 Reem Handklo-Jamal1
Reem Handklo-Jamal1 Eshcar Meisel1,2
Eshcar Meisel1,2 Daniel Yakubovich1,3Leonid Vysochek2Roy Beinart1,2Michael Glikson1,2†Julie R. McMullen4
Daniel Yakubovich1,3Leonid Vysochek2Roy Beinart1,2Michael Glikson1,2†Julie R. McMullen4 Nathan Dascal1Eyal Nof1,2
Nathan Dascal1Eyal Nof1,2 Shimrit Oz1,2*†
Shimrit Oz1,2*†- 1Sackler School of Medicine, Tel Aviv University, Tel Aviv, Israel
- 2Heart Center, Sheba Medical Center, Ramat-Gan, Israel
- 3Neonatology Department, Schneider Children’s Medical Center, Petah-Tikva, Israel
- 4Baker Heart and Diabetes Institute, Melbourne, VIC, Australia
Andersen–Tawil syndrome (ATS) type-1 is associated with loss-of-function mutations in KCNJ2 gene. KCNJ2 encodes the tetrameric inward-rectifier potassium channel Kir2.1, important to the resting phase of the cardiac action potential. Kir-channels’ activity requires interaction with the agonist phosphatidylinositol-4,5-bisphosphate (PIP2). Two mutations were identified in ATS patients, V77E in the cytosolic N-terminal “slide helix” and M307V in the C-terminal cytoplasmic gate structure “G-loop.” Current recordings in Kir2.1-expressing HEK cells showed that each of the two mutations caused Kir2.1 loss-of-function. Biotinylation and immunostaining showed that protein expression and trafficking of Kir2.1 to the plasma membrane were not affected by the mutations. To test the functional effect of the mutants in a heterozygote set, Kir2.1 dimers were prepared. Each dimer was composed of two Kir2.1 subunits joined with a flexible linker (i.e. WT-WT, WT dimer; WT-V77E and WT-M307V, mutant dimer). A tetrameric assembly of Kir2.1 is expected to include two dimers. The protein expression and the current density of WT dimer were equally reduced to ~25% of the WT monomer. Measurements from HEK cells and Xenopus oocytes showed that the expression of either WT-V77E or WT-M307V yielded currents of only about 20% compared to the WT dimer, supporting a dominant-negative effect of the mutants. Kir2.1 sensitivity to PIP2 was examined by activating the PIP2 specific voltage-sensitive phosphatase (VSP) that induced PIP2 depletion during current recordings, in HEK cells and Xenopus oocytes. PIP2 depletion induced a stronger and faster decay in Kir2.1 mutant dimers current compared to the WT dimer. BGP-15, a drug that has been demonstrated to have an anti-arrhythmic effect in mice, stabilized the Kir2.1 current amplitude following VSP-induced PIP2 depletion in cells expressing WT or mutant dimers. This study underlines the implication of mutations in cytoplasmic regions of Kir2.1. A newly developed calibrated VSP activation protocol enabled a quantitative assessment of changes in PIP2 regulation caused by the mutations. The results suggest an impaired function and a dominant-negative effect of the Kir2.1 variants that involve an impaired regulation by PIP2. This study also demonstrates that BGP-15 may be beneficial in restoring impaired Kir2.1 function and possibly in treating ATS symptoms.
Introduction
Andersen–Tawil syndrome (ATS, also known as long-QT7) is a channelopathy, typically characterized by a triad of symptoms: cardiac arrhythmias, periodic paralysis, and dysmorphic features (Tawil et al., 1994). However, variable symptom severity and incomplete penetrance are associated with the syndrome, in addition to a phenotypic mimicry of catecholaminergic polymorphic ventricular tachycardia (CPVT) characterized by bidirectional ventricular tachycardia (VT) (Barajas-Martinez et al., 2011).
KCNJ2 encodes the inwardly-rectifying potassium channel Kir2.1, a major component of the cardiac action potential repolarization phase. Pathogenic variants of KCNJ2 gene account for 60-70% of clinical ATS cases, termed type-1 ATS (Plaster et al., 2001). Most of the pathogenic variants cause Kir2.1 loss-of-function and exert a dominant-negative effect, attributed to trafficking or gating defects (Tristani-Firouzi et al., 2002; Hosaka et al., 2003; Ma et al., 2011). Type-2 ATS is caused by mutations in genes other than KCNJ2 (Kokunai et al., 2014).
Kir channels are homotetramers composed of four identical subunits that form a K+-selective transmembrane pore. Kir2.1 subunit topology includes two transmembrane domains (TMDs) with the outer and inner helices that form the transmembrane conduction pathway. Regulatory cytoplasmic regions are the N-terminal “slide helix” that lays in parallel to the plasma membrane, and a large cytosolic C-terminal domain (CTD). The assembled CTD structures form a tightening in the cytosolic ion permeation pathway through a gate termed the “G-loop,” aligned below the transmembrane ion permeation pathway (Figure 1C).
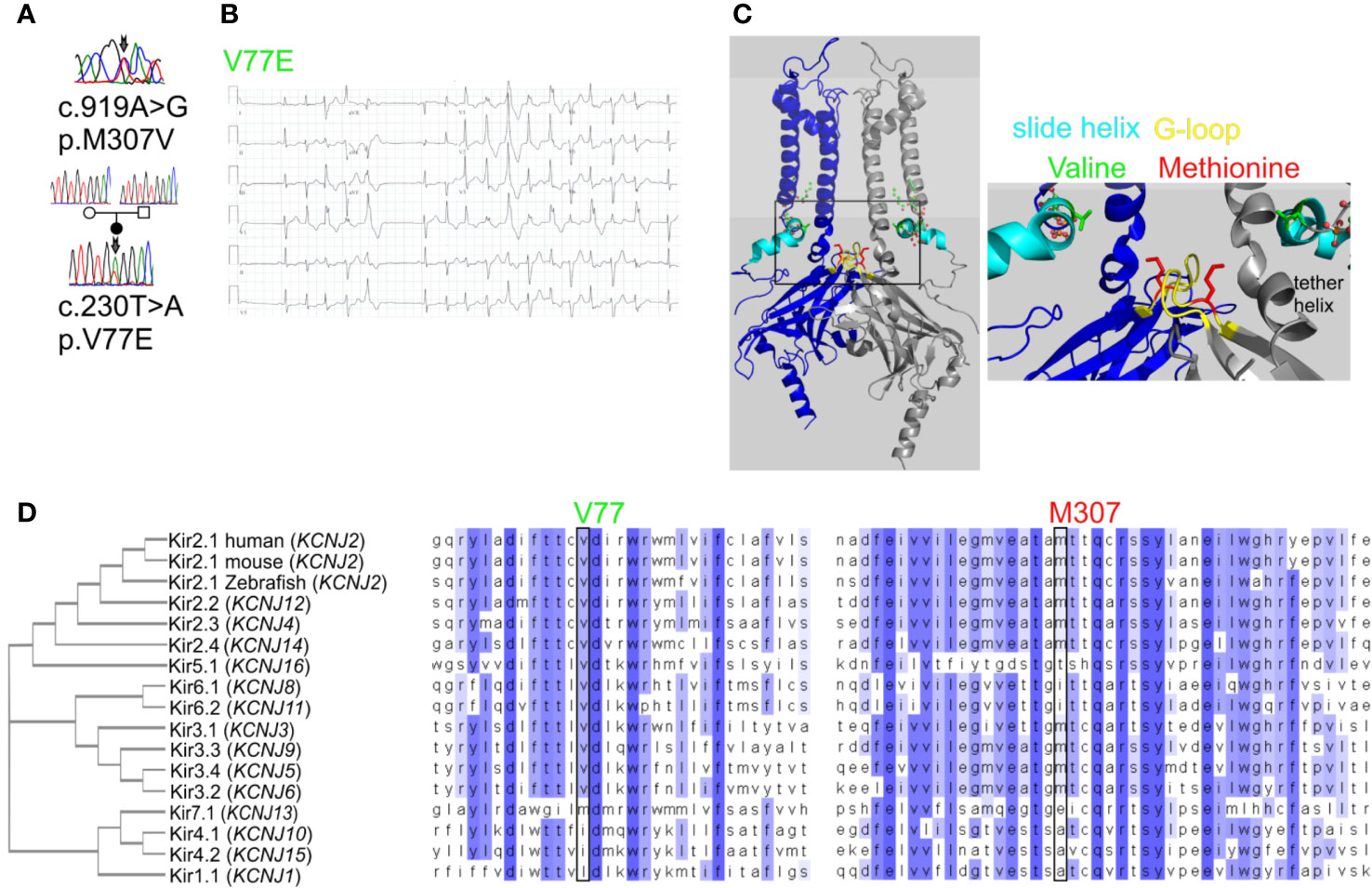
Figure 1 Mutations in Kir2.1 associated with Andersen–Tawil syndrome. (A) DNA sequencing of two patients revealed mutations in Kir2.1. (B) Representative ECG from the carrier of V77E variant showing bidirectional ventricular tachycardia, prolonged QT interval with ST and T wave abnormalities. (C) Left, cartoon presentation of two subunits (blue and gray) out of four, of the inwardly rectifying potassium channel protein (Kir2.2 with PIP2, PDB ID: 3SPI (Hansen et al., 2011)). PIP2 is in ball-and-stick (atom code: oxygen, red; carbon, green; phosphorous, orange). The N-terminal slide helix is in cyan and the cytoplasmic gate G-loop is in yellow. Side chains as sticks of V75 and M308 in Kir2.2, homologous to V77 and M307 in Kir2.1, are in green and red, respectively. The plasma membrane is in light gray. Right, zoom on the mutated residues’ location. (D) Kir channels phylogenetic tree. Amino acid sequence alignments of 15 human Kir proteins, in addition to mouse and Zebrafish Kir2.1.
Kir channels are activated by the phosphoinositide PI(4,5)P2 (PIP2) (Huang et al., 1998). ATS-associated mutations in Kir2.1 were linked to reduced PIP2 affinity (Lopes et al., 2002). The crystal structure of Kir2.2 showed that PIP2 interacts with an interface between the inner TMD and the CTD complex, inducing the formation of a cytoplasmic extension of the TMD: the tether helix. The tether helix is pivotal for both the direct binding of PIP2 and for the conformational gating transduction (Figure 1C) (Hansen et al., 2011; Lacin et al., 2017).
The hydroxylamine derivative BGP-15 is a small molecule that showed beneficial and cytoprotective effects in diabetes, cancer, cardiomyopathy, heart failure, atrial fibrillation (AF), and muscle dysfunction (Gehrig et al., 2012; Literati-Nagy et al., 2014; Sapra et al., 2014; Nascimento et al., 2018). BGP-15 has been well-tolerated in phase I–II human clinical trials as an insulin sensitizer. The putative target pathways include heat-shock proteins induction (Crul et al., 2013), poly ADP-ribose polymerase 1 (PARP-1) inhibition (Racz et al., 2002), and phosphorylation of IGF1R (Sapra et al., 2014). The exact mode of action is not clear, but several reports suggest that BGP-15 modulates plasma membrane composition, dynamics, and fluidity, involving lipid-rafts reorganization (Gombos et al., 2011; Sapra et al., 2014; Balogi et al., 2019).
In the current work, we report two ATS-associated Kir2.1 mutations. Using molecular and electrophysiological methods we show that these mutations are loss-of-function with a dominant-negative effect. We developed a calibrated protocol to asses Kir2.1 functional sensitivity to PIP2 using voltage-sensitive phosphatase (VSP) and investigated the effect of BGP-15 on Kir2.1 regulation.
Materials and Methods
Clinical and Genetic Assessment
The participants in this study provided written informed consent for both the clinical and genetic studies, which were approved by the Institutional Ethics Committee of the Sheba Medical Center, Tel-Hashomer (approval 0379-13-SMC). Genetic findings were obtained by a sequence screening of the following genes: KCNQ1, HERG, SCN5A, KCNE1, KCNE2, KCNJ2, and RYR2. Patients were clinically evaluated by 12-lead electrocardiogram (ECG), exercise testing, and 24-h Holter monitoring.
Cellular Expression Models
HEK cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 10 mg/ml streptomycin, and 2 mM L-glutamine (Biological Industries, Kibbutz Beit-Haemek, Israel) at 37°C with 5% CO2. Transfections for electrophysiological experiments were using Trans-ITx2 (Mirus, Madison, WI, USA) in 35-mm dishes and biotinylation experiments were using the calcium-phosphate method in 10-cm dishes. 90%–100% confluent dishes were split 1:2 on day 1 and transfected on day 2. On day 3, cells were split on coverslips for patch-clamp experiments and recorded on days 4-5. Biotinylation procedures started on day 4.
HL-1 cells were cultured on gelatin/fibronectin pre-coated dishes, in Claycomb media supplemented with 10% FBS, 100 μM norepinephrine (Sigma-Aldrich, St. Louis, Mo, USA), 100 U/ml penicillin, 10 mg/ml streptomycin, and 2 mM L-glutamine (Biological Industries). For imaging, cells were plated on gelatin/fibronectin-coated culture slides (Lab-Tek Nalge Nunc International, IL, USA) and transfected using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA).
Xenopus laevis oocytes experiments were approved by Tel-Aviv University Institutional Animal Care and Use Committee (permits M-08-081 and M-13-002). Maintenance and operation of female frogs were essentially as previously described (Oz et al., 2017). Briefly, frogs were maintained at 20 ± 2°C on a 10-h light/14-h dark cycle. Portions of ovary were removed from anesthetized frogs [0.17% procainemethanesulphonate (MS222)]. Oocytes were defolliculated by collagenase, injected with RNA and incubated for 2–3 days at 22°C in NDE solution (in mM: 96 NaCl, 2 KCl, 1 MgCl2, 1 CaCl2, 5 HEPES, 2.5 pyruvic acid; 50 mg l−1 gentamycin).
DNA Constructs
DNA of GFP was C-terminally fused to the human KCNJ2 coding sequence (accession number NM_000891.2), following the elimination of KCNJ2 stop codon and cloned into pcDNA3 vector using restriction enzymes (BamHI-KCNJ2-EcoRI-GFP-ApaI). This is the Kir2.1-GFP monomer. DNA sequence encoding the HA-tag (a.a. YPYDVPDYA) was inserted after alanine 115 in the extracellular domain, using a Q5 site-directed mutagenesis kit (New England Biolabs, Ipswich, MA, USA). Point mutations were introduced using PCR [KAPA HiFi HotStart (Kapa Biosciences, Wilmington, MA, USA)]. DNA constructs of Kir2.1 concatenated dimers were prepared using the HA-tagged KCNJ2 constructs. Kir2.1 WT-WT construct is the WT dimer and WT-V77E, WT-M307V, or WT-R67W construct are mutant dimers. The first Kir2.1 subunit was created with BamHI at the 5′ end, then the stop codon was eliminated and four codons for glycine were inserted, followed by the SalI site at the 3′ end. The second Kir2.1 subunit was WT or mutated, with SalI site at the 5′ end that followed by four codons for glycine, ending with EcoRI site at the 3′. Thus, the linker between two Kir2.1 subunits was composed of (a.a.) GGGGVDGGGG. Kir2.1 dimers between SalI and EcoRI sites were cloned into pcDNA3 vector for mammalian cell’s expression and pGSB vector [modified pGEMHE (Shistik et al., 1998)] for RNA in-vitro synthesis. For HEK cells expression danio rerio voltage-sensing phosphatase (dr-VSP) was in pcDNA vector, and for Xenopus oocytes expression dr-VSP was in pGEM vector (Hossain et al., 2008).
RNA was transcribed in vitro using a standard procedure, from a template cDNA in oocyte expression vectors containing 5′ and 3′ untranslated sequences of Xenopus β-globin (Dascal and Lotan, 1992).
Immunocytochemistry and Imaging
Cells expressing extracellular HA-tag on Kir2.1-GFP were fixed in 1% paraformaldehyde in phosphate buffer saline (PBS) for 10 min to retain GFP fluorescence, without permeabilization, then blocked with CAS blocking agent (Thermo Fisher Scientific) for 30 min at room temperature. Cells were incubated with mouse anti-HA antibody (Millipore, Burlington, MA, USA), washed with PBS, and incubated with Alexa-594 conjugated anti-mouse secondary antibody (ThermoFisher Scientific). Cell nuclei were counterstained with DAPI. Images were captured with an LSM 700 confocal microscope using Zen software (ZEISS, Oberkochen, Germany). Three channels (blue, green, red) were acquired sequentially.
Biotinylation Assay
Biotinylation of surface proteins was carried out essentially as described (Nof et al., 2019). Briefly, transfected HEK cells were incubated with 0.5 mg/ml EZ-Link Sulfo-NHS-SS-Biotin (Pierce, Rockford, IL, USA) in PBS pH 8, supplemented with 1 mM Ca2+ and 0.5 mM Mg2+, for 30 min at 4°C. Incubation in 50 mM glycine for 10 min ended the reaction. Cells were scraped, washed and lysed for 30 min at 4°C in (mM): 20 Tris–HCl pH 8, 150 NaCl, 1 EGTA, and 1% NP40 supplemented with 0.5 phenylmethylsulfonyl fluoride (PMSF), protease inhibitor cocktail (Roche, Basel, Switzerland), 25 β-glycerol phosphate, and 1 sodium orthovanadate (Sigma-Aldrich). Equal amounts of total protein were incubated with streptavidin-agarose beads (Pierce) for 2 h at 4°C in a rotating device. Proteins were eluted in sample buffer for 1 h at room temperature, followed by 5-min incubation at 65°C, then resolved by SDS-PAGE and Western blotted.
Electrophysiology
Whole-cell currents were recorded from HEK cells using the patch-clamp technique, at 23°C. Signals were amplified using an Axopatch 200B patch-clamp amplifier (Molecular Devices, San Jose, CA, USA), sampled at 1 kHz, and filtered with a low-pass Bessel filter at 10 kHz. Data were acquired with DigiData1440A (Molecular Devices). Patch pipettes (Harvard apparatus, Holliston, MA, USA) were pulled with a resistance of 3–5 MΩ. Series resistance was compensated by 80%–90%. The intracellular pipette solution contained (in mM): 130 KCl, 2 MgCl2, 4 MgATP, 10 EGTA, 10 HEPES, adjusted with KOH to pH 7.2 (295 mOsm). The external solution contained (in mM): 135 NaCl, 4 KCl, 10 glucose, 10 HEPES, 1.8 CaCl2, 1 MgCl2, adjusted with NaOH to pH 7.4 (305 mOsm). For Ba2+-sensitive currents (Figure 2), cells were additionally perfused with the Ba2+-containing external solution (in mM): 10 BaCl2, 120 NaCl, 4 KCl, 10 glucose, 10 HEPES, 1.8 CaCl2, 1 MgCl2. The net Ba2+-sensitive current was a subtraction of the current evoked in the presence of perfused BaCl2 from the current evoked in the standard external solution without BaCl2, using the same protocol in one cell.
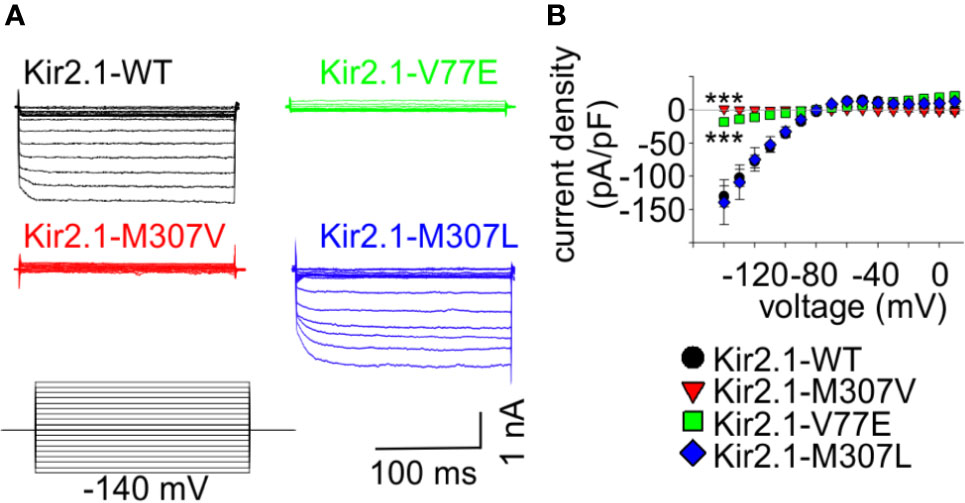
Figure 2 Mutations V77E and M307V induce Kir2.1 loss-of-function. Net Ba2+-sensitive currents recorded from HEK cells expressing Kir2.1-GFP (WT, V77E, M307V, M307L) elicited by a voltage-steps protocol. (A) Representative currents. (B) Average current density (WT, n = 12; V77E, n = 10; M307V, n = 6; M307L, n = 8). Statistical analysis in the voltage step to −140 mV using one-way ANOVA (Dunnett’s test, comparison to WT). M307L, not significant; V77E and M307V, ***p < 0.001.
HEK cells were held at −60 mV in the voltage step protocol (Figure 2) and at 0 mV in the voltage ramp protocol. Ramps were set from −120 to 0 mV in 1 s. In the double-ramp protocol (example in Figure 5A), a 10-s sweep was followed by 80 s at holding potential before the next sweep. Often, the maximal amplitude of the first ramp in sweeps II–IV was smaller than the maximal amplitude of the first ramp in the previous sweep (probably due to a partial replenishment of PIP2 between the sweeps). Using voltage ramps enabled to distinguish Kir2.1 from leak currents that may have developed due to deterioration of the cell during the protocol. When the ramp waveform did not have a typical Kir2.1 rectification the sweep was excluded. VSP effect was quantified from currents elicited by voltage ramps as the ratio between the maximal amplitude at −120 mV after (I2) and before (I1) the depolarizing step, and as % VSP effect according to ((1 − (I2/I1)) * 100). When the amplitude ratio was 1, the VSP effect was 0%, and if VSP activation abolished the current completely, I2/I1 was 0 and the VSP effect was 100%. Normalized ramp-elicited currents were divided by the maximal inward current at −120 mV and plotted against the voltage. The current density was calculated as the current amplitude divided by the capacitance of each cell. Normalized current density was compared to the mean current density of the control group at −120 mV. Cells transfected on the same day and recorded during a 2-day session were considered as one experiment. 50 µM BGP-15 (Cayman Chemical, Ann Arbor, MI, USA) were incubated overnight in cell culture medium and were added to the bath solution during recording.
Xenopus oocytes were injected with RNAs of Kir2.1 concatenated dimers (0.05, 0.1, or 1 ng of the WT dimer, as indicated, and 1 ng of the mutant dimers) and VSP (10 ng). Currents were recorded using the two-electrode voltage clamp (TEVC) technique. Oocytes were held at −30 mV and current–voltage relationships were produced by voltage ramps from −120 to 30 mV in 2 s. Bath solution contained (in mM): 24 KCl, 72 NaCl, 1 CaCl2, 1 MgCl2, and 5 HEPES, in pH = 7.4. Naïve cells’ endogenous, depolarization-induced, inward currents (possibly via sodium or stretch-activated ion channels) were partially blocked by 50 µM Gd3+ (Baud et al., 1982; Yang and Sachs, 1989). The net Kir2.1 currents were obtained by subtraction of the mean current obtained with the same VSP activation protocol from four naïve oocytes from the same batch of oocytes in the same experiment or in the presence of 50 µM Gd3+. VSP effect was quantified from currents elicited by voltage ramps as the ratio between the maximal amplitude at −80 mV. The reversal potential, Vrev, was the potential at which the net current was zero. The rectification index was calculated by the division of the current 50 mV positive to Vrev by the current 50 mV negative to Vrev.
Software and Data Analysis and Presentation
The structure of Kir2.2-V75E was generated by PyMOL (Molecular Graphics System, Version 2.0 Schrödinger) based on the crystal structure of Kir2.2 channel (PDB ID:3SPI). The rotamer with minimal steric clashes was selected. Subsequently, the energy minimization procedure was conducted by YASARA force field server default values (http://www.yasara.org) (Krieger et al., 2009).
DNA chromatogram was visualized by BioEdit. Multiple DNA sequence alignments were using CLUSTAL Omega 1.2.4. Western blot bands were quantified using ImageJ (NIH, United States). Figures were prepared using CorelDraw 2018 (Corel Corp., Ottawa, Canada). SigmaPlot 13 and 14 (Systat Software, CA, USA) and Prism (GraphPad, San Diego, CA, USA) were used for graph preparation and statistical assays. Currents were analyzed using Clampfit 10.7.
Data were presented as mean ± standard error or box plots indicating 25–75 percentiles (box borders), median (black line), mean (dotted red line), and 5–95 percentiles (whiskers). Statistical tests were performed on raw data. Two-group comparisons were performed using unpaired t-test if the data passed the Shapiro-Wilk normality test, otherwise, Mann–Whitney rank-sum test was used. Multiple group comparisons were done using one-way ANOVA, followed by Dunnet’s tests for normally distributed data and Kruskal-Wallis one-way ANOVA on ranks followed by Dunn’s test, otherwise. Two-way ANOVA with repeated measures was performed to compare current courses in voltage ramp protocol, between groups. ***, p < 0.001; **, p < 0.01; *, p < 0.05; ns, not significant.
Results
Kir2.1 Variants V77E and M307V Are Associated With ATS
We have previously described a case of familial almost incessant polymorphous and bidirectional VT. Association of the disease with mutations in several calcium-handling related genes was excluded (Nof et al., 2004). Years later, a larger gene screening detected a c.919A > G substitution in the KCNJ2 gene encoding the inward-rectifier potassium channel Kir2.1. This missense mutation led to a substitution of methionine at position 307 to valine (p.M307V) in the Kir2.1 protein (Figure 1A). Shortly after the report, one family member had a syncope while driving, and implantable cardioverter defibrillator (ICD) was implanted. Symptomatic arrhythmia events were not documented since then. Another family member was implanted with an ICD due to her ventricular arrhythmias. A few years later, she had a ventricular fibrillation (VF) storm which the ICD was able to terminate. All other family members described in the previous report (Nof et al., 2004) remained asymptomatic and were not implanted with ICD despite the heavy arrhythmic burden.
In another case, a 35-year-old woman with multiple premature ventricular contractions (PVCs) and non-sustained ventricular tachycardia (NSVT) was genetically and clinically evaluated. She reported a lower limb muscle weakness. A thigh-muscle biopsy, performed at another institute, was normal. ECG recording showed polymorphic and bidirectional VT at rest and exercise. Missense variation c.230T > A in KCNJ2 was detected, leading to substitution of valine 77 to glutamic acid (p.V77E) in Kir2.1. Both parents did not carry the mutation in KCNJ2, and kinship genetic test based on single-nucleotide polymorphism (SNP) confirmed parenthood. Thus, V77E mutation is a de-novo mutation in the proband (Figures 1A, B). She developed tachycardia-induced cardiomyopathy with 40% left ventricular ejection fraction (LVEF). An ablation attempt was aborted due to the polymorphic nature of her PVCs, and an ICD was implanted. She did not receive any ICD shocks during follow up.
Following the association of the KCNJ2 mutations with cardiac arrhythmia symptoms, ATS type-1 was diagnosed in the carriers of both mutations. The clinical manifestations included ventricular arrhythmias but without periodic paralysis or facial/skeletal dysmorphic features that have been previously reported, with a variable penetrance, in other ATS cases.
Residue M307 is located at the G-loop in Kir2.1 C-terminus. Residue V77 is located at the regulatory slide helix in Kir2.1 N-terminus (Figure 1C). Both residues are conserved among species and different inward-rectifier potassium channels. The hydrophobic valine 77 is highly conserved among 11 out of 15 KCNJ genes, and in all 15 KCNJ genes this position is occupied by hydrophobic residues. The hydrophobic methionine 307 is conserved in 8 out of 15 KCNJ genes (Figure 1D).
Both V77E and M307V Cause Kir2.1 Loss-of-Function Without Affecting Cell Surface Expression
In order to study the functional properties of the mutants, Kir2.1-GFP constructs were expressed in HEK cells, and currents were measured using whole-cell patch-clamp technique. During a voltage-step protocol, cells expressing Kir2.1-GFP WT showed large inward voltage-dependent currents at voltages negative to −80 mV that were blocked by 10 mM Ba2+. The net Ba2+-sensitive currents were inwardly rectifying with a reversal potential close to the calculated K+ equilibrium potential (according to the calculated Nernst potential of −90 mV and liquid junction potential of 3.9 mV, the calculated reversal potential of K+ is ~−86 mV), and small outward currents were recorded with voltage steps between −80 to −20 mV (Figure 2).
Expression of the Kir2.1-GFP V77E and M307V mutants produced almost no Ba2+-sensitive currents, indicating that these are loss-of-function mutations. The substitution of M307 with the hydrophobic residue leucine (M307L) did not affect the current density and rectification that were similar to that of Kir2.1-GFP WT (Figure 2).
To investigate whether protein synthesis and trafficking in these loss-of-function Kir2.1 variants were impaired, two methods were employed: imaging of immunofluorescent Kir2.1 and biochemical detection of cell-surface Kir2.1 by biotinylation (Figure 3).
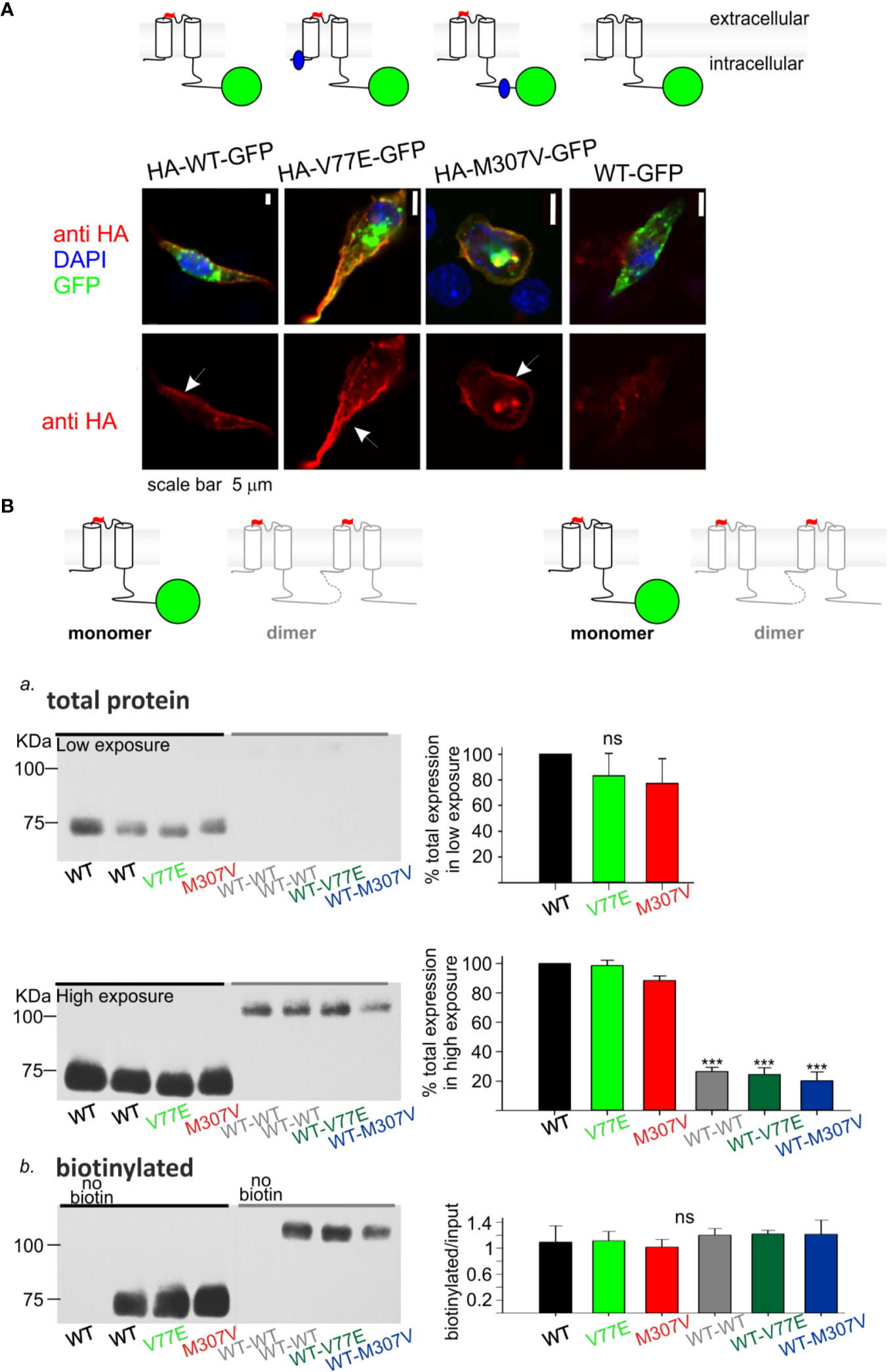
Figure 3 Loss-of-function Kir2.1 mutations express on the cell surface. (A) Immunostaining of HL-1 cells transfected with Kir2.1-GFP with and without an extracellular HA-tag. Top, an illustration of the Kir2.1 subunit constructs. Cylinders: Kir2.1 transmembrane domains; blue: mutation location; green: GFP; Red: HA-tag. Bottom, HA-tag was immunostained (red), expressed GFP was detected (green) and the nucleus was stained with DAPI (blue). The three optical channels are superimposed (upper panel) and the red channel is also shown separately (lower panel). Arrows point to the cell surface signal. (B) Biotinylation of HEK cells transfected with Kir2.1-GFP monomers and Kir2.1 dimers (WT and mutant) as illustrated above and detected with the HA-tag antibody. Left, representative blots from one experiment. Right, normalized signals. a. Total Kir2.1 protein level. Total expression was normalized to WT in the same experiment. Low film exposure was used to quantify the difference in Kir2.1 monomers’ level within a dynamic range (upper panel, N = 4). Statistical analysis: one-way ANOVA. The differences between Kir2.1 monomers and dimers were shown in high film exposure of the blot, where Kir2.1 monomer signal was saturated (lower panel, N = 3). Statistical analysis: one-way ANOVA (Dunnett’s test, comparison to WT Group). b. biotin-labeled Kir2.1 level. As a control for non-specific signals, Kir2.1-GFP WT and WT dimer expressing groups were not incubated with biotin. The amount of protein on the plasma membrane relative to total was quantified by a division of the biotinylated with input level in high exposure (N = 3). Statistical analysis: one-way ANOVA.
Cultured HL-1 atrial cells (Claycomb et al., 1998) were transfected with the WT and mutated Kir2.1-GFP that included an extracellular HA-tag. We used Kir2.1-GFP WT construct that did not include the extracellular HA-tag, as control. HL-1 cells were gently fixed and immunostained with HA-tag antibody. Green fluorescence following the expression of the Kir2.1-GFP was observed, mainly as aggregates in cytoplasmic organelles (probably the endoplasmic reticulum and Golgi apparatus), but also at the cell periphery. Following immunostaining, red fluorescence was detected at the plasma membrane of cells expressing Kir2.1-GFP WT, V77E, and M307V, but not when the HA-tag was not expressed. When the red and green signals were superimposed, a yellow signal was detected on the cell border, demonstrating the expression of Kir2.1-GFP WT, V77E, and M307V at the cell surface (Figure 3A).
Next, HEK cells were transfected with Kir2.1-GFP and incubated with biotin. Biotin-labeled cell-membrane proteins and total protein fractions were analyzed using Western blot. Kir2.1-GFP WT, V77E, and M307V were detected at the cell surface with no significant difference between groups, indicating that V77E and M307V did not affect Kir2.1 cellular expression, or cell surface expression that possibly reflects trafficking [Figures 3Ba (upper panel), Bb].
In order to investigate the functional and biochemical properties of each of the two variants, V77E and M307V, expressed in a heterozygotic context, we used a subunit concatenation strategy where two Kir2.1 subunits were fused as dimers. In the Kir2.1 dimers, the first Kir2.1 subunit was WT and the second subunit was either a WT subunit (“WT dimer”) or a mutated subunit (“mutant dimers”). Kir2.1 subunits were connected with a flexible 10 a.a. linker that included eight glycines. When the same amount of DNA was used for the transfection of Kir2.1-GFP monomers and Kir2.1 dimers, total expression levels of Kir2.1 dimers were 20%–30% of the monomers, with no significant difference between the WT and mutant dimers (Figure 3Ba, lower panel, Supplemental image 1). The expressed Kir2.1 dimers were exported to the cell surface so that the biotinylated fractions of WT and mutant dimers were not significantly different from each other (Figure 3Bb, Supplemental image 2). These experiments show that the expression of Kir2.1 dimer on the plasma membrane is reduced compared to Kir2.1-GFP monomers, but the mutations do not affect the surface expression of either Kir2.1 monomers or dimers.
A reduction in WT dimer function correlated with the reduction in its expression level: normalized current density at −120 mV of the WT dimer was 28% ± 5% of the Kir2.1-GFP WT monomer (Figure 4A). The normalized current waveform elicited by voltage ramp from −120 to 0 mV showed only a small change in current rectification and selectivity properties of the WT dimer compared to the Kir2.1-GFP WT monomer (Figure 4B). These results demonstrate that Kir2.1 subunit concatenation partially affected protein expression, but the cell-surface expressed Kir2.1 composed of the WT dimers had functional properties similar to the Kir2.1-GFP WT monomers.
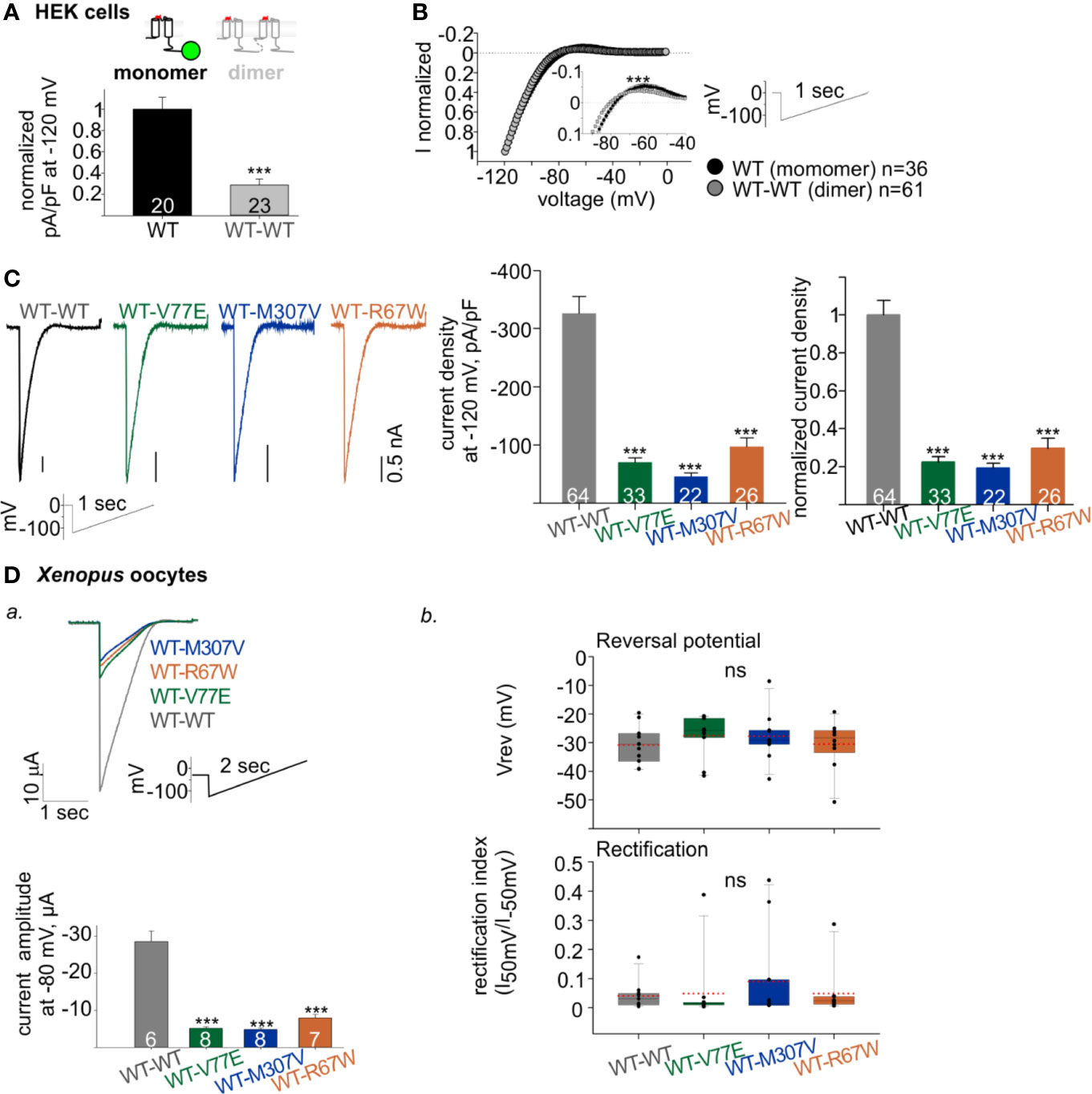
Figure 4 Functional properties of Kir2.1 dimers. (A) Normalized current densities of Kir2.1-GFP WT monomer and WT dimers, recorded in HEK cells transfected with equal amounts of DNA. Normalization was performed to the mean current density of Kir2.1-GFP from the same experiment (N = 3). Statistical analysis: t-test. (B) Normalized currents (left) elicited by a ramp protocol (right). Every 10th recorded point is presented. This is a summary of all cells from different experiments (N = 7 experiments for Kir2.1-GFP WT monomer and N = 12 for WT dimer). Inset, zoom on the reversal potential and the outward currents. Statistical analysis: two-way ANOVA with repeated measures, p < 0.001. (C) Left, representative currents elicited by ramp protocol of HEK cells expressing Kir2.1 dimers. Middle, current densities at −120 mV. Right, current density at −120 mV normalized to mean current density from cells expressing Kir2.1 WT dimers from the same experiment (WT-WT was compared to WT-V77E in N = 7, to WT-M307V in N = 4 and to WT-R67W in N = 6 experiments). Statistical analysis: one-way ANOVA (Dunn’s test, comparison to WT Group). (D) Kir2.1 dimers RNA (1 ng/oocyte) was injected in Xenopus oocytes and recorded in 24 mM K+ bath solution. a. Top, representative evoked currents by a voltage ramp protocol. Bottom, average current amplitudes at −80 mV. Statistical analysis: t-test compared to the WT-WT group. N = 1 experiment. b. Reversal potentials (Vrev) and rectification index calculated from the current elicited by voltage ramps, n = 10–11, N = 2 experiments. Statistical analysis: one-way ANOVA.
Both V77E and M307V Induce a Dominant-Negative Effect on Current Amplitude but Do Not Change the Selectivity and Rectification Properties
The construction of Kir2.1 dimers enabled functional investigation of loss-of-function mutant with a fixed stoichiometry of WT:mutant subunits within a Kir2.1 tetramer. Kir2.1 is expected to be an assembly of 2 dimers, therefore the expression of mutant dimers is expected to form Kir2.1 tetramer composed of two WT and two mutated subunits. As a control, we used the loss-of-function Kir2.1-R67W variant, previously reported as a dominant-negative mutant (Andelfinger et al., 2002) with no effect on cell surface expression (Kalscheur et al., 2014). When each of the mutant dimers were expressed (WT-V77E, WT-M307V, and WT-R67W), typical rectifying Kir2.1 currents were recorded in HEK cells (Figure 4C, left). However, when equal amounts of DNA were used for transfection of WT and mutant dimers, the normalized current densities of mutant dimers were 22% ± 3%, 19% ± 2%, and 30% ± 5%, for WT-V77E, WT-M307V, and WT-R67W, respectively, compared to WT dimer (Figure 4C, right). The prominent reduction in current indicates that V77E and M307V mutated subunits possibly exert a dominant-negative effect on the functional WT subunits since the reduction in current was beyond their expected relative expression (of 50%) in a Kir2.1 tetramer.
In order to confirm the dominant-negative effect of the mutants, RNA’s of Kir2.1 dimers were injected in Xenopus oocytes, and currents were measured. RNA injection in Xenopus oocytes enabled to control protein expression and to limit variability between cells, compared to DNA transfection in HEK cells. Similar to HEK cells, oocytes’ current amplitudes in mutants dimers were reduced to 18% ± 1%, 17% ± 1%, and 28% ± 3% of WT dimer, for WT-V77E, WT-M307V, and WT-R67W, respectively (Figure 4Da). These results in Xenopus oocytes corroborate the putative dominant-negative effect of the loss-of-function Kir2.1 variants M307V and V77E, as shown in HEK cells.
Although a dominant-negative effect is highly plausible, the subunits within the dimer may have additional features not seen in the native channel. Nonetheless, the previously reported dominant-negative variant R67W (Andelfinger et al., 2002) showed a similar reduction in amplitude within the dimer as the two novel mutations V77E and M307V.
Channel selectivity and rectification properties of the mutant dimers were analyzed in Xenopus oocytes. The calculation of these parameters in oocytes was reliable due to the large current amplitude compared to HEK cells. Oocytes were perfused with 24 mM K+ solution (the cytosolic K+ concentration in Xenopus oocytes is estimated ~100 mM (Dascal, 1987) and the calculated Nernst potential is ~−36 mV). The reversal potential and rectification index did not show a significant change when mutant dimers were compared to the WT dimer (Figure 4Db). Thus, although the dominant-negative mutants strongly reduced the whole-cell conductance in cells expressing Kir2.1 composed of the mutant dimers (WT-V77E, WT-M307V, and WT-R67W), the selectivity and rectification properties of the residual Kir2.1 currents were not affected, compared to Kir2.1 composed of the WT dimers.
Kir2.1 Mutations V77E, M307V, and R67W Increase Current Sensitivity to PIP2 Depletion
We next characterized the functional sensitivity of Kir2.1 to PIP2. We depleted PIP2 membrane levels by voltage activation of VSP phosphatase that dephosphorylates plasma membrane phosphoinositide at 5′ position upon depolarization (Murata et al., 2005). PIP2 depletion via VSPs results in a decay of Kir2.1 current (Murata and Okamura, 2007; Sakata and Okamura, 2014; Rjasanow et al., 2015).
We aimed to design a calibrated VSP activation protocol in order to quantify the VSP effect on Kir2.1 currents within a dynamic range. We coexpressed Kir2.1 and VSP in HEK cells and recorded the changes in Kir2.1 current amplitudes following a gradual increase in VSP activation time (Figures 5A, B) or voltage (Figure 5C). We employed a double-pulse voltage ramp protocol and measured the current amplitudes at −120 mV before VSP activation (I1) and after VSP activation (I2). We then calculated the I2/I1 ratio (left Y-axis, Figures 5B–D) and the corresponding % VSP effect (right Y-axis, Figures 5B–D). VSP activation by 5-s depolarization to 100 mV was chosen as the standard duration for the following analysis.
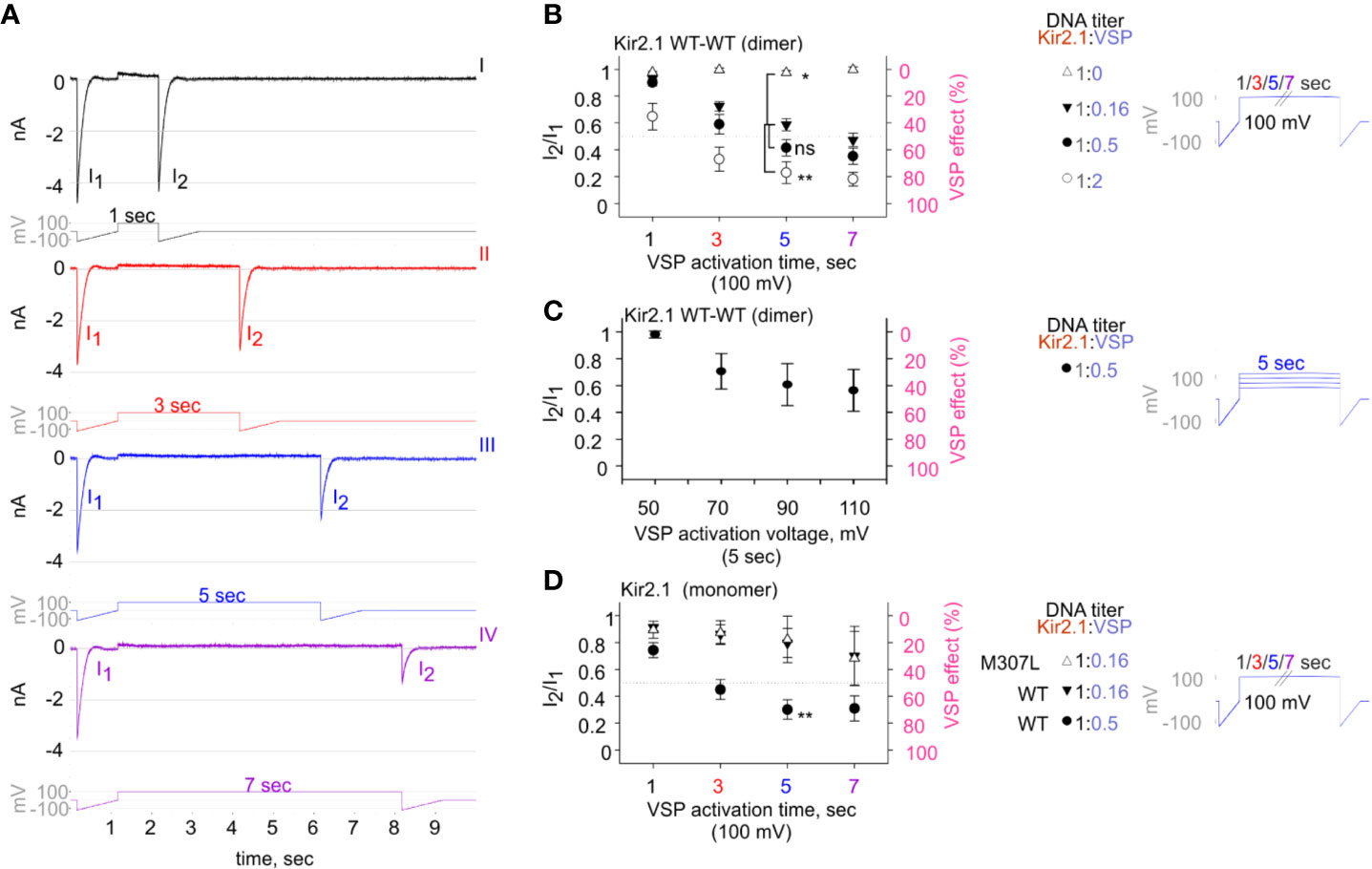
Figure 5 The calibrated VSP activation protocol testing Kir2.1 sensitivity to PIP2 depletion by VSP in HEK cells. (A) A representative four sweeps (I–IV)-protocol used to quantify Kir2.1 current amplitude reduction following VSP activation by depolarization. Each sweep started with a voltage ramp, followed by a 100-mV voltage step (for 1, 3, 5, or 7 s) and ended with a second voltage ramp. An example of currents elicited in a HEK cell transfected with Kir2.1 WT dimer and VSP in 1:0.16 DNA ratio. (B–D) Summary of the maximal amplitude ratio in each sweep after/before VSP activation (left Y-axis), also displayed as % VSP effect (right Y-axis, in pink). Voltage-protocol on the right. (B) At DNA ratio Kir2.1:VSP 1:0 (△), n = 10, 6, 4, 3; 1:0.16 (▼), n = 63, 63, 52, 44; 1:0.5 (•), n = 26, 22, 19, 18; 1:2 (○), n = 13, 10, 9, 8 for VSP activation at 100 mV for 1, 3, 5, and 7 s, respectively. Statistical analysis: one-way ANOVA at 5-s VSP activation [comparison to 1:0.16 (▼) group]. (C) VSP activation by steps to increasing voltages in each sweep (right). Currents amplitude ratio and the corresponding VSP effect from HEK cells transfected with WT dimer and VSP in 1:0.5 DNA ratio (left). n = 8, 8, 7, 6 for 5-s VSP activation at 50, 70, 90, and 110 mV, respectively. (D) Currents amplitude ratio and the corresponding VSP effect in Kir2.1-GFP monomers. At DNA ratio 1:0.16 of Kir2.1-GFP M307L:VSP (△), n = 5, 5, 4, 3; Kir2.1-GFP WT : VSP 1:0.16 (▼), n = 8, 9, 4, 4; Kir2.1-GFP WT : VSP 1:0.5 (•), n = 22, 19, 14, 9 for 1, 3, 5, and 7 s, respectively. Statistical analysis: t-test at 5-s VSP activation of the WT expressing groups, p = 0.004.
VSP activation protocol was additionally calibrated using variable Kir2.1:VSP DNA ratios for transfection. In the group that did not express VSP (Kir2.1:VSP DNA ratio 1:0), the depolarizing pulses did not change the amplitude ratio of I2/I1 and the VSP effect was nearly 0 (Figure 5B, empty triangle). Raising the DNA proportion of VSP-to-WT dimers exacerbated the reduction in current amplitudes following VSP activation. After 5 s of depolarization to 100 mV, the second ramp evoked a slight reduction of 2.6% ± 1.7% in the current, in the absence of expressed VSP. When VSP was expressed, current reductions of 41% ± 4.5%, 58% ± 6%, and 77% ± 8% were evoked when the WT dimer:VSP DNA ratios were 1:0.16, 1:0.5, and 1:2, respectively (Figure 5B). Augmenting the depolarizing voltage of the 5-s step gradually increased the VSP effect on WT dimer current. VSP effect increased from 2% ± 2.6% to 29.5% ± 13.3%, 39.3% ± 15.6%, and 43.64% ± 15.6% for 50, 70, 90, and 110 mV, respectively (Figure 5C).
Current reduction following the VSP activation protocol was also observed in Kir2.1-GFP composed of monomers. The VSP effect was dependent on the transfected DNA proportion Kir2.1:VSP. After 5 s at 100 mV, the VSP effect increased from 20% ± 10% to 70% ± 7% when Kir2.1-GFP WT : VSP DNA ratios were 1:0.16 and 1:0.5, respectively (Figure 5D). VSP effect on the functional variant Kir2.1-GFP M307L was similar to that of Kir2.1-GFP WT. After 5 s of VSP activation at 100 mV, the VSP effect on Kir2.1-GFP M307L monomer was 17% ± 17% (Kir2.1:VSP DNA ratio 1:0.16, Figure 5D).
We then employed the calibrated VSP activation protocol on Kir2.1 mutant dimers, with the parameters that allowed quantification of Kir2.1 current response within a dynamic range. We transfected HEK cells with Kir2.1 WT and mutant dimers, at a Kir2.1:VSP DNA ratio of 1:0.16, and measured the VSP effect on Kir2.1 currents. Using the calibrated VSP activation protocol, with the 5-s step to 100 mV, the VSP effect increased from 41% ± 4.5% in Kir2.1 WT dimers, to 82% ± 3.7%, 81% ± 4.5%, and 74% ± 5.8% in mutant dimers (Figures 6A, B), representing a 197% ± 9%, 192% ± 11%, and 179% ± 14% increase in the normalized VSP effect of WT-V77E, WT-M307V, and WT-R67W currents, respectively (Figure 6C).
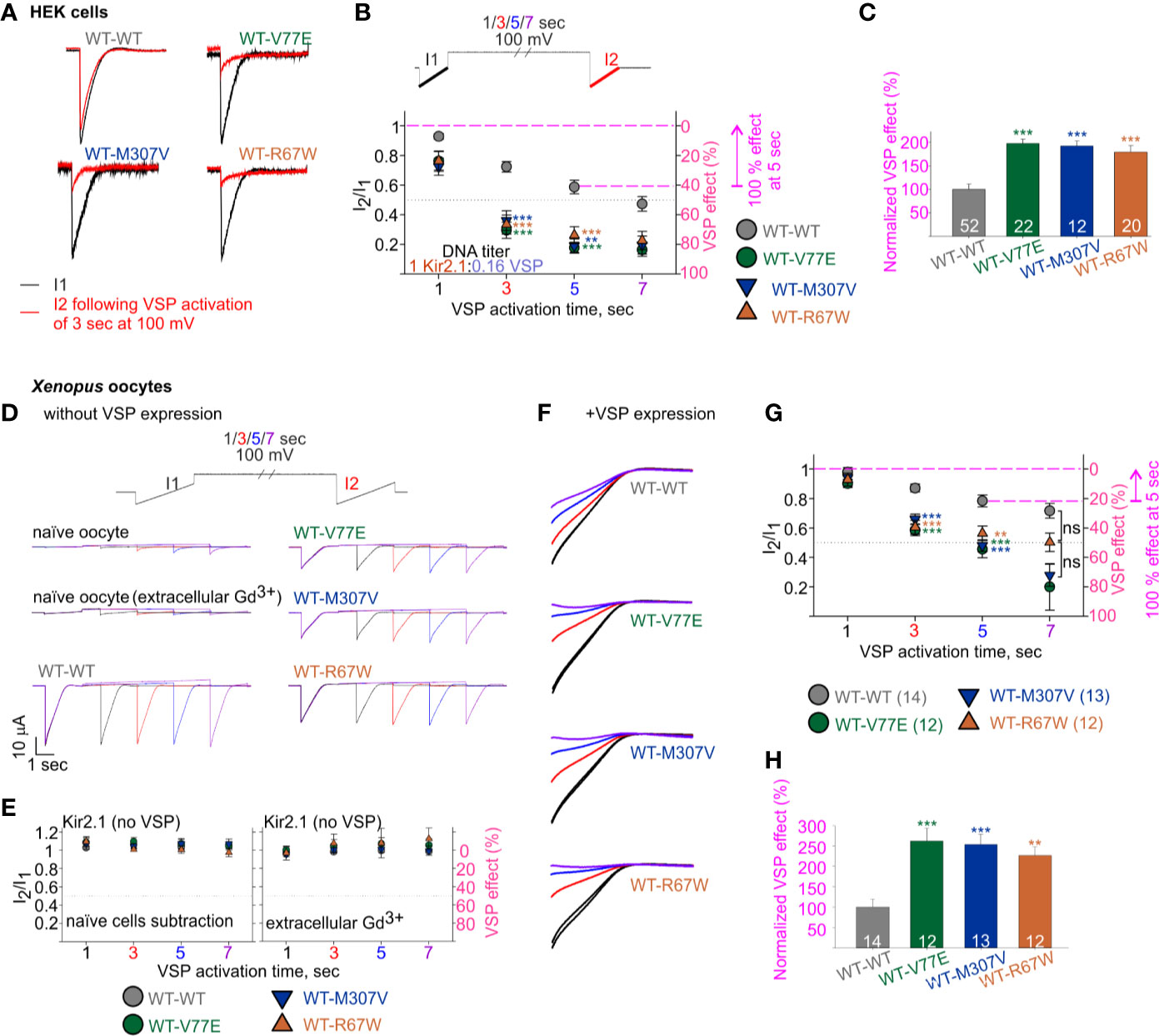
Figure 6 Increased sensitivity of mutant dimers to PIP2 depletion using the calibrated VSP activation protocol. (A–C) Currents in HEK cells expressing Kir2.1 dimers and VSP. (A) Representative ramp currents before (black) and after (red) VSP activation by a 3-s step to 100 mV. Traces of the WT dimer are the same as sweep II in Figure 5A. (B) Currents amplitude ratio and the corresponding VSP effect recorded from cells transfected with Kir2.1 dimer:VSP DNA ratio of 1:0.16. WT-WT is as summarized in Figure 5B (n = 63, 63, 52, 44). WT-V77E, n = 20, 20, 22, 17; WT-M307V, n = 16, 15, 12, 7; WT-R67W, n = 26, 24, 20, 15 for VSP activation by 100 mV step for 1, 3, 5, and 7 s, respectively. Statistical analysis: one-way ANOVA (Dunn’s test, comparison to WT-WT group). At 5-s VSP activation: WT-V77E and WT-R67W, p < 0.001; WT-M307V, p = 0.003. The dashed magenta lines in panel B illustrate the normalizing value for the VSP effect on panel C. (C) Normalized VSP effect at a 5-s VSP activation compared to WT dimer. Statistical analysis: one-way ANOVA (Dunn’s test, comparison to WT-WT Group). (D–H) Currents in Xenopus oocytes expressing Kir2.1 dimers without (D, E) and with VSP (F–H). Representative currents in naïve and Kir2.1 dimers expressing oocytes (D), and the average current amplitude ratio and the corresponding VSP effect (left, n = 7–8; right, n = 3–4) (E). (F) Representative currents elicited in oocytes using the VSP activation protocol, and following subtraction of the averaged current from naïve cells. I1 and I2 of sweep I are in black, I2 of the sweeps II in red, III in blue, and IV in purple. Current amplitude ratio and the corresponding VSP effect (G) and the normalized VSP effect per experiment, at 5-s VSP activation (H). N = 2 experiments. Statistical analysis at 3- and 5-s VSP activation was one-way ANOVA (Dunnett’s test, comparison to WT-WT group). In panel G, at 5-s VSP activation, WT-R67W, p = 0.005. At 7-s VSP activation: statistical analysis was using Kruskal–Wallis one-way ANOVA. In panel H, statistical analysis was using Kruskal–Wallis one-way ANOVA. WT-R67W, p = 0.003. The dashed magenta lines in panel G illustrate the normalizing value for the VSP effect in panel (H).
The current amplitude of the Kir2.1 mutant dimers WT-V77E and WT-M307V was 19%–22% of the WT-WT dimer (Figure 4C). This reduced current is expected to be the result of a reduced conductance and not due to low protein expression (Figure 3B). Thus, the Kir2.1:VSP expression ratio is expected to be similar in mutant compared to WT dimers, suggesting that the strong VSP effect reflects an increased sensitivity to PIP2 depletion in Kir2.1 mutant dimers.
We sought to test the calibrated VSP activation protocol in Xenopus oocytes (Figures 6D–H). We started by applying the VSP activation protocol in naïve Xenopus oocytes. We detected time- and voltage-dependent endogenous inward currents. The current amplitude evoked by the voltage ramp increased following depolarization steps, and at −120 mV it often reached a few µA following a 7-s depolarization to 100 mV (Figure 6D, a representative naïve oocyte out of six, and one oocyte out of nine in the presence of Gd3+). Thus, when we wanted to test VSP effect on Kir2.1 expressing oocytes, we either subtracted the mean current of naïve cells obtained by the VSP activation protocol or applied 50 µM Gd3+ in the external solution. Gd3+ partially inhibited the increase in the endogenous currents following the depolarization (Figure 6D), while Kir2.1 current amplitude and the VSP effect did not change (not shown). When VSP was not expressed in oocytes, the depolarizing pulses did not reduce the currents of WT and mutant dimers, when either method to avoid artifacts by endogenous currents was applied (Figure 6E).
We wanted to test whether the reduction in basal current amplitude itself in the mutant dimers’ current (Figure 4Da) underlies the difference in VSP effect. Thus, we adjusted RNA amounts of WT and mutant dimes by injecting 10- to 20-fold less RNA of the WT dimer than of mutant dimers, to attain similar amplitudes. When Kir2.1:VSP RNA ratio was 0.1:10 or 0.05:10 in WT dimer and 1:10 in mutant dimers, the average current amplitudes at −80 mV of WT dimer were −10 ± 1.1 µA for 0.1 ng RNA (n = 8) and −1.8 ± 0.11 µA for 0.05 ng RNA (n = 6), while the amplitudes of the mutant dimers were −3 ± 0.3 µA (n = 12), −2.3 ± 0.27 µA (n = 13), and −5 ± 1 µA (n = 12) for WT-V77E, WT-M307V, and WT-R67W, respectively.
Oocytes expressing both VSP and Kir2.1 dimers showed current amplitude decays during the VSP activation protocol (Figure 6F). The VSP effect was stronger in all mutant dimers compared to WT dimer, despite the lower Kir2.1:VSP RNA ratio used for WT dimer. The VSP effect, following a 5-s depolarization step to 100 mV, was 21% ± 4% in WT dimer and 54% ± 6%, 52% ± 64%, and 43% ± 65% in mutant dimers (Figure 6G), corresponding a normalized VSP effect of 262% ± 631%, 254% ± 625%, and 226% ± 621% in WT-V77E, WT-M307V, and WT-R67W, respectively (Figure 6H). These results demonstrate that the mutant dimers WT-V77E, WT-M307V, and WT-R67W have an increased sensitivity to PIP2 depletion (and to a similar extent), compared to WT dimers of Kir2.1.
BGP-15 Decreases Kir2.1 Sensitivity to VSP-Induced PIP2 Depletion
We studied the effect of BGP-15 on Kir2.1 current. BGP-15 has been previously shown to have cardio-protective properties and to reduce cardiac arrhythmias, possibly via lipid regulation (Gombos et al., 2011; Zhang et al., 2011; Gungor et al., 2014; Sapra et al., 2014). We hypothesized that BGP-15 affects lipid-regulated ion channels’ function, consequently leading to an improvement in cardiac muscle function and reducing cardiac arrhythmia. HEK cells expressing Kir2.1 dimers and VSP were incubated overnight with BGP-15 (50 µM) and currents were recorded on the following day. Kir2.1 current density was not affected by BGP-15 incubation (Figure 7A, left). Selectivity and rectification of Kir2.1 current were barely affected by BGP-15 incubation, as shown by the normalized current waveform elicited by a voltage-ramp (Figure 7A, right).
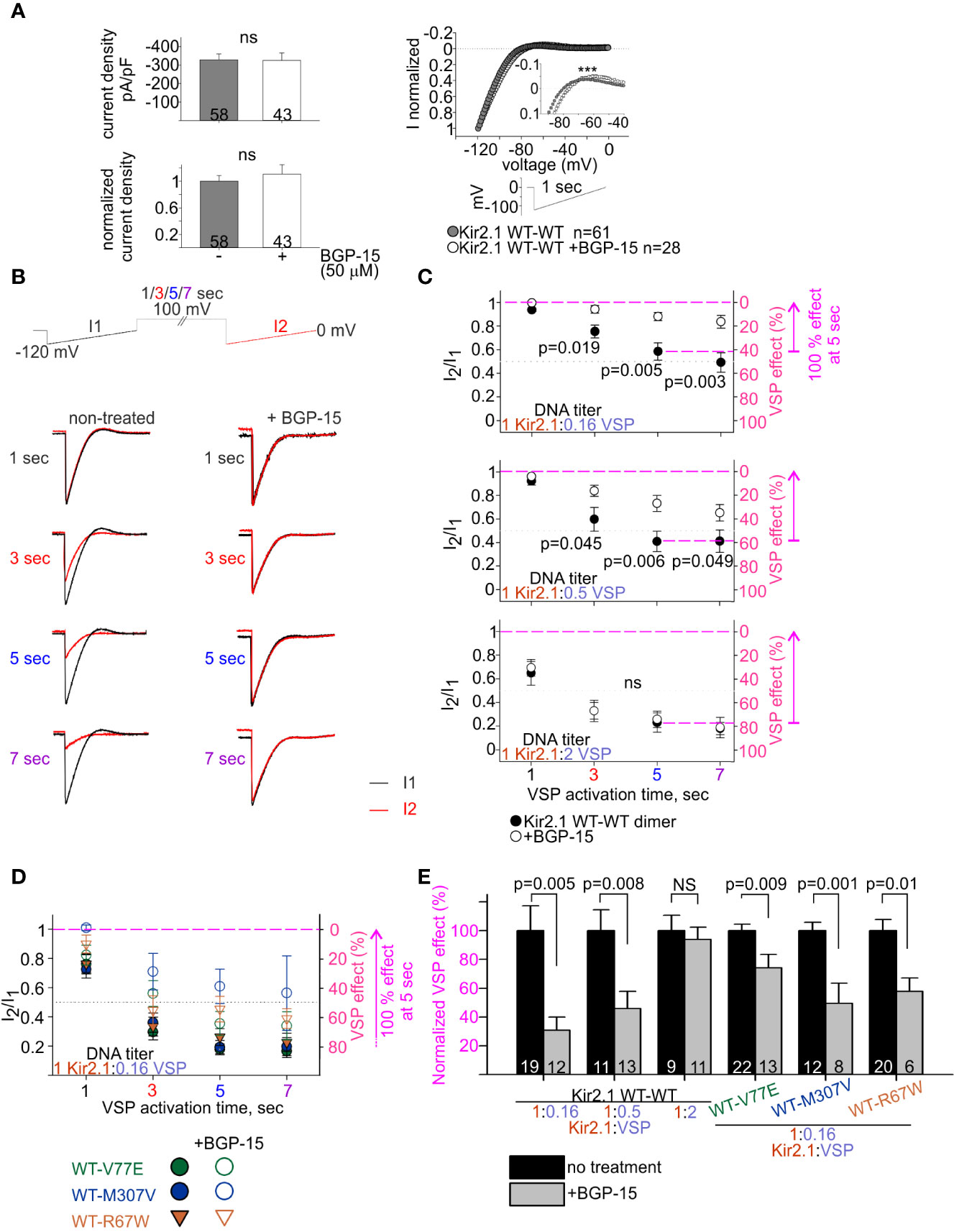
Figure 7 BGP-15 reduces the sensitivity of mutant dimers to PIP2 depletion using the calibrated VSP activation protocol. (A) Left, averaged current densities at −120 mV from HEK cells expressing Kir2.1 WT dimers with and without BGP-15 treatment. Top, current densities, bottom, current densities in cells that were incubated with BGP-15 and normalized to the mean current density in non-treated cells from the same experiment. Statistical analysis: Mann–Whitney rank-sum test. Right, ramp current waveform normalized to the current at −120 mV, from non-treated cells (same as WT dimer in Figure 4B), and BGP-15 treated cells. Statistical analysis: two-way ANOVA with repeated measures, p < 0.001. (B) Representative currents elicited in non-treated and 50 µM BGP-15 incubated HEK cells. Example in cells transfected with Kir2.1 WT dimer and VSP in 1:0.5 DNA ratio. (C) Average current amplitude ratio and the corresponding VSP effect of WT dimer and VSP expressing cells, following BGP-15 treatment. Top, n = 13, 12, 12, 11 for BGP-15-treated and n = 25, 21, 19, 15 for non-treated cells. N = 3 experiments. Middle, n = 16, 13, 13, 13 for BGP-15-treated and n = 17, 14, 11, 10 for non-treated cells. N = 2 experiments. Bottom, n = 12, 11, 11, 5 for BGP-15-treated and n = 13, 10, 9, 7 for non-treated cells. N = 2 experiments. Statistical analysis: t-test comparing treated with non-treated groups in the same protocol. (D) Average current amplitude ratio and the corresponding VSP effect of mutant dimer and VSP expressing cells with and without BGP-15 treatment. Kir2.1:VSP DNA ratio was 1:0.16. VSP effect from non-treated mutant dimers is the same as in Figure 6B. For BGP-15 treated groups, WT-V77E n = 18,16,13,7 from N = 3 experiments, WT-M307V n = 5,7,8,3 from N = 1 experiment, WT-R67W n = 7, 8, 6, 6 from N = 2 experiments. (E) Normalized VSP effect on Kir2.1 currents following a 5-s depolarization step to 100 mV. The mean VSP effect in non-treated cells (represented by dashed magenta lines in (C, D), of each experiment, is the normalizing value for the VSP effect in panel (E). Statistical analysis: t-test.
We then examined the effect of BGP-15 incubation on the PIP2 sensitivity of Kir2.1 WT dimers, using the VSP activation protocol (Figure 7B). Within the dynamic range of VSP action, BGP-15 incubation reduced the VSP effect. After 5 s of depolarization to 100 mV, VSP effect in BGP-15 treated cells decreased from 42% ± 7% to 12% ± 3% at 1:0.16 DNA ratio (Figure 7C, top) and from 59% ± 8% to 27% ± 7% at 1:0.5 DNA ratio (Figure 7C, middle). Basically, when normalized to the VSP effect without BGP-15 from the same experiment, VSP effect in BGP-15 treated cells was reduced to 30% ± 9% and 45% ± 12%, in cells transfected with Kir2.1:VSP DNA ratios of 1:0.16 and 1:0.5, respectively (Figure 7E). However, when the VSP proportion was raised to a DNA ratio of 1:2 Kir2.1:VSP, BGP-15 incubation did not reduce the VSP effect. After 5-s depolarization to 100 mV, VSP effect was 77% ± 8% in untreated cells, and 74% ± 7% in BGP-15 treated cells (Figure 7C, bottom), and the normalized VSP effect following BGP-15 incubation was 95% ± 6% (Figure 7E). To summarize, within a dynamic response range, BGP-15 incubation reduced the VSP effect and stabilized Kir2.1 amplitudes in the calibrated VSP activation protocol.
The VSP activation protocol was then used to test the effect of BGP-15 incubation on the PIP2-sensitivity of Kir2.1 mutant dimers. Within the dynamic Kir2.1:VSP DNA ratio of 1:0.16 determined for WT dimers (Figure 7C), BGP-15 treatment reduced the VSP effect and increased its variability in the three mutant dimers, compared to the non-treated cells (Figure 7D). After 5-s depolarization to 100 mV, the normalized VSP effect following BGP-15 incubation was 74% ± 9%, 49% ± 14%, and 58% ± 9% of the corresponding non treated groups, for the mutant dimers WT-V77E, WT-M307V, and WT-R67W, respectively (Figure 7E).
In all, in this set of experiments BGP-15 incubation reduced Kir2.1 current susceptibility to PIP2 depletion and stabilized Kir2.1 amplitudes in the calibrated VSP activation protocol, of both Kir2.1 WT and the ATS-associated mutant dimers.
Discussion
In this study, we report on ATS associated with two mutations in KCNJ2, both induce a loss-of-function and exert a dominant-negative effect on the Kir2.1 tetramer. The major outcomes and findings include 1) development of a calibrated functional assay to assess PIP2 sensitivity of Kir2.1 current that was tested by VSP activation, and 2) Kir2.1 mutants V77E, M307V, and R67W showed high sensitivity to PIP2 depletion, which we interpret as lability in the PIP2-Kir2.1 interaction, 3) BGP-15 incubation decreased the sensitivity to PIP2 depletion, of both WT and mutant Kir2.1, possibly stabilizing the Kir2.1 open state.
Phenotypic Variability in ATS
ATS is a rare genetic disease, with a prevalence of 0.08–0.1:100,000 (Horga et al., 2013; Stunnenberg et al., 2018). Cardiac ventricular arrhythmias may include prolonged QT interval, prominent U-wave or ventricular ectopy. Bidirectional VTs were also reported with ATS, although clinical distinction from CPVT is challenging in the absence of other characteristics (Kimura et al., 2012; Chakraborty et al., 2015; Nguyen and Ferns, 2018).
Mutations in Ca2+-handling proteins contribute to the etiology of CPVT, while impaired activity of inward-rectifier K+-channels is the main cause of ATS (Napolitano et al., 2004; Veerapandiyan et al., 2004). In our report, identification of loss-of-function KCNJ2 variants established an ATS-1 diagnosis. Although ATS-1 is considered a relatively benign channelopathy, some affected members had malignant ventricular arrhythmias that necessitated an ICD implantation.
The mode of inheritance of the familial M307V mutation was autosomal dominant. Expression was variable and gender-dependent: from six affected family members, three females reported palpitations and presented with incessant bidirectional VT. Two of them had symptomatic arrhythmia events and were implanted with an ICD. In contrast, the three males presented with PVCs, all with normal QT interval, and remained asymptomatic (Nof et al., 2004). The variant M307V was previously reported a de novo KCNJ2 mutation in a 15-year-old male with ATS manifestations including muscular weakness and dysmorphic features, although his ECG findings were within a normal range (Liu et al., 2015). The V77E mutation occurred de-novo, as reported in 30-50% of KCNJ2 mutations (Tristani-Firouzi et al., 2002; Hasegawa et al., 2015).
Structural Analysis of the Mutated Kir2.1
The mutation V77E, located in the amphipathic slide helix of Kir2.1 N-terminus, is a substitution of a non-polar, hydrophobic, residue with a negatively charged residue. Most disease-associated variants in Kir2.1 slide helix were detected in polar residues facing the CTD (Table 1, Figure 8A). Inter and intra-subunit interactions in the interface of CTD and polar residues in the slide helix were suggested to transduce Kir2.1 opening (Decher et al., 2007; Bavro et al., 2012). The position of V77 side chain, in vicinity to the PIP2 docking site and particularly the switch to a charged residue, may be implicated in the formation of a new molecular interaction that may have altered the channel interaction network and affected the activation dynamics or Kir2.1-PIP2 docking, directly or indirectly. Positively charged residues K182, K185 and K187 on Kir2.1 inner TMD and the tether helix structure (equivalent to K183, R186, and K188 in Kir2.2), are candidates for such interaction. These residues directly interact with 5′ phosphate in the inositol ring of PIP2 in a Kir2.2-PIP2 structural model (Hansen et al., 2011; Lu et al., 2016).
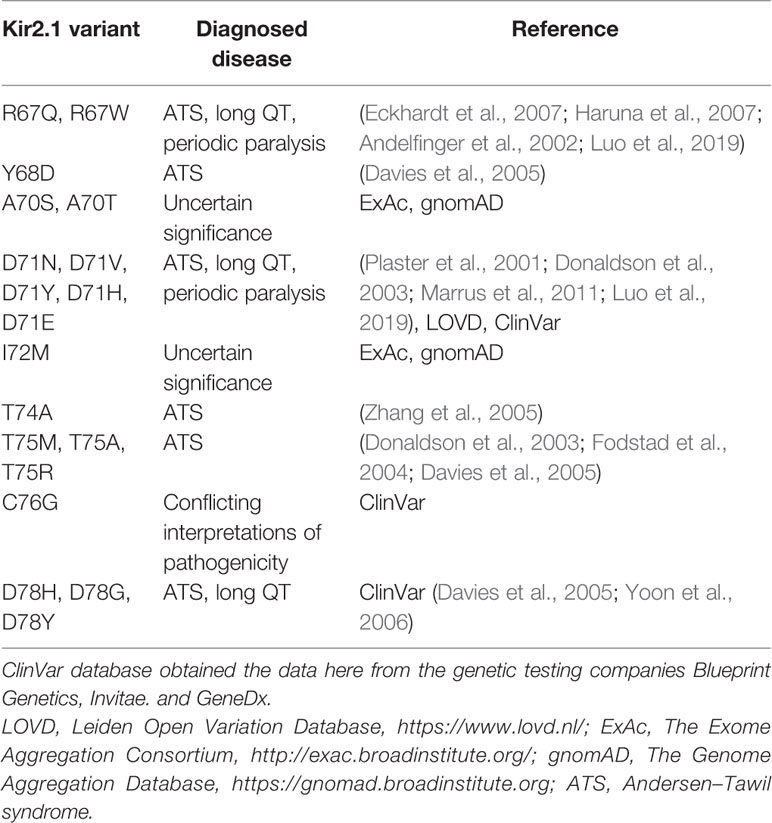
Table 1 Missense variants in Kir2.1 slide helix.
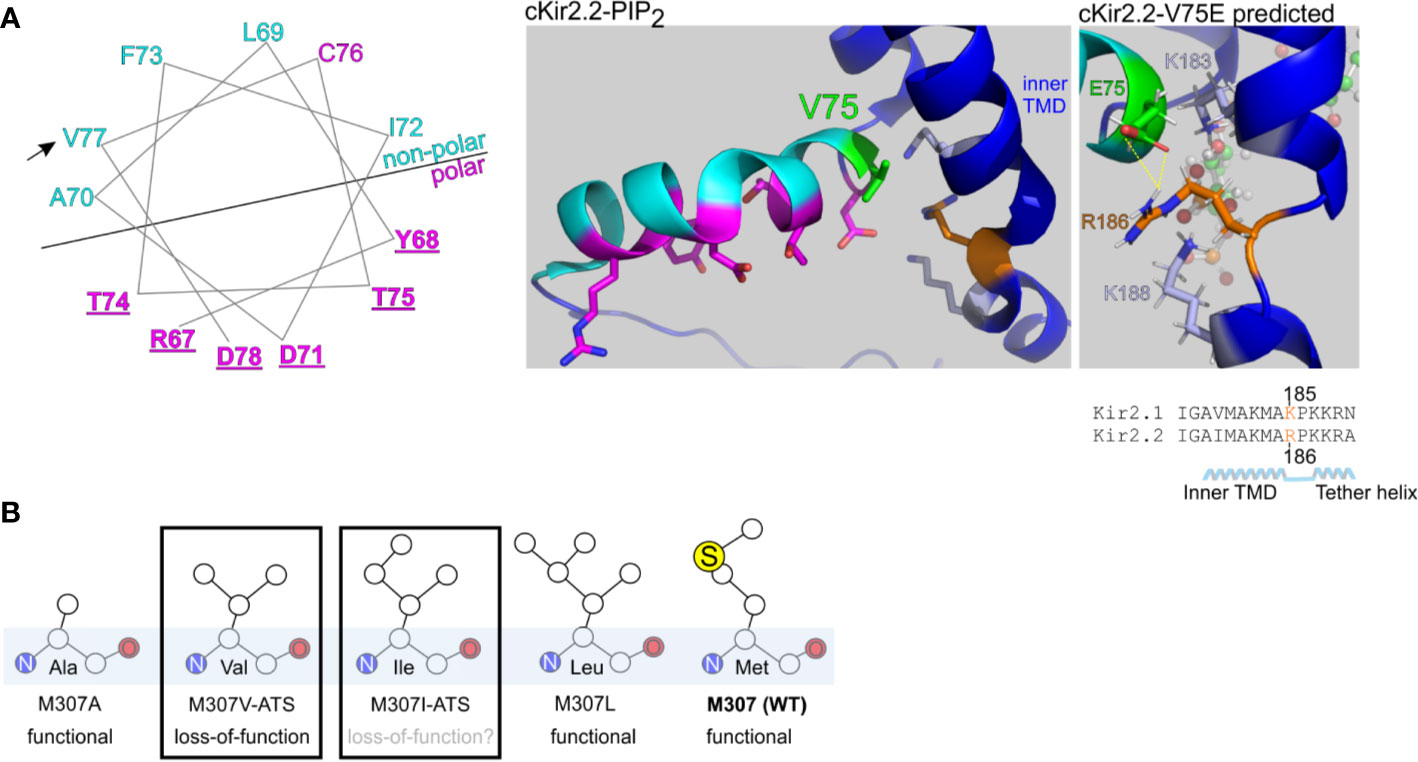
Figure 8 Structural predictions of ATS-associated Kir2.1 variants. (A) Left, wheel plot illustration of the residues in Kir2.1 N-terminal slide helix. Polar residues in magenta, non-polar residues in cyan. Mutations in the underlined residues are disease-associated (Table 1). Arrow points on the ATS-associated residue in this report. Middle, polar (magenta) and non-polar (cyan) residues in chicken Kir2.2 + PIP2 structure (PDB ID:3SPI). V75 in Kir2.2 (green) is the homolog of V77 in Kir2.1. Side chains of K183, K188 (light blue) and R186 (orange) are illustrated. Right, V75E mutation was introduced in Kir2.2 + PIP2 structure and underwent a structural refinement. Putative interaction with R186 is shown in yellow. (B) Functional properties of Kir2.1 variants in position 307.
We performed a limited structural analysis of Kir2.1 valine 77 substitution to glutamic acid, based on Kir2.2-PIP2 crystal structure (PDB ID: 3SPI) where the corresponding residue is valine 75. The mutated residue E75 formed a putative polar contact with the residue R186 in Kir2.2, which is homologous to K185 in Kir2.1, a residue between the inner TMD and the tether helix (Figure 8A, right). Following the energy minimization simulation, we have seen that the R186 side chain is expected to rotate and face the E75 side chain, with a minimal distance of 1.9 Å between the charged residues. This interaction may reduce the flexibility of the tether helix or destabilize the interaction with the agonist PIP2, consequently leading to the observed loss-of-function in Kir2.1-V77E.
Methionine 307 is located at the G-loop, a cytoplasmic gating structure in the apex of the CTD (Pegan et al., 2005) (Figure 1C). The functional role of the G-loop, its conformation in different gating states and its reciprocated modulation by Kir ligands remain incompletely understood (Pegan et al., 2006; Nishida et al., 2007; Whorton and Mackinnon, 2011; Whorton and Mackinnon, 2013). However, alterations in the pore diameter, shape and biochemical properties conveyed by the G-loop residues’ side chains are expected to lead to functional impairment (Ma et al., 2007). Our functional results show that unlike in the loss-of-function variant Kir2.1-M307V, currents measured in Kir2.1-M307L were indistinguishable from Kir2.1-WT (Figure 2), like in the previously reported Kir2.1 variant M307A (Pegan et al., 2005). However, the mutation M307I was found in ATS patients, presumably inducing Kir2.1 loss-of-function (Choi et al., 2007). Remarkably, the size of the hydrophobic side chain did not reflect a structural/functional tolerance (Figure 8B), and the potential of methionine to form disulfide bonds seems redundant. Further investigation is needed to shed light on the role of M307 residue, and G-loop in general, in the transition to the open state of Kir2.1.
Applying the Calibrated VSP Activation Protocol to Study PIP2 Regulation of Kir2.1
PIP2 is Kir2 primary agonist (Rohacs et al., 2003) and VSP activation reduces Kir2.1 current amplitude by depleting available PIP2 molecules at the plasma membrane. We designed a protocol that correlated the changes in Kir2.1 amplitude ratios induced by a calibrated VSP activation, in two heterologous expression systems. We propose that the rate of Kir2.1 current reduction following VSP-induced PIP2 depletion reflects Kir2.1 sensitivity to PIP2 interaction so that an accelerated response rate is the consequence of a lower gating efficiency due to low PIP2 affinity to Kir2.1.
In the calibrated VSP activation protocol we compared functional properties of dimer-composed Kir2.1 channels: the homomeric WT channel and the three heteromeric Kir2.1 mutants. We assumed that two dimers assembled into a tetrameric protein, but in a less-likely scenario four WT subunits originated from four mutated dimers might have assembled into one functional homomeric WT channel. This scenario cannot explain the increase in the VSP effect that occurred in mutant dimers but not in WT dimers (Figure 6).
The three Kir2.1 mutants putatively represent three mechanisms of Kir2.1 loss-of-function: V77E that destabilizes the tether helix, M307V that affects G-loop structure and R67W that is involved in inter-subunit interaction with the tether helix and the CTD. Yet, the three Kir2.1 mutant dimers showed a similar reduction in the amplitude compared to the WT dimer (Figure 4), and the calibrated VSP activation protocol did not reveal a difference in the VSP effect of the three mutant dimers (Figure 6), in two expression systems. We propose that the mechanisms of the pathogenic variants V77E, M307V, and R67W converge onto a similar impairment in the gating mechanism. One potential mechanism is a reduced affinity to the agonist PIP2 in mutated Kir2.1 subunits and a total reduction in PIP2 interaction in the heteromeric channel that destabilizes an open state. Based on available crystal structures, we propose a direct impairment of PIP2 interaction in the V77E variant (Figure 8) and an allosteric effect in R67W and M307V variants that may impair an open state conformational transduction.
Finally, we tested the effect of BGP-15 on Kir2.1 currents and its therapeutic potential in ATS-associated Kir2.1 mutants. In a previous report, the oral administration of BGP-15 reduced AF episodes in genetically-engineered mice (Sapra et al., 2014). The AF was induced by age-dependent electrical remodeling that accompanied atrial enlargement in two transgenic mouse models with a dilated and failing heart. The AF mouse model had reduced ion channel expression, including KCNJ3 (Kir3.1) and KCNA5 (Kv1.5) genes (Sapra et al., 2014). Normalization of the fibrillation episodes by BGP-15 may have been related to the stabilization of the resting potential by an increased activity of potassium channels. In this study, following BGP-15 incubation, Kir2.1 currents were more resistant to PIP2 depletion by VSP activation. We propose that BGP-15 is associated with the regulation of PIP2 level, availability or localization, resulting in stabilization of Kir2.1 open-state. An indirect effect on other lipid regulators of Kir2.1 is plausible, e.g. via molulation of the interaction with Kir2.1 suppresor cholesterol, as BGP-15 was shown to remodel cholesterol-enriched lipid platforms (Romanenko et al., 2004; Gombos et al., 2011).
To note, Kir2.1 is predominantly expressed in the cardiac ventricles and ATS is an electrophysiologic disorder of the ventricle (Melnyk et al., 2002). However, other PIP2-dependent inward rectifier potassium channels are expressed in the atria, mainly Kir2.2, Kir2.3, and acetylcholine-activated Kir3.1 and Kir3.4, in addition to other potassium channels such as Kv11.1 (hERG), Kv7.1 (KCNQ1), and Kir6.2 (Schram et al., 2002). Further investigation will shed light on whether the BGP-15 effect is shared by other PIP2-dependent ion channels or is unique to Kir2.1.
Conclusion
In this study, two cases of ATS with cardiac arrhythmias were described, highlighting ATS phenotypic variability and resemblance with CPVT. The ATS-associated mutations, V77E and M307V, expand the spectrum of known KCNJ2 mutations. Mutations were functionally and biochemically studied and showed loss-of-function with a dominant-negative effect. A calibrated protocol quantitated Kir2.1 current amplitude sensitivity to PIP2 depletion and was tested on ATS-associated variants. The Kir2.1 variants showed increased PIP2 sensitivity, which was partially restored by BGP-15.
Data Availability Statement
The datasets generated for this study can be found in the LOVD [https://databases.lovd.nl/shared/variants/0000630833].
Author Contributions
RH-J performed TEVC experiments. EM designed the study and prepared DNA constructs. DY performed simulations. LV performed biochemical studies. RB, MG, and EN evaluated the patients. ND, MG, JM, and SO acquired financial support for the project. ND, JM, and SO conceived and planned the experiments. SO designed the study, performed patch-clamp experiments, and wrote the manuscript with input from all authors.
Funding
This work was supported by Dizengoff Trading Company (1952) Ltd. (MG), Israel Science Foundation (1282/18; ND), and Melbourne Biomedical Precinct Grant, Victorian Government’s Trade and Investment Office for Israel Funding Program (JM and SO).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declare that this study received funding from Dizengoff Trading Company (1952) Ltd. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
Acknowledgments
We thank the patients and their families. We also thank Dr. Ronit S. Cherki for preliminary data and Prof. Yasushi Okamura (Osaka University, Japan) for dr-VSP construct.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2020.00672/full#supplementary-material
References
Andelfinger, G., Tapper, A. R., Welch, R. C., Vanoye, C. G., George, A. L., Jr., Benson, D. W. (2002). KCNJ2 mutation results in Andersen syndrome with sex-specific cardiac and skeletal muscle phenotypes. Am. J. Hum. Genet. 71, 663–668. doi: 10.1086/342360
Balogi, Z., Multhoff, G., Jensen, T. K., Lloyd-Evans, E., Yamashima, T., Jaattela, M., et al. (2019). Hsp70 interactions with membrane lipids regulate cellular functions in health and disease. Prog. Lipid Res. 74, 18–30. doi: 10.1016/j.plipres.2019.01.004
Barajas-Martinez, H., Hu, D., Ontiveros, G., Caceres, G., Desai, M., Burashnikov, E., et al. (2011). Biophysical and molecular characterization of a novel de novo KCNJ2 mutation associated with Andersen-Tawil syndrome and catecholaminergic polymorphic ventricular tachycardia mimicry. Circ. Cardiovasc. Genet. 4, 51–57. doi: 10.1161/CIRCGENETICS.110.957696
Baud, C., Kado, R. T., Marcher, K. (1982). Sodium channels induced by depolarization of the Xenopus laevis oocyte. Proc. Natl. Acad. Sci. U. S. A. 79, 3188–3192. doi: 10.1073/pnas.79.10.3188
Bavro, V. N., De Zorzi, R., Schmidt, M. R., Muniz, J. R., Zubcevic, L., Sansom, M. S., et al. (2012). Structure of a KirBac potassium channel with an open bundle crossing indicates a mechanism of channel gating. Nat. Struct. Mol. Biol. 19, 158–163. doi: 10.1038/nsmb.2208
Chakraborty, P., Kaul, B., Mandal, K., Isser, H. S., Bansal, S., Subramanian, A. (2015). Bidirectional ventricular tachycardia of unusual etiology. Indian Pacing Electrophysiol. J. 15, 296–299. doi: 10.1016/j.ipej.2016.02.007
Choi, B. O., Kim, J., Suh, B. C., Yu, J. S., Sunwoo, I. N., Kim, S. J., et al. (2007). Mutations of KCNJ2 gene associated with Andersen-Tawil syndrome in Korean families. J. Hum. Genet. 52, 280–283. doi: 10.1007/s10038-006-0100-7
Claycomb, W. C., Lanson, N. A., Jr., Stallworth, B. S., Egeland, D. B., Delcarpio, J. B., Bahinski, A., et al. (1998). HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. U. S. A 95, 2979–2984. doi: 10.1073/pnas.95.6.2979
Crul, T., Toth, N., Piotto, S., Literati-Nagy, P., Tory, K., Haldimann, P., et al. (2013). Hydroximic acid derivatives: pleiotropic HSP co-inducers restoring homeostasis and robustness. Curr. Pharm. Des. 19, 309–346. doi: 10.2174/138161213804143716
Dascal, N., Lotan, I. (1992). “Expression of exogenous ion channels and neurotransmitter receptors in RNA-injected Xenopus oocytes,” in Protocols in Molecular Neurobiology. Eds. Longstaff, A., Revest, P. (Totowa, NJ: Humana Press), 205–225.
Dascal, N. (1987). The use of Xenopus oocytes for the study of ion channels. CRC Crit. Rev. Biochem. 22, 317–387. doi: 10.3109/10409238709086960
Davies, N. P., Imbrici, P., Fialho, D., Herd, C., Bilsland, L. G., Weber, A., et al. (2005). Andersen-Tawil syndrome: new potassium channel mutations and possible phenotypic variation. Neurology 65, 1083–1089. doi: 10.1212/01.wnl.0000178888.03767.74
Decher, N., Renigunta, V., Zuzarte, M., Soom, M., Heinemann, S. H., Timothy, K. W., et al. (2007). Impaired interaction between the slide helix and the C-terminus of Kir2.1: a novel mechanism of Andersen syndrome. Cardiovasc. Res. 75, 748–757. doi: 10.1016/j.cardiores.2007.05.010
Donaldson, M. R., Jensen, J. L., Tristani-Firouzi, M., Tawil, R., Bendahhou, S., Suarez, W. A., et al. (2003). PIP2 binding residues of Kir2.1 are common targets of mutations causing Andersen syndrome. Neurology 60, 1811–1816. doi: 10.1212/01.WNL.0000072261.14060.47
Eckhardt, L. L., Farley, A. L., Rodriguez, E., Ruwaldt, K., Hammill, D., Tester, D. J., et al. (2007). KCNJ2 mutations in arrhythmia patients referred for LQT testing: a mutation T305A with novel effect on rectification properties. Heart Rhythm. 4, 323–329. doi: 10.1016/j.hrthm.2006.10.025
Fodstad, H., Swan, H., Auberson, M., Gautschi, I., Loffing, J., Schild, L., et al. (2004). Loss-of-function mutations of the K(+) channel gene KCNJ2 constitute a rare cause of long QT syndrome. J. Mol. Cell Cardiol. 37, 593–602. doi: 10.1016/j.yjmcc.2004.05.011
Gehrig, S. M., Van Der Poel, C., Sayer, T. A., Schertzer, J. D., Henstridge, D. C., Church, J. E., et al. (2012). Hsp72 preserves muscle function and slows progression of severe muscular dystrophy. Nature 484, 394–398. doi: 10.1038/nature10980
Gombos, I., Crul, T., Piotto, S., Gungor, B., Torok, Z., Balogh, G., et al. (2011). Membrane-lipid therapy in operation: the HSP co-inducer BGP-15 activates stress signal transduction pathways by remodeling plasma membrane rafts. PloS One 6, e28818. doi: 10.1371/journal.pone.0028818
Gungor, B., Gombos, I., Crul, T., Ayaydin, F., Szabo, L., Torok, Z., et al. (2014). Rac1 participates in thermally induced alterations of the cytoskeleton, cell morphology and lipid rafts, and regulates the expression of heat shock proteins in B16F10 melanoma cells. PloS One 9, e89136. doi: 10.1371/journal.pone.0089136
Hansen, S. B., Tao, X., Mackinnon, R. (2011). Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature 477, 495–498. doi: 10.1038/nature10370
Haruna, Y., Kobori, A., Makiyama, T., Yoshida, H., Akao, M., Doi, T., et al. (2007). Genotype-phenotype correlations of KCNJ2 mutations in Japanese patients with Andersen-Tawil syndrome. Hum. Mutat. 28, 208. doi: 10.1002/humu.9483
Hasegawa, K., Ohno, S., Kimura, H., Itoh, H., Makiyama, T., Yoshida, Y., et al. (2015). Mosaic KCNJ2 mutation in Andersen-Tawil syndrome: targeted deep sequencing is useful for the detection of mosaicism. Clin. Genet. 87, 279–283. doi: 10.1111/cge.12357
Horga, A., Raja Rayan, D. L., Matthews, E., Sud, R., Fialho, D., Durran, S. C., et al. (2013). Prevalence study of genetically defined skeletal muscle channelopathies in England. Neurology 80, 1472–1475. doi: 10.1212/WNL.0b013e31828cf8d0
Hosaka, Y., Hanawa, H., Washizuka, T., Chinushi, M., Yamashita, F., Yoshida, T., et al. (2003). Function, subcellular localization and assembly of a novel mutation of KCNJ2 in Andersen’s syndrome. J. Mol. Cell Cardiol. 35, 409–415. doi: 10.1016/S0022-2828(03)00046-4
Hossain, M. I., Iwasaki, H., Okochi, Y., Chahine, M., Higashijima, S., Nagayama, K., et al. (2008). Enzyme domain affects the movement of the voltage sensor in ascidian and zebrafish voltage-sensing phosphatases. J. Biol. Chem. 283, 18248–18259. doi: 10.1074/jbc.M706184200
Huang, C. L., Feng, S., Hilgemann, D. W. (1998). Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature 391, 803–806. doi: 10.1038/35882
Kalscheur, M. M., Vaidyanathan, R., Orland, K. M., Abozeid, S., Fabry, N., Maginot, K. R., et al. (2014). KCNJ2 mutation causes an adrenergic-dependent rectification abnormality with calcium sensitivity and ventricular arrhythmia. Heart Rhythm. 11, 885–894. doi: 10.1016/j.hrthm.2014.02.015
Kimura, H., Zhou, J., Kawamura, M., Itoh, H., Mizusawa, Y., Ding, W. G., et al. (2012). Phenotype variability in patients carrying KCNJ2 mutations. Circ. Cardiovasc. Genet. 5, 344–353. doi: 10.1161/CIRCGENETICS.111.962316
Kokunai, Y., Nakata, T., Furuta, M., Sakata, S., Kimura, H., Aiba, T., et al. (2014). A Kir3.4 mutation causes Andersen-Tawil syndrome by an inhibitory effect on Kir2.1. Neurology 82, 1058–1064. doi: 10.1212/WNL.0000000000000239
Krieger, E., Joo, K., Lee, J., Lee, J., Raman, S., Thompson, J., et al. (2009). Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 77 (Suppl 9), 114–122. doi: 10.1002/prot.22570
Lacin, E., Aryal, P., Glaaser, I. W., Bodhinathan, K., Tsai, E., Marsh, N., et al. (2017). Dynamic role of the tether helix in PIP2-dependent gating of a G protein-gated potassium channel. J. Gen. Physiol. 149 (8), 799–811. doi: 10.1085/jgp.201711801
Literati-Nagy, B., Tory, K., Peitl, B., Bajza, A., Koranyi, L., Literati-Nagy, Z., et al. (2014). Improvement of insulin sensitivity by a novel drug candidate, BGP-15, in different animal studies. Metab. Syndr. Relat. Disord. 12, 125–131. doi: 10.1089/met.2013.0098
Liu, X. L., Huang, X. J., Luan, X. H., Zhou, H. Y., Wang, T., Wang, J. Y., et al. (2015). Case report: A Chinese child with Andersen-Tawil syndrome due to a de novo KCNJ2 mutation. J. Neurol. Sci. 352, 105–106. doi: 10.1016/j.jns.2015.02.027
Lopes, C. M., Zhang, H., Rohacs, T., Jin, T., Yang, J., Logothetis, D. E. (2002). Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies. Neuron 34, 933–944. doi: 10.1016/S0896-6273(02)00725-0
Lu, S., An, H., Zhang, H., Long, M. (2016). Structural Basis for Differences in Dynamics Induced by Leu Versus Ile Residues in the CD Loop of Kir Channels. Mol. Neurobiol. 53, 5948–5961. doi: 10.1007/s12035-015-9466-x
Luo, S., Xu, M., Sun, J., Qiao, K., Song, J., Cai, S., et al. (2019). Identification of gene mutations in patients with primary periodic paralysis using targeted next-generation sequencing. BMC Neurol. 19, 92. doi: 10.1186/s12883-019-1322-6
Ma, D., Tang, X. D., Rogers, T. B., Welling, P. A. (2007). An andersen-Tawil syndrome mutation in Kir2.1 (V302M) alters the G-loop cytoplasmic K+ conduction pathway. J. Biol. Chem. 282, 5781–5789. doi: 10.1074/jbc.M608776200
Ma, D., Taneja, T. K., Hagen, B. M., Kim, B. Y., Ortega, B., Lederer, W. J., et al. (2011). Golgi export of the Kir2.1 channel is driven by a trafficking signal located within its tertiary structure. Cell 145, 1102–1115. doi: 10.1016/j.cell.2011.06.007
Marrus, S. B., Cuculich, P. S., Wang, W., Nerbonne, J. M. (2011). Characterization of a novel, dominant negative KCNJ2 mutation associated with Andersen-Tawil syndrome. Channels (Austin) 5, 500–509. doi: 10.4161/chan.5.6.18524
Melnyk, P., Zhang, L., Shrier, A., Nattel, S. (2002). Differential distribution of Kir2.1 and Kir2.3 subunits in canine atrium and ventricle. Am. J. Physiol. Heart Circ. Physiol. 283, H1123–H1133. doi: 10.1152/ajpheart.00934.2001
Murata, Y., Okamura, Y. (2007). Depolarization activates the phosphoinositide phosphatase Ci-VSP, as detected in Xenopus oocytes coexpressing sensors of PIP2. J. Physiol. 583, 875–889. doi: 10.1113/jphysiol.2007.134775
Murata, Y., Iwasaki, H., Sasaki, M., Inaba, K., Okamura, Y. (2005). Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature 435, 1239–1243. doi: 10.1038/nature03650
Napolitano, C., Priori, S. G., Bloise, R. (2004). Catecholaminergic Polymorphic Ventricular Tachycardia. (University of Washington, Seattle, USA: GeneReviews) Bookshelf ID: NBK1289.
Nascimento, T. L., Silva, M. T., Miyabara, E. H. (2018). BGP-15 improves contractile function of regenerating soleus muscle. J. Muscle Res. Cell Motil. 39, 25–34. doi: 10.1007/s10974-018-9495-y
Nguyen, D., Ferns, S. J. (2018). Asymptomatic ventricular tachycardia: diagnostic pitfalls of Andersen-Tawil syndrome-a case report. Eur. Heart J. Case Rep. 2, yty083. doi: 10.1093/ehjcr/yty083
Nishida, M., Cadene, M., Chait, B. T., Mackinnon, R. (2007). Crystal structure of a Kir3.1-prokaryotic Kir channel chimera. EMBO J. 26, 4005–4015. doi: 10.1038/sj.emboj.7601828
Nof, E., Lahat, H., Constantini, N., Luria, D., Rosenfeld, G., Eldar, M., et al. (2004). A novel form of familial bidirectional ventricular tachycardia. Am. J. Cardiol. 93, 231–234. doi: 10.1016/j.amjcard.2003.09.049
Nof, E., Vysochek, L., Meisel, E., Burashnikov, E., Antzelevitch, C., Clatot, J., et al. (2019). Mutations in NaV1.5 Reveal Calcium-Calmodulin Regulation of Sodium Channel. Front. Physiol. 10, 700. doi: 10.3389/fphys.2019.00700
Oz, S., Pankonien, I., Belkacemi, A., Flockerzi, V., Klussmann, E., Haase, H., et al. (2017). Protein kinase A regulates C-terminally truncated CaV 1.2 in Xenopus oocytes: roles of N- and C-termini of the alpha1C subunit. J. Physiol. 595, 3181–3202. doi: 10.1113/JP274015
Pegan, S., Arrabit, C., Zhou, W., Kwiatkowski, W., Collins, A., Slesinger, P. A., et al. (2005). Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat. Neurosci. 8, 279–287. doi: 10.1038/nn1411
Pegan, S., Arrabit, C., Slesinger, P. A., Choe, S. (2006). Andersen’s syndrome mutation effects on the structure and assembly of the cytoplasmic domains of Kir2.1. Biochemistry 45, 8599–8606. doi: 10.1021/bi060653d
Plaster, N. M., Tawil, R., Tristani-Firouzi, M., Canun, S., Bendahhou, S., Tsunoda, A., et al. (2001). Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell 105, 511–519. doi: 10.1016/S0092-8674(01)00342-7
Racz, I., Tory, K., Gallyas, F., Jr., Berente, Z., Osz, E., Jaszlits, L., et al. (2002). BGP-15 - a novel poly(ADP-ribose) polymerase inhibitor - protects against nephrotoxicity of cisplatin without compromising its antitumor activity. Biochem. Pharmacol. 63, 1099–1111. doi: 10.1016/S0006-2952(01)00935-2
Rjasanow, A., Leitner, M. G., Thallmair, V., Halaszovich, C. R., Oliver, D. (2015). Ion channel regulation by phosphoinositides analyzed with VSPs-PI(4,5)P2 affinity, phosphoinositide selectivity, and PI(4,5)P2 pool accessibility. Front. Pharmacol. 6, 127. doi: 10.3389/fphar.2015.00127
Rohacs, T., Lopes, C. M., Jin, T., Ramdya, P. P., Molnar, Z., Logothetis, D. E. (2003). Specificity of activation by phosphoinositides determines lipid regulation of Kir channels. Proc. Natl. Acad. Sci. U. S. A 100, 745–750. doi: 10.1073/pnas.0236364100
Romanenko, V. G., Fang, Y., Byfield, F., Travis, A. J., Vandenberg, C. A., Rothblat, G. H., et al. (2004). Cholesterol sensitivity and lipid raft targeting of Kir2.1 channels. Biophys. J. 87, 3850–3861. doi: 10.1529/biophysj.104.043273
Sakata, S., Okamura, Y. (2014). Phosphatase activity of the voltage-sensing phosphatase, VSP, shows graded dependence on the extent of activation of the voltage sensor. J. Physiol. 592, 899–914. doi: 10.1113/jphysiol.2013.263640
Sapra, G., Tham, Y. K., Cemerlang, N., Matsumoto, A., Kiriazis, H., Bernardo, B. C., et al. (2014). The small-molecule BGP-15 protects against heart failure and atrial fibrillation in mice. Nat. Commun. 5, 5705. doi: 10.1038/ncomms6705
Schram, G., Pourrier, M., Melnyk, P., Nattel, S. (2002). Differential distribution of cardiac ion channel expression as a basis for regional specialization in electrical function. Circ. Res. 90, 939–950. doi: 10.1161/01.RES.0000018627.89528.6F
Shistik, E., Ivanina, T., Blumenstein, Y., Dascal, N. (1998). Crucial role of N terminus in function of cardiac L-type Ca2+ channel and its modulation by protein kinase C. J. Biol. Chem. 273, 17901–17909. doi: 10.1074/jbc.273.28.17901
Stunnenberg, B. C., Raaphorst, J., Deenen, J. C. W., Links, T. P., Wilde, A. A., Verbove, D. J., et al. (2018). Prevalence and mutation spectrum of skeletal muscle channelopathies in the Netherlands. Neuromuscul. Disord. 28, 402–407. doi: 10.1016/j.nmd.2018.03.006
Tawil, R., Ptacek, L. J., Pavlakis, S. G., Devivo, D. C., Penn, A. S., Ozdemir, C., et al. (1994). Andersen’s syndrome: potassium-sensitive periodic paralysis, ventricular ectopy, and dysmorphic features. Ann. Neurol. 35, 326–330. doi: 10.1002/ana.410350313
Tristani-Firouzi, M., Jensen, J. L., Donaldson, M. R., Sansone, V., Meola, G., Hahn, A., et al. (2002). Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome). J. Clin. Invest. 110, 381–388. doi: 10.1172/JCI15183
Veerapandiyan, A., Statland, J. M., Tawil, R. (2004). Andersen-Tawil Syndrome. (University of Washington, Seattle, USA: GeneReviews) Bookshelf ID: NBK1264.
Whorton, M. R., Mackinnon, R. (2011). Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell 147, 199–208. doi: 10.1016/j.cell.2011.07.046
Whorton, M. R., Mackinnon, R. (2013). X-ray structure of the mammalian GIRK2-betagamma G-protein complex. Nature 498, 190–197. doi: 10.1038/nature12241
Yang, X. C., Sachs, F. (1989). Block of stretch-activated ion channels in Xenopus oocytes by gadolinium and calcium ions. Science 243, 1068–1071. doi: 10.1126/science.2466333
Yoon, G., Oberoi, S., Tristani-Firouzi, M., Etheridge, S. P., Quitania, L., Kramer, J. H., et al. (2006). Andersen-Tawil syndrome: prospective cohort analysis and expansion of the phenotype. Am. J. Med. Genet. A 140, 312–321. doi: 10.1002/ajmg.a.31092
Zhang, L., Benson, D. W., Tristani-Firouzi, M., Ptacek, L. J., Tawil, R., Schwartz, P. J., et al. (2005). Electrocardiographic features in Andersen-Tawil syndrome patients with KCNJ2 mutations: characteristic T-U-wave patterns predict the KCNJ2 genotype. Circulation 111, 2720–2726. doi: 10.1161/CIRCULATIONAHA.104.472498
Zhang, D., Ke, L., Mackovicova, K., Van Der Want, J. J., Sibon, O. C., Tanguay, R. M., et al. (2011). Effects of different small HSPB members on contractile dysfunction and structural changes in a Drosophila melanogaster model for Atrial Fibrillation. J. Mol. Cell Cardiol. 51, 381–389. doi: 10.1016/j.yjmcc.2011.06.008
Keywords: Andersen–Tawil syndrome, cardiac arrhythmia, PIP2, inward-rectifier potassium channel, slide helix, G-loop, voltage-sensitive phosphatase, BGP-15
Citation: Handklo-Jamal R, Meisel E, Yakubovich D, Vysochek L, Beinart R, Glikson M, McMullen JR, Dascal N, Nof E and Oz S (2020) Andersen–Tawil Syndrome Is Associated With Impaired PIP2 Regulation of the Potassium Channel Kir2.1. Front. Pharmacol. 11:672. doi: 10.3389/fphar.2020.00672
Received: 28 November 2019; Accepted: 23 April 2020;
Published: 15 May 2020.
Edited by:
Avi Priel, Hebrew University of Jerusalem, IsraelReviewed by:
Bernard Attali, Tel Aviv University, IsraelYael Stern-Bach, Hebrew University of Jerusalem, Israel
Copyright © 2020 Handklo-Jamal, Meisel, Yakubovich, Vysochek, Beinart, Glikson, McMullen, Dascal, Nof and Oz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shimrit Oz, c2hpbXJpdC5vei5mckBnbWFpbC5jb20=
†Present address: Michael Glikson, Shaare Zedek Medical Center, Jerusalem, Israel
Shimrit Oz, Rappaport Faculty of Medicine, Technion, Haifa, Israel