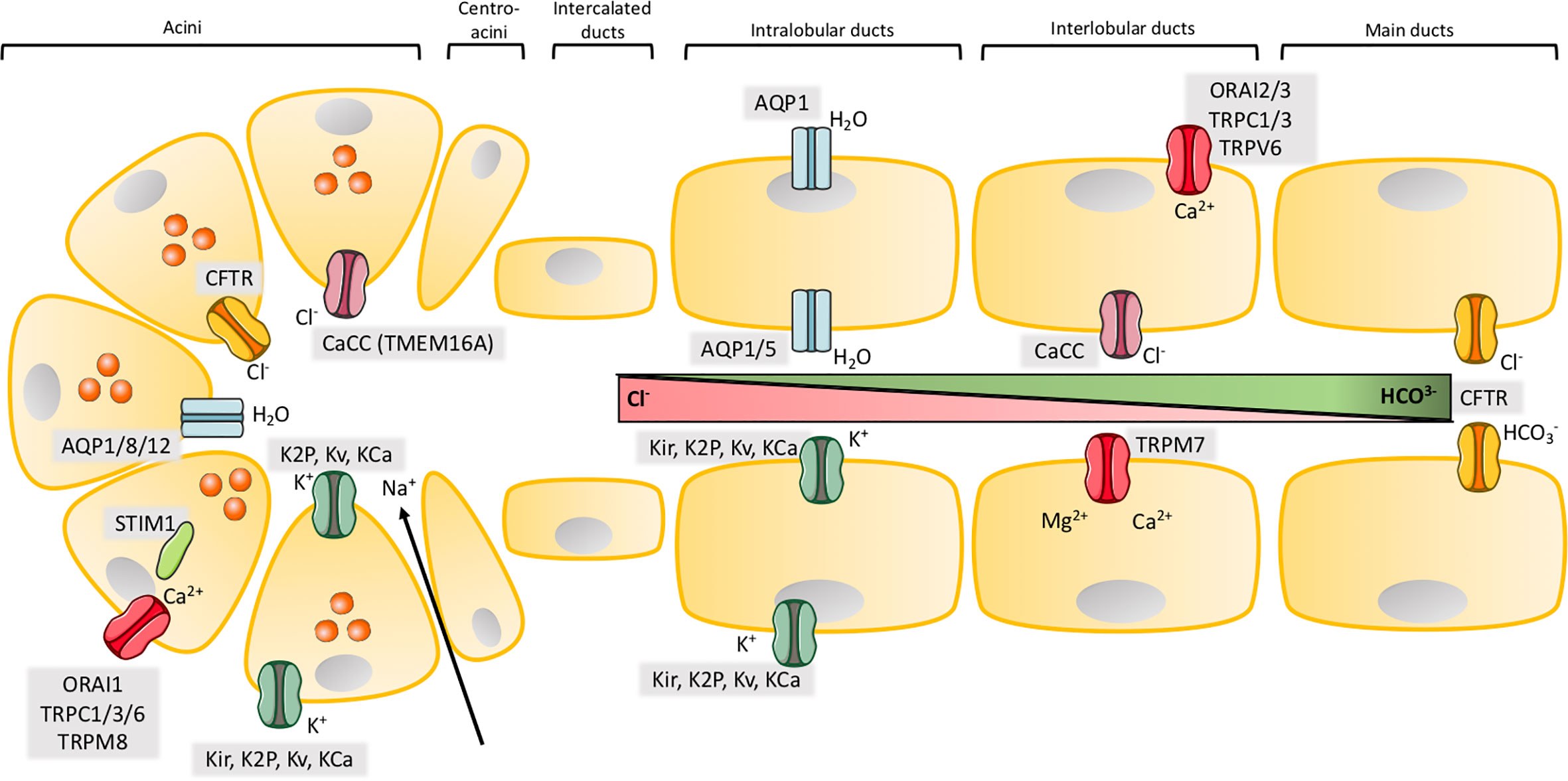
Figure 1 Ion channels in exocrine pancreas. Illustration of the structure of acinar and major ductal segments of secretory glands in pancreas. Acinar cells secrete digestive enzymes (orange circles in acini) and an isotonic NaCl rich fluid which transports the enzymes to the ducts. Fluid secretion in acini cells is regulated by a Cl- secretion process. Cl- secretion is activated by [Ca2+]i, from a Ca2+ influx through SOCs in the basolateral membrane, where Cl- channels, Ca2+ activated Cl- channels (CaCC) and different types of K+ channels are activated to provide the efflux of their respected ions. K+ channels also create a driving force by maintaining a negative membrane potential. The negative charge mediated by a high concentration of Cl- ions results in transport of Na+ through tight junctions to the luminal space. NaCl makes the driving force for water to efflux through aquaporins and a cell shrinkage. This cell shrinkage reduces [Ca2+]i, which inhibits Cl- and K+ efflux through their channels and in parallel activates basolateral transporters and pumps to restore both Cl- and K+. The digestive enzymes are transported in the NaCl isotonic fluid to the ducts, which is low in HCO3- concentration in the proximal ducts, but this concentration increases through the transport to the distal duct cells. The ductal fluid becomes rich in HCO3-, by a two-step process. The first step takes place in the proximal ducts, where Cl-/HCO3- exchangers secretes HCO3- and absorb Cl- and Cl- channels recycle Cl-. As in the acinar cells an osmotic reaction happens, where efflux of negative HCO3- and Na+ drives water flow through aquaporins. This results in high concentration of HCO3- (~100 mM), a low concentration of Cl- (~25 mM) and a high fraction of water in the pancreatic juice. The second step takes place in the distal part of the ducts, where the specific Cl- channel CFTR changes selectivity to HCO3- and function as a HCO3- efflux channel to determine the final concentration of the HCO3- rich fluid (~140 mM). K+ channels may, as in acini, take part in the secretion of K+ and regulation of anion transport by maintaining the membrane potential in both the basolateral and luminal membrane. SOCs ensure the influx of Ca2+ which takes part in regulation of ion channels through [Ca2+]i as in acini. Activation or inhibition of P2 receptors by Ca2+ signaling also regulate anion secretion through K+ and Cl- channels.
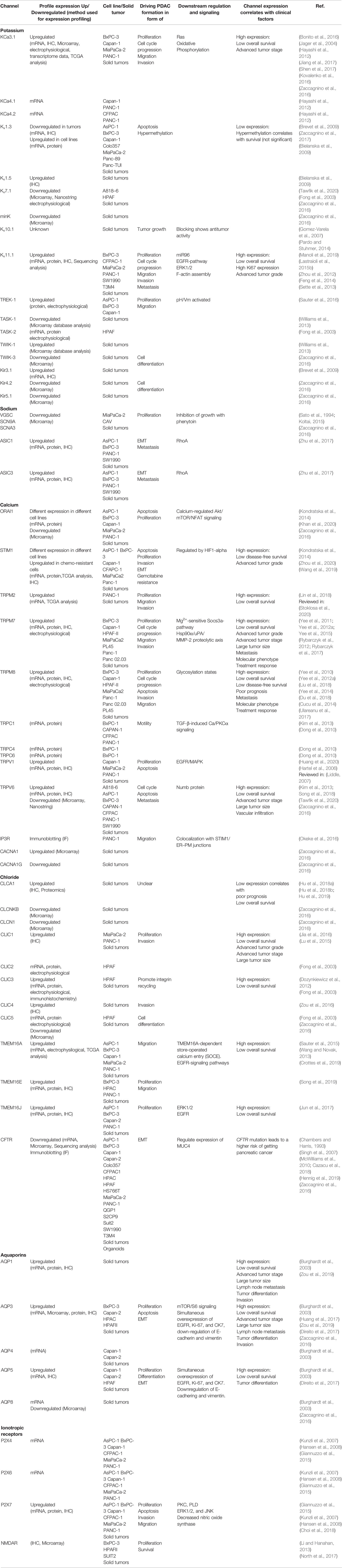
Table 2 Profile expression of ion channels in pancreatic ductal adenocarcinoma (PDAC) cell lines and tissue and how they are involved in driving PDAC formation and how channel expression correlate with clinical factors.

Figure 2 Ion channels in pancreatic ductal adenocarcinoma (PDAC). Illustration of ion channels, which have been shown to have a role in hallmarks of cancer, thereby PDAC development and progression. As cancer cells lose their polarity, the localization of the channels is unknown, and on the illustration, it should be considered that the channels have no particular localization. The aberrant expression in PDAC cells, are shown for; Store-operated channels (SOCs) and transient receptor potential (TRP) channels, K+ channels, Cl- channels, aquaporins (AQP), Na+ channels and P2X7R. These channels are known to be involved in PDAC development and progression through proliferation, cell cycle progression, differentiation, migration, invasion, metastasis, and apoptosis. The known pathways and mechanism, which have been shown to be involved in these processes are shown in a grey box next to the channel and are mentioned in Table 2. The channels shown to be expressed in PDAC, but where the role is unknown are also shown in Table 2.
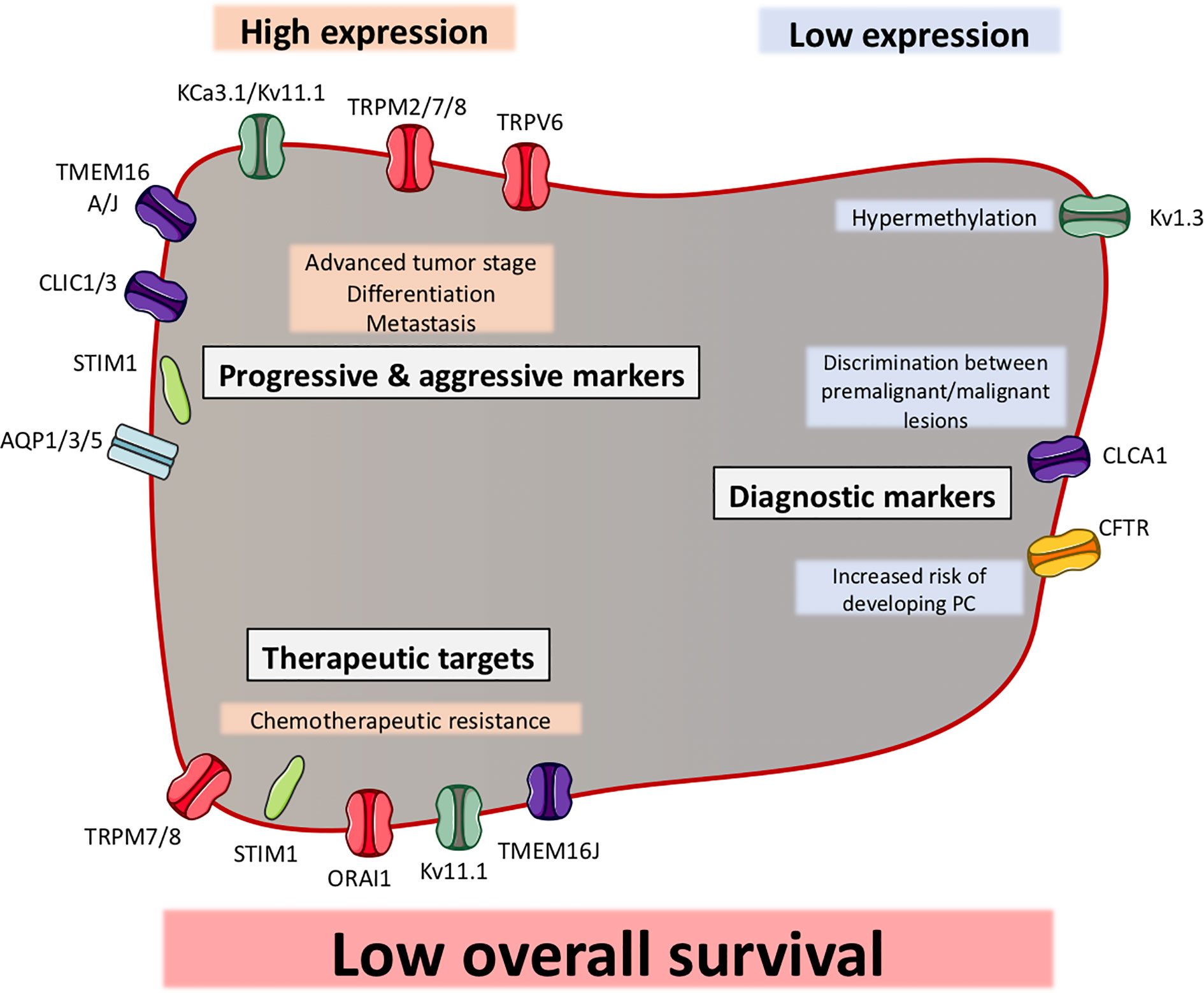
Figure 3 Ion channels can function as biomarkers in pancreatic ductal adenocarcinoma (PDAC). Illustration of ion channels, where the expression has been shown to be correlated with clinical factors. Most of the ion channels show a high expression in PDAC, which correlates with clinical factors (indicated in grey boxes). Some ion channels have shown to be to have a low expression in PDAC, which correlates with other clinical factors. The ion channels are grouped as progression and aggressiveness markers, diagnostic markers or therapeutic targets. Among all ion channels, their expression (except CFTR) have been shown to be correlated with a low overall survival.
Expression, Localization, and Role of Ion Channels in Healthy Pancreatic Epithelial Cells
Potassium Channels
The relevance of K+ channels in the exocrine pancreas received great attention in the 1970s–1980s, notably by Petersen’s team, thanks to electrophysiological studies of ion channels on acinar pancreatic epithelial cells dissociated from the pancreas of different animal species. While several studies have shown the expression of different families of K+ channels on both acinar and duct pancreatic cells (Petersen and Findlay, 1987; Bleich and Warth, 2000; Thevenod, 2002; Hayashi and Novak, 2013; Venglovecz et al., 2015), few studies have shown their physiological role in exocrine secretion.
Two excellent reviews have summarized the role of these channels in physiological process of ductal fluid secretion, likely by contributing to maintain the membrane potential and thereby providing driving forces for anion transport (Hayashi and Novak, 2013; Venglovecz et al., 2015). Among these channels the voltage- and Ca2+-activated K+, big conductance (BK, maxi-K), and the intermediate (IK, KCa3.1) Ca2+-activated K+ channels have been intensely studied in pancreatic ductal cells. The BK which is activated by cAMP and PKA is found on the basolateral membrane of rat pancreatic duct cells (Gray et al., 1990a). The authors suggest its role in pancreatic bicarbonate secretion. BK is also found mainly expressed in the apical membrane of guinea-pig non-transformed pancreatic duct epithelial cells (PDEC) (Venglovecz et al., 2011) where it regulates the bicarbonate secretion stimulated by the bile acid chenodeoxycholate likely through changes of the membrane potential. KCa3.1 was first characterized in cultured PDEC where it is expressed on the basolateral membrane of duct epithelial cells (Nguyen and Moody, 1998). Activation of P2Y2R induced an increase of free intracellular calcium ([Ca2+]i) that activates KCa3.1, which in turn hyperpolarized the membrane potential, leading to a Cl–dependent bicarbonate secretion (Jung et al., 2006). KCa3.1 was also found located on the basolateral and luminal membrane of pancreatic mouse and human duct cells (Hayashi et al., 2012). The same authors demonstrated that both luminal and basolateral KCa3.1 channels were involved in the regulation of membrane potential.
In acinar cells, the membrane potential created by K+ channels, and waves of [Ca2+]i provide the necessary driving force for Cl- efflux through the luminal membrane, which is a key step in initiating fluid and electrolyte secretion (Lee et al., 2012). The activation of K+ channels located on the basolateral membrane hyperpolarizes the resting membrane potential, promoting the driving force for luminal Cl- efflux through Cl- channels (Petersen, 2005). A Ca2+-dependent maxi- K+ channel (200 pS) has been characterized upon stimulation with acetylcholine (ACh), cholecystokinin (CCK), and bombesin in pancreatic acinar cells (Maruyama et al., 1983; Iwatsuki and Petersen, 1985a; Iwatsuki and Petersen, 1985b; Petersen et al., 1985; Suzuki et al., 1985; Petersen and Findlay, 1987). Moreover, Pearson et al. (1984) showed, on isolated pancreas acinar pig cells, that neural and hormonal (ACh, bombesin and pentagastrin) stimulation evokes a Ca2+-dependent cell hyperpolarization by causing an increase in membrane K+ conductance (Pearson et al., 1984). An intermediate Ca2+-activated K+ channel is also expressed in both the basolateral and the apical membranes of acinar cells (Thompson-Vest et al., 2006), but its role has not been studied.
KCNQ1 (KVLQT1, Kv7.1) and KCNE1 (IsK, minK) have been found in abundance in pancreatic acinar cells (Kottgen et al., 1999; Bleich and Warth, 2000; Demolombe et al., 2001; Warth and Barhanin, 2002; Warth et al., 2002; Hayashi et al., 2012). By using mouse models associated with electrophysiological studies, Warth et al. (2002) showed that KCNQ1 was predominantly located at the basolateral membrane and its co-assemblage with KCNE1 leads to a voltage-dependent K+ current that was increased by cholinergic stimulation and inhibited by the KCNQ1 blocker (Kim and Greger, 1999; Kottgen et al., 1999; Warth et al., 2002). The fact that inhibition of KCNQ1 channels diminishes intestinal Cl- secretion, made the authors suggest its involvement in pancreatic electrolyte secretion process.
K+ inwardly rectifying channels (Kir) channels are expressed in exocrine pancreas. Kir 2.1, Kir2.3, Kir7.1, Kir5.1, and Kir4.2 were detected in rat pancreatic acini (Kim et al., 2000; Pessia et al., 2001). In-situ hybridization analysis confirmed the expression of Kir5.1 in human pancreatic acinar and ductal cells (Liu et al., 2000). Moreover, it has been suggested that Kir5.1 forms heteromeric channels with Kir4.2 in rat pancreas and is involved in the pH-dependent regulation of K+ flux (Pessia et al., 2001). Kir1.3 was also detected by northern blot analysis, in human pancreas (Shuck et al., 1997). The 2-Pore K+ channel (K2P) family has also been found in human exocrine pancreas; however, their localization and function are still unknown. For example, TALK-1 and TALK-2 are very specifically expressed in exocrine pancreas where they are activated by NOS and ROS (Girard et al., 2001; Duprat et al., 2005), while TASK-2 is expressed in both exocrine and endocrine pancreas (Duprat et al., 1997; Duprat et al., 2005).
Calcium Channels
As Petersen and co-workers showed the relevance of K+ channels in exocrine pancreas, they have also described the role of Ca2+ signaling, in pancreatic acinar cells (Petersen, 2014). In the early 70’s they showed that movements of Ca2+ was evoked upon ACh stimulation released Ca2+ from intracellular stores and that only a small part of Ca2+ was taken up from the extracellular solution (Case and Clausen, 1973; Matthews et al., 1973). This Ca2+ signaling is involved in exocrine pancreatic fluid secretion as both acinar and duct cells in pancreas are regulated by receptors that change [Ca2+]i, which activates epithelial Ca2+-dependent K+ and Cl- ion channels, thereby enzyme and fluid secretion (Petersen, 2014). The Ca2+ signal is initiated by ACh or CCK, binding to specific receptors (Case and Clausen, 1973; Matthews et al., 1973; Petersen and Ueda, 1976), which generates specific Ca2+ signals. These signals start by Ca2+ activating phospholipase C, which hydrolyzes PIP2, hence generating IP3 and diacylglycerol. IP3 binds to IP3 receptors located in the ER at the apical pole of the acinar cells mediating a Ca2+ wave to the basal pole (Mogami et al., 1997; Hong et al., 2011). This evokes a Ca2+ ER store depletion that results in clustering of the ER Ca2+ sensor STIM1, which activates store-operated channels (SOCs) and transient receptor potential (TRP) channels, leading to Ca2+ influx (Petersen and Tepikin, 2008). Members and regulators of SOCs are the SOC channel pore-forming ORAI proteins (ORAI1-3) and their regulators STIM (STIM1-2) (Hoth and Niemeyer, 2013). ORAI1 is the best described among these and are found to be expressed at the apical membrane of pancreatic acinar cells where it colocalizes with IP3R (Hong et al., 2011; Lur et al., 2011) and at the basolateral membrane where it colocalizes with STIM1 (Lur et al., 2011). Recently, it has been shown that inhibition of ORAI1 in pancreatic acinar cells abolished SOC entry upon stimulation with thapsigargin, CCK, and the bile acid taurolithocholic acid 3-sulfate, indicating that ORAI1 mediates SOC entry in pancreatic acinar cells (Gerasimenko et al., 2013; Wen et al., 2015).
TRPC channels have also been found to participate or influence store-dependent Ca2+ influx in pancreatic acinar cells. TRPC1 was found to localize both at the apical and lateral regions of the basolateral membrane, and pancreatic acinar cells isolated from TRPC1-/- mice showed reduced Ca2+ influx and Ca2+ oscillation frequency (Hong et al., 2011). The role of TRPC1 in pancreatic acinar cells is not yet known, but it is suggested to have a similar role as in salivary glands, where they regulate fluid secretion and Ca2+ activated K+ channels (Liu et al., 2007). TRPC3 was found in the junctional site of the apical pole and the basolateral membrane of pancreatic acini cells and in TRPC3-/- mice a reduction of Ca2+ influx was seen (Kim et al., 2006). Furthermore, TRPC6 seemed to be expressed in the pancreatic acini cells, but its localization and role are unknown (Kim et al., 2006). These data suggest that TRPC channels are involved in the SOC entry of pancreatic acini cells and could contribute to fluid secretion. Other TRP channels have been found to be expressed in exocrine pancreas; TRPV6 (Kim et al., 2013), TRPM7 (Yee et al., 2011) and TRPM8 (Yee et al., 2010). However, only the role of TRPM7 is described. In a zebra fish model, it has been found that TRPM7 is involved in the developmental processes of exocrine pancreas, which was linked to Mg2+ signaling (Yee et al., 2011). Diminish of cell cycle progression and cell growth in TRPM7-mutated zebra fish models attenuated proliferation of exocrine pancreatic epithelia. This was partially rescued by adding extra Mg2+ to the embryo medium (Yee et al., 2011). Furthermore, the proliferation was also regulated by suppressor of cytokine signaling 3a (socs3a), indicating that TRPM7 plays a role in the development of exocrine pancreas (Yee et al., 2011), but the physiological role in fluid secretion is yet to be determined.
In duct cells, HCO3- secretion is mediated by cAMP/Ca2+ signaling systems. Through specific Ca2+ channels and Ca2+ activated ion channels (Ca2+-activated K+ and Cl- channels), Ca2+ can act as key player in regulation and secretion of pancreatic juices (Lee M. G. et al., 2012). The localization of SOCs in duct cells, due to HCO3- fluid secretion, is not well studied. However, it has been found that SOC-mediated Ca2+ influx can be a driving force for exocytosis, evoked by trypsin (Kim et al., 2008) in dog PDEC. The same authors have shown the function of SOCs in dog PDEC where the typical inward rectifying current was found, as for other types of epithelial cells (Kim et al., 2013). Furthermore, it was found that STIM1, STIM2, ORAI1, ORAI2, and ORAI3 as well as TRPC1 and TRPV6 are all expressed in dog PDEC, where ORAI3 was shown to be the dominant expressing type (Kim et al., 2013). Moreover, STIM1 and ORAI3 are colocalized in both single cell PDEC and polarized monolayers upon thapsigargin treatment (Kim et al., 2013). Using thapsigargin, the same authors showed an increased [Ca2+]i only at the basolateral membrane, indicating that SOCs are mainly located at this site of the plasma membrane (Kim et al., 2013). It might be hypothesized that the localization of SOCs and Ca2+-activated ion channels are the same in pancreatic duct cells as in acinar cells, and that they play a role in HCO3- secretion, as they play a role in enzyme and fluid secretion in acinar cells (Maleth and Hegyi, 2014).
Aquaporin Channels
Aquaporins (AQPs) are activated by Ca2+ and mediate a water flow through the luminal membrane. The role of some AQP types in physiological and pathophysiological processes of exocrine pancreas has already been reviewed (Burghardt et al., 2006; Delporte, 2014; Arsenijevic et al., 2019). AQP1 is expressed at the apical and basolateral membrane of centro-acinar cells and intercalated ductal cells (Burghardt et al., 2003; Burghardt et al., 2006) and is also expressed in capillary endothelial cells and at the pancreatic zymogen granule membrane (Cho et al., 2002; Burghardt et al., 2003). AQP5 has been found to be co-localized with AQP1 in the apical membrane of centro-acinar cells and intercalated ductal cells (Burghardt et al., 2003). Otherwise, AQP8 is expressed only in acinar cells in the apical membrane (Isokpehi et al., 2009). In the two-step process of pancreatic fluid secretion, AQP8 in the pancreatic acinar cells ensures the water flow across the plasma membrane, where NaCl makes the driving force. In the pancreatic ductal cells the driving force is maintained by HCO3- and Na+ through AQP1 and AQP5 (Burghardt et al., 2006). However, this theory is not well explained, since pancreatic fluid secretion was not found to be altered in AQP1, AQP5, AQP8, or AQP12 knockout mice (Ma et al., 2001; Yang et al., 2005; Ohta et al., 2009). Recently, the role of AQP1 has been confirmed to be involved in pancreatic fluid and bicarbonate secretion in an AQP1-knockout mouse model (Venglovecz et al., 2018).
Chloride Channels
In the early 80’s the evidence for Ca2+ activated Cl- channels (CaCC) were presented by whole-cell patch clamp and single-channel currents in rat lacrimal acinar. Marty and co-workers showed that Cl- currents were evoked by muscarinic receptor activation and Ca2+, as previously demonstrated for the K+ current (Marty et al., 1984; Petersen, 1992). Shortly after, following investigations confirmed this Ca2+ activated Cl- current in rat pancreatic acinar cells (Randriamampita et al., 1988). The localization of these Ca2+-dependent channels was proposed to be both on the basolateral and the luminal site, but speculations and further studies revealed that the localization of CaCC was found in the luminal membrane of pancreatic acinar cells, where an early activation of Cl- currents was seen upon ACh stimulation (Kasai and Augustine, 1990; Zdebik et al., 1997). A small delayed current was found after Ca2+ has spread to the basal pole of the cell, suggesting that CaCC are highly located at the luminal membrane and to some extent in the basolateral of pancreatic acinar cells (Kasai and Augustine, 1990). New evidence shows clearly that CaCC are exclusively localized to the apical membrane and regulate pancreatic fluid secretion (Marty et al., 1984; Kasai and Augustine, 1990; Park et al., 2001).
Gray and his team have investigated the properties and roles of Cl- channels in pancreatic duct epithelial cells. They and others, found two types of Cl- channels in pancreatic ducts cells; cystic fibrosis transmembrane conductance regulator (CFTR), regulated by rises in [cAMP]I and CaCC, regulated by an increase in [Ca2+]i (Gray et al., 1989; Riordan et al., 1989; Gray et al., 1990b; Gray et al., 1993; al-Nakkash and Cotton, 1997; Nguyen et al., 1997). Both types of channels have been found in several species and to be localized in the apical membrane of duct cells (Gray et al., 1990b; Marino et al., 1991; Ashton et al., 1993; Evans et al., 1996; al-Nakkash and Cotton, 1997; Zeng et al., 1997; Ishiguro et al., 2002; Wang and Novak, 2013; Yokoyama et al., 2019). CaCC have been found in rodent pancreatic ducts. Here, it was shown that increases in [Ca2+]i, evoked by either ionomycin or ACh activated the Cl- channels (Gray et al., 1990b; Gray et al., 1995). Furthermore, Cl- currents were detected in mouse pancreatic ducts with no detectable function of CFTR, which indicates that these currents are carried by an ion channel that is distinct from CFTR (Gray et al., 1994; Winpenny et al., 1995).
Until now, it has been shown that mammalian TMEM16 proteins have different physiological functions. TMEM16A and B are suggested to be CaCC, where both of TMEM16E and F are suggested to have scramblase and channel activities. TMEM16D, G, and J are suggested to only have a scramblase activity. Therefore, the channel nature of all TMEM16 proteins is still not clearly identified [Reviewed in (Falzone et al., 2018)]. Recently, it has been suggested that TMEM16A, of the TMEM16/Anoctamin family, is the CaCC gene candidate for Cl- secretion (Caputo et al., 2008; Schroeder et al., 2008; Yang et al., 2008). In rodent pancreatic acinar cells and intercalated ducts, expression of TMEM16A was found by immunostaining and RT-PCR (Huang et al., 2009; Yokoyama et al., 2019). The biophysical properties of the channel agreed with Ca2+-dependent Cl- currents, described elsewhere (Yang et al., 2008). Another study demonstrated that Ca2+-dependent Cl- secretion was defective in acinar cells from TMEM16A-null mice, indicating that TMEM16A has a physiological role in pancreatic fluid secretion (Ousingsawat et al., 2009).
The model of how Cl- is secreted through channels in exocrine pancreas is described as a two-step process, starting by the activation by ACh or CCK, which trigger an IP3-mediated rise of cytosolic Ca2+ (Iwatsuki and Petersen, 1977; Reubi et al., 2003; Gautam et al., 2005; Wang and Cui, 2007). In response to this stimulation, the NaCl rich fluid starts to be produced (Hegyi and Petersen, 2013). At the basolateral membrane the Na+-K+-2Cl- cotransporters (NKCC), Cl-/HCO3- exchangers and Na+/K+ pumps are activated, to function together to establish the Cl- uptake mechanism. The increased [Ca2+]i enhances the Cl- conductance of the luminal membrane and a K+ channel-mediated hyperpolarization of the basolateral membrane creates the driving force for Cl- efflux to the luminal space. At the apical membrane, Cl- ions pass through the Cl- channels. This hormonal stimulation by ACh and CCK, leading to increased [Ca2+]i, plays the central role in activating enzyme release and electrogenic Cl- secretion (Petersen and Gallacher, 1988; Mogami et al., 1997; Giovannucci et al., 2002; Petersen, 2005). While Cl- passes through the acinar cells a negative charge in the luminal space arises, which moves Na+ from the interstitial space to the acinar lumen via the paracellular pathway through leaky tight junctions, resulting in NaCl secretion. In physiological circumstances the acinar luminal Cl- concentration contains 135 mM Cl and 25 mM HCO3- (Park et al., 2010). The second step in pancreatic fluid secretion occurs in the duct cells and depends on the high concentration of luminal Cl- as it activates HCO3- efflux through Cl-/HCO3- exchangers, which elevates the luminal HCO3- concentration and thereby activates CFTR functioning to secrete Cl- and to some extend HCO3- (Ishiguro et al., 2009; Wilschanski and Novak, 2013). The HCO3- concentration in the fluid increases along the ducts, while the Cl- concentration reciprocally decreases. By the time the pancreatic fluid leaves the ducts the ratio is inverse, with the HCO3- concentration around 140 mM and the Cl- concentration around 20 mM (Park et al., 2010). These specific concentrations will inhibit CFTR and Cl-/HCO3- exchangers to prevent HCO3- reabsorption (Wright et al., 2004).
Sodium Channels
The efflux of Na+ through tight junctions in both the acinar and ductal cells is a part of regulating the HCO3- rich fluid to be isotonic and to keep the cell osmolarity (Lee M. G. et al., 2012). The expression and function of Na+ channels in normal pancreatic tissue are controversial. Some studies have shown functional expression of amiloride sensitive epithelial sodium channels (ENaC) in interlobular ducts from mice (Zeiher et al., 1995; Pascua et al., 2009). Moreover, transcripts of different subunits of ENaC have been also detected in human pancreas (McDonald et al., 1994; Waldmann et al., 1995; Novak and Hansen, 2002). Other studies have shown no functional activity of ENaC in isolated small ducts from rats or in PDAC cell lines Capan-1 and HPAF (Novak and Hansen, 2002; Fong et al., 2003; Wang and Novak, 2013), which is in accordance with the secretory nature of pancreatic ducts.
Expression of Ion Channels in PDAC Cells and Human Tissues, Function, and Associated Signaling Pathways in Cell Lines
Potassium Channels in PDAC
Kv Channels
It is widely accepted that Kv channels participate in cancer development and progression and their expression has shown to be aberrant in several types of tumor tissue, also in PDAC (Serrano-Novillo et al., 2019; Teisseyre et al., 2019). It has been shown that Kv1.3 is expressed in different human PDAC cell lines, harboring mutation in p53 (Zaccagnino et al., 2017). The authors demonstrate that the inhibition of Kv1.3 by clofazimine, induces apoptosis in-vitro and reduces tumor weight in-vivo (Zaccagnino et al., 2017). Another study has reported a remodeling of Kv1.3 and Kv1.5 on a large cohort of human tissue samples (Bielanska et al., 2009). In fact, they showed that protein expression of Kv1.3 was lower in PDAC tissue, while Kv1.5 had a higher protein expression in PDAC tissue compared to healthy tissue (Bielanska et al., 2009; Comes et al., 2013). The low expression of Kv1.3 in PDAC can be explained by a hypermethylation of the KCNA3 gene promoter (Brevet et al., 2009). Similar to Kv1.3 the expression of Kv7.1 has recently been shown to be down-regulated in PDAC (Zaccagnino et al., 2016; Tawfik et al., 2020). KCNQ1 (gene coding for Kv7.1) was downregulated in PDAC tissue, compared to normal tissue. In addition, downregulation of KCNQ1 was found in a system comparing PDAC A818–6 cells grown as a highly malignant undifferentiated monolayer (ML) or as three-dimensional (3D) single layer hollow spheres (HS). Database analysis showed that KCNQ1 was involved in the enrichment of pancreatic secretion in normal pancreatic epithelium and HS, suggesting that a downregulation of KCNQ1 might impair fluid secretion in PDAC and ML cells, while being maintained in normal pancreas and HS cells (Tawfik et al., 2020). Another comprehensive study has been investigating the gene-expression levels of the transportome in PDAC and normal specimens (Zaccagnino et al., 2016). The authors showed the downregulation of five different K+ channels, including the K+ voltage-gated channels; KCNQ1 and KCNE1. Moreover, their results showed a downregulation of genes coding for the Kir4.2 (KCNJ15), Kir5.1 (KCNJ16), and the K2P channel TWIK-3 (KCNK7). In addition, the expression of KCNJ15 and KCNK7 was associated with the expression of EMT transcription factors (Zaccagnino et al., 2016). The authors also suggested that the higher expression of K+ channels in normal pancreatic epithelium takes part in setting the resting membrane potential, which generates the driving force of fluid and ion secretion in the pancreatic ducts (Zaccagnino et al., 2016).
Kv10.1 is another Kv channel that has been reported in pancreatic cancer. The expression of Kv10.1 in peripheral tissues is very restricted (Hemmerlein et al., 2006), including pancreatic tissue (Pardo et al., 1999). A xenograft mouse model of pancreatic cancer showed that monoclonal antibodies blocking the Kv10.1 current exerts antitumor activity (Gomez-Varela et al., 2007). Because Kv10.1 is nearly absent in normal tissue, there is a certain tumor selectivity for Kv10.1 expression, which gives rise to the possibility that Kv10.1 can be used as a targeting channel for the delivery of cytotoxic compounds (Pardo and Stuhmer, 2014). However, the expression and function of Kv10.1 in PDAC must be further investigated.
Interestingly, another Kv channel, Kv11.1 has been implicated as an oncogene in various cancers, including PDAC (Arcangeli et al., 2014; Lastraioli et al., 2015a). In contrast, to the Kv10.1 expression, Kv11.1 is ubiquitously expressed in normal human tissues including heart where it is mainly expressed (Sanguinetti et al., 1995; McDonald et al., 1997; Pond et al., 2000; Camacho, 2006; Comes et al., 2015). KCNH2 (gene coding for Kv11.1) was identified as a gene with somatic mutations that could drive the metastatic process of PDAC (Zhou et al., 2012). Here, exome sequencing analysis showed that KCNH2 clustered into a single network related to cancer development. To investigate the involvement of KCNH2 in PDAC progression, the authors showed that knockdown of Kv11.1 reduced proliferation, colony formation and migration in PDAC cell lines. Immunohistochemical analysis of Kv11.1 expression showed expression in 8 out of 38 (21%) PDAC tissues, versus one out of 37 (2.7%) in normal tissues (Zhou et al., 2012). Another study further investigated the expression and role of Kv11.1 in PDAC (Feng et al., 2014). Here, immunohistochemical analysis confirmed a strong expression in PDAC tissues, with highest expression in the cytoplasm and membrane. In contrary, normal tissue showed only weak expression. The expression was confirmed in PDAC cell lines (Feng et al., 2014). Knockdown of Kv11.1 showed a significant decreased proliferation rate, higher number of cells undergoing apoptosis, cell cycle arrest in G1 phase and a reduction of migration and invasion, suggesting that Kv11.1 has a role in different aspects of PDAC progression (Feng et al., 2014). This was confirmed in a xenograft mouse model, were a knockdown of Kv11.1 in CFPAC-1 cells showed reduced tumor growth and fewer metastatic nodules, compared to tumors in mice injected with control cells (Feng et al., 2014). Furthermore, it was found that miR-96 was downregulated in tumor tissue and PDAC cells. The overexpression of miR-96 reduced cell proliferation, migration, and invasion in-vitro and reduced the Kv11.1 expression, tumor growth, and formation of metastasis in-vivo (Feng et al., 2014). This indicates that Kv11.1 could function as an oncogene in PDAC and be a potential target of miR-96 (Feng et al., 2014). Further investigations showed that Kv11.1 promotes pancreatic cancer cell migration, by modulation of F-actin organization and dynamics (Lastraioli et al., 2015b) suggesting its involvement in cancer metastasis (Arcangeli et al., 2014; Manoli et al., 2019).
KCa Channels/KCa3.1/IK
IK (KCa3.1) channels are the K+ channels most frequently studied among this family in PDAC. Even though, transcripts of KCa4.1 and KCa4.2 also have been shown in some cell lines (Hayashi et al., 2012). Investigation of the KCa3.1 mRNA expression in primary pancreatic cancer tumors show that 8 of 9 tumors (89%) contain a 6- to 66-fold higher expression, compared to normal pancreatic tissue (Jager et al., 2004). KCa3.1 is also found overexpressed in several PDAC cell lines (Jager et al., 2004). The over-expression of KCa3.1 was associated with an increased Ca2+-activated K+-current. Pharmacological inhibition (by TRAM-34, Clotrimazole) of KCa3.1 completely suppressed cell proliferation of MiaPaCa-2 and BxPC-3 cells but not PANC-1 cells (Jager et al., 2004). Moreover, application of [Ca2+]o while inhibiting with TRAM-34 or Clotrimazole rescued the MiaPaCa-2 and BxPC-3 cell proliferation but did not affect this of PANC-1 suggesting that PANC-1 cell line grows independently of functional KCa3.1 channels (Jager et al., 2004). Bonito and co-workers have also reported the role of KCa3.1 in PDAC cell proliferation and migration (Bonito et al., 2016). They showed a significant mRNA upregulation of KCa3.1 in MiaPaCa-2 and BxPC-3, but not in Capan-1 and PANC-1 cells. In addition, Patch clamp measurements revealed a Ca2+-activated K+ current, which was reduced by TRAM-34 and clotrimazole. Interestingly, a transient gene silencing of KCa3.1 in MiaPaCa-2 cells completely abolished the Ca2+ current (Bonito et al., 2016). MiaPaca-2 cell proliferation was inhibited with TRAM-34 and 1% FBS, whereas no effect was found by application of TRAM-34 and 10% FBS in the culture media, as shown before (Jager et al., 2004). Silencing of KCa3.1 removed the ability of MiaPaCa-2 cells to proliferate, and attenuated their cell invasion and migration. Surprisingly treatment upon TRAM-34 or clotrimazole increased cell migration. It was hypothesized that this could be due to Ca2+ homeostasis, which was investigated by Ca2+ imaging that confirmed that TRAM-34 evoked an increase of [Ca2+]i (Bonito et al., 2016) possibly leading to promotion of cell migration (Lotz et al., 2004). This indicates that KCa3.1 expression and function are important for cell proliferation, migration and invasion (Bonito et al., 2016). Another study has identified KCa3.1 as a regulator of oxidative phosphorylation in MiaPaCa-2 cells as silencing and inhibition of KCa3.1 determined the effect of channel dependent-oxidative phosphorylation in proliferation and ATP generation (Kovalenko et al., 2016). In addition, MiaPaCa-2 cells showed mRNA and protein levels in mitochondria, suggesting that KCa3.1 is involved in proliferation through metabolic processes (Kovalenko et al., 2016). Three other studies have identified KCNN4 (gene coding for KCa3.1) as a gene related to PDAC as its transcripts and gene-level were upregulated compared to normal pancreatic tissue (Zaccagnino et al., 2016; Jiang et al., 2017; Shen et al., 2017). The upregulation of KCNN4 was associated with the gene expression of different EMT transcription factors (Zaccagnino et al., 2016).
Two-Pore K+ Channels (K2P)
The outward conducting, pH and membrane potential activated K2P channels have an impact on physiological processes. They can regulate the cell volume, the membrane potential in form of being pH sensitive, modulate ion transport and Ca2+ homeostasis. They are involved in cancer progression due to their impact on cell growth survival and migration, as it has been shown in different types of cancer (Mu et al., 2003; Kim et al., 2004; Voloshyna et al., 2008; Alvarez-Baron et al., 2011; Lee G. W. et al., 2012; Nagy et al., 2014; Sauter et al., 2016). A broad data base analysis of K2P expression in different cancers revealed an aberrant expression of different K2P in PDAC (Williams et al., 2013). mRNA expression of KCNK1 (gene coding for TWIK-1) was upregulated in PDAC compared to normal tissue, and KCNK3 (gene coding for TASK-1) were downregulated (Williams et al., 2013). One study has found the functional mRNA and protein expression of KCNK5 (gene coding for TASK-2) in PDAC cell lines HPAF, but the role in cancer progression was not further studied (Fong et al., 2003). In another study a pH sensitive K+ current was identified in BxPC-3 cells and was probably mediated by TREK-1 (Sauter et al., 2016). TREK-1 protein expression was shown in PDAC cell lines where it was shown that TREK-1 was involved in proliferation (Sauter et al., 2016). A similar pattern was shown in a scratch wound healing assay were activation of TREK-1 lead to decreased migration. These results indicate that TREK-1 has a potential inhibiting role in PDAC proliferation and migration (Sauter et al., 2016). Very few studies have been done on the role of K2P channels in PDAC. However, it can be suggested from other types of cancer that these channels can be related to cancer progression (Comes et al., 2015).
Calcium Channels in PDAC
ORAI and STIM
It is well known that physiological Ca2+ signaling has many effects in the exocrine pancreas, and takes part in stimulating secretion of HCO3- and other ions (Hegyi and Petersen, 2013; Maleth and Hegyi, 2014). In non-excitable cells, such as cancer cells, Ca2+ entry occurs mainly through SOCs (Mo and Yang, 2018) but also through transient receptor potential channels (TRP), which are selective for both Ca2+ and Na+ (Worley et al., 2007).
There is increasing evidence of dysregulated Ca2+ signaling in cancer. This evidence is based on the implication of SOCs and TRP in key hallmarks of cancer progression and as prognostic markers in several types of cancers (Prevarskaya et al., 2007; Shapovalov et al., 2016; Chen et al., 2019). Some members of SOCs and TRP have been studied in PDAC, even though knowledge is less pronounced compared to other types of cancer, such as breast-, cervical-, and colorectal cancer (Chen et al., 2019).
The complex of ORAI1 and STIM1 has been shown to play a role in carcinogenesis and to be involved in regulation of proliferation, migration, invasion and apoptosis in different types of cancer (Chen et al., 2019). Only two studies have been performed on PDAC showing that ORAI1 and STIM1 mediate SOC entry and that they are involved in proliferation, survival and apoptosis (Kondratska et al., 2014; Khan et al., 2020). It has been shown that both ORAI1 and STIM1 were expressed in several PDAC cell lines at mRNA and protein levels, with PANC-1 showing the highest levels of both. Knockdown of ORAI1 and STIM1 with siRNA showed a significant reduction of Ca2+ entry. This was confirmed in PANC-1, AsPC-1, MiaPaCa-2, and Capan-1 cells, indicating that SOC entry is mediated by ORAI1 and STIM1 in different PDAC cell lines. A recent study has revealed the involvement of Calcium Release-Activated calcium (CRAC) channel (ORAI1) in proliferation of PDAC (Khan et al., 2020). An inhibition with CRAC channel inhibitor, RP4010, showed a significant reduction of cell proliferation and colony formation in MiaPaCa-2 cells and in L3.6pl (a pancreatic adenosquamous carcinoma derivated cell line). The influx of calcium was also inhibited upon treatment with RP4010, suggesting that cell proliferation is mediated by regulation of Ca2+ entry through CRAC channel (Khan et al., 2020). It was proposed that cell proliferation was calcium-regulated through the AKT/mTOR signaling pathway as RP4010 inhibition decreased the mRNA levels and protein expression of phosphorylated AKT, modulated the expression of proteins important for downstream AKT/mTOR signaling. Furthermore, RP4010 or ORAI1 knockdown showed a decrease in mRNA levels and in nuclear translocation of NFAT1, suggesting that CRAC channel takes part in modulating calcium signaling associated with NFAT translocation and that PDAC proliferation is regulated through the calcium-activated AKT/mTOR/NFAT signaling (Khan et al., 2020). To test if RP4010 could enhance anticancer activity of standard used treatments gemcitabine and Nab-Paclitaxel, a combination of the three drugs were used to treat PDAC cell lines. The results showed a decrease in proliferation. A synergistic effect of certain dose combinations of RP4010 with gemcitabine/Nab-Paclitaxel was found to inhibit cell growth. In addition, this synergistic treatment downregulated the expression of NFATC1 and mTOR mRNA and NFAT1, NF-κB, and phosphorylated S6K proteins, suggesting that inhibition of cell proliferation through CRAC channel are mediated by a downregulation of mTOR, NFAT and NF-κB signaling (Khan et al., 2020). The anticancer activity and the synergistic effect of RP4010/Gemcitabine/Nab-Paclitaxel were tested in-vivo. In a patient-derived xenograft mouse model, it was shown that Ki-67 expression decreased with the treatment of RP4010 or by the triple combination treatment (Khan et al., 2020). The overexpression found by Kondratska and co-workers can explain increased [Ca2+]i levels in PDAC cell lines, and that this is a mechanism for survival (Kondratska et al., 2014). In contrast, another study has found decreased gene expression levels of ORAI1 (Zaccagnino et al., 2016).
A recent study has been investigating the role of STIM1 in PDAC progression (Wang et al., 2019). shRNA knockdown of STIM1 showed decreased proliferation, invasion, and upregulation of E-cadherin protein levels and downregulation of vimentin levels, suggesting that STIM1 is involved in carcinogenesis of PDAC and in some way involved in Epithelial-Mesenchymal transition (EMT). Even though, E-cadherin levels have shown to be upregulated, in contrary to what is usually seen in cells undergoing EMT where E-cadherin decrease in favor of N-cadherin (Gheldof and Berx, 2013). Furthermore, tissue microarray analysis showed that the STIM1 expression positively correlated with HIF-1α (Wang et al., 2019). It was further shown that similar protein expression levels of STIM1 and HIF-1α were expressed in different PDAC cell lines. STIM1 and HIF-1α protein levels were also upregulated in some PDAC tumor samples compared to non-tumor samples. Knockdown of HIF-1α in PANC-1 cells revealed a significantly lower mRNA and protein expression of STIM1. The co-upregulation of both proteins and the downregulation of STIM1 upon knockdown of HIF-1α indicate that STIM1 is regulated by HIF-1α on the transcriptional level. STIM1 promoter activity was tested in PANC-1 cells upon normoxia or hypoxia, where HIF-1α binding sites, under hypoxic conditions reduced STIM1 promoter activity (Wang et al., 2019). These results indicate that HIF-1α probably regulates STIM1 transcription and that STIM1 overexpression, in a hypoxic environment, can promote PDAC progression and invasion (Wang et al., 2019).
The EMT process is stimulated upon loss of cell-cell contact and occurs in migrating cancer cells (Gheldof and Berx, 2013). It has been shown in disconnected individual PANC-1 cells that ER/Plasma membrane junctions containing STIM1, together with the IP3Rs, redistribute to the leading edge of focal adhesions (Okeke et al., 2016). An inhibition of IP3Rs and SOC entry reduced the migrating capacity of PANC-1 cells. This mechanism indicates the importance of Ca2+ signaling in migration through SOC entry and intracellular calcium channels (Okeke et al., 2016).
TRP Channels
TRP form an adaptable family of ion channel proteins where the majority are calcium permeable and show regulatory patterns that are sensitive to different environmental factors (Shapovalov et al., 2016). The role of TRP has been reported in different types of cancer (Prevarskaya et al., 2007). It has been proposed that TRPC1 can regulate PDAC cell proliferation through TGF-β signaling, as TGF-β has been shown to be one of the key modulators of EMT in mammary epithelial cells (Radisky and LaBarge, 2008). In PDAC cell line BxPC-3, TGF-β has shown to induce [Ca2+]i increase leading to activation of the Ca2+-dependent protein kinase Cα (PKC-α) and its translocation to the plasma membrane. PKC-α activation by TGF-β initiates the motility and migration by inhibiting tumor suppressor PTEN (Chow et al., 2008). Further on, it has been shown that there is a high expression of TRPC1, TRPC4 and TRPC6 in BxPC-3 cells (Dong et al., 2010). Here, it was confirmed that TGF-β induces cytosolic Ca2+ concentrations through TRPC1, followed by a PKC-α activation, thus initiating motility and migration. This was shown by a pharmacological inhibition of SOC entry pathways with 2-APB and La3+, which abolishes the TGF-β induced cytosolic Ca2+ increase. Furthermore, blocking of PKC-α with selective PKC-α inhibitors inhibited the TGF-β mediated Ca2+ entry. In addition, knockdown of TRPC1 with siRNA reversed the effect of TGF-β on cell motility, although, knockdown of TRPC4 and TRPC6 did not have an effect on motility of TGF-β mediated BxPC-3 cell motility (Dong et al., 2010). These observations suggest that dysregulated Ca2+ entry through TRPC1 could be involved in EMT, and thereby invasion and metastasis of PDAC.
TRPV channels function as sensors in the central and peripheral nervous system where the majority is sensitive to voltage and temperature (Premkumar and Abooj, 2013). TRPV1 has shown to be related to oncogenesis and is expressed in different types of cancer (Domotor et al., 2005; Lazzeri et al., 2005; Sanchez et al., 2005; Miao et al., 2008; Morelli et al., 2014; Vercelli et al., 2014). TRPV1 can be activated by multiple pathways, which can promote pancreatic inflammation and pain, but also pancreatic cancer (Hartel et al., 2006; Huang et al., 2020). TRPV1 was shown to be upregulated at the mRNA and protein level in PDAC tissue compared to normal pancreatic tissue (Hartel et al., 2006). TRPV1 staining has been shown in both normal acini and ducts but with highest intensity in nerves of inflamed tissue surrounding the cancer. The elevated TRPV1 expression in infiltrating nerves was associated with pain in patients with PDAC. The same authors showed that inhibition of TRPV1 with resiniferatoxin induces apoptosis by targeting mitochondrial respiration and decreases cell growth in some PDAC cell lines (Hartel et al., 2006).
Recently, it has been shown that TRPV1 regulates the Epidermal Growth Factor Receptor (EGFR) in PANC-1 cell line (Huang et al., 2020). In this study, an overexpression of TRPV1 has been associated with a decrease in protein expression of EGFR in PANC-1. Vice versa, the downregulation and inhibition of TRPV1 increases the protein expression of EGFR. In addition, an overexpression of TRPV1 showed increased levels of ubiquitinated EGFR. The membranous fractions of EGFR were reduced, while the cytoplasmic were increased compared to the control (Huang et al., 2020). This indicates that TRPV1 promotes EGFR ubiquitination and thereby a downregulation of EGFR activity, resulting in EGFR cytoplasmic translocation and degradation, which was found to be mainly through the lysosomal pathway (Huang et al., 2020). Furthermore, it was shown that TRPV1 overexpression inhibited proliferation, probably through the MAPK signaling pathway. Overexpression of TRPV1 resulted in decreased mRNA levels of KRAS and AKT2 and a treatment with EGF reduced the protein expression of ERK, JNK, and CREB, suggesting that a TRPV1 overexpression decreases EGFR/MAPK dependent proliferation in PANC-1 cells (Huang et al., 2020). The two above mentioned studies show contrary results in form of how the expression of TRPV1 is related to proliferation. Hartel et al., demonstrated that inhibition of TRPV1 terminate cell growth and induced apoptosis, where Huang et al., found that an overexpression of TRPV1 leads to a reduced proliferation rate (Hartel et al., 2006; Huang et al., 2020).
Another member of the TRPV family, TRPV6, was also found to be overexpressed in the primary pancreatic cancer tissues at both protein and mRNA levels. Moreover, by immunohistochemical analysis, it was found that TRPV6 is mainly localized in the cytoplasm in both tumor and normal tissue (Song et al., 2018). In-vitro, the highest level of TRPV6 was found in two pancreatic cell lines, Capan-2 and SW1990. The knockdown of TRPV6, by siRNA, resulted in reduced proliferation, cell cycle arrest in G0/G1 phase, promotion of apoptosis, and suppression of cell migration and invasion (Song et al., 2018). Furthermore, the silencing of TRPV6 resulted in a significant increase of sensitivity to the chemotherapeutic reagent oxaliplatin (Song et al., 2018). In contrast to this finding, Zaccagnino et al., 2016 showed a downregulation of TRPV6 in PDAC tissue, compared to normal pancreatic epithelium. Moreover, Tawfik et al., 2020 found also a downregulation of TRPV6 expression in a PDAC cell line A818–6 grown in a highly malignant undifferentiated monolayer. The authors suggest that a lower expression of TRPV6 could contribute to an inhibited epithelial fluid secretion in PDAC (Tawfik et al., 2020).
The TRPM family is also constituted with several members, which have been found to be implicated in carcinogenesis. One of the most studied in PDAC is TRPM7, which is a particular channel having an intrinsic kinase, together with its closest homolog TRPM6 (Yee et al., 2012a). TRMP7 is ubiquitously expressed and controls cellular homeostasis of ions, especially Mg2+ and Ca2+. Interpreting that the developmental role of TRPM7 in zebrafish could be the same in humans, the role of TRPM7 has been studied in the development and progression of PDAC. Here, it has been shown that there was an overexpression of TRPM7 protein in PDAC tissue compared to normal tissue, and that TRPM7 is required for Mg2+-regulated proliferation. Knockdown of TRPM7 with siRNA showed that this channel is necessary to prevent cell cycle arrest in G0/G1 phases. Furthermore, the proliferation of TRPM7-deficient PDAC cells was rescued by adding Mg2+ to the cell culture medium (Yee et al., 2011). Another study confirmed the overexpression of TRPM7 both at mRNA and protein levels in PDAC tissue (Rybarczyk et al., 2012). Furthermore, it was shown that TRPM7 silenced BxPC-3 cells decreased [Mg2+]i, suggesting that TRPM7 takes part in regulating Mg2+ uptake in PDAC cells. In contrary to previous findings, these authors demonstrated that the silencing of TRPM7 had no effect on cell viability or proliferation, but a significant decrease of BxPC-3 cell migration (Rybarczyk et al., 2012). TRPM7 has also been found to be involved in cell invasion in both MiaPaCa2- and PANC-1 cells. In the two last cell lines, TRPM7 regulates constitutive cation currents, the influx and homeostasis of Mg2+, and cell invasion through the Hsp90α/uPA/MMP-2 proteolytic pathway (Rybarczyk et al., 2017).
Besides TRPM7, also other TRPM channels are found to be expressed in pancreatic cancer (Yee et al., 2010; Yee et al., 2014). TRPM8 is expressed in different types of adult human tissue and has also been found to be expressed in PDAC. In a panel of PDAC cell lines, mRNA TRPM8 was consistently overexpressed compared to the control cell line (H6c7) (Yee et al., 2010). This pattern has further been confirmed by immunohistochemistry in human PDAC tumors, compared to normal pancreatic tissue (Yee et al., 2010; Yee et al., 2014). Here, it was found that TRPM8 has a role in carcinogenesis in form of proliferation, migration and senescence. TRPM8 is required for proliferation by promoting cell cycle progression in PANC-1 and BxPC-3 cells, as a knockdown of TRPM8 showed a significant decrease in proliferation rate and a cell cycle arrest in G0/G1 phase (Yee et al., 2010). In another study, the knockdown of TRPM8 showed the opposite effect on proliferation. Here, the proliferation increased by 30% in PANC-1 cells and in contrary the proliferation was suppressed in HEK/M8 cells. It was found that TRPM8 is expressed in a non-glycosylated form in different PDAC cell lines, and that the channel in this form might have a protective role in PDAC (Ulareanu et al., 2017). Concerning the involvement of TRPM8 in migration and invasion, two studies show opposite results. One study demonstrated that TRPM8 also is required for cell migration, as a knockdown of the channel impaired migration of BxPC-3 and MiaPaCa-2 by 60% and 45%, respectively (Yee et al., 2014). Where another study found that it enhanced the motility of PANC-1 cells (Cucu et al., 2014).
Recently, it has been shown that a third member of the TRPM family, also plays a role in PDAC progression (Lin et al., 2018). An overexpression of TRPM2 enhanced the proliferative, migrative and invasive abilities of PANC-1 cells, compared to the control cells and the results were inversed when TRPM2 was silenced in PANC-1 cells. It should be noted, that the study does not mention the application of a proliferation inhibitor during the Scratch wound-healing assay, which investigates the migratory role of TRPM2. Therefore, one can speculate if the wound-healing could be caused by proliferation, and not migration. Nevertheless, these results suggest that TRPM2 is involved at least in cell growth and invasion (Lin et al., 2018).
Voltage-Dependent Calcium Channels
The expression of two voltage dependent Ca2+ channels have been found to be dysregulated in PDAC, namely, CaV2.1 (CACNA1A) and CaV3.1 (CACNA1G) are upregulated and downregulated, respectively (Zaccagnino et al., 2016). These sparse data indicate that voltage-dependent Ca2+ channels might have a role in PDAC progression.
Chloride Channels in PDAC
Ca2+-Activated Chloride Channel (CaCC) and TMEM Proteins
Aberrant expression and dysregulated function of Cl- channels have shown to be involved in carcinogenesis, especially their role in cell volume regulation has shown to be important for cancer cell migration and infiltration (Duran et al., 2010; Prevarskaya et al., 2010; Anderson et al., 2019). In Capan-1 cells, CaCC are expressed at the apical membrane, as shown for normal pancreatic acinar and ductal cells (Park et al., 2001; Wang et al., 2013; Wang and Novak, 2013).
The functional role of TMEM16A has been found to vary between different types of cancer (Ayoub et al., 2010; Liu et al., 2012; Ruiz et al., 2012; Britschgi et al., 2013). While a pro-proliferative role was found in breast and prostate cancer, the role in pancreatic cancer has been found to be contradictory. An anti-proliferative effect was found by a knockdown and overexpression strategy in CFPAC-1 cells (Ruiz et al., 2012), where another study found that inhibition with the TMEM16A specific inhibitor T16Ainh-A01 decreased the proliferation rate in CFPAC-1 cells (Mazzone et al., 2012). Both studies lack the comparison of PDAC cell lines with a normal pancreatic epithelial control cell line. This was considered in a recent study, where the role of TMEM16A was investigated in PDAC cell lines and compared to a normal pancreatic epithelial control cell line (Sauter et al., 2015). The mRNA expression of TMEM16A was upregulated, with a 1,450-fold, in AsPC-1, BxPC-3, and especially in Capan-1 cells (Sauter et al., 2015). The upregulation was confirmed by an increase in TMEM16A protein expression for all three cell lines. Furthermore, it was shown that TMEM16A carries the major component of CaCC current in these cell lines (Sauter et al., 2015). Moreover, the authors found that knockdown of TMEM16A had no effect on proliferation. Inhibition, by T16Ainh-A01 or other CaCC inhibitors, failed to affect PDAC cell lines proliferation, while T16Ainh-A01 had a significant effect on the control cell line cell proliferation, which almost completely lack the expression of TMEM16A. These results suggest that the inhibition by T16Ainh-A01 is unspecific for TMEM16A, and that this channel has no implication in proliferation, at least in these three PDAC cell lines (Sauter et al., 2015). According to the role of TMEM16A in migration, gene silencing reduced the migratory capability of AsPC-1 and BxPC-3 cells, where the inhibition with T16Ainh-A01 was ineffective (Sauter et al., 2015). Other CaCC inhibitors caused a decrease in migration of BxPC-3 cells. Nevertheless, Capan-1 cells showed the highest expression of TMEM16A, the migration was very slow, suggesting that TMEM16A is not implicated in the role of migration in Capan-1 cells and supporting that TMEM16A has different roles in carcinogenesis of PDAC cells (Sauter et al., 2015).
Another recent study has performed a database investigation on the expression of TMEM16A and found that mRNA TMEM16A expression is upregulated in pancreatic cancer (Crottes et al., 2019). The authors found that extracellular application of EGF increased [Ca2+]i and the outward-rectifying Cl- current, which were both inhibited by different TMEM16A inhibitors. The regulation of Cl- currents and the Ca2+ response were probably due to SOC entry. Furthermore, silencing of TMEM16A in AsPC-1 cells reduced migration even under EGF treatments, while EGF induced migration in the control cell line. This indicates that TMEM16A is involved in EGF-induced PDAC migration and progression, probably through Ca2+ signaling. In addition, this study investigated the possible role of TMEM16A to classify PDAC patients (Crottes et al., 2019). They found 10 genes involved in EGF-induced TMEM16A-dependent Ca2+ signaling, which could distinguish neuro-endocrine tumors from other pancreatic cancers. In PDAC, these genes formed three clusters with different genetic profiles that could reflect different molecular characterizations (Crottes et al., 2019).
Another TMEM16 protein expressed in pancreatic cancer is the TMEM16J protein, which also has been found to be overexpressed (Jun et al., 2017). TMEM16J is not a well characterized protein, but it is proposed that it might function as a cation channel activated by the cAMP/PKA signaling pathway (Falzone et al., 2018; Kim et al., 2018). An upregulation of TMEM16J gene-, mRNA-, and protein overexpression were found in AsPC-1, BxPC-3, and Capan-2 cell lines and a small overexpression in PANC-1 cells. An overexpression of TMEM16J in PANC-1 cells resulted in phosphorylated ERK1/2 levels, but not total ERK1/2 levels. Furthermore, both EGFR and phosphorylated EGFR levels were upregulated in PANC-1 cells overexpressing TMEM16J and an immunoprecipitation assay revealed that both TMEM16A and TMEM16J formed protein complexes with EGFR, but the binding affinity for TMEM16J was 132% higher, than for the one of TMEM16A (Jun et al., 2017), suggesting that TMEM16J are involved in upregulation and activation of EGFR. In contrary, a knockdown of TMEM16J in AsPC-1 cells resulted in inhibition of phosphorylated ERK1/2, EGFR and phosphorylated EGFR and a decreased proliferation rate. These results were confirmed in-vivo, were a xenograft mouse model was made by implanting PANC-1 cells stably overexpressing TMEM16J. It was shown that tumor growth was significantly increased and immunohistochemistry of these tumors confirmed the TMEM16J overexpression (Jun et al., 2017). These results indicate that TMEM16J is implicated in cell proliferation and tumor growth. Another member of the TMEM16 family, TMEM16E, has been shown to be implicated in PDAC. It is not yet clear whether the TMEM16E protein function as an ion channel or scramblase (Falzone et al., 2018). It has been shown, by immunohistochemical analysis, that TMEM16E is entirely expressed in PDAC but not in normal pancreatic tissue (Song et al., 2019). The highest expression of both mRNA and protein of TMEM16E was found in PANC-1 cells. The impact of TMEM16E on migration was investigated by a wound-scratch assay and a siRNA knockdown of TMEM16E showed a significant decrease in PANC-1 cell migration (Song et al., 2019). Even though, it should be mentioned that the authors do not account for the possible effect of proliferation in this assay. The migration was in some ways confirmed by the downregulation of vimentin protein expression, compared to the control, which showed a higher expression of vimentin, suggesting that TMEM16E is implicated in migration of PANC-1 cells. In addition, the proliferation of PANC-1 cells was significantly decreased upon knockdown of TMEM16E suggesting its role in proliferation (Song et al., 2019). This assay supports the speculation on the TMEM16E role in migration.
Besides being activated by Ca2+, CaCC can also be activated and regulated by specific proteins, namely Calcium-activated Chloride channel regulators (CLCAs) also called Calcium Chloride channel accessory proteins. CLCAs are expressed in different types of cancer and have been implicated in regulation of proliferation, migration and metastasis (Yurtsever et al., 2012; Lang and Stournaras, 2014; Stock and Schwab, 2015). CLCA1 has been shown to be overexpressed in pancreatic cancer (Hu et al., 2018a; Hu et al., 2018b). However, the expression pattern and underlying molecular mechanism of its role in PDAC is less known. Finally, low gene expression of Chloride Channel Kb (CLCNKB) and Chloride Voltage-Gated Channel 1 (CLCN1) have been reported in human PDAC tissue compared to normal pancreatic epithelium (Zaccagnino et al., 2016).
CFTR
It has been shown that CFTR is expressed in some PDAC cell lines. An early study showed that CFTR only was expressed in Capan-1 cells among nine different pancreatic cell lines and that the expression varied as a function of confluence (Chambers and Harris, 1993). Singh et al., confirmed the almost non-existent expression of CFTR in PDAC cell lines. Indeed, mRNA levels were detectable in normal pancreatic tissue and three (Capan-1, Suit2 and SW1990) out of 16 pancreatic cell lines (Singh et al., 2007). Furthermore, Zaccagnino et al., reported the downregulation of CFTR, at gene-level, in human PDAC tissue compared to normal pancreatic epithelium. This downregulation was associated with gene expression of EMT transcription factors (Zaccagnino et al., 2016). Furthermore, Singh et al., showed that wild type CFTR negatively regulated MUC4 expression while silencing of CFTR upregulated MUC4 expression. As MUC4 is a protein involved in tumor migration and metastasis, the negative regulation by CFTR indicates a protective role and a tumor suppressing function by inhibiting MUC4 and hence pancreatic cancer progression (Singh et al., 2007).
A recent study has investigated CFTR expression in patient derivated PDAC organoids, in order to enable routine organoid subtyping for personalized treatment (Hennig et al., 2019). It has been suggested that subtyping could be based on the expression of cytokeratin 81 (KRT81) and hepatocyte nuclear factor 1A (HNF1A). As the antibody for HNF1A was no longer available, the authors permitted CFTR to replace it as a potential marker instead of HNF1A (Hennig et al., 2019). Organoids can be categorized into the established quasi-mesenchymal, exocrine-like, and classical subtypes. Immunofluorescence staining showed a mutual expression pattern where exocrine-like organoids were CFTR+/KRT81- and quasi-mesenchymal CFTR-/KRT81+. The protein expression revealed by IF was compared to mRNA levels of CFTR, which matched in 8 out of 10 cases (Hennig et al., 2019). In addition, it was confirmed, by immunohistochemical analysis, that both CFTR and KRT81 were preserved in 6 out of 7 tumors, indicating that the organoids had the same subtype as their primary tumor (Hennig et al., 2019). These results suggest that CFTR could be a supplement marker for HFN1A and that CFTR/KRT81 together might be a suitable way to evaluate subtype organoids for personal treatments (Hennig et al., 2019).
Cl- Intracellular Channel Proteins (CLICs)
CLICs are ubiquitously expressed and have been identified in several types of cancer, where they are either overexpressed or downregulated compared to the normal tissue (Peretti et al., 2015). In PDAC, CLICs are mostly found upregulated, even though their specific role in PDAC progression and development is not yet understood. CLIC2, CLIC3, and CLIC5 have been shown to be expressed at mRNA and protein levels in a HPAF cell line. By an electrophysiological study, the authors revealed that there was no single channel/conductance for apical Cl- secretion, but that these CLICs rather contributed to provide a constant net conductance across the plasma membrane (Fong et al., 2003). Another study has shown the importance of CLIC3 in PDAC, as immunohistochemical analysis and mRNA levels showed an overexpression of CLIC3 in PDAC tissue compared to normal pancreatic tissue (Dozynkiewicz et al., 2012). It was also found that CLIC3 in collaboration with Rab25 promoted cancer cell invasion and migration by integrin recycling from late endosomes/lysosomes (Dozynkiewicz et al., 2012). CLIC1 was overexpressed in primary tumors compared to normal pancreatic tissue, and strongly expressed in MiaPaCa-2 and PANC-1 cells (Lu et al., 2015). Silencing of CLIC1 showed a significant decrease in the proliferation rate, colony formation and the invasive abilities of both MiaPaCa-2 and PANC-1 cells, suggesting that CLIC1 contributes to the aggressive role of these PDAC cells (Lu et al., 2015). In addition, gene expression levels of CLIC5 has been found to be downregulated in PDAC tissue, compared to normal pancreatic epithelium and to be associated with gene expression of transcription factors related to cell differentiation (Zaccagnino et al., 2016).
Aquaporins (AQPs) in PDAC
AQPs are expressed in various types of cancers and are predicted to be key regulators in tumor development and progression (Papadopoulos and Saadoun, 2015). The expression and role of AQPs in PDAC is poorly studied, yet few studies have described their involvement in PDAC progression (Burghardt et al., 2003; Direito et al., 2017; Huang et al., 2017; Zou et al., 2019). Burghardt and co-workers have found mRNA expression of AQP1, AQP3, AQP4, AQP5, and AQP8 in PDAC. All subtypes were expressed in solid tumors, where only AQP3, AQP4, and AQP5 were expressed in PDAC cell lines (Burghardt et al., 2003). Further studies have found an upregulation of AQP1 and AQP3 in PDAC tissue compared to normal pancreatic tissue (Zou et al., 2019). The expression of AQP1 correlated with the expression of AQP3, suggesting that these two channels cooperate during PDAC development. Another study has shown the overexpression of AQP3 and AQP5 in PDAC tissue (Direito et al., 2017). AQP5 localization in PDAC was found to be in the entire plasma membrane and in the cytoplasm of ducts cells, where in normal pancreas the localization is in the apical membrane. Furthermore, AQP5 and AQP3 were suggested to be involved in proliferation and tumor transformation as a simultaneous overexpression was found to be correlated with an increased expression of EGFR, Ki-67, CK7, and a decrease of E-cadherin and increase of Vimentin (Direito et al., 2017). Another study investigating AQP has, through TCGA analysis, revealed that AQP3 shows the highest expression among AQPs in PDAC (Huang et al., 2017). The authors investigated the role of AQP3 further, with a focus on how microRNA (miR-874) regulates gene expression and post-translational events in PDAC. In a panel of eight pancreatic cell lines, they detected that cell lines with high AQP3 mRNA levels had lower miR-874 levels, where cell lines with high miR-874 had lower AQP3 levels suggesting that AQP3 expression is regulated by miR-874 (Huang et al., 2017). It was found that both modulation of AQP3 and miR-874 altered the expression and activity of mTOR and its downstream target S6, suggesting that an overexpression of AQP3 is associated with proliferation and cell survival by mTOR signaling in PDAC. In contrast to other studies, Zaccagnino et al. (2016) showed a downregulation of AQP3 and AQP8 expression in PDAC tissue compared to normal pancreatic one. Furthermore, they showed that AQP3 expression was associated with several cell differentiation related transcription factors.
Sodium Channels in PDAC
ASIC
It has recently been shown that acid-sensing ion channels (ASICs), an H+-gated subgroup of ENaC, are expressed in PDAC cell lines and tissue (Zhu et al., 2017). ASIC1 and ASIC3 were found functionally expressed and mRNA and protein expression were also found in PDAC cell lines. In all cases, the expression was upregulated compared to the normal control cell line. These results were confirmed in PDAC tissue where immunohistochemical analysis and qPCR revealed the overexpression compared to non-cancerous pancreatic tissue, suggesting that ASIC1 and ASIC3 have a pathophysiological role in PDAC (Zhu et al., 2017). Separate inhibition or knockdown of ASIC1 and ASIC3 decreased the acidity-promoted invasion and migration capacity of PDAC cell lines, but did not decrease the proliferation rate, suggesting that ASIC1 and ASIC3 are involved in the metastatic process of PDAC, but not tumor cell growth (Zhu et al., 2017). Furthermore, it was shown that ASIC1 and ASIC3 are involved in acidity-promoted EMT, as silencing or inhibition of ASIC1 or ASIC3 in PDAC cells showed decreased protein expression of mesenchymal markers Vimentin, N-cadherin, Snail, and ZEB1, while the epithelial marker E-cadherin showed increased protein expression. In contrary, PDAC cells overexpressing ASIC1 and ASIC3 showed an increase in mesenchymal markers and a decrease in epithelial markers, under acidic conditions. This was confirmed in human PDAC tissue samples by IF analysis. It was further investigated whether this mechanism was regulated by [Ca2+]i, where it was found that inhibition of ASIC1 or ASIC3 resulted in a decrease of [Ca2+]i upon acidification. In addition, the removal of [Ca2+]i upon acidic conditions decreased mesenchymal markers and increased the epithelial ones. It was determined that the RhoA pathway, which is involved in cytoskeleton re-arrangement and cell migration, was a major effector of EMT induced by ASIC1/3-[Ca2+]i activation in acidic conditions (Zhu et al., 2017). The role of ASIC1 and ASIC3 was further confirmed in-vivo, where a xenograft mouse model injected with BxPC-3 cells with a stable knockdown of ASIC1 and ASIC3 showed a significant decrease in lung and liver metastasis, but no obvious effect on tumor growth (Zhu et al., 2017).
VGSCs
Another subfamily of Na+ channels, namely voltage gated sodium channels (VGSCs), has shown to be implicated in cancer progression (Angus and Ruben, 2019). An early study has shown that Ca2+ blockers Phenytoin and Verapamil inhibited the growth of pancreatic cancer cell lines MiaPaCa-2 and CAV, both in-vitro and in-vivo (Sato et al., 1994). Phenytoin and Verapamil were chosen because they appeared to be blocking different Ca2+ channels; T-type and L-type voltage dependent Ca2+ channels, respectively (Sato et al., 1994). It has been suggested that this growth-inhibition of pancreatic cancer cell was rather due to the block of VGSC than the block of Ca2+ channels, as both Phenytoin and Verapamil show high affinity for VGSC in the inactivated state of the channel (Ragsdale et al., 1991; Koltai, 2015). In addition, the expression of VGSC (SCN9A and SCN3A) was downregulated in PDAC (Zaccagnino et al., 2016).
Ionotropic Receptors in PDAC
Purinergic Receptors (P2XR) and N-Methyl-D-Aspartate Receptors (NMDAR)
Different types of ionotropic receptors including P2XR and NMDAR have been reported to be expressed in PDAC (Kunzli et al., 2007; Hansen et al., 2008; Burnstock and Novak, 2012; Li and Hanahan, 2013; North et al., 2017). Among P2XR, P2X7R is the most well described (Kunzli et al., 2007; Hansen et al., 2008; Burnstock and Novak, 2012).This ionotropic receptor has shown to be overexpressed in PDAC cell lines and tissue (Kunzli et al., 2007; Giannuzzo et al., 2015), and to be implicated in the proliferating, apoptotic, migrating, and invading processes of PDAC (Kunzli et al., 2007; Hansen et al., 2008; Giannuzzo et al., 2015; Giannuzzo et al., 2016; Choi et al., 2018). In addition, the expression of NMDAR was found in both PDAC cell lines and PDAC tumors, and their inhibition and blocking resulted in reduced different PDAC cell lines viability and survival (Li and Hanahan, 2013; North et al., 2017). Furthermore, an inhibition of NMDAR prevented growth of tumor xenografts (Li and Hanahan, 2013; North et al., 2017).
Ion Channels as PDAC Biomarkers
A growing number of studies have investigated ion channel expression in pancreatic cell lines and human tissues, showing modulation of mRNA and/or protein expression between normal and cancer cells. Among all the studied channels, only CFTR has lower expression in cancer cell lines compared to normal cells (Singh et al., 2007), while Kv1.3, Kv7.1, and TASK-1 were downregulated in PDAC tissue compared to healthy tissue (Brevet et al., 2009; Williams et al., 2013; Tawfik et al., 2020), suggesting a protective role and tumor suppressive function for these channels. Although most of the ion channels are overexpressed in PDAC, studies on ion channel expression patterns in correlation with clinical parameters are more limited.
Diagnostic Markers
Some attention has been given to the connection between pancreatic cancer risk and CFTR deficiency. Mutations in the CFTR gene cause the hereditary life shortening disease cystic fibrosis (CF). Severe clinical manifestations occur upon CF in secretory epithelial tissues and in pancreas, mutations causing loss of function lead to pancreatic insufficiency (Wilschanski and Novak, 2013; Castellani and Assael, 2017). Different cohort studies have investigated how different variants of CFTR affect the risk of pancreatic cancer (Sheldon et al., 1993; Neglia et al., 1995). It has been shown that CF patients present an elevated risk to develop pancreatic cancer, even though the overall risk of developing cancer is the same as for the general population (Sheldon et al., 1993; Neglia et al., 1995). Furthermore, studies also indicate that patients who are CFTR mutant carriers develop pancreatic cancer in a younger age, compared to patients carrying a wildtype form of CFTR (McWilliams et al., 2010; Hamoir et al., 2013), and patients carrying a germline mutation to some degree have an increased risk of developing PDAC (Cazacu et al., 2018). One mechanism of which a CFTR mutation could cause pancreatic cancer is by the defect of CFTR and ion transport leading to dysregulated mucus secretion and obstruction of the pancreatic ducts, which all are events that could result in pancreatitis (McWilliams et al., 2010). Patients with chronic pancreatitis have a 26-fold higher risk for developing pancreatic cancer compared to the general population (Lowenfels et al., 1993; Kirkegard et al., 2017), suggesting that CFTR mutation could be considered as a new risk factor for developing PDAC.
In order to discriminate pancreatic premalignant/malignant lesions from benign lesions, an explorative proteomic approach was performed on a cohort of 24 patients using targeting mass spectrometry analysis of different biomarkers (Jabbar et al., 2018). This study proposed CLCA1 to be a supportive marker, which together with mucin-5AC (MUC5AC) and prostate stem-cell antigen (PSCA) could distinguish cystic precursor lesions from PDAC, suggesting that CLCA1 is a potential biomarker in PDAC diagnosis.
Prognostic Markers of Cancer Progression and Aggressiveness
Different studies have investigated ion channels as potential biomarkers of PDAC development and progression. Using immunohistochemical analysis, high KCa3.1 expression in PDAC tissue was correlated with TNM stages III and IV (Jiang et al., 2017), and high expression of STIM1 was correlated with tumor grade (Wang et al., 2019). Upregulation of Kv11.1 expression was associated with advanced tumor grade and high expression of Ki67 proliferative marker (Lastraioli et al., 2015b), whereas TRPV6, TRPM8 and AQP1/AQP3 channels were positively correlated with tumor stages III and IV and large tumor size (Yee et al., 2014; Du et al., 2018; Liu et al., 2018; Song et al., 2018; Zou et al., 2019). Finally, TRPM7 and Cl- intracellular channel proteins (CLIC1-3). CLIC1 overexpression was shown to be correlated with the three clinical parameters: advanced tumor grade, advanced tumor stage and large tumor size (Rybarczyk et al., 2012; Lu et al., 2015; Yee et al., 2015; Jia et al., 2016). These results suggest that all the ion channels cited above are associated with pancreatic tumor growth.
Regarding the metastatic status, immunohistochemistry experiments showed that TRPV6 expression was higher in cases where PDAC was infiltrating (Song et al., 2018). The same results were observed at mRNA and protein levels for CLIC3, with a highly detectable expression in regions where the tumor was invading normal pancreatic tissue (Dozynkiewicz et al., 2012). Other studies revealed higher TRPM7 and TRPM8 staining in metastatic tumors than in non-metastatic tumors (Yee et al., 2015; Liu et al., 2018), which was confirmed by qPCR for TRPM8 (Du et al., 2018). AQP1 and AQP3 were also more expressed in PDAC patients with lymph node metastasis and invasion, than in non-invasive cancers (Zou et al., 2019), whereas overexpression of Kir3.1 potassium channel (GIRK1) was not found to be correlated with metastatic status (Brevet et al., 2009).
Gene expression correlation analysis demonstrated that TRPM2 is strongly correlated with different genes including toll-like receptor 7 (TLR7) (Lin et al., 2018), which has already been associated with PDAC progression (Ochi et al., 2012; Grimmig et al., 2015; Wang et al., 2016). Moreover, AQP1 and AQP3 protein expression was highest in poorly differentiated tumors (Direito et al., 2017; Zou et al., 2019), whereas AQP5 is more expressed in moderately differentiated tumors (Direito et al., 2017), suggesting that AQPs are associated with tumor aggressiveness.
Finally, most of the studied ion channels in PDAC tissue were associated with overall survival of the patients. The authors usually used immunohistochemical staining on large cohorts and Kaplan-Meier survival analysis, to observe a correlation between high channel expression and short patient survival. This is the case for KCa3.1 (Jiang et al., 2017), STIM1 (Wang et al., 2019), TRPM8 (Liu et al., 2018), TRPV6 (Song et al., 2018), TMEM16J (Jun et al., 2017), CLIC1 (Lu et al., 2015; Jia et al., 2016), and AQP1/AQP3 (Zou et al., 2019). The same correlation was obtained on gene expression using qPCR for TRPM8 (Du et al., 2018) or TCGA for TRPM2 (Lin et al., 2018) and TMEM16A (Crottes et al., 2019). Furthermore, the mutation status of TRPM2 was also analyzed using Kaplan-Meier in 10 patients out of 159, and the mutated TRPM2 gene revealed a negative correlation with patient survival, compared to patients expressing wildtype TRPM2 (Lin et al., 2018). Studies on shorter cohorts also revealed that high protein expression of Kv11.1 and TRPM7 channels are inversely correlated with overall survival using Pearson correlation on 18 patients, and multivariate overall survival analysis on 44 samples, respectively (Rybarczyk et al., 2012; Lastraioli et al., 2015a) Comparison of ion channels expression level in 9 patients with short survival (<12 months) and 10 patients with long survival (>45 months) showed that short survival was correlated with high expression of CLIC3 and low expression of CLCA1 (Hu et al., 2018b). This low CLCA1 expression correlated with shorter disease-free survival was confirmed using tissue microarrays, immunohistochemistry and Kaplan-Meier analysis in 140 patients (Hu et al., 2018a). Except for CLCA1 which could be proposed as a good prognostic marker, all the other studied ion channels could be proposed as poor prognostic markers.
These studies on human tissues highlighted the major clinical relevance of ion channels expression in pancreatic cancer development (Table 2 and Figure 3). Indeed, the expression of potassium, calcium, chloride channels, and aquaporins is mainly associated with aggressiveness and invasiveness and inversely correlated to patient survival, suggesting that they may be potential markers of poor prognosis.
Therapeutic Targets
In general, PDAC cells are resistant to pro-apoptotic reagents, and overexpression of ion channels was shown to be involved in this resistance. Knockdown of TRPM7 in combination with gemcitabine treatment enhanced cytotoxicity in PANC-1 cells even though the precise mechanisms are not yet determined (Yee et al., 2012b), whereas the silencing of TRPV6 in Capan-2 PDAC cells resulted in a significant increase of sensitivity to the chemotherapeutic reagent oxaliplatin, but had little effect on gemcitabine and cisplatin treatments (Song et al., 2018). Another study has shown that silencing of TRPM8 in combination with gemcitabine suppressed the proliferation and invasion properties of PANC-1 and BxPC-3 cells. In addition, gemcitabine-sensitivity depended on TRPM8 silencing in these cell lines, where mRNA level of multi-drug related proteins was decreased, and expression of apoptosis-related proteins was also affected, suggesting that TRPM8 is involved in multi-drug resistance and apoptosis of PDAC cells (Liu et al., 2018). PANC-1 cells apoptosis was also increased after treatment with chemotherapeutic reagents 5-fluorouracil or gemcitabine in combination with a knockdown of ORAI1, STIM1, or both (Kondratska et al., 2014). Furthermore, it was shown that cells treated with either 5-fluorouracil or gemcitabine increased ORAI1 and STIM1 expression as well as SOC entry suggesting that ORAI1 and STIM1 confer resistance to chemotherapy, probably through the increase of SOC entry (Kondratska et al., 2014). More recently, STIM1 was found to be involved in gemcitabine resistance in PDAC (Zhou et al., 2020). The transcriptome sequencing analysis in established gemcitabine resistant PDAC cell lines, showed that STIM1 was significantly upregulated in the gemcitabine resistant cell lines, compared to the parental cell line (Zhou et al., 2020). Among the chloride channels, knockdown of TMEM16J provided an additive effect on inhibiting proliferation upon treatment with gemcitabine and erlotinib, suggesting that a TMEM16J inhibitor can help to prevent gemcitabine resistance associated with the prolonged use of gemcitabine (Jun et al., 2017). The team of Arcangeli has been investigating another therapeutic perspective with the development of a novel anti-Kv11.1 antibody-conjugated PEG-TiO2 nanoparticles for targeting PDAC cells (Sette et al., 2013).
Conclusion
Increasing evidence indicates that ion channels are involved in the regulation of cancer proliferation, apoptosis, chemo-resistance, migration, and invasion. The field of ion channels in PDAC still constitutes a novel area of research and even studies conclude their involvement in the malignancy and aggressiveness of PDAC, only relatively few studies provide the complete signaling pathways. Moreover, the majority of the studies cited in this review were carried out on 2D cultured cell lines. It appears thus necessary to develop and/or increase better approaches (organoids, 3D culture, and/or animal models) to investigate the candidate channel(s) as well as its (their) function and associated signaling pathways in PDAC. However, recently, an increasing number of publications on signaling in pancreatic cancer take the tumor microenvironment into-account. This reflects the interest in ionic channels and their potential promising use as therapeutic targets in the fight against pancreatic cancer.
Author Contributions
JS, ID-D, AA, and HO-A: design and manuscript preparation. All authors contributed to the article and approved the submitted version.
Funding
JS is grateful for the funding by the Marie Skłodowska-Curie Innovative Training Network (ITN) Grant Agreement number: 813834 - pHioniC - H2020-MSCA-ITN-2018. HO-A is grateful for the funding by the Ministère de l’Enseignement Supérieur et de la Recherche, the Région Hauts-de-France (Picardie), the FEDER (Fonds Européen de Développement Économique Régional), the Université Picardie Jules Verne, and the Ligue Contre le Cancer (Septentrion).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aichler, M., Seiler, C., Tost, M., Siveke, J., Mazur, P. K., Da Silva-Buttkus, P., et al. (2012). Origin of pancreatic ductal adenocarcinoma from atypical flat lesions: a comparative study in transgenic mice and human tissues. J. Pathol. 226 (5), 723–734. doi: 10.1002/path.3017
al-Nakkash, L., Cotton, C. U. (1997). Bovine pancreatic duct cells express cAMP- and Ca(2+)-activated apical membrane Cl- conductances. Am. J. Physiol. 273 (1 Pt 1), G204–G216. doi: 10.1152/ajpgi.1997.273.1.G204
Alvarez-Baron, C. P., Jonsson, P., Thomas, C., Dryer, S. E., Williams, C. (2011). The two-pore domain potassium channel KCNK5: induction by estrogen receptor alpha and role in proliferation of breast cancer cells. Mol. Endocrinol. 25 (8), 1326–1336. doi: 10.1210/me.2011-0045
Anderson, K. J., Cormier, R. T., Scott, P. M. (2019). Role of ion channels in gastrointestinal cancer. World J. Gastroenterol. 25 (38), 5732–5772. doi: 10.3748/wjg.v25.i38.5732
Angus, M., Ruben, P. (2019). Voltage gated sodium channels in cancer and their potential mechanisms of action. Channels (Austin) 13 (1), 400–409. doi: 10.1080/19336950.2019.1666455
Arcangeli, A., Crociani, O., Bencini, L. (2014). Interaction of tumour cells with their microenvironment: ion channels and cell adhesion molecules. A focus on pancreatic cancer. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 369 (1638), 20130101. doi: 10.1098/rstb.2013.0101
Arsenijevic, T., Perret, J., Van Laethem, J. L., Delporte, C. (2019). Aquaporins Involvement in Pancreas Physiology and in Pancreatic Diseases. Int. J. Mol. Sci. 20 (20). doi: 10.3390/ijms20205052
Ashton, N., Evans, R. L., Elliott, A. C., Green, R., Argent, B. E. (1993). Regulation of fluid secretion and intracellular messengers in isolated rat pancreatic ducts by acetylcholine. J. Physiol. 471, 549–562. doi: 10.1113/jphysiol.1993.sp019915
Ayoub, C., Wasylyk, C., Li, Y., Thomas, E., Marisa, L., Robe, A., et al. (2010). ANO1 amplification and expression in HNSCC with a high propensity for future distant metastasis and its functions in HNSCC cell lines. Br. J. Cancer 103 (5), 715–726. doi: 10.1038/sj.bjc.6605823
Bergmann, F., Andrulis, M., Hartwig, W., Penzel, R., Gaida, M. M., Herpel, E., et al. (2011). Discovered on gastrointestinal stromal tumor 1 (DOG1) is expressed in pancreatic centroacinar cells and in solid-pseudopapillary neoplasms–novel evidence for a histogenetic relationship. Hum. Pathol. 42 (6), 817–823. doi: 10.1016/j.humpath.2010.10.005
Bielanska, J., Hernandez-Losa, J., Perez-Verdaguer, M., Moline, T., Somoza, R., Ramon, Y. C. S., et al. (2009). Voltage-dependent potassium channels Kv1.3 and Kv1.5 in human cancer. Curr. Cancer Drug Targets 9 (8), 904–914. doi: 10.2174/156800909790192400
Bleich, M., Warth, R. (2000). The very small-conductance K+ channel KvLQT1 and epithelial function. Pflugers Arch. 440 (2), 202–206. doi: 10.1007/s004240000257
Bonito, B., Sauter, D. R., Schwab, A., Djamgoz, M. B., Novak, I. (2016). KCa3.1 (IK) modulates pancreatic cancer cell migration, invasion and proliferation: anomalous effects on TRAM-34. Pflugers Arch. 468 (11-12), 1865–1875. doi: 10.1007/s00424-016-1891-9
Brevet, M., Fucks, D., Chatelain, D., Regimbeau, J. M., Delcenserie, R., Sevestre, H., et al. (2009). Deregulation of 2 potassium channels in pancreas adenocarcinomas: implication of KV1.3 gene promoter methylation. Pancreas 38 (6), 649–654. doi: 10.1097/MPA.0b013e3181a56ebf
Britschgi, A., Bill, A., Brinkhaus, H., Rothwell, C., Clay, I., Duss, S., et al. (2013). Calcium-activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc. Natl. Acad. Sci. U. S. A. 110 (11), E1026–E1034. doi: 10.1073/pnas.1217072110
Burghardt, B., Elkaer, M. L., Kwon, T. H., Racz, G. Z., Varga, G., Steward, M. C., et al. (2003). Distribution of aquaporin water channels AQP1 and AQP5 in the ductal system of the human pancreas. Gut 52 (7), 1008–1016. doi: 10.1136/gut.52.7.1008
Burghardt, B., Nielsen, S., Steward, M. C. (2006). The role of aquaporin water channels in fluid secretion by the exocrine pancreas. J. Membr. Biol. 210 (2), 143–153. doi: 10.1007/s00232-005-0852-6
Burnstock, G., Novak, I. (2012). Purinergic signalling in the pancreas in health and disease. J. Endocrinol. 213 (2), 123–141. doi: 10.1530/JOE-11-0434
Camacho, J. (2006). Ether a go-go potassium channels and cancer. Cancer Lett. 233 (1), 1–9. doi: 10.1016/j.canlet.2005.02.016
Caputo, A., Caci, E., Ferrera, L., Pedemonte, N., Barsanti, C., Sondo, E., et al. (2008). TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 322 (5901), 590–594. doi: 10.1126/science.1163518
Case, R. M., Clausen, T. (1973). The relationship between calcium exchange and enzyme secretion in the isolated rat pancreas. J. Physiol. 235 (1), 75–102. doi: 10.1113/jphysiol.1973.sp010379
Castellani, C., Assael, B. M. (2017). Cystic fibrosis: a clinical view. Cell Mol. Life Sci. 74 (1), 129–140. doi: 10.1007/s00018-016-2393-9
Cazacu, I. M., Farkas, N., Garami, A., Balasko, M., Mosdosi, B., Alizadeh, H., et al. (2018). Pancreatitis-Associated Genes and Pancreatic Cancer Risk: A Systematic Review and Meta-analysis. Pancreas 47 (9), 1078–1086. doi: 10.1097/MPA.0000000000001145
Chambers, J. A., Harris, A. (1993). Expression of the cystic fibrosis gene and the major pancreatic mucin gene, MUC1, in human ductal epithelial cells. J. Cell Sci. 105 ( Pt 2), 417–422.
Chen, C., Vincent, J. D., Clarke, I. J. (1994). Ion channels and the signal transduction pathways in the regulation of growth hormone secretion. Trends Endocrinol. Metab. 5 (6), 227–233. doi: 10.1016/1043-2760(94)p3080-q
Chen, Y. F., Lin, P. C., Yeh, Y. M., Chen, L. H., Shen, M. R. (2019). Store-Operated Ca(2+) Entry in Tumor Progression: From Molecular Mechanisms to Clinical Implications. Cancers (Basel) 11 (7), 899. doi: 10.3390/cancers11070899
Cho, S. J., Sattar, A. K., Jeong, E. H., Satchi, M., Cho, J. A., Dash, S., et al. (2002). Aquaporin 1 regulates GTP-induced rapid gating of water in secretory vesicles. Proc. Natl. Acad. Sci. U. S. A. 99 (7), 4720–4724. doi: 10.1073/pnas.072083499
Choi, J. H., Ji, Y. G., Ko, J. J., Cho, H. J., Lee, D. H. (2018). Activating P2X7 Receptors Increases Proliferation of Human Pancreatic Cancer Cells via ERK1/2 and JNK. Pancreas 47 (5), 643–651. doi: 10.1097/MPA.0000000000001055
Chow, J. Y., Dong, H., Quach, K. T., Van Nguyen, P. N., Chen, K., Carethers, J. M. (2008). TGF-beta mediates PTEN suppression and cell motility through calcium-dependent PKC-alpha activation in pancreatic cancer cells. Am. J. Physiol. Gastrointest. Liver Physiol. 294 (4), G899–G905. doi: 10.1152/ajpgi.00411.2007
Comes, N., Bielanska, J., Vallejo-Gracia, A., Serrano-Albarras, A., Marruecos, L., Gomez, D., et al. (2013). The voltage-dependent K(+) channels Kv1.3 and Kv1.5 in human cancer. Front. Physiol. 4, 283. doi: 10.3389/fphys.2013.00283
Comes, N., Serrano-Albarras, A., Capera, J., Serrano-Novillo, C., Condom, E., Ramon, Y. C. S., et al. (2015). Involvement of potassium channels in the progression of cancer to a more malignant phenotype. Biochim. Biophys. Acta 1848 (10 Pt B), 2477–2492. doi: 10.1016/j.bbamem.2014.12.008
Coradini, D., Casarsa, C., Oriana, S. (2011). Epithelial cell polarity and tumorigenesis: new perspectives for cancer detection and treatment. Acta Pharmacol. Sin. 32 (5), 552–564. doi: 10.1038/aps.2011.20
Crottes, D., Lin, Y. T., Peters, C. J., Gilchrist, J. M., Wiita, A. P., Jan, Y. N., et al. (2019). TMEM16A controls EGF-induced calcium signaling implicated in pancreatic cancer prognosis. Proc. Natl. Acad. Sci. U. S. A. 116 (26), 13026–13035. doi: 10.1073/pnas.1900703116
Cucu, D., Chiritoiu, G., Petrescu, S., Babes, A., Stanica, L., Duda, D. G., et al. (2014). Characterization of functional transient receptor potential melastatin 8 channels in human pancreatic ductal adenocarcinoma cells. Pancreas 43 (5), 795–800. doi: 10.1097/MPA.0000000000000106
Delporte, C. (2014). Aquaporins in salivary glands and pancreas. Biochim. Biophys. Acta 1840 (5), 1524–1532. doi: 10.1016/j.bbagen.2013.08.007
Demolombe, S., Franco, D., de Boer, P., Kuperschmidt, S., Roden, D., Pereon, Y., et al. (2001). Differential expression of KvLQT1 and its regulator IsK in mouse epithelia. Am. J. Physiol. Cell Physiol. 280 (2), C359–C372. doi: 10.1152/ajpcell.2001.280.2.C359
Direito, I., Paulino, J., Vigia, E., Brito, M. A., Soveral, G. (2017). Differential expression of aquaporin-3 and aquaporin-5 in pancreatic ductal adenocarcinoma. J. Surg. Oncol. 115 (8), 980–996. doi: 10.1002/jso.24605
Djamgoz, M. B., Coombes, R. C., Schwab, A. (2014). Ion transport and cancer: from initiation to metastasis. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 369 (1638), 20130092. doi: 10.1098/rstb.2013.0092
Domotor, A., Peidl, Z., Vincze, A., Hunyady, B., Szolcsanyi, J., Kereskay, L., et al. (2005). Immunohistochemical distribution of vanilloid receptor, calcitonin-gene related peptide and substance P in gastrointestinal mucosa of patients with different gastrointestinal disorders. Inflammopharmacology 13 (1-3), 161–177. doi: 10.1163/156856005774423737
Dong, H., Shim, K. N., Li, J. M., Estrema, C., Ornelas, T. A., Nguyen, F., et al. (2010). Molecular mechanisms underlying Ca2+-mediated motility of human pancreatic duct cells. Am. J. Physiol. Cell Physiol. 299 (6), C1493–C1503. doi: 10.1152/ajpcell.00242.2010
Dozynkiewicz, M. A., Jamieson, N. B., Macpherson, I., Grindlay, J., van den Berghe, P. V., von Thun, A., et al. (2012). Rab25 and CLIC3 collaborate to promote integrin recycling from late endosomes/lysosomes and drive cancer progression. Dev. Cell 22 (1), 131–145. doi: 10.1016/j.devcel.2011.11.008
Du, J. D., Zheng, X., Chen, Y. L., Huang, Z. Q., Cai, S. W., Jiao, H. B., et al. (2018). Elevated Transient Receptor Potential Melastatin 8 (TRPM8) Expression Is Correlated with Poor Prognosis in Pancreatic Cancer. Med. Sci. Monit. 24, 3720–3725. doi: 10.12659/MSM.909968
Duprat, F., Lesage, F., Fink, M., Reyes, R., Heurteaux, C., Lazdunski, M. (1997). TASK, a human background K+ channel to sense external pH variations near physiological pH. EMBO J. 16 (17), 5464–5471. doi: 10.1093/emboj/16.17.5464
Duprat, F., Girard, C., Jarretou, G., Lazdunski, M. (2005). Pancreatic two P domain K+ channels TALK-1 and TALK-2 are activated by nitric oxide and reactive oxygen species. J. Physiol. 562 (Pt 1), 235–244. doi: 10.1113/jphysiol.2004.071266
Duran, C., Thompson, C. H., Xiao, Q., Hartzell, H. C. (2010). Chloride channels: often enigmatic, rarely predictable. Annu. Rev. Physiol. 72, 95–121. doi: 10.1146/annurev-physiol-021909-135811
Evans, R. L., Ashton, N., Elliott, A. C., Green, R., Argent, B. E. (1996). Interactions between secretin and acetylcholine in the regulation of fluid secretion by isolated rat pancreatic ducts. J. Physiol. 496 (Pt 1), 265–273. doi: 10.1113/jphysiol.1996.sp021683
Falzone, M. E., Malvezzi, M., Lee, B. C., Accardi, A. (2018). Known structures and unknown mechanisms of TMEM16 scramblases and channels. J. Gen. Physiol. 150 (7), 933–947. doi: 10.1085/jgp.201711957
Feng, J., Yu, J., Pan, X., Li, Z., Chen, Z., Zhang, W., et al. (2014). HERG1 functions as an oncogene in pancreatic cancer and is downregulated by miR-96. Oncotarget 5 (14), 5832–5844. doi: 10.18632/oncotarget.2200
Fong, P., Argent, B. E., Guggino, W. B., Gray, M. A. (2003). Characterization of vectorial chloride transport pathways in the human pancreatic duct adenocarcinoma cell line HPAF. Am. J. Physiol. Cell Physiol. 285 (2), C433–C445. doi: 10.1152/ajpcell.00509.2002
Furuya, S., Naruse, S., Ko, S. B., Ishiguro, H., Yoshikawa, T., Hayakawa, T. (2002). Distribution of aquaporin 1 in the rat pancreatic duct system examined with light- and electron-microscopic immunohistochemistry. Cell Tissue Res. 308 (1), 75–86. doi: 10.1007/s00441-002-0527-x
Gabbi, C., Kim, H. J., Hultenby, K., Bouton, D., Toresson, G., Warner, M., et al. (2008). Pancreatic exocrine insufficiency in LXRbeta-/- mice is associated with a reduction in aquaporin-1 expression. Proc. Natl. Acad. Sci. U. S. A. 105 (39), 15052–15057. doi: 10.1073/pnas.0808097105
Gallacher, D. V., Maruyama, Y., Petersen, O. H. (1984). Patch-clamp study of rubidium and potassium conductances in single cation channels from mammalian exocrine acini. Pflugers Arch. 401 (4), 361–367. doi: 10.1007/BF00584336
Gautam, D., Han, S. J., Heard, T. S., Cui, Y., Miller, G., Bloodworth, L., et al. (2005). Cholinergic stimulation of amylase secretion from pancreatic acinar cells studied with muscarinic acetylcholine receptor mutant mice. J. Pharmacol. Exp. Ther. 313 (3), 995–1002. doi: 10.1124/jpet.105.084855
Gerasimenko, J. V., Gryshchenko, O., Ferdek, P. E., Stapleton, E., Hebert, T. O., Bychkova, S., et al. (2013). Ca2+ release-activated Ca2+ channel blockade as a potential tool in antipancreatitis therapy. Proc. Natl. Acad. Sci. U. S. A. 110 (32), 13186–13191. doi: 10.1073/pnas.1300910110
Gheldof, A., Berx, G. (2013). Cadherins and epithelial-to-mesenchymal transition. Prog. Mol. Biol. Transl. Sci. 116, 317–336. doi: 10.1016/B978-0-12-394311-8.00014-5
Giannuzzo, A., Pedersen, S. F., Novak, I. (2015). The P2X7 receptor regulates cell survival, migration and invasion of pancreatic ductal adenocarcinoma cells. Mol. Cancer 14, 203. doi: 10.1186/s12943-015-0472-4
Giannuzzo, A., Saccomano, M., Napp, J., Ellegaard, M., Alves, F., Novak, I. (2016). Targeting of the P2X7 receptor in pancreatic cancer and stellate cells. Int. J. Cancer 139 (11), 2540–2552. doi: 10.1002/ijc.30380
Giovannucci, D. R., Bruce, J. I., Straub, S. V., Arreola, J., Sneyd, J., Shuttleworth, T. J., et al. (2002). Cytosolic Ca(2+) and Ca(2+)-activated Cl(-) current dynamics: insights from two functionally distinct mouse exocrine cells. J. Physiol. 540 (Pt 2), 469–484. doi: 10.1113/jphysiol.2001.013453
Girard, C., Duprat, F., Terrenoire, C., Tinel, N., Fosset, M., Romey, G., et al. (2001). Genomic and functional characteristics of novel human pancreatic 2P domain K(+) channels. Biochem. Biophys. Res. Commun. 282 (1), 249–256. doi: 10.1006/bbrc.2001.4562
Gomez-Varela, D., Zwick-Wallasch, E., Knotgen, H., Sanchez, A., Hettmann, T., Ossipov, D., et al. (2007). Monoclonal antibody blockade of the human Eag1 potassium channel function exerts antitumor activity. Cancer Res. 67 (15), 7343–7349. doi: 10.1158/0008-5472.CAN-07-0107
Gouaux, E., Mackinnon, R. (2005). Principles of selective ion transport in channels and pumps. Science 310 (5753), 1461–1465. doi: 10.1126/science.1113666
Gray, M. A., Greenwell, J. R., Argent, B. E. (1988). Secretin-regulated chloride channel on the apical plasma membrane of pancreatic duct cells. J. Membr. Biol. 105 (2), 131–142. doi: 10.1007/BF02009166
Gray, M. A., Harris, A., Coleman, L., Greenwell, J. R., Argent, B. E. (1989). Two types of chloride channel on duct cells cultured from human fetal pancreas. Am. J. Physiol. 257 (2 Pt 1), C240–C251. doi: 10.1152/ajpcell.1989.257.2.C240
Gray, M. A., Greenwell, J. R., Garton, A. J., Argent, B. E. (1990a). Regulation of maxi-K+ channels on pancreatic duct cells by cyclic AMP-dependent phosphorylation. J. Membr. Biol. 115 (3), 203–215. doi: 10.1007/bf01868636
Gray, M. A., Pollard, C. E., Harris, A., Coleman, L., Greenwell, J. R., Argent, B. E. (1990b). Anion selectivity and block of the small-conductance chloride channel on pancreatic duct cells. Am. J. Physiol. 259 (5 Pt 1), C752–C761. doi: 10.1152/ajpcell.1990.259.5.C752
Gray, M. A., Plant, S., Argent, B. E. (1993). cAMP-regulated whole cell chloride currents in pancreatic duct cells. Am. J. Physiol. 264 (3 Pt 1), C591–C602. doi: 10.1152/ajpcell.1993.264.3.C591
Gray, M. A., Winpenny, J. P., Porteous, D. J., Dorin, J. R., Argent, B. E. (1994). CFTR and calcium-activated chloride currents in pancreatic duct cells of a transgenic CF mouse. Am. J. Physiol. 266 (1 Pt 1), C213–C221. doi: 10.1152/ajpcell.1994.266.1.C213
Gray, M. A., Winpenny, J. P., Verdon, B., McAlroy, H., Argent, B. E. (1995). Chloride channels and cystic fibrosis of the pancreas. Biosci. Rep. 15 (6), 531–541. doi: 10.1007/BF01204355
Grimmig, T., Matthes, N., Hoeland, K., Tripathi, S., Chandraker, A., Grimm, M., et al. (2015). TLR7 and TLR8 expression increases tumor cell proliferation and promotes chemoresistance in human pancreatic cancer. Int. J. Oncol. 47 (3), 857–866. doi: 10.3892/ijo.2015.3069
Hamoir, C., Pepermans, X., Piessevaux, H., Jouret-Mourin, A., Weynand, B., Habyalimana, J. B., et al. (2013). Clinical and morphological characteristics of sporadic genetically determined pancreatitis as compared to idiopathic pancreatitis: higher risk of pancreatic cancer in CFTR variants. Digestion 87 (4), 229–239. doi: 10.1159/000348439
Hanahan, D., Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144 (5), 646–674. doi: 10.1016/j.cell.2011.02.013
Hansen, M. R., Krabbe, S., Novak, I. (2008). Purinergic receptors and calcium signalling in human pancreatic duct cell lines. Cell Physiol. Biochem. 22 (1-4), 157–168. doi: 10.1159/000149793
Hartel, M., di Mola, F. F., Selvaggi, F., Mascetta, G., Wente, M. N., Felix, K., et al. (2006). Vanilloids in pancreatic cancer: potential for chemotherapy and pain management. Gut 55 (4), 519–528. doi: 10.1136/gut.2005.073205
Hayashi, M., Novak, I. (2013). Molecular basis of potassium channels in pancreatic duct epithelial cells. Channels (Austin) 7 (6), 432–441. doi: 10.4161/chan.26100
Hayashi, M., Wang, J., Hede, S. E., Novak, I. (2012). An intermediate-conductance Ca2+-activated K+ channel is important for secretion in pancreatic duct cells. Am. J. Physiol. Cell Physiol. 303 (2), C151–C159. doi: 10.1152/ajpcell.00089.2012
Hede, S. E., Amstrup, J., Christoffersen, B. C., Novak, I. (1999). Purinoceptors evoke different electrophysiological responses in pancreatic ducts. P2Y inhibits K(+) conductance, and P2X stimulates cation conductance. J. Biol. Chem. 274 (45), 31784–31791. doi: 10.1074/jbc.274.45.31784
Hede, S. E., Amstrup, J., Klaerke, D. A., Novak, I. (2005). P2Y2 and P2Y4 receptors regulate pancreatic Ca(2+)-activated K+ channels differently. Pflugers Arch. 450 (6), 429–436. doi: 10.1007/s00424-005-1433-3
Hegyi, P., Petersen, O. H. (2013). The exocrine pancreas: the acinar-ductal tango in physiology and pathophysiology. Rev. Physiol. Biochem. Pharmacol. 165, 1–30. doi: 10.1007/112_2013_14
Hemmerlein, B., Weseloh, R. M., Mello de Queiroz, F., Knotgen, H., Sanchez, A., Rubio, M. E., et al. (2006). Overexpression of Eag1 potassium channels in clinical tumours. Mol. Cancer 5:41. doi: 10.1186/1476-4598-5-41
Hennig, A., Wolf, L., Jahnke, B., Polster, H., Seidlitz, T., Werner, K., et al. (2019). CFTR Expression Analysis for Subtyping of Human Pancreatic Cancer Organoids. Stem Cells Int. 2019:1024614. doi: 10.1155/2019/1024614
Hong, J. H., Li, Q., Kim, M. S., Shin, D. M., Feske, S., Birnbaumer, L., et al. (2011). Polarized but differential localization and recruitment of STIM1, Orai1 and TRPC channels in secretory cells. Traffic 12 (2), 232–245. doi: 10.1111/j.1600-0854.2010.01138.x
Hoth, M., Niemeyer, B. A. (2013). The neglected CRAC proteins: Orai2, Orai3, and STIM2. Curr. Top. Membr. 71, 237–271. doi: 10.1016/B978-0-12-407870-3.00010-X
Hu, D., Ansari, D., Pawlowski, K., Zhou, Q., Sasor, A., Welinder, C., et al. (2018a). Proteomic analyses identify prognostic biomarkers for pancreatic ductal adenocarcinoma. Oncotarget 9 (11), 9789–9807. doi: 10.18632/oncotarget.23929
Hu, D., Ansari, D., Zhou, Q., Sasor, A., Hilmersson, K. S., Bauden, M., et al. (2018b). Calcium-activated chloride channel regulator 1 as a prognostic biomarker in pancreatic ductal adenocarcinoma. BMC Cancer 18 (1), 1096. doi: 10.1186/s12885-018-5013-2
Hu, D., Ansari, D., Bauden, M., Zhou, Q., Andersson, R. (2019). The Emerging Role of Calcium-activated Chloride Channel Regulator 1 in Cancer. Anticancer Res. 39 (4), 1661–1666. doi: 10.21873/anticanres.13271
Huang, F., Rock, J. R., Harfe, B. D., Cheng, T., Huang, X., Jan, Y. N., et al. (2009). Studies on expression and function of the TMEM16A calcium-activated chloride channel. Proc. Natl. Acad. Sci. U. S. A. 106 (50), 21413–21418. doi: 10.1073/pnas.0911935106
Huang, X., Huang, L., Shao, M. (2017). Aquaporin 3 facilitates tumor growth in pancreatic cancer by modulating mTOR signaling. Biochem. Biophys. Res. Commun. 486 (4), 1097–1102. doi: 10.1016/j.bbrc.2017.03.168
Huang, J., Liu, J., Qiu, L. (2020). Transient receptor potential vanilloid 1 promotes EGFR ubiquitination and modulates EGFR/MAPK signalling in pancreatic cancer cells. Cell Biochem. Funct. 38 (4), 401–408. doi: 10.1002/cbf.3483
Hurley, P. T., Ferguson, C. J., Kwon, T. H., Andersen, M. L., Norman, A. G., Steward, M. C., et al. (2001). Expression and immunolocalization of aquaporin water channels in rat exocrine pancreas. Am. J. Physiol. Gastrointest. Liver Physiol. 280 (4), G701–G709. doi: 10.1152/ajpgi.2001.280.4.G701
Ishiguro, H., Naruse, S., Kitagawa, M., Mabuchi, T., Kondo, T., Hayakawa, T., et al. (2002). Chloride transport in microperfused interlobular ducts isolated from guinea-pig pancreas. J. Physiol. 539 (Pt 1), 175–189. doi: 10.1113/jphysiol.2001.012490
Ishiguro, H., Steward, M. C., Naruse, S., Ko, S. B., Goto, H., Case, R. M., et al. (2009). CFTR functions as a bicarbonate channel in pancreatic duct cells. J. Gen. Physiol. 133 (3), 315–326. doi: 10.1085/jgp.200810122
Ishiguro, H., Yamamoto, A., Nakakuki, M., Yi, L., Ishiguro, M., Yamaguchi, M., et al. (2012). Physiology and pathophysiology of bicarbonate secretion by pancreatic duct epithelium. Nagoya J. Med. Sci. 74 (1-2), 1–18.
Ishii, T. M., Silvia, C., Hirschberg, B., Bond, C. T., Adelman, J. P., Maylie, J. (1997). A human intermediate conductance calcium-activated potassium channel. Proc. Natl. Acad. Sci. U. S. A. 94 (21), 11651–11656. doi: 10.1073/pnas.94.21.11651
Isokpehi, R. D., Rajnarayanan, R. V., Jeffries, C. D., Oyeleye, T. O., Cohly, H. H. (2009). Integrative sequence and tissue expression profiling of chicken and mammalian aquaporins. BMC Genomics 10 Suppl 2, S7. doi: 10.1186/1471-2164-10-S2-S7
Itoh, T., Rai, T., Kuwahara, M., Ko, S. B., Uchida, S., Sasaki, S., et al. (2005). Identification of a novel aquaporin, AQP12, expressed in pancreatic acinar cells. Biochem. Biophys. Res. Commun. 330 (3), 832–838. doi: 10.1016/j.bbrc.2005.03.046
Iwatsuki, N., Petersen, O. H. (1977). Pancreatic acinar cells: localization of acetylcholine receptors and the importance of chloride and calcium for acetylcholine-evoked depolarization. J. Physiol. 269 (3), 723–733. doi: 10.1113/jphysiol.1977.sp011925
Iwatsuki, N., Petersen, O. H. (1985a). Action of tetraethylammonium on calcium-activated potassium channels in pig pancreatic acinar cells studied by patch-clamp single-channel and whole-cell current recording. J. Membr. Biol. 86 (2), 139–144. doi: 10.1007/BF01870780
Iwatsuki, N., Petersen, O. H. (1985b). Inhibition of Ca2+-activated K+ channels in pig pancreatic acinar cells by Ba2+, Ca2+, quinine and quinidine. Biochim. Biophys. Acta 819 (2), 249–257. doi: 10.1016/0005-2736(85)90180-4
Jabbar, K. S., Arike, L., Verbeke, C. S., Sadik, R., Hansson, G. C. (2018). Highly Accurate Identification of Cystic Precursor Lesions of Pancreatic Cancer Through Targeted Mass Spectrometry: A Phase IIc Diagnostic Study. J. Clin. Oncol. 36 (4), 367–375. doi: 10.1200/JCO.2017.73.7288
Jager, H., Dreker, T., Buck, A., Giehl, K., Gress, T., Grissmer, S. (2004). Blockage of intermediate-conductance Ca2+-activated K+ channels inhibit human pancreatic cancer cell growth in vitro. Mol. Pharmacol. 65 (3), 630–638. doi: 10.1124/mol.65.3.630
Jia, N., Dong, S., Zhao, G., Gao, H., Li, X., Zhang, H. (2016). CLIC1 overexpression is associated with poor prognosis in pancreatic ductal adenocarcinomas. J. Cancer Res. Ther. 12 (2), 892–896. doi: 10.4103/0973-1482.154057
Jiang, S., Zhu, L., Yang, J., Hu, L., Gu, J., Xing, X., et al. (2017). Integrated expression profiling of potassium channels identifys KCNN4 as a prognostic biomarker of pancreatic cancer. Biochem. Biophys. Res. Commun. 494 (1-2), 113–119. doi: 10.1016/j.bbrc.2017.10.072
Joiner, W. J., Wang, L. Y., Tang, M. D., Kaczmarek, L. K. (1997). hSK4, a member of a novel subfamily of calcium-activated potassium channels. Proc. Natl. Acad. Sci. U. S. A. 94 (20), 11013–11018. doi: 10.1073/pnas.94.20.11013
Jun, I., Park, H. S., Piao, H., Han, J. W., An, M. J., Yun, B. G., et al. (2017). ANO9/TMEM16J promotes tumourigenesis via EGFR and is a novel therapeutic target for pancreatic cancer. Br. J. Cancer 117 (12), 1798–1809. doi: 10.1038/bjc.2017.355
Jung, S. R., Kim, K., Hille, B., Nguyen, T. D., Koh, D. S. (2006). Pattern of Ca2+ increase determines the type of secretory mechanism activated in dog pancreatic duct epithelial cells. J. Physiol. 576 (Pt 1), 163–178. doi: 10.1113/jphysiol.2006.114876
Kalman, K., Nguyen, A., Tseng-Crank, J., Dukes, I. D., Chandy, G., Hustad, C. M., et al. (1998). Genomic organization, chromosomal localization, tissue distribution, and biophysical characterization of a novel mammalian Shaker-related voltage-gated potassium channel, Kv1.7. J. Biol. Chem. 273 (10), 5851–5857. doi: 10.1074/jbc.273.10.5851
Kasai, H., Augustine, G. J. (1990). Cytosolic Ca2+ gradients triggering unidirectional fluid secretion from exocrine pancreas. Nature 348 (6303), 735–738. doi: 10.1038/348735a0
Khan, H. Y., Mpilla, G. B., Sexton, R., Viswanadha, S., Penmetsa, K. V., Aboukameel, A., et al. (2020). Calcium Release-Activated Calcium (CRAC) Channel Inhibition Suppresses Pancreatic Ductal Adenocarcinoma Cell Proliferation and Patient-Derived Tumor Growth. Cancers (Basel) 12 (3), 750. doi: 10.3390/cancers12030750
Kim, S. J., Greger, R. (1999). Voltage-dependent, slowly activating K+ current (I(Ks)) and its augmentation by carbachol in rat pancreatic acini. Pflugers Arch. 438 (5), 604–611. doi: 10.1007/s004249900071
Kim, S. J., Kerst, G., Schreiber, R., Pavenstadt, H., Greger, R., Hug, M. J., et al. (2000). Inwardly rectifying K+ channels in the basolateral membrane of rat pancreatic acini. Pflugers Arch. 441 (2-3), 331–340. doi: 10.1007/s004240000427
Kim, C. J., Cho, Y. G., Jeong, S. W., Kim, Y. S., Kim, S. Y., Nam, S. W., et al. (2004). Altered expression of KCNK9 in colorectal cancers. APMIS 112 (9), 588–594. doi: 10.1111/j.1600-0463.2004.apm1120905.x
Kim, J. Y., Zeng, W., Kiselyov, K., Yuan, J. P., Dehoff, M. H., Mikoshiba, K., et al. (2006). Homer 1 mediates store- and inositol 1,4,5-trisphosphate receptor-dependent translocation and retrieval of TRPC3 to the plasma membrane. J. Biol. Chem. 281 (43), 32540–32549. doi: 10.1074/jbc.M602496200
Kim, M. H., Choi, B. H., Jung, S. R., Sernka, T. J., Kim, S., Kim, K. T., et al. (2008). Protease-activated receptor-2 increases exocytosis via multiple signal transduction pathways in pancreatic duct epithelial cells. J. Biol. Chem. 283 (27), 18711–18720. doi: 10.1074/jbc.M801655200
Kim, M. S., Hong, J. H., Li, Q., Shin, D. M., Abramowitz, J., Birnbaumer, L., et al. (2009). Deletion of TRPC3 in mice reduces store-operated Ca2+ influx and the severity of acute pancreatitis. Gastroenterology 137 (4), 1509–1517. doi: 10.1053/j.gastro.2009.07.042
Kim, M. H., Seo, J. B., Burnett, L. A., Hille, B., Koh, D. S. (2013). Characterization of store-operated Ca2+ channels in pancreatic duct epithelia. Cell Calcium 54 (4), 266–275. doi: 10.1016/j.ceca.2013.07.002
Kim, H., Kim, H., Lee, J., Lee, B., Kim, H. R., Jung, J., et al. (2018). Anoctamin 9/TMEM16J is a cation channel activated by cAMP/PKA signal. Cell Calcium 71, 75–85. doi: 10.1016/j.ceca.2017.12.003
Kirkegard, J., Mortensen, F. V., Cronin-Fenton, D. (2017). Chronic Pancreatitis and Pancreatic Cancer Risk: A Systematic Review and Meta-analysis. Am. J. Gastroenterol. 112 (9), 1366–1372. doi: 10.1038/ajg.2017.218
Ko, S. B., Naruse, S., Kitagawa, M., Ishiguro, H., Furuya, S., Mizuno, N., et al. (2002). Aquaporins in rat pancreatic interlobular ducts. Am. J. Physiol. Gastrointest. Liver Physiol. 282 (2), G324–G331. doi: 10.1152/ajpgi.00198.2001
Koltai, T. (2015). Voltage-gated sodium channel as a target for metastatic risk reduction with re-purposed drugs. F1000Res 4, 297. doi: 10.12688/f1000research.6789.1
Kondratska, K., Kondratskyi, A., Yassine, M., Lemonnier, L., Lepage, G., Morabito, A., et al. (2014). Orai1 and STIM1 mediate SOCE and contribute to apoptotic resistance of pancreatic adenocarcinoma. Biochim. Biophys. Acta 1843 (10), 2263–2269. doi: 10.1016/j.bbamcr.2014.02.012
Kong, S. C., Giannuzzo, A., Novak, I., Pedersen, S. F. (2014). Acid-base transport in pancreatic cancer: molecular mechanisms and clinical potential. Biochem. Cell Biol. 92 (6), 449–459. doi: 10.1139/bcb-2014-0078
Kottgen, M., Hoefer, A., Kim, S. J., Beschorner, U., Schreiber, R., Hug, M. J., et al. (1999). Carbachol activates a K+ channel of very small conductance in the basolateral membrane of rat pancreatic acinar cells. Pflugers Arch. 438 (5), 597–603. doi: 10.1007/s004249900070
Kovalenko, I., Glasauer, A., Schockel, L., Sauter, D. R., Ehrmann, A., Sohler, F., et al. (2016). Identification of KCa3.1 Channel as a Novel Regulator of Oxidative Phosphorylation in a Subset of Pancreatic Carcinoma Cell Lines. PloS One 11 (8), e0160658. doi: 10.1371/journal.pone.0160658
Koyama, Y., Yamamoto, T., Kondo, D., Funaki, H., Yaoita, E., Kawasaki, K., et al. (1997). Molecular cloning of a new aquaporin from rat pancreas and liver. J. Biol. Chem. 272 (48), 30329–30333. doi: 10.1074/jbc.272.48.30329
Kunzelmann, K. (2005). Ion channels and cancer. J. Membr. Biol. 205 (3), 159–173. doi: 10.1007/s00232-005-0781-4
Kunzelmann, K. (2016). Ion channels in regulated cell death. Cell Mol. Life Sci. 73 (11-12), 2387–2403. doi: 10.1007/s00018-016-2208-z
Kunzli, B. M., Berberat, P. O., Giese, T., Csizmadia, E., Kaczmarek, E., Baker, C., et al. (2007). Upregulation of CD39/NTPDases and P2 receptors in human pancreatic disease. Am. J. Physiol. Gastrointest. Liver Physiol. 292 (1), G223–G230. doi: 10.1152/ajpgi.00259.2006
Lang, F., Stournaras, C. (2014). Ion channels in cancer: future perspectives and clinical potential. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 369 (1638), 20130108. doi: 10.1098/rstb.2013.0108
Lastraioli, E., Lottini, T., Bencini, L., Bernini, M., Arcangeli, A. (2015a). hERG1 Potassium Channels: Novel Biomarkers in Human Solid Cancers. BioMed. Res. Int. 2015:896432. doi: 10.1155/2015/896432
Lastraioli, E., Perrone, G., Sette, A., Fiore, A., Crociani, O., Manoli, S., et al. (2015b). hERG1 channels drive tumour malignancy and may serve as prognostic factor in pancreatic ductal adenocarcinoma. Br. J. Cancer 112 (6), 1076–1087. doi: 10.1038/bjc.2015.28
Lazzeri, M., Vannucchi, M. G., Spinelli, M., Bizzoco, E., Beneforti, P., Turini, D., et al. (2005). Transient receptor potential vanilloid type 1 (TRPV1) expression changes from normal urothelium to transitional cell carcinoma of human bladder. Eur. Urol. 48 (4), 691–698. doi: 10.1016/j.eururo.2005.05.018
Lee, M. G., Ahn, W., Lee, J. A., Kim, J. Y., Choi, J. Y., Moe, O. W., et al. (2001). Coordination of pancreatic HCO3- secretion by protein-protein interaction between membrane transporters. JOP 2 (4 Suppl), 203–206.
Lee, G. W., Park, H. S., Kim, E. J., Cho, Y. W., Kim, G. T., Mun, Y. J., et al. (2012). Reduction of breast cancer cell migration via up-regulation of TASK-3 two-pore domain K+ channel. Acta Physiol. (Oxf) 204 (4), 513–524. doi: 10.1111/j.1748-1716.2011.02359.x
Lee, M. G., Ohana, E., Park, H. W., Yang, D., Muallem, S. (2012). Molecular mechanism of pancreatic and salivary gland fluid and HCO3 secretion. Physiol. Rev. 92 (1), 39–74. doi: 10.1152/physrev.00011.2011
Lemstrova, R., Soucek, P., Melichar, B., Mohelnikova-Duchonova, B. (2014). Role of solute carrier transporters in pancreatic cancer: a review. Pharmacogenomics 15 (8), 1133–1145. doi: 10.2217/pgs.14.80
Li, L., Hanahan, D. (2013). Hijacking the neuronal NMDAR signaling circuit to promote tumor growth and invasion. Cell 153 (1), 86–100. doi: 10.1016/j.cell.2013.02.051
Liddle, R. A. (2007). The role of Transient Receptor Potential Vanilloid 1 (TRPV1) channels in pancreatitis. Biochim. Biophys. Acta 1772 (8), 869–878. doi: 10.1016/j.bbadis.2007.02.012
Lin, R., Wang, Y., Chen, Q., Liu, Z., Xiao, S., Wang, B., et al. (2018). TRPM2 promotes the proliferation and invasion of pancreatic ductal adenocarcinoma. Mol. Med. Rep. 17 (6), 7537–7544. doi: 10.3892/mmr.2018.8816
Litan, A., Langhans, S. A. (2015). Cancer as a channelopathy: ion channels and pumps in tumor development and progression. Front. Cell Neurosci. 9, 86. doi: 10.3389/fncel.2015.00086
Liu, Y., McKenna, E., Figueroa, D. J., Blevins, R., Austin, C. P., Bennett, P. B., et al. (2000). The human inward rectifier K(+) channel subunit kir5.1 (KCNJ16) maps to chromosome 17q25 and is expressed in kidney and pancreas. Cytogenet. Cell Genet. 90 (1-2), 60–63. doi: 10.1159/000015662
Liu, X., Cheng, K. T., Bandyopadhyay, B. C., Pani, B., Dietrich, A., Paria, B. C., et al. (2007). Attenuation of store-operated Ca2+ current impairs salivary gland fluid secretion in TRPC1(-/-) mice. Proc. Natl. Acad. Sci. U. S. A. 104 (44), 17542–17547. doi: 10.1073/pnas.0701254104
Liu, W., Lu, M., Liu, B., Huang, Y., Wang, K. (2012). Inhibition of Ca(2+)-activated Cl(-) channel ANO1/TMEM16A expression suppresses tumor growth and invasiveness in human prostate carcinoma. Cancer Lett. 326 (1), 41–51. doi: 10.1016/j.canlet.2012.07.015
Liu, J., Hu, G., Gong, Y., Yu, Q., He, B., Li, W., et al. (2018). Silencing of TRPM8 inhibits aggressive tumor phenotypes and enhances gemcitabine sensitivity in pancreatic cancer. Pancreatology 18 (8), 935–944. doi: 10.1016/j.pan.2018.08.011
Lotz, M. M., Wang, H., Song, J. C., Pories, S. E., Matthews, J. B. (2004). K+ channel inhibition accelerates intestinal epithelial cell wound healing. Wound Repair Regener. 12 (5), 565–574. doi: 10.1111/j.1067-1927.2004.012509.x
Lowenfels, A. B., Maisonneuve, P., Cavallini, G., Ammann, R. W., Lankisch, P. G., Andersen, J. R., et al. (1993). Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N. Engl. J. Med. 328 (20), 1433–1437. doi: 10.1056/NEJM199305203282001
Lu, J., Dong, Q., Zhang, B., Wang, X., Ye, B., Zhang, F., et al. (2015). Chloride intracellular channel 1 (CLIC1) is activated and functions as an oncogene in pancreatic cancer. Med. Oncol. 32 (6), 616. doi: 10.1007/s12032-015-0616-9
Lur, G., Haynes, L. P., Prior, I. A., Gerasimenko, O. V., Feske, S., Petersen, O. H., et al. (2009). Ribosome-free terminals of rough ER allow formation of STIM1 puncta and segregation of STIM1 from IP(3) receptors. Curr. Biol. 19 (19), 1648–1653. doi: 10.1016/j.cub.2009.07.072
Lur, G., Sherwood, M. W., Ebisui, E., Haynes, L., Feske, S., Sutton, R., et al. (2011). InsP(3)receptors and Orai channels in pancreatic acinar cells: co-localization and its consequences. Biochem. J. 436 (2), 231–239. doi: 10.1042/BJ20110083
Ma, T., Jayaraman, S., Wang, K. S., Song, Y., Yang, B., Li, J., et al. (2001). Defective dietary fat processing in transgenic mice lacking aquaporin-1 water channels. Am. J. Physiol. Cell Physiol. 280 (1), C126–C134. doi: 10.1152/ajpcell.2001.280.1.C126
Maleth, J., Hegyi, P. (2014). Calcium signaling in pancreatic ductal epithelial cells: an old friend and a nasty enemy. Cell Calcium 55 (6), 337–345. doi: 10.1016/j.ceca.2014.02.004
Manoli, S., Coppola, S., Duranti, C., Lulli, M., Magni, L., Kuppalu, N., et al. (2019). The Activity of Kv 11.1 Potassium Channel Modulates F-Actin Organization During Cell Migration of Pancreatic Ductal Adenocarcinoma Cells. Cancers (Basel) 11 (2), 135. doi: 10.3390/cancers11020135
Marino, C. R., Matovcik, L. M., Gorelick, F. S., Cohn, J. A. (1991). Localization of the cystic fibrosis transmembrane conductance regulator in pancreas. J. Clin. Invest. 88 (2), 712–716. doi: 10.1172/JCI115358
Marty, A., Tan, Y. P., Trautmann, A. (1984). Three types of calcium-dependent channel in rat lacrimal glands. J. Physiol. 357, 293–325. doi: 10.1113/jphysiol.1984.sp015501
Maruyama, Y., Petersen, O. H., Flanagan, P., Pearson, G. T. (1983). Quantification of Ca2+-activated K+ channels under hormonal control in pig pancreas acinar cells. Nature 305 (5931), 228–232. doi: 10.1038/305228a0
Matthews, E. K., Petersen, O. H., Williams, J. A. (1973). Pancreatic acinar cells: acetylcholine-induced membrane depolarization, calcium efflux and amylase release. J. Physiol. 234 (3), 689–701. doi: 10.1113/jphysiol.1973.sp010367
Mazzone, A., Eisenman, S. T., Strege, P. R., Yao, Z., Ordog, T., Gibbons, S. J., et al. (2012). Inhibition of cell proliferation by a selective inhibitor of the Ca(2+)-activated Cl(-) channel, Ano1. Biochem. Biophys. Res. Commun. 427 (2), 248–253. doi: 10.1016/j.bbrc.2012.09.022
McDonald, F. J., Snyder, P. M., McCray, P. B., Jr., Welsh, M. J. (1994). Cloning, expression, and tissue distribution of a human amiloride-sensitive Na+ channel. Am. J. Physiol. 266 (6 Pt 1), L728–L734. doi: 10.1152/ajplung.1994.266.6.L728
McDonald, T. V., Yu, Z., Ming, Z., Palma, E., Meyers, M. B., Wang, K. W., et al. (1997). A minK-HERG complex regulates the cardiac potassium current I(Kr). Nature 388 (6639), 289–292. doi: 10.1038/40882
McWilliams, R. R., Petersen, G. M., Rabe, K. G., Holtegaard, L. M., Lynch, P. J., Bishop, M. D., et al. (2010). Cystic fibrosis transmembrane conductance regulator (CFTR) gene mutations and risk for pancreatic adenocarcinoma. Cancer 116 (1), 203–209. doi: 10.1002/cncr.24697
Miao, X., Liu, G., Xu, X., Xie, C., Sun, F., Yang, Y., et al. (2008). High expression of vanilloid receptor-1 is associated with better prognosis of patients with hepatocellular carcinoma. Cancer Genet. Cytogenet. 186 (1), 25–32. doi: 10.1016/j.cancergencyto.2008.05.011
Mo, P., Yang, S. (2018). The store-operated calcium channels in cancer metastasis: from cell migration, invasion to metastatic colonization. Front. Biosci. (Landmark Ed.) 23, 1241–1256. doi: 10.2741/4641
Mogami, H., Nakano, K., Tepikin, A. V., Petersen, O. H. (1997). Ca2+ flow via tunnels in polarized cells: recharging of apical Ca2+ stores by focal Ca2+ entry through basal membrane patch. Cell 88 (1), 49–55. doi: 10.1016/s0092-8674(00)81857-7
Morelli, M. B., Amantini, C., Nabissi, M., Liberati, S., Cardinali, C., Farfariello, V., et al. (2014). Cross-talk between alpha1D-adrenoceptors and transient receptor potential vanilloid type 1 triggers prostate cancer cell proliferation. BMC Cancer 14, 921. doi: 10.1186/1471-2407-14-921
Mu, D., Chen, L., Zhang, X., See, L. H., Koch, C. M., Yen, C., et al. (2003). Genomic amplification and oncogenic properties of the KCNK9 potassium channel gene. Cancer Cell 3 (3), 297–302. doi: 10.1016/s1535-6108(03)00054-0
Nagy, D., Gonczi, M., Dienes, B., Szoor, A., Fodor, J., Nagy, Z., et al. (2014). Silencing the KCNK9 potassium channel (TASK-3) gene disturbs mitochondrial function, causes mitochondrial depolarization, and induces apoptosis of human melanoma cells. Arch. Dermatol. Res. 306 (10), 885–902. doi: 10.1007/s00403-014-1511-5
Neglia, J. P., FitzSimmons, S. C., Maisonneuve, P., Schoni, M. H., Schoni-Affolter, F., Corey, M., et al. (1995). The risk of cancer among patients with cystic fibrosis. Cystic Fibrosis and Cancer Study Group. N. Engl. J. Med. 332 (8), 494–499. doi: 10.1056/NEJM199502233320803
Nguyen, T. D., Moody, M. W. (1998). Calcium-activated potassium conductances on cultured nontransformed dog pancreatic duct epithelial cells. Pancreas 17 (4), 348–358. doi: 10.1097/00006676-199811000-00005
Nguyen, T. D., Koh, D. S., Moody, M. W., Fox, N. R., Savard, C. E., Kuver, R., et al. (1997). Characterization of two distinct chloride channels in cultured dog pancreatic duct epithelial cells. Am. J. Physiol. 272 (1 Pt 1), G172–G180. doi: 10.1152/ajpgi.1997.272.1.G172
Niemeyer, B. A., Mery, L., Zawar, C., Suckow, A., Monje, F., Pardo, L. A., et al. (2001). Ion channels in health and disease. 83rd Boehringer Ingelheim Fonds International Titisee Conference. EMBO Rep. 2 (7), 568–573. doi: 10.1093/embo-reports/kve145
North, W. G., Liu, F., Lin, L. Z., Tian, R., Akerman, B. (2017). NMDA receptors are important regulators of pancreatic cancer and are potential targets for treatment. Clin. Pharmacol. 9, 79–86. doi: 10.2147/CPAA.S140057
Novak, I., Hansen, M. R. (2002). Where have all the Na+ channels gone? In search of functional ENaC in exocrine pancreas. Biochim. Biophys. Acta 1566 (1-2), 162–168. doi: 10.1016/s0005-2736(02)00598-9
Novak, I., Wang, J., Henriksen, K. L., Haanes, K. A., Krabbe, S., Nitschke, R., et al. (2011). Pancreatic bicarbonate secretion involves two proton pumps. J. Biol. Chem. 286 (1), 280–289. doi: 10.1074/jbc.M110.136382
Novak, I., Haanes, K. A., Wang, J. (2013). Acid-base transport in pancreas-new challenges. Front. Physiol. 4, 380. doi: 10.3389/fphys.2013.00380
Novak, I. (2000). Keeping up with bicarbonate. J. Physiol. 528 Pt 2, 235. doi: 10.1111/j.1469-7793.2000.00235.x
Ochi, A., Graffeo, C. S., Zambirinis, C. P., Rehman, A., Hackman, M., Fallon, N., et al. (2012). Toll-like receptor 7 regulates pancreatic carcinogenesis in mice and humans. J. Clin. Invest. 122 (11), 4118–4129. doi: 10.1172/JCI63606
Ohta, E., Itoh, T., Nemoto, T., Kumagai, J., Ko, S. B., Ishibashi, K., et al. (2009). Pancreas-specific aquaporin 12 null mice showed increased susceptibility to caerulein-induced acute pancreatitis. Am. J. Physiol. Cell Physiol. 297 (6), C1368–C1378. doi: 10.1152/ajpcell.00117.2009
Okeke, E., Parker, T., Dingsdale, H., Concannon, M., Awais, M., Voronina, S., et al. (2016). Epithelial-mesenchymal transition, IP3 receptors and ER-PM junctions: translocation of Ca2+ signalling complexes and regulation of migration. Biochem. J. 473 (6), 757–767. doi: 10.1042/BJ20150364
Ousingsawat, J., Martins, J. R., Schreiber, R., Rock, J. R., Harfe, B. D., Kunzelmann, K. (2009). Loss of TMEM16A causes a defect in epithelial Ca2+-dependent chloride transport. J. Biol. Chem. 284 (42), 28698–28703. doi: 10.1074/jbc.M109.012120
Pallagi, P., Hegyi, P., Rakonczay, Z., Jr. (2015). The Physiology and Pathophysiology of Pancreatic Ductal Secretion: The Background for Clinicians. Pancreas 44 (8), 1211–1233. doi: 10.1097/MPA.0000000000000421
Pandiri, A. R. (2014). Overview of exocrine pancreatic pathobiology. Toxicol. Pathol. 42 (1), 207–216. doi: 10.1177/0192623313509907
Papadopoulos, M. C., Saadoun, S. (2015). Key roles of aquaporins in tumor biology. Biochim. Biophys. Acta 1848 (10 Pt B), 2576–2583. doi: 10.1016/j.bbamem.2014.09.001
Pardo, L. A., Stuhmer, W. (2014). The roles of K(+) channels in cancer. Nat. Rev. Cancer 14 (1), 39–48. doi: 10.1038/nrc3635
Pardo, L. A., del Camino, D., Sanchez, A., Alves, F., Bruggemann, A., Beckh, S., et al. (1999). Oncogenic potential of EAG K(+) channels. EMBO J. 18 (20), 5540–5547. doi: 10.1093/emboj/18.20.5540
Park, M. K., Lomax, R. B., Tepikin, A. V., Petersen, O. H. (2001). Local uncaging of caged Ca(2+) reveals distribution of Ca(2+)-activated Cl(-) channels in pancreatic acinar cells. Proc. Natl. Acad. Sci. U. S. A. 98 (19), 10948–10953. doi: 10.1073/pnas.181353798
Park, H. W., Nam, J. H., Kim, J. Y., Namkung, W., Yoon, J. S., Lee, J. S., et al. (2010). Dynamic regulation of CFTR bicarbonate permeability by [Cl-]i and its role in pancreatic bicarbonate secretion. Gastroenterology 139 (2), 620–631. doi: 10.1053/j.gastro.2010.04.004
Pascua, P., Garcia, M., Fernandez-Salazar, M. P., Hernandez-Lorenzo, M. P., Calvo, J. J., Colledge, W. H., et al. (2009). Ducts isolated from the pancreas of CFTR-null mice secrete fluid. Pflugers Arch. 459 (1), 203–214. doi: 10.1007/s00424-009-0704-9
Pearson, G. T., Flanagan, P. M., Petersen, O. H. (1984). Neural and hormonal control of membrane conductance in the pig pancreatic acinar cell. Am. J. Physiol. 247 (5 Pt 1), G520–G526. doi: 10.1152/ajpgi.1984.247.5.G520
Pedersen, S. F., Stock, C. (2013). Ion channels and transporters in cancer: pathophysiology, regulation, and clinical potential. Cancer Res. 73 (6), 1658–1661. doi: 10.1158/0008-5472.CAN-12-4188
Pedersen, S. F., Novak, I., Alves, F., Schwab, A., Pardo, L. A. (2017). Alternating pH landscapes shape epithelial cancer initiation and progression: Focus on pancreatic cancer. Bioessays 39 (6). doi: 10.1002/bies.201600253
Peretti, M., Angelini, M., Savalli, N., Florio, T., Yuspa, S. H., Mazzanti, M. (2015). Chloride channels in cancer: Focus on chloride intracellular channel 1 and 4 (CLIC1 AND CLIC4) proteins in tumor development and as novel therapeutic targets. Biochim. Biophys. Acta 1848 (10 Pt B), 2523–2531. doi: 10.1016/j.bbamem.2014.12.012
Pessia, M., Imbrici, P., D’Adamo, M. C., Salvatore, L., Tucker, S. J. (2001). Differential pH sensitivity of Kir4.1 and Kir4.2 potassium channels and their modulation by heteropolymerisation with Kir5.1. J. Physiol. 532 (Pt 2), 359–367. doi: 10.1111/j.1469-7793.2001.0359f.x
Petersen, O. H., Findlay, I. (1987). Electrophysiology of the pancreas. Physiol. Rev. 67 (3), 1054–1116. doi: 10.1152/physrev.1987.67.3.1054
Petersen, O. H., Gallacher, D. V. (1988). Electrophysiology of pancreatic and salivary acinar cells. Annu. Rev. Physiol. 50, 65–80. doi: 10.1146/annurev.ph.50.030188.000433
Petersen, O. H., Tepikin, A. V. (2008). Polarized calcium signaling in exocrine gland cells. Annu. Rev. Physiol. 70, 273–299. doi: 10.1146/annurev.physiol.70.113006.100618
Petersen, O. H., Ueda, N. (1976). Pancreatic acinar cells: the role of calcium in stimulus-secretion coupling. J. Physiol. 254 (3), 583–606. doi: 10.1113/jphysiol.1976.sp011248
Petersen, O. H., Findlay, I., Iwatsuki, N., Singh, J., Gallacher, D. V., Fuller, C. M., et al. (1985). Human pancreatic acinar cells: studies of stimulus-secretion coupling. Gastroenterology 89 (1), 109–117. doi: 10.1016/0016-5085(85)90751-6
Petersen, O. H. (1992). Stimulus-secretion coupling: cytoplasmic calcium signals and the control of ion channels in exocrine acinar cells. J. Physiol. 448, 1–51. doi: 10.1113/jphysiol.1992.sp019028
Petersen, O. H. (2005). Ca2+ signalling and Ca2+-activated ion channels in exocrine acinar cells. Cell Calcium 38 (3-4), 171–200. doi: 10.1016/j.ceca.2005.06.024
Petersen, O. H. (2014). Calcium signalling and secretory epithelia. Cell Calcium 55 (6), 282–289. doi: 10.1016/j.ceca.2014.01.003
Pond, A. L., Scheve, B. K., Benedict, A. T., Petrecca, K., Van Wagoner, D. R., Shrier, A., et al. (2000). Expression of distinct ERG proteins in rat, mouse, and human heart. Relation to functional I(Kr) channels. J. Biol. Chem. 275 (8), 5997–6006. doi: 10.1074/jbc.275.8.5997
Premkumar, L. S., Abooj, M. (2013). TRP channels and analgesia. Life Sci. 92 (8-9), 415–424. doi: 10.1016/j.lfs.2012.08.010
Prevarskaya, N., Zhang, L., Barritt, G. (2007). TRP channels in cancer. Biochim. Biophys. Acta 1772 (8), 937–946. doi: 10.1016/j.bbadis.2007.05.006
Prevarskaya, N., Skryma, R., Shuba, Y. (2010). Ion channels and the hallmarks of cancer. Trends Mol. Med. 16 (3), 107–121. doi: 10.1016/j.molmed.2010.01.005
Prevarskaya, N., Skryma, R., Shuba, Y. (2018). Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 98 (2), 559–621. doi: 10.1152/physrev.00044.2016
Radisky, D. C., LaBarge, M. A. (2008). Epithelial-mesenchymal transition and the stem cell phenotype. Cell Stem Cell 2 (6), 511–512. doi: 10.1016/j.stem.2008.05.007
Ragsdale, D. S., Scheuer, T., Catterall, W. A. (1991). Frequency and voltage-dependent inhibition of type IIA Na+ channels, expressed in a mammalian cell line, by local anesthetic, antiarrhythmic, and anticonvulsant drugs. Mol. Pharmacol. 40 (5), 756–765.
Randriamampita, C., Chanson, M., Trautmann, A. (1988). Calcium and secretagogues-induced conductances in rat exocrine pancreas. Pflugers Arch. 411 (1), 53–57. doi: 10.1007/BF00581646
Reubi, J. C., Waser, B., Gugger, M., Friess, H., Kleeff, J., Kayed, H., et al. (2003). Distribution of CCK1 and CCK2 receptors in normal and diseased human pancreatic tissue. Gastroenterology 125 (1), 98–106. doi: 10.1016/s0016-5085(03)00697-8
Riordan, J. R., Rommens, J. M., Kerem, B., Alon, N., Rozmahel, R., Grzelczak, Z., et al. (1989). Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245 (4922), 1066–1073. doi: 10.1126/science.2475911
Rodriguez-Boulan, E., Nelson, W. J. (1989). Morphogenesis of the polarized epithelial cell phenotype. Science 245 (4919), 718–725. doi: 10.1126/science.2672330
Roux, B. (2017). Ion channels and ion selectivity. Essays Biochem. 61 (2), 201–209. doi: 10.1042/EBC20160074
Ruiz, C., Martins, J. R., Rudin, F., Schneider, S., Dietsche, T., Fischer, C. A., et al. (2012). Enhanced expression of ANO1 in head and neck squamous cell carcinoma causes cell migration and correlates with poor prognosis. PloS One 7 (8), e43265. doi: 10.1371/journal.pone.0043265
Rybarczyk, P., Gautier, M., Hague, F., Dhennin-Duthille, I., Chatelain, D., Kerr-Conte, J., et al. (2012). Transient receptor potential melastatin-related 7 channel is overexpressed in human pancreatic ductal adenocarcinomas and regulates human pancreatic cancer cell migration. Int. J. Cancer 131 (6), E851–E861. doi: 10.1002/ijc.27487
Rybarczyk, P., Vanlaeys, A., Brassart, B., Dhennin-Duthille, I., Chatelain, D., Sevestre, H., et al. (2017). The Transient Receptor Potential Melastatin 7 Channel Regulates Pancreatic Cancer Cell Invasion through the Hsp90alpha/uPA/MMP2 pathway. Neoplasia 19 (4), 288–300. doi: 10.1016/j.neo.2017.01.004
Sanchez, M. G., Sanchez, A. M., Collado, B., Malagarie-Cazenave, S., Olea, N., Carmena, M. J., et al. (2005). Expression of the transient receptor potential vanilloid 1 (TRPV1) in LNCaP and PC-3 prostate cancer cells and in human prostate tissue. Eur. J. Pharmacol. 515 (1-3), 20–27. doi: 10.1016/j.ejphar.2005.04.010
Sanguinetti, M. C., Jiang, C., Curran, M. E., Keating, M. T. (1995). A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 81 (2), 299–307. doi: 10.1016/0092-8674(95)90340-2
Sato, K., Ishizuka, J., Cooper, C. W., Chung, D. H., Tsuchiya, T., Uchida, T., et al. (1994). Inhibitory effect of calcium channel blockers on growth of pancreatic cancer cells. Pancreas 9 (2), 193–202. doi: 10.1097/00006676-199403000-00009
Sauter, D. R. P., Novak, I., Pedersen, S. F., Larsen, E. H., Hoffmann, E. K. (2015). ANO1 (TMEM16A) in pancreatic ductal adenocarcinoma (PDAC). Pflugers Arch. 467 (7), 1495–1508. doi: 10.1007/s00424-014-1598-8
Sauter, D. R., Sorensen, C. E., Rapedius, M., Bruggemann, A., Novak, I. (2016). pH-sensitive K(+) channel TREK-1 is a novel target in pancreatic cancer. Biochim. Biophys. Acta 1862 (10), 1994–2003. doi: 10.1016/j.bbadis.2016.07.009
Schneider, G., Siveke, J. T., Eckel, F., Schmid, R. M. (2005). Pancreatic cancer: basic and clinical aspects. Gastroenterology 128 (6), 1606–1625. doi: 10.1053/j.gastro.2005.04.001
Schroeder, B. C., Cheng, T., Jan, Y. N., Jan, L. Y. (2008). Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 134 (6), 1019–1029. doi: 10.1016/j.cell.2008.09.003
Serrano-Novillo, C., Capera, J., Colomer-Molera, M., Condom, E., Ferreres, J. C., Felipe, A. (2019). Implication of Voltage-Gated Potassium Channels in Neoplastic Cell Proliferation. Cancers (Basel) 11 (3), 287. doi: 10.3390/cancers11030287
Sette, A., Spadavecchia, J., Landoulsi, J., Casale, S., Haye, B., Crociani, O., et al. (2013). Development of novel anti-Kv 11.1 antibody-conjugated PEG-TiO2 nanoparticles for targeting pancreatic ductal adenocarcinoma cells. J. Nanopart Res. 15, 2111. doi: 10.1007/s11051-013-2111-6
Shapovalov, G., Ritaine, A., Skryma, R., Prevarskaya, N. (2016). Role of TRP ion channels in cancer and tumorigenesis. Semin. Immunopathol. 38 (3), 357–369. doi: 10.1007/s00281-015-0525-1
Sheldon, C. D., Hodson, M. E., Carpenter, L. M., Swerdlow, A. J. (1993). A cohort study of cystic fibrosis and malignancy. Br. J. Cancer 68 (5), 1025–1028. doi: 10.1038/bjc.1993.474
Shen, S., Gui, T., Ma, C. (2017). Identification of molecular biomarkers for pancreatic cancer with mRMR shortest path method. Oncotarget 8 (25), 41432–41439. doi: 10.18632/oncotarget.18186
Shuck, M. E., Piser, T. M., Bock, J. H., Slightom, J. L., Lee, K. S., Bienkowski, M. J. (1997). Cloning and characterization of two K+ inward rectifier (Kir) 1.1 potassium channel homologs from human kidney (Kir1.2 and Kir1.3). J. Biol. Chem. 272 (1), 586–593. doi: 10.1074/jbc.272.1.586
Singh, A. P., Chauhan, S. C., Andrianifahanana, M., Moniaux, N., Meza, J. L., Copin, M. C., et al. (2007). MUC4 expression is regulated by cystic fibrosis transmembrane conductance regulator in pancreatic adenocarcinoma cells via transcriptional and post-translational mechanisms. Oncogene 26 (1), 30–41. doi: 10.1038/sj.onc.1209764
Song, H., Dong, M., Zhou, J., Sheng, W., Li, X., Gao, W. (2018). Expression and prognostic significance of TRPV6 in the development and progression of pancreatic cancer. Oncol. Rep. 39 (3), 1432–1440. doi: 10.3892/or.2018.6216
Song, H. Y., Zhou, L., Hou, X. F., Lian, H. (2019). Anoctamin 5 regulates cell proliferation and migration in pancreatic cancer. Int. J. Clin. Exp. Pathol. 12 (12), 4263–4270.
Steward, M. C., Ishiguro, H., Case, R. M. (2005). Mechanisms of bicarbonate secretion in the pancreatic duct. Annu. Rev. Physiol. 67, 377–409. doi: 10.1146/annurev.physiol.67.031103.153247
Stock, C., Schwab, A. (2015). Ion channels and transporters in metastasis. Biochim. Biophys. Acta 1848 (10 Pt B), 2638–2646. doi: 10.1016/j.bbamem.2014.11.012
Stoklosa, P., Borgstrom, A., Kappel, S., Peinelt, C. (2020). TRP Channels in Digestive Tract Cancers. Int. J. Mol. Sci. 21 (5), 1877. doi: 10.3390/ijms21051877
Suzuki, K., Petersen, C. C., Petersen, O. H. (1985). Hormonal activation of single K+ channels via internal messenger in isolated pancreatic acinar cells. FEBS Lett. 192 (2), 307–312. doi: 10.1016/0014-5793(85)80131-9
Tawfik, D., Zaccagnino, A., Bernt, A., Szczepanowski, M., Klapper, W., Schwab, A., et al. (2020). The A818-6 system as an in-vitro model for studying the role of the transportome in pancreatic cancer. BMC Cancer 20 (1), 264. doi: 10.1186/s12885-020-06773-w
Teisseyre, A., Palko-Labuz, A., Sroda-Pomianek, K., Michalak, K. (2019). Voltage-Gated Potassium Channel Kv1.3 as a Target in Therapy of Cancer. Front. Oncol. 9, 933. doi: 10.3389/fonc.2019.00933
Thevenod, F. (2002). Ion channels in secretory granules of the pancreas and their role in exocytosis and release of secretory proteins. Am. J. Physiol. Cell Physiol. 283 (3), C651–C672. doi: 10.1152/ajpcell.00600.2001
Thompson-Vest, N., Shimizu, Y., Hunne, B., Furness, J. B. (2006). The distribution of intermediate-conductance, calcium-activated, potassium (IK) channels in epithelial cells. J. Anat. 208 (2), 219–229. doi: 10.1111/j.1469-7580.2006.00515.x
Tolon, R. M., Sanchez-Franco, F., Lopez Fernandez, J., Lorenzo, M. J., Vazquez, G. F., Cacicedo, L. (1996). Regulation of somatostatin gene expression by veratridine-induced depolarization in cultured fetal cerebrocortical cells. Brain Res. Mol. Brain Res. 35 (1-2), 103–110. doi: 10.1016/0169-328x(95)00188-x
Ulareanu, R., Chiritoiu, G., Cojocaru, F., Deftu, A., Ristoiu, V., Stanica, L., et al. (2017). N-glycosylation of the transient receptor potential melastatin 8 channel is altered in pancreatic cancer cells. Tumour Biol. 39 (8):1010428317720940. doi: 10.1177/1010428317720940
Venglovecz, V., Hegyi, P., Rakonczay, Z., Jr., Tiszlavicz, L., Nardi, A., Grunnet, M., et al. (2011). Pathophysiological relevance of apical large-conductance Ca(2)+-activated potassium channels in pancreatic duct epithelial cells. Gut 60 (3), 361–369. doi: 10.1136/gut.2010.214213
Venglovecz, V., Rakonczay, Z., Jr., Gray, M. A., Hegyi, P. (2015). Potassium channels in pancreatic duct epithelial cells: their role, function and pathophysiological relevance. Pflugers Arch. 467 (4), 625–640. doi: 10.1007/s00424-014-1585-0
Venglovecz, V., Pallagi, P., Kemeny, L. V., Balazs, A., Balla, Z., Becskehazi, E., et al. (2018). The Importance of Aquaporin 1 in Pancreatitis and Its Relation to the CFTR Cl(-) Channel. Front. Physiol. 9, 854. doi: 10.3389/fphys.2018.00854
Vercelli, C., Barbero, R., Cuniberti, B., Racca, S., Abbadessa, G., Piccione, F., et al. (2014). Transient receptor potential vanilloid 1 expression and functionality in mcf-7 cells: a preliminary investigation. J. Breast Cancer 17 (4), 332–338. doi: 10.4048/jbc.2014.17.4.332
Voloshyna, I., Besana, A., Castillo, M., Matos, T., Weinstein, I. B., Mansukhani, M., et al. (2008). TREK-1 is a novel molecular target in prostate cancer. Cancer Res. 68 (4), 1197–1203. doi: 10.1158/0008-5472.CAN-07-5163
Waldmann, R., Champigny, G., Bassilana, F., Voilley, N., Lazdunski, M. (1995). Molecular cloning and functional expression of a novel amiloride-sensitive Na+ channel. J. Biol. Chem. 270 (46), 27411–27414. doi: 10.1074/jbc.270.46.27411
Wang, B. J., Cui, Z. J. (2007). How does cholecystokinin stimulate exocrine pancreatic secretion? From birds, rodents, to humans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292 (2), R666–R678. doi: 10.1152/ajpregu.00131.2006
Wang, J., Novak, I. (2013). Ion transport in human pancreatic duct epithelium, Capan-1 cells, is regulated by secretin, VIP, acetylcholine, and purinergic receptors. Pancreas 42 (3), 452–460. doi: 10.1097/MPA.0b013e318264c302
Wang, J., Haanes, K. A., Novak, I. (2013). Purinergic regulation of CFTR and Ca(2+)-activated Cl(-) channels and K(+) channels in human pancreatic duct epithelium. Am. J. Physiol. Cell Physiol. 304 (7), C673–C684. doi: 10.1152/ajpcell.00196.2012
Wang, F., Jin, R., Zou, B. B., Li, L., Cheng, F. W., Luo, X., et al. (2016). Activation of Toll-like receptor 7 regulates the expression of IFN-lambda1, p53, PTEN, VEGF, TIMP-1 and MMP-9 in pancreatic cancer cells. Mol. Med. Rep. 13 (2), 1807–1812. doi: 10.3892/mmr.2015.4730
Wang, J., Shen, J., Zhao, K., Hu, J., Dong, J., Sun, J. (2019). STIM1 overexpression in hypoxia microenvironment contributes to pancreatic carcinoma progression. Cancer Biol. Med. 16 (1), 100–108. doi: 10.20892/j.issn.2095-3941.2018.0304
Warth, R., Barhanin, J. (2002). The multifaceted phenotype of the knockout mouse for the KCNE1 potassium channel gene. Am. J. Physiol. Regul. Integr. Comp. Physiol. 282 (3), R639–R648. doi: 10.1152/ajpregu.00649.2001
Warth, R., Garcia Alzamora, M., Kim, J. K., Zdebik, A., Nitschke, R., Bleich, M., et al. (2002). The role of KCNQ1/KCNE1 K(+) channels in intestine and pancreas: lessons from the KCNE1 knockout mouse. Pflugers Arch. 443 (5-6), 822–828. doi: 10.1007/s00424-001-0751-3
Wen, L., Voronina, S., Javed, M. A., Awais, M., Szatmary, P., Latawiec, D., et al. (2015). Inhibitors of ORAI1 Prevent Cytosolic Calcium-Associated Injury of Human Pancreatic Acinar Cells and Acute Pancreatitis in 3 Mouse Models. Gastroenterology 149481-492 (2), e487. doi: 10.1053/j.gastro.2015.04.015
Williams, S., Bateman, A., O’Kelly, I. (2013). Altered expression of two-pore domain potassium (K2P) channels in cancer. PloS One 8 (10), e74589. doi: 10.1371/journal.pone.0074589
Wilschanski, M., Novak, I. (2013). The cystic fibrosis of exocrine pancreas. Cold Spring Harb. Perspect. Med. 3 (5):a009746. doi: 10.1101/cshperspect.a009746
Winpenny, J. P., Verdon, B., McAlroy, H. L., Colledge, W. H., Ratcliff, R., Evans, M. J., et al. (1995). Calcium-activated chloride conductance is not increased in pancreatic duct cells of CF mice. Pflugers Arch. 430 (1), 26–33. doi: 10.1007/BF00373836
Worley, P. F., Zeng, W., Huang, G. N., Yuan, J. P., Kim, J. Y., Lee, M. G., et al. (2007). TRPC channels as STIM1-regulated store-operated channels. Cell Calcium 42 (2), 205–211. doi: 10.1016/j.ceca.2007.03.004
Wright, A. M., Gong, X., Verdon, B., Linsdell, P., Mehta, A., Riordan, J. R., et al. (2004). Novel regulation of cystic fibrosis transmembrane conductance regulator (CFTR) channel gating by external chloride. J. Biol. Chem. 279 (40), 41658–41663. doi: 10.1074/jbc.M405517200
Yamaguchi, M., Steward, M. C., Smallbone, K., Sohma, Y., Yamamoto, A., Ko, S. B., et al. (2017). Bicarbonate-rich fluid secretion predicted by a computational model of guinea-pig pancreatic duct epithelium. J. Physiol. 595 (6), 1947–1972. doi: 10.1113/JP273306
Yang, B., Song, Y., Zhao, D., Verkman, A. S. (2005). Phenotype analysis of aquaporin-8 null mice. Am. J. Physiol. Cell Physiol. 288 (5), C1161–C1170. doi: 10.1152/ajpcell.00564.2004
Yang, Y. D., Cho, H., Koo, J. Y., Tak, M. H., Cho, Y., Shim, W. S., et al. (2008). TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 455 (7217), 1210–1215. doi: 10.1038/nature07313
Yee, N. S., Zhou, W., Lee, M. (2010). Transient receptor potential channel TRPM8 is over-expressed and required for cellular proliferation in pancreatic adenocarcinoma. Cancer Lett. 297 (1), 49–55. doi: 10.1016/j.canlet.2010.04.023
Yee, N. S., Zhou, W., Liang, I. C. (2011). Transient receptor potential ion channel Trpm7 regulates exocrine pancreatic epithelial proliferation by Mg2+-sensitive Socs3a signaling in development and cancer. Dis. Model Mech. 4 (2), 240–254. doi: 10.1242/dmm.004564
Yee, N. S., Chan, A. S., Yee, J. D., Yee, R. K. (2012a). TRPM7 and TRPM8 Ion Channels in Pancreatic Adenocarcinoma: Potential Roles as Cancer Biomarkers and Targets. Sci. (Cairo) 2012, 415158. doi: 10.6064/2012/415158
Yee, N. S., Zhou, W., Lee, M., Yee, R. K. (2012b). Targeted silencing of TRPM7 ion channel induces replicative senescence and produces enhanced cytotoxicity with gemcitabine in pancreatic adenocarcinoma. Cancer Lett. 318 (1), 99–105. doi: 10.1016/j.canlet.2011.12.007
Yee, N. S., Li, Q., Kazi, A. A., Yang, Z., Berg, A., Yee, R. K. (2014). Aberrantly Over-Expressed TRPM8 Channels in Pancreatic Adenocarcinoma: Correlation with Tumor Size/Stage and Requirement for Cancer Cells Invasion. Cells 3 (2), 500–516. doi: 10.3390/cells3020500
Yee, N. S., Kazi, A. A., Li, Q., Yang, Z., Berg, A., Yee, R. K. (2015). Aberrant over-expression of TRPM7 ion channels in pancreatic cancer: required for cancer cell invasion and implicated in tumor growth and metastasis. Biol. Open 4 (4), 507–514. doi: 10.1242/bio.20137088
Yokoyama, T., Takemoto, M., Hirakawa, M., Saino, T. (2019). Different immunohistochemical localization for TMEM16A and CFTR in acinar and ductal cells of rat major salivary glands and exocrine pancreas. Acta Histochem. 121 (1), 50–55. doi: 10.1016/j.acthis.2018.10.013
Yurtsever, Z., Sala-Rabanal, M., Randolph, D. T., Scheaffer, S. M., Roswit, W. T., Alevy, Y. G., et al. (2012). Self-cleavage of human CLCA1 protein by a novel internal metalloprotease domain controls calcium-activated chloride channel activation. J. Biol. Chem. 287 (50), 42138–42149. doi: 10.1074/jbc.M112.410282
Zaccagnino, A., Pilarsky, C., Tawfik, D., Sebens, S., Trauzold, A., Novak, I., et al. (2016). In silico analysis of the transportome in human pancreatic ductal adenocarcinoma. Eur. Biophys. J. 45 (7), 749–763. doi: 10.1007/s00249-016-1171-9
Zaccagnino, A., Manago, A., Leanza, L., Gontarewitz, A., Linder, B., Azzolini, M., et al. (2017). Tumor-reducing effect of the clinically used drug clofazimine in a SCID mouse model of pancreatic ductal adenocarcinoma. Oncotarget 8 (24), 38276–38293. doi: 10.18632/oncotarget.11299
Zdebik, A., Hug, M. J., Greger, R. (1997). Chloride channels in the luminal membrane of rat pancreatic acini. Pflugers Arch. 434 (2), 188–194. doi: 10.1007/s004240050382
Zeiher, B. G., Eichwald, E., Zabner, J., Smith, J. J., Puga, A. P., McCray, P. B., Jr., et al. (1995). A mouse model for the delta F508 allele of cystic fibrosis. J. Clin. Invest. 96 (4), 2051–2064. doi: 10.1172/JCI118253
Zeng, W., Lee, M. G., Yan, M., Diaz, J., Benjamin, I., Marino, C. R., et al. (1997). Immuno and functional characterization of CFTR in submandibular and pancreatic acinar and duct cells. Am. J. Physiol. 273 (2 Pt 1), C442–C455. doi: 10.1152/ajpcell.1997.273.2.C442
Zhou, B., Irwanto, A., Guo, Y. M., Bei, J. X., Wu, Q., Chen, G., et al. (2012). Exome sequencing and digital PCR analyses reveal novel mutated genes related to the metastasis of pancreatic ductal adenocarcinoma. Cancer Biol. Ther. 13 (10), 871–879. doi: 10.4161/cbt.20839
Zhou, J., Zhang, L., Zheng, H., Ge, W., Huang, Y., Yan, Y., et al. (2020). Identification of chemoresistance-related mRNAs based on gemcitabine-resistant pancreatic cancer cell lines. Cancer Med. 9 (3), 1115–1130. doi: 10.1002/cam4.2764
Zhu, S., Zhou, H. Y., Deng, S. C., Deng, S. J., He, C., Li, X., et al. (2017). ASIC1 and ASIC3 contribute to acidity-induced EMT of pancreatic cancer through activating Ca(2+)/RhoA pathway. Cell Death Dis. 8 (5), e2806. doi: 10.1038/cddis.2017.189
Zou, Q., Yang, Z., Li, D., Liu, Z., Yuan, Y. (2016). Association of chloride intracellular channel 4 and Indian hedgehog proteins with survival of patients with pancreatic ductal adenocarcinoma. Int. J. Exp. Pathol. 97 (6), 422–429. doi: 10.1111/iep.12213
Keywords: ion channels, exocrine pancreas, pancreatic ductal adenocarcinoma, signaling pathways, biomarkers
Citation: Schnipper J, Dhennin-Duthille I, Ahidouch A and Ouadid-Ahidouch H (2020) Ion Channel Signature in Healthy Pancreas and Pancreatic Ductal Adenocarcinoma. Front. Pharmacol. 11:568993. doi: 10.3389/fphar.2020.568993
Received: 02 June 2020; Accepted: 16 September 2020;
Published: 16 October 2020.
Edited by:
Sébastien Roger, Université de Tours, FranceReviewed by:
David Crottès, University of California, San Francisco, United StatesNelson Shu-Sang Yee, Penn State Milton S. Hershey Medical Center, United States
Copyright © 2020 Schnipper, Dhennin-Duthille, Ahidouch and Ouadid-Ahidouch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Halima Ouadid-Ahidouch, aGFsaW1hLmFoaWRvdWNoLW91YWRpZEB1LXBpY2FyZGllLmZy