
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 02 September 2020
Sec. Translational Pharmacology
Volume 11 - 2020 | https://doi.org/10.3389/fphar.2020.01328
This article is part of the Research Topic Inspired by Nature: Towards Novel Anti-infective Agents View all 5 articles
 Wilian C. Rosa1
Wilian C. Rosa1 Inaiá O. Rocha1Melissa B. Rodrigues1
Inaiá O. Rocha1Melissa B. Rodrigues1 Helena S. Coelho2,3
Helena S. Coelho2,3 Laura B. Denardi2Pauline C. Ledur2Nilo Zanatta1Thiago V. Acunha4
Laura B. Denardi2Pauline C. Ledur2Nilo Zanatta1Thiago V. Acunha4 Bernardo A. Iglesias4*
Bernardo A. Iglesias4* Helio G. Bonacorso1*
Helio G. Bonacorso1*The synthesis, antimicrobial activity evaluations, biomolecule-binding properties (DNA), and absorption and emission properties of a new series of (Z)-1,1,1-trichloro-4-alkyl(aryl)amino-4-arylbut-3-en-2-ones (4, 5) and 2,2-difluoro-3-alkyl(aryl)amino-4-aryl-6-(trichloromethyl)-2H-1,3,2-oxazaborinin-3-ium-2-uides (6, 7) in which 3(4)-alkyl(aryl) = H, Me, iso-propyl, n-butyl, C6H5, 4-CH3C6H4, 4-CH3OC6H4, 4-NO2C6H4, 4-FC6H4, 4-BrC6H4, 2-naphthyl, is reported. A series of β-enaminoketones (4, 5) is synthesized from the O,N-exchange reaction of some amines (3) with (Z)-1,1,1-trichloro-4-methoxy-4-aryl-but-3-en-2-ones (1, 2) at 61–90% yields. Subsequently, reactions of the resulting β-enaminoketones with an appropriate source of boron (BF3.OEt2) gave the corresponding oxazaborinine derivatives (6, 7) at 50–91% yields. UV-Vis and emission properties of biomolecule-binding properties for the DNA of these new BF2-β-enamino containing CCl3 units were also evaluated. Some compounds from the present series also exhibited potent antimicrobial effects on various pathogenic microorganisms at concentrations below those that showed cytotoxic effects. Compounds 4d, 4e, 6e, and 6f showed the best results and are very significant against P. zopfii, which causes diseases in humans and animals.
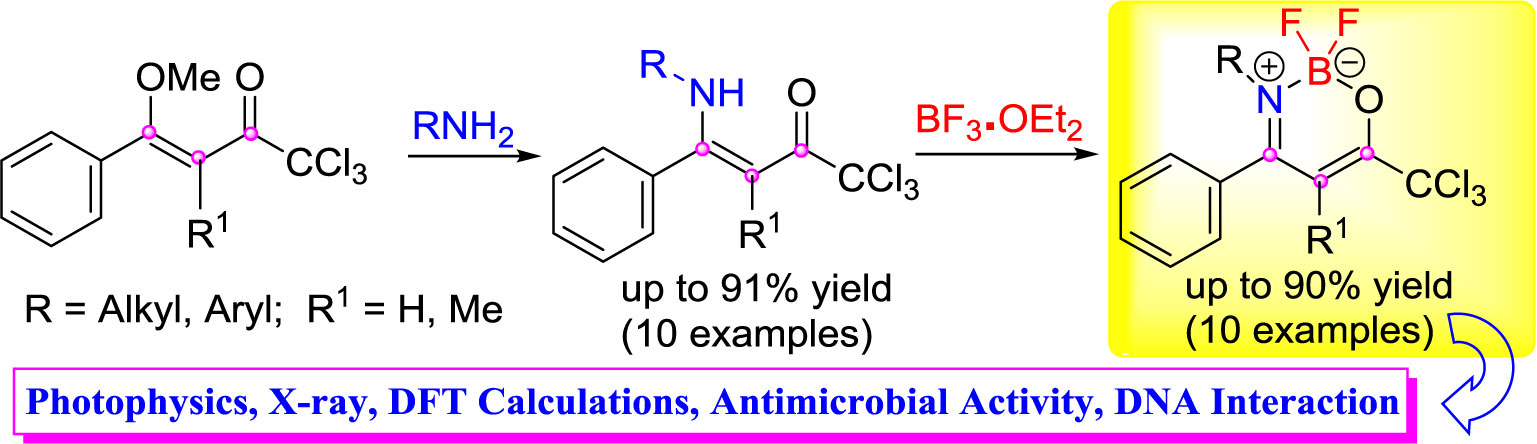
Graphical Abstract
Unsaturated heterocyclic compounds containing a boron atom have increased interest in recent years as a result of their potential in basic research and applications (Loudet and Burgess, 2007). Stands out BODIPY core (4,4-difluoro-4-bora-3a-azonia-4a-aza-s-indacenes) due to your small absorption, sharp emission bands with strong peak intensities, high values fluorescent quantum yields (Buyukcakir et al., 2009; Nagai and Chujo, 2010; Cakmak et al., 2011; He et al., 2011; Niu et al., 2011; Sozmen et al., 2014) and antimicrobial activity (Tekdaş et al., 2016) (Scheme 1).
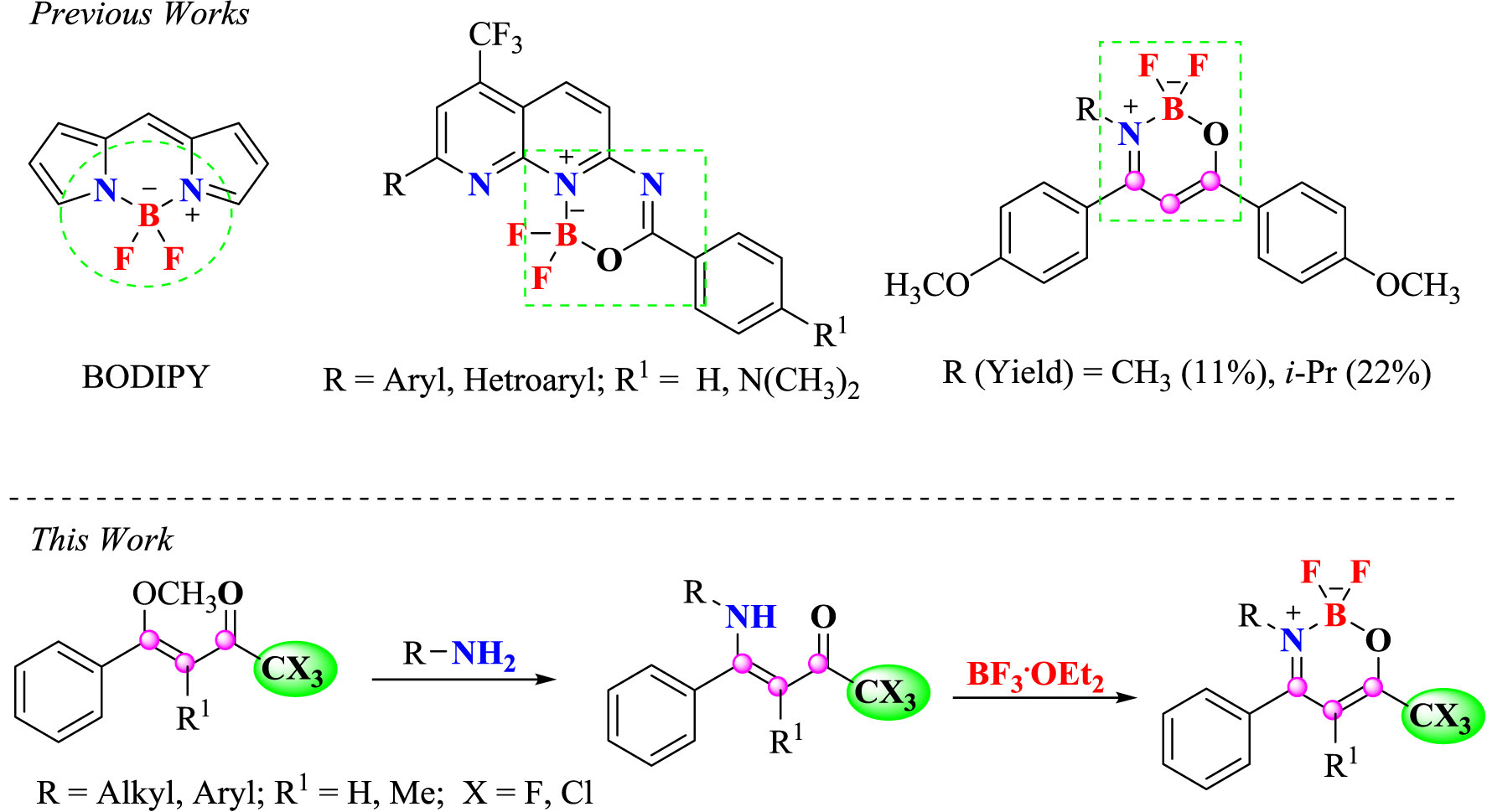
Scheme 1 Structure of BODIPY and nitrogenated heterocyclic containing a BF2 core, examples of β-iminoenolate boron complexes and the synthetic route for β-enamino trihalomethyl ketones and the corresponding oxazaborinine derivatives.
Indeed, these properties promote the use of these structures in several applications such as, light emitting diodes (Wang et al., 2013) and solar cells (Kolemen et al., 2010). Nevertheless, besides structure of the BODIPY have high planarity, also shows very small Stokes’ shifts (Zhang et al., 2008; Kubota et al., 2010). So, synthesis novel boron analogous compounds with designed structural variations are relevant for interesting fluorescence materials (Poon et al., 2010).
Recently, our research group developed several difluoro-organoboron analogues. Nitrogenated heterocyclic scaffolds such as pyridines, (Bonacorso et al., 2018a). pyrimidines (Bonacorso et al., 2019), naphthyridines (Bonacorso et al., 2016), quinoxalines (Calheiro, 2018) have been studied for the synthesis of analogous novel structures (Scheme 1).
On the other hand, β-enaminoketones are the isosteric analogues of 1,3-enolic ketones and have been used in recent years as N,O double-dentate ligands to form 1,3,2-oxazaborines. These difluoroboron complexes of β-enaminones, which belong to the family of β-iminoenolate boron complexes (Kubota et al., 2011), have attracted attention as a promising class of fluorophores (Josefik et al., 2012).
Shankarling et al. described the synthesis and spectral and electrochemical characterization of some boron difluoride complexes of benzoindoline-based β-enaminones (Kumbhar et al., 2015). In 2008, Xia et al. reported excellent solution-state fluorescence for some heterocyclic β-iminoenolates (Xia et al., 2008). Later in 2013, Yoshii et al. described the synthesis of boron-ketoiminate derivatives that showed aggregation-induced emission properties and were prepared by the reactions of β-enaminoketones derivatives with boron trifluoride–diethyl etherate (BF3.Et2O) (Scheme 1) (Yoshii et al., 2013).
On the other hand, β-enaminoketones are interesting and easy obtainable synthetic intermediary. Enaminoketones have received attention due to their ambident nucleophilicity characteristics of amines, else with the ambident electrophilicity of enones (Bonacorso et al., 2002a; Bonacorso et al., 2010; Bonacorso et al., 2013).
β-enaminoketones and β-enaminoketo-esters are synthesized through the condensation reactions between carbonyl compounds and primary or secondary amines acid-catalysed (Ferraz and Pereira, 2004; Ferraz and Gonçalo, 2007). Due to its acidic nature, citrus juice is considered a good catalyst. Recently, Marvi et al. reported an efficient and green method using citrus juice as natural catalyst (Marvi and Fekri, 2018).
β-enaminones they have been also used to prepare different important antibacterial, (Mahmud et al., 2011). anti-inflammatories (Michael et al., 2001), and antitumor agents (Boger et al., 1989). Moreover, β-enaminones are widely used in the preparation of γ-amino-alcohols, which are structural units present in various compounds with pharmacological properties and natural products (Harris and Braga, 2004). There are many protocols well-established for the preparation of the γ-amino-alcohols. Nevertheless, reduction of 1,3-difunctionalized unsaturated structures [β-enaminoketones (Barluenga et al., 1992; Bartoli et al., 1994; Bartoli et al., 2002), or β-aminoketones (Katritzky and Harris, 1990; Pilli et al., 1990)] in which they present nitrogen and oxygen are more frequently.
Moreover, trihalomethyl groups in heterocyclic structures may drastically amend their chemicals, physical and pharmacological characteristics. The construction of trihalomethylated structures is done through already halogenated building blocks (Martins et al., 1999; Bonacorso et al., 2000; Martins et al., 2004). Trihalomethylated β-enaminoketones have been synthetized from the reaction of β-alkoxyvinyl trihalomethyl ketones (Hojo et al., 1986; Gerus et al., 1991) or acetylenes (Linderman and Kirollos, 1990) with amines.
On this line, fluorinated natural compounds are present in several drug classes. The main progress refers to research in the area of steroids, alkaloids, nucleosides, macrolides, prostaglandins, and amino acids (Kumar et al., 2008). Quinine isolated from chinchona bark led successive and innovative class of antimalarials (Kumar et al., 2003) such as chloroquine, mefloquine, and primaquine. Another example is the fluorocorticoid and fluorouracil derivatives, these drugs are still clinically used (Bégué and Bonnet-Delpon, 2006).
Following our studies in this area of interest, we decided to investigate the synthesis of novel boron complexes from β-enaminoketones trihalomethyl that contain the CF3 or CCl3 group bonded at the 6-position, N-alkyl(aryl) substituents with electron-donating or electron-withdrawing effect at the 3-position and a phenyl group at the 4-position. Furthermore, in view of the biological potential of molecules containing trihalomethyl groups, the aim of this paper was also to report the antimicrobial screening and cytotoxicity analysis of the compounds synthetized here (Scheme 1). Moreover, UV-Vis and emission spectroscopy was employed and DNA-binding properties were investigated of these new BF2-β-enamino derivatives in the present work.
Unless otherwise indicated, all common reagents and solvents were used as obtained from commercial suppliers and without further purification. 1H and 13C NMR spectra were acquired in Bruker Avance III 400 MHz or Bruker Avance III 600 MHz spectrometers for one-dimensional experiments, with 5 mm sample tubes, 298 K, and digital resolution of 0.01 ppm, in CDCl3 as solvent, and using TMS as the internal reference. The 19F and 11B-NMR spectra were acquired in a Bruker Avance III (19F at 564 MHz and 11B at 192 MHz) equipped with a 5-mm PABBO probe, 5-mm sample tubes at 298 K, and digital resolution of 0.01 ppm, in CDCl3, and using CFCl3 and BF3·OEt2, respectively, as the external reference. All spectra can be found at the Supplementary Information File—Figures S1–S30.
All results were reported with the chemical shift (δ), multiplicity, integration, and coupling constant (Hz). The following abbreviations were used to explain multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, and dd = double doublet. All NMR chemical shifts were reported in parts per million and relative to the internal reference.
All melting points were determined using coverslips in a Microquímica MQAPF-302 apparatus and are uncorrected.
The CHN elemental analyses were performed in a Perkin–Elmer 2400 CHN elemental analyzer (University of São Paulo, Brazil).
High resolution mass spectra (HRMS) were obtained for all compounds in an LTQ Orbitrap Discovery mass spectrometer (Thermo Fisher Scientific). This hybrid system combines the LTQ XL linear ion trap mass spectrometer and an Orbitrap mass analyzer. Experiments were performed via direct infusion of the sample (flow: 10 µl min-1) in positive-ion mode using electrospray ionization. Elemental composition calculations for comparison were executed using the specific tool included in the Qual Browser module of Xcalibur (Thermo Fisher Scientific, release 2.0.7) software. The spectra can be found in the Supplementary Information File—Figures S30–S33.
Single crystals of compounds 4g and 6e were obtained by slow evaporation of EtOH at 25°C. Compounds 4g and 6e was measured using a Bruker D8 QUEST diffractometer using Cu Ka radiation (l = 1.54080 Å) with a KAPPA four-circle goniometer equipped with a PHOTON II CPAD area detector. Absorption corrections were performed using the multi-scan method. Anisotropic displacement parameters for non-hydrogen atoms were applied. Most hydrogen atom positions were calculated geometrically and refined using the riding model, although some hydrogen atoms were refined freely. The structure was solved and refined using the WinGX software package (Farrugia, 2012).
The structures were refined based on the full-matrix least-squares method using the SHELXL program (Sheldrick, 2008). The ORTEP projections of the molecular structures were generated using the ORTEP-3 program. Crystallographic information files (CIFs) for the novel structures were deposited at the Cambridge Crystallographic Data Centre (CCDC) under identification number 1810806 (4g) and 1855153 (6e). Crystallographic data can be observed in the ESI (Supplementary Information File).
Ultraviolet/visible absorption spectra were recorded using Shimadzu UV2600 spectrophotometer and dichloromethane (DCM), methanol (MeOH), and dimethyl sulfoxide (DMSO) as solvents and concentration solution at 10–5 M range. Steady-state emission fluorescence analysis of samples in the same solvents used in absorption analysis were measured with a Varian Cary 50 fluorescence spectrophotometer (emission; 300–700 nm range; slit 2.0 mm). Due to the absence of emission intensity of all derivatives in all solvents, it was not possible to determine fluorescence quantum yield values.
For DNA interactions, boron complexes titrations with calf-thymus DNA (CT-DNA) were performed by UV-vis analysis at room temperature in DMSO (2%)-Tris-HCl buffer mixture (pH 7.2) using DMSO stock solution of derivatives (10-4 M range). The DNA pair base concentrations of low molecular weight DNA from calf thymus (CT-DNA) were determined by UV-vis absorption spectroscopy using ϵ = 6.600 M−1cm−1 (per base pair) at λmax = 260 nm. Derivatives solutions in DMSO with Tris-HCl were titrated with increasing concentrations of CT-DNA (ranging from 0 to 100 μM). The absorption spectra of the compounds were acquired in the wavelength range of 250–700 nm. The binding constants (Kb) of derivatives were calculated according to the decay of the absorption bands of compounds using Equation 1 through a plot of [DNA]/(ϵa − ϵf) versus [DNA],
where [DNA] is the concentration of DNA in the base pairs, ϵa is the extinction coefficient (Aobs/[compound]), and ϵb and ϵf are the extinction coefficients of free and fully bound forms, respectively. In the plots of [DNA]/(ϵa − ϵf) versus [DNA], Kb is given by the ratio of the slope for the interception.
Competitive EB-binding studies were performed using steady-state emission fluorescence assay with CT-DNA. Compounds were dissolved in DMSO and through the gradual addition of the stock solution of the boron complexes to the quartz cuvette (1.0 cm path length) containing ethidium bromide dye (EB, 2.0 × 10-7 M) and DNA (1.0 × 10-5 M) in a Tris-HCl pH 7.4 buffer solution. Derivative concentration ranged from 0 to 100 µM. All samples were excited at λexc = 510 nm and emission fluorescence spectra were recorded at the range of 550–800 nm, 5 min after each addition of the complex solution in order to allow incubation to occur (incubation time). The fluorescence quenching Stern-Volmer constants (KSV) of compounds were calculated according to the decay of the emission bands of EB-DNA using Equation 2 through a plot of F0/F versus [DNA],
where F0 and F are the steady-state fluorescence intensities of EB-DNA adduct in the absence and presence of each derivative, respectively. Ksv and kq are the Stern-Volmer quenching constant and the bimolecular quenching rate constant, respectively. The [Q] and τo are the derivative concentration and fluorescence lifetime of EB-DNA without the quencher (23.0 × 10-9 s) (Nafisi et al., 2007).
The standard Gibbs free energy (ΔG°) of derivative-DNA complex was calculated from the values of binding constant (Kb) using Equation 3:
To magnetically stirred solutions of (Z)-1,1,1-trichloro-4-methoxy-4-phenylbut-3-en-2-one (1) (5 mmol) in ethanol (20 ml), the respective amines (3a-i, 5e) (1.5 eq, 7.5 mmol) were added and the resulting mixtures were refluxed for 24 h. After that, the solutions were cooled at –10°C, resulting in yellow or white solids. The solids were filtered under atmospheric pressure, washed with cold ethanol, and dried under reduced pressure.
Physical aspect: white solid. Yield: 61%. Melting point: 84–87°C.
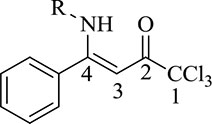
1H NMR (400 MHz, CDCl3) δ: 10.65 (s, 1H, NH), 7.50–7.45 (m, 3H, Ph), 7.38–7.36 (m, 2H, Ph), 5.68 (s, 1H, H–3), 3.77–3.65 (m, 1H, CH), 1.23 (s, 3H, CH3), 1.21 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ: 180.5 (C-2), 169.1 (C-4), 134.9 (Ph), 130.0 (Ph), 128.7 (Ph), 127.1 (Ph), 97.2 (CCl3), 87.3 (C-3), 46.8 (CH), 23.9 (CH3). MS (EI, 70 eV), m/z (%) = 305 (M+, 5), 270 (1), 242 (9), 188 (100), 146 (19), 103 (14), 77 (7). Anal. Cal. for C13H14Cl3NO (306.62): C, 50.92; H, 4.60; N, 4.57. Found: C, 50.89; H, 4.40; N, 4.59.
Physical aspect: brown oil. Yield: 90%.
1H NMR (400 MHz, CDCl3) δ: 10.74 (s, 1H, NH), 7.49–7.45 (m, 3H, Ph), 7.39–7.36 (m, 2H, Ph), 5.75 (s, 1H, H–3), 3.26 (q, J = 13.0 Hz, 2H, CH2–N), 1.57 (quint, J = 14.7 Hz, 2H, CH2), 1.37 (sext, J = 14.5 Hz, 2H, CH2), 0.88 (t, J = 7.3 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ: 180.6 (C-2), 170.2 (C-4), 134.6 (Ph), 130.1 (Ph), 128.7 (Ph), 127.4 (Ph), 97.2 (CCl3), 87.4 (C-3), 45.0 (CH2), 32.3 (CH2), 19.8 (CH2), 13.5 (CH3). MS (EI, 70 eV), m/z (%) = 319 (M+, 4), 256 (7), 202 (100), 184 (6), 103 (9). Anal. Cal. for C14H16Cl3NO (320.64): C, 52.44; H, 5.03; N, 4.37. Found: C, 52.46; H, 5.03; N, 4.44.
Physical aspect: yellow solid. Yield: 76%. Melting point: 123–124°C.
1H NMR (400 MHz, CDCl3) δ: 12.02 (s, 1H, NH), 7.41–7.39 (m, Ph), 7.36–7.33 (m, Ph), 7.16 (tt, J = 7.6, J = 2 Hz, Ph), 7.07 (tt, J = 7.3, J = 1.3 Hz, Ph), 6.81 (dd, J = 8.5, J = 1.1 Hz, Ph), 6.04 (s, 1H, H–3). 13C NMR (101 MHz, CDCl3) δ: 181.7 (C-2), 165.7 (C-4), 138.3 (Ph), 134.6 (Ph), 130.4 (Ph), 128.9 (Ph), 128.7 (Ph), 128.4 (Ph), 125.5 (Ph), 123.7 (Ph), 96.9 (CCl3), 90.9 (C-3). MS (EI, 70 eV), m/z (%) = 339 (M+, 4), 269 (12), 222 (100), 180 (5), 77 (16). HRMS (ESI-TOF): C16H12Cl3NO (M + H), requires 340.0057. Found 340.0069.
Physical aspect: yellow solid. Yield: 85%. Melting point: 112–113°C.
1H NMR (400 MHz, CDCl3) δ: 12.04 (s, 1H, NH), 7.42–7.32 (m, 4H, Ph), 6.95 (d, J = 8.2 Hz, Ph), 6.70 (d, J = 8.3 Hz, Ph), 6.02 (s, 1H, H–3), 2.25 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ: 181.5 (C-2), 165.8 (C-4), 135.6 (Ph), 135.4 (Ph), 134.7 (Ph), 130.3 (Ph), 129.5 (Ph), 128.6 (Ph), 128.4 (Ph), 123.6 (Ph), 97.0 (CCl3), 90.4 (C-3), 20.8 (CH3). MS (EI, 70 eV), m/z (%) = 353 (M+, 4), 319 (1), 236 (100), 91 (6). HRMS (ESI-TOF): C17H14Cl3NO (M + H), requires 354.0214. Found 354.0226.
Physical aspect: yellow solid. Yield: 88%. Melting point: 144–145°C.
1H NMR (400 MHz, CDCl3) δ: 12.06 (s, 1H, NH), 7.41–7.32 (m, 5H, Ph), 6.76 (d, J = 9.0 Hz, 1H, Ph), 6.69 (d, J = 9.1 Hz, 1H, Ph), 6.68 (s, 1H, H–3), 3.73 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ: 181.5 (C-2), 166.0 (C-4), 157.5 (Ph), 134.7 (Ph), 131.2 (Ph), 130.3 (Ph), 128.6 (Ph), 128.4 (Ph), 125.3 (Ph), 114.2 (Ph), 97.0 (CCl3), 90.0 (C-3), 55.4 (CH3). MS (EI, 70 eV), m/z (%) = 369 (M+, 9), 334 (2), 252 (100), 209 (26). HRMS (ESI-TOF): C17H14Cl3NO2 (M + H), requires 370.0163. Found 370.0173.
Physical aspect: yellow solid. Yield: 65%. Melting point: 107–109°C.
1H NMR (400 MHz, CDCl3) δ: 11.83 (s, 1H, NH), 8.03 (d, J = 9.1 Hz, 1H, Ph), 7.53–7.49 (m, 1H, Ph), 7.44–7.37 (m, 4H, Ph), 6.85 (d, J = 9.0 Hz, 1H, Ph), 6.16 (s, 1H, H–3). 13C NMR (101 MHz, CDCl3) δ: 182.6 (C-2), 163.8 (C-4), 144.4 (Ph), 144.0 (Ph), 133.9 (Ph), 131.2 (Ph), 129.3 (Ph), 128.2 (Ph), 124.8 (Ph), 122.4 (Ph), 96.5 (CCl3), 94.1 (C-3). Anal. Cal. for C16H11Cl3N2O3 (385.63): C, 49.83; H, 2.88; N, 7.26. Found: C, 49.78; H, 2.85; N, 7.26.
Physical aspect: solid beige. Yield: 87%. Melting point: 115–116°C.
1H NMR (400 MHz, CDCl3) δ: 11.95 (s, 1H, NH), 7.42–7.30 (m, 5H, Ph), 6.88–6.77 (m, 4 H, Ph), 6.04 (s, 1 H, H–3). 13C NMR (101 MHz, CDCl3) δ: 181.9 (C-2), 165.8 (C-4), 160.3 (d, J = 246.4 Hz, Ph), 134.4 (d, J = 3.2 Hz, Ph), 134.4 (Ph), 130.5 (Ph), 128.8 (Ph), 128.4 (Ph), 125.5 (d, J = 8.2 Hz, Ph), 115.9 (d, J = 22.9 Hz, Ph), 96.9 (CCl3), 90.8 (C-3). MS (EI, 70 eV), m/z (%) = 358 (M+, 3), 294 (4), 269 (13), 240 (100), 198 (4), 95 (8). HRMS (ESI-TOF): C16H11Cl3FNO (M + H), requires 357.9963. Found 357.9976.
Physical aspect: yellow solid. Yield: 80%. Melting point: 142–144°C.
1H NMR (400 MHz, CDCl3) δ: 11.92 (s, 1H, NH), 7.46–7.42 (m, 1H, Ph), 7.39–7.32 (m, 4H, Ph), 7.28 (d, J = 8.8 Hz, 2H, Ph), 6.67 (d, J = 8.7 Hz, 2H, Ph), 6.06 (s, 1H, H–3). 13C NMR (101 MHz, CDCl3) δ: 181.9 (C-2), 165.2 (C-4), 137.4 (Ph), 134.1 (Ph), 132.0 (Ph), 130.7 (Ph), 128.9 (Ph), 128.3 (Ph), 125.0 (Ph), 118.7 (Ph), 96.7 (CCl3), 91.5 (C-3).
MS (EI, 70 eV), m/z (%) = 418 (M+, 10), 301 (M+, 100), 301 (M+2, 20), 302 (M+3, 97), 269 (27), 271 (7), 234 (14), 221 (51), 193 (41), 192 (7), 165 (14), 102 (15), 76 (16). HRMS (ESI-TOF): C16H11BrCl3NO (M + H), C, 45.81; H, 2.64; N, 3.34. Found: C, 45.53; H, 2.54; N, 3.27.
Physical aspect: yellow solid. Yield: 85%. Melting point: 149–151°C.
1H NMR (400 MHz, CDCl3) δ: 12.39, 8.28 (d, J = 8.6 Hz, 1H), 7.85 (d, J = 8.1 Hz, 1H), 7.62 (ddd, J = 6.9, 4.7, 1.4 Hz, 2H), 7.55 (ddd, J = 8.0, 6.9, 1.2 Hz, 1H), 7.34–7.20 (m, 5 H, Ph), 7.12 (t, J = 7.9 Hz, 1H), 6.71 (d, J = 7.5 Hz, 1H), 6.19 (s, 1H, H–3). 13C NMR (101 MHz, CDCl3) δ: 182.1 (C-2), 167.1 (C-4), 134.7 (Ph), 134.3 (Ph), 134.0 (Ph), 130.3 (Ph), 128.6 (Ph), 128.4 (Ph), 128.0 (Ph), 127.2 (Ph), 126.7 (Ph), 126.5 (Ph), 124.9 (Ph), 123.2 (Ph), 121.9 (Ph), 96.9 (CCl3), 91.2 (C-3). MS (EI, 70 eV), m/z (%) = 391 (M+, 7), 355 (2), 290 (3), 272 (100), 273 (21), 244 (9), 166 (4), 127 (14), 77 (5). HRMS (ESI-TOF): C20H14Cl3NO (M + H), requires 390.0217. Found 390.0214.
Physical aspect: yellow solid. Yield: 80%. Melting point: 115–116°C.
1H NMR (400 MHz, CDCl3) δ: 13.11 (s, 1H, NH), 7.14 (d, J = 7.9 Hz, 1H), 7.05 (d, J = 8.0 Hz, 1H), 6.66 (d, J = 9.0 Hz, 1H), 6.61 (d, J = 9.1 Hz, 1H), 3.70 (s, 3H, OCH3), 2.34 (s, 3H, PhCH3), 1.95 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3) δ: 179.7 (C-2), 168.2 (C-4), 156.9 (Ph), 139.1 (Ph), 131.9 (Ph), 131.3 (Ph), 129.3 (Ph), 128.6 (Ph), 125.4 (Ph), 113.9 (Ph), 98.8 (CCl3), 94.8 (C-3), 55.3 (OCH3), 21.3 (CH3), 16.8 (CH3). Anal. Cal. for C19H18Cl3NO2 (385.63): C, 57.24; H, 4.55; N, 3.51. Found: C, 57.18; H, 4.57; N, 3.51.
In 50 ml round bottom flasks equipped with reflux condenser and drying tube, the respective (Z)-1,1,1-trichloro-4-alkyl(aryl)amino-4-phenylbut-3-en-2-ones (4a-i, 5e) (1 mmol) were dissolved in anhydrous CHCl3 (15 ml), followed by the addition of BF3·OEt2 (1 ml of 48% ether sol.) and anhydrous Et3N (1 ml). The mixtures were stirred at temperature of reflux for 18 h, then diluted with CHCl3, washed with water (3× 20 ml) and the organic phase was dried with Na2SO4, filtered, and then the solvent (CHCl3) was removed under reduced pressure. The resulting oils were solubilized in EtOH (15 ml) at ambient temperature and stored overnight in the freezer for precipitation of the products 6a-i and 7e. The resulting solids were filtered under reduced pressure and washed with cold ethanol. The pure products 6 and 7 were obtained at 50–91% yields.
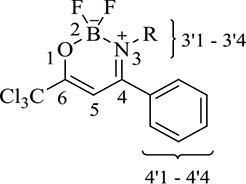
Physical aspect: brown solid. Yield: 74%. Melting point: 121–123°C.
1H NMR (400 MHz, CDCl3) δ: 7.59–7.57 (m, 3H, Ph), 7.34–7.32 (m, 2H, Ph), 6.23 (s, 1H, H–5), 4.19–4.14 (m, 1H, CH), 1.44 (t, J = 1.3 Hz, 1H, CH3), 1.42 (t, J = 1.3 Hz, 1H, CH3). 13C NMR (101 MHz, CDCl3) δ: 172.30 (C-4), 165.39 (C-6), 134.20 (C-4’1), 130.79 (C-4’4), 129.41 (C-4’2), 125.74 (C-4’3), 96.86 (C-5), 91.70 (CCl3), 54.20 (CH), 21.83 (CH3). 11B NMR (193 MHz, CDCl3) δ: 1.24 (t, J = 17.5 Hz). 19F NMR (565 MHz, CDCl3) δ: −134.64 – −134.99 (m). Anal. Cal. For C13H13BCl3F2NO (354.42): C, 44.06; H, 3.70; N, 3.95. Found: C, 44.08; H, 3.85; N, 4.02.
Physical aspect: brown solid. Yield: 80%. Melting point: 85–86°C.
1H NMR (400 MHz, CDCl3) δ: 7.60–7.55 (m, 3H, Ph), 7.38–7.36 (m, 2H, Ph), 6.29 (s, 1H, H–5), 3.58 (t, 2H CH2), 1.73 (quint, 2H, CH2), 1.19 (sext, 2H, CH2), 0.79 (t, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ: 172.7 (C-4), 165.7 (C-6), 133.3 (C-4’1), 131.1 (C-4’4), 129.3 (C-4’2), 126.3 (C-4’3), 96.4 (C-5), 91.8 (CCl3), 48.8 (NCH2), 32.3 (CH2), 20.1 (CH2), 13.4 (CH3). 11B NMR (193 MHz, CDCl3) δ: 1.13 (t, J = 15.8 Hz). 19F NMR (565 MHz, CDCl3) δ: −138.13 – −138.36 (m). Anal. Cal. For C14H15BCl3F2NO (368.44): C, 45.64; H, 4.10; N, 3.80. Found: C, 46.16; H, 3.84; N, 3.93.
Physical aspect: yellow solid. Yield: 65%. Melting point: 198–199°C.
1H NMR (400 MHz, CDCl3) δ: 7.41–7.14 (m, 10H, Ph), 6.60 (s, 1H, H–5). 13C NMR (101 MHz, CDCl3): 172.3 (C-4), 167.9 (C-6), 139.6 (C-3’1), 133.5 (C-4’1), 131.5 (C-4’4), 129.0 (C-4’2),128.7 (Ph), 128.6 (Ph), 128.2 (Ph), 126.4 (Ph), 96.3 (C-5), 91.8 (CCl3). 11B NMR (193 MHz, CDCl3) δ: 1.34 (t, J = 12.7 Hz). 19F NMR (565 MHz, CDCl3) δ: −132.97 – −134.25 (m). Anal. Cal. For C16H11BCl3F2NO (388.43): C, 49.47; H, 2.85; N, 3.61. Found: C, 49.41; H, 2.96; N, 3.61.
Physical aspect: yellow solid. Yield: 70%. Melting point: 173–175°C.
1H NMR (600 MHz, CDCl3) δ: 7.39 (t, J = 7.4 Hz, 1H, Ph), 7.31 (t, J = 7.7 Hz, 2H, Ph), 7.23 (d, J = 7.7 Hz, 2H, Ph), 7.04 (q, J = 8.4 Hz, 4H, Ph), 6.58 (s, 1H, H–5), 2.28 (s, 3H, CH3). 13C NMR (151 MHz, CDCl3) δ: 172.0 (C-4), 167.7 (C-6), 138.3 (C-3’1), 137.1 (C-3’4), 133.7 (C-4’1), 131.4 (C-4’4), 129.6 (C-4’2), 128.7 (Ph), 128.6 (Ph), 126.1 (C-4’3), 96.3 (C-5), 91.9 (CCl3), 21.0 (CH3). 11B NMR (193 MHz, CDCl3) δ: 1.31 (t, J = 12.4 Hz). 19F NMR (565 MHz, CDCl3) δ: −133.84 – −133.90 (m). Anal. Cal. For C17H13BCl3F2NO (402.46): C, 50.73; H, 3.26; N, 3.48. Found: C, 50.57; H, 3.32; N, 3.52.
Physical aspect: yellow solid. Yield: 91%. Melting point: 152–153°C.
1H NMR (400 MHz, CDCl3) δ: 7.42–7.38 (m, 1H, Ph), 7.35–7.30 (m, 2H, Ph), 7.25–7.22 (m, 2H, Ph), 7.06 (d, J = 9.0 Hz, 1H, Ph), 6.76 (d, J = 9.1 Hz, 1H, Ph), 6.58 (s, 1H, H–5), 3.75 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ: 171.8 (C-4), 167.5 (C-6), 159.1 (C-3’4), 133.7 (C-4’1), 132.4 (C-3’1), 131.3 (C-4’4), 128.8 (C-4’2), 128.6 (C-3’2), 127.4 (C-4’3), 114.2 (C-3’3), 96.3 (C-5), 91.8 (CCl3), 55.4 (OCH3). 11B NMR (193 MHz, CDCl3) δ: 1.29 (t, J = 12.2 Hz). 19F NMR (565 MHz, CDCl3) δ: −131.70 – −136.45 (m). Anal. Cal. For C17H13BCl3F2NO2 (418.46): C, 48.79; H, 3.13; N, 3.35. Found: C, 48.64; H, 3.22; N, 3.24.
Physical aspect: yellow solid. Yield: 60%. Melting point: 201–202°C.
1H NMR (400 MHz, CDCl3) δ: 8.13 (d, J = 9.2 Hz, 1H, Ph), 7.48–7.43 (m, 1H, Ph), 7.38–7.33 (m, 4H, Ph), 7.23 (dd, J = 8.4, 1.3 Hz, 1H), 6.66 (s, 1H, H–5). 13C NMR (101 MHz, CDCl3) δ: 173.6 (C-4), 169.7 (C-6), 147.1 (C-3’1), 145.3 (C-3’4), 132.9 (C-4’1), 132.3 (C-4’4), 129.3 (C-4’3), 128.6 (C-3’2), 127.8 (C-4’2), 124.4 (C-3’3), 96.5 (C-5), 91.6 (CCl3). 11B NMR (193 MHz, CDCl3) δ: 1.30. 19F NMR (565 MHz, CDCl3) δ −132.18 – −132.59 (m). Anal. Cal. For C16H10BCl3F2N2O3 (433.43); C, 44.34; H, 2.33; N, 6.46. Found: C, 44.33; H, 2.20; N, 6.38.
Physical aspect: yellow solid. Yield: 67%. Melting point: 169–170°C.
1H NMR (400 MHz, CDCl3) δ: 7.45–7.40 (m, 1H, Ph), 7.36–7.32 (m, 2H, Ph), 7.23–7.21 (m, 2H, Ph), 7.15–7.12 (m, 2H, Ph), 6.99–6.94 (m, 2H, Ph), 6.60 13C NMR (101 MHz, CDCl3) δ: 172.7 (C-4), 168.1 (C-6), 161.9 (d, J = 249.4 Hz, C-3’4), 135.6 (d, J = 3.1 Hz, C-3’1), 133.3 (C-4’1), 131.6 (C-4’4), 128.9 (C-4’2), 128.5 (C-4’3), 128.2 (d, J = 8.6 Hz, C-3’2), 116.1 (d, J = 23.1 Hz, C-3’3), 96.3 (C-5), 91.7 (CCl3). 11B NMR (193 MHz, CDCl3) δ: 1.28 (t, J = 12.5 Hz). 19F NMR (565 MHz, CDCl3) δ: −112.39 (s); −133,37 – −133,43 (m). Anal. Cal. For C16H10BCl3F3NO (406.42): C, 47.28; H, 2.48; N, 3.45. Found: C, 46.97; H, 2.30; N, 3.59.
Physical aspect: yellow solid. Yield: 50%. Melting point: 174–175°C.
1H NMR (400 MHz, CDCl3) δ: 7.46–7.33 (m, 5H, Ph), 7.23 (d, J = 7.3 Hz, 1H, Ph), 7.03 (d, J = 8.6 Hz, 1H, Ph), 6.61 (s, 1H, H–5). 13C NMR (101 MHz, CDCl3) δ: 172.6 (C-4), 168.4 (C-6), 138.7 (C-3’1), 133.2 (C-4’1), 132.3 (C-4’4), 131.8 (C-3’4), 129.0 (C-4’2), 128.6 (C-4’3), 128.0 (C-3’2), 122.3 (C-3’3), 94.7 (C-5), 91.7 (CCl3). 11B NMR (193 MHz, CDCl3) δ 1.25 (t, J = 12.0 Hz). 19F NMR (565 MHz, CDCl3) δ: −133.21 (s). Anal. Cal. For C16H10BBrCl3F2NO (467.33): C, 41.12; H, 2.16; N, 3.00. Found; C, 41.29; H, 2.00; N, 3.05.
Physical aspect: yellow solid. Yield: 74%. Melting point: 197–198°C.
1H NMR (400 MHz, CDCl3) δ: 7.77–7.74 (m, 3H, Ph), 7.49–7.37 (m, 4H, Ph), 7.21–7.17 (m, 3H, Ph), 7.10–7.06 (m, 2H, Ph), 6.69 (s, 1H, H–5). 13C NMR (101 MHz, CDCl3) δ: 174.9 (C-4), 168.5 (C-6), 135.9 (Ph), 133.8 (Ph), 133.5 (Ph), 131.4 (Ph), 129.0 (Ph), 128.6 (Ph), 128.3 (Ph), 128.2 (Ph), 127.4 (Ph), 127.2 (Ph), 126.5 (Ph), 125.0 (Ph), 124.7 (Ph), 122.8 (Ph), 96.2 (C-5), 91.9 (CCl3). 11B NMR (193 MHz, CDCl3) δ: 1.75–1.16 (m). 19F NMR (565 MHz, CDCl3) δ: −132.66 – −137.64 (m). Anal. Cal. For C20H13BCl3F2NO (438.49): C, 54.78; H, 2.99; N, 3.19. Found; C, 54.72; H, 3.17; N, 3.31.
Physical aspect: yellow solid. Yield: 69%. Melting point: 171–173°C.
1H NMR (400 MHz, CDCl3) δ: 7.10 (d, J = 8.0 Hz, 2H, Ar), 6.95–6.91 (m, 4H, Ar), 6.69 (d, J = 8.9 Hz, 2H, Ar), 3.72 (s, 3H, OCH3), 2.30 (s, 3H, PhCH3), 1.96 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3) δ: 177.0 (C-4), 164.1 (C-6), 158.7 (C-3’4), 140.3 (C-3’1), 133.0 (C-4’1), 130.0 (C-4’4), 129.3 (C-4’2), 127.9 (C-4’3), 127.2 (C-3’2), 113.9 (C-3’3), 103.4 (C-5), 94.0 (CCl3), 55.3 (q, J = 6.6 Hz, OCH3), 21.3 (q, J = 4.9 Hz, PhCH3). 16.7 (CH3). 11B NMR (193 MHz, CDCl3) δ: 0.66 – 0.53 (m). 19F NMR (565 MHz, CDCl3) δ: −138.63 – −138.74 (m). Anal. Cal. For C19H17BCl3F2NO2 (446.51): C, 51.11; H, 3.84; N, 3.14. Found; C, 51.11, H, 3.87, N, 3.12.
The starting materials (Z)-1,1,1-trichloro-4-methoxy-4-phenylbut-3-en-2-one (1) and (Z)-1,1,1-trichloro-4-methoxy-3-methyl-4-(p-tolyl)but-3-en-2-one (2) were synthetized according to the methodology reported by our group in previous works. Compounds 1 and 2 were synthesized in two steps from reactions of acetophenone or propiophenone using an excess amount of trimethyl orthoformate. Finally, the acylation reaction of the dimethyl acetal intermediates with trichloroacetyl chloride conducted to the compounds 1 and 2 (Colla et al., 1999; Bonacorso et al., 1999; Martins et al., 2004).
In order to obtain a novel series of (Z)-1,1,1-trichloro-4-alkyl(aryl)amino)-4-phenylbut-3-en-2-ones (4a-e and 5e), the methodology described in the literature was employed because the O,N-exchange reaction was already well established by our research group. In the present work, we reacted (Z)-1,1,1-trichloro-4-methoxy-4-phenylbut-3-en-2-one (1) (5 mmol) and several amines (3a-i) (7.5 mmol) at a molar ratio of 1:1.5, in ethanol (20 ml) for 24 h at reflux temperature (Scheme 2), according to similar procedures described in the literature (Bonacorso et al., 2002b; Martins et al., 2007).
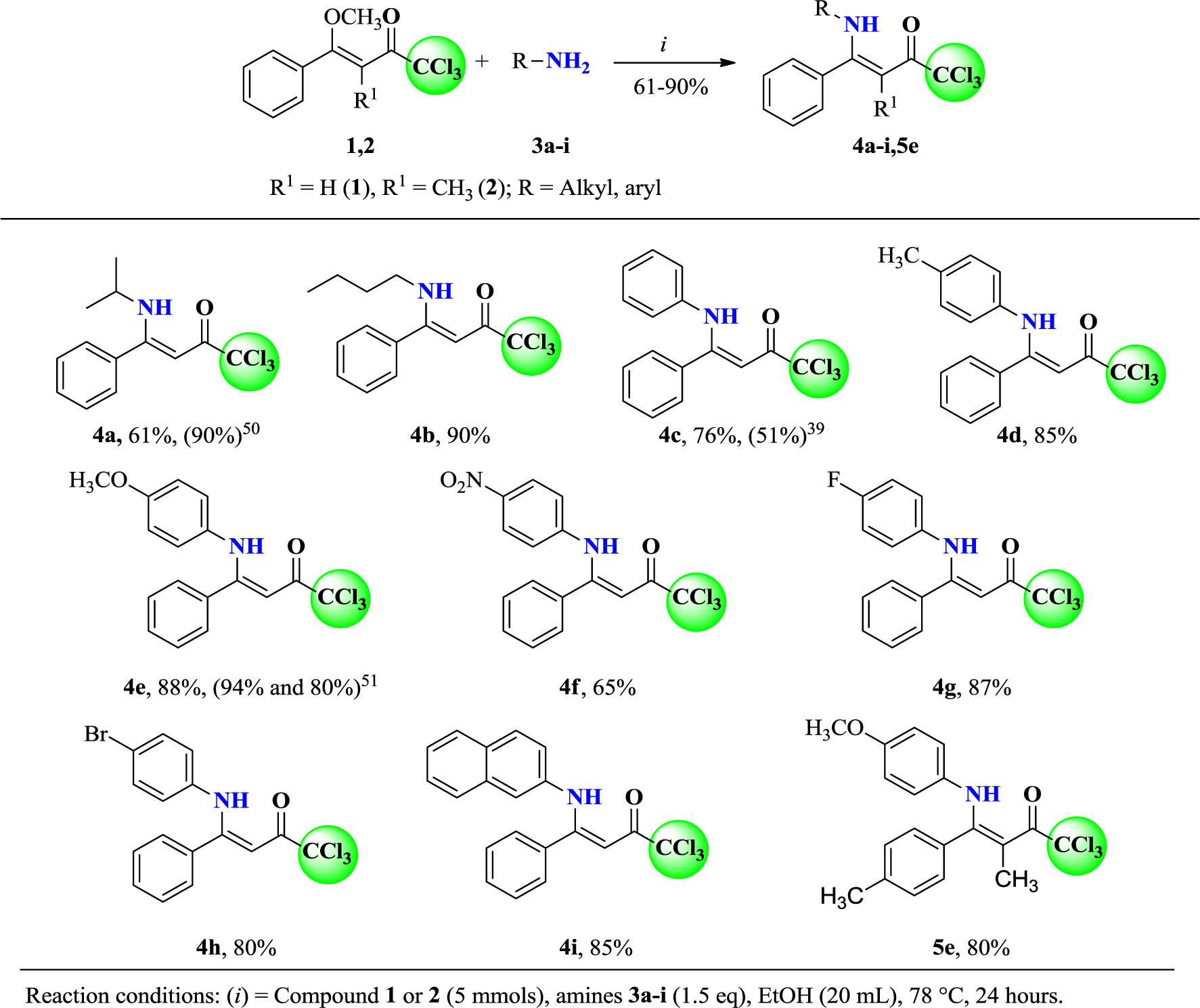
Scheme 2 Synthesis of (Z)-1,1,1-trichloro-4-alkyl(aryl)amino-4-phenylbut-3-en-2-ones (4a-i and 5e).
The products 4 and 5 were obtained by simple precipitation at low temperature. Subsequently, the products were filtered, washed with cold ethanol, and dried under reduced pressure. This procedure allowed to isolate 4a-i and 5e in 61 to 90% of yield (Scheme 2). Compounds 4a (Sosnovskikh et al., 2002), 4c (Hojo et al., 1986), and 4e (Toshiaki and Minoru, 1977)). are already described in the literature, but they were obtained through different precursors and procedures.
In order to explore the synthetic potential of 4a-i and 5e, (Z)-1,1,1-trichloro-4-phenyl-4-(p-tolylamino)but-3-en-2-one (4d) was employed to evaluate the influence of different reaction conditions such as solvent, temperature, time, and volume of BF3.OEt2 or Et3N to obtain the oxazaboron complexes 6, 7 (Table 1).
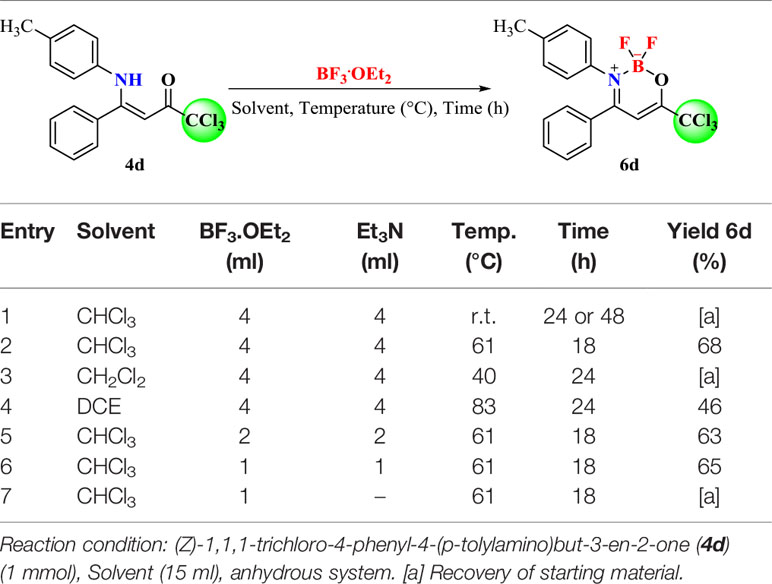
Table 1 Optimization of the reaction condition for the synthesis of 2,2-difluoro-3-p-tolyl-4-phenyl-6-(trichloromethyl)-2H-1,3,2-oxazaborinin-3-ium-2-uide (6d).
Optimization of the reaction began based on the methodology described by Bonacorso et al. in 2016. The reaction employs 1 mmol of the ligand precursor, 15 ml of anhydrous CHCl3, 4 ml of BF3.OEt2 (~16 mmol), and 4 ml of Et3N (~28 mmol) at room temperature, but this attempt did not lead to product formation after 24–48 h (Table 1 – Entry 1). The reactions were monitored by TLC. However, under the same conditions at the reflux temperature of CHCl3, product 6d was formed in 68% yield after 18 h of reaction (Table 1 – Entry 2). Based on the work described by Yoshii et al. in 2013 (Yoshii et al., 2013), the reaction was refluxed in anhydrous CH2Cl2 and the desired product was not obtained after 24 h of reaction (Table 1 – Entry 3). In this condition, a higher reflux temperature solvent was evaluated, which was ClCH2CH2Cl (DCE), although the result was only 46% yield (Table 1 – Entry 4).
From these results, the volume of BF3.OEt2 and Et3N for the reaction was also evaluated. By reducing the volume of BF3.OEt2 and Et3N from 4 to 2 ml, 63% yield of 6d was observed after 18 h (Table 1 – Entry 5). The reaction was still efficient and led to 65% yield (Table 1 – Entry 6) when the volumes of BF3.OEt2 and Et3N were reduced to 1 ml each. No formation of product was observed in the absence of triethylamine as base (Table 1 – Entry 7).
Thus, the results obtained by the optimization study showed that the best condition for the synthesis of the boron complexes (6a-i and 7e) was with 1 mmol of β-enaminoketone, 1 ml of Et3N (~7 mmol), and 1 ml of BF3.OEt2 (~4 mmol) in 15 ml of anhydrous CHCl3 under reflux for 18 h (Table 1 – Entry 6).
After the reaction optimization, we investigate the substrate scope of (Z)-1,1,1-trichloro-4-alkyl(aryl)amino-4-phenylbut-3-en-2-ones (4a-i and 5e) in order to expand the structural diversity of the corresponding boron complexes 6a-i, 7e (Scheme 3). As shown in Scheme 3, the (Z)-1,1,1-trichloro-4-alkyl(aryl)amino-4-phenylbut-3-en-2-ones (4a-i and 5e) were submitted to target BF2-coordination, where the alkyl and aryl derivatives furnished the desired products 6a-i and 7e in moderate to excellent yields (50–91%).
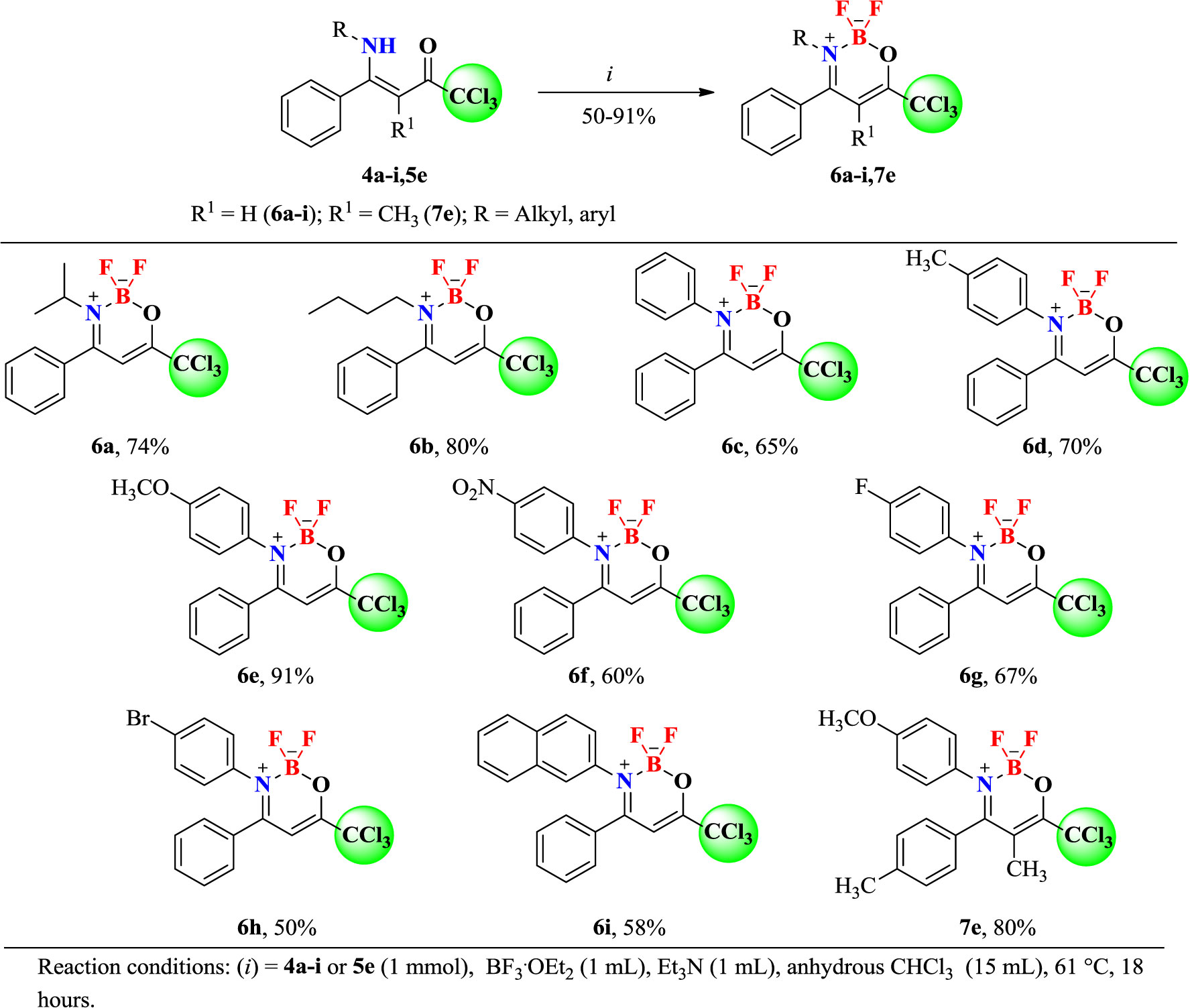
Scheme 3 Synthesis of 2,2-difluoro-3-alkyl(aryl)amino-4-phenyl-6-(trichloromethyl)-2H-1,3,2-oxazaborinin-3-ium-2-uide (6a-i and 7e).
Furthermore, 4-N-alkyl-substituted β-enaminoketones such as N-iso-propyl 6a (74%) and N-butyl 6b (80%), which were isolated in good yields, were also successfully included in the reaction scope. Similar conditions were also found for 4-substituted aryl-amines, including aniline 6c (65%) and 2-naphthylamine 6i (58%). Electron-donating 4-methylphenylamino and 4-methoxyphenylamino substituents from anilines 3d and 3e performed very well with this procedure and the desired complexes 6d and 6e were obtained in 70 and 91% yields, respectively. Electron-withdrawing groups, such as 4-nitrophenylamino from aniline 3f, also afforded the respective coordination complex 6f in 60% yield. 4-phenylhalogenated β-enaminoketones were also submitted to the target BF2-coordination. Thus, derivatives of 4-fluoro- and 4-bromo-substituted anilines presented good reactivities and allowed also the isolation of complexes 6g (67%) and 6h (50%).
In view of the differentiated properties that the trifluoromethyl substituent confers in various molecules in both chemical materials and medicinal chemistry, we also synthesized an example of substituted trifluoromethyl β-enaminoketone 9c to study its reactivity against BF3.OEt2 (Scheme 4). Compound 9c was synthesized using the methodology described using (Z)-1,1,1-trifluoro-4-methoxy-4-phenylbut-3-en-2-one (8) with aniline (3c). The compound 9c was obtained after recrystallization in 85% yield.
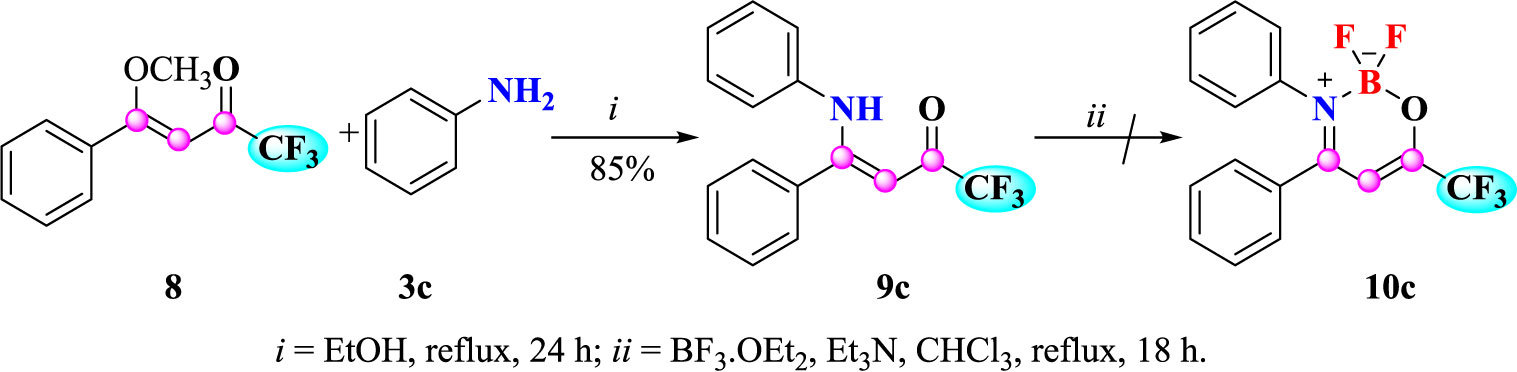
Scheme 4 Attempt to synthesize 2,2-difluoro-3,4-diphenyl-6-(trifluoromethyl)-2H-1,3,2-oxazaborinin-3-ium-2-uide (10c).
In order to investigate the synthetic reactivity of trifluoromethyl β-enaminoketone 9c against BF2.OEt2 complexation, the optimized reaction condition carried out for the (Z)-1,1,1-trichloro-4-alkyl(aryl)amino-4-phenylbut-3-en-2-ones (4a-i and 5e) was attempted for 9c. However, the formation of trifluoromethylated complex 10c was not possible because the high electron-withdrawing effect of the trifluoromethyl group at position 6, which hinders a stable N-B-O coordination.
The new structures of 4a-i, 5e, 6a-i, and 7e were confirmed and characterized by 1H-, 13C-, 11B-, and 19F-NMR spectroscopy, CHN elemental analysis or high-resolution mass spectra (HRMS).
The structures of compounds 4a-i, 5e were deduced on the basis of the NMR data of other β-enaminones previously synthesized in literature.50,51 The 1H NMR chemical shifts of the β-enaminoketone hydrogens (NH) for 4a-i, 5e showed chemical shifts at an average of 11.87 ppm, which suggests to us that the compounds 4a-i, 5e are in the Z,Z-configuration in solution (CDCl3), which is favored by an hydrogen interaction (N–H···OC). The vinyl hydrogen (H-3) was observed in the form of a singlet at an average of 6.06 ppm for all compounds 4, 5. The 13C NMR spectra of all compounds 4a-i and 5e showed, on average, chemical shifts at 181.4 ppm C-2, 166.7 ppm for C-4, 90.8 ppm for C-3, and 97.1 ppm for CCl3.
A comparison between the 1H-NMR data of compounds 4a-i, 5e and 6a-i, 7e showed clearly that complexes 6a-i and 7e do not show chemical shift signals (N–H) around at 12 ppm. Furthermore, an alteration in the chemical shifts of the H-3 signals from 6.06 ppm in average for the β-enaminones 4, 5 to 6.53 ppm in the boron complexes 6, 7 was also observed.
Upon comparing the 13C NMR data of the β-enaminone precursors 4a-i, 5e and the boron complexes 6a-i, 7e, the major evidence for the formation of 6a-i, 7e was the alteration in the chemical shifts at average of the carbons C-4, C-6, CCl3, and C-5 for 173.1, 167.3, 96.9, and 92.0 ppm, respectively.
Moreover, in order to demonstrate boron complex formation, 19F- and 11B-NMR experiments were also performed. 19F-NMR spectra of compounds 6a-i, 7e were performed using chloroform-d and, the signals were observed in the form of a multiplet for the compounds in the range of -131.7 ppm to -138.7 ppm. For the 11B-NMR spectra, the chemical shifts were observed in the form of a triplet at 1.26 ppm on average for each compound due to the coupling of both 19F nuclei. However, for compounds 6a-i and 7e, the 19F-NMR spectra showed a multiplet for each compound at -134.1ppm and -135.59 ppm, respectively.
The molecular structure of the compounds of series 4a-i, 5e, 6a-i, and 7e was determined and confirmed by single-crystal X-ray diffraction of the representative β-enaminone 4g and the boron complex 6e (Figure 1). For the compound 6e was observed that crystallization occurred at the monoclinic solvates-free form (Figure 1). The boron-atom has a tetrahedral geometry where the B-F, B-N, and B-O distances are 1.372, 1.590, and 1.452 Å, respectively and the bond angles around the B-atom are the 108.7° (F11-B1-N9) to 111.0° (F11-B1-F12). As observed, the O-B bond length is significantly shorter than the N-B one. The complete data of the X-ray diffraction are found in the Supplementary Information File.
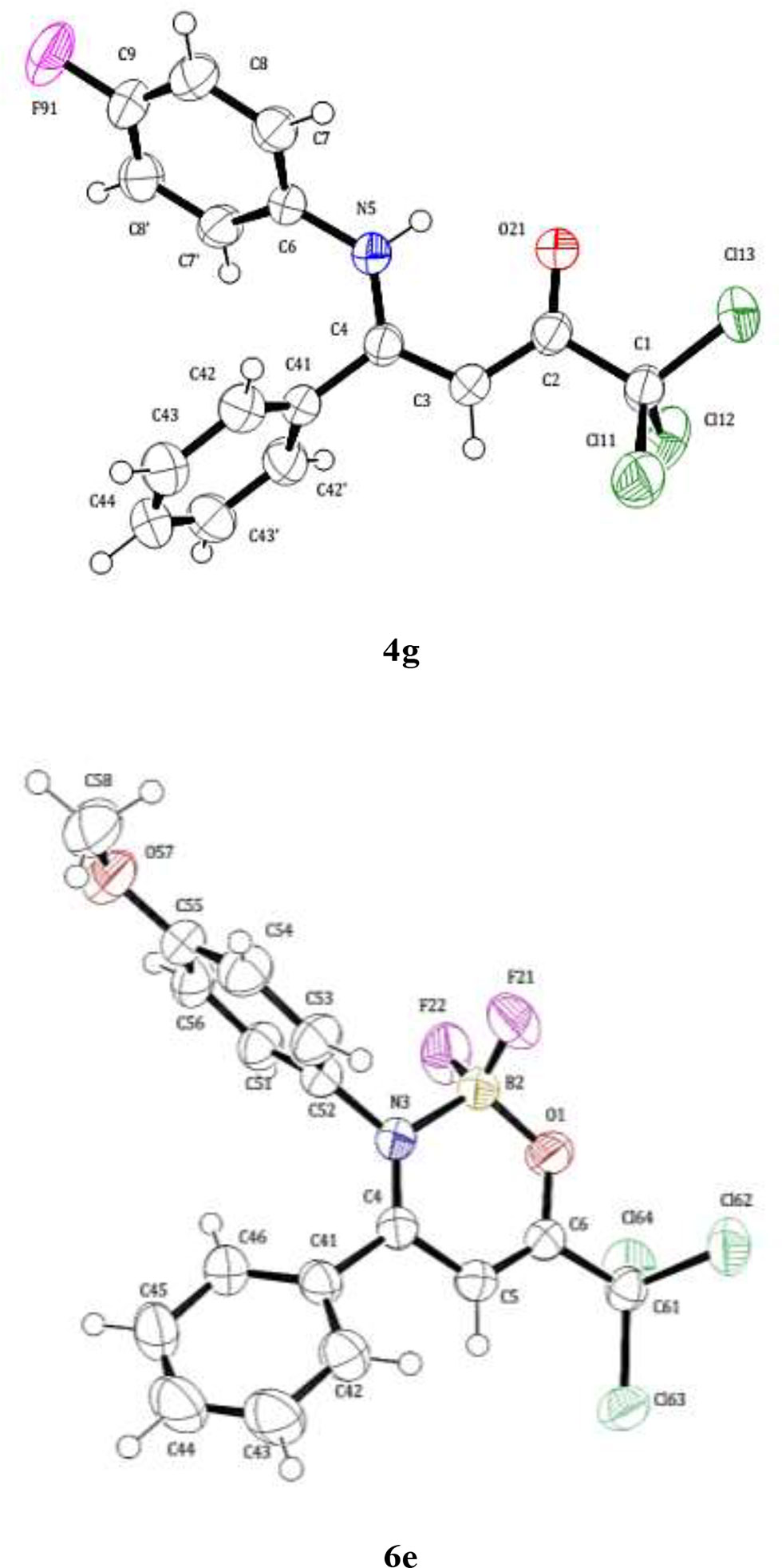
Figure 1 ORTEP of the crystal structure of (Z)-1,1,1-trichloro-4-((4-fluorophenyl)amino)-4-phenylbut-3-en-2-one (4g) (CCDC 1810806) and ORTEP of the crystal structure of 2,2-difluoro-3-(4-methoxyphenyl)-4-phenyl-6-(trichloromethyl)-2H-1,3,2-oxazaborinin-3-ium-2-uide (6e) (CCDC 1855153). Displacement ellipsoids are drawn at the 50% probability level and the H atoms are represented by circles with arbitrary radii.
The UV-Vis analyses of the compounds use dichloromethane, dimethyl sulfoxide, and methanol as solvents are showed in the Figure 2. All absorption spectra are listed in the Supplementary Information File (Figures S35–37). The UV-Vis data are presented in Table 2, where the solvents are presented according to polarity parameters (Moyon and Mitra, 2011).
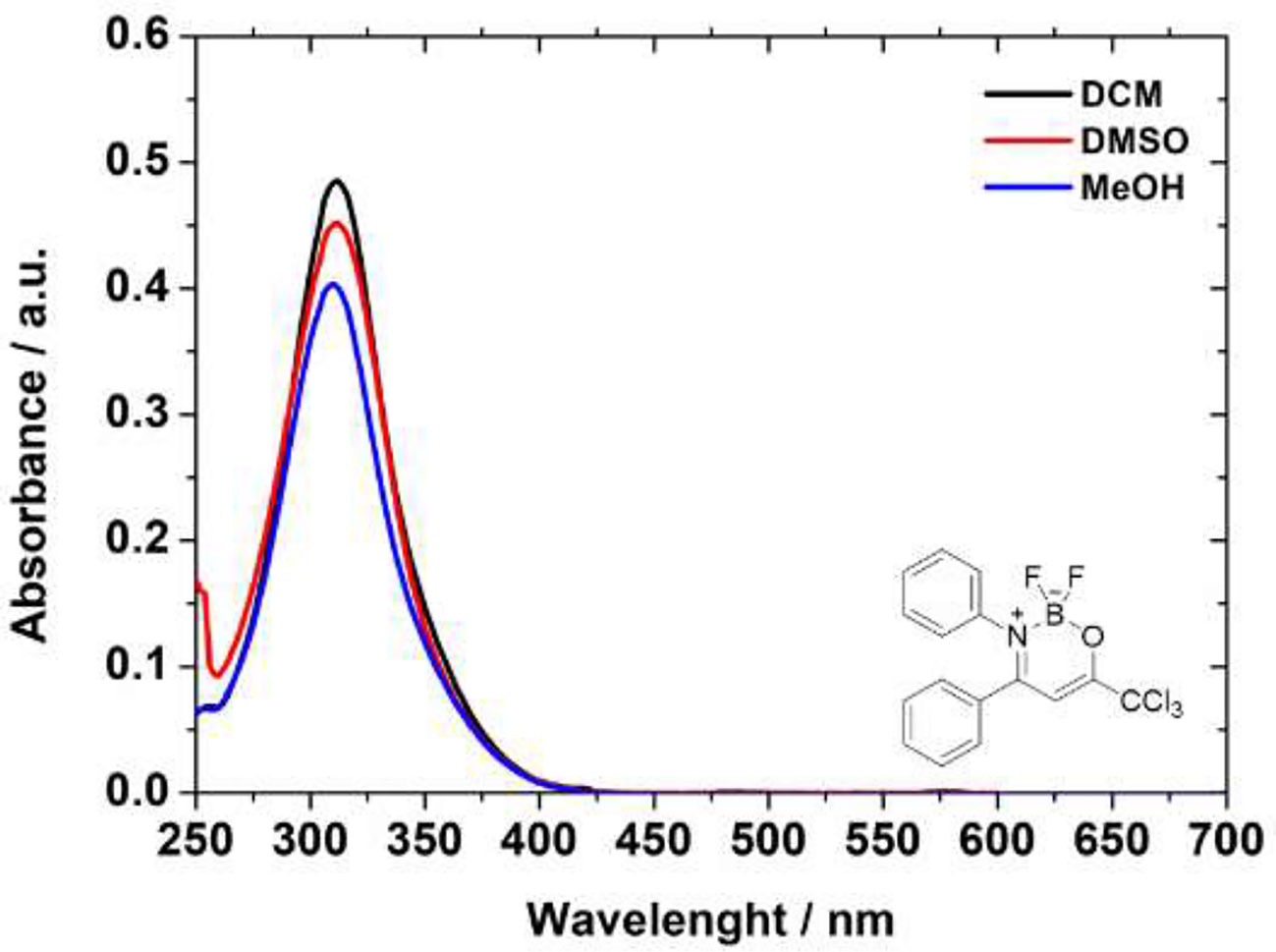
Figure 2 Comparative absorption spectra in solution of the BF2-complex compound 6c in DCM (black solid line), DMSO (red solid line), and MeOH (blue solid line), respectively.
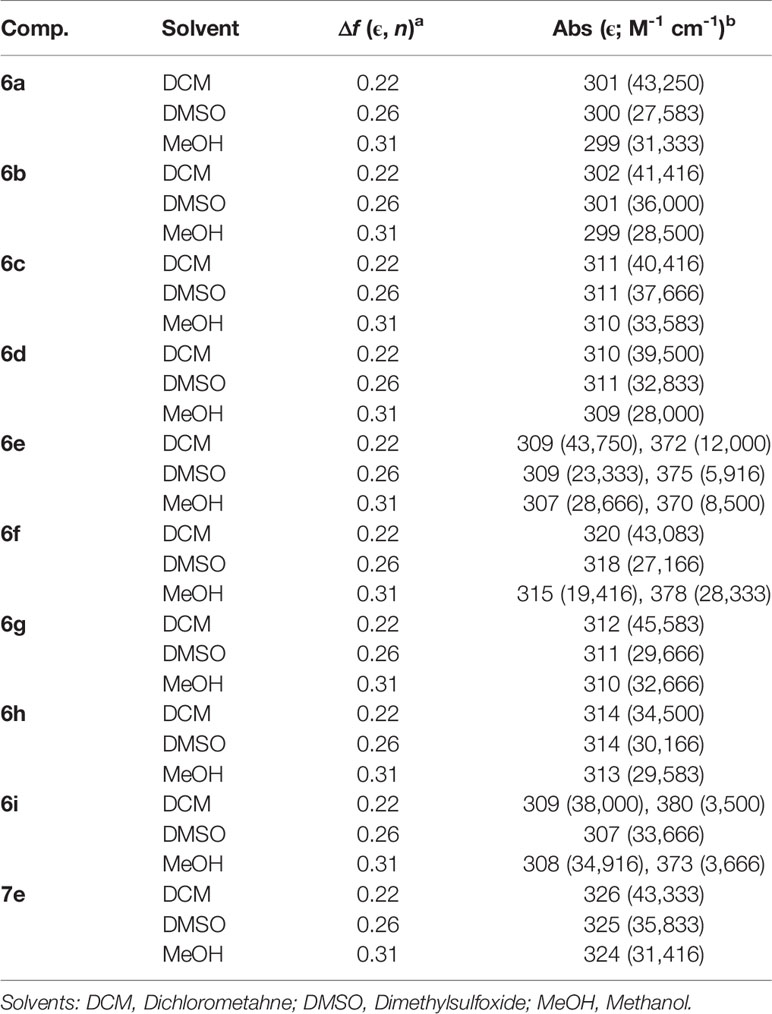
Table 2 UV-Vis absorption data and solvent parameters.
In general, all BF2-derivatives have absorption bands at UV nm region, with transitions according to the structure of these heterocycles. From the relatively large absorption coefficient (Table 2) it is plausible that this transition is S1(π) → S0(π) in nature. Yoshii et al. also reported and attributed the same nature transition in their compounds, although the authors measured in THF solution (Yoshii et al., 2013). Additionally, slightly solvatochromic behavior indicates no dependence of solvent properties. Unfortunately, all the BF2-derivatives studied here did not have luminescent properties in organic solution (Yoshii et al., 2013), regardless of the solvent used in the experiments (Supplementary Information File—Figure S38).
For a better insight into the frontier orbitals and the observed spectroscopic properties of compounds 6a-i and 7e, TD-DFT (Time-Dependent Density-Functional Theory) theoretical calculations were performed for compounds of series 6a-i and 7e using the Gaussian 09 package of programs (Frisch et al., 2016). All geometrical structures were optimized at the SCRF(PCM)-B3LYP/cc-pVTZ level of theory. The values calculate for the maximum absorption wavelengths were closed and in agreement with the experimental results in DCM, DMSO, and MeOH (solvent effect). As example, Table 3 highlights the electron distribution of the HOMO and LUMO for compound 6c. It is found that the HOMO and LUMO densities in 6c were delocalized over the whole molecule. The same behavior could be observed for all compounds studied (Supplementary Information File—Figures S58–S88).
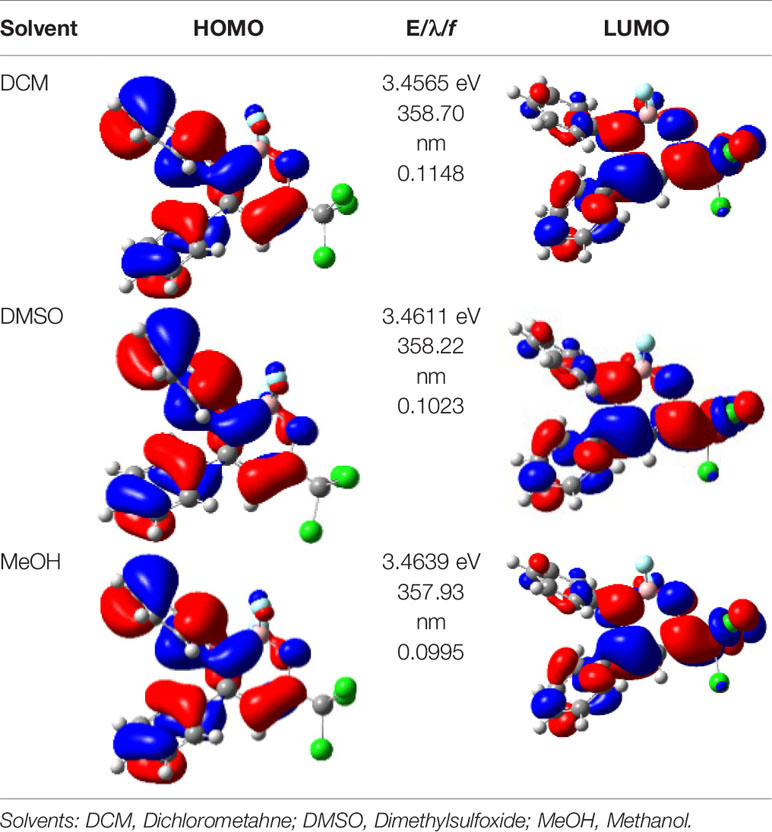
Table 3 Molecular orbital amplitude plots (generated with 0.02 au isovalue), excitation energy (E), and oscillator strengths (f) for HOMO-LUMO orbitals, calculated at the TD-DFT (SCRF(PCM))-B3LYP/cc-pVTZ level in DCM, DMSO, and MeOH for 2,2-difluoro-3,4-diphenyl-6-(trichloromethyl)-2H-1,3,2-oxazaborinin-3-ium-2-uide (6c).
To investigate the mentioned interactions, UV-Vis and emission fluorescence analysis are preferred because small molecule-DNA interactions may be experimentally monitored by changes in the intensity and position of the spectroscopic peak responses or changes in the dynamic viscosity of DNA (Chen et al., 1999; Ni et al., 2006).
In the present study, the interaction of the boron complexes 6a-i and 7e against CT-DNA (Calf Thymus DNA) was studied by UV-Vis at 250–600 nm region using DMSO (2%)/Tris-HCl pH 7.2 buffer mixture solution. The effect of different concentrations of CT-DNA on the UV-Vis spectra of derivative 6a is presented in Figure 3. Other BF2-derivatives absorption spectra are shown in Figures S39–S47 in the Supplementary Information File and the binding parameters are listed in Table 4.
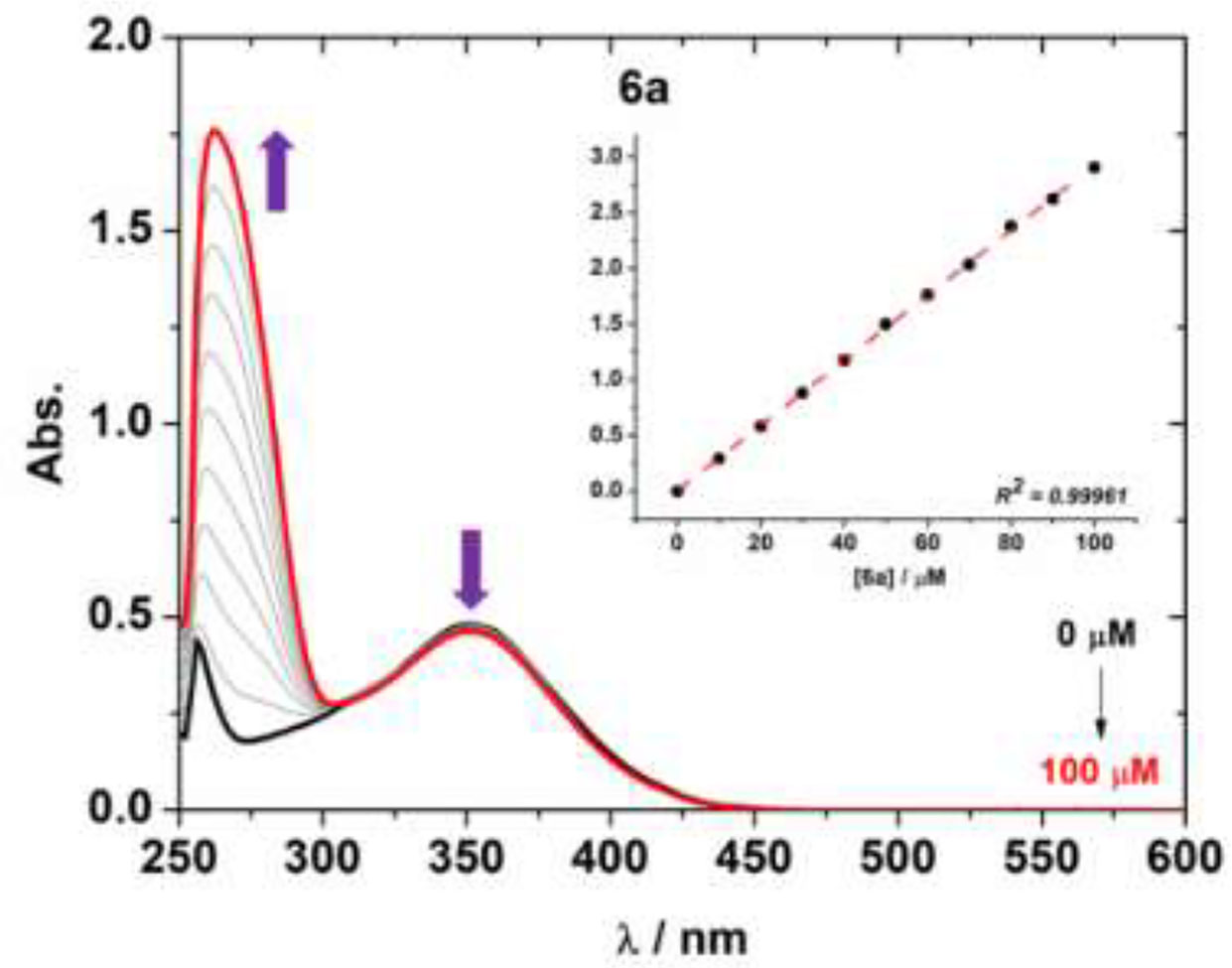
Figure 3 UV-Vis absorption spectra for compound 6a and the effect of successive additions of CT-DNA solution in the presence of a fixed concentration of 6a, in a DMSO (2%)/Tris-HCl pH 7.2 buffer mixture. Insert graph shows the plot [DNA]/(ϵa – ϵf) versus [DNA]. The concentration of CT-DNA ranged from 0 to 100 μM.
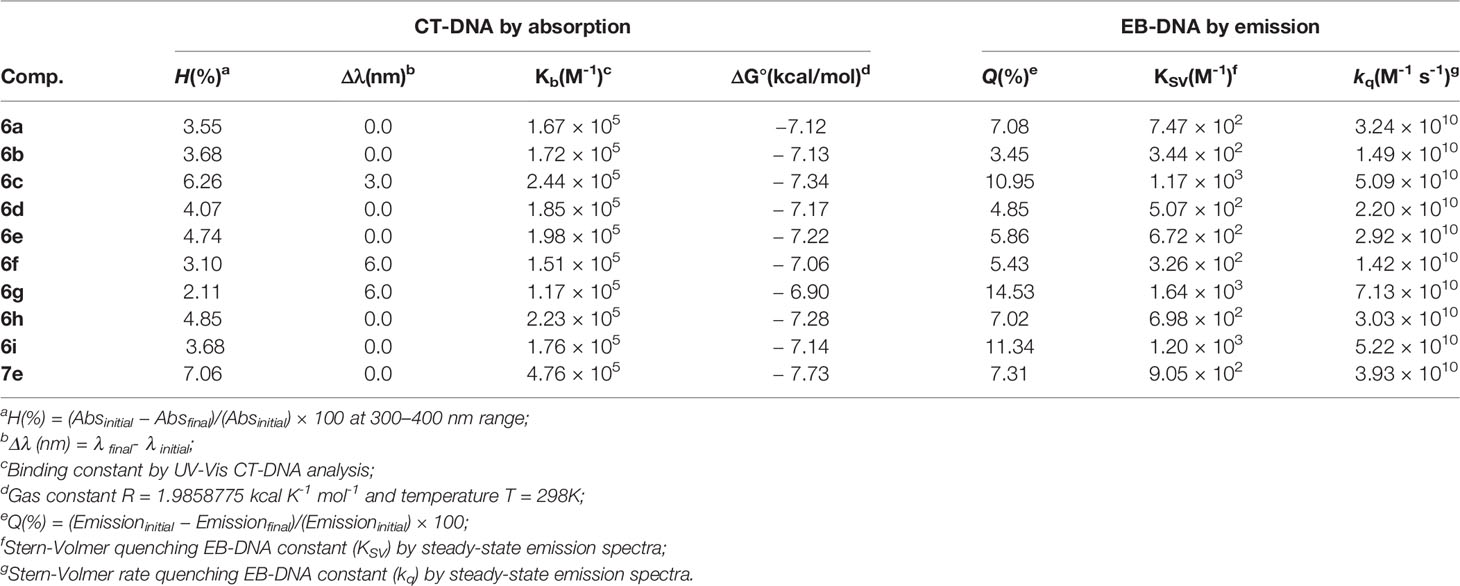
Table 4 CT-DNA-binding data from UV-Vis absorption and fluorescence emission of boron complexes.
Generally, the absorption spectra of organoboron compounds can be changed at 300–400 nm range upon several additions of CT-DNA (hypochromicity properties). A slight bathochromic shift in some cases was also observed, indicating a weak or non-electrostatic interaction observed between the cited compounds and CT-DNA (Table 4). The transition change of the derivatives may be a result of the interaction of the BF2 moiety with the nucleobase residues of the DNA (Figure 3), which are possible via hydrophobic forces, as previously in the literature (Bonacorso et al., 2018b).
Furthermore, the binding constant (Kb) data for the BF2-compounds were listed in Table 4. These Kb values are associated to the BF2-DNA complex stability, while the free energy indicates the spontaneity/non-spontaneity of derivative-DNA binding process, indicating the spontaneity of the interaction between the organoboron complexes and DNA.
In the emission assays, EB-DNA (Ethidium Bromide-DNA) analysis were also conducted to determine the displacement of the intercalating agent ethidium bromide (EB) dye from CT-DNA. As example, the fluorescence emission of compound 6a was monitored by increasing the compound concentration at a fixed concentration of CT-DNA pre-treated with EB dye (Figure 4). The EB-DNA adduct emission fluorescence spectra of other derivatives are listed in Figures S48–S56 in the Supplementary Information File.
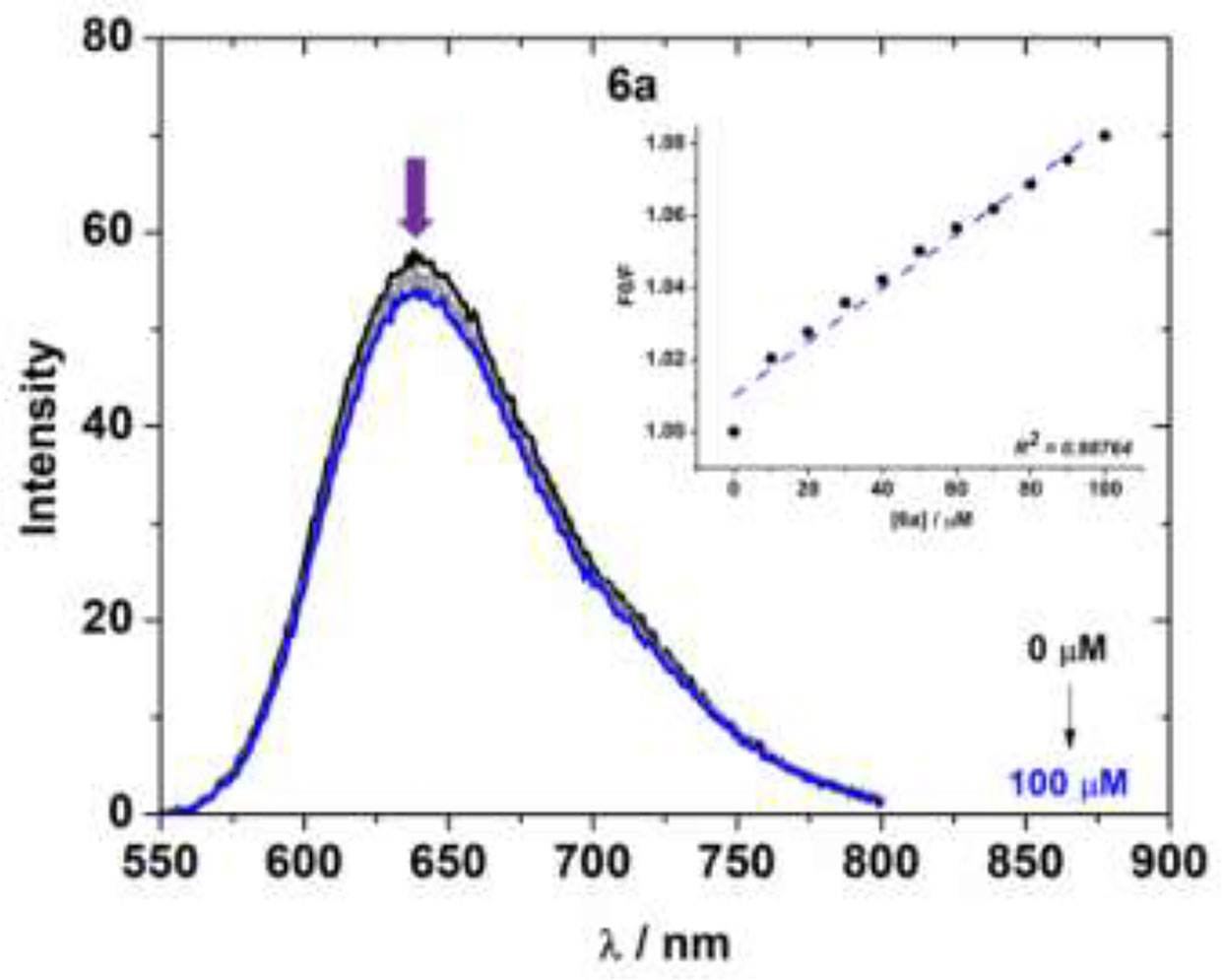
Figure 4 Fluorescence emission spectra of EB bound to CT-DNA in the presence of 6a in a DMSO (2%)/Tris-HCl pH 7.2 mixture at λexc = 510 nm. The inset shows the plot of F0/F versus the concentration of compound 6a according to the Stern-Volmer equation.
The EB-DNA competition experiment shows an intense emission band located at λem = 649 nm by excitation at λexc = 510 nm. After adding derivative 6a to the EB-DNA solution, the EB-DNA emission appeared to decrease in emission intensity. This fact demonstrates a weak fluorescence quenching of the EB-DNA adduct, as estimated by the KSV values (Table 4). This behavior can be assigned to the competition of the BF2 complexes with EB over binding to the base pair of CT-DNA (intercalation mode). High values were obtained for the quenching constant rate (kq), which were higher than the diffusion rate constant (kdiff ≈ 7.40 × 109 L mol-1 s-1 at 298K) (Montalti et al., 2006). These observed values indicate a possible static interaction mechanism between the BF2-complexes and EB-DNA (Montalti et al., 2006), which takes place via ground state association.
Twenty of the newly synthesized compounds 4a-i, 5e, 6a-i, and 7e were evaluated for them in vitro antimicrobial activity against a panel of microorganisms including yeasts, filamentous fungi, bacteria, and alga by determining their MIC and minimal fungicidal/bactericidal/algacidal concentrations using broth microdilution methods according to CLSI standard protocols (CLINICAL AND LABORATORY STANDARDS INSTITUTE (CLSI), 2008; CLINICAL AND LABORATORY STANDARDS INSTITUTE (CLSI), 2012; CLINICAL AND LABORATORY STANDARDS INSTITUTE (CLSI), 2015).
In order to classify the antimicrobial activity, antibacterial and two antifungal agents currently employed in therapeutics were compared (Supplementary Information File—Table S13). Therefore, the range of MICs until 10 μg/ml was considered significant activity for yeast-like fungi, with MIC-ranges between 20 and 40 μg/ml considered moderate activity and concentrations beyond not being considered. For filamentous fungi and P. zopfii, the MICs until 1.0 μg/ml were considered strong activity, with MIC ranges from >1 to 4 μg/ml considered moderate activity and above this range not being considered. For bacteria, all MICs under 4.0 μg/ml were considered as active and above they were not considered (Supplementary Information File—Table S13). The comparisons among MICs and MCC (minimal “cidal” concentrations) revealed that they were similar in 66% of cases (48/73) and showed that the MCC were higher by one or more concentration in 34% (25/73). The comparisons are important because they show the differences between compounds that are only inhibitory from those able to inhibit and kill pathogenic microorganisms.
Considering the antimicrobial activity of the synthesized compounds, the algaecidal action against P. zopfii stands out. Compounds 4a, 4b, 4d, 4e, 6c, 6e, 6f, and 6h were strongly effective in both inhibiting growth and causing algae death at a concentration below 1.0 µg/ml, while the other compounds shows moderate activity against this alga (except 4i and 5e). These results are encouraging, since P. zopfii is an environmental agent of bovine mastitis that causes many losses resulting from the compromised quality and production of milk and, in addition, it presents high antimicrobial resistance and the optimal treatment strategy for this infection has not yet been well established. In addition, P. zopfii can cause cutaneous and serious systemic infections in humans (Lass-Flörl and Mayr, 2007). The compounds that showed higher inhibitory and algaecidal activity were 4d, 4e, 6e, and 6f (MIC = 0.31 µg/ml). These results showed that changes in the basic structure as well as in the substituent slightly increased activity against P. zopfii.
In relation to the evaluated pathogenic yeasts (Candida spp. and Cryptococcus gattii) only compound 6i showed moderate activity for both, the other compounds of series 6 (6b-h) were moderately effective in inhibiting only the growth of C. gattii, as well as compounds 4a, 4b, 5e, and 7e. Compounds 4a-i and 5e have the same substituents as compounds 6a-i and 7e although the basic structure is comprised only by a β-enaminoketone for the former, whereas for compounds 6a-i and 7e a basic structure is constituted by a boron complex. Compounds 6a-j and 7e showed moderate activity against C. neoformans, and remained inactive against S. cerevisiae. Considering the difficulty in treating meningitis caused by C. gattii, which is normally resistant to one of the main antifungal drugs used in this pathology, fluconazole, increasing the mortality rate, potential treatment alternatives are extremely necessary.
Among filamentous fungi selected to study the activity of β-enaminoketone boron complex, the four most frequent agents of the life-threatening disease aspergillosis (A. fumigatus, A. flavus, A. niger, and A. terreus) were chosen. It is important to note that the antifungal therapy for these microorganisms is difficult and therapeutic failures are frequent (Leimann et al., 2004). Immunocompromised and neutropenic patients require fungicidal agents to treat their infections due to immunologic system failures (Dignani et al., 2003; Nucchi et al., 2004; Walsh et al., 2004).
Significant results were observed for compounds 4c, 4g, and 4h, which exhibited the best activities against Aspergillus. A. niger growth was completely inhibited at the concentration of 2.5 µg/ml. For boron complexes (6a-i and 7e), concentrations two or more times higher were required to inhibit A. niger growth. Compound 4g showed moderate activity against A. flavus (MIC = 20 µg/ml), which is the second most important agent of aspergillosis.
Another interesting point is the lack of activity of 4i against all the filamentous fungi studied when compared with 6i. The incorporation of BF2 resulted in the acquisition of antifungal activity. This shows that substituents in the boron complexes can bring significant differences in antimicrobial activities.
The activity of the series of compounds against a panel of bacteria clinically important was poor, with the best activities observed with 6c (MIC = 5.0 µg/ml) and 6i (MIC = 5.0 µg/ml), which were both against K. pneumoniae, and an opportunistic gram-negative rod. This is curious because, in general, the gram-positive cocci, which is represented here by S. aureus, is more sensible than gram negative rods (Kiska and Gilligan, 1999; Pfaller and Diekema, 2004).
The cytotoxicity of the β-enaminoketones and boron complexes were assessed using in vitro cell-based assay with 3T3 fibroblasts as the cell model, and MTT as endpoints to determine cell viability. As shown in Table S13 (Supplementary Information File), all compounds tested at concentrations between 1 and 100 µg/ml exhibited low or negligible cellular toxicity, as determined by MTT assay.
In summary, the aim of this study was to evaluate the synthetic potential of β-methoxyvinyl trichloromethyl ketones for the synthesis of trichloromethyl substituted β-enaminoketones (4a-i, 5e) (61–90%), which may be used to obtain a new series of 2,2-difluoro-3-alkyl(aryl)-4-phenyl-6-(trichloromethyl)-2H-1,3,2-oxazaborinin-3-ium-2-uides (6a-i, 7e) in moderate to very good yields (50–91%). Unfortunately, it was not possible to obtain the trifluoromethyl-substituted analogs.
The results of the DNA-binding studies by spectroscopic analysis of the new BF2-β-enaminoketone compounds indicated that the greatest interaction with nucleic acids.
Multidrug-resistant microorganisms are increasingly common. These types of microorganisms can resist the effects of conventional antimicrobial drugs, increasing the mortality rates in both humans and animals affected by infections that are difficult to treat. In this context, new and more potent compounds are required. The series presented here showed to be able to inhibit the growth of several tested microorganisms, with outstanding algaecidal activity, however weak or no antibacterial activity, and moderate antifungal activity. We believe that these compounds could be chemically modified to improve their antimicrobial activity. Some compounds from the present series exhibited potent antimicrobial effects on various pathogenic microorganisms at concentrations below those that showed cytotoxic effects. Notably 4d, 4e, 6e, and 6f showed the best results and were very significant against P. zopfii, which is an agent that causes diseases in humans and animals.
Finally, the new molecules presented here open good prospects for the development of analogue structures with possible application in microbiology and studies involving interactions with biomolecules. The introduction of other known chromophore substituents in similar chelates to those reported here is under initial development and will be published in due course.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
WR, IR, MR, NZ, and HB: These researchers and students were responsible for the development of the part concerning the synthesis, purification, NMR, GC-MS and X-ray diffractometric data, and TD-DFT calculations for the new compounds. HC, LD, and PL: These researches were responsible for the development of the part related to the evaluation of the minimum inhibitory concentration (MIC) of the series of compounds 5–7. Twenty of the newly synthesized compounds 4a-i, 5e, 6a-i, and 7e were evaluated for their in vitro antimicrobial activity against a panel of microorganisms including yeasts, filamentous fungi, bacteria, and alga by determining their MIC and minimal fungicidal/bactericidal/algaecidal concentrations using broth microdilution methods according to CLSI standard protocols. TA and BI: This researcher and the student were responsible for the development of the part concerning to the study of the optical properties of the new compounds (UV-vis, fluorescence, quantum yield calculations, Stokes shift) and the interaction of the boron complexes 6a-i and 7e against CT-DNA, which was studied by UV–Vis absorption spectroscopy.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank: Coordination for Improvement of Higher Education Personnel (CAPES/PROEX – Finance Code 001) for the fellowships; and the National Council for Scientific and Technological Development (CNPq) Proc. nr. 304711/2018-7 (grants) and Proc. Nr. 402.075/2016-1 and 409.150/2018-5 CNPq/Universal) and the Research Support Foundation of the State of Rio Grande do Sul (FAPERGS) (PqG, Proc. nr. 17/2551-0001275-5) for the financial support. The authors acknowledge especially João Pedro Pereira Copetti for TD-DFT calculations and kind assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2020.01328/full#supplementary-material
Barluenga, J., Aguilar, E., Fustero, S., Olano, B., Viado, A. L. (1992). Stereoselective synthesis of 1,3-smino alcohols and 1,3-amino ketones. J. Org. Chem. 57, 1219. doi: 10.1021/jo00030a033
Bartoli, G., Cimarelli, C., Palmieri, G. (1994). Convenient procedure for the reduction of β-enamino ketones: synthesis of γ-amino alcohols and tetrahydro-1,3-oxazines. J. Chem. Soc Perkin Trans. 1, 537–543. doi: 10.1039/P19940000537
Bartoli, G., Cupone, G., Dalpozzo, R., De Nino, A., Maiuolo, L., Procopio, A., et al. (2002). Stereoselective reduction of enaminones to syn γ-amino alcohols. Tetrahedron Lett. 43, 7441. doi: 10.1016/S0040-4039(02)01545-9
Bégué, J., Bonnet-Delpon, D. (2006). Recent advances (1995–2005) in fluorinated pharmaceuticals based on natural products. J. Fluor. Chem. 127, 992. doi: 10.1016/j.jfluchem.2006.05.006
Boger, D. L., Ishizaki, T., Wysocki, J. R. J., Munk, S. A., Kitos, P. A., Suntornwat, O. (1989). Total synthesis and evaluation of(.+-.)-N-(tert-butoxycarbonyl)-CBI, (.+-.)-CBI-CDPI1, and (.+-.)-CBI-CDPI2: CC-1065 functionalagents incorporating the equivalent 1,2,9,9a-tetrahydrocyclopropa[1,2-c]benz[1,2-e]indol-4-one (CBI) left-hand subunit. J. Am. Chem. Soc 111, 6461. doi: 10.1021/ja00198a089
Bonacorso, H. G., Martins, M. A. P., Bittencourt, S. R. T., Lourega, R. V., Zanatta, N., Flores, A. F. C. (1999). Trifluoroacetylation of unsymmetrical ketoneacetals. A convenient route to obtain alkyl side chain trifluoromethylatedheterocycles. J. Fluor. Chem. 99, 177. doi: 10.1016/S0022-1139(99)00146-3
Bonacorso, H. G., Bittencourt, S. R. T., Lourega, R. V., Flores, A. F. C., Zanatta, N., Martins, M. A. P. (2000). A Convenient synthetic method for fullyconjugated 3-alkyl- and 3-aryl-5-trifluoromethyl-1-methyl-1,2-thiazine 1-oxide from β-alkoxyvinyltrifluoromethyl ketones. Synthesis 10, 1431. doi: 10.1055/s-2000-7104
Bonacorso, H. G., Lourega, R. V., Wastowski, A. D., Flores, N., Martins, M. A. P. (2002a). β-Alkoxyvinyl trichloromethyl ketones as Nheterocyclic acylating agent. A new access to 5H-thiazolo[3,2-a]pyrimidin-5-ones. Tetrahedron Lett. 43, 9315. doi: 10.1016/S0040-4039(02)02337-7
Bonacorso, H. G., Wentz, A. P., Bittencourt, S. R. T., Marques, L. M. L., Zanatta, N., Martins, M. A. P. (2002b). Synthesis of some N-[1-alkyl(aryl)-3-oxo-4,4,4-trichloro(trifluoro)-1-buten-1-yl]-o-aminophenols and o-phenylenediamines as potential anticancer agents. Synth. Commun. 23, 335. doi: 10.1081/SCC-120002116
Bonacorso, H. G., Andrighetto, R., Zanatta, N., Martins, M. A. P. (2010). The unexpected cyclization routes ofN,N′-bis(oxotrifluoroalkenyl)-1,3-phenylenediamines in polyphosphoric acid medium. Tetrahedron Lett. 51, 3752. doi: 10.1016/j.tetlet.2010.05.041
Bonacorso, H. G., Andrighetto, R., Krüger, N., Navarini, J., Flores, A. F. C., Martins, M. A. P., et al. (2013). Cycloaromatization Reaction of4‐Alkoxy‐1,1,1‐trifluoroalk‐3‐en‐2‐ones with 2,6‐Diaminotoluene: The Unexpected Regioselective Synthesis of 2,4,7,8‐Tetrasubstituted Quinolines. J. Heterocycl. Chem. 50, E193. doi: 10.1002/jhet.1552
Bonacorso, H. G., Calheiro, T. P., Iglesias, B. A., Berni, I. R. C., Júnior, E. N. S., Rocha, J. B. T., et al. (2016). Synthesis, 11B- and 19F NMR spectroscopy, and optical and electrochemical properties of novel 9-aryl-3-(aryl/heteroaryl)-1,1-difluoro-7-(trifluoromethyl)-1H-[1,3,5,2]oxadiazaborinino[3,4-a][1,8]naphthyridin-11-ium-1-uide complexes. Tetrahedron Lett. 57, 5017. doi: 10.1016/j.tetlet.2016.09.068
Bonacorso, H. G., Calheiro, T. P., Iglesias, B. A., Acunha, T. V., Franceschini, S. Z., Ketzer, A., et al. (2018a). 1,1-Difluoro-3-aryl(heteroaryl)-1H-pyrido[1,2-c][1,3,5,2]oxadiazaborinin-9-ium-1-uides: synthesis; structure; and photophysical, electrochemical, and BSA-binding studies. New J. Chem. 42, 1913. doi: 10.1039/C7NJ04032F
Bonacorso, H. G., Calheiro, T. P., Iglesias, B. A., Silveira, S. H., Júnior, E. N. S., Ketzer, A., et al. (2018b). Multinuclear NMR spectroscopy, photophysical, electrochemical and DNA-binding properties of fluorinated 1,8-naphthyridine-based boron heterocycles. J. Fluor. Chem. 205, 8. doi: 10.1016/j.jfluchem.2017.11.006
Bonacorso, H. G., Calheiro, T. P., Acunha, T. V., Iglesias, B. A., Franceschini, S. Z., Ketzer, A., et al. (2019). Novel aryl(heteroaryl)-substituted(pyrimidyl)benzamide-based BF2 complexes: Synthesis, photophysical properties, BSA-binding, andmolecular docking analysis. Dyes Pigm. 161, 396. doi: 10.1016/j.dyepig.2018.09.076
Buyukcakir, O., Bozdemir, O. A., Kolemen, S., Erbas, S., Akkaya, E. U. (2009). Tetrastyryl-bodipy dyes: Convenient synthesis and characterization of elusive near IR fluorophores. Org. Lett. 11, 4644. doi: 10.1021/ol9019056
Cakmak, Y., Kolemen, S., Duman, S., Dede, Y., Dolen, Y., Kilic, B., et al. (2011). Designing excited states: Theory‐guided access to efficient photosensitizers for photodynamic action. Angew. Chem. Int. Ed. 50, 11937. doi: 10.1002/anie.201105736
Chen, Q. Y., Hi, D. H., Zhao, Y., Guo, J. X. (1999). Interaction of a novel red-region fluorescent probe, Nile Blue, with DNA and its application to nucleic acids assay. Analyst 124, 901. doi: 10.1039/a901174i
CLINICAL AND LABORATORY STANDARDS INSTITUTE (CLSI) (2008). Document M38-A2. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Filamentous Fungi; Approved Standard. 2nd ed (Wayne, Pennsylvania, USA: Clinical and Laboratory Standards Institute).
CLINICAL AND LABORATORY STANDARDS INSTITUTE (CLSI) (2012). Document M27-S4. Reference Method for Broth Dilution Antifungal Susceptibility Testing of yeasts; Approved Standard. 4th ed (Wayne, Pennsylvania, USA: informational supplement. Clinical and Laboratory Standards Institute).
CLINICAL AND LABORATORY STANDARDS INSTITUTE (CLSI) (2015). Document M07-A10. Reference Method for Dilution Antimicrobial Susceptibility Test of bacteria that grow aerobically; Approved Standard. 10th ed (Wayne, Pennsylvania, USA: Clinical and Laboratory Standards Institute).
Colla, A., Martins, M. A. P., Clar, G., Krimmer, S., Fischer, P. (1999). Synthesis 1991, 483; Martins, M. A. P.; Bastos, G. P.; Bonacorso, H. G.; Zanatta, N.; Flores, A. F. C.; Siqueira, G. M. Tetrahedron Lett. 40, 430.
Colla, A., Martins, M. A. P., Clar, G., Krimmer, S., Fischer, P. (1991). Trihaloacetylated Enol Ethers - General synthetic procedure and heterocyclic ring closure reactions with hydroxylamine. Synthesis 1991 (6), 483–486. doi: 10.1055/s-1991-26501
Dignani, M. C., Kiwan, E. N., Anaissie, E. J., Anaissie, E. J., McGinnis, M. R., Pfaller, M. A. (2003). Hyalohyphomycoses Vol. 309 (New York: Clinical Mycology; Churchil Livingstone).
Farrugia, L. J. (2012). WinGX and ORTEP for Windows: an update. J. Appl. Crystallogr. 45, 849. doi: 10.1107/S0021889812029111
Ferraz, H. M. C., Gonçalo, E. R. S. (2007). Preparações e aplicações sintéticas recentes de enaminonas. Quim. Nova 30, 957.
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R. (2016). Gaussian 09, Revision A.02 (Wallingford, CT: Gaussian, Inc.).
Gerus, I. I., Vdovenko, S. I., Gorbunova, M. G., Kukhar', V. P. (1991) 4-Trifluoromethylpyrimidines. Chem. Heterocycl. Compd. 27, 398. doi: 10.1007/BF00480838
Harris, M., II, Braga, A. C. H. (2004). An easy and efficient method to produce γ-amino alcohols by reduction of β-enamino ketones. J. Braz. Chem. Soc 15, 971. doi: 10.1590/S0103-50532004000600027
He, H., Lo, P. C., Yeung, S. L., Fong, W. P., Ng, D. K. P. (2011). Preparation of unsymmetrical distyryl BODIPY derivatives and effects of the styryl substituents on their in vitro photodynamic properties. Chem. Commun. 47, 4748. doi: 10.1039/C1CC10727E
Hojo, M., Masuda, R., Okada, E. (1986). A useful one-step synthesis of β-trihaloacetylvinyl ethers and trihaloacetylketene acetals. Synthesis 12, 1013. doi: 10.1055/s-1986-31852
Josefik, F., Svobodova, M., Bertolasi, V., Simunek, P., Machacek, V., Almonasy, N., et al. (2012). A new bicyclic oxazaborines with a bridged nitrogen atom, their thermic rearrangement and fluorescence properties. Organomet. Chem. 699, 75. doi: 10.1016/j.jorganchem.2011.11.004
Katritzky, A., Harris, P. A. (1990). Benzotriazole-assisted synthesis of novel Mannich bases from ketones and diverse aldehydes. Tetrahedron 46, 987. doi: 10.1016/S0040-4020(01)81378-8
Kiska, D. L., Gilligan, P. H. (1999). “–Pseudomonas,” in Manual of Clinical Microbiology, 7ed, vol. 517 . Eds. Murray, P. R., Baron, E. J., Pfaller, M. A., Tenover, F. C., Yolken, R. H. (Washington: ASM).
Kolemen, S., Cakmak, Y., Erten-Ela, S., Altay, Y., Brendel, J., Thelakkat, M., et al. (2010). Solid-state dye-sensitized solar cells using red and near-IR absorbing BODIPY sensitizers. Org. Lett. 12, 3812. doi: 10.1021/ol1014762
Kubota, Y., Uehara, J., Funabiki, K., Matsui, M. (2010). Strategy for the increasing the solid-state fluorescence intensity of pyrromethene–BF2 complexes. Tetrahedron Lett. 51, 6195. doi: 10.1016/j.tetlet.2010.09.106
Kubota, Y., Hara, H., Tanaka, K., Funabiki, K., Matsui, M. (2011). Synthesis and fluorescence properties of novel pyrazine–boron complexes bearing a β-iminoketone ligand. Org. Lett. 13, 6544. doi: 10.1021/ol202819w
Kumar, A. S., Katiyar, B., Agarwal, A., Chauhan, P. M. S. (2003). Current trends in antimalarial chemotherapy. Drugs Future 28, 243. doi: 10.1358/dof.2003.028.03.720299
Kumar, S., Misra, N., Raj, K., Srivastava, K., Puri, S. K. (2008). Novel class of hybrid natural products derived from lupeol as antimalarial agents. Nat. Prod. Res. 22, 305. doi: 10.1080/14786410701766349
Kumbhar, H. S., Gadilohar, B. L., Shankarling, G. S. (2015). Synthesis and spectroscopic study of highly fluorescent β-enaminone based boron complexes. Spectrochim. Acta A 146, 80. doi: 10.1016/j.saa.2015.03.044
Lass-Flörl, C., Mayr, A. (2007). Human Protothecosis. Hum. Prothothecosis Clin. Microbiol. Rev. 20, 230. doi: 10.1128/CMR.00032-06
Leimann, B. C. Q., Monteiro, P. C. F., Lazéra, M., Candanoza, E. R. U., Wanke, B. (2004). Protothecosis. Med. Mycol. 42, 95. doi: 10.1080/13695780310001653653
Linderman, R. J., Kirollos, K. S. (1990). Regioselective synthesis of trifluoromethyl substituted quinolines from trifluoroacetyl acetylenes. Tetrahedron Lett. 31, 2689. doi: 10.1016/S0040-4039(00)94673-2
Loudet, A., Burgess, K. (2007). BODIPY dyes and their derivatives: Syntheses and spectroscopic properties. Chem. Rev. 107, 4891. doi: 10.1021/cr078381n
Mahmud, T., Rehman, R., Hussain, R., Shafique, U., Anwar, J., Zaman, W., et al. (2011). Synthesis, Characterization and Antibacterial Activity of some Enaminones and their Complexes with Nickel (II) and Antimony (III). J. Chem. Soc. Pakistan. 33, 426.
Martins, M. A. P., Flores, A. F. C., Bastos, G. P., Zanatta, N., Bonacorso, H. G. (1999). Haloacetylated enol ethers. 14 [6]. Reaction of β‐alkoxyvinyl trifluoromethyl ketones with N‐methylhydroxylamine. J. Heterocycl. Chem. 36, 837. doi: 10.1002/jhet.5570360402
Martins, M. A. P., Cunico, W., Pereira, C. M. P., Flores, A. F. C., Bonacorso, H. G., Zanatta, N. (2004). 4-Alkoxy-1,1,1-trichloro-3-alken-2-ones: Preparation and applications in heterocyclic synthesis. Curr. Org. Synth. 1, 391. doi: 10.2174/1570179043366611
Martins, M. A. P., Emmerich, D. J., Pereira, C. M. P., Cunico, W., Rossato, M., Zanatta, N., et al. (2004). Synthesis of new halo-containing acetylenes and their application to the synthesis of azoles. Tetrahedron Lett. 45, 4935. doi: 10.1016/j.tetlet.2004.04.106
Martins, M. A. P., Peres, R. L., Fiss, G. F., Dimer, F. A., Mayer, R., Frizzo, C. P., et al. (2007). A solvent-free synthesis of β-enamino trihalomethyl ketones. J. Braz. Chem. Soc 18, 1486. doi: 10.1590/S0103-50532007000800006
Marvi, O., Fekri, L. Z. (2018). Citrus Juice: Green and natural catalyst for the solvent-free silica supported synthesis of β-enaminones using grindstone technique. Comb. Chem. High Throughput Screen 21, 19. doi: 10.2174/1386207321666180102115733
Michael, J. P., Koning, C. B., Hosken, G. D., Stanbury, T. V. (2001). Reformatsky reactions with N-arylpyrrolidine-2-thiones: synthesis of tricyclic analogues of quinolone antibacterial agents. Tetahedron 57, 9635. doi: 10.1016/S0040-4020(01)00964-4
Montalti, M., Credi, A., Prodi, L., Gandolfi, M. T. (2006). Handbook of Photochemistry. 3rd ed (Boca Raton, USA: CRC Press, Taylor & Francis).
Moyon, N. S., Mitra, S. (2011). Fluorescence solvatochromism in lumichrome and excited-state tautomerization: A combined experimental and DFT study. J. Phys. Chem. A. 115, 2456. doi: 10.1021/jp1102687
Nafisi, S., Saboury, A. A., Francois, N. K. J., Riahi, N. H. A. T. (2007). Stability and structural features of DNA intercalation with ethidium bromide, acridine orange and methylene blue. J. Mol. Struct. 827, 35. doi: 10.1016/j.molstruc.2006.05.004
Nagai, A., Chujo, Y. (2010). Aromatic ring-fused BODIPY-based conjugated polymers exhibiting narrow near-infrared emission bands. Macromolecules 43, 193. doi: 10.1021/ma901449c
Ni, Y., Lin, D., Kokat, S. (2006). Synchronous fluorescence, UV–visible spectrophotometric, and voltammetric studies of the competitive interaction of bis(1,10-phenanthroline) copper (II) complex and neutral red with DNA. Anal. Biochem. 352, 231. doi: 10.1016/j.ab.2006.02.031
Niu, S., Ulrich, G., Retailleau, P., Ziessel, R. (2011). Regioselective synthesis of 5-monostyryl and 2-tetracyanobutadiene BODIPY dyes. Org. Lett. 13, 4996. doi: 10.1021/ol201600s
Nucchi, M., Marr, K. A., Queiroz-Telles, F., Martins, C. A., Trabasso, P., Costa, S., et al. (2004). Fusarium infection in hematopoietic stem cell transplant recipients. Clin. Infect. 38, 1237. doi: 10.1086/383319
Pfaller, M. A., Diekema, D. J. (2004). Rare and Emerging Opportunistic Fungal Pathogens: Concern for Resistance beyond Candida albicans and Aspergillus fumigatus. J. Clin. Microbiol. 4419–4431. doi: 10.1128/JCM.42.10.4419-4431.2004
Pilli, R. A., Russowsky, D., Dias, L. C. (1990). Diastereoselective reduction of acyclic N-aryl-β-amino ketones. J. Chem. Soc. Perkin Trans. 1, 1213. doi: 10.1039/P19900001213
Poon, C. T., Lam, W. H., Wong, H. L., Yam, V. W. W. (2010). A Versatile photochromic dithienylethene-containing β-diketonate ligand: Near-infrared photochromic behavior and photoswitchable luminescence properties upon incorporation of a boron (III) center. J. Am. Chem. Soc 132, 13992. doi: 10.1021/ja105537j
Sheldrick, G. M. A. (2008). A short history of SHELX. Acta Crystallogr. Sect. A: Found. Crystallogr. 64, 112. doi: 10.1107/S0108767307043930
Sosnovskikh, V. Y., Usachev, B., II, Roschenthaler, G. V. (2002). Reactions of aromatic methyl ketimines with halonitriles as a new route to pyrimidines with two polyhaloalkyl groups. Tetrahedron 58, 1375. doi: 10.1016/S0040-4020(01)01231-5
Sozmen, F., Kolemen, S., Kumada, H., Ono, M., Sajib, H., Akkaya, E. U. (2014). Designing BODIPY-based probes for fluorescence imaging of β-amyloid plaques. RSC Adv. 4, 51032. doi: 10.1039/C4RA07754G
Tekdaş, D. A., Viswanathan, G., Topal, S. Z., Looi, C. Y., Wong, W. ,. F., Tan, G. M. Y., et al. (2016). Antimicrobial activity of a quaternized BODIPY against Staphylococcus strains. Org. Biomol. Chem. 14, 2665. doi: 10.1039/C5OB02477C
Toshiaki, M., Minoru, S. (1977). Reactions of α-alkyl- and α-aryl-substituted N-benzylideneamines with trihaloacetylating agents. Chem. Pharm. Bull. 25, 1230. doi: 10.1248/cpb.25.1230
Walsh, T. J., Groll, A., Hiemenz, J., Flemming, R., Roilides, E., Anaissie, E. (2004). Infections due to emerging and uncommon medically important fungal pathogens. Clin. Microbiol. Infect. 10, 48. doi: 10.1111/j.1470-9465.2004.00839.x
Wang, B., Li, P., Yu, F., Song, P., Sun, X., Yang, S., et al. (2013). A reversible fluorescence probe based on Se–BODIPY for the redox cycle between HClO oxidative stress and H2S repair in living cells. Chem. Commun. 49, 1014. doi: 10.1039/C2CC37803E
Xia, X., Wu, B., Xiang, G. (2008). Synthesis, structure and spectral study of two types of novel fluorescent BF2 complexes with heterocyclic 1,3-enaminoketone ligands. J. Fluor. Chem. 129, 402. doi: 10.1016/j.jfluchem.2008.01.019
Yoshii, R., Nagai, A., Tanaka, K., Chujo, Y. (2013). Highly emissive boron ketoiminate derivatives as a new class of aggregation‐induced emission fluorophores. Chem. Eur. J. 19, 4506. doi: 10.1002/chem.201203703
Keywords: β-enaminoketones, difluoro-organoboron complexes, antimicrobial agents, DNA-binding assays, photophysical properties
Citation: Rosa WC, Rocha IO, Rodrigues MB, Coelho HS, Denardi LB, Ledur PC, Zanatta N, Acunha TV, Iglesias BA and Bonacorso HG (2020) Novel Alkyl(aryl)-Substituted 2,2-Difluoro-6-(trichloromethyl)-2H-1,3,2-oxazaborinin-3-ium-2-uides: Synthesis, Antimicrobial Activity, and CT-DNA Binding Evaluations. Front. Pharmacol. 11:1328. doi: 10.3389/fphar.2020.01328
Received: 19 April 2020; Accepted: 10 August 2020;
Published: 02 September 2020.
Edited by:
Daniel Silva, Unilever, United KingdomReviewed by:
Stephen Alan Westcott, Mount Allison University, CanadaCopyright © 2020 Rosa, Rocha, Rodrigues, Coelho, Denardi, Ledur, Zanatta, Acunha, Iglesias and Bonacorso. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bernardo A. Iglesias, YmVybmFyZG9wZ3FAZ21haWwuY29t; Helio G. Bonacorso, aGVsaW8uYm9uYWNvcnNvQHVmc20uYnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.