 Brunilda Alushi1,2*†
Brunilda Alushi1,2*† Lavinia Curini1,3†
Lavinia Curini1,3† Mary Roxana Christopher1
Mary Roxana Christopher1 Herko Grubitzch4,5Ulf Landmesser1,4
Herko Grubitzch4,5Ulf Landmesser1,4 Amedeo Amedei3,6*†
Amedeo Amedei3,6*† Alexander Lauten1,2†
Alexander Lauten1,2†- 1Department of Cardiology, Charite´ Universitätsmedizin Berlin and German Centre for Cardiovascular Research (DZHK), Berlin, Germany
- 2Department of General and Interventional Cardiology, Helios Klinikum Erfurt, Erfurt, Germany
- 3Department of Experimental and Clinical Medicine, University of Florence, Firenze, Italy
- 4Berlin Institute of Health, Berlin, Germany
- 5Department of Cardiology, German Heart Centre Berlin (DHZB), Berlin, Germany
- 6Sod of Interdisciplinary Internal Medicine, Azienda Ospedaliera Universitaria Careggi (AOUC), Florence, Italy
Calcific aortic valve disease (CAVD) is the most frequent heart valve disorder. It is characterized by an active remodeling process accompanied with valve mineralization, that results in a progressive aortic valve narrowing, significant restriction of the valvular area, and impairment of blood flow.The pathophysiology of CAVD is a multifaceted process, involving genetic factors, chronic inflammation, lipid deposition, and valve mineralization. Mineralization is strictly related to the inflammatory process in which both, innate, and adaptive immunity are involved. The underlying pathophysiological pathways that go from inflammation to calcification and, finally lead to severe stenosis, remain, however, incompletely understood. Histopathological studies are limited to patients with severe CAVD and no samples are available for longitudinal studies of disease progression. Therefore, alternative routes should be explored to investigate the pathogenesis and progression of CAVD.Recently, increasing evidence suggests that epigenetic markers such as non-coding RNAs are implicated in the landscape of phenotypical changes occurring in CAVD. Furthermore, the microbiome, an essential player in several diseases, including the cardiovascular ones, has recently been linked to the inflammation process occurring in CAVD. In the present review, we analyze and discuss the CAVD pathophysiology and future therapeutic strategies, focusing on the real and putative role of inflammation, calcification, and microbiome.
Introduction
Calcific aortic valve disease (CAVD) is the most common valve disease worldwide (Nkomo et al., 2006). Epidemiological studies show that 2.8% of adults over 75 years old have some CAVD degree, and as many as 25% of adults over 65 years old have at least valvular sclerosis (Miller et al., 2011).
CAVD is a chronic process characterized by progressive fibrotic tissue remodeling and mineralization (Mathieu et al., 2015). Over the years, there is a disease continuum from sclerosis to chronic inflammation and finally leaflet calcification, culminating with severe stenosis. Human pathologic samples have shown that key features in the CAVD development include pathological concentrations of inflammatory cells and lipid species (Otto et al., 1994; O’Brien et al., 1996).
Several risk factors have been identified as relevant in the CAVD progression. Among others, male gender, high triglyceride levels, and smoking have been independently associated with early aortic valve replacement in the presence of CAVD. Levels of oxidative stress, higher in patients that are more exposed to certain risk factors than others, could be responsible for the starting of the sclerotic CAVD phase (O’Brien, 2006; Ferreira-Gonzalez et al., 2013).
Calcification is recognized as an active disease process driven by the native valvular interstitial cells (VICs) (Sun et al., 2013). These cells acquire an osteogenic and pro-calcific profile in response to different pathological stimuli, such as inflammatory mediators, endothelial damage, low-density lipoprotein (LDL) accumulation, reactive oxygen species (ROS), increase calcium/phosphate (Ca/Pi) levels, modified lipids, and cyclic stretch (Liao et al., 2008; Rajamannan et al., 2011; Rattazzi et al., 2014). The underlying process that ends with the ectopic mineralization of the aortic valve is still not entirely understood. Treatment options include operative valve replacement and percutaneous implantation of valve prosthesis (Lauten et al., 2013; Lauten et al., 2014; Daubert et al., 2016; Alushi et al., 2019; Wernly et al., 2019).
To date, there is no medical treatment available to prevent or reverse calcium deposition within the valve leaflets. Conventional cardiovascular drugs studied in clinical trials failed to influence the disease progression or reduce adverse outcomes, therefore additional research is required to understand the mechanisms of disease progression and, identify novel therapeutic targets (Cowell et al., 2005; Freeman and Otto, 2005; Rossebo et al., 2008; Chan et al., 2010).
CAVD Biology
Histological Structure of the Aortic Valve
The aortic heart valve consists of three leaflets that allow blood flow from the left ventricle to the aorta without regurgitation (Liao et al., 2008). Each leaflet has a trilaminar structure that is vital for the biomechanical properties of the aortic valve (Tseng and Grande-Allen, 2011; Lindman et al., 2016) as shown in Figure 1. Histologically, the leaflets are composed of three distinct layers: fibrosa, spongiosa, and ventricularis. Fibrosa and ventricularis are the external layers, facing the aorta and the left ventricle respectively. Fibrosa consists largely of collagen fibers with dispersed VICs, that are thought to be responsible for reinforcing the valvular structure (Dweck et al., 2012a).
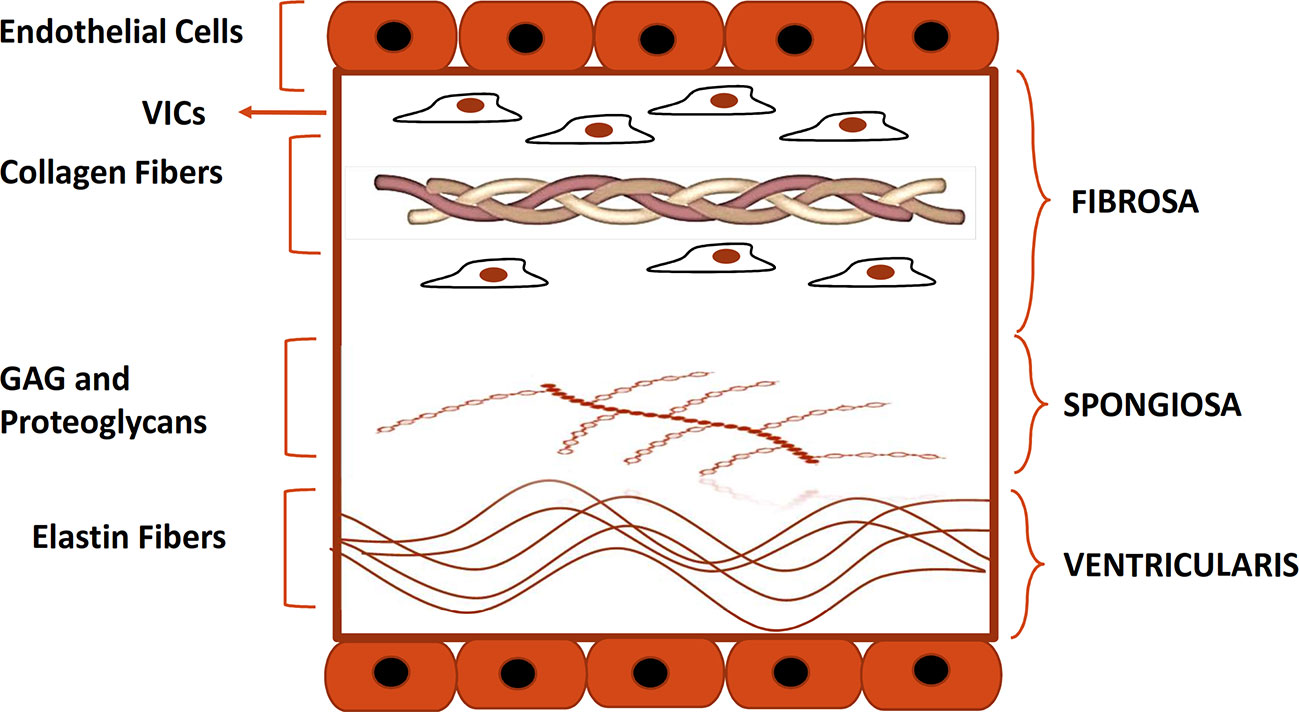
Figure 1 Histological structure of the aortic valve. Depicted is the histological structure of the healthy aortic valve. Fibrosa, spongiosa, and, ventricularis are the three layers that make up the structure of a normal aortic valve. The Fibrosa layer is composed of type I and III collagen fibers and contains also VICs. Spongiosa and ventricularis layers are respectively composed of GAG and proteoglycans and elastin fibers. Endothelial cells form a monolayer on each side of the cusp. GAG, glycosaminoglycans; VIC, valve interstitial cell.
The central layer, spongiosa, is rich in glycosaminoglycans (GAGs) and is responsible for absorbing some of the mechanical stress generated during the cardiac cycle.
The ventricularis is localized on the ventricular side of the leaflets and consists of collagen and elastin fibers (Chen and Simmons, 2011; Mathieu et al., 2015). The tissue composition of ventricularis provides more compliance and grants the apposition of free edge leaflet regions, thus preventing the backward blood flow into the left ventricle during diastole (Lindman et al., 2016). The normal valve leaflet is avascular and free of infiltrating lymphocytes or monocytes.
Pathobiology of CAVD
The CAVD pathophysiology is schematically depicted in Figure 2. The process of progressive fibrocalcific remodeling of the aortic valve is multifactorial, involving genetic predisposition, endothelial shear stress, chronic inflammation, lipid deposition, and valve calcification (Freeman and Otto, 2005; Perrot et al., 2019; Thériault et al., 2019). In its early stages, CAVD resembles atherosclerosis. The existence of shared risk factors for the disease development and the correlation between the severity of CAVD and that of coronary calcification suggest a shared disease pathway, at least in the initial phases of both diseases.
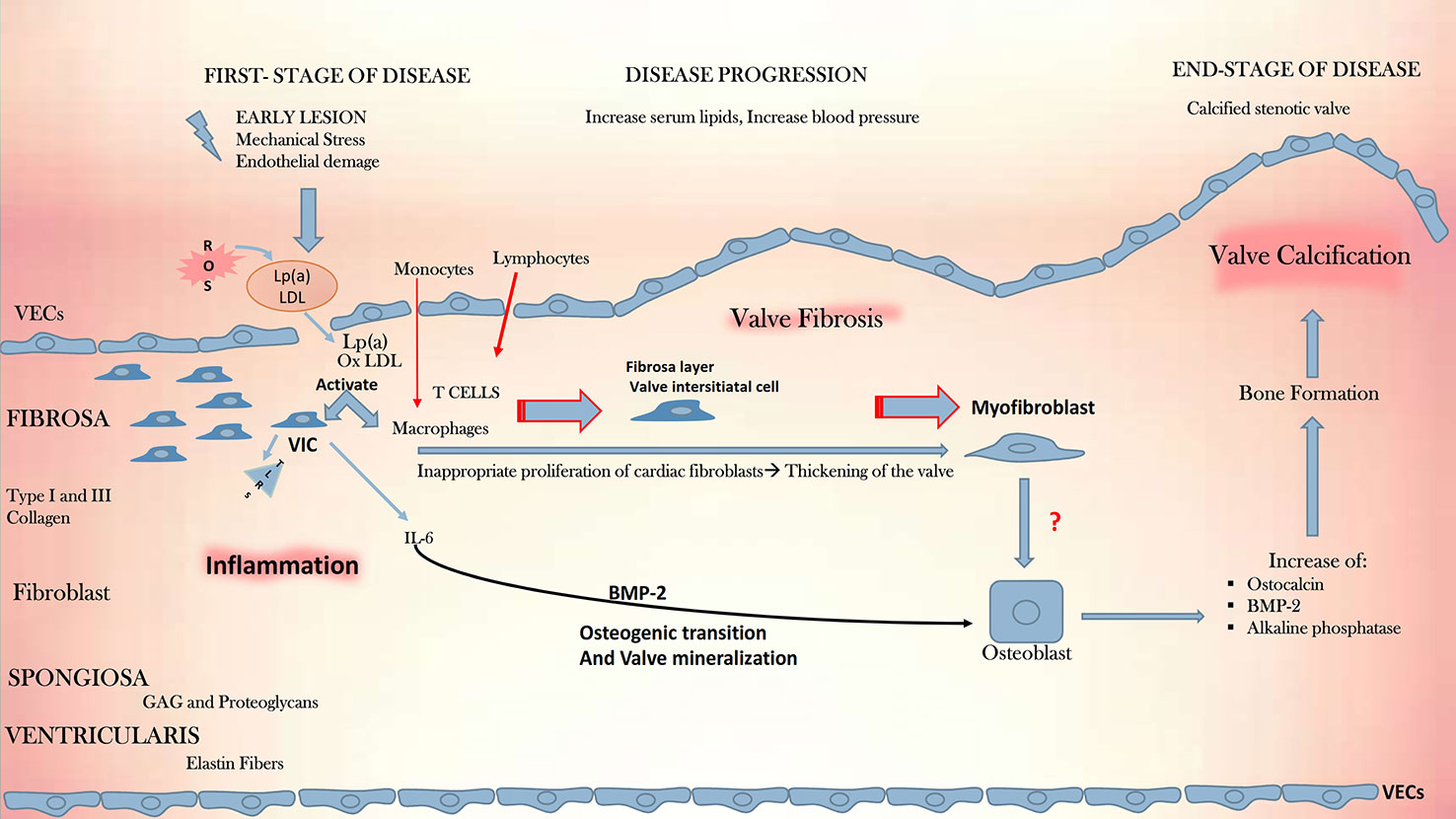
Figure 2 CAVD development. Depicted is the pathophysiologic cascade leading to CAVD. Initial lesions, such as mechanical stress, endothelial damage and, production of ROS, initiate an ox-LDL-mediated inflammation in the endothelium and promote the activation of macrophages and quiescent VICs. These start to express TLRs and the cytokine pro -inflammatory IL-6, which plays a fundamental role in inflammation, together with macrophages and T cells. IL-6 mediates mineralization through the BMP-2 signal, driving the osteogenic transition and culminating with the aortic valve calcification. CAVD, calcific aortic valve disease; GAG, glycosaminoglycans; IL-6, interleukin 6; Lp(a), lipoprotein a; ox-LDL, oxidized low-density lipoprotein; ROS, reactive oxygen species; TLRs, toll-like receptors, VECs, valve endothelial cells; VICs, vascular interstitial cells, BMP-2, bone morphogenetic protein 2.
Alike atherosclerosis, the triggering event in CAVD is endothelial damage resulting from increased mechanical stress and reduced shear stress. Physiologic fluid shear stress (FSS) contributes to valve homeostasis, whereas altered shear stress, on the contrary, stimulates the endothelial upregulation of vascular cell adhesion molecule-1 (VCAM-1) and intracellular adhesion molecule-1 (ICAM-1) (Sucosky et al., 2009; Sun et al., 2013). The endothelial damage following the altered shear stress allows the infiltration of lipids, especially LDL and lipoprotein (a) [Lp(a)], starting the recruitment of inflammatory cells into the leaflets. Later appears the formation of focal subendothelial plaque-like lesions constituted from LDL, Lp(a) and infiltrates of inflammatory cells which interact in processes that release ROS causing LDL oxidation. In time, the expansion of this inflammatory process triggers the release of factors that cause VICs to acquire an osteogenic profile, with the formation of microcalcifications initiating leaflet mineralization (Otto et al., 1994; O’Brien et al., 1996). The two disease processes differ in the later disease phases: while in atherosclerosis, smooth muscle cells are the active players in the chronic inflammation within the plaque, in CAVD fibroblasts are involved in the prominent mineralization process and only with a small number of smooth muscle cells is present.
The hallmark of the early CAVD stages is inflammation, characterized by the activation of the leaflet endothelium via enhanced expression of cell adhesion molecules (VCAM-1, ICAM-1) (Sun et al., 2013; de Sousa et al., 2017). Disease initiation involves the activation of VICs, recruitment of immune cells, and subsequent sclerosis of the valve leaflets owing to fibrosis and formation of calcific nodules. All these phenomena start and interest more the fibrosa layer within the valve. The first macroscopic change in the leaflets, seen as microcalcifications, or focal thickening with preserved valve function, is nominated aortic valve sclerosis, nonetheless, the initiating events likely occur much earlier. The activation degree of the immune system seems to be different on different examined valve areas. Interestingly, the thickening process and the formation of calcium nodules accompanied by neo-angiogenesis, are both localized near the aortic surface of the leaflets (Cote et al., 2013).
The final disease, namely calcific aortic stenosis, is characterized by large calcified noduli on the leaflet surface, that protrude into the sinuses of Valsalva, hindering the leaflet mobility (Rajamannan et al., 2011). In this final phase the leaflets are infiltrated by immune cells and concomitantly angiogenesis occurs, along with deposition of lipids, proteoglycans, and cell debris (Chen and Simmons, 2011). Finally, the calcification of the valvular matrix leads to an increased stiffness and obstruction of the blood flow (Figure 2). With an orifice under 1 cm2, versus 2.5–4.5 cm2 observed in a normal valve, the stenotic valve generates a pressure gradient of over 40 mmHg, categorized as severe stenosis with an indication to valve replacement (Zigelman and Edelstein, 2009; Rutkovskiy et al., 2017).
Impact of Inflammation in CAVD Remodeling
Inflammation is the primary response of innate immunity and occurs after endothelial damage with its activation and lipid deposition. The innate immune response, with both its components, cellular and humoral responses are implicated in this process. As response to an injury induced by foreign organisms, dead cells or physical irritants, the innate immune system represents the first response to external or internal triggers and initiates the process of tissue regeneration (Hulin et al., 2018; De Almeida et al., 2019). Once the inflammation process has been triggered, it will proceed along a certain course of events until the inflammatory stimulus is eradicated and the healing mechanism can begin. However, if the inflammatory source cannot be eliminated, inflammation will progress, varying in intensity over time (Hakansson and Molin, 2011).
Histological samples of human CAVD valves are characterized by calcified areas rich in lymphocytes, macrophages, and osteoblast-like cells (Hulin et al., 2018). The inflammatory process can be acute or chronic (Pahwa and Jialal, 2019). Cote´ et al. showed that chronic inflammatory infiltrates, composed of the CD45+ leukocytes, CD68+ macrophages, and a few scattered CD3+ T cells, were present near the calcified areas. Moreover, the chronic inflammatory infiltrates in the aortic valve were independently associated with several indices of remodeling, suggesting that inflammation may participate in mineralization and the fibrotic process (Cote et al., 2013). A significant association between the degree of aortic valve inflammation and the development of calcification has been previously reported (and recently confirmed) using 18F-fluorodeoxyglucose positron emission tomography. In these studies, a high degree of inflammation and calcification was documented in patients with severe CAVD, with the latter being the predominant pathogenic process (Marincheva-Savcheva et al., 2011; New and Aikawa, 2011; Dweck et al., 2012b). This could in part explain the failure of statin therapy in slowing the CAVD progression (Cowell et al., 2005; Rossebo et al., 2008).
Previously thought to be a passive disorder, CAVD is now recognized as an active process, whereby endothelial progenitor cells (EPCs) and inflammatory cells promote tissue remodeling (Yip and Simmons, 2011). In summary, mechanical shear stress and atherogenic factors activate VICs and initiate the recruitment of inflammatory cells. The following remodeling of extracellular matrix with leaflet stiffening and valvular dysfunction causes further mechanical stress that maintains the self-perpetuating cycle of endothelial dysfunction. As suggested by studies with molecular imaging, the continuous maintenance of this shear stress-inflammation cycle could result irreversibly to calcium deposition and finally to severe CAVD (New and Aikawa, 2011).
Role of Lipids
Over recent years, several studies have highlighted the central role of ox-LDL as an activator of inflammation, both in atherosclerosis and in CAVD (Cote et al., 2008). In atherosclerotic plaques, ox-LDL activates the inflammatory cells and the production of cytokines promoting tissue remodeling and disease progression (Hansson, 2005). Existing evidence of valve infiltration by ox-LDL in cases of CAVD, supports the concept that the development of valve calcification may be, at least in part, influenced by ox-LDL and, that an association exists between high plasma levels of ox-LDL and CAVD (Mohty et al., 2008). Although having some similarities with atherosclerosis, CAVD differs from it since the aortic valve has some inherent properties that differ from the vascular wall. Indeed, the cellular organization, as well as the hemodynamic stress imposed upon the aortic valve, vary from that of the arteries. Furthermore, the phases that characterize the pathological process occurring within the valve, from early inflammation stages to the calcification ones, are more numerous and qualitatively different from atherosclerosis, as shown in Figure 2. Most importantly, statins, although efficient in reducing adverse events in patients with atherosclerosis, are inefficient in CAVD (Cowell et al., 2005; Rossebo et al., 2008; Chan et al., 2010).
Role of the Immune Response in CAVD
The immune system, with innate and adaptive responses, plays a central role in the development of different chronic disorders, including atherosclerosis and CAVD. Evidence suggests that inflammation, as the primary response of the innate immunity, promotes the mineralization of VICs in response to several factors. Adaptive immunity, on the other hand, could play a role in instrumenting the immune response (Cote et al., 2013). In this regard, experimental studies have shown that the increased leukocyte density in mineralized aortic valves correlates with faster disease progression (Mathieu et al., 2015).
Innate Response in CAVD
The innate immune response in CAVD |+|}_–be initially triggered by several oxidized lipid species, through both the toll-like receptors (TLRs) and the nuclear factor-κ B (NF-κB) pathway. VICs express TLRs, known to play a key role in inflammation and initiation of antigen-specific adaptive immune responses. TLRs can be activated by several lipid species, especially ox-LDL, with the signal passing through the recruitment of specific adaptor molecules to the activation of the transcription factor NF-κB (Kawasaki and Kawai, 2014; Garcia-Rodriguez et al., 2018). In this way, TLRs signal inflammation through the NF-κB pathway and are vital for maintaining tissue homeostasis.
NF-Kappa B Pathway
The NF-κB plays an important role in signal integration by responding to mediators of endothelial injury. VICs actively participate in the regulation of inflammation by producing a high level of cytokines, and the NF-κB pathway contributes to a stimulus-dependent and cell-type-specific manner. The NF-κB activation (canonical and non-canonical) pathway is mediated by different extracellular signals including angiotensin II, ox-LDL, CD40 ligand, advanced glycation end-products, and inflammatory cytokines. The canonic pathway is activated, among others, by tumor necrosis factor alpha (TNF-α) and interleukin-1β (IL-1β). Angiotensin II-induced ROS generated also on mineralized aortic valves, are involved in the activation of the NF-κB canonic pathway (Jamaluddin et al., 2007). The non-canonical pathway is activated, among others, by vasoactive peptides, ox-LDL, activated CD40 receptor, B cell-activating factor (BAFF), lymphotoxin β (LTβ) monocyte released cytokines, or advanced glycation end-products (Xiao et al., 2004; Morrison et al., 2005). In summary, activated NF-κB controls the initiation of vascular inflammation, through “leucocyte adherence” and “chemotaxis” and is a crucial signaling integrator of vascular injury.
Cytokines
One important target of the NF-kB pathway is IL-6. Highly expressed in valves with severe CAVD, IL-6 is a pleiotropic cytokine secreted by multiple vascular cell types, such as macrophages, lymphocytes, fibroblasts, endothelial cells, and smooth muscle cells. It mediates local vascular monocyte activation and protection from ROS-induced cellular stress via the downstream transcription effector signal transducer and activator of transcription 3 (STAT3) (Brasier, 2010; El Husseini et al., 2014; Rahat et al., 2016; Huang et al., 2019). By controlling the monocyte activation vial the IL-6 pathway, NF-κB mediates the systemic acute phase response and plays a central role in the initiation and maintaining of the vascular inflammation (Brasier, 2010; Perez et al., 2019).
IL-1β is another intriguing cytokine involved in the valve calcification process. Its levels are indeed increased in the stenotic aortic valve, making IL-1β subject to numerous research studies in the last decade (Isoda et al., 2010; Nadlonek et al., 2013; Lee and Choi, 2018). The IL-1β enhances the expression of matrix metalloproteinases (MMPs), a group of enzymes responsible for the degradation of extracellular matrix proteins. The MMPs exacerbate the process of valvular stenosis and activate the NF-κB pathway, leading to the production of several cytokines, such as IL-6, IL-8, and MCP-1 (monocyte chemoattractant protein-1) involved in both pathological processes, namely atherosclerosis and CAVD (Deshmane et al., 2009).
Finally, the IL-1 receptor agonist (IL-1Ra) plays a protective role in valvular disease, as its deficiency is closely associated with inflammatory cell infiltration and valve thickening (Isoda et al., 2010). Using in vitro and in vivo models, Zeng et al. elucidated the role of IL-37, an anti-inflammatory member of the IL-1 family. IL-37 attenuates the expression of bone morphogenetic protein 2 (BMP2) and alkaline phosphatase (ALP) enzyme, inhibiting the osteogenic process. In CAVD, the expression levels of IL-37 are very low, consequently, BMP2 is free to promote VICs calcification by ALP expression leading subsequently to aortic valvular thickening. Confirming the protective role of IL-37, in vivo experiments showed that mice expressing this human cytokine, displays significantly lower BMP-2 levels and a lower degree of aortic valve thickening (Zeng et al., 2017).
Adaptive Response in CAVD
The key role of lymphocytes in CAVD and the presence of T-lymphocyte clonal expansion in stenotic valves has been highlighted by various studies (Wallby et al., 2002; Mazzone et al., 2004; Mazur et al., 2018). In a study involving patients with severe CAVD, Wu et al. observed the presence of CD8 +/CD28− T cells near the mineralized nodules of the aortic valve and a higher prevalence of circulating CD3+ T cells, namely the subset of CD8+ and CD57+ T cells expressing HLA-DR, in subjects with CAVD (Wu et al., 2007). CD8+/CD28− T cells play a crucial role in CAVD, as demonstrated by the correlation between the degree of clonal expansion and the severity of valve calcification. Furthermore, these cells play a crucial role in both activation and differentiation of memory effector status among circulating T cells. Winchester et al., investigated the composition of the lymphocytic infiltration in patients with (bi-and tri-cuspid CAVD). They showed an aggregate of infiltrating lymphocytes containing CD4+ and CD8+ T cells, with a preponderance of CD4 T cells. The clones shared between blood and the valve were found in the memory-effector CD8+CD28− T cell subset, demonstrating the trafficking of members of the same T cell clone between the peripheral circulation and the valve. Importantly, the proportion of activation of peripheral blood T-lymphocytes strongly correlated with the degree of valve calcification, indicating that the extent of these events is somehow related to CAVD severity (Winchester et al., 2011).
This data suggests that in patients with CAVD, an adaptive systemic immune response is occurring, coupled with the valvular lymphocytic infiltration, probably triggered by recognition of antigens expressed in the valve. The valvular events that lead to an immune response, such as intracellular pathogens, or self-antigens, remain unclear. One hypothesis is that in response to shear stress the VICs express stress-induced molecules which could be perceived as antigens and generate an immune response.
Mineralization
At some point during the CAVD progression, VICs start to produce a calcified matrix and enter an osteogenic phase. The mechanisms of this switch are still poorly understood. One hypothetical pathway involves IL-6 and the tumor necrosis factor ligand superfamily member 11 (also called receptor activator of NF-κB ligand—RANKL). IL-6 has been shown to induce the expression of RANKL in bone cells, which activates its cognate receptor RANK that finally activates VICs to produce extracellular matrix and might therefore promote matrix calcification (Wada et al., 2006). This could be a hypothetical pathway, which through IL-6-RANKL overexpression promotes the osteogenic reprogramming of VICs (Kaden et al., 2004). Another pathway involving the expression of BMP2 via inflammatory cytokines and oxidized lipid derivatives was shown to induce osteogenic reprogramming in several cell types including VICs (Shao et al., 2005).
Genetic Factors Implicated in CAVD
Numerous genetic factors are implicated in the CAVD pathogenesis (LaHaye et al., 2014). Two recent genome-wide association (GWAS) studies showed that a genetic variation in the LPA locus, mediated by levels of Lp(a), is involved in both atherosclerotic disease and CAVD and that several pathways are shared by both conditions. Lp(a) and non-high-density lipoprotein cholesterol as shared risk factors contributed to the frequent co-existence of these disorders (Thanassoulis et al., 2013; Helgadottir et al., 2018).
A recent transcriptome-wide association study (TWAS) identified PALMD (palmdelphin), a gene that promotes myoblast differentiation and muscle regeneration, as an additional key gene in CAVD. The study showed that lowered expression levels of PALMD mRNA in valve tissues are associated with risk alleles for CAVD and higher disease severity (Nie et al., 2017; Theriault et al., 2018). Recently, Theriault et al. identified some additional susceptibility genes in patients with CAVD. Using both techniques, GWAS and TWAS, IL-6, ALPL (alkaline phosphatase), and NAV1 (neuron navigator 1) emerged as important contributors involved not only in pulse and blood pressure modulation—two important factors associated with blood flow turbulence, already identified as risk factors for CAVD development—but also in the valve mineralization process. Moreover, the ALPL gene codes for tissue-nonspecific alkaline phosphatase, a crucial enzyme involved in mineralization. This gene was present in calcified aortic valves contrary to non-calcified valves (Thériault et al., 2019).
All these findings taken together constitute a new step toward the further elucidation of the CAVD pathogenesis and may open new paths for the development of innovative therapeutic approaches.
Gender as a Risk Factor for CAVD
The role of gender as a biological variable, especially that of the male sex as a risk factor for cardiovascular diseases, including CAVD, has been long established (Carroll et al., 1992; Institute of Medicine Board on Population H and Public Health P, 2012). However, data regarding the significance of male sex in CAVD is lacking. Indeed, most studies addressing sex-specific differences in CAVD focus on conditions caused by CAVD, such as ventricular dysfunction, rather than CAVD itself (Dobson et al., 2016). It is mostly unclear whether the onset and progression of CAVD are inherently different in men and women, due to genetic and cellular-scale sex differences, or whether the initial disease process is common in both sexes but undergoes subsequent differentiation after (Porras et al., 2017). Aggarwal et al. reported that male gender correlates with an higher incidence of CAVD, as men displayed higher levels of calcification for the same degree of aortic stenosis when compared to women (Aggarwal et al., 2013). Another study showed significant differences between stenotic valves in males and females, where the latter were more prone to fibrosis. (Sharma and Eghbali, 2014) Evidence points towards genetic and epigenetic differences, specifically the differential production of miRNAs, being the origin of these sex-specific differences (Sharma and Eghbali, 2014).
Epigenetic Regulation of CAVD
The remarkable developments in the field of epigenetics could offer answers regarding the link between environmental and proatherogenic factors, induction and perpetuation of inflammation, and CAVD development. The four major epigenetic mechanisms, collectively known as “epigenome”, include non-coding regulatory RNAs (ncRNA), DNA methylation, histone modifications, and chromatin remodeling (Murr, 2010). Studying the link between epigenome and CAVD could be clinically relevant since epigenetic markers could represent non-invasive biomarkers for monitoring CAVD initiation and progression and predicting prognosis.
ncRNAs, a group of single-stranded cellular RNAs made up of 18–25 nucleotides, represent a new area of interest in the field of human biology. These molecules, normally not coding for proteins, were previously regarded as “dark matter” since their function was largely unknown. Increasingly discovered to play a central role in the regulation of various molecular pathways and cell differentiation, ncRNAs are categorized in short, medium, or long (lncRNAs) based on their transcript size (Aryal et al., 2014). MicroRNAs (miRNAs) are the most well-studied ncRNAs, involved in cell development, proliferation, lipid metabolism, cancer metabolism, and angiogenesis (Palazzo and Lee, 2015). They regulate gene expression after transcription, by translational repression or transcript degradation. The role of miRNAs and especially lncRNAs in regulating atherosclerotic processes has been investigated in several studies suggesting that some miRNAs may be important inducers of proinflammatory pathways and others stimulators or inhibitors of calcification in the human aortic VICs (Gosev et al., 2017). Among others, miRNA-30b, miRNA-138, and miRNA-204 can suppress the differentiation of mesenchymal cells into osteoblasts whereas miRNA-29b and miRNA-214 promote calcification (Zhang et al., 2014; Wang et al., 2015; Fang et al., 2018; Lu et al., 2019; Zheng et al., 2019). miRNA-138 was indicated as a suppressor of osteoblastic differentiation of human aortic VICs by targeting FOXC1, representing, therefore, an inhibitor of VIC osteogenic differentiation during the development of CAVD (Lu et al., 2019). In 2017, the role of miRNA-29b as a promoter of valvular calcification, through the TGF-ß3/Smad3 and wnt3/ß-catenin pathways was reported (Fang et al., 2018) whereas in 2019, miRNA-214 was identified as a promoter of the human aortic VICs calcification by accelerating inflammatory chain reactions through MyD88/NF-κB signaling (Zheng et al., 2019).
Another epigenetic mark, the alteration of DNA methylation, could have a role in the osteogenic differentiation of aortic VICs by downregulating Notch/drosophila/homolog 1/translocation-associated (NOTCH1), a regulator of calcification-related gene networks in human valvular endothelium (Theodoris et al., 2015; Hadji et al., 2016). Markers of histone modification have also been reported to have a pro-inflammatory and osteogenic role in CAVD development. They participate in the shear-stress induced proinflammatory pathways via alteration of the silent information regulator-two (SIRT) gene expression (Roos et al., 2014).
In summary, increasing evidence supports the role of epigenetic factors as regulators of key processes underlying valvular tissue remodeling and participators in the landscape of phenotypical changes occurring in CAVD. Understanding the epigenetic mechanisms involved in the initiation and progression of CAVD will not only help to enlighten the pathology, but since these markers are potentially reversible, it could offer important targets for diagnostic and therapeutic interventions.
The Putative Role of the Microbiome
Herein we hypothesize a putative role of the microbiome in the development and progression of CAVD, either via stimulation of immune response and valve calcification or via direct valvular damage caused by specific bacterial taxa. Different hints point to a role microbiome in the promotion of CAVD and its deterioration to severe calcified aortic stenosis. Some known risk factors for CAVD, such as age, male sex, hypertension, and diabetes mellitus, have a known relationship to the microbiome. In fact, patients with type II diabetes mellitus (T2DM), which is associated with the gut microbiota dysbiosis, are at higher risk for developing CAVD (Harsch and Konturek, 2018). Tissue histopathological studies demonstrated more calcification in tissue samples from patients with T2DM compared to non-diabetic patients (Mosch et al., 2017). Furthermore, the presence of metabolic syndrome is associated with faster disease progression and worse outcome in patients with aortic stenosis (Katz et al., 2009). Here we discuss in detail the involvement of oral and gut microbiome (GM) in pathophysiological processes that promote cardiovascular diseases, including CAVD.
Oral Microbiome
The oral microbiome represents, after the GM, the second-largest microbial community in humans and plays a fundamental role in maintaining the oral cavity homeostasis and preventing the development of several diseases (Jia et al., 2018; Deo and Deshmukh, 2019). The involvement of the oral microbiome in pathologies such as obesity, diabetes, cancer, and cardiovascular diseases is well documented (Gao et al., 2016; Eberhard et al., 2017; Long et al., 2017; Yang et al., 2019). Different studies suggest that oral pathologies with oral microbiome alteration, have a tight link with heart diseases; for example, the gingival ulceration in periodontitis causes bacteremia and could induce the formation of atherosclerotic plaques (DeStefano et al., 1993; Bartova et al., 2014). Increasing evidence suggests a close association between oral microbiome alteration and CAVD, demonstrating the presence of oral bacteria in the valvular tissue by using a polymerase chain reaction (PCR) (Nakano et al., 2006). Nomura et al., investigated the mechanisms through which Streptococcus mutans (S. mutants), the major pathogen responsible for dental caries, colonizes heart valves constituting a potential virulence factor for the development of infective endocarditis. They demonstrated that specific strains of S. mutants have selective virulence for infectious endocarditis (Nomura et al., 2014; Oliveira et al., 2015). There is an increasingly accepted hypothesis that bacterial endocarditis is one of the causes of aortic valve calcification, supported by in vivo studies showing larger calcifications in animals inoculated with endocarditis-related pathogens.(Cohen et al., 2004)
Gut Microbiome
The composition of the human microbiome is specific to each individual and the study of the GM is increasingly focusing on its role in the physiology of the host organism in during phases of health and disease (Blander et al., 2017). Changes in the composition of GM are involved in several diseases including atherosclerosis, hypertension, heart failure, chronic kidney disease, obesity, diabetes, inflammatory bowel disease, and colon cancer (Amedei and Barcelo-Coblijn, 2019). Growing evidence suggests that intestinal bacteria play an essential role in cardiovascular diseases. GM metabolites such as choline, betaine, and trimethylamine N-oxide (TMAO), are the subject of research studies investigating the correlation between the intestinal microbiome and the calcification of the aortic valve. Diets based on red meat, eggs, and dairy products are rich in choline, lecithin, and carnitine, constituting, therefore, a potential source of TMAO. Recent metabolomic studies indicate that fasting TMAO, choline, and phosphatidylcholine plasma levels are related to high risk of cardiovascular diseases. Furthermore, carnitine has been indicated as an independent predictor of major adverse cardiovascular events, with TMAO as the main driver behind the risk association (Velasquez et al., 2016; Canyelles et al., 2018). The idea that gut microbiota could contribute to CAVD is new. Very recently Kocyigit et al. reported a significant association between gut microbiota metabolites and CAVD (Kocyigit et al., 2017) whereas Liu et al. showed that patients with CAVD and those with coronary artery disease suffer from different gut microbial dysbiosis (Liu et al., 2019). The next section discusses the putative link between the GM and CAVD development.
The Role of the GM in Adaptive Immunity
Several studies indicate a very close connection between the GM and adaptive immunity. Bacterial metabolites function as a link between the commensal microbiome and the immune system, by affecting the shifting of the immune response between pro- and anti-inflammatory pathways. Hypothetically, the gap between the microbiome and immune system could be filled by short-chain fatty acids (SCFAs), generated by bacterial fermentation of dietary fiber in the intestinal lumen (Russo et al., 2016). SCFAs such as acetate, propionate, and butyrate act as important inductors of regulatory T (Treg) cell differentiation and reinforce the gut barrier function by expressing the transcription factor Foxp3 and consequently suppressing inflammatory responses in the intestine (Arpaia et al., 2013). A diet rich in SCFAs might antagonize several immunological defects, therefore GM-derived metabolites could offer a therapeutic advantage for patients suffering from various immunological conditions (Luu et al., 2019).
Significant links between fungal- and bacterial-induced cytokine responses and specific gut bacterial species were identified through the Human Functional Genomics Project. For instance, the production of interferon-γ (IFN-γ) and TNF-α is strongly associated with the GM (Schirmer et al., 2016). The fermentation of bacterial SCFAs acts through activation of G-protein coupled receptors by modulating the activity of intestinal epithelial cells and leukocyte life cycle. Furthermore, due to their direct effect on lymphocytes and by activating macrophages and dendric cells, SCFAs can induce a T-lymphocyte tolerogenic profile, acting as a link between the microbiome and the immune system (Correa-Oliveira et al., 2016).
In summary, the role of adaptive immunity in CAVD has been explored, but the valvular events that lead to an immune response, such as intracellular pathogens or self-antigens, remain largely undetermined. Alike atherosclerosis, one could hypothesize that the presence of a certain type of gut or mouth microbiome could lead to the presence of bacterial DNA inducing valvular specific antigens that could trigger an immune response. The putative interaction between the microbiome and immune response leading to CAVD development is schematized in Figure 3.
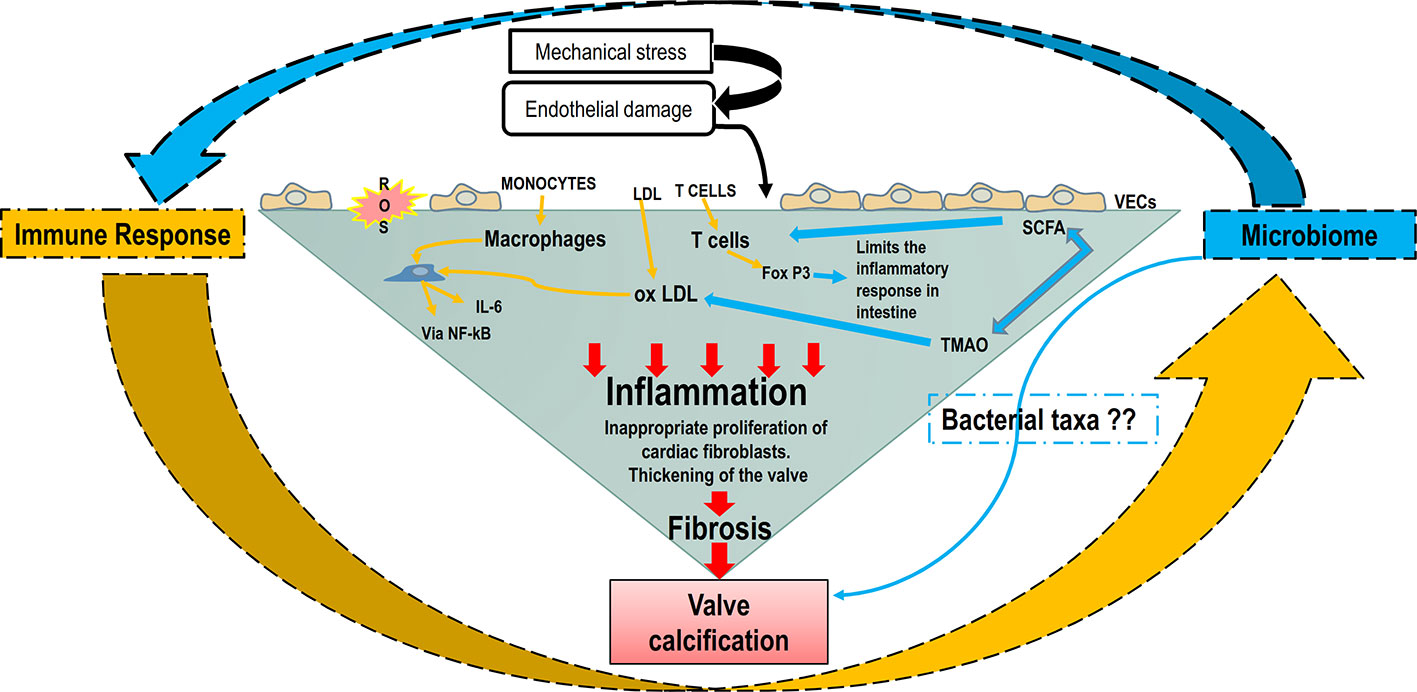
Figure 3 The putative interaction between the microbiome and immune response in CAVD development. Increased shear stress, genetic factors, and oxygen reactive species cause the endothelial damage that allows ox-LDLs, immune and inflammatory cells to infiltrate the valve. Macrophages and ox-LDLs stimulate VICs inflammation through NF-κB/IL-6 signaling pathway. The microbiome and microbial metabolites can modulate the immune system through the intense effect of SFCAs on T cells’ differentiation. Furthermore, the bacterial metabolite TMAO can induce ox-LDL and maintain the inflammatory loop. IL-6, interleukin 6; NF-κB, nuclear factor κ-light-chain enhancer of activated B cells; ox-LDL, oxidized low-density lipoprotein; SCFA, short-chain fatty acid; TMAO-Trimethylamine-N oxide; VECs, valve endothelial cells; VICs, vascular interstitial cells.
Novel Potential Therapeutic Strategies
Currently, no drug strategies can prevent or reverse CAVD and valve replacement remains the only treatment option. Patients with severe CAVD undergo valve replacement either with surgery or via percutaneous transcatheter procedures. Drugs already used to treat vascular complications might also improve CAVD outcomes, but the mechanisms of valve calcification differ from other vascular conditions (Hutcheson et al., 2014).
A randomized trial of intensive lipid-lowering therapy failed to stop ore reverse the progression of CAVD. However, the possibility that this specific pharmacological treatment induces a small reduction in disease progression or in the main clinical endpoints cannot be excluded (Cowell et al., 2005; Rossebo et al., 2008). Anti-inflammatory agents represent a promising therapeutic strategy that requires additional research in CAVD patients (Hurle et al., 2013). Due to the role of angiotensin II in CAVD development, inhibitors of angiotensin-converting enzyme (ACE-I) have also been explored. Notably, no significant difference in disease progression was observed in a retrospective cohort of more than 200 patients. The benefits of ACE-I are mostly related to their effect on left ventricular remodeling.
Human monoclonal antibodies have been used successfully in many conditions. In 2015 Lerman et al. found that denosumab, a human monoclonal antibody targeting the receptor activator of nuclear factor-κB ligand, may reduce valvular calcium deposition to basal levels (Lerman et al., 2015). Furthermore, denosumab reduced calcium deposition in the aorta, although the mechanisms by which it affected ectopic calcification, must be further examined. (Helas et al., 2009) Nonetheless, this field remains attractive and promising.
After GWAS and TWAS studies discovering several genes implicated in the CAVD pathogenesis, genetic therapy could also be a possible therapeutic development. A growing body of experimental evidence supports the role of epigenetic factors such as miRNAs and markers of DNA methylation, as regulators of key processes underlying valvular tissue remodeling and participators in the landscape of phenotypical changes occurring in CAVD. Since these markers are potentially reversible, they could offer important targets for diagnostic and therapeutic interventions.
The recently introduced, tissue-engineered heart valves [tissue engineered heart valves (TEHVs)] may constitute a valid alternative to traditional strategies of valve replacement. In TEHVs, a decellularized scaffold is seeded with patient’s cells subjected to an appropriate series of stimuli able to promote cellular activity. TEHVs could offer the advantages of biocompatibility and longevity, while preventing pathological tissue responses and regurgitation. Additionally, TEHVs could allow VICs to produce their own extracellular matrix, thus displaying the capacity for growth and remodeling, which is critical for pediatric patients. TEHVs can be produced from synthetic or natural materials (Berry et al., 2010).
Conclusions
Despite its high prevalence and associated mortality, there are no drugs to prevent or cure CAVD. Therefore, the development of new therapeutic strategies capable of counteracting the CAVD development is crucial.
Being a multifactorial disease, CAVD is characterized by a complex pathobiology that involves environmental and genetic factors, immune-molecular pathways, hemodynamic factors, and shear stress, all intertwined in the systemic district. Several of the many steps of disease progression have been clarified but some others, like the critical switch from inflammatory-fibrotic to osteogenic program, or the putative role of microbiota, remain still unclear. The quickly evolving field of epigenetic regulation of CAVD, involving especially miRNAs and lncRNAs, could offer novel potential biomarkers for the development of new diagnostic and therapeutic interventions. Furthermore, the microbiome could play a role in promoting and perpetuating CAVD by inducing the production of endogenous or exogenous antigens via the activation of inflammatory pathways. The elucidation of these conundrums will help identify new disease targets, host or microbial, and support the design of new potential therapeutic strategies to prevent or reverse the effects of CAVD.
Author Contributions
BA, LC, AL, and AA were involved in the study concept and design. BA, LC, and MC drafted the manuscript. AL, UL, HG, and AA revised the manuscript for important intellectual contents. All authors had access to all the data, including figures, and approved the manuscript for final submission.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aggarwal, S. R., Clavel, M. A., Messika-Zeitoun, D., Cueff, C., Malouf, J., Araoz, P. A., et al. (2013). Sex differences in aortic valve calcification measured by multidetector computed tomography in aortic stenosis. Circ. Cardiovasc. Imaging 6 (1), 40–47. doi: 10.1161/CIRCIMAGING.112.980052
Alushi, B., Beckhoff, F., Leistner, D., Franz, M., Reinthaler, M., Stahli, B. E., et al. (2019). Pulmonary Hypertension in Patients With Severe Aortic Stenosis: Prognostic Impact After Transcatheter Aortic Valve Replacement: Pulmonary Hypertension in Patients Undergoing TAVR. JACC Cardiovasc. Imaging 12 (4), 591–601. doi: 10.1016/j.jcmg.2018.02.015
Amedei, A., Barcelo-Coblijn, G. (2019). Editorial of Special Issue “The Interplay of Microbiome and Immune Response in Health and Diseases”. Int. J. Mol. Sci. 20 (15), 3708. doi: 10.3390/ijms20153708
Arpaia, N., Campbell, C., Fan, X., Dikiy, S., van der Veeken, J., deRoos, P., et al. (2013). Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504 (7480), 451–455. doi: 10.1038/nature12726
Aryal, B., Rotllan, N., Fernandez-Hernando, C. (2014). Noncoding RNAs and atherosclerosis. Curr. Atheroscl. Rep. 16 (5), 407. doi: 10.1007/s11883-014-0407-3
Bartova, J., Sommerova, P., Lyuya-Mi, Y., Mysak, J., Prochazkova, J., Duskova, J., et al. (2014). Periodontitis as a risk factor of atherosclerosis. J. Immunol. Res. 2014, 636893. doi: 10.1155/2014/636893
Berry, J. L., Steen, J. A., Koudy Williams, J., Jordan, J. E., Atala, A., Yoo, J. J. (2010). Bioreactors for development of tissue engineered heart valves. Ann. BioMed. Eng. 38 (11), 3272–3279. doi: 10.1007/s10439-010-0148-6
Blander, J. M., Longman, R. S., Iliev, I. D., Sonnenberg, G. F., Artis, D. (2017). Regulation of inflammation by microbiota interactions with the host. Nat. Immunol. 18 (8), 851–860. doi: 10.1038/ni.3780
Brasier, A. R. (2010). The nuclear factor-kappaB-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc. Res. 86 (2), 211–218. doi: 10.1093/cvr/cvq076
Canyelles, M., Tondo, M., Cedo, L., Farras, M., Escola-Gil, J. C., Blanco-Vaca, F. (2018). Trimethylamine N-Oxide: A Link among Diet, Gut Microbiota, Gene Regulation of Liver and Intestine Cholesterol Homeostasis and HDL Function. Int. J. Mol. Sci. 19 (10), 3228. doi: 10.3390/ijms19103228
Carroll, J. D., Carroll, E. P., Feldman, T., Ward, D. M., Lang, R. M., McGaughey, D., et al. (1992). Sex-associated differences in left ventricular function in aortic stenosis of the elderly. Circulation 86 (4), 1099–1107. doi: 10.1161/01.CIR.86.4.1099
Chan, K. L., Teo, K., Dumesnil, J. G., Ni, A., Tam, J., Investigators, A. (2010). Effect of Lipid lowering with rosuvastatin on progression of aortic stenosis: results of the aortic stenosis progression observation: measuring effects of rosuvastatin (ASTRONOMER) trial. Circulation. 121 (2), 306–314. doi: 10.1161/CIRCULATIONAHA.109.900027
Chen, J. H., Simmons, C. A. (2011). Cell-matrix interactions in the pathobiology of calcific aortic valve disease: critical roles for matricellular, matricrine, and matrix mechanics cues. Circ. Res. 108 (12), 1510–1524. doi: 10.1161/CIRCRESAHA.110.234237
Cohen, D. J., Malave, D., Ghidoni, J. J., Iakovidis, P., Everett, M. M., You, S., et al. (2004). Role of oral bacterial flora in calcific aortic stenosis: an animal model. Ann. Thorac. Surg. 77 (2), 537–543. doi: 10.1016/S0003-4975(03)01454-1
Correa-Oliveira, R., Fachi, J. L., Vieira, A., Sato, F. T., Vinolo, M. A. (2016). Regulation of immune cell function by short-chain fatty acids. Clin. Transl. Immunol. 5 (4), e73. doi: 10.1038/cti.2016.17
Cote, C., Pibarot, P., Despres, J. P., Mohty, D., Cartier, A., Arsenault, B. J., et al. (2008). Association between circulating oxidised low-density lipoprotein and fibrocalcific remodelling of the aortic valve in aortic stenosis. Heart 94 (9), 1175–1180. doi: 10.1136/hrt.2007.125740
Cote, N., Mahmut, A., Bosse, Y., Couture, C., Page, S., Trahan, S., et al. (2013). Inflammation is associated with the remodeling of calcific aortic valve disease. Inflammation 36 (3), 573–581. doi: 10.1007/s10753-012-9579-6
Cowell, S. J., Newby, D. E., Prescott, R. J., Bloomfield, P., Reid, J., Northridge, D. B., et al. (2005). A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N Engl. J. Med. 352 (23), 2389–2397. doi: 10.1056/NEJMoa043876
Daubert, M. A., Weissman, N. J., Hahn, R. T., Pibarot, P., Parvataneni, R., Mack, M. J., et al. (2016). Long-Term Valve Performance of TAVR and SAVR: A Report From the PARTNER I Trial. JACC Cardiovasc. Imaging. S1936-878X(16)30895-6. doi: 10.1016/j.jcmg.2016.11.004
De Almeida, C. V., de Camargo, M. R., Russo, E., Amedei, A. (2019). Role of diet and gut microbiota on colorectal cancer immunomodulation. World J. Gastroenterol. 25 (2), 151–162. doi: 10.3748/wjg.v25.i2.151
de Sousa, J., Sousa Aarao, T. L., Rodrigues de Sousa, J., Hirai, K. E., Silva, L. M., Dias, L. B., Jr., et al. (2017). Endothelium adhesion molecules ICAM-1, ICAM-2, VCAM-1 and VLA-4 expression in leprosy. Microb Pathog 104, 116–124. doi: 10.1016/j.micpath.2017.01.021
Deo, P. N., Deshmukh, R. (2019). Oral microbiome: Unveiling the fundamentals. J. Maxillofac. Pathol. 23 (1), 122–128. doi: 10.4103/jomfp.JOMFP_304_18
Deshmane, S. L., Kremlev, S., Amini, S., Sawaya, B. E. (2009). Monocyte chemoattractant protein-1 (MCP-1): an overview. J. Interferon. Cytokine Res. 29 (6), 313–326. doi: 10.1089/jir.2008.0027
DeStefano, F., Anda, R. F., Kahn, H. S., Williamson, D. F., Russell, C. M. (1993). Dental disease and risk of coronary heart disease and mortality. BMJ. 306 (6879), 688–691. doi: 10.1136/bmj.306.6879.688
Dobson, L. E., Fairbairn, T. A., Musa, T. A., Uddin, A., Mundie, C. A., Swoboda, P. P., et al. (2016). Sex-related differences in left ventricular remodeling in severe aortic stenosis and reverse remodeling after aortic valve replacement: A cardiovascular magnetic resonance study. Am. Heart J. 175, 101–111. doi: 10.1016/j.ahj.2016.02.010
Dweck, M. R., Boon, N. A., Newby, D. E. (2012a). Calcific aortic stenosis: a disease of the valve and the myocardium. J. Am. Coll. Cardiol. 60 (19), 1854–1863. doi: 10.1016/j.jacc.2012.02.093
Dweck, M. R., Jones, C., Joshi, N. V., Fletcher, A. M., Richardson, H., White, A., et al. (2012b). Assessment of valvular calcification and inflammation by positron emission tomography in patients with aortic stenosis. Circulation 125 (1), 76–86. doi: 10.1161/CIRCULATIONAHA.111.051052
Eberhard, J., Stumpp, N., Winkel, A., Schrimpf, C., Bisdas, T., Orzak, P., et al. (2017). Streptococcus mitis and Gemella haemolysans were simultaneously found in atherosclerotic and oral plaques of elderly without periodontitis-a pilot study. Clin. Invest. 21 (1), 447–452. doi: 10.1007/s00784-016-1811-6
El Husseini, D., Boulanger, M. C., Mahmut, A., Bouchareb, R., Laflamme, M. H., Fournier, D., et al. (2014). P2Y2 receptor represses IL-6 expression by valve interstitial cells through Akt: implication for calcific aortic valve disease. J. Mol. Cell Cardiol. 72, 146–156. doi: 10.1016/j.yjmcc.2014.02.014
Fang, M., Wang, C. G., Zheng, C., Luo, J., Hou, S., Liu, K., et al. (2018). Mir-29b promotes human aortic valve interstitial cell calcification via inhibiting TGF-beta3 through activation of wnt3/beta-catenin/Smad3 signaling. J. Cell. Biochem. 119 (7), 5175–5185. doi: 10.1002/jcb.26545
Ferreira-Gonzalez, I., Pinar-Sopena, J., Ribera, A., Marsal, J. R., Cascant, P., Gonzalez-Alujas, T., et al. (2013). Prevalence of calcific aortic valve disease in the elderly and associated risk factors: a population-based study in a Mediterranean area. Eur. J. Prev. Cardiol. 20 (6), 1022–1030. doi: 10.1177/2047487312451238
Freeman, R. V., Otto, C. M. (2005). Spectrum of calcific aortic valve disease: pathogenesis, disease progression, and treatment strategies. Circulation. 111 (24), 3316–3326. doi: 10.1161/CIRCULATIONAHA.104.486738
Gao, S., Li, S., Ma, Z., Liang, S., Shan, T., Zhang, M., et al. (2016). Presence of Porphyromonas gingivalis in esophagus and its association with the clinicopathological characteristics and survival in patients with esophageal cancer. Infect. Agent Cancer 11, 3. doi: 10.1186/s13027-016-0049-x
Garcia-Rodriguez, C., Parra-Izquierdo, I., Castanos-Mollor, I., Lopez, J., San Roman, J. A., Sanchez Crespo, M. (2018). Toll-Like Receptors, Inflammation, and Calcific Aortic Valve Disease. Front. Physiol. 9, 201. doi: 10.3389/fphys.2018.00201
Gosev, I., Zeljko, M., Duric, Z., Nikolic, I., Gosev, M., Ivcevic, S., et al. (2017). Epigenome alterations in aortic valve stenosis and its related left ventricular hypertrophy. Clin. Epigenet. 9, 106. doi: 10.1186/s13148-017-0406-7
Hadji, F., Boulanger, M. C., Guay, S. P., Gaudreault, N., Amellah, S., Mkannez, G., et al. (2016). Altered DNA Methylation of Long Noncoding RNA H19 in Calcific Aortic Valve Disease Promotes Mineralization by Silencing NOTCH1. Circulation 134 (23), 1848–1862. doi: 10.1161/CIRCULATIONAHA.116.023116
Hakansson, A., Molin, G. (2011). Gut microbiota and inflammation. Nutrients. 3 (6), 637–682. doi: 10.3390/nu3060637
Hansson, G. K. (2005). Inflammation, atherosclerosis, and coronary artery disease. N Engl. J. Med. 352 (16), 1685–1695. doi: 10.1056/NEJMra043430
Harsch, I. A., Konturek, P. C. (2018). The Role of Gut Microbiota in Obesity and Type 2 and Type 1 Diabetes Mellitus: New Insights into “Old” Diseases. Med. Sci. (Basel). 6 (2), 32. doi: 10.3390/medsci6020032
Helas, S., Goettsch, C., Schoppet, M., Zeitz, U., Hempel, U., Morawietz, H., et al. (2009). Inhibition of receptor activator of NF-kappaB ligand by denosumab attenuates vascular calcium deposition in mice. Am. J. Pathol. 175 (2), 473–478. doi: 10.2353/ajpath.2009.080957
Helgadottir, A., Thorleifsson, G., Gretarsdottir, S., Stefansson, O. A., Tragante, V., Thorolfsdottir, R. B., et al. (2018). Genome-wide analysis yields new loci associating with aortic valve stenosis. Nat. Commun. 9 (1), 987. doi: 10.1038/s41467-018-03252-6
Huang, J., Xiong, T., Zhang, Z., Tan, Y., Guo, L. (2019). Inhibition of the receptor for advanced glycation inhibits lipopolysaccharide-mediated High mobility group protein B1 and Interleukin-6 synthesis in human gingival fibroblasts through the NF-kappaB signaling pathway. Arch. Biol. 105, 81–87. doi: 10.1016/j.archoralbio.2019.06.006
Hulin, A., Hego, A., Lancellotti, P., Oury, C. (2018). Advances in Pathophysiology of Calcific Aortic Valve Disease Propose Novel Molecular Therapeutic Targets. Front. Cardiovasc. Med. 5, 21. doi: 10.3389/fcvm.2018.00021
Hurle, M. R., Yang, L., Xie, Q., Rajpal, D. K., Sanseau, P., Agarwal, P. (2013). Computational drug repositioning: from data to therapeutics. Clin. Pharmacol. Ther. 93 (4), 335–341. doi: 10.1038/clpt.2013.1
Hutcheson, J. D., Aikawa, E., Merryman, W. D. (2014). Potential drug targets for calcific aortic valve disease. Nat. Rev. Cardiol. 11 (4), 218–231. doi: 10.1038/nrcardio.2014.1
Institute of Medicine Board on Population H, Public Health P (2012). The National Academies Collection: Reports funded by National Institutes of Health. Sex-Specific Reporting of Scientific Research: A Workshop Summary (Washington (DC): National Academies Press (US)National Academy of Sciences;).
Isoda, K., Matsuki, T., Kondo, H., Iwakura, Y., Ohsuzu, F. (2010). Deficiency of interleukin-1 receptor antagonist induces aortic valve disease in BALB/c mice. Arterioscler. Thromb. Vasc. Biol. 30 (4), 708–715. doi: 10.1161/ATVBAHA.109.201749
Jamaluddin, M., Wang, S., Boldogh, I., Tian, B., Brasier, A. R. (2007). TNF-alpha-induced NF-kappaB/RelA Ser(276) phosphorylation and enhanceosome formation is mediated by an ROS-dependent PKAc pathway. Cell Signal. 19 (7), 1419–1433. doi: 10.1016/j.cellsig.2007.01.020
Jia, G., Zhi, A., Lai, P. F. H., Wang, G., Xia, Y., Xiong, Z., et al. (2018). The oral microbiota - a mechanistic role for systemic diseases. Br. Dent. J. 224 (6), 447–455. doi: 10.1038/sj.bdj.2018.217
Kaden, J. J., Bickelhaupt, S., Grobholz, R., Haase, K. K., Sarikoc, A., Kilic, R., et al. (2004). Receptor activator of nuclear factor kappaB ligand and osteoprotegerin regulate aortic valve calcification. J. Mol. Cell Cardiol. 36 (1), 57–66. doi: 10.1016/j.yjmcc.2003.09.015
Katz, R., Budoff, M. J., Takasu, J., Shavelle, D. M., Bertoni, A., Blumenthal, R. S., et al. (2009). Relationship of metabolic syndrome with incident aortic valve calcium and aortic valve calcium progression: the Multi-Ethnic Study of Atherosclerosis (MESA). Diabetes 58 (4), 813–819. doi: 10.2337/db08-1515
Kawasaki, T., Kawai, T. (2014). Toll-like receptor signaling pathways. Front. Immunol. 5, 461. doi: 10.3389/fimmu.2014.00461
Kocyigit, D., Gurses, K. M., Stahlman, M., Boren, J., Soyal, M. F., Canpinar, H., et al. (2017). Abstract 12134: Gut Microbiota Play a Role in Calcific Aortic Valve Stenosis. Circulation 136 (suppl_1), A12134–A1213A.
LaHaye, S., Lincoln, J., Garg, V. (2014). Genetics of valvular heart disease. Curr. Cardiol. Rep. 16 (6), 487. doi: 10.1007/s11886-014-0487-2
Lauten, A., Figulla, H. R., Ferrari, M. (2013). Transfemoral TAVI and aortic calcification: the destructive impact of endogenous calcium. Heart. 99 (13), 971. doi: 10.1136/heartjnl-2012-303482
Lauten, A., Figulla, H. R., Mollmann, H., Holzhey, D., Kotting, J., Beckmann, A., et al. (2014). TAVI for low-flow, low-gradient severe aortic stenosis with preserved or reduced ejection fraction: a subgroup analysis from the German Aortic Valve Registry (GARY). EuroIntervention 10 (7), 850–859. doi: 10.4244/EIJV10I7A145
Lee, S. H., Choi, J. H. (2018). Involvement of inflammatory responses in the early development of calcific aortic valve disease: lessons from statin therapy. Anim. Cells Syst. (Seoul). 22 (6), 390–399. doi: 10.1080/19768354.2018.1528175
Lerman, D. A., Prasad, S., Alotti, N. (2015). Calcific Aortic Valve Disease: Molecular Mechanisms and Therapeutic Approaches. Eur. Cardiol. 10 (2), 108–112. doi: 10.15420/ecr.2015.10.2.108
Liao, J., Joyce, E. M., Sacks, M. S. (2008). Effects of decellularization on the mechanical and structural properties of the porcine aortic valve leaflet. Biomaterials. 29 (8), 1065–1074. doi: 10.1016/j.biomaterials.2007.11.007
Lindman, B. R., Clavel, M. A., Mathieu, P., Iung, B., Lancellotti, P., Otto, C. M., et al. (2016). Calcific aortic stenosis. Nat. Rev. Dis. Primers 2, 16006. doi: 10.1038/nrdp.2016.6
Liu, Z., Li, J., Liu, H., Tang, Y., Zhan, Q., Lai, W., et al. (2019). The intestinal microbiota associated with cardiac valve calcification differs from that of coronary artery disease. Atherosclerosis 284, 121–128. doi: 10.1016/j.atherosclerosis.2018.11.038
Long, J., Cai, Q., Steinwandel, M., Hargreaves, M. K., Bordenstein, S. R., Blot, W. J., et al. (2017). Association of oral microbiome with type 2 diabetes risk. J. Periodontal. Res. 52 (3), 636–643. doi: 10.1111/jre.12432
Lu, P., Yin, B., Liu, L. (2019). MicroRNA-138 Suppresses Osteoblastic Differentiation of Valvular Interstitial Cells in Degenerative Calcific Aortic Valve Disease. Int. Heart J. 60 (1), 136–144. doi: 10.1536/ihj.18-086
Luu, M., Pautz, S., Kohl, V., Singh, R., Romero, R., Lucas, S., et al. (2019). The short-chain fatty acid pentanoate suppresses autoimmunity by modulating the metabolic-epigenetic crosstalk in lymphocytes. Nat. Commun. 10 (1), 760. doi: 10.1038/s41467-019-08711-2
Marincheva-Savcheva, G., Subramanian, S., Qadir, S., Figueroa, A., Truong, Q., Vijayakumar, J., et al. (2011). Imaging of the aortic valve using fluorodeoxyglucose positron emission tomography increased valvular fluorodeoxyglucose uptake in aortic stenosis. J. Am. Coll. Cardiol. 57 (25), 2507–2515. doi: 10.1016/j.jacc.2010.12.046
Mathieu, P., Bouchareb, R., Boulanger, M. C. (2015). Innate and Adaptive Immunity in Calcific Aortic Valve Disease. J. Immunol. Res. 2015, 851945. doi: 10.1155/2015/851945
Mazur, P., Mielimonka, A., Natorska, J., Wypasek, E., Gaweda, B., Sobczyk, D., et al. (2018). Lymphocyte and monocyte subpopulations in severe aortic stenosis at the time of surgical intervention. Cardiovasc. Pathol. 35, 1–7. doi: 10.1016/j.carpath.2018.03.004
Mazzone, A., Epistolato, M. C., De Caterina, R., Storti, S., Vittorini, S., Sbrana, S., et al. (2004). Neoangiogenesis, T-lymphocyte infiltration, and heat shock protein-60 are biological hallmarks of an immunomediated inflammatory process in end-stage calcified aortic valve stenosis. J. Am. Coll. Cardiol. 43 (9), 1670–1676. doi: 10.1016/j.jacc.2003.12.041
Miller, J. D., Weiss, R. M., Heistad, D. D. (2011). Calcific aortic valve stenosis: methods, models, and mechanisms. Circ. Res. 108 (11), 1392–1412. doi: 10.1161/CIRCRESAHA.110.234138
Mohty, D., Pibarot, P., Despres, J. P., Cote, C., Arsenault, B., Cartier, A., et al. (2008). Association between plasma LDL particle size, valvular accumulation of oxidized LDL, and inflammation in patients with aortic stenosis. Arterioscler. Thromb. Vasc. Biol. 28 (1), 187–193. doi: 10.1161/ATVBAHA.107.154989
Morrison, M. D., Reiley, W., Zhang, M., Sun, S. C. (2005). An atypical tumor necrosis factor (TNF) receptor-associated factor-binding motif of B cell-activating factor belonging to the TNF family (BAFF) receptor mediates induction of the noncanonical NF-kappaB signaling pathway. J. Biol. Chem. 280 (11), 10018–10024. doi: 10.1074/jbc.M413634200
Mosch, J., Gleissner, C. A., Body, S., Aikawa, E. (2017). Histopathological assessment of calcification and inflammation of calcific aortic valves from patients with and without diabetes mellitus. Histol. Histopathol. 32 (3), 293–306. doi: 10.14670/HH-11-797
Murr, R. (2010). Interplay between different epigenetic modifications and mechanisms. Adv. Genet. 70, 101–141. doi: 10.1016/B978-0-12-380866-0.60005-8
Nadlonek, N., Lee, J. H., Reece, T. B., Weyant, M. J., Cleveland, J. C., Jr., Meng, X., et al. (2013). Interleukin-1 Beta induces an inflammatory phenotype in human aortic valve interstitial cells through nuclear factor kappa Beta. Ann. Thorac. Surg. 96 (1), 155–162. doi: 10.1016/j.athoracsur.2013.04.013
Nakano, K., Inaba, H., Nomura, R., Nemoto, H., Takeda, M., Yoshioka, H., et al. (2006). Detection of cariogenic Streptococcus mutans in extirpated heart valve and atheromatous plaque specimens. J. Clin. Microbiol. 44 (9), 3313–3317. doi: 10.1128/JCM.00377-06
New, S. E., Aikawa, E. (2011). Molecular imaging insights into early inflammatory stages of arterial and aortic valve calcification. Circ. Res. 108 (11), 1381–1391. doi: 10.1161/CIRCRESAHA.110.234146
Nie, Y., Chen, H., Guo, C., Yuan, Z., Zhou, X., Zhang, Y., et al. (2017). Palmdelphin promotes myoblast differentiation and muscle regeneration. Sci. Rep. 7, 41608. doi: 10.1038/srep41608
Nkomo, V. T., Gardin, J. M., Skelton, T. N., Gottdiener, J. S., Scott, C. G., Enriquez-Sarano, M. (2006). Burden of valvular heart diseases: a population-based study. Lancet. 368 (9540), 1005–1011. doi: 10.1016/S0140-6736(06)69208-8
Nomura, R., Otsugu, M., Naka, S., Teramoto, N., Kojima, A., Muranaka, Y., et al. (2014). Contribution of the interaction of Streptococcus mutans serotype k strains with fibrinogen to the pathogenicity of infective endocarditis. Infect. Immun. 82 (12), 5223–5234. doi: 10.1128/IAI.02164-14
O’Brien, K. D., Reichenbach, D. D., Marcovina, S. M., Kuusisto, J., Alpers, C. E., Otto, C. M. (1996). Apolipoproteins B, (a), and E accumulate in the morphologically early lesion of ‘degenerative’ valvular aortic stenosis. Arterioscler. Thromb. Vasc. Biol. 16 (4), 523–532. doi: 10.1161/01.ATV.16.4.523
O’Brien, K. D. (2006). Pathogenesis of calcific aortic valve disease: a disease process comes of age (and a good deal more). Arterioscler. Thromb. Vasc. Biol. 26 (8), 1721–1728. doi: 10.1161/01.ATV.0000227513.13697.ac
Oliveira, F. A., Forte, C. P., Silva, P. G., Lopes, C. B., Montenegro, R. C., Santos, A. K., et al. (2015). Molecular Analysis of Oral Bacteria in Heart Valve of Patients With Cardiovascular Disease by Real-Time Polymerase Chain Reaction. Med. (Baltimore) 94 (47), e2067. doi: 10.1097/MD.0000000000002067
Otto, C. M., Kuusisto, J., Reichenbach, D. D., Gown, A. M., O’Brien, K. D. (1994). Characterization of the early lesion of ‘degenerative’ valvular aortic stenosis. Histological and immunohistochemical studies. Circulation. 90 (2), 844–853. doi: 10.1161/01.cir.90.2.844
Palazzo, A. F., Lee, E. S. (2015). Non-coding RNA: what is functional and what is junk? Front. Genet. 6 (2), 2. doi: 10.3389/fgene.2015.00002
Perez, C., Mondejar, R., Garcia-Diaz, N., Cereceda, L., Leon, A., Montes, S., et al. (2019). Advanced-stage Mycosis Fungoides. Role of STAT3, NFKB and NFAT pathways. Br. J. Dermatol182 (6). doi: 10.1111/bjd.18098
Perrot, N., Theriault, S., Dina, C., Chen, H. Y., Boekholdt, S. M., Rigade, S., et al. (2019). Genetic Variation in LPA, Calcific Aortic Valve Stenosis in Patients Undergoing Cardiac Surgery, and Familial Risk of Aortic Valve Microcalcification. JAMA Cardiol. 4 (7), 620–627. doi: 10.1001/jamacardio.2019.1581
Porras, A. M., McCoy, C. M., Masters, K. S. (2017). Calcific Aortic Valve Disease: A Battle of the Sexes. Circ. Res. 120 (4), 604–606. doi: 10.1161/CIRCRESAHA.117.310440
Rahat, M. A., Coffelt, S. B., Granot, Z., Muthana, M., Amedei, A. (2016). Macrophages and Neutrophils: Regulation of the Inflammatory Microenvironment in Autoimmunity and Cancer. Mediators Inflamm. 2016, 5894347. doi: 10.1155/2016/5894347
Rajamannan, N. M., Evans, F. J., Aikawa, E., Grande-Allen, K. J., Demer, L. L., Heistad, D. D., et al. (2011). Calcific aortic valve disease: not simply a degenerative process: A review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: Calcific aortic valve disease-2011 update. Circulation 124 (16), 1783–1791. doi: 10.1161/CIRCULATIONAHA.110.006767
Rattazzi, M., Bertacco, E., Iop, L., D’Andrea, S., Puato, M., Buso, G., et al. (2014). Extracellular pyrophosphate is reduced in aortic interstitial valve cells acquiring a calcifying profile: implications for aortic valve calcification. Atherosclerosis 237 (2), 568–576. doi: 10.1016/j.atherosclerosis.2014.10.027
Roos, C. M., Zhang, B., Verzosa, G., Oehler, E. A., Hagler, M. A., Zhang, H., et al. (2014). Abstract 27: Role of Sirtuin 6 in the Initiation and Progression of Calcific Aortic Valve Disease. Arterioscler. Thromb. Vasc. Biol. 34 (suppl_1), A27–A2A.
Rossebo, A. B., Pedersen, T. R., Boman, K., Brudi, P., Chambers, J. B., Egstrup, K., et al. (2008). Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl. J. Med. 359 (13), 1343–1356. doi: 10.1056/NEJMoa0804602
Russo, E., Taddei, A., Ringressi, M. N., Ricci, F., Amedei, A. (2016). The interplay between the microbiome and the adaptive immune response in cancer development. Therap. Adv. Gastroenterol. 9 (4), 594–605. doi: 10.1177/1756283X16635082
Rutkovskiy, A., Malashicheva, A., Sullivan, G., Bogdanova, M., Kostareva, A., Stenslokken, K. O., et al. (2017). Valve Interstitial Cells: The Key to Understanding the Pathophysiology of Heart Valve Calcification. J. Am. Heart Assoc. 6 (9), e006339. doi: 10.1161/JAHA.117.006339
Schirmer, M., Smeekens, S. P., Vlamakis, H., Jaeger, M., Oosting, M., Franzosa, E. A., et al. (2016). Linking the Human Gut Microbiome to Inflammatory Cytokine Production Capacity. Cell 167 (4), 1125–1136 e8. doi: 10.1016/j.cell.2016.11.046
Shao, J. S., Cheng, S. L., Pingsterhaus, J. M., Charlton-Kachigian, N., Loewy, A. P., Towler, D. A. (2005). Msx2 promotes cardiovascular calcification by activating paracrine Wnt signals. J. Clin. Invest. 115 (5), 1210–1220. doi: 10.1172/JCI24140
Sharma, S., Eghbali, M. (2014). Influence of sex differences on microRNA gene regulation in disease. Biol. Sex Dif. 5 (1), 3. doi: 10.1186/2042-6410-5-3
Sucosky, P., Balachandran, K., Elhammali, A., Jo, H., Yoganathan, A. P. (2009). Altered shear stress stimulates upregulation of endothelial VCAM-1 and ICAM-1 in a BMP-4- and TGF-beta1-dependent pathway. Arterioscler. Thromb. Vasc. Biol. 29 (2), 254–260. doi: 10.1161/ATVBAHA.108.176347
Sun, L., Rajamannan, N. M., Sucosky, P. (2013). Defining the role of fluid shear stress in the expression of early signaling markers for calcific aortic valve disease. PloS One 8 (12), e84433. doi: 10.1371/journal.pone.0084433
Thériault, S., Dina, C., Messika-Zeitoun, D., Scouarnec, S. L., Capoulade, R., Gaudreault, N., et al. (2019). Genetic Association Analyses Highlight IL6, ALPL, and NAV1 As 3 New Susceptibility Genes Underlying Calcific Aortic Valve Stenosis. Circ.: Genomic Precis. Med. 12 (10), 431–441. doi: 10.1161/CIRCGEN.119.002617
Thanassoulis, G., Campbell, C. Y., Owens, D. S., Smith, J. G., Smith, A. V., Peloso, G. M., et al. (2013). Genetic associations with valvular calcification and aortic stenosis. N Engl. J. Med. 368 (6), 503–512. doi: 10.1056/NEJMoa1109034
Theodoris, C. V., Li, M., White, M. P., Liu, L., He, D., Pollard, K. S., et al. (2015). Human disease modeling reveals integrated transcriptional and epigenetic mechanisms of NOTCH1 haploinsufficiency. Cell 160 (6), 1072–1086. doi: 10.1016/j.cell.2015.02.035
Theriault, S., Gaudreault, N., Lamontagne, M., Rosa, M., Boulanger, M. C., Messika-Zeitoun, D., et al. (2018). A transcriptome-wide association study identifies PALMD as a susceptibility gene for calcific aortic valve stenosis. Nat. Commun. 9 (1), 988. doi: 10.1038/s41467-018-03260-6
Tseng, H., Grande-Allen, K. J. (2011). Elastic fibers in the aortic valve spongiosa: a fresh perspective on its structure and role in overall tissue function. Acta Biomater. 7 (5), 2101–2108. doi: 10.1016/j.actbio.2011.01.022
Velasquez, M. T., Ramezani, A., Manal, A., Raj, D. S. (2016). Trimethylamine N-Oxide: The Good, the Bad and the Unknown. Toxins (Basel). 8 (11), 326. doi: 10.3390/toxins8110326
Wada, T., Nakashima, T., Hiroshi, N., Penninger, J. M. (2006). RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol. Med. 12 (1), 17–25. doi: 10.1016/j.molmed.2005.11.007
Wallby, L., Janerot-Sjoberg, B., Steffensen, T., Broqvist, M. (2002). T lymphocyte infiltration in non-rheumatic aortic stenosis: a comparative descriptive study between tricuspid and bicuspid aortic valves. Heart. 88 (4), 348–351. doi: 10.1136/heart.88.4.348
Wang, Y., Chen, S., Deng, C., Li, F., Wang, Y., Hu, X., et al. (2015). MicroRNA-204 Targets Runx2 to Attenuate BMP-2-induced Osteoblast Differentiation of Human Aortic Valve Interstitial Cells. J. Cardiovasc. Pharmacol. 66 (1), 63–71. doi: 10.1097/FJC.0000000000000244
Wernly, B., Zappe, A. K., Unbehaun, A., Sinning, J. M., Jung, C., Kim, W. K., et al. (2019). Transcatheter valve-in-valve implantation (VinV-TAVR) for failed surgical aortic bioprosthetic valves. Clin. Res. Cardiol. 108 (1), 83–92. doi: 10.1007/s00392-018-1326-z
Winchester, R., Wiesendanger, M., O’Brien, W., Zhang, H. Z., Maurer, M. S., Gillam, L. D., et al. (2011). Circulating activated and effector memory T cells are associated with calcification and clonal expansions in bicuspid and tricuspid valves of calcific aortic stenosis. J. Immunol. 187 (2), 1006–1014. doi: 10.4049/jimmunol.1003521
Wu, H. D., Maurer, M. S., Friedman, R. A., Marboe, C. C., Ruiz-Vazquez, E. M., Ramakrishnan, R., et al. (2007). The lymphocytic infiltration in calcific aortic stenosis predominantly consists of clonally expanded T cells. J. Immunol. 178 (8), 5329–5339. doi: 10.4049/jimmunol.178.8.5329
Xiao, G., Fong, A., Sun, S. C. (2004). Induction of p100 processing by NF-kappaB-inducing kinase involves docking IkappaB kinase alpha (IKKalpha) to p100 and IKKalpha-mediated phosphorylation. J. Biol. Chem. 279 (29), 30099–30105. doi: 10.1074/jbc.M401428200
Yang, Y., Cai, Q., Zheng, W., Steinwandel, M., Blot, W. J., Shu, X. O., et al. (2019). Oral microbiome and obesity in a large study of low-income and African-American populations. J. Microbiol. 11 (1), 1650597. doi: 10.1080/20002297.2019.1650597
Yip, C. Y., Simmons, C. A. (2011). The aortic valve microenvironment and its role in calcific aortic valve disease. Cardiovasc. Pathol. 20 (3), 177–182. doi: 10.1016/j.carpath.2010.12.001
Zeng, Q., Song, R., Fullerton, D. A., Ao, L., Zhai, Y., Li, S., et al. (2017). Interleukin-37 suppresses the osteogenic responses of human aortic valve interstitial cells in vitro and alleviates valve lesions in mice. Proc. Natl. Acad. Sci. U. S. A 114 (7), 1631–1636. doi: 10.1073/pnas.1619667114
Zhang, M., Liu, X., Zhang, X., Song, Z., Han, L., He, Y., et al. (2014). MicroRNA-30b is a multifunctional regulator of aortic valve interstitial cells. J. Thorac. Cardiovasc. Surg. 147 (3), 1073–1080. doi: 10.1016/j.jtcvs.2013.05.011
Zheng, D., Zang, Y., Xu, H., Wang, Y., Cao, X., Wang, T., et al. (2019). MicroRNA-214 promotes the calcification of human aortic valve interstitial cells through the acceleration of inflammatory reactions with activated MyD88/NF-kappaB signaling. Clin. Res. Cardiol. 108 (6), 691–702. doi: 10.1007/s00392-018-1398-9
Keywords: severe aortic stenosis, calcific aortic valve, aortic valve replacement, TAVR, microbiome, immune system, inflammation
Citation: Alushi B, Curini L, Christopher MR, Grubitzch H, Landmesser U, Amedei A and Lauten A (2020) Calcific Aortic Valve Disease-Natural History and Future Therapeutic Strategies. Front. Pharmacol. 11:685. doi: 10.3389/fphar.2020.00685
Received: 17 November 2019; Accepted: 27 April 2020;
Published: 13 May 2020.
Edited by:
Heinfried H. Radeke, University Hospital Frankfurt, GermanyReviewed by:
Luigi Sironi, University of Milan, ItalyCarole L. Wilson, Medical University of South Carolina, United States
Copyright © 2020 Alushi, Curini, Christopher, Grubitzch, Landmesser, Amedei and Lauten. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brunilda Alushi, YnJ1bmlsZGEuYWx1c2hpQGNoYXJpdGUuZGU=; Amedeo Amedei, YWFtZWRlaUB1bmlmaS5pdA==
†These authors have contributed equally to this work