
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 08 May 2020
Sec. Experimental Pharmacology and Drug Discovery
Volume 11 - 2020 | https://doi.org/10.3389/fphar.2020.00640
This article is part of the Research Topic Metalloenzymes: Potential Drug Targets View all 10 articles
 Jean-Louis Charli*
Jean-Louis Charli* Adair Rodríguez-Rodríguez
Adair Rodríguez-Rodríguez Karina Hernández-Ortega
Karina Hernández-Ortega Antonieta Cote-Vélez
Antonieta Cote-Vélez Rosa María Uribe
Rosa María Uribe Lorraine Jaimes-Hoy
Lorraine Jaimes-Hoy Patricia Joseph-Bravo
Patricia Joseph-BravoThyrotropin releasing hormone (TRH: Glp-His-Pro-NH2) is a peptide mainly produced by brain neurons. In mammals, hypophysiotropic TRH neurons of the paraventricular nucleus of the hypothalamus integrate metabolic information and drive the secretion of thyrotropin from the anterior pituitary, and thus the activity of the thyroid axis. Other hypothalamic or extrahypothalamic TRH neurons have less understood functions although pharmacological studies have shown that TRH has multiple central effects, such as promoting arousal, anorexia and anxiolysis, as well as controlling gastric, cardiac and respiratory autonomic functions. Two G-protein-coupled TRH receptors (TRH-R1 and TRH-R2) transduce TRH effects in some mammals although humans lack TRH-R2. TRH effects are of short duration, in part because the peptide is hydrolyzed in blood and extracellular space by a M1 family metallopeptidase, the TRH-degrading ectoenzyme (TRH-DE), also called pyroglutamyl peptidase II. TRH-DE is enriched in various brain regions but is also expressed in peripheral tissues including the anterior pituitary and the liver, which secretes a soluble form into blood. Among the M1 metallopeptidases, TRH-DE is the only member with a very narrow specificity; its best characterized biological substrate is TRH, making it a target for the specific manipulation of TRH activity. Two other substrates of TRH-DE, Glp-Phe-Pro-NH2 and Glp-Tyr-Pro-NH2, are also present in many tissues. Analogs of TRH resistant to hydrolysis by TRH-DE have prolonged central efficiency. Structure-activity studies allowed the identification of residues critical for activity and specificity. Research with specific inhibitors has confirmed that TRH-DE controls TRH actions. TRH-DE expression by β2-tanycytes of the median eminence of the hypothalamus allows the control of TRH flux into the hypothalamus-pituitary portal vessels and may regulate serum thyrotropin secretion. In this review we describe the critical evidences that suggest that modification of TRH-DE activity in tanycytes, and/or in other brain regions, may generate beneficial consequences in some central and metabolic disorders and identify potential drawbacks and missing information needed to test these hypotheses.
Thyrotropin releasing hormone (TRH; Glp-His-Pro-NH2) is a small peptide expressed mainly in the brain, secreted by neurons. TRH interacts with plasma membrane G-protein-coupled receptors; one TRH receptor (TRH-R1) is detected in humans, whereas an additional one (TRH-R2) in rodents (Sun et al., 2003) and a total of three in teleosts and frogs (Bidaud et al., 2004; Saito et al., 2011). In mammals, TRH-R1 and TRH-R2 arise from different genes (Sun et al., 2003); the maps of their brain expression do not coincide (Heuer et al., 2000); they possess distinct ligand-independent activities, with TRH-R2 showing a stronger constitutive activity (Wang and Gershengorn, 1999). These distinct characteristics and the differential behaviors of KO mice for each receptor (Rabeler et al., 2004; Zeng et al., 2007; Sun et al., 2009) suggest that each type of receptor fulfills distinct functions in response to TRH. However, data in mice indicate that central TRH effects may depend mainly on TRH-R1 expression (Thirunarayanan et al., 2013).
The best-known function of TRH is the control of the hypothalamus–pituitary–thyroid (HPT) axis; TRH is synthesized by processing of a protein precursor in neurons of the paraventricular nucleus of the hypothalamus (PVN) that project their terminal boutons into the median eminence (hypophysiotropic neurons). Once released into the extracellular space TRH diffuses into the hypothalamic-pituitary portal vessels from where it controls secretion of thyrotropin (TSH) from the anterior pituitary. In turn, TSH controls the synthesis and secretion of thyroid hormones (TH) in the thyroid. This link adjusts TH secretion according to energy balance information, which is integrated by the hypophysiotropic PVN TRH neurons. TRH may also control prolactin secretion from the anterior pituitary (Martinez de la Escalera and Weiner, 1992; Joseph-Bravo et al., 2015b), although hypophysiotropic PVN TRH neurons regulating prolactin may differ from those controlling TSH secretion (Sánchez et al., 2001).
Various additional hypothalamic and extrahypothalamic neuronal populations express Trh (Lechan et al., 1986; Segerson et al., 1987; Hökfelt et al., 1989; Heuer et al., 2000), but their physiological role is not well understood. Soon after the discovery of extrahypothalamic TRH, several studies revealed central pharmacological effects of TRH that were independent of HPT axis control in laboratory animals, including increased locomotion, arousal, improvement of depression-like behaviors, reduction of epileptic seizures, and neuroprotection (Horita, 1998; Gary et al., 2003; Daimon et al., 2013; Fröhlich and Wahl, 2019). These observations led to attempts to use TRH in therapeutic contexts, for example in some neurodegenerative diseases (Gary et al., 2003; Daimon et al., 2013; Fröhlich and Wahl, 2019). Moreover, TRH is also present in selected loci outside the central nervous system whose importance is still poorly appreciated.
Early evidence that the half-life of TRH in rodent blood or in tissue extracts is 3–5 min (Redding and Schally, 1969; Prasad and Peterkofsky, 1976; Taylor and Dixon, 1976), and that TRH pharmacological effects are of short duration led to the synthesis of TRH analogues with improved stability, agonist potency and/or brain accessibility (Horita, 1998; Gary et al., 2003; Daimon et al., 2013; Fröhlich and Wahl, 2019). Other efforts were directed to elucidate the mechanism contributing to the rapid extracellular disappearance of TRH. TRH can be removed from the brain extracellular space by transport into brain cells; nevertheless, this event has a small Vmax and may have a very limited quantitative importance (Charli et al., 1984). The molecular entity that contributes to this transport has not been characterized; it may reflect TRH-R mediated endocytosis (Ashworth et al., 1995), or the action of a TRH transporter (Bagul et al., 2014).
Alternatively, TRH may be hydrolyzed by peptidases. Pyroglutamyl peptidase I (PPI; EC 3.4.19.3) is a soluble cysteine aminopeptidase with a wide specificity, that hydrolyses almost any Glp-X peptide, except if X is a proline (Awadé et al., 1994). This enzyme is present in all life kingdoms, and found in many tissues, including brain. Although PPI hydrolyses TRH in vitro (Morier et al., 1979; O'Leary and O'Connor, 1995; Charli et al., 1998), the only evidence that it contributes to TRH turnover in situ is that an inhibitor of PPI (and PE) enhances TRH levels and release in primary cultures of hypothalamic cells (Faivre-Bauman et al., 1986). Since PPI action is probably restricted to the cytoplasm, it may contribute to the intracellular turnover of TRH leaking from intracellular granules, but its subcellular localization is not compatible with a post-secretory role. In support of this idea, the specific inhibition of PPI activity in brain slices does not change TRH release and the intraperitoneal injection of a specific inhibitor of PPI that inhibits brain PPI activity does not change brain TRH levels (Charli et al., 1987). Furthermore, inhibition of enzyme activity does not protect TRH from degradation in rat serum (Friedman and Wilk, 1985).
Prolyl endopeptidase (PE; EC 3.4.21.26) is a serine endopeptidase with a wide specificity; it hydrolyses internal Pro-X peptide bonds, except if X is a proline. This enzyme is soluble, present in all life kingdoms, and many tissues, including the mammalian brain, albeit a membrane-bound isoform has also been identified. PE hydrolyzes the Pro-NH2 bond of TRH in vitro (Wilk, 1983; O'Leary and O'Connor, 1995; Charli et al., 1998; Tenorio-Laranga et al., 2008). PE role in TRH turnover in vivo is controversial; inhibition of PE enhances TRH levels and release in primary cultures of hypothalamic cells (Faivre-Bauman et al., 1986), the intraperitoneal injection of a PE inhibitor enhances TRH concentration in the cerebral cortex (Bellemere et al., 2005), and PE or a similar enzyme degrades TRH in rabbit seminal plasma (Siviter and Cockle, 1995). However, other experiments showed that PE inhibition in brain slices does not change TRH release into the medium and that the intraperitoneal injection of a specific inhibitor of PE does not change brain TRH levels in vivo (Charli et al., 1987). Furthermore, the intraseptal injection of a PE inhibitor does not change the effect of TRH on time of arousal from ethanol-induced narcosis in rats (Lazcano et al., 2012). Since PE regulates inositol polyphosphate phosphatase activities (Williams et al., 1999), the effect of PE inhibition on TRH levels may result from an alteration of the intracellular turnover of the secretory granules that contain TRH. Likewise, inhibition of PE does not protect TRH from degradation in rat serum (Friedman and Wilk, 1985). Thus, in general, PE may not control the extracellular turnover of TRH.
Another peptidase that may hydrolyze TRH is prolyl carboxypeptidase (PCP; EC 3.4.16.2) (Jeong et al., 2012), but there is no evidence that PCP coincides with TRH or alters TRH biological actions in vivo. Therefore, the quantitative importance of the previously described mechanisms of inactivation of TRH may be very limited in vivo, in particular for its extracellular inactivation, due in most cases to incompatible subcellular localizations of peptide and enzymes (Charli et al., 1998). In sum, contrary to what is sometimes mentioned in the literature, neither uptake nor soluble peptidases constitute relevant mechanisms for the extracellular inactivation of TRH.
The discovery of the thyrotropin-releasing hormone-degrading ectoenzyme (TRH-DE; EC 3.4.19.6) stems from the identification of an enzymatic activity that hydrolyses the Glp–His bond of TRH, initially in serum and called thyroliberinase, to underpin its narrow specificity (Bauer et al., 1981), and later in brain (Garat et al., 1985). The most striking property of this pyroglutamyl peptidase is a very narrow specificity (Lanzara et al., 1989; Wilk and Wilk, 1989; Elmore et al., 1990; Gallagher and O'Connor, 1998; Heuer et al., 1998a), a compelling argument in favor of looking at this peptidase as a therapeutic target, for example when enhancement of TRH action may be beneficial. TRH-DE was initially named pyroglutamyl peptidase II (PPII), to stress its clear biochemical difference with PPI; in contrast to PPI, TRH-DE/PPII is a metallopeptidase (Czekay and Bauer, 1993). A review describing the major biochemical characteristics of TRH-DE was previously published (Sánchez et al., 2013).
In the next sections, we briefly review the structure and proposed physiological roles of TRH-DE, focusing on recent advances, and we identify major challenges to translate this knowledge to clinical applications. Although the biochemical, topological and functional properties of the enzyme strongly argue in favor of its implication in the extracellular inactivation of TRH, its role may vary with cellular context, and might extend beyond hydrolysis of TRH. Contexts in which an intervention on TRH-DE may have therapeutic potential are delineated, and limitations are identified.
The sequence and genomic locus of the Trhde gene (we use the rodent abbreviations for gene names throughout the review) was initially described in humans (Schomburg et al., 1999); there is only one gene localized on chromosome 12. In mammals, the Trhde gene is adjacent to the tryptophan hydroxylase 2 gene (https://www.ensembl.org). In mouse, the gene localizes to chromosome 10 (http://www.informatics.jax.org/marker/MGI:2384311) while in rats, to chromosome 7. Trhde gene sequences and enzymatic activities have been detected in mammals, including activity in human serum and cerebrospinal fluid (Prasad and Jayaraman, 1986; Bundgaard and Møss, 1990; Charli et al., 1998). The gene is also detected in other vertebrate classes (Schomburg et al., 1999; Chávez-Gutiérrez et al., 2006). TRH-DE is not an essential gene in mice as TRH-DE KO animals are healthy, reproduce normally and their metabolic parameters are normal when bred in standard conditions (Tang et al., 2010).
Trhde transcription initiation sites have not been formally mapped. In mouse, three transcription start sites have been proposed (http://www.informatics.jax.org/marker/MGI:2384311); two are conserved in rat for which various transcripts differing widely in size have been detected (Schauder et al., 1994). Apart from alternative transcription start sites, there is little information about the mechanisms that lead to transcript diversity, except for a potential alternative splicing event in rats at exon 14-intron 14 boundary that leads to a truncated TRH-DE isoform (Chavez-Gutierrez et al., 2005). In mice three alternative splicing events may generate various TRH-DE isoforms (http://www.informatics.jax.org/). Bioinformatic approaches suggest that in mice response elements for multiple transcription factors exist along the 2 Kb region upstream from the most upstream of the predicted transcriptional start sites (Figure 1). However, promoter structure and genomic regulatory elements have not been experimentally mapped.
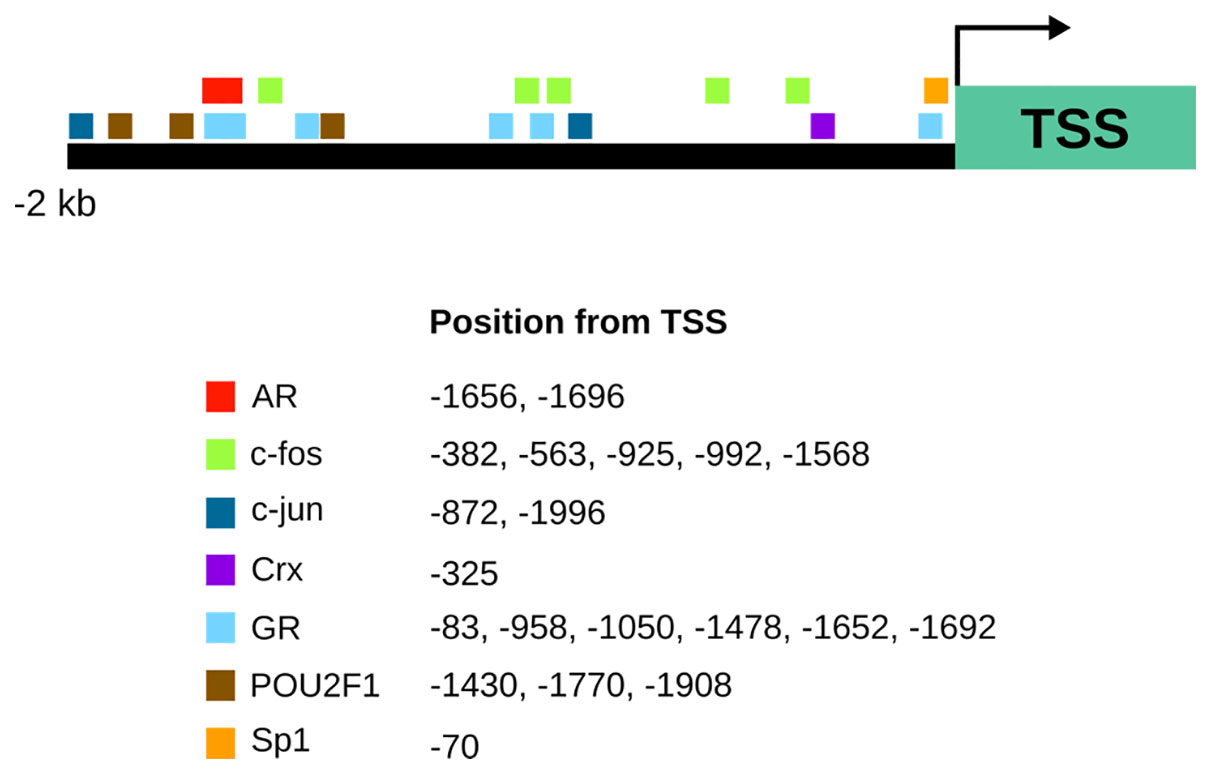
Figure 1 A map of candidates for transcriptional regulation of Trhde expression predicted by bioinformatic analysis. Transcription factor binding sites were identified 2 kb upstream of the most upstream of the 3 predicted transcriptional start sites (TSSR99301) of the mouse with PROMO (Messeguer et al., 2002; Farre, 2003). This site is conserved in rats. AR, androgen receptor; Crx, Cone-rod homeobox protein; GR, glucocorticoid receptor; NF-kappaB, nuclear factor kappa-light-chain-enhancer of activated B cells; POU2F1, POU domain, class 2, transcription factor 1; Sp1, specificity protein 1.
Another transcript derived from the human Trhde locus is Trhde antisense RNA 1 (Trhde-as1) (https://www.ncbi.nlm.nih.gov/nuccore/NR_026837). This long non-coding RNA is found in the cytoplasm and binds directly to miR-103 in samples of human lung, inhibiting parameters of human lung cancer progression (Zhuan et al., 2019). Individuals challenged with an injection of LPS show a significant drop of Trhde-as1 expression in adipose tissue samples (Ferguson et al., 2016). The relevance of Trhde-as1 expression for Trhde function is unknown.
The purification and cloning of the rat brain/pituitary isoform revealed that TRH-DE is 1,025 amino acids long (1,024 in human) (Bauer, 1994; Schauder et al., 1994). TRH-DE is a member of the M1 family of zinc-aminopeptidases, a family of 12 members (in humans) that includes aminopeptidases that have a wider specificity than TRH-DE, including aminopeptidase N (the closest relative of TRH-DE), which preferentially hydrolyses neutral aminoacids from the N-terminus (Sjöström et al., 2002). Although TRH-DE 3D structure is still unknown, analogy with other members of the family suggests it is a type II cell surface peptidase with a small intracellular domain, a single transmembrane domain, and a large extracellular part with a flexible stem followed by a catalytic domain, and an additional domain separated by a flexible loop (Chávez-Gutiérrez et al., 2006). Topology of the domains is consistent with biochemical evidence that demonstrated TRH-DE is an ectoenzyme (Charli et al., 1988), making it a prime candidate for primary hydrolysis of TRH in the extracellular space.
A large set of TRH derivatives has been tested to define TRH-DE specificity; TRH analogues modified on the C- or N-terminal residues are poor substrates of TRH-DE (Lanzara et al., 1989; Wilk and Wilk, 1989; Heuer et al., 1998a; Kelly et al., 2000). For example, gonadotropin-releasing hormone (GnRH), a decapeptide with a N-terminal Glp-His is not a substrate of TRH-DE (O'Connor and O'Cuinn, 1985; Elmore et al., 1990; Gallagher and O'Connor, 1998). Substitution of the histidyl residue of TRH by a series of amino acids influences the ability of porcine TRH-DE to catalyze the hydrolysis of the Glp-X bond in TRH-like peptides; thus, TRH-like peptides substituted with Gly, Asn, Gln, Trp, Glu, Asp, Pro or Cys are not substrates of TRH-DE, while low turnover rates are observed with Ala, Ser, Thr, Ile, Leu, Val. In contrast, TRH-like peptides substituted with Tyr or Phe have turnover rates approaching that of TRH (Kelly et al., 2000). This agrees with evidence that Glp-Phe-Pro-NH2 is a substrate of bovine brain TRH-DE (Kelly et al., 1997; Gallagher and O'Connor, 1998). The structural determinants of TRH-DE narrow specificity have been partially clarified; two amino-acid substitutions in the catalytic domain are critical and form a conserved signature that distinguishes it from other M1 family members (Chávez-Gutiérrez et al., 2006).
One of the two TRH-like peptides that are relatively good substrates of the enzyme in vitro, Gln-Phe-Pro-NH2, has been detected in peripheral organs including pancreas and thyroid gland (Khan et al., 1992; Gkonos et al., 1994; Kulkarni et al., 1995). Although it was initially suggested that TRH-like peptides, other than TRH, were not detectable in brain (Rondeel et al., 1995a; Rondeel et al., 1995b), it was later shown that, among other, Gln-Tyr-Pro-NH2, and Gln-Phe-Pro-NH2 are present in brain (Pekary et al., 2004; Pekary et al., 2005). To our knowledge, although these peptides are likely the product of posttranslational processing of precursor peptides (Linden et al., 1996), the precursors are still unknown. However, they may act as intercellular messengers, including in the brain, since their concentrations can vary rapidly in response to some stimuli, suggesting changes in synthesis and/or release (Pekary and Sattin, 2012; Pekary et al., 2015), and there is direct evidence that a peptide similar to Gln-Phe-Pro-NH2 can be released into hypothalamic extracellular space in vitro (Méndez et al., 1999). Bona fide receptors for Gln-Phe-Pro-NH2 and Gln-Tyr-Pro-NH2 have not been detected; they are weak agonists of TRH receptors (Hinkle et al., 2002).
Information about TRH-DE biosynthesis, transport and turnover is scant. The membrane isoform is a N-glycosylated protein; within its large extracellular domain, 12 putative N-glycosylation sites are found (Schauder et al., 1994). In its native form, TRH-DE can be partially deglycosylated (by about 50%) without affecting its activity (Bauer, 1994). The relevance of remaining glycosyl residues is not yet known. N-linked protein glycosylation starts in the endoplasmic reticulum and is a conserved process in eukaryotic cells. The subsequent details of processing that occur in Golgi compartments, according to protein-, cell- and species-specific clues (Aebi, 2013), have not been described for TRH-DE. Thyroliberinase, the soluble isoform secreted into blood, may be a product of alternative splicing or proteolysis (Schmitmeier et al., 2002). A membrane-bound truncated TRH-DE (TRH-DE*) is detected in many tissues, formed probably through an alternative splicing event; TRH-DE* does not hydrolyze TRH but it inhibits TRH-DE activity when it dimerizes with the full-length TRH-DE (Chavez-Gutierrez et al., 2005). In line with these results, correlative evidence suggests that the tissue-specific pattern of TRH-DE activity is defined in part by the Trhde/Trhde* expression ratio (Chavez-Gutierrez et al., 2005). Finally, little is known about TRH-DE interactions with other proteins. TRH-DE* may interact physically with Akt1 in mesenchymal-derived soft tissue sarcoma (STS) cell lines (Zhu et al., 2011) but an interpretation of the physiological relevance of this interaction awaits resolution.
To investigate the biological functions of TRH-DE, TRH-DE inhibitors have been synthesized. Protease inhibitors frequently include structural features that enable their interaction with the active site of the enzyme (Cushman et al., 1977; Kaysser, 2019). TRH-DE is weakly inhibited by the products of hydrolysis of TRH, Glp and His-Pro-NH2; combination of these molecules with zinc-binding groups results in poor TRH-DE inhibitors (Bauer et al., 1997). In general, TRH derivatives with modifications to the C- or N-terminal residues of TRH do not inhibit TRH-DE activity (Lanzara et al., 1989; Wilk and Wilk, 1989; Elmore et al., 1990; Gallagher and O'Connor, 1998; Kelly et al., 2000). However, GnRH, a Glp-His peptide C-terminally extended with hydrophobic residues does inhibit TRH-DE activity (O'Connor & O'Cuinn, 1985; Elmore et al., 1990; Gallagher and O'Connor, 1998). Furthermore, some analogues modified on the histidine residue have also shown some potential. Thus, the first specific inhibitor described was N-[1-carboxy-2-phenylethyl]N-imidazole benzyl histidyl-β-naphthylamide (CPHNA). With a Ki of 8 µM, CPHNA has no effect on TRH receptors and does not inhibit PE activity (Charli et al., 1989).
The systematic screening of a directed peptide library led to the finding that Glp-Asn-Pro-7-amido-4-methyl coumarin (Glp-Asn-Pro-AMC) has a Ki of 0.97 µM, making this peptide a relatively potent reversible TRH-DE inhibitor (Kelly et al., 2000). Improved potency was achieved by extension of Glp-Asn-ProNH2 with hydrophobic amino acids at the C-terminus. Thus, Glp-Asn-Pro-Tyr-Trp-Trp-AMC displays a Ki of 1 nM, which makes it the most potent competitive TRH-DE inhibitor described to date. Unfortunately, the in vivo potency of this peptide is poor (Kelly et al., 2005)
Another potent synthetic inhibitor of TRH-DE is the phosphinic analogue of TRH, GlpΨ[P(O)(OH)]HisProNH2 (Ψ-TRH) with a Ki of 170 nM, in which the scissile peptide bond of TRH has been replaced by the chemically stable phosphinic bond (Matziari et al., 2008).
Finally, a natural TRH-DE inhibitor named Hermodice carunculata protease inhibitor (HcPI) was isolated from the marine annelide H. carunculata. HcPI inhibits rat TRH-DE (a brain membrane extract with 7.5 mg protein/ml) with an IC50 of 4.8 μg/ml (Pascual et al., 2004). It is a small hydrophilic molecule very specific for TRH-DE, since it is at least 100 fold less potent at inhibiting the activity of the nearest homologue in the M1 family, aminopeptidase N (Cruz et al., 2008), and it is brain permeant (Pascual et al., 2004). HcPI structure is still unknown; solving it may lead to new tools.
TRH is a poor drug candidate. Although TRH is stable in the gastrointestinal tract, it has low intestinal permeability (Yokohama et al., 1984). The intranasal route of delivery presents a possible alternative route to access the brain (Veronesi et al., 2007; Zada et al., 2019). Furthermore, TRH has also a short plasma half-life, being rapidly hydrolyzed by peptidases (Redding and Schally, 1969; Prasad and Peterkofsky, 1976; Taylor and Dixon, 1976), and its cerebral absorption is low (Cornford et al., 1978). Finally, TRH has extensive neuroendocrine, central and autonomic/cardiac effects that may limit its therapeutic use (Gary et al., 2013; Khomane et al., 2011; Joseph-Bravo et al., 2015b).
Thus, TRH analogues have been developed to increase stability, delivery, specificity and potency. The properties of these analogs have been previously described extensively (Colson and Gershengorn, 2006; Monga et al., 2008; Khomane et al., 2011; Gary et al., 2003). Their stability in the presence of TRH-DE has seldom been directly tested or was tested against tissue homogenates that contain irrelevant peptidases. However, their stability in serum is a relatively good measure of their resistance to TRH-DE, since thyroliberinase is the main activity degrading TRH in serum. In this section, we describe TRH analogues that are resistant to TRH-DE.
TRH-like peptides in which the N-terminal Glp residue is replaced with a non-natural amino acid have promising pharmacological properties. Taltirelin ([1-methyl-(S)-4,5-dihydroorotyl)]-L-His-L-ProNH2) was synthesized by replacing the Glp residue of TRH with (S)-4,5, dihydroorotic acid (Suzuki et al., 1990). Taltirelin produces central nervous system (CNS) effects at about 100 times lower doses than TRH and has eight times longer duration of antagonistic action on pentobarbital-induced sleep than TRH (Yamamura et al., 1990; Yamamura et al., 1991a; Kinoshita et al., 1998). The differences in the activities of Taltirelin and TRH in the CNS have been attributed to the higher stability of Taltirelin in blood against thyroliberinase, and its higher lipophilicity than TRH that account for its increased penetration across the blood–brain barrier. Taltirelin resistance to hydrolysis by TRH-DE has been questioned, as Taltirelin was rapidly degraded in rat cerebral and pituitary homogenates (Kodama et al., 1997; Kobayashi et al., 2019a), but it is likely that in these homogenates enzymes distinct from TRH-DE were actively degrading Taltirelin.
Montirelin (6-methyl-5-oxothiomorpholinyl-3-carbonyl-His-Pro-NH2, CG-3703) is a derivative of the Gln moiety of TRH, which is an efficient CNS-stimulating agent (Horita, 1998). This TRH analog readily penetrates into the brain after its systemic administration in rats (Itoh et al., 1995) and inhibits specific Gln-[3methyl-His]-Pro-NH2 binding (Ki = 35 nM) to rat brain receptors as efficiently as TRH (Ki = 39.7 nM) (Urayama et al., 2001). Montirelin exhibits high stability in the blood (Sugimoto et al., 1996) and shows strong resistance to enzymatic degradation by pyroglutamyl aminopeptidase from brain or other related pyroglutamate aminopeptidases from liver and pituitary (Bauer, 1979; Griffiths et al., 1982).
Azetirelin (Nα-[[(5)-4-oxo2-azetidinyl]carbonyl]-His-Pro-NH2 dehydrate) has arousal actions 10–40 fold more potent and 8–36 times longer lasting than TRH (Yamamoto and Shimizu, 1987). This longer duration of action is probably because Azetirelin has a longer half-life in dogs and humans than TRH. However, Azetirelin has low oral bioavailability (Sasaki et al., 1994) and methods to improve its absorption have been evaluated (Sasaki et al., 1999).
Posatirelin (L-6-ketopiperidine-2-carbonyl-L-leucyl-proline amide) is a neutral analog with improved CNS activity and with cholinergic, catecholaminergic and neurotrophic properties (Oka et al., 1989; Amenta et al., 1997; Monga et al., 2008). Posatirelin has a stronger anticataleptic effect than TRH (Szirtes et al., 1986), and improves cognitive and motor disturbances in rats (Drago et al., 1996), while having poor influence on TSH release (Szirtes et al., 1984). Several human studies have also suggested beneficial effects in subjects with cognitive impairments (Cucinotta et al., 1994; Parnetti et al., 1995). Posatirelin is stable in human and rabbit serum, while it is slowly degraded in rat and mouse plasma (Terauchi et al., 1988).
Rovatirelin (1-{N-[(4S,5S)-(5-methyl-2-oxooxazolidin-4-yl)-carbonyl]-3-(thiazol-4-yl)-L-alanyl}-(2R)-2-methylpyrrolidine trihydrate) has a higher affinity for TRH-R1 (Ki = 702 nM) than Taltirelin (Ki = 3,880 nM) but still lower than TRH (Ki = 128 nM) in cell membrane preparation from CHO-K1 cells overexpressing the human TRH receptor (Ijiro et al., 2015). Rovatirelin is resistant to thyroliberinase in rat plasma and also to TRH-DE in pituitary (Kobayashi et al., 2018; Kobayashi et al., 2019a; Kobayshi et al., 2019b).
TRH-like peptides with simultaneous replacement of Glp by a panel of hetero ring-containing carboxylic acids and central histidine by 1-alkyl-L-histidines activate TRH-R2 with higher potencies than TRH-R1. One of these, with simultaneous substitution of Glp by pyrazine-2-carboxylic acid (2-Pyz) and central His with C3H7 (2-Pyz-L-His(1-alkyl)-L-Pro-NH2), exhibits high selectivity towards TRH-R2 but poor potency compared to TRH, high stability in rat blood plasma, antagonizes pentobarbital-induced sleeping time with a higher potency than TRH, and is devoid of adverse cardiovascular and CNS effects (Meena et al., 2015).
Another class of TRH derivative corresponds to those that have dual pharmacological activity, acting both as inhibitor of TRH-DE and as receptor agonist. Glp-Asn-Pro-AMC reversibly inhibits TRH-DE activity and binds preferentially to central TRH receptors (Kelly et al., 2000). The replacement of the hydrophobic L-amino acid residues with their D-isomers in C-terminally extended analogs of Glp-Asn-Pro-NH2 led to Glp-Asn-Pro-D-Tyr-D-Trp-NH2 (named JAK4D), which is effective at producing and potentiating some central actions of TRH without evoking TSH release in vivo. This peptide has high plasma stability and combined potent inhibition of TRH-DE (Ki 151 nM) with high affinity binding to central TRH receptors (Ki 6.8 nM). In addition, JAK4D has a high ability to cross the blood–brain barrier and shows a clean preliminary toxicology profile (Scalabrino et al., 2007; Kelly et al., 2015).
To conclude, TRH analogs incorporating resistance to hydrolysis by TRH-DE and/or inhibition of TRH-DE activity associate with enhanced pharmacological activity, compared to TRH.
Trhde expression is detected at relatively late phases of brain development. TRH-DE activity appears in the male rat brain a few days before birth, and peaks at post-natal day 8 in hypothalamus and posterior cerebral cortex, while it decreases afterwards to adult values. The pattern differs in the olfactory bulb, two peaks of specific activity being observed at the 3th and 22nd day (Vargas et al., 1992b). Trhde expression is at least 10-fold higher in brain than in nine other organs of male or female Fisher 344 rats and this ratio is stable from 2 to 104 weeks (Yu et al., 2014) (Figure 2). Thus, the brain is the region with the highest TRH-DE specific activity in adult animals (Vargas et al., 1992a; Lin and Wilk, 1998).
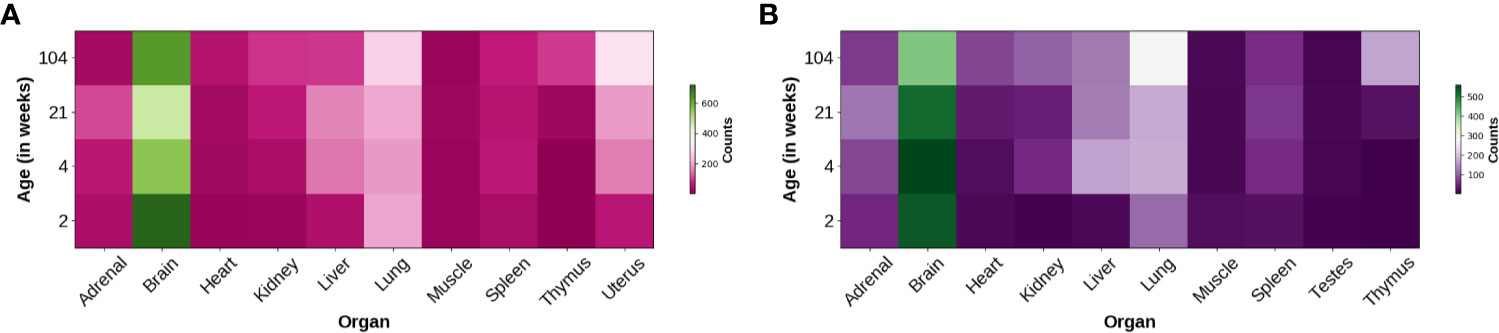
Figure 2 Distribution of Trhde expression in multiple rat organs. Heatmaps show the heterogeneous Trhde expression, predominant in the brain. Other differences can be observed along multiple developmental stages and between females (A) and males (B). Data were refined from a database obtained from a transcriptomic profiling study in Fischer 344 rats (Yu et al., 2014; Hicks et al., 2018).
Trhde transcript levels and activity vary widely among brain regions, with highest values detected in cerebral cortex and hippocampus, as well as in cerebellar hemispheres, regions with limited Trh expression (Vargas et al., 1987; Vargas et al., 1992a; Heuer et al., 1998b; Lin and Wilk, 1998). In the adult rat brain, the map of Trhde transcript distribution is generally consistent with the proposal that it is expressed by neurons (Heuer et al., 1998b). This is congruent with evidence that TRH-DE activity is enriched in synaptosomes (Torres et al., 1986) and that in primary culture of E17 hypothalamic cells, neurons are the major cell type expressing TRH-DE activity (Cruz et al., 1991). Furthermore, many rat cortical Trhde mRNA positive cells are Slc17a7 mRNA positive (express vesicular glutamate transporter 1), and some Gad mRNA positive (express glutamic acid decarboxylase), which directly confirms that neurons express Trhde (Rodríguez-Molina et al., 2014). Finally, single cell transcriptomic data of mouse CNS and peripheral nervous system (Zeisel et al., 2018) (Table 1) indicate that apart from many neuron types, the majority of non-neuronal cells have undetectable levels of Trhde mRNA.
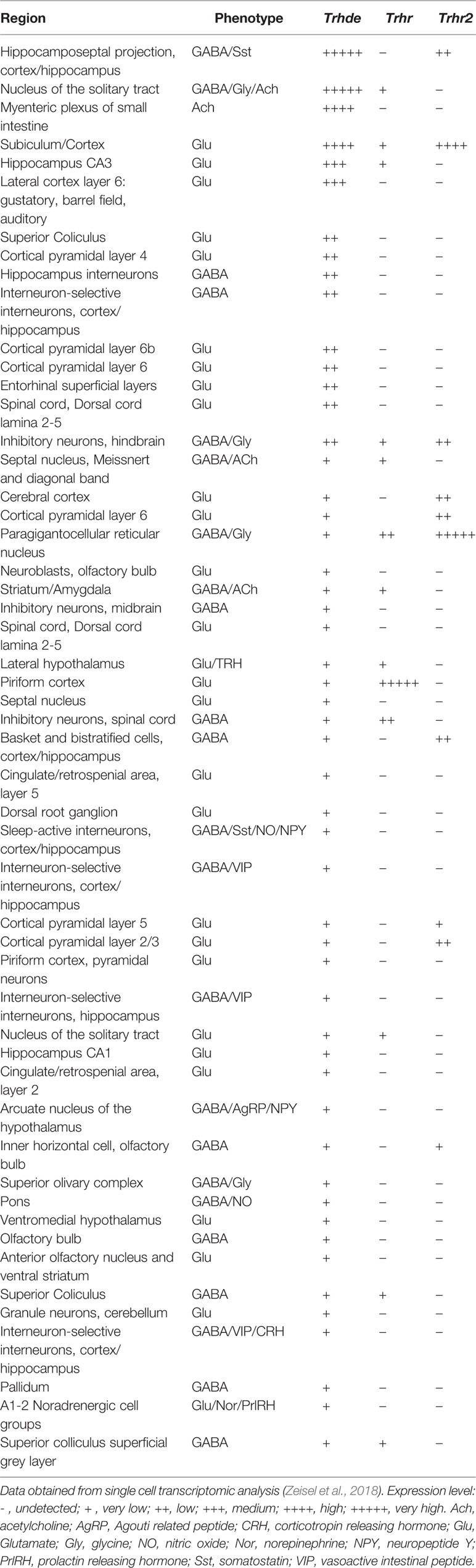
Table 1 Cells types along the mouse central and peripheral nervous system with the highest expression of Trhde, and relative expression of Trhr and Trhr2.
Many TRH neurons are glutamatergic (Zeisel et al., 2018). Although Trhde is expressed by multiple types of glutamatergic neurons, the regional distributions of Trh and Trhde mRNAs do not match (Heuer et al., 2000); in contrast, destruction of serotonin (5HT) neurons of the raphe nucleus, where TRH colocalizes with 5HT, decreases TRH content, but not TRH-DE activity, in their projections to the spinal cord (Joseph-Bravo et al., 1994), suggesting Trhde is not co-expressed with Trh in this pathway. However, the single cell transcriptomic map of the mouse brain (Zeisel et al., 2018) shows that while many Trhde neurons do not express Trh, most Trh cell types do express low levels of Trhde, while two hypothalamic Trh cell types are clearly positive for Trhde. An independent single-cell mouse transcriptomic study showed that in the medial hypothalamus there are 5 predominant clusters of Trh neurons, one being clearly positive for Trhde (Campbell et al., 2017).
Some TRH-DE neurons may be TRH-R1 or TRH-R2 positive, because the maps of the distribution of Trhde, Trhr and Trhr2 mRNAs in the brain reveal a partial co-expression of Trhde and one of its receptors (Heuer et al., 2000). Data at single cell level are consistent with coexpression in some neurons, but also indicate many mismatches (Zeisel et al., 2018). An example of a site of co-expression is the medial hypothalamus, where three of the five clusters of Trhde neurons do express Trhr (Campbell et al., 2017). Furthermore, in the medial septum a large fraction of the Trhde cells does not express Trhr (Lazcano et al., 2012). We have no clues about the relationship between the subcellular localizations of TRH-DE and TRH-Rs in the cells that do express both Trhde and Trhr or Trhr2, in part because of the lack of TRH-DE antibodies suitable for immunocytochemistry. Existing data thus suggest no systematic association of TRH-DE with TRH receptors. What is thus the relevance of TRH-DE for TRH communication?
In brain slices, exogenous TRH is rapidly hydrolyzed, a major product being His-Pro-NH2, one of the two catabolites of TRH hydrolysis by TRH-DE in vitro, suggesting indeed that TRH-DE hydrolyzes TRH in the brain extracellular fluid (Méndez et al., 1999). In brain slices, inhibition of TRH-DE activity with CPHNA increases TRH levels recovered in the extracellular medium, with a stronger effect in regions with higher TRH-DE activity (Charli et al., 1989). Glp-Asn-Pro-Tyr-Trp-Trp-AMC also enhanced recovery of TRH released from rat brain slices (Kelly et al., 2005). Furthermore, perfusion of cortical slices with a TRH-DE inhibitor mimics the effect of TRH on the shape of action potentials generated by pyramidal neurons, suggesting that endogenous TRH released in the extracellular fluid is hydrolyzed by TRH-DE (Rodríguez-Molina et al., 2014). Thus, extracellular turnover of TRH (either exogenous or endogenous) in brain slices is relatively rapid and occurs at least in part through hydrolysis by TRH-DE.
Besides these in vitro studies, there is indirect evidence that TRH may be hydrolyzed through TRH-DE activity in brain in vivo. If TRH is hydrolyzed by TRH-DE in the brain extracellular space, the reaction product His-Pro-NH2 could be degraded to His-Pro by dipeptidyl peptidase IV, a widely expressed ectoenzyme (Mentlein, 1999), and/or cyclize spontaneously to His-Pro-diketopiperazine (HPDKP), which may be metabolically stable and accumulate in the brain (Prasad, 1989). HPDKP is indeed found in the brain and although diet may be an external source, part of HPDKP most likely comes from TRH hydrolysis since its concentration is reduced in the hypothalamus and cortex of TRH knockout mice (Yamada et al., 1999).
A more direct evidence for the importance of TRH-DE in controlling the effects of TRH in the brain comes from a study on the effect of TRH on ethanol-induced narcosis. One of the pharmacological effects of TRH is its capacity to enhance alertness, and reverse narcosis, and one of the brain regions that mediate this last effect is the lateral septum. An intra-medial septum injection of the TRH-DE inhibitors pGlu-Asn-Pro-AMC, or Ψ-TRH which is not an agonist of TRH-R1, enhances the effect of exogenous TRH on the duration of ethanol-induced loss of righting reflex; the time of loss of the reflex is also decreased by the injection of Ψ-TRH alone (Lazcano et al., 2012). These data give further support to the proposal that TRH-DE regulates TRH turnover in vivo.
Although the relevance of TRH-DE for TRH inactivation in brain requires more extensive studies, it seems reasonable to conclude that its activity may at least in some anatomical contexts be present in neuron targets of TRH or adjacent neurons, and control the turnover of TRH, and thus duration and/or intensity of TRH effect. Although the primary products of TRH-DE activity (Glp and His-Pro-NH2) are inactive against TRH receptors, supporting an inactivation role, the product of His-Pro-NH2 cyclization, HPDKP, may mimic or oppose some TRH effects, making it possible to argue that TRH-DE bio-transforms TRH (Prasad, 1995). At pharmacological doses HPDKP may be neuroprotective and improve glucose metabolism in obese and/or diabetic animals (Minelli et al., 2008). However, the physiological relevance of TRH biotransformation to HPDKP in brain is currently unknown. Thus, it seems pertinent to conclude that a major function of TRH-DE is TRH inactivation in the brain. In contrast, it remains to be determined whether Gln-Phe-Pro-NH2 and Gln-Tyr-Pro-NH2 actions are indeed terminated by TRH-DE activity in vivo.
This section covers some of the major pharmacological properties of TRH, and of the two TRH-like peptides that are potential in vivo substrates of TRH-DE in the CNS, to illustrate the contexts in which TRH-DE targeting may be considered therapeutically beneficial.
Except for TRH neurons controlling the HPT axis, few experimental studies have addressed the in vivo physiological role of other TRH neurons. This is because of the absence of useful TRH-R antagonists in vivo, of the difficulty to manipulate the activity of specific TRH subtypes, and to limited information about the projections of various TRH neuron types. Some studies used strategies that correlate behavior with markers of TRH neuron activity, such as evaluation of the expression of Trh or cFOS, or region-specific manipulation of Trh expression. Along with tracing experiments, this research laid the ground to insert some TRH neurons in physiological contexts. Many studies have rather evaluated the impact of TRH or analogues on behavior and/or biochemical markers of neuronal activity. Peripheral or central (intracerebroventricular or nucleus-specific) administration of TRH or analogues produces in laboratory animals multiple behavioral changes, besides activation of the HPT axis. The mechanisms of TRH action are not fully understood, although advances have been made to characterize the electrophysiological/biochemical effects of TRH, and to identify target cells and circuits involved. Initial results led to early attempts to use TRH and analogues with improved stability, agonist potency and/or brain accessibility for experimental treatment of psychiatric or neurological diseases. A comprehensive analysis of TRH effects is beyond the scope of this review; the reader is referred to excellent reviews (Kelly, 1995; Horita, 1998; Gary et al., 2003; Daimon et al., 2013; Fröhlich and Wahl, 2019). As for other receptors, TRH or analogue action is not only limited by access and stability, but also by ligand-induced desensitization and down regulation (Sun et al., 2003; Hinkle et al., 2012). Reduced TRH receptor binding and behavioral tolerance is observed after repeated administration of TRH (Ogawa et al., 1983; Simasko and Horita, 1985), a fact that stresses one of the limits of the therapeutic use of any analogue. We will restrict the following paragraphs to some well-established effects of TRH or analogues, and whenever knowledge permits to discuss the targeting of TRH-DE.
TRH administration acutely increases arousal and locomotion; these effects are reproduced by direct injection of TRH into nucleus accumbens and septum (Horita, 1998). Other central regions are likely involved; for example, TRH increases the activity of lateral hypothalamus orexin neurons, which project to several areas involved in arousal (González et al., 2009; Hara et al., 2009). TRH-induced dopamine release has been observed in different paradigms and was proposed to be involved in TRH-induced motor activity (Kalivas et al., 1987; Yamamura et al., 1991b). The effect of TRH on arousal is also related with TRH-induced awakening from ethanol narcosis and coincides with the stimulatory effect of acute ethanol administration on Trh mRNA levels, accompanied with decreased Trhde mRNA levels and TRH-DE activity, and TRH tissue content, suggesting increased release of TRH in nucleus accumbens (de Gortari et al., 2005). In the septum, there is evidence that inhibition of TRH-DE activity promotes arousal from ethanol narcosis (Lazcano et al., 2012). Contrary to TRH, Gln-Phe-Pro-NH2 and Gln-Tyr-Pro-NH2 are not analeptic (Hinkle et al., 2002). Amplification of TRH action on arousal by TRH-DE inhibition may be therapeutically useful in niche applications.
TRH or analogue administration has generally a strong anorexic effect. The anorexic effect of TRH is probably mediated through multiple target regions; one of them is the nucleus accumbens, where TRH may interact with dopaminergic communication (Puga et al., 2016). Other regions potentially involved in the anorexic effect of TRH are various hypothalamic nuclei, such as the tuberomammillary nucleus, where TRH containing axons innervate histamine neurons (Gotoh et al., 2007; Sárvári et al., 2012), or the perifornical area/bed nucleus of stria terminalis region, from where TRH neurons may control the activity of proopiomelanocortin neurons of the arcuate nucleus (Péterfi et al., 2018). However, it was also noted that TRH injection into the brainstem promotes food intake through TRH-R1 activation (Ao et al., 2006), suggesting that control of food intake through TRH manipulation may require the optimization of the access route. There is not yet any experimental evidence showing that TRH-DE may regulate the anorexic (or orexigenic) effect of TRH.
TRH improves cognitive function (Bennett et al., 1997). TRH analogs administered to animals prevent the memory impairment caused by chronic ethanol administration or brain lesions (Bennett et al., 1997; Gary et al., 2003; Daimon et al., 2013; Meena et al., 2015). In vivo, TRH, or some analogs, activate the septo-hippocampal and basalis cortical cholinergic systems, eliciting acetylcholine release onto the hippocampus and cortex and restore cognitive deficits of animals with lesions in the medial septum (Itoh et al., 1994; Bennett et al., 1997; Horita, 1998; Prokai-Tatrai and Prokai, 2009; Daimon et al., 2013). The TRH analogue 2-Pyz-L-His (1-alkyl)-L-Pro-NH2, which exhibits high selectivity towards TRH-R2, and is more stable in rat blood plasma than TRH, possesses cognitive-enhancing activity when administered intravenously in the scopolamine-induced cognition impairment mice model, evaluated with the Morris water maze and the passive avoidance tests (Meena et al., 2015).
The hippocampus is critical in learning and memory. TRH immunoreactivity is detected in pyramidal and granular layers of CA1 and CA3, and in ventral dentate gyrus coincident with TRH binding (Hökfelt et al., 1989; Sharif et al., 1989) and with expression of Trhr (Calzá et al., 1992; Heuer et al., 2000). In support of the relevance of hippocampal TRH communication in learning, the status of TRH transmission is modified in the hippocampus of rats trained for 5 days in the water maze. TRH release is reduced, whereas expression of Trh and Trhr is upregulated in CA3, supporting plastic changes induced by the process of learning (Aguilar-Valles et al., 2007). TRH produces a time-dependent biphasic effect on NMDA receptor responses (Zarif et al., 2016), whereas it increases the excitability of GABA interneurons (Deng et al., 2006) in the hippocampus. Interestingly, pyramidal cells of the Ammon's horn express Trhde mRNA in abundance (Heuer et al., 1998b); Trhde may be expressed on the cell body and/or dendrites of the CA neurons. Blockade of NMDA receptors by MK801 decreases relatively rapidly TRH-DE activity in hippocampal slices, while blockade of AMPA or GABA-A receptors does not (Rodríguez-Molina et al., 2009). Additional cognitive effects of TRH may be direct onto cerebral cortex, where Trhde mRNA expression is also high (Heuer et al., 1998b) and TRH-DE regulates TRH action on pyramidal neurons action potential shape (Rodríguez-Molina et al., 2014). Thus, cognitive actions of TRH may be amplified by inhibition of TRH-DE activity.
Several reports show that endogenous or exogenous TRH is an anticonvulsant (Kubek et al., 1989; Sattin, 1999; Sah et al., 2011). In man, TRH is efficient for treatment of West syndrome, Lennox-Gastaut syndrome, and early infantile epileptic encephalopathy (Takeuchi et al., 2001). The electrical subthreshold stimulation produced during kindling in the amygdala or the hippocampus increases Trh expression or TRH content (Rosen et al., 1992; Kubek et al., 1993; de Gortari et al., 1995). Amygdala kindling increases Trh expression, but decreases that of Trhr, Trhr2, and Trhde in amygdala, and hippocampus, while only that of Trhr2 and Trhde in frontal cortex (de Gortari et al., 2006). TRH content increases as kindling progresses whereas the activity of TRH-DE increases in the initial stages and decreases once kindling is established (de Gortari et al., 1995). Decreased Trhde expression may facilitate the protective effects of TRH. These data suggest that inhibition of TRH-DE may be protective in some forms of epilepsy.
The peripheral or central administration of TRH produces an anxiolytic effect in several animal tests (Vogel et al., 1980; Thompson and Rosen, 2000; Gutiérrez-Mariscal et al., 2008). There is evidence that endogenous TRH can perform a similar function. Thus, in animals subjected to the defensive burying test, there is a specific inhibition of TRH release and Trh mRNA levels in amygdala but not in other limbic areas (Gutiérrez-Mariscal et al., 2008). Furthermore, amygdalar Trh expression and anxiety behavior are inversely correlated in the elevated plus maze and in the open field test (Gutiérrez-Mariscal et al., 2012). In adult male rats, exposure to an open field test during the inactive phase increases Trh expression in the cortical nucleus of the amygdala, an area involved in processing fear stimuli (Gutiérrez-Mariscal et al., 2008). Trhr KO mice are more anxious than wild type relatives (Zeng et al., 2007) while those deficient in Trhr2 exhibit reduced anxiety (Sun et al., 2009). It is warranted to test the status of Trhde KO mice.
Experimental evidence for an antidepressant effect of TRH in animal models and humans is controversial, although Trhr2-deficient mice are euthyroid and exhibit a depressive behavior (Sun et al., 2009). As an example of controversy, the injection of TRH into rats decreases the immobility time in the Porsolt test (Drago et al., 1990) whereas the injection of Taltirelin into the amygdala increases the immobility time in chronic-stress-induced depressed mice. In the depressed mice, Trh and Trhr expression in the basolateral amygdala is increased after 14 days of 2 h of daily restraint (Choi et al., 2015). This result seems to contradict the reported anxiolytic effect of TRH. Species and strain differences in stress responses and the previous stress history of the animal (Joseph-Bravo et al., 2015a) may explain some of these discrepant results. Finally, Gln-Tyr-Pro-NH2 is active in the Porsolt Swim Test (Pekary et al., 2005). Since mood stabilizing valproate acutely enhances, among other TRH-like peptides, Gln-Tyr-Pro-NH2 concentration in brain regions and most notably in pyriform cortex (Pekary et al., 2004), it will be relevant to take Gln-Tyr-Pro-NH2 into account if TRH-DE is targeted for an anti-depressant effect.
Several neurologic disorders improve with treatment with TRH and some of its stable analogs. This has been shown for scopolamine-induced memory loss, prolonged impaired consciousness (Horita, 1998; Meena et al., 2015), and neuronal death occurring after a transient ischemic attack (TIA) (Shishido et al., 1999). In rats, Trh expression increases in peri-ischemic tissue, while that of Trhde drops drastically. The response is independent of age of induction of TIA, and post-ischemia time. This may be a TRH protective effect, amplified by Trhde down regulation (Buga et al., 2012). Neuroprotective actions of TRH analogs include protection against neuronal injury induced by excitotoxicity and oxidative stress, and inflammation (Gary et al., 2003; Daimon et al., 2013; Fröhlich and Wahl, 2019). In the spinal cord, TRH may be involved in motor activity control and pain modulation; its administration promotes the recovery from acute spinal cord injury and has been used clinically with promising results (Fehlings and Baptiste, 2005). Taltirelin, which is degradation resistant, has been accepted in the treatment of spinocerebellar degeneration (SCD) (Kinoshita et al., 1994; Kinoshita et al., 1998). Taltirelin has anti-ataxic and neuroprotective actions (Urayama et al., 2002; Nakamura et al., 2005; Veronesi et al., 2007). Finally, JAK4D elicits large neuroprotective effects in neurodegenerative animal models. Systemic administration of JAK4D reduces cognitive deficits in a kainate (KA)-induced rat model of neurodegeneration, protects against free radical release and neuronal damage evoked by KA in rat, and reduces motor decline and lumbar spinal cord neuronal loss in a transgenic Amyotrophic Lateral Sclerosis mice model (Kelly et al., 2015). These data point to the importance of targeting TRH-DE to improve treatment of neurodegenerative diseases with TRH derivatives.
TRH analogs resistant to degradation by TRH-DE are promising for treating some neurological disorders. However, autonomic effects of these analogs need to be considered. Central TRH application has effects on the respiratory, cardiac and gastric systems. Most of these actions are due to targets of the TRH neurons present in brain stem. TRH is synthesized in raphé pallidus, obscurus, and para-pyramidal neurons that project to sympathetic preganglionic neurons of the intermediolateral (IML) cell column of the spinal cord and to vagal motor neurons located in the dorsal vagal complex (Taché et al., 2006). Some of TRH neurons that project to the IML use serotonin and substance P as co-transmitters; depending on the complement of receptors in target neurons, TRH participates in several aspects of respiratory network control (Hodges and Richerson, 2008).
In the vagal complex TRH activates neurons of the dorsal motor nucleus of the vagus that project to visceral organs such as pancreas regulating insulin secretion (Yang et al., 2002), or as stomach, stimulating gastric secretion (Tache, 2012). TRH-TRH-R1 signaling in brain stem, or TRH administered in low doses, have protective action against damaging agents in the gastric mucosa, whereas this is not the case in high doses as injections into the cisterna magna induce a dose-related activation of gastric vagal efferent discharges in rats. TRH applied in the hindbrain modulates gastric secretion and motility, blood pressure, hepatic blood flow, stimulates brown adipose tissue thermogenesis and respiratory output (Taché et al., 2006; Hou et al., 2012; Yang et al., 2012; Pilowsky, 2014). Although TRH-DE activity is lower in the brain stem than in other regions of the brain (Vargas et al., 1992a), the potential to alter autonomic functions if TRH-DE is affected cannot be underestimated.
In conclusion, at CNS level, many TRH effects, and some Gln-Phe-Pro-NH2 and Gln-Tyr-Pro-NH2 actions, may have a therapeutic potential (i.e. antiepilepsy, anxiolysis, improvement of learning, neuroprotection). The presence of TRH-DE in many of the sites of action of TRH suggests that inhibition of TRH-DE activity should increase the action of endogenous TRH. This may produce results that are better suited than those sought with TRH analogues.
This topic has been recently reviewed (Rodríguez-Rodríguez et al., 2019) and will only be briefly discussed here. Although a large part of CNS Trhde is expressed in neurons, a few years ago it was discovered that Trhde is expressed in tanycytes of the hypothalamus, a glial cell type localized at the base and ventrolateral walls of the third ventricle of the hypothalamus (Sánchez et al., 2009). All sub-types of tanycytes express Trhde; the β2 tanycytes, which send cytoplasmic extensions into the external layer of the median eminence where they are close to TRH terminals of the parvocellular TRHergic neurons of the PVN, in proximity of the portal capillaries that transport TRH to the anterior pituitary have the highest expression of Trhde (Sánchez et al., 2009). In median eminence explants, TRH-DE inhibition enhances TRH recovered from the incubation medium, suggesting that the enzyme actively hydrolyses TRH in the median eminence extracellular space (Sánchez et al., 2009). Thus, after release TRH may be partially hydrolyzed by TRH-DE before entry into the portal vessels. A physiological consequence would be that the amount of TRH reaching the anterior pituitary would not only depend on the activity of the TRH neurons, but also on median eminence TRH-DE activity, which would regulate the intensity of TRH-induced TSH secretion according to clues detected at median eminence level. Consistent with this idea, it was shown that although the intraperitoneal injection of HcPI in control animals does not change serum concentration of TSH, it does increase it in cold stressed animals (Sánchez et al., 2009). Although HcPI inhibits TRH-DE activity inside the blood–brain barrier, as well in the anterior pituitary and serum, Trhde expression is not detected in thyrotropes, and inhibition of TRH-DE expression or activity does not enhance TRH-induced TSH release (Bauer et al., 1990; Cruz et al., 2008); this evidence strongly suggests that anterior pituitary TRH-DE is not critical for the control of TSH release. Finally, the role of the serum enzyme, that is produced by the liver (Schmitmeier et al., 2002) cannot be discarded, since this isoform likely circulates through the portal vessels. Although a definitive demonstration for control of TRH entry into the portal capillaries by tanycyte TRH-DE is required, evidence about regulation of TRH-DE activity is also consistent with this proposal.
A major mode of control of the activity of the HPT axis is negative feedback at various levels, including inhibition of Trh mRNA synthesis in the hypophysiotropic neurons of the PVN (Fekete and Lechan, 2014). At the level of the tanycytes, Trhde expression and activity are rapidly (in a few hours) stimulated by peripheral injection of T4, thus putatively contributing to the negative feedback (Sánchez et al., 2009). The effect of T4 on Trhde depends on the local conversion of T4 to T3 by deiodinase 2 (Dio2) also expressed in tanycytes (Marsili et al., 2011). The mechanism by which TH induces Trhde expression is not known; thyroid hormone response elements have not been detected in the vicinity of the putative transcription initiation of Trhde (Figure 1), although many genes regulated by TH do not contain them (Chatonnet et al., 2013).
The HPT axis is regulated by energy balance (Fekete and Lechan, 2014; Joseph-Bravo et al., 2015b). Apart from down regulation of TRH neurons activity, a prolonged fast enhances Dio2 and Trhde expression and activity in the male rat median eminence (Diano et al., 1998; Lazcano et al., 2015). The transient induction of Trhde expression may contribute to prolong the downward adjustment of the HPT axis, along with an increase in thyroliberinase activity, in spite of a partial reactivation of Trh synthesis by the PVN at 72 h fasting (Lazcano et al., 2015). Other data show that diet-induced obesity (with unsaturated fat) enhances median eminence Trhde expression in male rats (Jaimes-Hoy et al., 2019), suggesting that the relationship between energy balance and activity of TRH-DE is complex. Finally, Trhde expression in the median eminence is programed by neonatal stress, along with other aspects of HPT axis function (Jaimes-Hoy et al., 2016). It is thus evident that long-term events shape median eminence TRH-DE activity.
In these models, there are few clues about the immediate regulators of Trhde expression/activity, and it has not been shown that Trhde adjustments are critical for HPT axis activity. The local thyroid status is probably involved (Lazcano et al., 2015) and besides hormonal signals, tanycytes are responsive to neuronal inputs. Recently it was shown that the release of TRH in the median eminence activates the TRH-R1 receptor on the surface of tanycytes, resulting in two rapid events: an increase of the surface of tanycyte end-feets covering the portal capillaries, and an increase in TRH-DE activity in the median eminence, both of which may reduce TRH access to the capillary bed. Furthermore, a long term (days) blockade of Gq transduction decreased TRH-DE activity. Thus, the firing of TRH neurons likely regulates TRH-DE activity and localization. This coordinated mechanism may help to shape the pulses of TRH that reach the anterior pituitary (Müller-Fielitz et al., 2017).
An intriguing question is the relevance of TRH-like peptides sensitive to hydrolysis by TRH-DE in the control of the HPT axis. Gln-Phe-Pro-NH2 is released from hypothalamic slices (Méndez et al., 1999), Gln-Phe-Pro-NH2, as well as TRH, is detected in rat thyroid (Rausell et al., 1999; Smyth et al., 1999) and Gln-Phe-Pro-NH2 enhances serum T3 concentration (Cremades et al., 1998). Thus, median eminence and thyroid gland may be sites where the functional relationship between Gln-Phe-Pro-NH2 and TRH-DE warrants further investigation.
Thyroid diseases are very common and a major one is sub-clinical or clinical hypothyroidism, which is mainly treated with T4 (and T3). Although this therapeutic option is successful, it is not perfect and there is room for improvement. Among the major problems of the therapeutic use of TH is the risk of producing unattended hyperthyroidism and thus cardiovascular or skeletal consequences. These problems have led to significant avenues of research, to limit unintended effects. One of these approaches has been the development of specific agonists of TH receptor β (Runfola et al., 2020). Another focus has been on the development of thyroid hormone-peptide complexes, to direct TH to the tissues that express the peptide receptor (Finan et al., 2016).
Peripheral inhibition of peptidase activity has been quite successful in various contexts (hypertension, type 2 diabetes). Although requiring confirmatory experiments, the weight of evidence strongly suggests that in rats median eminence TRH-DE activity controls the output of TRH and thus TSH secretion, at least during bouts of TRH neurons activity. This suggests that inhibition of this enzyme may transiently increase TRH-induced TSH secretion, enhancing the hypothalamic signal that promotes TSH secretion in conditions of insufficient drive. Because β2 tanycyte TRH-DE activity is outside the blood brain barrier, it should be possible to target this compartment and enhance the extracellular concentration of TRH in the portal vessels, without affecting TRH communication in the rest of the CNS. Furthermore, the narrow specificity of TRH-DE should facilitate the development of agents that have no impact on other peptides turnover. The final purpose of this manipulation would be to enhance thyroid hormone secretion without changing the natural pattern of control of the HPT axis and to produce self-regulated and small effects on TH concentration, reducing cardiovascular and bone effects.
Various milestones should be reached to target peripheral TRH-DE activity to improve thyroid status in hypothyroidism. One is to characterize the long-term impact of TRH-DE inhibition on HPT axis parameters, since the axis can self-regulate efficiently; for example, the long-term ablation of tanycytes does not change serum TSH concentration (Yoo et al., 2019). Another consideration is that changes in serum TSH concentration may have non thyroidal effects. Likewise, prolactin secretion would have to be monitored, to exclude alteration of this critical hormone. Finally, the distribution and role of TRH-DE activity in the periphery requires clarification.
In mammals, apart from the negative control of prolactin secretion by hypothalamic tuberoinfundibular dopamine neurons, hypothalamic prolactin releasing factors are also important (Grattan, 2015). TRH is not only a thyrotropin releasing factor but also a prolactin releasing factor. TRH is necessary to maintain maximal prolactin output in lactating mice (Yamada et al., 2006). In primary cultures of female rat anterior pituitary cells, Trhde expression is detected in some lactotropes, and inhibition of TRH-DE expression or activity enhances TRH-induced prolactin secretion (Bauer et al., 1990; Cruz et al., 2008). If the importance of Trhde for the control of prolactin secretion, which is contingent on many factors, is confirmed in relevant in vivo models, inhibition of anterior pituitary TRH-DE activity might increase prolactin secretion.
Although targeting of CNS and/or median eminence/pituitary TRH-DE activity has experimental support, a much less understood aspect is the function of Trhde expressed in the periphery. This must be fully assessed for establishing the safety profile of the peripheral inhibition of TRH-DE activity or expression. Figures 2 and 3 illustrate that although the brain is a major source of Trhde expression, developmentally regulated expression of Trhde is also detected in multiple rat tissues, of both sexes. However, it should be noted that in rats and rabbits, the peripheral tissues have low levels of TRH-DE activity (Vargas et al., 1992a). Human transcriptomic data indicate baseline expression of Trhde in various peripheral organs, being more abundant in kidney, spleen, ovaries, uterus, adipose tissue, small intestine Peyer's patch, duodenum, pancreas, and lung, and lower in colon, prostate, stomach and testis. In these tissues, Trhde expression is detected in adults. For kidney, lung, stomach and testis, Trhde expression has also been reported at early stages of embryonic development (“ensembl.org: TRHDE—ENSG00000072657,” n.d. and “bgee.org: TRH-DE—ENSG00000072657”) (Figure 3). We review below the state of knowledge about TRH-DE in the best-studied tissues.
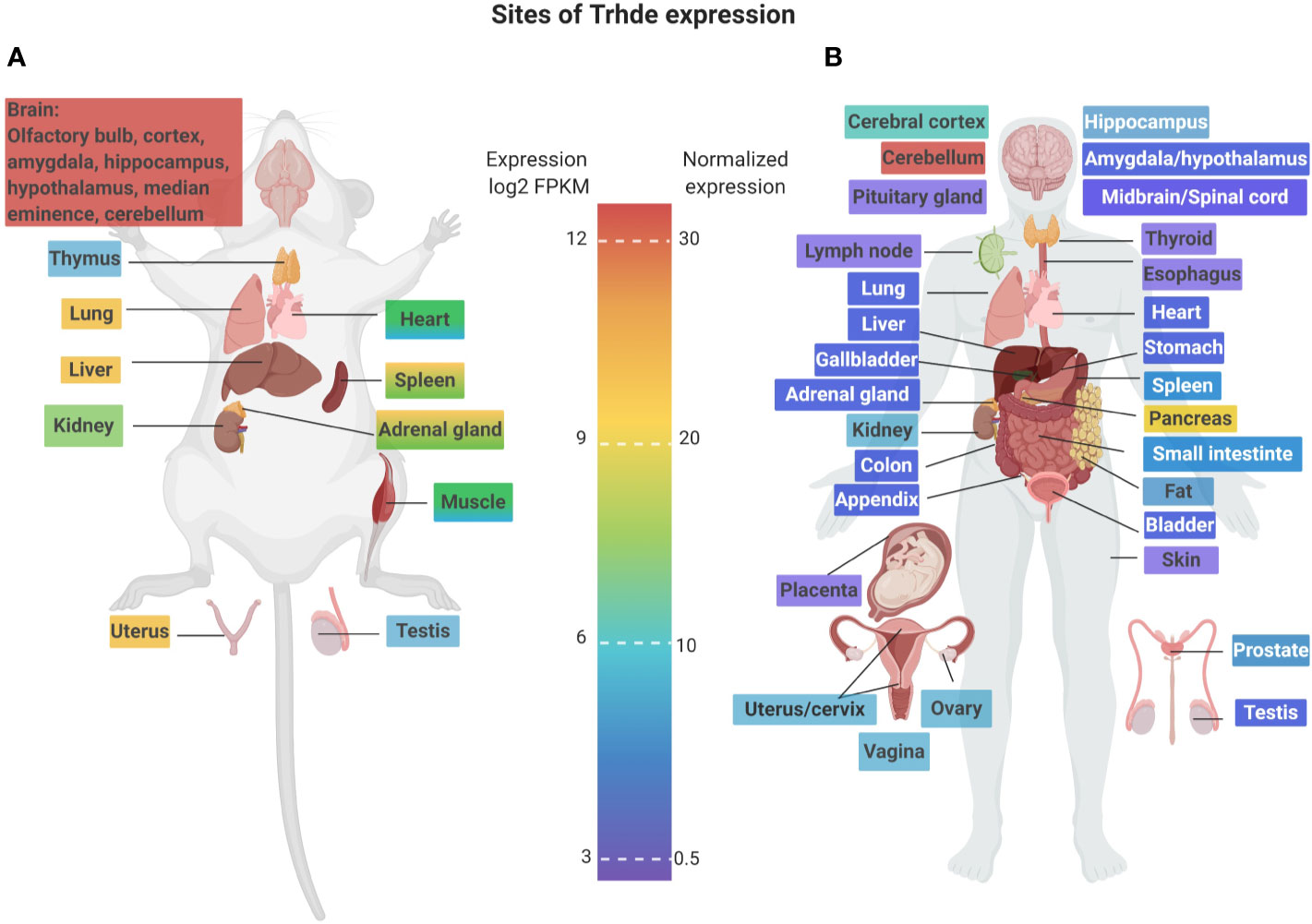
Figure 3 Potential sites of targeting of TRH-DE in rats and humans. Trhde is mainly expressed in a few major organs; in rats, gene expression is higher in brain followed by lung, liver and uterus; in humans, major sites of expression are cerebellum and pancreas. Panel (A) illustrates tissue Trhde expression distribution in rat (log2 FPKM, fragments per kilobase of transcript per million mapped reads) based on the database of the rat body map available from http://pgx.fudan.edu.cn/ratbodymap/index.html (Yu et al., 2014); some of the brain regions which express medium (i.e. hypothalamus) to high mRNA levels are also indicated (Heuer et al., 1998b). Panel (B) illustrates tissue Trhde expression distribution in humans based on the Human Protein Atlas available from http://www.proteinatlas.org (Uhlén et al., 2015). Colors on organ/tissue names represent expression levels of Trhde. Information regarding sex and age of the donor, can be found at http://www.proteinatlas.org/about/cellines. Figure was created with Biorender.com.
Thyroliberinase is a liver specific isoform of TRH-DE (Schmitmeier et al., 2002) that is secreted into the general circulation. It is the only enzyme that degrades TRH in rat serum (Friedman and Wilk, 1985). Its role is puzzling; it probably reaches the hypothalamic-pituitary portal vessels, but its specific relevance for anterior pituitary control is untested. Thyroliberinase may also reduce the buildup of TRH concentration in the peripheral tissues where TRH may act as a paracrine effector (Morley, 1979). Finally, it should contribute significantly to the limited efficiency of the parenteral injection of TRH, being an interesting target to enhance the efficacy of parenteral TRH analogues.
One of the peripheral tissues in which TRH presence was noticed early is the rat pancreas, where it is concentrated in the islets of Langerhans (Martino et al., 1978). A transient prenatal expression of Trh is detected in pancreatic β-cells, which coincides with development of insulin secretory activity (Yamaoka and Itakura, 1999; Basmaciogullari et al., 2000; Štrbák, 2018). In rodents, Trhr is expressed in the pancreas, including in β-cells (Yamada et al., 2000; Luo and Yano, 2004); a potential effect of TRH-TRH-R1 is the promotion of programmed cell death during development (Mulla et al., 2009). Trh KO mice have an increased glycemia, which may be the result of reduced secretion of insulin in response to high glucose concentration (Yamada et al., 1997). On the other hand, Trhr KO mice are hypothyroid and hyperglycemic (Zeng et al., 2007), while Trhr2 KO mice are euthyroid and normoglycemic (Sun et al., 2009). Gln-Phe-Pro-NH2 is detected in pancreas, where it attenuates TRH effect on insulin secretion in perifused islets (Kulkarni et al., 1995). Although pancreatic Trhde expression is significant in human (Figure 3), there is no evidence for TRH-DE activity in developing or adult rat (Vargas et al., 1992b). Trhde KO mice are normoglycemic (Tang et al., 2010). The relevance of TRH-DE in human pancreatic function requires an urgent investigation; is expression leading to TRH-DE activity, or to non-active isoforms?
Trh, and Trhr are expressed in the heart, in fibroblasts and in cardiomyocytes (Carnell et al., 1992; Shi et al., 1996); TRH, as well as Gln-Phe-Pro-NH2 and Gln-Tyr-Pro-NH2, are detected in heart tissue, where values vary along the circadian cycle (Pekary and Sattin, 2017). Various evidences suggest that heart TRH might be relevant in cardiovascular disease. TRH induces hypertrophic and fibrotic markers in heart cells (Schuman et al., 2014). Overexpression of Trh in left ventricular cells promotes heart hypertrophy in rats (Jin et al., 2004). Spontaneously hypertensive rats overexpress Trh in the left ventricle, and reduction of Trh expression inhibits fibrosis and cardiomyocyte enlargement (Schuman et al., 2011). Furthermore, angiotensin II–induced heart hypertrophy depends on Trh expression (Peres Diaz et al., 2018). Thus, TRH may have a substantial role in cardiac hypertrophy. Trhde is expressed in the heart of Fischer-344 male and female rats, with noticeable developmental increases (Figure 2) (Yu et al., 2014). In the human heart, Trhde transcripts can be detected at 10 weeks (Szabo et al., 2015). It will be important to clarify whether TRH-DE activity is present in heart and controls local TRH and/or Gln-Phe-Pro-NH2 and Gln-Tyr-Pro-NH2 actions.
TRH-like immunoreactivity is detected in various organs of the digestive system (Leppäluoto et al., 1978). TRH levels are high in gastric juice and mucosa (Nishio et al., 1999). In the digestive system, while TRH-R1 presence in the stomach is scarce (Nishio et al., 1990), TRH-R1 has been detected by immunohistochemistry in the small intestine (Mitsuma et al., 1995), where TRH excites submucus neurons and enhances heat shock protein 60 expression (Zafirov et al., 1991; Sasahara et al., 1998). One subtype of cholinergic neuron, in the myenteric plexus of the mouse small intestine, is characterized by a very high expression of Trhde (Zeisel et al., 2018) (Table 1), but these neurons, which may receive vagal afferents, do not express Trhr or Trhr2. In this system, TRH-DE may inactivate TRH produced and released locally. However, as for the heart, there is yet no evidence that TRH-DE activity is present in the intestinal mucosa. Because of the high relative expression of Trhde in neurons of the enteric system, potential adverse effects of TRH-DE manipulation should be considered. In humans, intestinal Trhde expression initiates around 10–20 weeks of age (Szabo et al., 2015), being significant in adults. An accumulation of neuropeptides, including TRH, in the lumen of the colon of patients with inflammatory bowel disease (Yamamoto et al., 1996) is noticeable; animal models of the disease might shed light about TRH-DE role in this pathology, if any.
TRH has significant effects over the function of immune system, some of which dependent on pituitary control (Kamath et al., 2009). Hypophysiotropic TRH neurons are involved in inflammation responses dependent and independent of T-cell activation. LPS administration doesn't induce T-cell activation and is linked with an initial reduction of Trh expression in the PVN (Kamath et al., 2009). However, T-cell dependent inflammation induces the opposite, with the response dependent on hypophysiotropic TRH neurons and prolactin secretion from the pituitary (Perez Castro et al., 1999). Although the site of TRH production acting directly on immune cells is unknown, TRH-R1 can be detected in lymphoid tissues, including the thymus (Montagne et al., 1999) and bone marrow (Matre et al., 2003). Administration of TRH in rats increases the proliferation of cells of the thymus (Pawlikowski et al., 1992) and protects against its involution and reduction of the count of lymphocytes in peripheral blood, induced by lateral hypothalamus lesion (Lesnikov et al., 1992). TRH may thus be of therapeutic value in diseases dependent on immune system activation. Administration of TRH protects against pathogens such as encephalomyelitis virus (Pierpaoli and Yi, 1990), and candidiasis (Blaszkowska et al., 2004). Transcriptomic analysis reveals that Trhde is expressed in thymus, with a significant drop of expression in aging rats (Figure 2) (Yu et al., 2014). Although TRH-DE activity and function have not been determined in lymphoid organs, TRH-DE inhibitors could be interesting alternatives to TRH treatment in inflammatory diseases.
In the reproductive system, Leydig cells express both Trh and Trhr (Pekary and Sattin, 2017). Repeated administration of TRH counters aging-induced alteration of rat testicular structure and function (Pierpaoli, 2013). Gln-Phe-Pro-NH2 is also expressed by the prostate and detected in seminal fluid (Bilek et al., 1992; Khan et al., 1992; Gkonos et al., 1994); however, its local effect is unknown. In females, TRH increases the contractility of urethral and vaginal muscle (Zacur et al., 1985) and Gln-Phe-Pro-NH2 is detected in the mammary gland (Ghilchik et al., 2000). Since low levels of expression of Trhde occur in various reproductive organs (Figure 3), its relevance for TRH or Gln-Phe-Pro-NH2 actions may not be important but should nevertheless be investigated.
In conclusion, there is little evidence that, apart from pituitary and liver, other peripheral tissues which express Trhde give rise to significant TRH-DE activity. It remains possible that in some tissues expression might be mainly attributed to the mRNA coding for the truncated version of TRH-DE (Chavez-Gutierrez et al., 2005) and that TRH-DE* is not only a dominant negative isoform of TRH-DE but is also a non-catalytic protein. However, Trhde KO mice do not show any obvious problem in standard conditions, except for a small decrease of body weight (Tang et al., 2010). Finally, it will be critical to clarify the extent of TRH-DE activity in human tissues.
With the available information of the wide distribution of TRH receptors, and the multiple functions TRH displays, the pharmacological use of agonists of the TRH-R1 receptors that resist degradation by TRH-DE might impact on many functions.
Albeit alternatives are not completely excluded yet, it appears that hydrolysis by the TRH degrading ectoenzyme in the serum and the extracellular space of the brain, including the median eminence, is a critical pathway for TRH inactivation. The relevance of this enzyme for Gln-Phe-Pro-NH2 and Gln-Tyr-Pro-NH2 inactivation remains to be investigated. Since most central pharmacological effects of peptidase resistant analogues of TRH are beneficial in rodents, and there is some consistent evidence in humans, targeting central TRH-DE with specific inhibitors may have significant advantages over TRH agonists in some central diseases, in particular to produce more focused effects according to the natural activity of the TRH neurons. On the other hand, the limited span of the distribution of TRH-DE activity in the periphery opens the possibility that inhibition of median eminence TRH-DE activity may be an interesting target for treatment of hypothyroidism and may have few non-intended targets, if a compound that does not pass the blood-brain barrier is chosen. However, it will be necessary to understand the full extent of the distribution of peripheral TRH-DE activity and function in humans before this idea proceeds further.
Targeting the inactivation process in the brain will increase ligand concentration in the synaptic space and increase TRH effect, alike the mechanism of action of drugs that inhibit neurotransmitter uptake. However, an intrinsic problem lies on the stringent specificity of TRH-DE for the TRH structure. Thus, it is not surprising that some of the TRH-DE inhibitors function also as TRH-R agonists (Scalabrino et al., 2007) and may thus promote receptor down regulation or, undesirable side effects. For example, even for a more stable analog of TRH as Taltirelin, hyperprolactinemia is induced (Kanasaki et al., 2011). Even if the analog does not produce endocrine effects in humans, other peripheral effects such as induction of gastric erosion and hypermotility of the intestine may occur. The challenge to design a specific inhibitor to TRH-DE not recognized by TRH-R1 has been overcome with Ψ-TRH design, although better chemistries and affinities are required. In theory, this should be possible, as has been done for other proteases (Patchett and Cordes, 1985; Traube et al., 2014; Lai et al., 2015).
Even if better inhibitors are found, unknowns remain. Are Gln-Phe-Pro-NH2 and Gln-Tyr-Pro-NH2 physiologically relevant substrates of TRH-DE? Does inhibition of TRH-DE activity has concrete effects in preclinical models? These, and other milestones, need to be passed to determine if manipulation of TRH-DE expression or activity has therapeutic potential.
J-LC conceived and wrote the manuscript. PJ-B conceived and wrote the manuscript. AR-R generated figures and table and edited the manuscript. KH-O, AC-V, RU, and LJ-H wrote specific parts of the manuscript.
Supported by grants from DGAPA-UNAM to J-LC (PAPITT IN206712), and AC-V (PAPITT IN 206416 and 212719), and from CONACYT to JLC (PN562; CB254960).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This manuscript is dedicated to the memory of Dr. Milagros Méndez who was our first PhD student and contributed to delineate the in vivo role of the multiple peptidases that hydrolyze TRH. The research described in this paper has benefited from the talented help of many. Besides the individuals that are included in the reference list, we thank Elizabeth Mata, Sergio González and Graciela Cabeza Pérez, for providing animals, to Shirley Elizabeth Ainsworth Gore, Jesús Omar Arriaga Pérez, Arturo Ocádiz Ramírez, Juan Manuel Hurtado Ramírez, Servando Aguirre Cruz, Roberto Pablo Rodríguez Bahena, and David Santiago Castañeda Carreón for computing, communication and library support, and to Fidelia Romero, Mariana Gutierrez-Mariscal, Miguel Cisneros and Manuel Villa for technical help.
Aebi, M. (2013). N-linked protein glycosylation in the ER. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 1833, 2430–2437. doi: 10.1016/j.bbamcr.2013.04.001
Aguilar-Valles, A., Sánchez, E., de Gortari, P., García-Vazquez, A. I., Ramírez-Amaya, V., Bermúdez-Rattoni, F., et al. (2007). The expression of TRH, its receptors and degrading enzyme is differentially modulated in the rat limbic system during training in the Morris water maze. Neurochem. Int. 50, 404–417. doi: 10.1016/j.neuint.2006.09.009
Amenta, F., Sabbatini, M., Coppi, G., Maggioni, A., Olgiati, V., Panocka, I. (1997). Effect of treatment with the neuroactive peptide posatirelin on microanatomical changes of frontal cortex and hippocampus caused by lesions of the locus coeruleus. Drugs Exp. Clin. Res. 23, 77–88.
Ao, Y., Go, V. L. W., Toy, N., Li, T., Wang, Y., Song, M. K., et al. (2006). Brainstem Thyrotropin-Releasing Hormone regulates food intake through vagal-dependent cholinergic stimulation of ghrelin Secretion. Endocrinology 147, 6004–6010. doi: 10.1210/en.2006-0820
Ashworth, R., Yu, R., Nelson, E. J., Dermer, S., Gershengorn, M. C., Hinkle, P. M. (1995). Visualization of the thyrotropin-releasing hormone receptor and its ligand during endocytosis and recycling. Proc. Natl. Acad. Sci. 92, 512–516. doi: 10.1073/pnas.92.2.512
Awadé, A. C., Cleuziat, P. H., GonzalèS, T. H., Robert-Baudouy, J. (1994). Pyrrolidone carboxyl peptidase (Pcp): An enzyme that removes pyroglutamic acid (pGlu) from pGlu-peptides and pGlu-proteins. Proteins 20, 34–51. doi: 10.1002/prot.340200106
Bagul, P., Khomane, K. S., Kesharwani, S. S., Pragyan, P., Nandekar, P. P., Meena, C. L., et al. (2014). Intestinal transport of TRH analogs through PepT1: the role of in silico and in vitro modeling: role of structural modifications on PepT1-mediated transport. J. Mol. Recognit. 27, 609–617. doi: 10.1002/jmr.2385
Basmaciogullari, A., Cras-Meneur, C., Czernichow, P., Scharfmann, R. (2000). Pancreatic pattern of expression of thyrotropin-releasing hormone during rat embryonic development. J. Endocrinol. 166, 481–488. doi: 10.1677/joe.0.1660481
Bauer, K., Nowak, P., Kleinkauf, H. (1981). Specificity of a serum peptidase hydrolyzing thyroliberin at the pyroglutamyl-histidine bond. Eur. J. Biochem. 118, 173–176. doi: 10.1111/j.1432-1033.1981.tb05501.x
Bauer, K., Carmeliet, P., Schulz, M., Baes, M., Denef, C. (1990). Regulation and cellular localization of the membrane bound Thyrotropin-Releasing Hormone-degrading enzyme in primary cultures of neuronal, glial and adenohypophyseal cells. Endocrinology 127, 1224–1233. doi: 10.1210/endo-127-3-1224
Bauer, K., Heuer, H., Ifflander, F., Peters, A., Schmitmeier, S., Shomburg, L., et al. (1997). ““Inactivation of thyrotropin-releasing hormone (TRH) by a TRH-specific ectoenzyme,”,” in Cell-Surface Peptidases in Health and Disease. Eds. Kenny, B., AJ, C. M. (UK: Oxford: BIOS Scientific Publishers), 239–248.
Bauer, K. (1979). Thyroliberin analogues as competitive inhibitors of thyroliberin degradation by brain enzymes. Hoppe-Seyler's Z Physiol. Chem. 360, 1126.
Bauer, K. (1994). Purification and Characterization of the Thyrotropin-releasing-hormone-degrading Ectoenzyme. Eur. J. Biochem. 224, 387–396. doi: 10.1111/j.1432-1033.1994.00387.x
Bellemere, G., Vaudry, H., Morain, P., Jegou, S. (2005). Effect of prolyl endopeptidase inhibition on arginine-vasopressin and Thyrotrophin-Releasing Hormone catabolism in the rat brain. J. Neuroendocrinol. 17, 306–313. doi: 10.1111/j.1365-2826.2005.01308.x
Bennett, G. W., Ballard, T. M., Watson, C. D., Fone, K. C. F. (1997). Effect of neuropeptides on cognitive function. Exp. Gerontol. 32, 451–469. doi: 10.1016/S0531-5565(96)00159-3
Bidaud, I., Galas, L., Bulant, M., Jenks, B. G., Ouwens, D. T. W. M., Jégou, S., et al. (2004). Distribution of the mRNAs encoding the thyrotropin-releasing hormone (TRH) precursor and three TRH receptors in the brain and pituitary of Xenopus laevis: Effect of background color adaptation on TRH and TRH receptor gene expression: proTRH and x TRHR mRNAs in Brain and Pituitary of Xenopus. J. Comp. Neurol. 477, 11–28. doi: 10.1002/cne.20235
Bilek, R., Gkonos, P. J., Tavianini, M. A., Smyth, D. G., Roos, B. A. (1992). The thyrotrophin-releasing hormone (TRH)-like peptides in rat prostate are not formed by expression of the TRH gene but are suppressed by thyroid hormone. J. Endocrinol. 132, 177–184. doi: 10.1677/joe.0.1320177
Blaszkowska, J., Pawlikowski, M., Komorowski, J., Kurnatowski, P. (2004). Effect of thyroliberin on the course of experimental candidosis in mice. Thyroliberin-Wirkung auf den Verlauf der experimentellen Candidose der Maus. Mycoses 47, 115–120. doi: 10.1111/j.1439-0507.2004.00961.x
Buga, A.-M., Scholz, C. J., Kumar, S., Herndon, J. G., Alexandru, D., Cojocaru, G. R., et al. (2012). Identification of new therapeutic targets by genome-wide analysis of gene expression in the ipsilateral cortex of aged rats after stroke. PloS One 7, e50985. doi: 10.1371/journal.pone.0050985
Bundgaard, H., Møss, J. (1990). Effect of thyroliberin on the course of experimental candidosis in mice. Pharmaceut. Res. 07, 885–892. doi: 10.1023/A:1015933504191
Calzá, L., Giardino, L., Ceccatelli, S., Zanni, M., Elde, R., Hökfelt, T. (1992). Distribution of thyrotropin-releasing hormone receptor messenger RNA in the rat brain: An in situ hybridization study. Neuroscience 51, 891–909. doi: 10.1016/0306-4522(92)90528-A
Campbell, J. N., Macosko, E. Z., Fenselau, H., Pers, T. H., Lyubetskaya, A., Tenen, D., et al. (2017). A molecular census of arcuate hypothalamus and median eminence cell types. Nat. Neurosci. 20, 484–496. doi: 10.1038/nn.4495
Carnell, N. E., Feng, P., Kim, U. J., Wilber, J. F. (1992). Preprothyrotropin-releasing hormone mRNA and TRH are present in the rat heart. Neuropeptides 22, 209–212. doi: 10.1016/0143-4179(92)90047-Z
Chávez-Gutiérrez, L., Matta-Camacho, E., Osuna, J., Horjales, E., Joseph-Bravo, P., Maigret, B., et al. (2006). Homology modeling and site-directed mutagenesis of pyroglutamyl peptidase II. Insights into omega-versus aminopeptidase specificity in the M1 family. J. Biol. Chem. 281, 18581–18590. doi: 10.1074/jbc.M601392200
Charli, J.-L., Ponce, G., McKelvy, J. F., Joseph-Bravo, P. (1984). Accumulation of Thyrotropin Releasing Hormone by rat hypothalamic slices. J. Neurochem. 42, 981–986. doi: 10.1111/j.1471-4159.1984.tb12700.x
Charli, J. L., Mendez, M., Joseph-Bravo, P., Wilk, S. (1987). Specific inhibitors of pyroglutamyl peptidase I and prolyl endopeptidase do not change the in vitro release of TRH or its content in rodent brain. Neuropeptides 9, 373–378. doi: 10.1016/0143-4179(87)90010-2
Charli, J.-L., Cruz, C., Vargas, M.-A., Joseph-Bravo, P. (1988). The narrow specificity pyroglutamate amino peptidase degrading TRH in rat brain is an ectoenzyme. Neurochem. Int. 13, 237–242. doi: 10.1016/0197-0186(88)90060-5
Charli, J.-L., Mendez, M., Vargas, M.-A., Cisneros, M., Assai, M., Joseph-Bravo, P., et al. (1989). Pyroglutamyl peptidase II inhibition specifically increases recovery of TRH released from rat brain slices. Neuropeptides 14, 191–196. doi: 10.1016/0143-4179(89)90044-9
Charli, J. L., Vargas, M. A., Cisneros, M., de Gortari, P., Baeza, M. A., Jasso, P., et al. (1998). TRH inactivation in the extracellular compartment: role of pyroglutamyl peptidase II. Neurobiol. (Bp) 6, 45–57.
Chatonnet, F., Guyot, R., Benoit, G., Flamant, F. (2013). Genome-wide analysis of thyroid hormone receptors shared and specific functions in neural cells. Proc. Natl. Acad. Sci. 110, E766–E775. doi: 10.1073/pnas.1210626110
Chavez-Gutierrez, L., Bourdais, J., Aranda, G., Vargas, M. A., Matta-Camacho, E., Ducancel, F., et al. (2005). A truncated isoform of pyroglutamyl aminopeptidase II produced by exon extension has dominant-negative activity. J. Neurochem. 92, 807–817. doi: 10.1111/j.1471-4159.2004.02916.x
Choi, J., Kim, J., Kim, T.-K., Park, J.-Y., Lee, J.-E., Kim, H., et al. (2015). TRH and TRH receptor system in the basolateral amygdala mediate stress-induced depression-like behaviors. Neuropharmacology 97, 346–356. doi: 10.1016/j.neuropharm.2015.03.030
Colson, A., Gershengorn, M. (2006). Thyrotropin-Releasing Hormone analogs. MRMC 6, 221–226. doi: 10.2174/138955706775476019
Cornford, E. M., Braun, L. D., Crane, P. D., Oldendorf, W. H. (1978). Blood-brain barrier restriction of peptides and the low uptake of enkephalins. Endocrinology 103, 1297–1303. doi: 10.1210/endo-103-4-1297
Cremades, A., Peñafiel, R., Rausell, V., Del Rio-Garcia, J., Smyth, D. G. (1998). The thyrotropin-releasing hormone-like peptides pGlu-Phe-Pro amide and pGlu-Glu-Pro amide increase plasma triiodothyronine levels in the mouse; the activity is sensitive to testosterone. Eur. J. Pharmacol. 358, 63–67. doi: 10.1016/S0014-2999(98)00593-7
Cruz, C., Charli, J.-L., Vargas, M. A., Joseph-Bravo, P. (1991). Neuronal localization of pyroglutamate aminopeptidase II in primary cultures of fetal mouse brain. J. Neurochem. 56, 1594–1601. doi: 10.1111/j.1471-4159.1991.tb02056.x
Cruz, R., Vargas, M. A., Uribe, R. M., Pascual, I., Lazcano, I., Yiotakis, A., et al. (2008). Anterior pituitary pyroglutamyl peptidase II activity controls TRH-induced prolactin release. Peptides 29, 1953–1964. doi: 10.1016/j.peptides.2008.07.011
Cucinotta, D., Senin, U., Girardello, R., Crepaldi, G. (1994). Posatirelin effect on patients with senile dementia of Alzheimer type (SDAT): a double-blind multicentre trial vs ascorbic acid and citicoline. J. Neurol. 241, S129.
Cushman, D. W., Cheung, H. S., Sabo, E. F., Ondetti, M. A. (1977). Design of potent competitive inhibitors of angiotensin-converting enzyme. Carboxyalkanoyl and mercaptoalkanoyl amino acids. Biochemistry 16, 5484–5491. doi: 10.1021/bi00644a014
Czekay, G., Bauer, K. (1993). Identification of the thyrotropin-releasing-hormone-degrading ectoenzyme as a metallopeptidase. Biochem. J. 290, 921–926. doi: 10.1042/bj2900921
Daimon, C., Chirdon, P., Maudsley, S., Martin, B. (2013). The role of Thyrotropin Releasing Hormone in aging and neurodegenerative diseases. AJAD 29–59. doi: 10.7726/ajad.2013.1003
de Gortari, P., Fernández-Guardiola, A., Martinez, A., Cisneros, M., Joseph-Bravo, P. (1995). Changes in TRH and its degrading enzyme pyroglutamyl peptidase II, during the development of amygdaloid kindling. Brain Res. 679, 144–150. doi: 10.1016/0006-8993(95)00237-K
de Gortari, P., Romero, F., Cisneros, M., Joseph-bravo, P. (2005). Acute administration of alcohol modulates pyroglutamyl amino peptidase II activity and mRNA levels in rat limbic regions. Neurochem. Int. 46, 347–356. doi: 10.1016/j.neuint.2004.11.002
de Gortari, P., Uribe, R. M., García-Vázquez, A., Aguilar-Valles, A., Martínez, A., Valdés, A., et al. (2006). Amygdala kindling differentially regulates the expression of the elements involved in TRH transmission. Neurochem. Int. 48, 31–42. doi: 10.1016/j.neuint.2005.08.003
Deng, P.-Y., Porter, J. E., Shin, H.-S., Lei, S. (2006). Thyrotropin-releasing hormone increases GABA release in rat hippocampus: TRH modulation of hippocampal GABAergic transmission. J. Physiol. 577, 497–511. doi: 10.1113/jphysiol.2006.118141
Diano, S., Naftolin, F., Goglia, F., Horvath, T. L. (1998). Fasting-induced increase in type II iodothyronine deiodinase activity and messenger ribonucleic acid levels is not reversed by thyroxine in the rat hypothalamus. Endocrinology 139, 2879–2884. doi: 10.1210/endo.139.6.6062
Drago, F., Pulvirenti, L., Spadaro, F., Pennisi, G. (1990). Effects of TRH and prolactin in the behavioral despair (swim) model of depression in rats. Psychoneuroendocrinology 15, 349–356. doi: 10.1016/0306-4530(90)90060-M
Drago, F., Coppi, G., Antonuzzo, P. A., Valerio, C., Genazzani, A. A., Grassi, M., et al. (1996). Effects of RGH 2202 on cognitive and motor behavior of the rat. Neurobiol. Aging 17, 67–71. doi: 10.1016/0197-4580(95)02006-3
Elmore, M. A., Griffiths, E. C., O'Connor, B., O'Cuinn, G. (1990). Further characterization of the substrate specificity of a TRH hydrolysing pyroglutamate aminopeptidase from guinea-pig brain. Neuropeptides 15, 31–36. doi: 10.1016/0143-4179(90)90157-T
Ensemble release 99. - January 2020. EMBL-EBI. http://Jan2020.archive.ensembl.org/Homo_sapiens/Gene/ExpressionAtlas?g=ENSG00000072657;r=12:72087266-72670758 [accessed march 21, 2020].
Faivre-Bauman, A., Loudes, C., Barret, A., Tixier-Vidal, A., Bauer, K. (1986). Possible role of neuropeptide degrading enzymes on thyroliberin secretion in fetal hypothalamic cultures grown in serum free medium. Neuropeptides 7, 125–138. doi: 10.1016/0143-4179(86)90088-0
Farre, D. (2003). Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res. 31, 3651–3653. doi: 10.1093/nar/gkg605
Fehlings, M. G., Baptiste, D. C. (2005). Current status of clinical trials for acute spinal cord injury. Injury 36, S113–S122. doi: 10.1016/j.injury.2005.06.022
Fekete, C., Lechan, R. M. (2014). Central regulation of hypothalamic-pituitary-thyroid axis under physiological and pathophysiological conditions. Endocrine Rev. 35, 159–194. doi: 10.1210/er.2013-1087
Ferguson, J. F., Xue, C., Hu, Y., Li, M., Reilly, M. P. (2016). Adipose tissue RNASeq reveals novel gene–nutrient interactions following n-3 PUFA supplementation and evoked inflammation in humans. J. Nutr. Biochem. 30, 126–132. doi: 10.1016/j.jnutbio.2015.12.010
Finan, B., Clemmensen, C., Zhu, Z., Stemmer, K., Gauthier, K., Müller, L., et al. (2016). Chemical hybridization of glucagon and thyroid hormone optimizes therapeutic impact for metabolic disease. Cell 167, 843–857.e14. doi: 10.1016/j.cell.2016.09.014
Fröhlich, E., Wahl, R. (2019). The forgotten effects of thyrotropin-releasing hormone: Metabolic functions and medical applications. Front. Neuroendocrinol. 52, 29–43. doi: 10.1016/j.yfrne.2018.06.006
Friedman, T. C., Wilk, S. (1985). The effect of inhibitors of prolyl endopeptidase and pyroglutamyl peptide hydrolase on TRH degradation in rat serum. Biochem. Biophys. Res. Commun. 132, 787–794. doi: 10.1016/0006-291X(85)91201-X
Gallagher, S. P., O'Connor, B. (1998). A study of a highly specific pyroglutamyl aminopeptidase type-II from the membrane fraction of bovine brain. Int. J. Biochem. Cell Biol. 30, 115–133. doi: 10.1016/S1357-2725(97)00074-5
Garat, B., Miranda, J., Charli, J.-L., Ioseph-Bravo, P. (1985). Presence of a membrane bound pyroglutamyl amino peptidase degrading thyrotropin releasing hormone in rat brain. Neuropeptides 6, 27–40. doi: 10.1016/0143-4179(85)90128-3
Gary, K. A., Sevarino, K. A., Yarbrough, G. G., Prange, A. J., Winokur, A. (2003). The Thyrotropin-Releasing Hormone (TRH) hypothesis of homeostatic regulation: implications for TRH-based therapeutics. J. Pharmacol. Exp. Ther. 305, 410–416. doi: 10.1124/jpet.102.044040
Ghilchik, M. W., Tobaruela, M., del Rio-Garcia, J., Smyth, D. G. (2000). Characterization of neutral TRH-like peptides in mammary gland, mammary tumors and milk. Biochim. Biophys. Acta (BBA) Gen. Subj. 1475, 55–60. doi: 10.1016/S0304-4165(00)00043-X
Gkonos, P. J., Kwok, C. K., Block, N. L., Roos, B. A. (1994). Identification of the human seminal TRH-like peptide pGlu-Phe-Pro-NH2 in normal human prostate. Peptides 15, 1281–1283. doi: 10.1016/0196-9781(94)90154-6
González, J. A., Horjales-Araujo, E., Fugger, L., Broberger, C., Burdakov, D. (2009). Stimulation of orexin/hypocretin neurones by thyrotropin-releasing hormone: TRH activates central orexin neurones. J. Physiol. 587, 1179–1186. doi: 10.1113/jphysiol.2008.167940
Gotoh, K., Fukagawa, K., Fukagawa, T., Noguchi, H., Kakuma, T., Sakata, T., et al. (2007). Hypothalamic neuronal histamine mediates the thyrotropin-releasing hormone-induced suppression of food intake. J. Neurochem. 103, 1102–1110. doi: 10.1111/j.1471-4159.2007.04802.x
Grattan, D. R. (2015). 60 YEARS OF NEUROENDOCRINOLOGY: The hypothalamo-prolactin axis. J. Endocrinol. 226, T101–T122. doi: 10.1530/JOE-15-0213
Griffiths, E. C., McDermott, J. R., Smith, A. I. (1982). Mechanisms of brain inactivation of centrally-acting thyrotrophin-releasing hormone (TRH) analogues: a high-performance liquid chromatography study. Regul. Peptides 5, 1–11. doi: 10.1016/0167-0115(82)90070-2
Gutiérrez-Mariscal, M., de Gortari, P., López-Rubalcava, C., Martínez, A., Joseph-Bravo, P. (2008). Analysis of the anxiolytic-like effect of TRH and the response of amygdalar TRHergic neurons in anxiety. Psychoneuroendocrinology 33, 198–213. doi: 10.1016/j.psyneuen.2007.11.002
Gutiérrez-Mariscal, M., Sánchez, E., Rebolledo-Solleiro, D., García-Vázquez, A. I., Cote-Vélez, A., Acasuso-Rivero, C., et al. (2012). The acute response of the amygdalar TRH system to psychogenic stressors varies dependent on the paradigm and circadian condition. Brain Res. 1452, 73–84. doi: 10.1016/j.brainres.2012.02.071
Hökfelt, T., Tsuruo, Y., Ulfhake, B., Cullheim, S., Arvidsson, U., Foster, G. A., et al. (1989). SECTION II. SYNAPTIC ROLE OF TRH: Distribution of TRH-like immunoreactivity with special reference to coexistence with other neuroactive compounds. Ann. NY Acad. Sci. 553, 76–105. doi: 10.1111/j.1749-6632.1989.tb46633.x
Hara, J., Gerashchenko, D., Wisor, J. P., Sakurai, T., Xie, X., Kilduff, T. S. (2009). Thyrotropin-Releasing Hormone increases behavioral arousal through modulation of hypocretin/orexin neurons. J. Neurosci. 29, 3705–3714. doi: 10.1523/JNEUROSCI.0431-09.2009
Heuer, H., Ehrchen, J., Bauer, K., Schäfer, M. K. H. (1998a). Region-specific expression of thyrotrophin-releasing hormone-degrading ectoenzyme in the rat central nervous system and pituitary gland: Distribution of the TRH-degrading ectoenzyme. Eur. J. Neurosci. 10, 1465–1478. doi: 10.1046/j.1460-9568.1998.00158.x
Heuer, H., Schäfer, M. K.-H., Bauer, K. (1998b). The Thyrotropin-Releasing Hormone-degrading ectoenzyme: the third element of the Thyrotropin-Releasing Hormone-signaling system. Thyroid 8, 915–920. doi: 10.1089/thy.1998.8.915
Heuer, H., Schäfer, M. K. H., O'Donnell, D., Walker, P., Bauer, K. (2000). Expression of thyrotropin-releasing hormone receptor 2 (TRH-R2) in the central nervous system of rats. J. Comp. Neurol. 428, 319–336. doi: 10.1002/1096-9861(20001211)428:2<319::AID-CNE10>3.0.CO;2-9
Hicks, S. C., Okrah, K., Paulson, J. N., Quackenbush, J., Irizarry, R. A., Bravo, H. C. (2018). Smooth quantile normalization. Biostatistics 19, 185–198. doi: 10.1093/biostatistics/kxx028
Hinkle, P. M., Pekary, A. E., Senanayaki, S., Sattin, A. (2002). Role of TRH receptors as possible mediators of analeptic actions of TRH-like peptides. Brain Res. 935, 59–64. doi: 10.1016/S0006-8993(02)02454-X
Hinkle, P. M., Gehret, A. U., Jones, B. W. (2012). Desensitization, trafficking, and resensitization of the pituitary Thyrotropin-Releasing Hormone receptor. Front. Neurosci. 6, 180. doi: 10.3389/fnins.2012.00180
Hodges, M. R., Richerson, G. B. (2008). Contributions of 5-HT neurons to respiratory control: Neuromodulatory and trophic effects. Respiratory Physiol. Neurobiol. 164, 222–232. doi: 10.1016/j.resp.2008.05.014
Horita, A. (1998). An update on the CNS actions of TRH and its analogs. Life Sci. 62, 1443–1448. doi: 10.1016/S0024-3205(98)00087-3
Hou, L., Zhou, X., Chen, Y., Qiu, D., Zhu, L., Wang, J. (2012). Thyrotropin-releasing hormone causes a tonic excitatory postsynaptic current and inhibits the phasic inspiratory inhibitory inputs in inspiratory-inhibited airway vagal preganglionic neurons. Neuroscience 202, 184–191. doi: 10.1016/j.neuroscience.2011.12.003
Ijiro, T., Nakamura, K., Ogata, M., Inada, H., Kiguchi, S., Maruyama, K., et al. (2015). Effect of rovatirelin, a novel thyrotropin-releasing hormone analog, on the central noradrenergic system. Eur. J. Pharmacol. 761, 413–422. doi: 10.1016/j.ejphar.2015.05.047
Itoh, Y., Ogasawara, T., Mushiroi, T., Yamazaki, A., Ukai, Y., Kimura, K. (1994). Effect of NS-3, a thyrotropin-releasing hormone analog, on in vivo acetylcholine release in rat brain: regional differences and its sites of action. J. Pharmacol. Exp. Ther. 271, 884–890.
Itoh, Y., Sugimoto, T., Ukai, Y., Morino, A., Kimura, K. (1995). Permeability of NS-3, a Thyrotropin-releasing Hormone analogue, into the brain after its systemic administration in rats: a microdialysis study. J. Pharm. Pharmacol. 47, 833–836. doi: 10.1111/j.2042-7158.1995.tb05750.x
Jaimes-Hoy, L., Gutiérrez-Mariscal, M., Vargas, Y., Pérez-Maldonado, A., Romero, F., Sánchez-Jaramillo, E., et al. (2016). Neonatal maternal separation alters, in a sex-specific manner, the expression of TRH, of TRH-degrading ectoenzyme in the rat hypothalamus, and the response of the thyroid axis to starvation. Endocrinology 157, 3253–3265. doi: 10.1210/en.2016-1239
Jaimes-Hoy, L., Romero, F., Charli, J.-L., Joseph-Bravo, P. (2019). Sex dimorphic responses of the hypothalamus–pituitary–thyroid axis to maternal separation and palatable diet. Front. Endocrinol. 10, 445. doi: 10.3389/fendo.2019.00445
Jeong, J. K., Szabo, G., Kelly, K., Diano, S. (2012). Prolyl carboxypeptidase regulates energy expenditure and the thyroid axis. Endocrinology 153, 683–689. doi: 10.1210/en.2011-1399
Jin, H., Fedorowicz, G., Yang, R., Ogasawara, A., Peale, F., Pham, T., et al. (2004). Thyrotropin-Releasing Hormone is induced in the left ventricle of rats with heart failure and can provide inotropic support to the failing heart. Circulation 109, 2240–2245. doi: 10.1161/01.CIR.0000127951.13380.B4
Joseph-Bravo, P., Fresán, M. E., Cisneros, M., Vargas, M. A., Charli, J.-L. (1994). Pyroglutamyl peptidase II activity is not in the processes of bulbospinal TRHergic neurons. Neurosci. Lett. 178, 243–246. doi: 10.1016/0304-3940(94)90769-2
Joseph-Bravo, P., Jaimes-Hoy, L., Charli, J.-L. (2015a). Regulation of TRH neurons and energy homeostasis-related signals under stress. J. Endocrinol. 224, R139–R159. doi: 10.1530/JOE-14-0593
Joseph-Bravo, P., Jaimes-Hoy, L., Uribe, R.-M., Charli, J.-L. (2015b). 60 YEARS OF NEUROENDOCRINOLOGY: TRH, the first hypophysiotropic releasing hormone isolated: control of the pituitary–thyroid axis. J. Endocrinol. 226, T85–T100. doi: 10.1530/JOE-15-0124
Kalivas, P. W., Stanley, D., Prange, A. J. (1987). Interaction between thyrotropinreleasing hormone and the mesolimbic dopamine system. Neuropharmacology 26, 33–38. doi: 10.1016/0028-3908(87)90041-4
Kamath, J., Yarbrough, G. G., Prange, A. J., Winokur, A. (2009). The thyrotropin-releasing hormone (TRH)–immune system homeostatic hypothesis. Pharmacol. Ther. 121, 20–28. doi: 10.1016/j.pharmthera.2008.09.004
Kanasaki, H., Oride, A., Mijiddorj, T., Purwana, I., Miyazaki, K. (2011). Secondary amenorrhea in a woman with spinocerebellar degeneration treated with thyrotropin-releasing hormone: a case report and in vitro analysis. J. Med. Case Rep. 5, 567. doi: 10.1186/1752-1947-5-567
Kaysser, L. (2019). Built to bind: biosynthetic strategies for the formation of small-molecule protease inhibitors. Nat. Prod. Rep. 36, 1654–1686. doi: 10.1039/C8NP00095F
Kelly, J. A., Loscher, C. E., Gallagher, S., O'Connor, B. (1997). Degradation of pyroglutamyl-phenylalanyl-proline amide by a pyroglutamyl aminopeptidase purified from membrane fractions of bovine brain. Biochem. Soc. Trans. 25, 114S–114S. doi: 10.1042/bst025114s
Kelly, J. A., Slator, G. R., Tipton, K. F., Williams, C. H., Bauer, K. (2000). Kinetic investigation of the specificity of porcine brain Thyrotropin-releasing Hormone-degrading ectoenzyme for Thyrotropin-releasing Hormone-like peptides. J. Biol. Chem. 275, 16746–16751. doi: 10.1074/jbc.M910386199
Kelly, J. A., Scalabrino, G. A., Slator, G. R., Cullen, A. A., Gilmer, J. F., Lloyd, D. G., et al. (2005). Structure–activity studies with high-affinity inhibitors of pyroglutamyl-peptidase II. Biochem. J. 389, 569–576. doi: 10.1042/BJ20041722
Kelly, J. A., Boyle, N. T., Cole, N., Slator, G. R., Colivicchi, M. A., Stefanini, C., et al. (2015). First-in-class thyrotropin-releasing hormone (TRH)-based compound binds to a pharmacologically distinct TRH receptor subtype in human brain and is effective in neurodegenerative models. Neuropharmacology 89, 193–203. doi: 10.1016/j.neuropharm.2014.09.024
Kelly, J. A. (1995). Thyrotropin-releasing hormone: basis and potential for its therapeutic use. Essays Biochem. 30, 133–149.
Khan, Z., Aitken, A., Garcia, J. R., Smyth, D. G. (1992). Isolation and identification of two neutral thyrotropin releasing hormone-like peptides, pyroglutamylphenylalanineproline amide and pyroglutamylglutamineproline amide, from human seminal fluid. J. Biol. Chem. 267, 7464–7469.
Khomane, K. S., Meena, C. L., Jain, R., Bansal, A. K. (2011). Novel thyrotropin-releasing hormone analogs: a patent review. Expert Opin. Ther. Patents 21, 1673–1691. doi: 10.1517/13543776.2011.623127
Kinoshita, K., Nagao, T., Ono, H. (1994). Effects of TA-0910, an orally active TRH analog, on the spinal reflex in spinal rats. Neuropharmacology 33, 1183–1188. doi: 10.1016/S0028-3908(05)80008-5
Kinoshita, K., Yamamura, M., Sugihara, J., Suzuki, M., Matsuoka, Y. (1998). Taltirelin Hydrate (TA-0910): an orally active Thyrotropin-Releasing Hormone mimetic agent with multiple actions. CNS Drug Rev. 4, 25–41. doi: 10.1111/j.1527-3458.1998.tb00039.x
Kobayashi, N., Sato, N., Fujimura, Y., Kihara, T., Sugita, K., Takahashi, K., et al. (2018). Discovery of the orally effective Thyrotropin-Releasing Hormone mimetic: 1-{N -[(4 S ,5 S )-(5-Methyl-2-oxooxazolidine-4-yl)carbonyl]-3-(thiazol-4-yl)- L -alanyl}-(2 R )-2-methylpyrrolidine Trihydrate (Rovatirelin Hydrate). ACS Omega 3, 13647–13666. doi: 10.1021/acsomega.8b01481
Kobayashi, K., Abe, Y., Harada, H., Oota, E., Endo, T., Takeda, H. (2019a). Non-clinical pharmacokinetic profiles of rovatirelin, an orally available thyrotropin-releasing hormone analogue. Xenobiotica 49, 106–119. doi: 10.1080/00498254.2017.1423130
Kobayshi, K., Abe, Y., Kawai, A., Furihata, T., Harada, H., Endo, T., et al. (2019b). Human mass balance, pharmacokinetics and metabolism of rovatirelin and identification of its metabolic enzymes in vitro. Xenobiotica 49, 1434–1446. doi: 10.1080/00498254.2019.1580796
Kodama, H., Furuuchi, S., Takahashi, M., Sugihara, J., Yoshikawa, M. (1997). Disposition of Taltirelin. (1): absorption, distribution, metabolism and excretion in rats and dogs. Drug Metab. Pharmacokinet. 12, 460–474. doi: 10.2133/dmpk.12.460
Kubek, M. J., Low, W. C., Sattin, A., Morzorati, S. L., Meyerhoff, J. L., Larsen, S. H. (1989). Role of TRH in seizure modulation. Ann. N. Y. Acad. Sci. 553, 286–303. doi: 10.1111/j.1749-6632.1989.tb46650.x
Kubek, M. J., Knoblach, S. M., Sharif, N. A., Burt, D. R., Buterbaugh, G. G., Fuson, K. S. (1993). Thyrotropin-Releasing hormone gene expression and receptors are differentially modified in limbic foci by seizures. Ann. Neurol. 33, 70–76. doi: 10.1002/ana.410330112
Kulkarni, R. N., Wang, Z. L., Akinsanya, K. O., Bennet, W. M., Wang, R. M., Smith, D. M., et al. (1995). Pyroglutamyl-phenylalanyl-proline amide attenuates thyrotropin-releasing hormone-stimulated insulin secretion in perifused rat islets and insulin-secreting clonal beta-cell lines. Endocrinology 136, 5155–5164. doi: 10.1210/endo.136.11.7588254
Lai, C.-T., Li, H.-J., Yu, W., Shah, S., Bommineni, G. R., Perrone, V., et al. (2015). Rational modulation of the induced-fit conformational change for slow-onset inhibition in Mycobacterium tuberculosis InhA. Biochemistry 54, 4683–4691. doi: 10.1021/acs.biochem.5b00284
Lanzara, R., Liebman, M., Wilk, S. (1989). The use of analogues of TRH to probe the specificity of pyroglutamyl peptidase II. Ann. NY Acad. Sci. 553, 559–562. doi: 10.1111/j.1749-6632.1989.tb46696.x
Lazcano, I., Uribe, R. M., Martínez-Chávez, E., Vargas, M. A., Matziari, M., Joseph-Bravo, P., et al. (2012). Pyroglutamyl peptidase II inhibition enhances the analeptic effect of Thyrotropin-Releasing Hormone in the rat medial septum. J. Pharmacol. Exp. Ther. 342, 222–231. doi: 10.1124/jpet.112.192278
Lazcano, I., Cabral, A., Uribe, R. M., Jaimes-Hoy, L., Perello, M., Joseph-Bravo, P., et al. (2015). Fasting enhances pyroglutamyl peptidase II activity in tanycytes of the mediobasal hypothalamus of male adult rats. Endocrinology 156, 2713–2723. doi: 10.1210/en.2014-1885
Lechan, R. M., Wu, P., Jackson, I. M. D. (1986). Immunolocalization of the Thyrotropin-Releasing Hormone prohormone in the rat central nervous system. Endocrinology 119, 1210–1216. doi: 10.1210/endo-119-3-1210
Leppäluoto, J., Koivusalo, F., Kraama, R. (1978). Thyrotropin-releasing factor: Distribution in neural and gastrointestinal tissues. Acta Physiol. Scandinavica 104, 175–179. doi: 10.1111/j.1748-1716.1978.tb06264.x
Lesnikov, V. A., Korneva, E. A., Dall'ara, A., Pierpaoli, W. (1992). The involvement of pineal gland and melatonin in immunity and aging: II. Thyrotropin-Releasing Hormone and melatonin forestall involution and promote reconstitution of the thymus in anterior hypothalamic area (Aha)-lesioned mice. Int. J. Neurosci. 62, 141–153. doi: 10.3109/00207459108999767
Lin, J., Wilk, S. (1998). Quantitation and regulation of pyroglutamyl peptidase II messenger RNA levels in rat tissues and GH3 cells. Neuroendocrinology 67, 197–208. doi: 10.1159/000054315
Linden, H., del Rio Garcia, J., Huber, A., Kreil, G., Smyth, D. (1996). The TRH-like peptides in rabbit testis are different from the TRH-like peptide in the prostate. FEBS Lett. 379, 11–14. doi: 10.1016/0014-5793(95)01468-3
Luo, L., Yano, N. (2004). Expression of thyrotropin-releasing hormone receptor in immortalized beta-cell lines and rat pancreas. J. Endocrinol. 181, 401–412. doi: 10.1677/joe.0.1810401
Méndez, M., Cisneros, M., Baez, A., Joseph-Bravo, P., Charli, J. L. (1999). Three TRH-like molecules are released from rat hypothalamus in vitro. Neurochem. Res. 24, 815–823. doi: 10.1023/a:1020993527602
Müller-Fielitz, H., Stahr, M., Bernau, M., Richter, M., Abele, S., Krajka, V., et al. (2017). Tanycytes control the hormonal output of the hypothalamic-pituitary-thyroid axis. Nat. Commun. 8, 484. doi: 10.1038/s41467-017-00604-6
Marsili, A., Sanchez, E., Singru, P., Harney, J. W., Zavacki, A. M., Lechan, R. M., et al. (2011). Thyroxine-induced expression of pyroglutamyl peptidase II and inhibition of TSH release precedes suppression of TRH mRNA and requires type 2 deiodinase. J. Endocrinol. 211, 73–78. doi: 10.1530/JOE-11-0248
Martinez de la Escalera, G., Weiner, R. I. (1992). Dissociation of dopamine from its receptor as a signal in the pleiotropic hypothalamic regulation of prolactin secretion. Endocr. Rev. 13, 241–255. doi: 10.1210/edrv-13-2-241
Martino, E., Lernmark, A., Seo, H., Steiner, D. F., Refetoff, S. (1978). High concentration of thyrotropin-releasing hormone in pancreatic islets. Proc. Natl. Acad. Sci. 75, 4265–4267. doi: 10.1073/pnas.75.9.4265
Matre, V., Høvring, P. I., Fjeldheim, Å.-K., Helgeland, L., Orvain, C., Andersson, K. B., et al. (2003). The human neuroendocrine thyrotropin-releasing hormone receptor promoter is activated by the haematopoietic transcription factor c-Myb. Biochem. J. 372, 851–859. doi: 10.1042/bj20030057
Matziari, M., Bauer, K., Dive, V., Yiotakis, A. (2008). Synthesis of the phosphinic analogue of Thyrotropin Releasing Hormone. J. Org. Chem. 73, 8591–8593. doi: 10.1021/jo8014215
Meena, C. L., Ingole, S., Rajpoot, S., Thakur, A., Nandekar, P. P., Sangamwar, A. T., et al. (2015). Discovery of a low affinity thyrotropin-releasing hormone (TRH)-like peptide that exhibits potent inhibition of scopolamine-induced memory impairment in mice. RSC Adv. 5, 56872–56884. doi: 10.1039/C5RA06935A
Mentlein, R. (1999). Dipeptidyl-peptidase IV (CD26)-role in the inactivation of regulatory peptides. Regul. Peptides 85, 9–24. doi: 10.1016/S0167-0115(99)00089-0
Messeguer, X., Escudero, R., Farre, D., Nunez, O., Martinez, J., Alba, M. M. (2002). PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 18, 333–334. doi: 10.1093/bioinformatics/18.2.333
Minelli, A., Bellezza, I., Grottelli, S., Galli, F. (2008). Focus on cyclo(His-Pro): history and perspectives as antioxidant peptide. Amino Acids 35, 283–289. doi: 10.1007/s00726-007-0629-6
Mitsuma, T., Rhue, N., Sobue, G., Hirooka, Y., Kayama, M., Yokoi, Y., et al. (1995). Distribution of thyrotropin releasing hormone receptor in rats: an immunohistochemical study. Endocr. Regul. 29, 129–134.
Monga, V., Meena, C., Kaur, N., Jain, R. (2008). Chemistry and biology of Thyrotropin-Releasing Hormone (TRH) and its analogs. CMC 15, 2718–2733. doi: 10.2174/092986708786242912
Montagne, J.-J., Ladram, A., Nicolas, P., Bulant, M. (1999). Cloning of Thyrotropin-Releasing Hormone precursor and receptor in rat thymus, adrenal gland, and testis. Endocrinology 140, 1054–1059. doi: 10.1210/endo.140.3.6558
Morier, E., Moreau, O., Masson, M. A., Han, K.-K., Rips, R. (1979). Evidence for the enzymic degradation of thyrotropin-releasing-hormone (trh) and pseudo-hormone (pyroglutamyl-histidyl-amphetamine) by calf liver pyroglutamine-amino-peptidase. Int. J. Biochem. 10, 769–783. doi: 10.1016/0020-711X(79)90155-1
Morley, J. E. (1979). Extrahypothalamic Thyrotropin Releasing Hormone (TRH) — Its distribution and its functions. Life Sci. 25, 1539–1550. doi: 10.1016/0024-3205(79)90435-1
Mulla, C. M., Geras-Raaka, E., Raaka, B. M., Gershengorn, M. C. (2009). High levels of Thyrotropin-Releasing Hormone receptors activate programmed cell death in human pancreatic precursors. Pancreas 38, 197–202. doi: 10.1097/MPA.0b013e31818d14a8
Nakamura, T., Honda, M., Kimura, S., Tanabe, M., Oda, S., Ono, H. (2005). Taltirelin improves motor ataxia independently of monoamine levels in rolling mouse Nagoya, a model of spinocerebellar atrophy. Biol. Pharm. Bull. 28, 2244–2247. doi: 10.1248/bpb.28.2244
Nishio, Y., Kusugami, K., Kaneko, H., Yamamoto, H., Konagaya, T., Nagai, H., et al. (1999). Intraluminal thyrotropin-releasing hormone affects gastric somatostatin and acid secretion through its specific receptor in rats. Scand. J. Gastroenterol. 34, 270–275. doi: 10.1080/00365529950173672
O'Connor, B., O'Cuinn, G. (1985). Purification of and kinetic studies on a narrow specificity synaptosomal membrane pyroglutamate aminopeptidase from guinea-pig brain. Eur. J. Biochem. 150, 47–52. doi: 10.1111/j.1432-1033.1985.tb08986.x
O'Leary, R., O'Connor, B. (1995). Thyrotropin-releasing hormone. J. Neurochem. 65, 953–963. doi: 10.1046/j.1471-4159.1995.65030953.x
Ogawa, N., Mizuno, S., Nukina, I., Tsukamoto, S., Mori, A. (1983). Chronic thyrotropin releasing hormone (TRH) administration on TRH receptors and muscarinic cholinergic receptors in CNS. Brain Res. 263, 348–350. doi: 10.1016/0006-8993(83)90328-1
Oka, M., Ochi, Y., Furukawa, K., Ito, T., Miura, Y., Karasawa, T., et al. (1989). L-6-ketopiperidine-2-carbonyl-L-leucyl-L-proline amide as a novel thyrotropin releasing hormone analogue with improving effects on impaired central nervous systems functions. Arzneimittelforschung 39, 297–303.
Péterfi, Z., Farkas, E., Nagyunyomi-Sényi, K., Kádár, A., Ottó, S., Horváth, A., et al. (2018). Role of TRH/UCN3 neurons of the perifornical area/bed nucleus of stria terminalis region in the regulation of the anorexigenic POMC neurons of the arcuate nucleus in male mice and rats. Brain Struct. Funct. 223, 1329–1341. doi: 10.1007/s00429-017-1553-5
Parnetti, L., Ambrosoli, L., Abate, G., Azzini, C., Balestreri, R., Bartorelli, L., et al. (1995). Posatirelin for the treatment of late-onset Alzheimer's disease: a double-blind multicentre study vs citicoline and ascorbic acid. Acta Neurol. Scand. 92, 135–140. doi: 10.1111/j.1600-0404.1995.tb01027.x
Pascual, I., Gil-Parrado, S., Cisneros, M., Joseph-Bravo, P., Díaz, J., Possani, L. D., et al. (2004). Purification of a specific inhibitor of pyroglutamyl aminopeptidase II from the marine annelide Hermodice carunculata. Int. J. Biochem. Cell Biol. 36, 138–152. doi: 10.1016/S1357-2725(03)00175-4
Patchett, A. A., Cordes, E. H. (1985). “The design and properties of N-Carboxyalkyldipeptide inhibitors of angiotensin-converting Enzyme,” in Advances in Enzymology - and Related Areas of Molecular Biology. Ed. Meister, A. (Hoboken, NJ, USA: John Wiley & Sons, Inc.), 1–84. doi: 10.1002/9780470123034.ch1
Pawlikowski, M., Żerek-Mełeń, G., Winczyk, K. (1992). Thyroliberin (TRH) increases thymus cell proliferation in rats. Neuropeptides 23, 199–202. doi: 10.1016/0143-4179(92)90123-E
Pekary, A. E., Sattin, A. (2012). Rapid modulation of TRH and TRH-like peptide release in rat brain and peripheral tissues by ghrelin and 3-TRP-ghrelin. Peptides 36, 157–167. doi: 10.1016/j.peptides.2012.04.021
Pekary, A. E., Sattin, A. (2017). TRH and TRH-Like peptide levels co-vary with reproductive and metabolic rhythms. Horm. Metab. Res. 49, 86–94. doi: 10.1055/s-0042-111012
Pekary, A. E., Sattin, A., Meyerhoff, J. L., Chilingar, M. (2004). Valproate modulates TRH receptor, TRH and TRH-like peptide levels in rat brain. Peptides 25, 647–658. doi: 10.1016/j.peptides.2004.01.016
Pekary, A. E., Faull, K. F., Paulson, M., Lloyd, R. L., Sattin, A. (2005). TRH-like antidepressant peptide, pyroglutamyltyroslyprolineamide, occurs in rat brain. J. Mass Spectrom 40, 1232–1236. doi: 10.1002/jms.904
Pekary, A. E., Sattin, A., Lloyd, R. L. (2015). Ketamine modulates TRH and TRH-like peptide turnover in brain and peripheral tissues of male rats. Peptides 69, 66–76. doi: 10.1016/j.peptides.2015.04.003
Peres Diaz, L. S., Schuman, M. L., Aisicovich, M., Toblli, J. E., Pirola, C. J., Landa, M. S., et al. (2018). Angiotensin II requires an intact cardiac thyrotropin-releasing hormone (TRH) system to induce cardiac hypertrophy in mouse. J. Mol. Cell. Cardiol. 124, 1–11. doi: 10.1016/j.yjmcc.2018.09.009
Perez Castro, C., Peñalva, R., Páez Pereda, M., Renner, U., Reul, J. M., Stalla, G. K., et al. (1999). Early activation of thyrotropin-releasing-hormone and prolactin plays a critical role during a T cell-dependent immune response. Endocrinology 140, 690–697. doi: 10.1210/endo.140.2.6482
Pierpaoli, W., Yi, C. (1990). The involvement of pineal gland and melatonin in immunity and aging I. Thymus-mediated, immunoreconstituting and antiviral activity of thyrotropin-releasing hormone. J. Neuroimmunol. 27, 99–109. doi: 10.1016/0165-5728(90)90059-V
Pierpaoli, W. (2013). Aging-reversing properties of Thyrotropin-Releasing Hormone. CAS 6, 92–98. doi: 10.2174/1874609811306010012
Pilowsky, P. M. (2014). ““Peptides, serotonin, and breathing,”,” in Progress in Brain Research (Oxford, UK:Elsevier), 169–189. doi: 10.1016/B978-0-444-63274-6.00009-6
Prasad, C., Jayaraman, A. (1986). Metabolism of thyrotropin-releasing hormone in human cerebrospinal fluid. Isolation and characterization of pyroglutamate aminopeptidase activity. Brain Res. 364, 331–337. doi: 10.1016/0006-8993(86)90845-0
Prasad, C., Peterkofsky, A. (1976). Demonstration of pyroglutamylpeptidase and amidase activities toward thyrotropin-releasing hormone in hamster hypothalamus extracts. J. Biol. Chem. 251, 3229–3234.
Prasad, C. (1989). Neurobiology of Cyclo(His-Pro). Ann. NY Acad. Sci. 553, 232–251. doi: 10.1111/j.1749-6632.1989.tb46646.x
Prasad, C. (1995). Bioactive cyclic dipeptides. Peptides 16, 151–164. doi: 10.1016/0196-9781(94)00017-Z
Prokai-Tatrai, K., Prokai, L. (2009). Prodrugs of Thyrotropin-Releasing Hormone and related peptides as central nervous system agents. Molecules 14, 633–654. doi: 10.3390/molecules14020633
Puga, L., Alcántara-Alonso, V., Coffeen, U., Jaimes, O., de Gortari, P. (2016). TRH injected into the nucleus accumbens shell releases dopamine and reduces feeding motivation in rats. Behav. Brain Res. 306, 128–136. doi: 10.1016/j.bbr.2016.03.031
Rabeler, R., Mittag, J., Geffers, L., Rüther, U., Leitges, M., Parlow, A. F., et al. (2004). Generation of Thyrotropin-Releasing Hormone receptor 1-deficient mice as an animal model of central hypothyroidism. Mol. Endocrinol. 18, 1450–1460. doi: 10.1210/me.2004-0017
Rausell, V., Fraser, H. M., Tobaruela, M., del Rio-Garcia, J., Smyth, D. G. (1999). Identification of the TRH-like peptides pGlu–Glu–Pro amide and pGlu–Phe–Pro amide in rat thyroid: regulation by thyroid status. Regul. Peptides 81, 55–60. doi: 10.1016/S0167-0115(99)00017-8
Redding, T. W., Schally, A. V. (1969). Studies on the Thyrotropin-Releasing Hormone (TRH) activity in peripheral blood. Exp. Biol. Med. 131, 420–425. doi: 10.3181/00379727-131-33892
Rodríguez-Molina, V., Vargas, M.Á., Joseph-Bravo, P., Charli, J.-L. (2009). NMDA receptor up-regulates pyroglutamyl peptidase II activity in the rat hippocampus. Neurosci. Lett. 449, 211–214. doi: 10.1016/j.neulet.2008.11.005
Rodríguez-Molina, V., Patiño, J., Vargas, Y., Sánchez-Jaramillo, E., Joseph-Bravo, P., Charli, J.-L. (2014). TRH regulates action potential shape in cerebral cortex pyramidal neurons. Brain Res. 1571, 1–11. doi: 10.1016/j.brainres.2014.05.015
Rodríguez-Rodríguez, A., Lazcano, I., Sánchez-Jaramillo, E., Uribe, R. M., Jaimes-Hoy, L., Joseph-Bravo, P., et al. (2019). Tanycytes and the control of Thyrotropin-Releasing Hormone flux into portal capillaries. Front. Endocrinol. 10, 401. doi: 10.3389/fendo.2019.00401
Rondeel, J. M. M., Klootwijk, W., Linkels, E., Jeucken, W., H. M, P., Hofland, L. J., et al. (1995a). Further studies on the regulation, localization and function of the TRH-like peptide pyroglutamyl-glutamyl-prolineamide in the rat anterior pituitary gland. J. Endocrinol. 146, 293–300. doi: 10.1677/joe.0.1460293
Rondeel, J. M. M., Klootwijk, W., Linkels, E., van Haasteren, G. A. C., de Greef, W. J., Visser, T. J. (1995b). Regulation of the TRH-like peptide pyroglutamyl-glutamyl-prolineamide in the rat anterior pituitary gland. J. Endocrinol. 145, 43–49. doi: 10.1677/joe.0.1450043
Rosen, J. B., Cain, C. J., Weiss, S. R. B., Post, R. M. (1992). Alterations in mRNA of enkephalin, dynorphin and thyrotropin releasing hormone during amygdala kindling: an in situ hybridization study. Mol. Brain Res. 15, 247–255. doi: 10.1016/0169-328X(92)90115-R
Runfola, M., Sestito, S., Bellusci, L., La Pietra, V., D'Amore, V. M., Kowalik, M. A., et al. (2020). Design, synthesis and biological evaluation of novel TRβ selective agonists sustained by ADME-toxicity analysis. Eur. J. Med. Chem. 188, 112006. doi: 10.1016/j.ejmech.2019.112006
Sánchez, E., Uribe, R. M., Corkidi, G., Zoeller, R. T., Cisneros, M., Zacarias, M., et al. (2001). Differential responses of Thyrotropin-Releasing Hormone (TRH) neurons to cold exposure or suckling indicate functional heterogeneity of the TRH system in the paraventricular nucleus of the rat hypothalamus. Neuroendocrinology 74, 407–422. doi: 10.1159/000054707
Sánchez, E., Vargas, M. A., Singru, P. S., Pascual, I., Romero, F., Fekete, C., et al. (2009). Tanycyte Pyroglutamyl Peptidase II contributes to regulation of the hypothalamic-pituitary-thyroid axis through glial-axonal associations in the median eminence. Endocrinology 150, 2283–2291. doi: 10.1210/en.2008-1643
Sánchez, E., Charli, J.-L., Lechan, R. M. (2013). ““Pyroglutamyl-Peptidase II,”,” in Handbook of Proteolytic Enzymes (Oxford, UK:Elsevier), 414–419. doi: 10.1016/B978-0-12-382219-2.00083-1
Sárvári, A., Farkas, E., Kádár, A., Zséli, G., Füzesi, T., Lechan, R. M., et al. (2012). Thyrotropin-releasing hormone-containing axons innervate histaminergic neurons in the tuberomammillary nucleus. Brain Res. 1488, 72–80. doi: 10.1016/j.brainres.2012.10.010
Sah, N., Rajput, S. K., Singh, J. N., Meena, C. L., Jain, R., Sikdar, S. K., et al. (2011). l-pGlu-(2-propyl)-l-His-l-ProNH2 attenuates 4-aminopyridine-induced epileptiform activity and sodium current: a possible action of new thyrotropin-releasing hormone analog for its anticonvulsant potential. Neuroscience 199, 74–85. doi: 10.1016/j.neuroscience.2011.10.008
Saito, Y., Mekuchi, M., Kobayashi, N., Kimura, M., Aoki, Y., Masuda, T., et al. (2011). Molecular cloning, molecular evolution and gene expression of cDNAs encoding thyrotropin-releasing hormone receptor subtypes in a teleost, the sockeye salmon (Oncorhynchus nerka). Gen. Comp. Endocrinol. 174, 80–88. doi: 10.1016/j.ygcen.2011.07.011
Sasahara, H., Otaka, M., Itoh, S., Iwabuchi, A., Odashima, M., Wada, I., et al. (1998). Effect of preinduction of heat-shock proteins on acetic acid-induced small intestinal lesions in rats. Dig. Dis. Sci. 43, 2117–2130. doi: 10.1023/a:1018827802462
Sasaki, I., Fujita, T., Murakami, M., Yamamoto, A., Nakamura, E., Imasaki, H., et al. (1994). Intestinal absorption of azetirelin, a new Thyrotropin-Releasing Hormone (TRH) analogue. I. Possible factors for the low oral bioavailability in rats. Biol. Pharm. Bull. 17, 1256–1261. doi: 10.1248/bpb.17.1256
Sasaki, I., Tozaki, H., Matsumoto, K., Ito, Y., Fujita, T., Murakami, M., et al. (1999). Development of an oral formulation of azetirelin, a new thyrotropin-releasing hormone (TRH) analogue, using n-lauryl-beta-D-maltopyranoside as an absorption enhancer. Biol. Pharm. Bull. 22, 611–615. doi: 10.1248/bpb.22.611
Sattin, A. (1999). The role of TRH and related peptides in the mechanism of action of ECT. J. ECT 15, 76–92. doi: 10.1097/00124509-199903000-00007
Scalabrino, G. A., Hogan, N., O'Boyle, K. M., Slator, G. R., Gregg, D. J., Fitchett, C. M., et al. (2007). Discovery of a dual action first-in-class peptide that mimics and enhances CNS-mediated actions of thyrotropin-releasing hormone. Neuropharmacology 52, 1472–1481. doi: 10.1016/j.neuropharm.2007.02.003
Schauder, B., Schomburg, L., Kohrle, J., Bauer, K. (1994). Cloning of a cDNA encoding an ectoenzyme that degrades thyrotropin-releasing hormone. Proc. Natl. Acad. Sci. 91, 9534–9538. doi: 10.1073/pnas.91.20.9534
Schmitmeier, S., Thole, H., Bader, A., Bauer, K. (2002). Purification and characterization of the thyrotropin-releasing hormone (TRH)-degrading serum enzyme and its identification as a product of liver origin. Eur. J. Biochem. 269, 1278–1286. doi: 10.1046/j.1432-1033.2002.02768.x
Schomburg, L., Turwitt, S., Prescher, G., Lohmann, D., Horsthemke, B., Bauer, K. (1999). Human TRH-degrading ectoenzyme cDNA cloning, functional expression, genomic structure and chromosomal assignment. Eur. J. Biochem. 265, 415–422. doi: 10.1046/j.1432-1327.1999.00753.x
Schuman, M. L., Landa, M. S., Toblli, J. E., Peres Diaz, L. S., Alvarez, A. L., Finkielman, S., et al. (2011). Cardiac Thyrotropin-Releasing Hormone mediates left ventricular hypertrophy in spontaneously hypertensive rats. Hypertension 57, 103–109. doi: 10.1161/HYPERTENSIONAHA.110.161265
Schuman, M. L., Peres Diaz, L. S., Landa, M. S., Toblli, J. E., Cao, G., Alvarez, A. L., et al. (2014). Thyrotropin-releasing hormone overexpression induces structural changes of the left ventricle in the normal rat heart. Am. J. Physiol. Heart Circ. Physiol. 307, H1667–H1674. doi: 10.1152/ajpheart.00494.2014
Segerson, T. P., Hoefler, H., Childers, H., Wolfe, H. J., Wu, P., Jackson, I. M. D., et al. (1987). Localization of Thyrotropin-Releasing Hormone prohormone messenger ribonucleic acid in rat brain by in situ hybridization. Endocrinology 121, 98–107. doi: 10.1210/endo-121-1-98
Sharif, N. A., Towle, A. C., Burt, D. R., Mueller, R. A., Breese, G. R. (1989). Contransmitters: differential effects of serotonin (5-HT)-depleting drugs on levels of 5-HT and TRH and their receptors in rat brain and spinal cord. Brain Res. 480, 365–371. doi: 10.1016/0006-8993(89)90209-6
Shi, Z.-X., Xu, W., Mergner, W. J., Li, Q.-L., Cole, K. H., Wilber, J. F. (1996). Localization of Thyrotropin-Releasing Hormone mRNA expression in the rat heart by in situ hybridization histochemistry. Pathobiology 64, 314–319. doi: 10.1159/000164066
Shishido, Y., Furushiro, M., Tanabe, S., Shibata, S., Hashimoto, S., Yokokura, T. (1999). Effects of prolyl endopeptidase inhibitors and neuropeptides on delayed neuronal death in rats. Eur. J. Pharmacol. 372, 135–142. doi: 10.1016/S0014-2999(99)00185-5
Simasko, S. M., Horita, A. (1985). Treatment of rats with the TRH analog MK-771. Neuropharmacology 24, 157–165. doi: 10.1016/0028-3908(85)90175-3
Siviter, R. J., Cockle, S. M. (1995). Peptides related to thyrotrophin-releasing hormone are degraded in seminal plasma by an enzyme similar to prolyl endopeptidase. J. Endocrinol. 144, 61–66. doi: 10.1677/joe.0.1440061
Sjöström, H., Norén, O., Olsen, J. (2002). ““Structure and function of aminopeptidase N,”,” in Cellular Peptidases in Immune Functions and Diseases 2. Eds. Langner, J., Ansorge, S. ((Boston, MA: Springer US)), 25–34. doi: 10.1007/0-306-46826-3_2
Smyth, D. G., del Rio-Garcia, J., Wallnöfer, H., Gogl, H., Simma, W., Huber, A., et al. (1999). Protirelin (thyrotropin-releasing hormone) in thyroid gland: possible involvement in regulation of thyroid status. Zhongguo Yao Li Xue Bao 20, 289–291.
Štrbák, V. (2018). Pancreatic Thyrotropin Releasing Hormone and Mechanism of Insulin Secretion. Cell Physiol. Biochem. 50, 378–384. doi: 10.1159/000494013
Sugimoto, T., Hayashi, T., Okita, A., Morino, A. (1996). Pharmacokinetics of the new thyrotropin releasing hormone analogue montirelin hydrate. 1st communication: plasma concentrations, metabolism and excretion after a single intravenous administration to rats, dogs and monkeys. Arzneimittelforschung 46, 106–113.
Sun, Y., Lu, X., Gershengorn, M. (2003). Thyrotropin-releasing hormone receptors – similarities and differences. J. Mol. Endocrinol. 30, 87–97. doi: 10.1677/jme.0.0300087
Sun, Y., Zupan, B., Raaka, B. M., Toth, M., Gershengorn, M. C. (2009). TRH-receptor-type-2-deficient mice are euthyroid and exhibit increased depression and reduced anxiety phenotypes. Neuropsychopharmacol 34, 1601–1608. doi: 10.1038/npp.2008.217
Suzuki, M., Sugano, H., Matsumoto, K., Yamamura, M., Ishida, R. (1990). Synthesis and central nervous system actions of thyrotropin-releasing hormone analog containing a dihydroorotic acid moiety. J. Med. Chem. 33, 2130–2137. doi: 10.1021/jm00170a014
Szabo, L., Morey, R., Palpant, N. J., Wang, P. L., Afari, N., Jiang, C., et al. (2015). Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol. 16, 126. doi: 10.1186/s13059-015-0690-5
Szirtes, T., Kisfaludy, L., Palosi, E., Szporny, L. (1984). Synthesis of thyrotropin-releasing hormone analogs. 1. Complete dissociation of central nervous system effects from thyrotropin-releasing activity. J. Med. Chem. 27, 741–745. doi: 10.1021/jm00372a006
Szirtes, T., Kisfaludy, L., Palosi, E., Szporny, L. (1986). Synthesis of thyrotropin-releasing hormone analogs. 2. Tripeptides structurally greatly different from TRH with high central nervous system activity. J. Med. Chem. 29, 1654–1658. doi: 10.1021/jm00159a015
Taché, Y., Yang, H., Miampamba, M., Martinez, V., Yuan, P. Q. (2006). Role of brainstem TRH/TRH-R1 receptors in the vagal gastric cholinergic response to various stimuli including sham-feeding. Autonomic Neurosci. 125, 42–52. doi: 10.1016/j.autneu.2006.01.014
Tache, Y. (2012). Brainstem neuropeptides and vagal protection of the gastric mucosal against injury: role of prostaglandins, nitric oxide and calcitonin-gene related peptide in capsaicin afferents. CMC 19, 35–42. doi: 10.2174/092986712803414097
Takeuchi, Y., Takano, T., Abe, J., Takikita, S., Ohno, M. (2001). Thyrotropin-releasing hormone: role in the treatment of West syndrome and related epileptic encephalopathies. Brain Dev. 23, 662–667. doi: 10.1016/S0387-7604(01)00303-5
Tang, T., Li, L., Tang, J., Li, Y., Lin, W. Y., Martin, F., et al. (2010). A mouse knockout library for secreted and transmembrane proteins. Nat. Biotechnol. 28, 749–755. doi: 10.1038/nbt.1644
Taylor, W. L., Dixon, J. E. (1976). The inhibition of thyrotropin-releasing hormone deamidation in porcine hypothalamic tissues. Biochim. Biophys. Acta (BBA) Gen. Subj. 444, 428–434. doi: 10.1016/0304-4165(76)90386-X
Tenorio-Laranga, J., Venäläinen, J. I., Männistö, P. T., García-Horsman, J. A. (2008). Characterization of membrane-bound prolyl endopeptidase from brain: Membrane-bound prolyl oligopeptidase: mPOP. FEBS J. 275, 4415–4427. doi: 10.1111/j.1742-4658.2008.06587.x
Terauchi, Y., Ishikawa, S., Oida, S., Negoro, T., Kagemoto, A., Sekine, Y. (1988). Determination of L-6-keto-piperidine-2-carbonyl-L-leucyl-L-proline amide (RGH-2202), a new analog of thyrotropin-releasing hormone, in plasma by radioimmunoassay. J. Pharmacobio. Dynamics 11, 459–464. doi: 10.1248/bpb1978.11.459
Thirunarayanan, N., Nir, E. A., Raaka, B. M., Gershengorn, M. C. (2013). Thyrotropin-Releasing Hormone Receptor Type 1 (TRH-R1), not TRH-R2, Primarily Mediates Taltirelin Actions in the CNS of Mice. Neuropsychopharmacol 38, 950–956. doi: 10.1038/npp.2012.256
Thompson, B. L., Rosen, J. B. (2000). Effects of TRH on acoustic startle, conditioned fear and active avoidance in rats. Neuropeptides 34, 38–44. doi: 10.1054/npep.1999.0785
Torres, H., Charli, J., Gonzaleznoriega, A., Vargas, M., Joseph-Bravo, P. (1986). Subcellular distribution of the enzymes degrading thyrotropin releasing hormone and metabolites in rat brain. Neurochem. Int. 9, 103–110. doi: 10.1016/0197-0186(86)90038-0
Traube, T., Shokhen, M., Albeck, A. (2014). A new method for filtering of reactive “warheads” of transition-state analog protease inhibitors. Eur. J. Med. Chem. 77, 134–138. doi: 10.1016/j.ejmech.2014.02.059
Uhlén, M., Fagerberg, L., Hallstrom, B. M., Lindskog, C., Oksvold, P., Mardinoglu, A., et al. (2015). Tissue-based map of the human proteome. Science 347, 1260419–1260419. doi: 10.1126/science.1260419
Urayama, A., Yamada, S., Hirano, K., Deguchi, Y., Kimura, R. (2001). Brain receptor binding characteristics and pharmacokinetic-pharmacodynamic analysis of thyrotropin-releasing hormone analogues. Life Sci. 70, 647–657. doi: 10.1016/S0024-3205(01)01445-X
Urayama, A., Yamada, S., Kimura, R., Zhang, J., Watanabe, Y. (2002). Neuroprotective effect and brain receptor binding of taltirelin, a novel thyrotropin-releasing hormone (TRH) analogue, in transient forebrain ischemia of C57BL/6J mice. Life Sci. 72, 601–607. doi: 10.1016/S0024-3205(02)02268-3
Vargas, M., Mendez, M., Cisneros, M., Joseph-Bravo, P., Charli, J. L. (1987). Regional distribution of the membrane-bound pyroglutamate amino peptidase-degrading thyrotropin-releasing hormone in rat brain. Neurosci. Lett. 79, 311–314. doi: 10.1016/0304-3940(87)90450-2
Vargas, M. A., Cisneros, M., Herrera, J., Joseph-Bravo, P., Charli, J.-L. (1992a). Regional distribution of pyroglutamyl peptidase II in rabbit brain, spinal cord, and organs. Peptides 13, 255–260. doi: 10.1016/0196-9781(92)90105-C
Vargas, M. A., Herrera, J., Uribe, R. M., Charli, J.-L., Joseph-Bravo, P. (1992b). Ontogenesis of pyroglutamyl peptidase II activity in rat brain, adenohypophysis and pancreas. Dev. Brain Res. 66, 251–256. doi: 10.1016/0165-3806(92)90087-D
Veronesi, M. C., Kubek, D. J., Kubek, M. J. (2007). Intranasal delivery of a Thyrotropin-Releasing Hormone analog attenuates seizures in the amygdala-kindled rat. Epilepsia 48, 2280–2286. doi: 10.1111/j.1528-1167.2007.01218.x
Vogel, R. A., Frye, G. D., Wilson, J. H., Kuhn, C. M., Mailman, R. B., Mueller, R. A., et al. (1980). Attenuation of the effect of punishment by thyrotropin-releasing hormone: comparisons with chlordiazepoxide. J. Pharmacol. Exp. Ther. 212, 153–161.
Wang, W., Gershengorn, M. C. (1999). Rat TRH receptor type 2 exhibits higher basal signaling activity than TRH receptor type 1. Endocrinology 140, 4916–4919. doi: 10.1210/endo.140.10.7159
Wilk, S., Wilk, E. (1989). Pyroglutamyl peptidase II, a thyrotropin releasing hormone degrading enzyme: purification and specificity studies of the rabbit brain enzyme. Neurochem. Int. 15, 81–89. doi: 10.1016/0197-0186(89)90079-X
Williams, R. S. B., Eames, M., Ryves, W. J., Viggars, J., Harwood, A. J. (1999). Loss of a prolyl oligopeptidase confers resistance to lithium by elevation of inositol (1,4,5) trisphosphate. EMBO J. 18, 2734–2745. doi: 10.1093/emboj/18.10.2734
Yamada, M., Saga, Y., Shibusawa, N., Hirato, J., Murakami, M., Iwasaki, T., et al. (1997). Tertiary hypothyroidism and hyperglycemia in mice with targeted disruption of the thyrotropin-releasing hormone gene. Proc. Natl. Acad. Sci. 94, 10862–10867. doi: 10.1073/pnas.94.20.10862
Yamada, M., Shibusawa, N., Hashida, T., Satoh, T., Monden, T., Prasad, C., et al. (1999). Abundance of Cyclo (His-Pro)-like immunoreactivity in the brain of TRH-deficient mice. Endocrinology 140, 538–541. doi: 10.1210/endo.140.1.6607
Yamada, M., Shibusawa, N., Hashida, T., Ozawa, A., Monden, T., Satoh, T., et al. (2000). Expression of Thyrotropin-Releasing Hormone (TRH) receptor subtype 1 in mouse pancreatic islets and HIT-T15, an insulin-secreting clonal β cell line. Life Sci. 66, 1119–1125. doi: 10.1016/S0024-3205(00)00415-X
Yamada, M., Shibusawa, N., Ishii, S., Horiguchi, K., Umezawa, R., Hashimoto, K., et al. (2006). Prolactin secretion in mice with Thyrotropin-Releasing Hormone deficiency. Endocrinology 147, 2591–2596. doi: 10.1210/en.2005-1326
Yamamoto, M., Shimizu, M. (1987). Effects of a new TRH analogue, YM-14673 on the central nervous system. Naunyn-Schmiedeberg's Arch. Pharmacol. 336, 561–565. doi: 10.1007/BF00169314
Yamamoto, H., Morise, K., Kusugami, K., Furusawa, A., Konagaya, T., Nishio, Y., et al. (1996). Abnormal neuropeptide concentration in rectal mucosa of patients with inflammatory bowel disease. J. Gastroenterol. 31, 525–532. doi: 10.1007/BF02355052
Yamamura, M., Kinoshita, K., Nakagawa, H., Tanaka, T., Maeda, K., Ishida, R. (1990). Pharmacological study of TA-0910, a new thyrotropin-releasing hormone (TRH) analog. (I). Effects on the central nervous system by oral administration. Jpn. J. Pharmacol. 53, 451–461. doi: 10.1254/jjp.53.451
Yamamura, M., Kinoshita, K., Nakagawa, H., Ishida, R. (1991a). Pharmacological study of TA-0910, a new thyrotropin-releasing hormone (TRH) analog (II): Involvement of the DA system in the locomotor stimulating action of TA-0910. Jpn. J. Pharmacol. 55, 57–68. doi: 10.1254/jjp.55.57
Yamamura, M., Kinoshita, K., Nakagawa, H., Ishida, R. (1991b). Pharmacological study of TA-0910, a new thyrotropin-releasing hormone (TRH) analog (III): Inhibition of pentobarbital anesthesia. Jpn. J. Pharmacol. 55, 69–80. doi: 10.1254/jjp.55.69
Yamaoka, T., Itakura, M. (1999). Development of pancreatic islets (review). Int. J. Mol. Med. 247–261. doi: 10.3892/ijmm.3.3.247
Yang, H., Taché, Y., Ohning, G., Go, V. L. W. (2002). Activation of raphe pallidus neurons increases insulin through medullary Thyrotropin-Releasing Hormone (TRH)-vagal pathways. Pancreas 25, 301–307. doi: 10.1097/00006676-200210000-00014
Yang, H., Nyby, M. D., Ao, Y., Chen, A., Adelson, D. W., Smutko, V., et al. (2012). Role of brainstem thyrotropin-releasing hormone-triggered sympathetic overactivation in cardiovascular mortality in type 2 diabetic Goto–Kakizaki rats. Hypertens. Res. 35, 157–165. doi: 10.1038/hr.2011.154
Yokohama, S., Yoshioka, T., Yamashita, K., Kitamori, N. (1984). Intestinal absorption mechanisms of thyrotropin-releasing hormone. J. Pharmacobio. Dynamics 7, 445–451. doi: 10.1248/bpb1978.7.445
Yoo, S., Cha, D., Kim, S., Jiang, L., Adebesin, M., Wolfe, A., et al. (2019). Ablation of tanycytes of the arcuate nucleus and median eminence increases visceral adiposity and decreases insulin sensitivity in male mice. bioRxiv. doi: 10.1101/637587
Yu, Y., Zhao, C., Su, Z., Wang, C., Fuscoe, J. C., Tong, W., et al. (2014). Comprehensive RNA-Seq transcriptomic profiling across 11 organs, 4 ages, and 2 sexes of Fischer 344 rats. Sci. Data 1, 140013. doi: 10.1038/sdata.2014.13
Zacur, H. A., Genadry, R., Rock, J. A., King, T. M., Smith, B. R., Foster, G. V. (1985). Thyrotropin-Releasing Hormone-Induced contraction of urethral and vaginal muscle. J. Clin. Endocrinol. Metab. 61, 787–789. doi: 10.1210/jcem-61-4-787
Zada, M. H., Kubek, M., Khan, W., Kumar, A., Domb, A. (2019). Dispersible hydrolytically sensitive nanoparticles for nasal delivery of thyrotropin releasing hormone (TRH). J. Control Release 295, 278–289. doi: 10.1016/j.jconrel.2018.12.050
Zafirov, D. H., Cooke, H. J., Wood, J. D. (1991). Thyrotropin-releasing hormone excites submucous neurons an guinea-pig ileum. Eur. J. Pharmacol. 204, 109–112. doi: 10.1016/0014-2999(91)90843-F
Zarif, H., Petit-Paitel, A., Heurteaux, C., Chabry, J., Guyon, A. (2016). TRH modulates glutamatergic synaptic inputs on CA1 neurons of the mouse hippocampus in a biphasic manner. Neuropharmacology 110, 69–81. doi: 10.1016/j.neuropharm.2016.04.004
Zeisel, A., Hochgerner, H., Lönnerberg, P., Johnsson, A., Memic, F., van der Zwan, J., et al. (2018). Molecular architecture of the mouse nervous system. Cell 174, 999–1014.e22. doi: 10.1016/j.cell.2018.06.021
Zeng, H., Schimpf, B. A., Rohde, A. D., Pavlova, M. N., Gragerov, A., Bergmann, J. E. (2007). Thyrotropin-Releasing Hormone receptor 1-deficient mice display increased depression and anxiety-like behavior. Mol. Endocrinol. 21, 2795–2804. doi: 10.1210/me.2007-0048
Zhu, Q.-S., Rosenblatt, K., Huang, K.-L., Lahat, G., Brobey, R., Bolshakov, S., et al. (2011). Vimentin is a novel AKT1 target mediating motility and invasion. Oncogene 30, 457–470. doi: 10.1038/onc.2010.421
Keywords: thyroid hormone, thyrotropin, thyrotropin-releasing hormone, thyrotropin-releasing hormone-degrading ectoenzyme, anxiety, depression, mood
Citation: Charli J-L, Rodríguez-Rodríguez A, Hernández-Ortega K, Cote-Vélez A, Uribe RM, Jaimes-Hoy L and Joseph-Bravo P (2020) The Thyrotropin-Releasing Hormone-Degrading Ectoenzyme, a Therapeutic Target? Front. Pharmacol. 11:640. doi: 10.3389/fphar.2020.00640
Received: 04 February 2020; Accepted: 21 April 2020;
Published: 08 May 2020.
Edited by:
Jean Sévigny, Laval University, CanadaReviewed by:
Gabor Wittmann, Tufts Medical Center, United StatesCopyright © 2020 Charli, Rodríguez-Rodríguez, Hernández-Ortega, Cote-Vélez, Uribe, Jaimes-Hoy and Joseph-Bravo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean-Louis Charli, Y2hhcmxpQGlidC51bmFtLm14
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.