 Surabhi Shukla
Surabhi Shukla Babu L. Tekwani
Babu L. Tekwani
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 24 April 2020
Sec. Neuropharmacology
Volume 11 - 2020 | https://doi.org/10.3389/fphar.2020.00537
Histone deacetylases (HADC) are the enzymes that remove acetyl group from lysine residue of histones and non-histone proteins and regulate the process of transcription by binding to transcription factors and regulating fundamental cellular process such as cellular proliferation, differentiation and development. In neurodegenerative diseases, the histone acetylation homeostasis is greatly impaired, shifting towards a state of hypoacetylation. The histone hyperacetylation produced by direct inhibition of HDACs leads to neuroprotective actions. This review attempts to elaborate on role of small molecule inhibitors of HDACs on neuronal differentiation and throws light on the potential of HDAC inhibitors as therapeutic agents for treatment of neurodegenerative diseases. The role of HDACs in neuronal cellular and disease models and their modulation with HDAC inhibitors are also discussed. Significance of these HDAC inhibitors has been reviewed on the process of neuronal differentiation, neurite outgrowth and neuroprotection regarding their potential therapeutic application for treatment of neurodegenerative diseases.
Reversible histone acetylation and deacetylation functions play a key role in gene regulation. The process of acetylation and deacetylation are accelerated by enzymes Histone acetyl transferases (HATs) and Histone deacetylases (HDACs), respectively (Icardi et al., 2012; Rousseaux and Khochbin, 2015). Small molecule inhibitors of HDACs play important role in transcription regulation by changing the steady state of cells towards hyperacetylation. In neurodegenerative disease, the histone acetylation homeostasis is significantly compromised, shifting towards hypoacetylation (Saha and Pahan, 2005). The histone hyperacetylation produced by direct inhibition of HDACs leads to neuroprotective actions. This review presents a background on protein lysine acetylation and deacetylation functions, histone acetylation and deacetylation, HATs and HDACs and HDAC inhibitors (HDACi). The review particularly emphasizes details on HDAC inhibitors and their potential therapeutic application in various neurodegenerative diseases. The importance of HDAC inhibitors in neuronal differentiation and neuroprotection is also discussed.
Post-translational modifications are prominent, important and necessary processes for transformation of raw protein molecules into functional entities. The acetylation of proteins at specific lysine residues by acetyltransferases enzymes has emerged as a biologically related regulatory modification like phosphorylation (Kouzarides, 2000). The acetyl transferases catalyze the transfer of an acetyl group from acetyl Co-A to α-amino group of the amino terminal residues or to the ϵ-group of specific lysine residue (Allis et al., 2007). Deacetylation, the reverse reaction of removal of acetyl group from acetyl lysine residues, is accomplished by another group of enzymes named deacetylases. Lysine protein acetylation was first identified in histones many years ago. Lysine acetyl transferases (KATs) and lysine deacetylases (KDACs) are often referred to as histone acetyl transferases (HATs) and histone deacetylases (HDACs) (Yang and Seto, 2008). A variety of proteins undergo acetylation process including histones and non-histone proteins. More than 80 transcription factors are known for Lysine acetylation, including a large number of nuclear regulators and several cytoplasmic proteins (Glozak et al., 2005). Posttranslational modifications play a very significant role in a variety of cellular and biological process. Some of the prominent cellular process controlled by the lysine protein acetylation/deacetylation include chromatin modification and transcription, gene silencing, cellular differentiation, cell cycle progression, DNA replication, DNA repair (Choudhary et al., 2009; Aksnes et al., 2015) and apoptosis (Lebel et al., 2010). Most of the intermediate enzymes in the metabolic pathways are acetylated. Acetylation of these enzymes regulates cellular metabolism by directly affecting their stability and functions (Zhao et al., 2010). Lysine protein acetylation also plays a key role in p53 functions and interaction and stabilization of microtubules.
Chromatin is a complex structure in which DNA and histones are bundled within the nucleus of the cell. The vital component of the chromatin is the nucleosome, which is composed of octamers of the four essential histones (H3, H4, H2A, and H2B). Each histone octamer is wrapped around with 147 base pairs of DNA (Kouzarides, 2007). Chromatin is a very active structure as it responds to various signals and control functions of the DNA. Histone modifications are the main components that regulate DNA functions. Histones are well known to undergo a variety of post-translational modifications, namely phosphorylation, acetylation, methylation, and ubiquitination (Bannister and Kouzarides, 2011). Histone acetylation is a prominent and most important post-translational modifications that regulates gene expression. Acetylation and deacetylation are highly dynamic process and catalyzed by the interplay of two opposing enzymes histone acetyl transferases (HATs) and histone deacetylases (HDACs) (Figure 1). HATs catalyze the addition of an acetyl group at the lysine residue while HDACs catalyze removal of acetyl group from epsilon amino group of the lysine side chain of histone. Acetylation of histone tails neutralizes their positive charge resulting into the loosening of histones and the associated DNA, thereby providing the loose chromatin structure that is accessible to the transcription factors and thus promoting transcriptional activation (Parthun, 2007). Conversely, deacetylation of histone favors chromatin compaction. Generally, hyperacetylation of histone is associated with transcriptional activation, whereas hypoacetylation is associated with transcriptional repression (Hsieh et al., 2004).
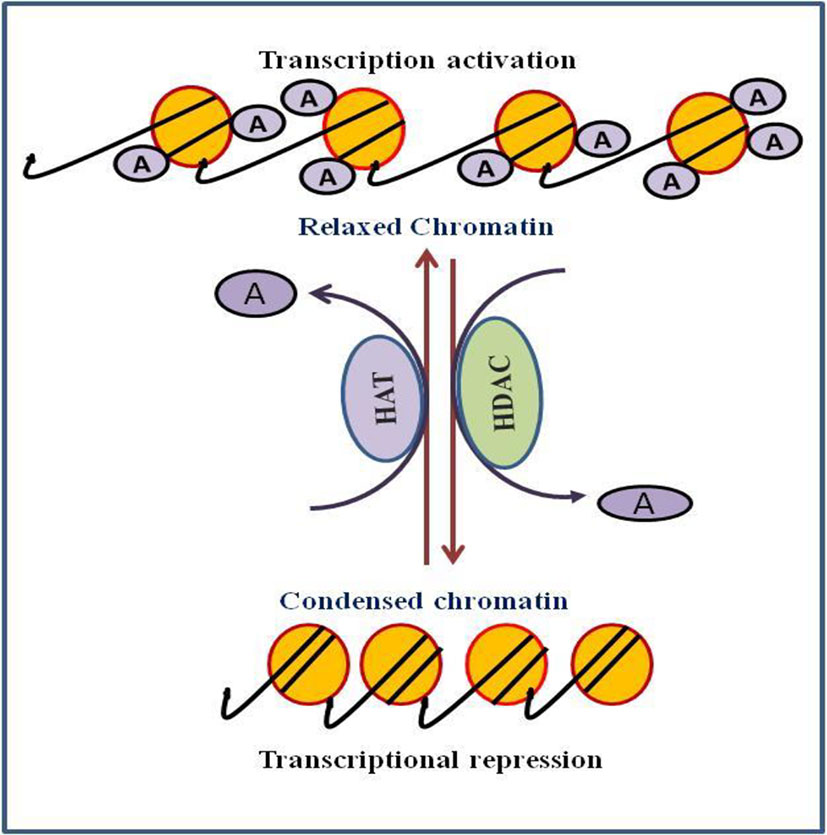
Figure 1 Role of histone acetylation and deacetylation in process of transcription regulation.
In humans, there are about 30 known HAT/KATs, which have been grouped into five distinct families based on the functional and structural similarity of their catalytic domains (Dekker and Haisma, 2009). The HATs arrange in distinct multi-subunit complexes, which provides them the ability to regulate their properties like substrate specificity and selectivity like targeting to specific loci and selectivity to access non-target proteins (Berndsen and Denu, 2008; Wapenaar and Dekker, 2016). GCN5 related N-Acetyl transferases (GNATs) have derived their name from bearing similarity to yeast GCN5 (general control nonderepressible-5) enzymes, having GCN5, and its relative and some distantly related HATs like Hat1, Elp3, and Hpa2. GNATs are cluster of four conserved motifs (A–D) within the catalytic domain HAT domain (Figure 2). Motif A is the most highly conserved among all four motifs, and contains an Arg/Gln-X-X-Gly-X-Gly/Ala sequence that is important for acetyl-CoA recognition (Roth et al., 2001). The chromodomain or bromodomain in GNATs are involved in their binding with methylated and acetylated lysine respectively (Neuwald, 1997). The MYST families of HATs are initially called after their pioneer family members MOZ, Ybf2, Sas2, and Tip60. The MYST HATs plays a very important role in posttranslational modification of histones and have a characteristic domain that contains acetyl CoA binding motif and a zinc finger motif (Avvakumov and Côté, 2007). The other families are global co-activators nuclear receptor co-activators and divers. The global co-activator includes p300 and CREB binding Protein. Nuclear receptor co-activators include steroid receptor co-activator (SRC-1), ACTR, and (Transcriptional intermediary factors) TIF2. The divers include TAFII 250 [TATA binding protein (TBP)—associated factor] and P160 (Sadoul et al., 2011).
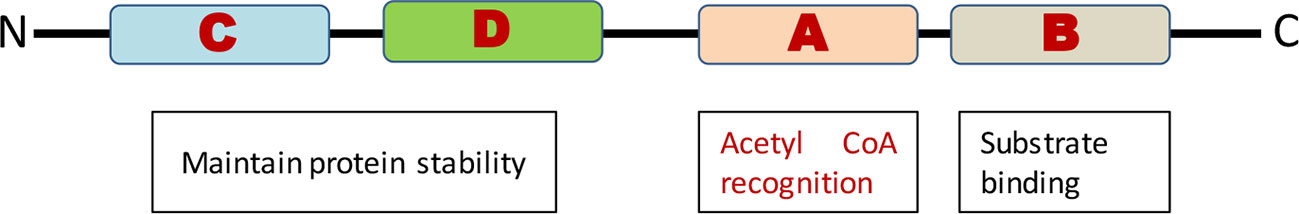
Figure 2 Conserved motifs of GNATs and their functions.
Histone deacetylases are enzymes that remove an acetyl group from histones leading to compact chromatin structures. HDACs are part of co-repressor complexes. The HDACs are well known to control multiple vital biological processes such as cell proliferation, differentiation and development by binding to many transcription factors and transcription co-regulators (Haberland et al., 2009).
Till date, total eighteen mammalian HDACs are well characterized and grouped into four major classes based upon their homology to yeast histone deacetylases (Figure 3). In Class 1, HDACs 1, 2, 3, and 8 are clustered based on their similarity to yeast transcriptional regulator, reduced potassium deficiency 3 (Rpd3) (De Ruijter et al., 2003). The Class I HDACs are predominantly localized in the nucleus. HDACs 1, 2, and 3 are shown to be involved in cell proliferation and death (Wiech et al., 2009) while HDAC 8 is found to be expressed in smooth muscle and plays a key role in muscle contractility. The Class II deacetylases share similarity with yeast Histone Deacetylase 1 (HD1) proteins and are further grouped into class IIa and Class IIb based on their structural similarity. The Class IIa includes HDACs 4,5, 7, and 9, while Class IIb includes HDAC 6 and 10. The classes II HDACs have been found to travel back and forth between the nucleus and cytoplasm and seem to have a role in tissue specific developmental activities (Bjerling et al., 2002). Class IV includes only one atypical HDAC isoform 11. It is well expressed in the kidneys, testis and brain. HDAC11 has been identified to deacetylate histones along with some non-histone proteins, namely a nuclear hormone receptor, transcription factors and cytoskeletal elements (Yang and Seto, 2008; Haberland et al., 2009). The Class I, II, and, IV HDACs are zinc-dependent as a cofactor and known as the classical HDACs. Class III HDACs, commonly called sirtuins (SIRT 1-7), are typically different from the classical HDACs. These NAD-dependent HDACs are similar to yeast SIR 2 (Silent Information Regulators 2) (Wiech et al., 2009). Sirtuins are involved in regulation of metabolism, stress response and aging (Michan and Sinclair, 2007).
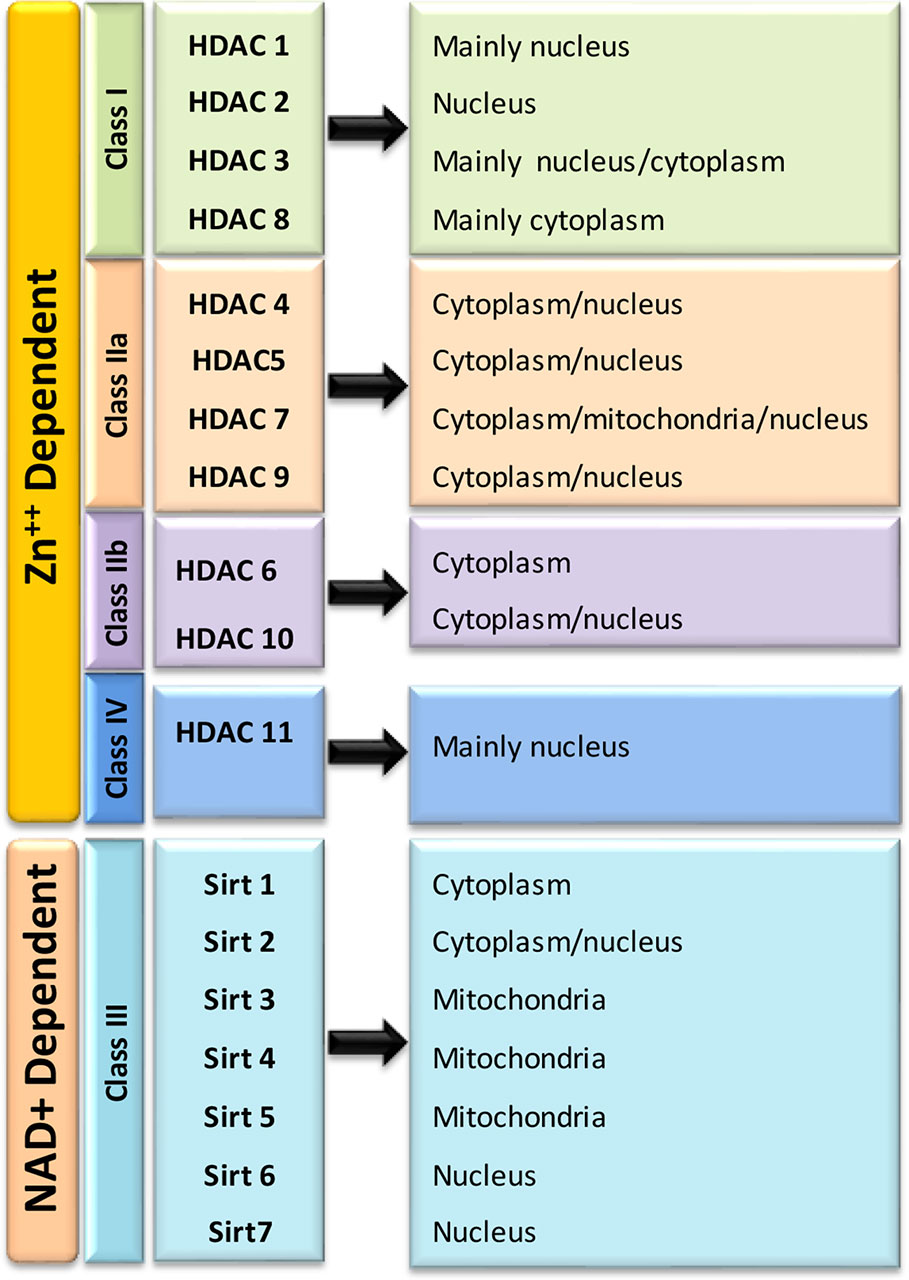
Figure 3 Classification of Histone Deacetylases (HDACs) and their cellular localization.
HDACi are small molecule natural or synthetic inhibitors of HDAC enzymes. These compounds vary in structures, selectivity and biological activities. HDACi pharmacophore is typically consisting of a metal-binding moiety or functional group, a capping group and a linker. The metal binding group, facilitates the catalytic metal binding to the active site of HDAC. The capping group interacts with the amino acids at lysine binding site while the linker having the structural similarity to the carbon chain present in the acetyl-lysine substrate, connects the metal binding and capping group for interaction with the HDAC active site (Finnin et al., 1999) (Figure 4). Recently, alternative strategies have been also suggested for achieving selective inhibition of HDAC that relies on perturbation of protein-protein interactions vital for the HDAC activity, rather than a traditional approach of the zinc ion chelation at the active site (Maolanon et al., 2016).
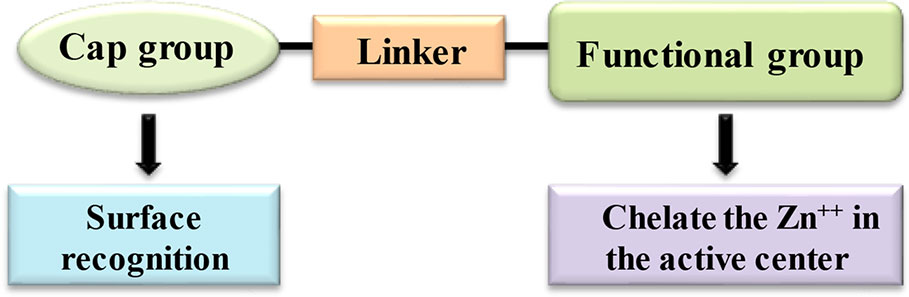
Figure 4 Basic pharmacophore structure of HDAC inhibitors.
The majority of HDAC inhibitors have been reported and their potential therapeutic applications have been discussed (Butler and Bates, 2006; Marks, 2010). HDACi have been classified into four major classes, based on the chemical nature of the inhibitor (Figure 5). A) Hydroxamates (e.g. Vorinostat (SAHA), Belinostat (PXD101), Panobinostat (LBH589) and Trichostatin A. The hydroxamate HDACi are non-specific and show activity against class I and class II HDACs. Trichostatin A and vorinostat are pan-HDAC inhibitors of all zinc-dependent HDACs and are reported to cross the blood brain barrier. B) The cyclic peptide HDACi includes depsipeptides and trapoxin. In in-vitro assays, cyclic peptides are active at nanomolar concentrations (Marks, 2010). C) The benzamide HDACi include entinostat (MS-275) mocetinostat, and D) Short chain fatty acids HDACi include sodium butyrate and valproic acid (Marks, 2010). The short chain fatty acids HDACi could also cross the blood brain barrier, however they are relatively moderate HDACi (Butler and Bates, 2006). The majority of HDACi available are non-specific inhibitors of all the HDAC isoforms. These are generally referred as pan-HDAC inhibitors. TSA and vorinostat are an example of canonical pan-HDAC inhibitors that inhibit HDACs 1–9 with equivalent potency. Selective HDACi can be classified into either class-specific (inhibiting several isoforms within a single class) or isoform-specific HDAC inhibitor (selectively inhibiting a specific HDAC isoform).
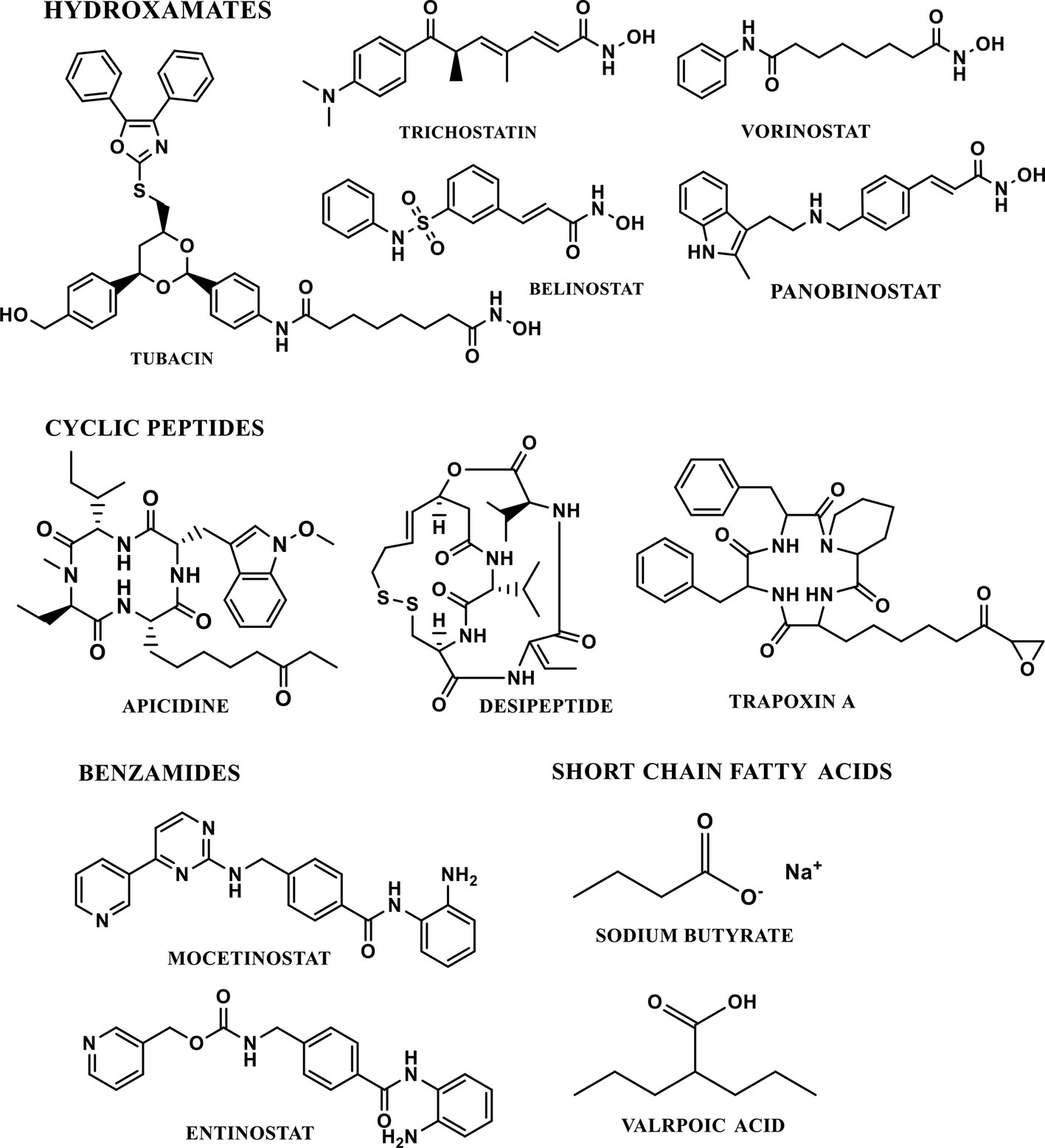
Figure 5 Chemical classes and structures of selected HDAC inhibitors.
Selective HDAC inhibitors are important for determining the molecular mechanism of functions of a specific HDAC isoform. Recently, significant attempts have been made towards the development of selective HDAC inhibitors. Tubacin, an HDAC inhibitor, selectively targets the HDAC6 and deacetylation of α-tubulin (Haggarty et al., 2003). Romidepsin (FK-228) is a cyclic tetrapeptide, which selectively inhibits HDAC 1 and 2 isoforms (Itoh et al., 2008). Apicidin, another cyclic tetrapeptide is a highly potent inhibitor of HDAC2 & 3(Khan et al., 2008). MS-275, a synthetic benzamide derivative, preferentially inhibits HDAC1 as compared to HDAC 2, 3 and 9. MS-275 exhibits little to no activity against HDAC 4, 6, 7, and 9 (Khan et al., 2008). Suramin inhibits human NAD+ dependent class III SIRT 1 and SIRT 2 activity (Trapp et al., 2007).
In recent years, inhibition of HDAC activity with HDACi has attracted significant therapeutic attention. Earlier interests were mainly based on the development of HDACi as anticancer agents (Kazantsev and Thompson, 2008; Tsilimigras et al., 2018). Some HDAC inhibitors, like butyrate, trichostatin A (TSA), suberoylanilide hydroxamic acid (SAHA), MS-275, inhibit cell growth and stimulate cell differentiation in various in-vitro cancer models, including leukemia. SAHA (vorinostat, brand name-Zolinza) become the first HDACi approved for cutaneous T-cell leukemia by the FDA. Several HDACi are now in Phase I and Phase II clinical trials for cancer therapeutics (Marks and Xu, 2009). Some HDACi used in clinical trial for neurological/cancer and other conditions are summarized in Table 1. In recent years, the therapeutic interests in HDACi have extended to non-malignant conditions impacting the nervous system (Kazantsev and Thompson, 2008; Konsoula and Barile, 2012; Ziemka-Nalecz et al., 2018). Targeting HDACs with HDACi might have potential for treatment of neurological disorders such as Huntington's disease, Alzheimer's disease, amyotrophic lateral sclerosis, seizure disorders, spinal muscular atrophy, Rett syndrome, stroke, Fragile X syndrome, and Rubinstein-Taybi syndrome (Chuang et al., 2009). The HDACi also seem promising for several psychiatric disorders therapeutics like depression, drug addiction, schizophrenia, and anxiety disorders (Butler and Bates, 2006; Guidotti et al., 2009). The levels and activities of HATs and HDACs are finely balanced in neuronal cells under normal conditions (Saha and Pahan, 2005). In neurodegenerative disease, the histone acetylation homeostasis is greatly impaired, shifting towards hypoacetylation (Saha and Pahan, 2005). The histone hyperacetylation produced by direct inhibition of HDACs leads to neuroprotective actions. Besides, inhibition of HDACs and histone hyperacetylation, the neuroprotective effect of HDACi may also involve multiple mechanisms of action, involving activation of the kinase pathway by extrinsic signals (Hao, 2004) the suppression of pro-apoptotic factors (Kim et al., 2007) or microglial–mediated inflammation (Peng et al., 2005), as reported earlier for valproic acid. Therefore, HDACs show great potential as a cellular target for the treatment of neurological and psychiatric disorders. In preclinical treatment models, the HDACi have exhibited neuroprotective effects and stimulation of neurogenesis in traumatic brain injury and ischemia (Kim et al., 2009), restoration of memory and learning in traumatic brain injury in neurodegenerative mice (Dash et al., 2009), enhanced neuronal differentiation and synaptic plasticity (Vecsey et al., 2007) and exerted antidepressant-like effects (Schroeder et al., 2007). Vorinostat significantly reduced IFN- λ induced neurotoxicity of human astrocytes (Hashioka et al., 2012).
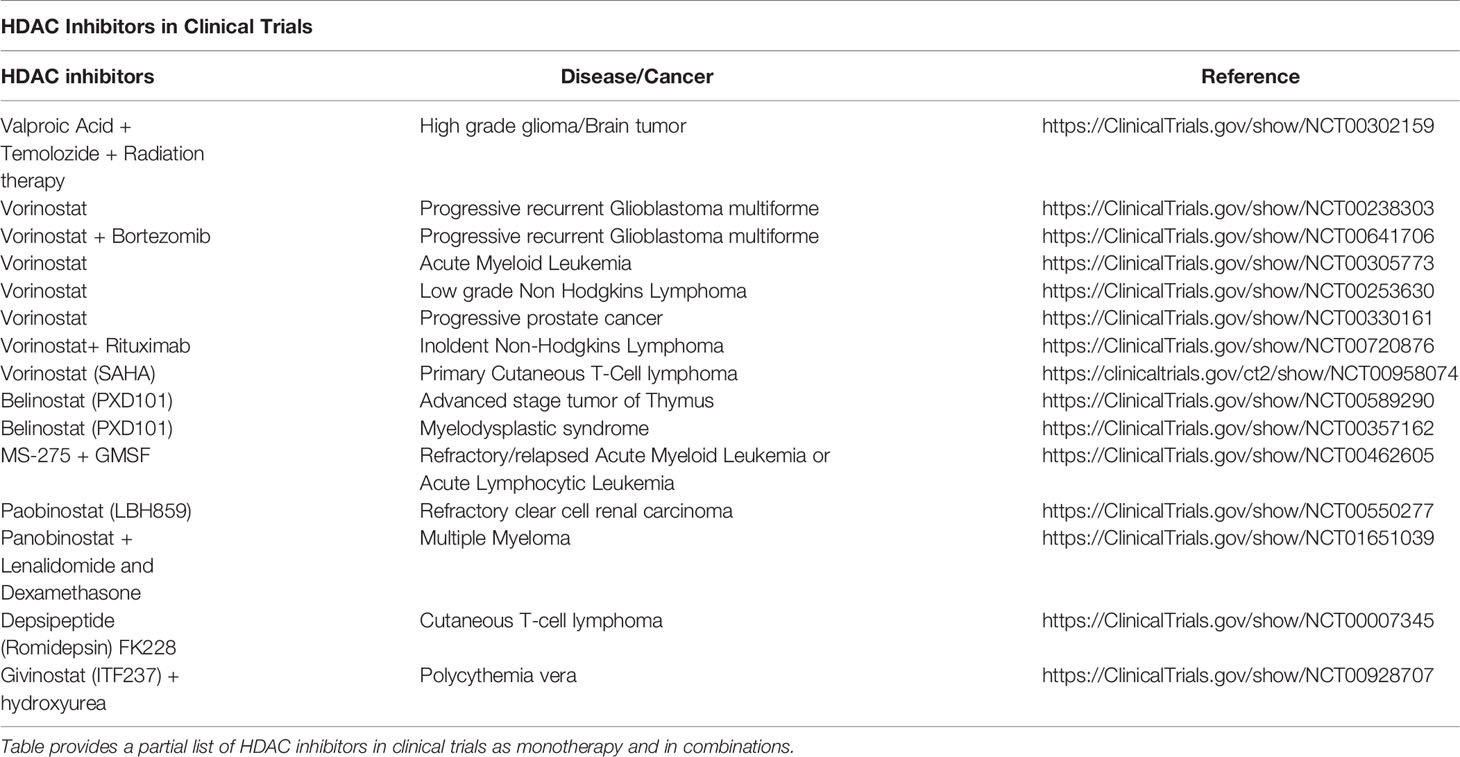
Table 1 Examples of HDAC Inhibitors in Clinical trials.
Treatment with HDAC inhibitors results in the hyperacetylation of chaperones including heat shock protein (HSP90, HSP70, HSP40, glucose-regulated protein 78 (GRP78) in turn affecting their function. Chaperones are proteins molecules that facilitate the folding and maturation of newly synthesized proteins and partially folded proteins in the cytosol and endoplasmic reticulum. Chaperons also play important role in preventing the aggregation of misfolded proteins (Rao et al., 2012). The mechanism of protection after cerebral ischemic events is not very clear; however, it has largely been attributed to HSP 70 chaperone functions that enhance cell survival by blocking protein aggregation (Giffard et al., 2004). There are other studies that show HSP 70 can also modulate inflammatory response. In the ischemic brain, treatment with HDAC inhibitors, markedly inhibited ischemia induced p53 overexpression and superinduced heat shock protein 70 (HSP70) (Ren et al., 2004; Faraco et al., 2006; Kim et al., 2007). Super induction of HSP 70 protein by HDAC inhibition may cause anti-inflammatory effects. A recent study, in a mouse transient middle artery cerebral occlusion model (tMCAO) model shows that HSP70 overexpression inactivates NF-κB by stabilizing a complex of HSP70-IκBα-NF-κB (Zheng et al., 2008).
The pathophysiology of Huntington's disease is closely linked to BDNF and HSP70 deficiency in affected brain regions. Class I and class II HDAC inhibitors regulate expression of BDNF and HSP70. Therefore, it is plausible that restoring BDNF and HSP70 to their normal levels is the molecular mechanism underlying the beneficial effects produced by HDAC inhibition in various HD models. (Zuccato et al., 2001; Hay et al., 2004; Tagawa et al., 2007).
The pathophysiology of Parkinson's disease is associated with aggregation of α-synuclein and progressive degeneration of dopaminergic neuron. In context to that, HDAC6 is reported to rescue polyglutamine-mediated neurodegeneration in an autophagy-dependent manner (Pandey et al., 2007). HDAC 6 senses ubiquitinated aggregates and subsequently activates the expression of chaperones, such as heat-shock protein Hsp70 and Hsp25 (Boyault et al., 2007). HDAC 6 plays important role in cellular function through its enzymatic and nonenzymatic actions (Seigneurin-Berny et al., 2001). The catalytic domains of HDAC6 promotes deacetylation of many non-histone proteins, such as HSP-90, a-tubulin, and cortactin, (Luxton and Gundersen, 2007; Valenzuela-Fernandez et al., 2008). The nonenzymatic action of HDAC6 is associated with ubiquitin binding domain that regulates the buildup of ubiquitinated misfolded proteins (Du et al., 2010; Du and Jiao, 2011). The results indicate that HDAC6 promotes cytoplasmic inclusion formation and facilitates autophagic degradation of these aggregates in the aggresome. Therefore, this action protects dopaminergic cells against a-synuclein toxicity and might have an important role in the suppression of Parkinson's disorder (Li et al., 2011). These findings were further confirmed by treatment of HDAC6 inhibitor tubacin to PC-12 cells over-expressing human mutant (A53T) α-synuclein (α-syn) and SH-SY5Y cells with MPP (+) that led to defects in aggresome formation accompanied by massive cell death in response to misfolded protein-induced stress (Su et al., 2011). A study in Alzheimer's disease models shows importance of HSP-70 and Glucose regulated protein 78 (GRP78) in bringing therapeutic benefits in AD models. In this study CM-695 a compound that selectively inhibits HDAC6 over class I HDAC isoforms and also inhibits phosphodiesterase 9 (PDE9) is used in chronic treatment of Tg2576 mice. Chronic treatment of TG2576 mice with CM-695 led to increase in chaperon protein HSP70 and GRP 78 which subsequently led to improvement in memory impairment and reduces brain Aβ (Cuadrado-Tejedor et al., 2019).
Spinal Muscular Atrophy (SMA) is a monogenic neuromuscular syndrome triggered due to mutations in Survival of motor neurone 1 (SMN1) gene (Maretina et al., 2018). The SMA patients maintain in situ copy of the paralog SMN2 gene, which produces reduced amounts of the SMN protein. Therefore, the insufficient levels of SMN protein in motor neurons results into SMA. The increasing expression of SMN2 is a primary therapeutic approach for treatment of SMA (Sumner, 2006). Several studies have demonstrated potential HDAC inhibitors in treatment of SAM (Mohseni et al., 2013) Histone acetylation has been shown to upregulate SMA 2 expression (Kernochan et al., 2005). An analysis of differential regulation of SMA2 gene by silencing of individual HDAC isoforms by shRNA identified HDAC 2 and 6 as potential regulators of SMA 2 expression (Evans et al., 2011) Screening of a library of cyclic peptide inhibitors of HDAC on a neuronal cell line derived from a SMA patient's induced pluripotent stem cells identified HDAC inhibitors, which induced the expression of SMA2 (Lai et al., 2017). Sodium butyrate and its analog was shown to increase the expression of SMA2 in SMA cultured cells (Butchbach et al., 2016). Sodium butyrate also attenuated neurological symptoms in the mouse model of SMA (Minamiyama et al., 2004) In SMA models, SAHA has been shown to improve motor abilities, by improving the number of motor neurons in the mice. SAHA treatment was shown to improve weight loss in SMA mice (Riessland et al., 2010). SAHA was also shown to activate the SMA2 gene expression (Hahnen et al., 2006). In another study, daily injection of TSA, in SMA mice after the onset of disease lead to HDAC inhibition, triggered the SMN2 gene expression and improved survival and motor pathology and attenuated weight loss (Avila et al., 2007). M344 a benzamide HDAC inhibitor showed highly strong (7 fold) upregulation of expression of SMA 2 gene in the fibroblast cells derived from a SMA patient (Riessland et al., 2006). A recent study reported that LBH589 treatment resulted in induction of H4 acetylation of SMN2 locus. A combinantion treatment of SMA cells with LBH589 and an antisense oligonucleotide that mimic Nusinersen (ASO_ISSN1) produced additive effects on SMN2 splicing and SMN protein expression (Pagliarini et al., 2019). Nusinersen, an anitisense oligonucleotide, has been recently approved by the US FDA for treatment of SMA (Hagenacker et al., 2020).
Huntington disease (HD) is a rare genetic autosomal dominant neurodegenerative syndrome triggered by expanded CAG repeats in the huntingtin gene, which leads to pathological elongation of huntingtin with a polyglutamine tract. The neurodegenerative conditions caused due to HD lead to cognitive, motor, and psychiatric symptoms, atrophy of the basal ganglia and the cerebral cortex (Wright et al., 2017; Illarioshkin et al., 2018). Transcriptional dysregulation has been suggested a prominent mechanism for pathogenesis of HD (Sharma and Taliyan, 2015b). Hypoacetylation of histones associated with genes which are downregulation due to HD have been suggested for this dysregulation (Sadri-Vakili et al., 2007). The therapeutic ability of HDAC inhibitors have been demonstrated in different models of HD (Sadri-Vakili and Jang-Ho, 2006; Sadri-Vakili et al., 2007). Also, a genome wide decrease in H3 acetylation was demonstrated in a mouse model of HD (McFarland et al., 2012). Selective inhibition of HDAC6 increased acetylation of microtubules, which compensated for the transport deficit in HD (Vaughan et al., 2008). Reversal of H3 hypoacetylation with pimelic diphenylamine, an HDAC inhibitor, in transgenic HD mice improved the motor deficits (Thomas et al., 2008). Treatment with HDAC inhibitors in R6/2 a HD mouse model showed improvement in rotarod performance, which indicates improvement in motor impairment (Ellrichmann et al., 2017). SAHA showed its ability to penetrate the blood brain barrier and escalate histone acetylation in the brain (Hockly et al., 2003). (Ferrante et al., 2003) conveyed that sodium butyrate treatment attenuated neurodegenerative phenotype and enhanced survival in R6/2 HD models. SAHA and sodium butyrate treatment has been shown to retard neurodegeneration and improves lethality in a Drosophila model of HD (Steffan et al., 2001). An impaired microtubule transport is also attributed to the neuronal toxicity observed in HD. Vorinostat and TSA have been shown to increase vesicular transport of BDNF by inhibiting HDAC6 and thus increasing the acetylation of α-tubulin at Lys 40. Acetylation of α-tubulin at Lys 40 regulate the binding and motility of kinesin-1 (Dompierre et al., 2007). Treatment with RGFP966, a benzamide HDAC3 inhibitor, improved motor deficits in rotarod and in open field in N171-82Q transgenic HD mice. RGFP966 treatment caused induction in the expression of macrophage migration inhibitory factor, which is associated with activation of glial cells (Jia et al., 2016). LBH589, a non-selective hydroxamate HDAC inhibitor, induced histone hyperacetylation and prevented striatal neuronal shrinkage in R6/2 HD mice (Chopra et al., 2016). Interventional treatment in early presymptomatic phenotypes of HD with LBH589, an HDAC inhibitor, yielded significant improvements in behavioral changes (Siebzehnrübl et al., 2018). Characterization of interactome of endogenous HDAC4 in brains of HD mouse models revealed interaction of HDAC4 with Wiskott-Aldrich Syndrome Protein and SCAR Homolog (WASH) complex. This study confirmed the role of HDAC4 in progression of HD (Federspiel et al., 2019).
Parkinson's disease is another neurodegenerative syndrome delineated by loss of dopaminergic neurons in the substantia nigra of the brain. HDACs and associated epigenetic functions have been suggested as potential therapeutic targets for PD (Harrison and Dexter, 2013; Sharma and Taliyan, 2015a; Hegarty et al., 2016b). A significant upregulation of HDAC 2 was reported in substantia nigra microglia of PD subjects (Tan et al., 2018).
In a study with a mouse model of PD treated with dopaminergic toxin MPTP, the administration of HDAC inhibitor phenyl butyrate significantly reduced the diminution of dopamine and loss of tyrosine hydroxylase-positive neurons in the substantia nigra (Gardian et al., 2004). Presynaptic α-synuclein mutations are linked with familial forms of PD (Gasser, 2001). The α-synuclein arbitrates neurotoxicity and targeting α-synuclein promotes toxicity, while cytoplasmic sequestration is protective in both cell culture and transgenic Drosophila (Kontopoulos et al., 2006). Toxicity caused by α-synuclein could be retrieved by the administration of HDAC inhibitors such as vorinostat and sodium butyrate in cell culture as well as transgenic flies. It was found that treatment with the HDACi lead to decreased neuronal death in response to α-synuclein (Kontopoulos et al., 2006).
In dopaminergic neuronal cells like MN9D, N27, and human SH-SY5Y cells, TSA treatment decreased survival rate and increased apoptosis, (Wang et al., 2009). This finding suggested that the effect of HDAC inhibitors might be complex. In some cases, the HDAC inhibitors could play a key role in pathogenesis of the neurodegenerative disease while in others these may act as feasible therapeutics for neurodegenerative disease. These contrasting effects of HDAC inhibitor may be attributed to varying epigenetic status, cell type and tissue specificity. HDAC inhibitors such as TSA and VPA, the pan-HDAC inhibitors of class I and class II are non-specific. Plausibly, this non-specificity might contribute to contradictory effects observed in different cell types (Wang et al., 2009). Neuron –restrictive silencer factor (NRSF) was suggested as a potential mediator for neuroprotective effects of TSA in MPTP induced model of PD (Suo et al., 2015). K560, a HDAC 1 and 2 isoform specific inhibitor, attenuated MPTP induced cell death in SH-SY5Y cells in vitro and prevented MPTP-induced loss of dopaminergic neurons in substantia nigra in mice in vivo (Choong et al., 2016). Similarly, M275 (HDAC 1 inhibitor) and tubastatin A (HDAC 6 inhibitor) improved sensorimotor reflexes and locomotor impairments (Pinho et al., 2016). The fibroblasts from PD patients with G2019S mutation in leucine rich repeat kinase 2 showed increased mitophagy and activation of class III HDACs (Yakhine-Diop et al., 2018). However, treatment with nicotinamide a class III HDAC inhibitor, though induced histone hyperacetylation and overexpression of neurotrophic factors but failed to provide neuroprotection. Nicotonamide treatment rather aggravated the PD pathology (Harrison et al., 2019). VPA was shown to protect SH-SY5Y dopaminergic neuronal cells from 6-OHDA-induced toxicity thorugh down regulation of apoptotic caspases namely, caspase-3, caspase-7, and caspase-9. VPA treatment also reduced the Bax/Bcl2 ratio in SH-SY5Y. This study suggested VPA treatment as a potential antidote for PD prevention (Hsu et al., 2020).
Most of the earlier reports on HDACi were focused on their anticancer, antiproliferative, and antiapoptotic properties. Hyperacetylation was associated with an elevation in expression of pro-apoptotic genes such as Bax and P21. Though, an escalation in acetylation also indorsed expression of anti-apoptotic or pro-survival genes like Bcl-2 and growth factors, which leads to protective effects in other cell types. This might be one mechanism by which HDAC inhibitors seem to be vindicatory in neurons and lethal in the cancerous cell (Saha and Pahan, 2005).
Amyotrophic Lateral Sclerosis (ALS) is a neurodegenerative syndrome leading to progressive damage of motor neurons in the brain, and spinal cord causing weakness, muscle atrophy and death (Rowland and Shneider, 2001). The familial form of ALS is due to gain of functional mutation in the gene encoding Cu/Zn superoxide dismutase1 (SOD1) an antioxidant enzyme (Rosen et al., 1993). As transcriptional regulation may be involved in the pathophysiology of ALS, role of HDAC inhibitors have been studied in transgenic ALS mouse models. Target specific as well as global changes in histones acetylation are associated with ALS (Bennett et al., 2019). Global proteomic analysis suggested lysine-hyperacetylation of glial fibrillary acidic protein in post-mortem ALS spinal cord (Liu et al., 2013). Muscle HDAC 4 and its regulator microRNA-206 are upregulated in transgenic ALS mice carrying human mutations in SOD gene (Bruneteau et al., 2013). Analysis of differential expression of HDAC isoforms showed increased HDAC2 levels in ALS brain and spinal cord related to controls (Boeselt et al., 2010). Studies done by (Ryu et al., 2005) have shown that administration of 4-phenylbutryrate starting before or shortly after the beginning of symptoms resulted in increased survival, enhanced pathological phenotype and reduction in clinical impairment. VPA injection in G86R SOD1 mutant mice significantly attenuated the death of motor neurons, maintained the normal level of acetylation of histones and restored the loss of HAT CBP (Rouaux et al., 2007). Co- treatment of 4-phenylbutyrate and AEOL10150 a catalytic antioxidant to ALS mice had a collective effect on survival time and there was a reduction in the markers of oxidative damage in the lumbar spinal cord (Petri et al., 2006). Treatment of SOD-G93A transgenic ALS mice with TSA ameliorated muscle atrophy and neuromuscular junction denervation (Yoo and Ko, 2011). Role of HDAC 4 to mediate nerve-skeletal muscle interaction has suggested it as apotential target for ALS. Genetic deletion of HDAC4 in skeletal muscles caused earlier onset of ALS in a mouse model, suggesting a potential risk in use of HDACi for treatment of ALS (Pigna et al., 2019). A combinantion treatment with SAHA (a pan HDACi), RGFP109 (HDAC1/3 inhibitor), and arimoclomol attenuated the loss of nuclear FUS, a hallmark of ALS. HDAC inhibition also rescued the DNA repair response in iPSC-derived motor neurons carrying the ALS associated FUSP525Lmutation. These observations suggest multiple mechanisms of neuroprotection by both HDAC inhibiting drugs and arimoclomol in ALS (Kuta et al., 2020).
Alzheimer's Disease (AD) is well known for gradual memory loss and personality changes eventually preceding to dementia (Burns and Iliffe, 2009). The elevated level of extracellular β-amyloid (Aβ) and neurofibrillary tangles developing from Tau protein hyperphosphorylation are the neuropathological hallmark of AD (Luca et al., 2018). Impairments in HDAC functions and associated pathways have been implicated in pathogenesis of AD and HDAC inhibitors have shown promising applications for treatment of AD (Yang et al., 2017; Dubey et al., 2018). Different HDAC isoforms have been implicated in AD pathogenesis, loss of HDAC 5 was shown to impair memory function, while little effect on pathogenesis of AD in a mouse model for amyloid pathology (Agis-Balboa et al., 2013). Similarly, a decrease in HDAC6 levels re-established learning and memory and α-tubulin acetylation in a mouse model of AD. Further analysis suggested that HDAC6 loss rendered neurons resistant to amyloid-β-mediated deterioration of mitochondrial trafficking, an prominent factor for AD (Govindarajan et al., 2013). The levels of HDAC2 were found to be altered in the basal forebrain region containing cholinergic neurons of the Nucleus Basalis of Meynert in AD patients (Mahady et al., 2018). Overexpression of HDAC3 in the hippocampus increased Aβ levels, activated microglia, and decreased dendritic spine density in APPswe/PS1dE9 mice (Zhu et al., 2017). These studies have led to the advancement of pan and isoform selective HDAC inhibitors as potential therapeutic agents for treatment of AD (Yang et al., 2017).
Treatments with tubastatin A and ACY-1215 (specific HDAC6 inhibitors) improved behavioral loss, suppressed tau hyperphosphorylation and reduced amyloid-β (Aβ) load in AD mice. The treatments also resulted in elevated tubulin acetylation and reduced production and facilitated autophagic clearance of Aβ and hyperphosphorylated tau (Zhang et al., 2014). The daily administration of 4-phenylbutyrate in Tg2576 AD model reversed spatial memory deficit by regulating hyperphosphorylation of Tau in the hippocampus. However, the Aβ levels were not affected due to treatment with 4-phenylbutyrate (Ricobaraza et al., 2009). In another study, daily injection of low doses of valproic acid in Tg2576 AD mice at the early onset of 27 months, significantly reduced Aβ plaque number and improved memory deficit. The mechanism of Aβ plaque reduction by valproic acid was because of inhibition of GSK-3β-mediated γ-secretase cleavage of APP (Qing et al., 2008). Treatment of 3xTg-AD mice with a class III HDAC inhibitor, nicotinamide, prohibited memory impairment and reduced Tau pathology without affecting Aβ production. There was also a prolonged but little increase in internal p25 level, which was associated with improved learning and memory (Green et al., 2008). Treatment of Tg6799 AD mice with valproic acid in different disease stages (pre-, early- and late-symptomatic) caused a suppression in expression of nuclear factor kappaB (NF-κB) and IL-1ß in the plasma and increased NGF levels in the hippocampus. The treatments with VPA also decreased escape latencies of Tg6799 mice at early and late-symptomatic stages of AD (Noh and Seo, 2014). Chronic treatment with HDAC inhibitors sodium butyrate, valproate, or vorinostat showed the profound restoration of contextual memory in APPswe/PS1dE9 transgenic mouse model of AD. The newly established memories were firmly sustained over a 2-week period in HDACi-treated transgenic AD mice (Kilgore et al., 2010). Recently, inhibition of phosphodiesterase 5 (PDE5) and HDAC has been suggested as a potent novel therapeutic approach for AD (Rabal et al., 2016; Sánchez-Arias et al., 2017; Rabal et al., 2018). An acridine-based HDAC inhbitor, designed as a multi-target agent against HDAC and acetylcholine esterase showed selective inhbition of HDAC6 and strong activity against Aβ-aggregation (Tseng et al., 2020). A new tacrine-hydroxamate pan HDACi exhibited inhibitory activity on Aβ1-42 self-aggregation as well as disaggregation activity on pre-formed Aβ fibrils, the pathological markers of AD (Xu et al., 2020). Knockout of HDAC2 in mice leads to cognitive enhancement, and inhbitors of HDAC2 have shown potential as therapeutics for restotation of memory affected due to AD (Poplawski et al., 2020).
Acetylation of tau protein has been strongly asoociated with pathology of AD. Acetylation of tau has been shown to cause inhbition of its degradation leading to accummultaion of tau. A careful evaluation of tau acetylation has been suggested during treatment with HDACi (Jeong et al., 2019).
Multiple Sclerosis (MS) is an overwhelming autoimmune disorder of CNS. Recent findings have indicated towards the potential therapeutic role of HDACi in this disorder (SG and Dangond, 2006; Faraco et al., 2011). A significant increase in lysine acetylation was reported in myelin basic protein in an autoimmune encephalomyelitis (EAE) model of MS (Lillico et al., 2018). A study by Camelo et al. (2005) on EAE, a model of multiple sclerosis, demonstrated that neuropathology of mice with EAE was improved by TSA treatment. The animals treated with TSA showed less spinal cord inflammatory infiltrates, demyelination and axonal loss. Interestingly, TSA treatment also increased the number of motor neurons in the ventral horn, suggesting the potential application of TSA in neuroprotection. TSA treatment increased activity of neuroprotective proteins such as insulin growth factor-2 (IGF-2), estrogen receptor-α, glutamate transporter EAAT2 and glutathione peroxidase, and decreased the expression of pro-apoptotic Bax, Bid, caspase-2, and apoptosis-inducing factor (Camelo et al., 2005).
Axonal damage is also an important cause of neurodegeneration, where histone deacetylases play an important role. A study with a specific wallerian degeneration mouse (WID) shows delayed wallerian degeneration due to increase in activity of NAD+ dependent histone deacetylase SIRT 1. These mice had an elevated activity of the enzyme that produces NAD+ leading to increased SIRT 1 activity. The neuronal impairment due to progressive degeneration after axonal transfection in this specific mouse was inhibited (Araki, 2004). A recent study reported amelioration of clinical symptoms in the mouse model of MS due to loss of HDAC11 and suggested potential use of HADC-11 specific inhibitors for treatment of chronic progressive MS (Sun et al., 2018).
Among the six animal models of neurodegenerative disorder described above treatments with different class of HDAC inhibitors shows beneficial effect of HDAC inhibitors in improving neurodegenerative conditions by improving survival of neurons and preventing neuronal death through different mechanism (Table 2).
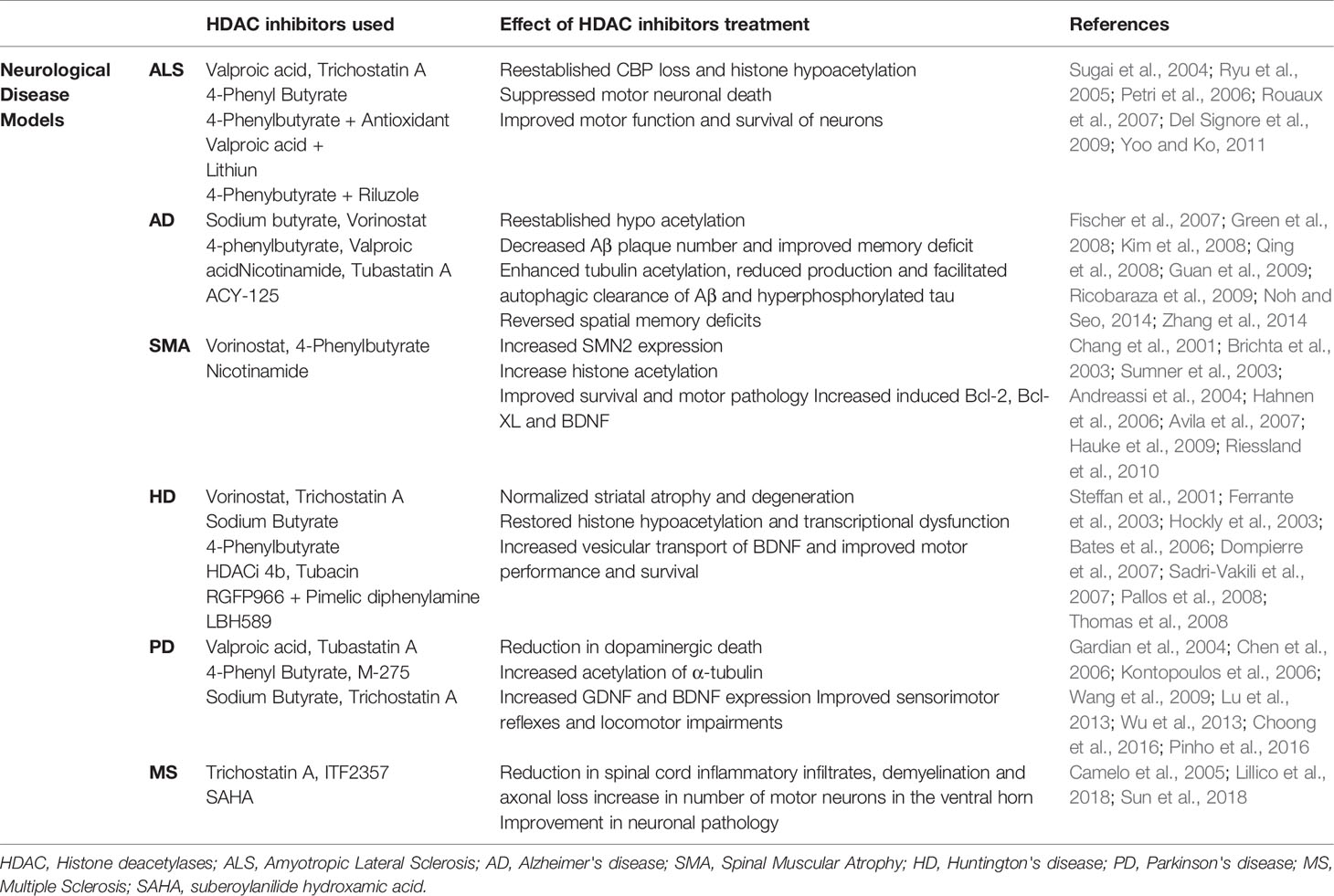
Table 2 HDAC Inhibitors used in neurodegenerative disease models and their effect.
Potential of therapeutic agents to generate neuroprotective actions has been considered as an important factor for their development for treatment of neurodegenerative disorders. Beside potential for treatment of neurogenerative diseases, the HDAC inhibitors have also been reported to produce prominent neuroprotective properties.
VPA has been shown to reduce the retinal neuronal death by optic nerve crush (ONC). VPA up- regulated levels of BDNF, Bcl-2, and TrkB in the retina post-injury (Zhang et al., 2012). VPA and Sodium butyrate treatment caused hyperacetylation of histone H3K14, reduced histone H3K9 hypermethylation in the BDNF promoter and increased transcriptional activity. These results suggest that VPA seemed to protect retinal ganglion cells (RGC) from ONC by hindering neuronal apoptosis, presumably, via the initiation of HDAC inhibition and BDNF-TrkB signaling (Zhang et al., 2012). BDNF gene expression has also been associated with mood-stabilizing effects in bipolar disorder (BD). BDNF plays an important role in promoting neuronal survival and synaptic plasticity and well-studied as a biomarker of illness activity in BD (Kuipers and Bramham, 2006). (Stertz et al., 2014) examined repeated exposure of D-amphetamine (AMPH) in a bipolar animal model and also investigated the effect of sodium butyrate. They found that repeated AMPH administration increased the HDAC function in the prefrontal cortex (PFC) without changing BDNF protein or mRNA levels, while the administration of sodium butyrate partially reversed the HDAC activity. HDAC inhibitors have shown potential for therapeutic application for treatment of neurodegenerative and psychiatric disease having neuroprotective and memory enhancing properties and increased expression of BDNF. (Koppel and Timmusk, 2013) compared the effect of class I, I/IIb, and II selective HDAC inhibitors on BDNF mRNA expression in rat primary neurons. The results indicated that inhibition of class II HDACs led to rapid elevation of BDNF mRNA levels, while class I HDAC inhibition created a distinctly delayed BDNF induction. Their finding suggested that class II HDAC inhibitors play an important part in regulating transcription of BDNF and may have potential application for treatment of neurological disorders. (Hasan et al., 2013) investigated the role of HDAC inhibitors in neuroprotection after ischemia. Their study demonstrated that VPA and TSA treatment after oxygen and glucose deprivation led to re-oxygenation in rat cortical neurons, promoted neuronal regeneration and neuronal protection through upregulation of BDNF expression. There was also a significant increase in three synaptic markers, namely PSD95, GAP 43, and synaptophysin in VPA and TSA treated rat cortical neurons. (Chen et al., 2006) have shown that VPA promoted dopaminergic neuron in midbrain neuron/glial culture by stimulating the release of neurotrophins like BDNF and GDNF. VPA pretreatment also protected midbrain dopaminergic (DA) neurons from LPS or 1-methyl-4-phenylpyridinium (MPP+)- induced neurotoxicity. LPS treatment impaired the uptake process of DA by 45% and this decrease was strongly hindered by VPA pretreatment in a dose-dependent manner. Similarly, treatment with MPP+ also reduced DA uptake by 70% and VPA significantly reduced the degeneration of DA neurons by MPP+ toxicity. A recent study by (Lu et al., 2013) with LB-205 (a pan HDAC inhibitor) in acute traumatic brain injury rat model showed that LB-205 treatment increased the survival of central nervous system cells after the injury by virtue of preservation of NGF expression and activation of tyrosine kinase A receptor (TrKA) pathway. The neuroprotective mechanism of HDACi was linked with upregulation of expression of NGF, activation of TrkA phosphorylation, phospho-protein kinase B (p-AKT), NFκB, and B-cell lymphoma 2 (Bcl-2) cell survival factors, while the downregulation of phospho-JNK, p75 neurotrophin receptor (NTR), and Bcl-2-associated X protein apoptosis factors (Wu et al., 2013) studied the effect of VPA, TSA, 4-phenyl butyric acid (4-PBA), and nicotinamide in astrocyte and dopamine neuron glial culture. TSA and VPA increased GDNF and BDNF transcripts in astrocytes. The neurotrophin transcript levels were increased due to increase in GDNF promoter activity leading to elevation in level of H3 acetylation. VPA, TSA, 4-PBA, and nicotinamide also protected dopamine neuron/glial culture from MPTP and LPS toxicity. Both 4-PBA and nicotinamide decreased the release of TNF-α and cell survivability in microglial culture stimulated with LPS (Wu et al., 2013).
Treatment of SH-SY 5Y cells with VPA, motivated neurite outgrowth and cell survival by triggering ERK pathway. VPA treatment enhanced norepinephrine uptake and also induced expression of GAP-43 and Bcl-2, the genes controlled by ERK pathway (Yuan, 2001). VPA has been shown to prolong the life span of cultured cortical neurons and promote neuronal growth (Hao, 2004). In vivo effects have also shown HDAC inhibitors to be neuroprotective in chronic neurodegenerative diseases (Siebzehnrubl et al., 2007). Increased histones acetylation and the transcription factors in neurons have been linked to shield from apoptosis in animal models of neurodegeneration and promote neuronal differentiation (Siebzehnrubl et al., 2007). Both, TSA and VPA have been linked to induce differentiation of hippocampal neural progenitor cells (Hsieh et al., 2004). HDAC inhibitors like MS-275, M334, and SAHA increased neuronal differentiation and inhibited oligodendrocytes production in adult subventricular zone precursor cells (Siebzehnrubl et al., 2007). (Zhou et al., 2011), showed that SAHA and sodium butyrate blocked cell cycle progression from G1 to S in the neurosphere formation of adult subventricular cells. Pro-neural transcription factors such as Neurog1 and Neurod1 were up regulated. SAHA has also been found to be neuroprotective in dopaminergic neurons from midbrain primary culture by induction of neurotrophic factors from astroglia through induction of histone acetylation (Chen et al., 2011). (Jung et al., 2008) have reported VPA to stimulate differentiation and reduce proliferation in rat cerebral cortex neural progenitor cells via the activation of ERK-P21 Cip/WAFI pathway. HDAC inhibitors have also shown roles in directing neuronal, cardiomyocytic and hepatic lineage differentiations (Kretsovali et al., 2012).
SAHA treatment increased survival of dopaminergic neurons in mesencephalic neuronal culture, which was evident by an increase in percentage of dopamine uptake (Chen et al., 2011). The neuroprotective quality of SAHA was attributed to astroglia. SAHA was also found to induce the release of neuronal survival factors from astroglia by increasing the release of neurotrophic factors GDNF and BDNF. SAHA induced hyperacetylation of histones and also the induced expression of GDNF and BDNF in mesencephalic neuronal culture (Chen et al., 2011). A study on the effect of various HDAC inhibitors on survival of rat retinal ganglion cells (RGC) led to the finding that sodium butyrate and VPA delayed the spontaneous cell death in RGC. However, TSA did not have the same effect in these cells (Biermann et al., 2011).
HDAC inhibitors induce histone acetylation in diseased neurons and may reinstate transcriptional stability and hence delay or prevent cell degeneration (Sterner and Berger, 2000). The neurotrophic activities of HDAC inhibitors may be epigenetically controlled by acetylation of histones along with non-histone proteins such as transcription factors (Sterner and Berger, 2000). The regulation of transcription by epigenetic modifications has been established to be significant for neurite outgrowth and neuroprotection in the peripheral nervous system during neuronal development. The increased acetylation of both transcription factors and histones in neurons has been revealed to stimulate neuronal differentiation and outgrowth (Gaub et al., 2010). TSA has been shown to produce differentiation of retinal ganglion Cells (RGC-5 cells). The differentiation of RGC-5 cells induced by TSA was transcription dependent, as the neurite outgrowth was blocked in presence of a RNA polymerase inhibitor alpha amanitin (Schwechter et al., 2007b). The differentiation of RGC-5 cells with HDAC inhibitors might be due to elimination of neuron gene-specific transcription repressor—neuron restrictive silencing factor (NRSF) from NRSE. It is critical process, however, not essentially adequate component of in vivo neuronal differentiation (Schwechter et al., 2007b). (Tomioka et al., 2014) have shown that TSA induced neurite outgrowth in PC12 cells by an expression of nur77, which was upregulated due to acetylation of Lys 14 on histone H3. (Wang et al., 2014) have shown that TSA improved the expression of neurotrophic factors in the Schwan cells (SC) and repressed the expression of genes associated with myelin production. TSA indicated to play a key role in the growth and biological functions such as proliferation, survival, migration, and myelination of SCs (Wang et al., 2014). In cultured cortical granule neuron (CGN), hyperacetylation stimulates neuronal outgrowth and growth cone remodeling through inhibition of HDAC I and HDAC II by a transcription-dependent mechanism. It was found that TSA induced a significant increase in expression of HATs such as p300, CBP and P/CAF in cultured CGN. Acetylation dependent p53 signaling functions are also important events, which allow physiological pro-outgrowth signal in primary neurons (Gaub et al., 2010). Transcriptional control, chromatin remodeling, and epigenetic modifications regulate the self-renewal and differentiation of neuronal stem cells (NSC) (Temple, 2001). Throughout the growth of the central nervous system in vertebrates, the fate of NSC is firmly controlled under temporal and regional manners and is also regulated by epigenetic controls (Temple, 2001). Reports have indicated that the HDACs play key role in regulation of proliferation and differentiation of neural stem cells (Sun et al., 2011). HDAC 1, 3, 5, and 7 are expressed in neural stem cells. However, the expression of these HDACs is significantly reduced after differentiation (Sun et al., 2007). HDAC2 is expressed in proliferating neural stem cells and is up regulated as neurons differentiate (MacDonald and Roskams, 2008). A study on the effect of TSA on multipotent neural stem cells, that have the ability for self- renewal and differentiation into neurons astrocytes and oligodendrocytes, demonstrated that TSA dose-dependently, promoted neurite outgrowth in multipotent neural stem cells by significantly increasing branching points and dendritic area. However, TSA inhibited the differentiation of NSC into astrocytes. TSA also mediated cholinergic differentiation of NSC as evident by a 10% increase in cholinergic neurons after TSA treatment (Balasubramaniyan et al., 2006). HDAC inhibitors have also been shown to increase neuronal differentiation of embryonic mouse neural stem cells (Siebzehnrubl et al., 2007). VPA treatment, at the progenitor stage of hippocampal treatment, led to robust inhibition of cell proliferation and stimulation of neuronal differentiation due to an increase in expression of proneural transcription factor. The treatment with HDAC inhibitors increased expression of Ngn, Math1, and P15 and shifted the cells towards neuronal fate. Also, the increased expression of proneural transcription factor was linked to acetylation of histone H4 (Yu et al., 2009). Similar to neural stem cells, the pluripotent stem cells have also shown the potential for the treatment of neurodegenerative disease owing to their competency to self-renew and differentiate into all cell types (Thomson, 1998). Pluripotent stem cells (iPSCs) are alike embryonic stem cells and are derived from epigenetic reprogramming of somatic cells by the exogenous expression of pluripotency-related transcription factors (Hochedlinger and Plath, 2009). HDAC inhibitors improve the reprogramming efficacy of the somatic cells into pluripotent cells (iPSC). HDAC inhibitors like, SAHA, TSA and VPA have shown an important role in reprogramming of mouse embryonic fibroblast to iPSCs (Huangfu et al., 2008). HDAC inhibitors have also been shown to play an important role in survival, proliferation, and differentiation of rat neural precursor cells (NPC). TSA treatment of NPC derived from Sprague Dawley rats E14.5 brain has shown decreased proliferation and increased differentiation of neurons along with decreased differentiation of astrocytes. Increased neuronal differentiation and proliferation of NPC was attributed to reduction in transcription of class II HDAC, while the transcription of class I HDAC was not changed (Liu et al., 2012). Hisahara et al. (2008) have reported important role of SIRT1, a NAD+ dependent class III HDAC in the differentiation of NPC. The SIRT1 was found predominantly in cytoplasm. However, when cultured NPCs were transferred to differentiation conditions, SIRT1 was expressed in the nucleus. This implicated modulation of neuronal differentiation by SIRT1 nuclear translocation. SIRT1 plays an important role in neuronal differentiation as evident by decrease in neuronal differentiation by inhibition of SIRT 1 by sirtinol, a SIRT1 inhibitor as well as use of siRNA against SIRT1. Overexpression of SIRT1 promoted neuronal differentiation (Hisahara et al., 2008). VPA induced neuronal differentiation in adult hippocampal neural progenitor cells (Hsieh et al., 2004). VPA treatment also increased the level of acetylated histones. The level of acetylated histones was elevated in the neuronal cells as compared to astrocytes and oligodendrocytes. The acetylation of histones promotes the differentiation of adult hippocampal progenitor cells; however, deacetylation of histones or deacetylase activity enables differentiation of oligodendrocytes and astrocytes. The VPA induced neuronal differentiation of neural progenitor cells was linked to upregulation of a neuron specific gene NeuroD, a bHLH transcription factor (Hsieh et al., 2004). In another study, VPA produced differentiation of cortical neurons by activation of ERK Pathway. Valproate induced phosphorylation of P44/42 ERK were MEK and REF dependent, which was similar to action of neurotrophins (Hao, 2004). VPA treatment in rat cerebral cortex induced differentiation and reduced the proliferation of neural progenitor cells via activation of ERK-P21 Cip/WAFI pathway mediated by β-catenin pathway (Jung et al., 2008). (Marin-Husstege et al., 2002) developed two pronged approaches to investigate the role of chromatin remodeling in oligodendrocyte differentiation. The first approach assumes chromatin compaction on negative regulatory sites of a differentiation gene. While, the second or alternative approach proposes chromatic compaction in the promoter of a gene encoding inhibitors of differentiation and in turn blocking the access to transcription start site. Histone deacetylation has also been shown to be important for oligodendrocyte differentiation. Deacetylation followed by methylation of histones promoted the neural stem cells differentiation into neurons and glial cells. The HDAC activity was vital for differentiation of oligodendrocyte from rat cortical progenitor cells (Marin-Husstege et al., 2005). Investigation on the effect of VPA on neuronal differentiation with human bone marrow-mesenchymal stromal cells (hBM-MSCs) showed that VPA treated MSCs differentiated on neuronal media and showed an increase in number/length of neurites as compared to differentiated MSCs. The VPA treated MSCs exhibited cell morphology similar to regular neurons. There was a significant increase in expression of neuronal marker such as Nestin, Musashi, CD133, and GFAP (Jeong et al., 2013). Sodium butyrate enhanced neuronal differentiation of medulloblastoma (MB) cells, the malignant brain tumors of childhood. Sodium butyrate triggered the Gria2 expression, a neuronal differentiation marker in human medulloblastoma cell lines, D283 and DAOY. HDAC inhibitors have shown to reduce MB stem cells survival and proliferation (Nör et al., 2013). VPA precisely escalated neuronal differentiation in human FGF1 gene 1B promoter (-540 to +31) driven green fluorescence F1BGFP (+) embryonic stem cells and Neural stem/progenitor cells (NSPCs) rather than GFP(-) cells (Kao et al., 2013). These NSPCs were crucial for VPA-induced neuronal differentiation than GFP (-) cells. VPA activated human FGF1 gene promoter by dissuasion of HDAC and GSK-3 functions. (Her et al., 2009) have shown that HDAC inhibitors, namely TSA, sodium butyrate, and VPA, promoted differentiation of rat C6 glioma cells by producing neuroactive 5α-reduce neurosteroid, which stimulated expression of serotonin stimulated brain derived neurotrophic factor transcription. These HDAC inhibitors also stimulated production of glial fibrillary acid (GFAP).
Niemann–Pick type C disease (NPC) is a kind of neurodegenerative condition associated with lipid storage disorder (Liscum and Klansek, 1998). In NPC1-/-, mice self-renewal, and differentiation of neural stem cells (NSC) are impaired. Treatment of NPC1-/- mice with VPA enhanced neuronal differentiation and restored impaired astrocytes in NSCs from NPC1−/− mice. There was an upregulation of vital neurotrophic genes (BDNF, MnSoD, TrkB, and NeuroD), by suppression of the REST/NRSF and HDAC complex by VPA treatment. Up-regulated neurotrophic genes enhanced neural differentiation in neural stem cells from NPC1−/− mice (Kim et al., 2007).
HDAC1 has also been shown to have an important player in regulating the switch between proliferation and differentiation in the retina of zebra fish. The ratio of differentiating cells to that of proliferating cells was increased proportionate to HDAC activity due to treatment of zebrafish with TSA, which suggests that HDAC might control the retinal neurogenesis in zebrafish (Yamaguchi, 2005).
Neuroregeneration is a new notion that includes neuroplasticity, neurogenesis, and neurorestoration (Enciu et al., 2011). Earlier, neuronal regeneration in the mammalian central nervous system (CNS) was considered to be untreatable, but recent reports have suggested that damaged neurons can be restored with stimulatory substances for example nerve growth factor (NGF) and brain derived neurotrophic factor (BDNF) (Diaz Brinton et al., 1998; Filbin, 2000; Fournier and Strittmatter, 2001; Pesavento et al., 2002). Neurite outgrowth, an important cellular process for neurogenesis, is a complex three stage process 1) sprouting; neurite formation initiation, 2) axon elongation, and 3) dendrites branching after the formation of synapses (Kiryushko et al., 2004). Neuritogenesis is a key cellular differentiation process for formation of new neurons. The cellular and molecular mechanisms of neuritogenesis is important, as it is essential for proper brain wiring and nerve regeneration, and along with various neurodegenerative diseases (Jones et al., 2001). Recent reports have emphasized the importance of neurite outgrowth in neuroregeneration and damaged neuronal repair (Tomioka et al., 2014). The physiological substances, which trigger the process of neuritogenesis, are commonly referred as neurotrophic factors. Neurotrophic factors are proteinaceous growth factors that stimulate neurite outgrowth and repair damaged neurons. It may also play a key role in regression and reestablishment of the damaged neurons to normal neuronal functions (Li et al., 2002). These factors are produced and released by the target neuronal tissues and initiate the growth and differentiation of developing neurons. The NGF, BDNF, neurotrophin-3 (NT-3), and neurotrophic factor-4/5 (NT-4/5) are commonly referred as neurotrophins. Use of natural neurotrophins appears to be an obvious choice for neuronal regeneration. However, regardless of their definite positive effects, therapeutic applications of natural neurotrophins like NGF and BDNF, for treatment of neurodegenerative diseases, are limited due to their inability to cross blood brain barrier and also susceptibility to degradation by peripheral peptidases (Tomioka et al., 2014). Recently, there has been significant surge in identification of small molecular weight neurotrophic compounds, which can promote growth, differentiation, survival of developing and damaged neurons (Longo and Massa, 2013; Obianyo and Ye, 2013). Several studies have been performed using neurotrophins as a probable therapy for neuronal degeneration and trauma causing severing of nerve connections. The neurotrophins may facilitate the survival of physically damaged neurons like injury by motorcycle accidents or sports-related injuries or in neurodegenerative diseases like Huntington's, Alzhiemer's, and Parkinson's disease. The therapeutic use these neurotrophins offered hope to make a more widely accepted clinical possibility for neuroregeneration. Several recent finding indicates that neuroprotective and neurotrophic role of small molecular weight inhibitors of HDACs and their possible use in treatment of neurological disorders (Camelo et al., 2005; Xu et al., 2011). HDAC inhibitors by chromatin modification and epigenetic regulation provides unique neuronal acclimatization like transcription-dependent neuroplasticity and learning or axon and dendrite regeneration (Kandel, 2001; Wu et al., 2007). Vorinostat exhibited independent neurotrophic action on NS-1 cells, clone of PC12 cells. Interrogation of intracellular neurotrophin signaling pathways with selective inhibitors of MEK1/2, phosphoinositide 3-kinase (PI3K) and tyrosine kinase A (TrkA) suggested the role of ERK pathway in vorinostat-induced neurotrophic action also involved the activation of upstream extracellular kinase TrkA. Vorinostat (SAHA) stimulated hyperacetylation of α-tubulin and histones H3/H4 in NS-1 cells (Shukla et al., 2016). CTPB [N-(4-chloro-3-trifluoromethyl-phenyl)-2-ethoxy-6-pentadecyl-benzamide], an activator of the histone acetyltransferase p300/CBP also showed neurotrophic action on in the SH-SY5Y neuronal cells. This report confirms the role of proteins hyperacetylation in induction of neurotrophic signaling pathways (Hegarty et al., 2016a). Treatment with VPA caused hyperacetylation of histones and elevated levels of neurotrophic/neuroprotective factors, which provided neuroprotective and neurorestorative phenotype in an in-vitro model of PD (Harrison et al., 2015). VPA treatment also induce neurotrophic factors in neuronal stem cells (Almutawaa et al., 2014). The neurotrophic action of TSA in a rat cultured cerebellar granule neurons and in cerebral cortical neurons involved acetylation of p53 and also protected neurons against glutathione depletion-induced oxidative stress (Maruoka et al., 2011). Treatment with sodium butyrate stimulated neurogenesis in the ischemic adult rat brain and also elevated protein levels of phospho-CREB, BDNF, and glial fibrillary acidic protein. The neurotrophic actions of sodium butyrate were blocked by intraventricular injection of K252a, a tyrosine kinase B receptor antagonist. (Kim et al., 2009). In vitro treatment of the retinal ganglion cell line (RGC-5) with TSA showed significant RNA transcription-dependent neurotrophic action (Schwechter et al., 2007a). The prominent neurotrophic potential of HDAC inhibitors strengthens their therapeutic application for treatment of neurodegenerative disorders.
This review presents the current status of knowledge about the role of various HDAC inhibitors in neuronal differentiation and neuroprotection in different cell lines, in vivo preclinical animal models and limited information on their clinical evaluations. The potential therapeutic application of several HDAC inhibitors for neurodegenerative diseases has also been discussed. HDAC inhibitors have significant therapeutic potential for many of the neurodegenerative diseases such as PD, AD, SMN, ALS, TBI, MS, and HD. HDAC inhibitors engender neuronal differentiation in neural stem cells (NSC), neural progenitor cells (NPCs), rat retinal ganglion cells (RGCs), dopaminergic neurons, SHSY5Y neuroblastoma cells, cortical granule neurons (CGN) and experimental cell lines such as the PC12 cells. HDAC inhibitors also possess neuroprotective and neurotrophic potential. However, potential limitations regarding safety of these HDAC inhibitors cannot be ignored. Reports indicate that HDAC inhibitors in some instances could play a role in pathogenesis of the neurodegenerative disease, while in others could act as potential therapeutics for the neurodegenerative disease. These opposing effects of HDAC inhibitors could be attributed to the varying epigenetic status of the targets, cell type and tissue specificity. On occasion, the HDAC inhibitors have been shown to be highly neurotoxic rather than being neuroprotective. The major problem associated with HDAC inhibitors is their lack of specificity and in particular their lack of isoform selectivity. Another issue is incomplete knowledge about the off-target actions of HDAC inhibitors. There is a great surge for learning about the mechanism of action and the various ways that HDAC inhibitors affect gene expression. Future research is expected to elucidate the mechanism of action HDAC inhibitors in order to design novel reagents with increased effectiveness and specificity. Research on specific/selective HDAC inhibitors would be beneficial from the therapeutic point of view and would likely overcome the problems currently associated with HDAC inhibitors. Hopefully, the rapid interest in the study of HDAC inhibitors will shed some light on the mechanism of action of these inhibitors and give us directions to combat neurodegenerative diseases.
SS and BT conceived the article. SS compiled the review and prepared the draft of the manuscript. BT reviewed and edited the manuscript. All authors read and approved the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Agis-Balboa, R. C., Pavelka, Z., Kerimoglu, C., Fischer, A. (2013). Loss of HDAC5 Impairs Memory Function: Implications for Alzheimer's Disease. J. Alzheimer's Dis. 33 (1), 35–44. doi: 10.3233/JAD-2012-121009
Aksnes, H., Hole, K., Arnesen, T. (2015). “Chapter Seven - Molecular, Cellular, and Physiological Significance of N-Terminal Acetylation,” in International Review of Cell and Molecular Biology. Ed. Jeon, K. W. (Academic Press)), 267–305.
Allis, C. D., Berger, S. L., Cote, J., Dent, S., Jenuwien, T., Kouzarides, T., et al. (2007). New Nomenclature for Chromatin-Modifying Enzymes. Cell 131 (4), 633–636. doi: 10.1016/j.cell.2007.10.039
Almutawaa, W., Kang, N. H., Pan, Y., Niles, L. P. (2014). Induction of Neurotrophic and Differentiation Factors in Neural Stem Cells by Valproic Acid. Basic Clin. Pharmacol. Toxicol. 115 (2), 216–221. doi: 10.1111/bcpt.12201
Araki, T. (2004). Increased Nuclear NAD Biosynthesis and SIRT1 Activation Prevent Axonal Degeneration. Science 305 (5686), 1010–1013. doi: 10.1126/science.1098014
Andreassi, C., Angelozzi, C., Tiziano, F. D., Vitali, T., De Vincenzi, E., Boninsegna, A., et al (2004). Phenylbutyrate increases SMN expression in vitro: relevance for treatment of spinal muscular atrophy. Eur. J. Hum. Genet. 12 (1), 59–65. doi: 10.1038/sj.ejhg.5201102
Avila, A. M., Burnett, B. G., Taye, A. A., Gabanella, F., Knight, M. A., Hartenstein, P., et al. (2007). Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J. Clin. Invest. 117 (3), 659–671. doi: 10.1172/jci29562
Avvakumov, N., Côté, J. (2007). The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene 26 (37), 5395–5407. doi: 10.1038/sj.onc.1210608
Balasubramaniyan, V., Boddeke, E., Bakels, R., Küst, B., Kooistra, S., Veneman, A., et al. (2006). Effects of histone deacetylation inhibition on neuronal differentiation of embryonic mouse neural stem cells. Neuroscience 143 (4), 939–951. doi: 10.1016/j.neuroscience.2006.08.082
Bannister, A. J., Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Cell Res. 21 (3), 381–395. doi: 10.1038/cr.2011.22
Bates, E. A., Victor, M., Jones, A. K., Shi, Y, Hart, A. C. (2006). Differential contributions of Caenorhabditis elegans histone deacetylases to huntingtin polyglutamine toxicity. J. Neurosci. 26 (10), 2830–2838. doi: 10.1523/jneurosci.3344-05.2006
Bennett, S. A., Tanaz, R., Cobos, S. N., Torrente, M. P. (2019). Epigenetics in amyotrophic lateral sclerosis: a role for histone post-translational modifications in neurodegenerative disease. Trans. Res. 204, 19–30. doi: 10.1016/j.trsl.2018.10.002
Berndsen, C. E., Denu, J. M. (2008). Catalysis and substrate selection by histone/protein lysine acetyltransferases. Curr. Opin. Struct. Biol. 18 (6), 682–689. doi: 10.1016/j.sbi.2008.11.004
Biermann, J., Boyle, J., Pielen, A., Lagreze, W. A. (2011). Histone deacetylase inhibitors sodium butyrate and valproic acid delay spontaneous cell death in purified rat retinal ganglion cells. Mol. Vis. 17, 395–403.
Bjerling, P., Silverstein, R. A., Thon, G., Caudy, A., Grewal, S., Ekwall, K. (2002). Functional Divergence between Histone Deacetylases in Fission Yeast by Distinct Cellular Localization and In Vivo Specificity. Mol. Cell. Biol. 22 (7), 2170–2181. doi: 10.1128/mcb.22.7.2170-2181.2002
Boeselt, S., Janssen, C., Dengler, R., Petri, S., Schmalbach, S., Sarlette, A. (2010). Differential Histone Deacetylase mRNA Expression Patterns in Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 69 (6), 573–581. doi: 10.1097/NEN.0b013e3181ddd404
Boyault, C., Zhang, Y., Fritah, S., Caron, C., Gilquin, B., Kwon, S. H., et al. (2007). HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 21 (17), 2172–2181. doi: 10.1101/gad.436407
Brichta, L., Hofmann, Y., Hahnen, E., Siebzehnrubl, F. A., Raschke, H., Blumcke, I., et al (2003). Valproic acid increases the SMN2 protein level: a well-known drug as a potential therapy for spinal muscular atrophy. Hum. Mol. Genet. 12 (19), 2481–2489. doi: 10.1093/hmg/ddg256
Bruneteau, G., Simonet, T., Bauché, S., Mandjee, N., Malfatti, E., Girard, E., et al. (2013). Muscle histone deacetylase 4 upregulation in amyotrophic lateral sclerosis: potential role in reinnervation ability and disease progression. Brain 136 (8), 2359–2368. doi: 10.1093/brain/awt164
Butchbach, M. E. R., Lumpkin, C. J., Harris, A. W., Saieva, L., Edwards, J. D., Workman, E., et al. (2016). Protective effects of butyrate-based compounds on a mouse model for spinal muscular atrophy. Exp. Neurol. 279, 13–26. doi: 10.1016/j.expneurol.2016.02.009
Butler, R., Bates, G. P. (2006). Histone deacetylase inhibitors as therapeutics for polyglutamine disorders. Nat. Rev. Neurosci. 7 (10), 784–796. doi: 10.1038/nrn1989
Camelo, S., Iglesias, A. H., Hwang, D., Due, B., Ryu, H., Smith, K., et al. (2005). Transcriptional therapy with the histone deacetylase inhibitor trichostatin A ameliorates experimental autoimmune encephalomyelitis. J. Neuroimmunol. 164 (1-2), 10–21. doi: 10.1016/j.jneuroim.2005.02.022
Chen, P. S., Peng, G. S., Li, G., Yang, S., Wu, X., Wang, C. C., et al. (2006). Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol. Psychiatry 11 (12), 1116–1125. doi: 10.1038/sj.mp.4001893
Chang, J. G., Hsieh-Li, H. M., Jong, J., Wang, N. M., Tsai, C. H., Li, H. (2001). Treatment of spinal muscular atrophy by sodium butyrate. Proc. Natl. Acad. Sci. U. S. A. 98 (17), 9808–9813. doi: 10.1073/pnas.171105098
Chen, S. H., Wu, H. M., Ossola, B., Schendzielorz, N., Wilson, B. C., Chu, C. H., et al. (2011). Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, protects dopaminergic neurons from neurotoxin-induced damage. Br. J. Pharmacol. 165 (2), 494–505. doi: 10.1111/j.1476-5381.2011.01575.x
Choong, C.-J., Sasaki, T., Hayakawa, H., Yasuda, T., Baba, K., Hirata, Y., et al. (2016). A novel histone deacetylase 1 and 2 isoform-specific inhibitor alleviates experimental Parkinson's disease. Neurobiol. Aging 37, 103–116. doi: 10.1016/j.neurobiolaging.2015.10.001
Chopra, V., Quinti, L., Khanna, P., Paganetti, P., Kuhn, R., Young, A. B., et al. (2016). LBH589, A Hydroxamic Acid-Derived HDAC Inhibitor, is Neuroprotective in Mouse Models of Huntington's Disease. J. Huntington's Dis. 5 (4), 347–355. doi: 10.3233/JHD-160226
Choudhary, C., Kumar, C., Gnad, F., Nielsen, M. L., Rehman, M., Walther, T. C., et al. (2009). Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science 325 (5942), 834–840. doi: 10.1126/science.1175371
Chuang, D.-M., Leng, Y., Marinova, Z., Kim, H.-J., Chiu, C.-T. (2009). Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 32 (11), 591–601. doi: 10.1016/j.tins.2009.06.002
Cuadrado-Tejedor, M., Perez-Gonzalez, M., Garcia-Munoz, C., Muruzabal, D., Garcia-Barroso, C., Rabal, O., et al. (2019). Taking Advantage of the Selectivity of Histone Deacetylases and Phosphodiesterase Inhibitors to Design Better Therapeutic Strategies to Treat Alzheimer's Disease. Front. Aging Neurosci. 11, 149. doi: 10.3389/fnagi.2019.00149
Dash, P. K., Orsi, S. A., Moore, A. N. (2009). Histone deactylase inhibition combined with behavioral therapy enhances learning and memory following traumatic brain injury. Neuroscience 163 (1), 1–8. doi: 10.1016/j.neuroscience.2009.06.028
De Ruijter, A. J. M., van Gennip, A. H., Caron, H. N., Kemp, S., van Kuilenburg, A. B. P. (2003). Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 370 (3), 737–749. doi: 10.1042/bj20021321
Dekker, F. J., Haisma, H. J. (2009). Histone acetyl transferases as emerging drug targets. Drug Discovery Today 14 (19), 942–948. doi: 10.1016/j.drudis.2009.06.008
Del Signore, S. J., Amante, D. J., Kim, J., Stack, E. C., Goodrich, S., Cormier, K., et al (2009). Combined riluzole and sodium phenylbutyrate therapy in transgenic amyotrophic lateral sclerosis mice. Amyotroph Lateral Scler 10 (2), 85–94. doi: 10.1080/17482960802226148
Diaz Brinton, R., Yamazaki, R., Gonzalez, C. M., O'Neill, K., Schreiber, S. S. (1998). Vasopressin-induction of the immediate early gene, NGFI-A, in cultured hippocampal glial cells. Mol. Brain Res. 57 (1), 73–85. doi: 10.1016/s0169-328x(98)00069-2
Dompierre, J. P., Godin, J. D., Charrin, B. C., Cordelieres, F. P., King, S. J., Humbert, S., et al. (2007). Histone Deacetylase 6 Inhibition Compensates for the Transport Deficit in Huntington's Disease by Increasing Tubulin Acetylation. J. Neurosci. 27 (13), 3571–3583. doi: 10.1523/jneurosci.0037-07.2007
Du, G., Jiao, R. (2011). To prevent neurodegeneration: HDAC6 uses different strategies for different challenges. Commun. Integr. Biol. 4 (2), 139–142. doi: 10.4161/cib.4.2.14272
Du, G., Liu, X., Chen, X., Song, M., Yan, Y., Jiao, R., et al. (2010). Drosophila Histone Deacetylase 6 Protects Dopaminergic Neurons against α-Synuclein Toxicity by Promoting Inclusion Formation. Mol. Biol. Cell 21 (13), 2128–2137. doi: 10.1091/mbc.e10-03-0200
Dubey, H., Gulati, K., Ray, A. (2018). ““Recent studies on cellular and molecular mechanisms in Alzheimer's disease: focus on epigenetic factors and histone deacetylase”,” in Reviews in the Neurosciences (De Gruyter).
Ellrichmann, G., Blusch, A., Fatoba, O., Brunner, J., Hayardeny, L., Hayden, M., et al. (2017). Laquinimod treatment in the R6/2 mouse model. Sci. Rep. 7 (1), 4947. doi: 10.1038/s41598-017-04990-1
Enciu, A. M., Nicolescu, M. I., Manole, C. G., Mureşanu, D. F., Popescu, L. M., Popescu, B. O. (2011). Neuroregeneration in neurodegenerative disorders. BMC Neurol. 11 (1), 75. doi: 10.1186/1471-2377-11-75
Evans, M. C., Cherry, J. J., Androphy, E. J. (2011). Differential regulation of the SMN2 gene by individual HDAC proteins. Biochem. Biophys. Res. Commun. 414 (1), 25–30. doi: 10.1016/j.bbrc.2011.09.011
Faraco, G., Pancani, T., Formentini, L., Mascagni, P., Fossati, G., Leoni, F., et al. (2006). Pharmacological inhibition of histone deacetylases by suberoylanilide hydroxamic acid specifically alters gene expression and reduces ischemic injury in the mouse brain. Mol. Pharmacol. 70 (6), 1876–1884. doi: 10.1124/mol.106.027912
Faraco, G., Cavone, L., Chiarugi, A. (2011). The therapeutic potential of HDAC inhibitors in the treatment of multiple sclerosis. Mol. Med. (Cambridge Mass ) 17 (5-6), 442–447. doi: 10.2119/molmed.2011.00077
Federspiel, J. D., Greco, T. M., Lum, K. K., Cristea, I. M. (2019). Hdac4 Interactions in Huntington's Disease Viewed Through the Prism of Multiomics. Mol. Cell Proteomics 18 (8 suppl. 1), S92–s113. doi: 10.1074/mcp.RA118.001253
Ferrante, R. J., Kubilus, J. K., Lee, J., Ryu, H., Beesen, A., Zucker, B., et al. (2003). Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J. Neurosci. 23 (28), 9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003
Filbin, M. T. (2000). Axon regeneration: Vaccinating against spinal cord injury. Curr. Biol. 10 (3), R100–R103. doi: 10.1016/s0960-9822(00)00302-x
Finnin, M., Donigian, J., Cohen, A., Richon, V. M., Rifkind, R. A., Marks, P. A., et al. (1999). Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 401, 188–193. doi: 10.1038/43710
Fischer, A., Sananbenesi, F., Wang, X., Dobbin, M., Tsai, L. H. (2007). Recovery of learning and memory is associated with chromatin remodelling. Nature 447 (7141), 178–182. doi: 10.1038/nature05772
Fournier, A. E., Strittmatter, S. M. (2001). Repulsive factors and axon regeneration in the CNS. Curr. Opin. Neurobiol. 11 (1), 89–94. doi: 10.1016/s0959-4388(00)00178-1
Gasser, T. (2001). Genetics of Parkinson's disease. J. Neurol. 248, 833–840. doi: 10.1007/s004150170066
Gardian, G., Yang, L., Cleren, C., Calingasan, N. Y., Klivenyi, P., Beal, M. F. (2004). Neuroprotective Effects of Phenylbutyrate Against MPTP Neurotoxicity. NMM 5 (3), 235–242. doi: 10.1385/nmm:5:3:235
Gaub, P., Tedeschi, A., Puttagunta, R., Nguyen, T., Schmandke, A., Di Giovanni, S. (2010). HDAC inhibition promotes neuronal outgrowth and counteracts growth cone collapse through CBP/p300 and P/CAF-dependent p53 acetylation. Cell Death Differ. 17 (9), 1392–1408. doi: 10.1038/cdd.2009.216
Giffard, R. G., Xu, L., Zhao, H., Carrico, W., Ouyang, Y., Qiao, Y., et al. (2004). Chaperones, protein aggregation, and brain protection from hypoxic/ischemic injury. J. Exp. Biol. 207 (Pt 18), 3213–3220. doi: 10.1242/jeb.01034
Glozak, M. A., Sengupta, N., Zhang, X., Seto, E. (2005). Acetylation and deacetylation of non-histone proteins. Gene 363, 15–23. doi: 10.1016/j.gene.2005.09.010
Govindarajan, N., Rao, P., Burkhardt, S., Sananbenesi, F., Schlüter, O. M., Bradke, F., et al. (2013). Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer's disease. EMBO Mol. Med. 5 (1), 52–63. doi: 10.1002/emmm.201201923
Green, K. N., Steffan, J. S., Martinez-Coria, H., Sun, X., Schreiber, S. S., Thompson, L. M., et al. (2008). Nicotinamide Restores Cognition in Alzheimer's Disease Transgenic Mice via a Mechanism Involving Sirtuin Inhibition and Selective Reduction of Thr231-Phosphotau. J. Neurosci. 28 (45), 11500–11510. doi: 10.1523/jneurosci.3203-08.2008
Guan, J. S., Haggarty, S. J., Giacometti, E., Dannenberg, J. H., Joseph, N., Gao, J., et al. (2008). HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459 (7243), 55–60. doi: 10.1038/nature07925
Guidotti, A., Dong, E., Kundakovic, M., Satta, R., Grayson, D. R., Costa, E. (2009). Characterization of the action of antipsychotic subtypes on valproate-induced chromatin remodeling. Trends Pharmacol. Sci. 30 (2), 55–60. doi: 10.1016/j.tips.2008.10.010
Haberland, M., Montgomery, R. L., Olson, E. N. (2009). The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 10 (1), 32–42. doi: 10.1038/nrg2485
Hagenacker, T., Wurster, C. D., Gunther, R., Schreiber-Katz, O., Osmanovic, A., Petri, S., et al. (2020). Nusinersen in adults with 5q spinal muscular atrophy: a non-interventional, multicentre, observational cohort study. Lancet Neurol. 19 (4), 317–325. doi: 10.1016/s1474-4422(20)30037-5
Haggarty, S. J., Koeller, K. M., Wong, J. C., Grozinger, C. M., Schreiber, S. L. (2003). Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. 100 (8), 4389–4394. doi: 10.1073/pnas.0430973100
Hahnen, E., Eyüpoglu, I. Y., Brichta, L., Haastert, K., Tränkle, C., Siebzehnrübl, F. A., et al. (2006). In vitro and ex vivo evaluation of second-generation histone deacetylase inhibitors for the treatment of spinal muscular atrophy. J. Neurochem. 98 (1), 193–202. doi: 10.1111/j.1471-4159.2006.03868.x
Hao, Y. (2004). Mood Stabilizer Valproate Promotes ERK Pathway-Dependent Cortical Neuronal Growth and Neurogenesis. J. Neurosci. 24 (29), 6590–6599. doi: 10.1523/jneurosci.5747-03.2004
Harrison, I. F., Dexter, D. T. (2013). Epigenetic targeting of histone deacetylase: Therapeutic potential in Parkinson's disease? Pharmacol. Ther. 140 (1), 34–52. doi: 10.1016/j.pharmthera.2013.05.010
Harrison, I. F., Crum, W. R., Vernon, A. C., Dexter, D. T. (2015). Neurorestoration induced by the HDAC inhibitor sodium valproate in the lactacystin model of Parkinson's is associated with histone acetylation and up-regulation of neurotrophic factors. Br. J. Pharmacol. 172 (16), 4200–4215. doi: 10.1111/bph.13208
Harrison, I. F., Powell, N. M., Dexter, D. T. (2019). The histone deacetylase inhibitor nicotinamide exacerbates neurodegeneration in the lactacystin rat model of Parkinson's disease. J. Neurochem. 148 (1), 136–156. doi: 10.1111/jnc.14599
Hasan, M. R., Kim, J.-H., Kim, Y. J., Kwon, K. J., Shin, C. Y., Kim, H. Y., et al. (2013). Effect of HDAC Inhibitors on Neuroprotection and Neurite Outgrowth in Primary Rat Cortical Neurons Following Ischemic Insult. Neurochem. Res. 38 (9), 1921–1934. doi: 10.1007/s11064-013-1098-9
Hashioka, S., Klegeris, A., McGeer, P. L. (2012). The histone deacetylase inhibitor suberoylanilide hydroxamic acid attenuates human astrocyte neurotoxicity induced by interferon-γ. J. Neuroinflammation 9 (1), 113. doi: 10.1186/1742-2094-9-113
Hauke, J., Riessland, M., Lunke, S., Eyupoglu, I. Y., Blumcke, I., El-Osta, A., et al. (2009). Survival motor neuron gene 2 silencing by DNA methylation correlates with spinal muscular atrophy disease severity and can be bypassed by histone deacetylase inhibition. Hum. Mol. Genet. 18 (2), 304–317. doi: 10.1093/hmg/ddn357
Hay, D. G., Sathasivam, K., Tobaben, S., Stahl, B., Marber, M., Mestril, R., et al. (2004). Progressive decrease in chaperone protein levels in a mouse model of Huntington's disease and induction of stress proteins as a therapeutic approach. Hum. Mol. Genet. 13 (13), 1389–1405. doi: 10.1093/hmg/ddh144
Hegarty, S. V., O'Leary, E., Solger, F., Stanicka, J., Sullivan, A. M., O'Keeffe, G. W. (2016a). A Small Molecule Activator of p300/CBP Histone Acetyltransferase Promotes Survival and Neurite Growth in a Cellular Model of Parkinson's Disease. Neurotoxic. Res. 30 (3), 510–520. doi: 10.1007/s12640-016-9636-2
Hegarty, S. V., Sullivan, A. M., O'Keeffe, G. W. (2016b). The Epigenome as a therapeutic target for Parkinson's disease. Neural Regeneration Res. 11 (11), 1735–1738. doi: 10.4103/1673-5374.194803
Her, S., Lee, M.-S., Morita, K. (2009). Trichostatin A Stimulates Steroid 5α-Reductase Gene Expression in Rat C6 Glioma Cells via a Mechanism Involving Sp1 and Sp3 Transcription Factors. J. Mol. Neurosci. 41 (2), 252–262. doi: 10.1007/s12031-009-9284-6
Hisahara, S., Chiba, S., Matsumoto, H., Tanno, M., Yagi, H., Shimohama, S., et al. (2008). Histone deacetylase SIRT1 modulates neuronal differentiation by its nuclear translocation. Proc. Natl. Acad. Sci. 105 (40), 15599–15604. doi: 10.1073/pnas.0800612105
Hochedlinger, K., Plath, K. (2009). Epigenetic reprogramming and induced pluripotency. Development 136 (4), 509–523. doi: 10.1242/dev.020867
Hockly, E., Richon, V. M., Woodman, B., Smith, D. L., Zhou, X., Rosa, E., et al. (2003). Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc. Natl. Acad. Sci. 100 (4), 2041–2046. doi: 10.1073/pnas.0437870100
Hsieh, J., Nakashima, K., Kuwabara, T., Mejia, E., Gage, F. H. (2004). Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. Proc. Natl. Acad. Sci. 101 (47), 16659–16664. doi: 10.1073/pnas.0407643101
Hsu, S. W., Hsu, P. C., Chang, W. S., Yu, C. C., Wang, Y. C., Yang, J. S., et al. (2020). Protective effects of valproic acid on 6-hydroxydopamine-induced neuroinjury. Environ. Toxicol. doi: 10.1002/tox.22920
Huangfu, D., Maehr, R., Guo, W., Eijkelenboom, A., Snitow, M., Chen, A. E., et al. (2008). Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat. Biotechnol. 26 (7), 795–797. doi: 10.1038/nbt1418
Icardi, L., De Bosscher, K., Tavernier, J. (2012). The HAT/HDAC interplay: Multilevel control of STAT signaling. Cytokine Growth Factor Rev. 23 (6), 283–291. doi: 10.1016/j.cytogfr.2012.08.002
Illarioshkin, S. N., Klyushnikov, S. A., Vigont, V. A., Seliverstov, Y. A., Kaznacheyeva, E. V. (2018). Molecular Pathogenesis in Huntington's Disease. Biochem. (Moscow) 83 (1030), 1030–1039. doi: 10.1134/S0006297918090043
Itoh, Y., Suzuki, T., Miyata, N. (2018). Isoform-selective histone deacetylase inhibitors. Curr. Pharm. Des. 14 (6), 529–544. doi: 10.2174/138161208783885335
Jeong, S.-G., Ohn, T., Kim, S. H., Cho, G.-W. (2013). Valproic acid promotes neuronal differentiation by induction of neuroprogenitors in human bone-marrow mesenchymal stromal cells. Neurosci. Lett. 554, 22–27. doi: 10.1016/j.neulet.2013.08.059
Jeong, H., Shin, S., Lee, J. S., Lee, S. H., Baik, J. H., Lim, S., et al. (2019). Pan-HDAC Inhibitors Promote Tau Aggregation by Increasing the Level of Acetylated Tau. Int. J. Mol. Sci. 20 (17). doi: 10.3390/ijms20174283
Jia, H., Wang, Y., Morris, C. D., Jacques, V., Gottesfeld, J. M., Rusche, J. R., et al. (2016). The Effects of Pharmacological Inhibition of Histone Deacetylase 3 (HDAC3) in Huntington's Disease Mice. PloS One 11 (3), e0152498–e0152498. doi: 10.1371/journal.pone.0152498
Jones, L. L., Oudega, M., Bunge, M. B., Tuszynski, M. H. (2001). Neurotrophic factors, cellular bridges and gene therapy for spinal cord injury. J. Physiol. 533 (1), 83–89. doi: 10.1111/j.1469-7793.2001.0083b.x
Jung, G.-A., Yoon, J.-Y., Moon, B.-S., Yang, D.-H., Kim, H.-Y., Lee, S.-H., et al. (2008). Valproic acid induces differentiation and inhibition of proliferation in neural progenitor cells via the beta-catenin-Ras-ERK-p21Cip/WAF1 pathway. BMC Cell Biol. 9 (1), 66. doi: 10.1186/1471-2121-9-66
Kandel, E. R. (2001). The Molecular Biology of Memory Storage: A Dialogue Between Genes and Synapses. Science 294 (5544), 1030–1038. doi: 10.1126/science.1067020
Kao, C.-Y., Hsu, Y.-C., Liu, J.-W., Lee, D.-C., Chung, Y.-F., Chiu, I.-M. (2013). The mood stabilizer valproate activates human FGF1 gene promoter through inhibiting HDAC and GSK-3 activities. J. Neurochem. 126 (1), 4–18. doi: 10.1111/jnc.12292
Kazantsev, A. G., Thompson, L. M. (2008). Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat. Rev. Drug Discovery 7 (10), 854–868. doi: 10.1038/nrd2681
Kernochan, L. E., Avila, A. M., Sumner, C. J., Fischbeck, K. H., Russo, M. L., Woodling, N. S., et al. (2005). The role of histone acetylation in SMN gene expression. Hum. Mol. Genet. 14 (9), 1171–1182. doi: 10.1093/hmg/ddi130
Khan, N., Jeffers, M., Kumar, S., Hackett, C., Boldog, F., Khramtsov, N., et al. (2008). Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 409 (2), 581–589. doi: 10.1042/bj20070779
Kilgore, M., Miller, C. A., Fass, D. M., Hennig, K. M., Haggarty, S. J., Sweatt, J. D., et al. (2010). Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 35 (4), 870–880. doi: 10.1038/npp.2009.197
Kim, H. J., Rowe, M., Ren, M., Hong, J. S., Chen, P. S., Chuang, D. M. (2007). Histone Deacetylase Inhibitors Exhibit Anti-Inflammatory and Neuroprotective Effects in a Rat Permanent Ischemic Model of Stroke: Multiple Mechanisms of Action. J. Pharmacol. Exp. Ther. 321 (3), 892–901. doi: 10.1124/jpet.107.120188
Kim, D., Frank, C. L., Dobbin, M. M., Tsunemoto, R. K., Tu, W., Peng, P. L., et al (2008). Deregulation of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron 60 (5), 803–817. doi: 10.1016/j.neuron.2008.10.015
Kim, H. J., Leeds, P., Chuang, D.-M. (2009). The HDAC inhibitor, sodium butyrate, stimulates neurogenesis in the ischemic brain. J. Neurochem. 110 (4), 1226–1240. doi: 10.1111/j.1471-4159.2009.06212.x
Kiryushko, D., Berezin, V., Bock, E. (2004). Regulators of Neurite Outgrowth: Role of Cell Adhesion Molecules. Ann. N. Y. Acad. Sci. 1014 (1), 140–154. doi: 10.1196/annals.1294.015
Konsoula, Z., Barile, F. A. (2012). Epigenetic histone acetylation and deacetylation mechanisms in experimental models of neurodegenerative disorders. J. Pharmacol. Toxicolog. Methods 66 (3), 215–220. doi: 10.1016/j.vascn.2012.08.001
Kontopoulos, E., Parvin, J. D., Feany, M. B. (2006). -synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet. 15 (20), 3012–3023. doi: 10.1093/hmg/ddl243
Koppel, I., Timmusk, T. (2013). Differential regulation of Bdnf expression in cortical neurons by class-selective histone deacetylase inhibitors. Neuropharmacology 75, 106–115. doi: 10.1016/j.neuropharm.2013.07.015
Kouzarides, T. (2000). Acetylation: a regulatory modification to rival phosphorylation? EMBO J. 19 (6), 1176–1179. doi: 10.1093/emboj/19.6.1176
Kouzarides, T. (2007). Chromatin Modifications and Their Function. Cell 128 (4), 693–705. doi: 10.1016/j.cell.2007.02.005
Kretsovali, A., Hadjimichael, C., Charmpilas, N. (2012). Histone Deacetylase Inhibitors in Cell Pluripotency, Differentiation, and Reprogramming. Stem Cells Int. 2012, 1–10. doi: 10.1155/2012/184154
Kuipers, S. D., Bramham, C. R. (2006). Brain-derived neurotrophic factor mechanisms and function in adult synaptic plasticity: new insights and implications for therapy. Curr. Opin. Drug Discov. Devel. 9 (5), 580–586.
Kuta, R., Larochelle, N., Fernandez, M., Pal, A., Minotti, S., Tibshirani, M., et al. (2020). Depending on the stress, histone deacetylase inhibitors act as heat shock protein co-inducers in motor neurons and potentiate arimoclomol, exerting neuroprotection through multiple mechanisms in ALS models. Cell Stress Chaperones 25 (1), 173–191. doi: 10.1007/s12192-019-01064-1
Lai, J.-I., Leman, L. J., Ku, S., Vickers, C. J., Olsen, C. A., Montero, A., et al. (2017). Cyclic tetrapeptide HDAC inhibitors as potential therapeutics for spinal muscular atrophy: Screening with iPSC-derived neuronal cells. Bioorg. Med. Chem. Lett. 27 (15), 3289–3293. doi: 10.1016/j.bmcl.2017.06.027
Lebel, E. A., Boukamp, P., Tafrov, S. T. (2010). Irradiation with heavy-ion particles changes the cellular distribution of human histone acetyltransferase HAT1. Mol. Cell. Biochem. 339 (1-2), 271–284. doi: 10.1007/s11010-010-0390-0
Li, G., Jiang, H., Chang, M., Xie, H., Hu, L. (2011). HDAC6 alpha-tubulin deacetylase: a potential therapeutic target in neurodegenerative diseases. J. Neurol. Sci. 304 (1-2), 1–8. doi: 10.1016/j.jns.2011.02.017
Li, P., Matsunaga, K., Yamakuni, T., Ohizumi, Y. (2002). Picrosides I and II, selective enhancers of the mitogen-activated protein kinase-dependent signaling pathway in the action of neuritogenic substances on PC12D cells. Life Sciences 71 (15), 1821–1835. doi: 10.1016/S0024-3205(02)01949-5
Lillico, R., Zhou, T., Khorshid Ahmad, T., Stesco, N., Gozda, K., Truong, J., et al. (2018). Increased Post-Translational Lysine Acetylation of Myelin Basic Protein Is Associated with Peak Neurological Disability in a Mouse Experimental Autoimmune Encephalomyelitis Model of Multiple Sclerosis. J. Proteome Res. 17 (1), 55–62. doi: 10.1021/acs.jproteome.7b00270
Liscum, L., Klansek, J. J. (1998). Niemann–Pick disease type C. Curr. Opin. Lipidol. 9 (2), 131–135. doi: 10.1097/00041433-199804000-00009
Liu, H., Wu, H., Wang, Y., Wang, Y., Wu, X., Ju, S., et al. (2012). Inhibition of class II histone deacetylase blocks proliferation and promotes neuronal differentiation of the embryonic rat neural progenitor cells. Acta Neurobiol. Exp. (Wars) 72 (4), 365–376.
Liu, D., Liu, C., Li, J., Azadzoi, K., Yang, Y., Fei, Z., et al. (2013). Proteomic analysis reveals differentially regulated protein acetylation in human amyotrophic lateral sclerosis spinal cord. PloS One 8 (12), e80779–e80779. doi: 10.1371/journal.pone.0080779
Longo, F. M., Massa, S. M. (2013). Small-molecule modulation of neurotrophin receptors: a strategy for the treatment of neurological disease. Nat. Rev. Drug Discovery 12, 507. doi: 10.1038/nrd4024 . https://www.nature.com/articles/nrd4024#supplementary-information.
Lu, J., Frerich, J. M., Turtzo, L. C., Li, S., Chiang, J., Yang, C., et al. (2013). Histone deacetylase inhibitors are neuroprotective and preserve NGF-mediated cell survival following traumatic brain injury. Proc. Natl. Acad. Sci. 110 (26), 10747–10752. doi: 10.1073/pnas.1308950110
Luca, A., Calandra, C., Luca, M. (2018). Molecular Bases of Alzheimer's Disease and Neurodegeneration: The Role of Neuroglia. Aging Dis. 9 (6), 1134–1152. doi: 10.14336/AD.2018.0201
Luxton, G. W., Gundersen, G. G. (2007). HDAC6-pack: cortactin acetylation joins the brew. Dev. Cell 13 (2), 161–162. doi: 10.1016/j.devcel.2007.07.014
MacDonald, J. L., Roskams, A. J. (2008). Histone deacetylases 1 and 2 are expressed at distinct stages of neuro-glial development. Dev. Dyn. 237 (8), 2256–2267. doi: 10.1002/dvdy.21626
Mahady, L., Nadeem, M., Malek-Ahmadi, M., Chen, K., Perez, S. E., Mufson, E. J. (2018). HDAC2 dysregulation in the nucleus basalis of Meynert during the progression of Alzheimer's disease. Neuropathol. Appl. Neurobiol. 0 (0), 380–397. doi: 10.1111/nan.12518
Maolanon, A. R., Madsen, A. S., Olsen, C. A. (2016). Innovative Strategies for Selective Inhibition of Histone Deacetylases. Cell Chem. Biol. 23 (7), 759–768. doi: 10.1016/j.chembiol.2016.06.011
Maretina, M. A., Zheleznyakova, G. Y., Lanko, K. M., Egorova, A. A., Baranov, V. S., Kiselev, A. V. (2018). Molecular Factors Involved in Spinal Muscular Atrophy Pathways as Possible Disease-modifying Candidates. Curr. Genomics 19 (5), 339–355. doi: 10.2174/1389202919666180101154916
Marin-Husstege, M., Muggironi, M., Liu, A., Casaccia-Bonnefil, P. (2002). Histone Deacetylase Activity Is Necessary for Oligodendrocyte Lineage Progression. J. Neurosci. 22 (23), 10333. doi: 10.1523/JNEUROSCI.22-23-10333.2002
Marin-Husstege, M., Muggironi, M., Raban, D., Skoff, R. P., Casaccia-Bonnefil, P. (2005). Oligodendrocyte Progenitor Proliferation and Maturation Is Differentially Regulated by Male and Female Sex Steroid Hormones. Dev. Neurosci. 26 (2-4), 245–254. doi: 10.1159/000082141
Marks, P., Xu, W.-S. (2009). Histone deacetylase inhibitors: Potential in cancer therapy. J. Cell. Biochem 107, 600–608. doi: 10.1002/jcb.2218
Marks, P. A. (2010). The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin. Investigational Drugs 19 (9), 1049–1066. doi: 10.1517/13543784.2010.510514
Maruoka, H., Sasaya, H., Sugihara, K., Shimoke, K., Ikeuchi, T. (2011). Low-molecular-weight compounds having neurotrophic activity in cultured PC12 cells and neurons. J. Biochem. 150 (5), 473–475. doi: 10.1093/jb/mvr113
McFarland, K. N., Das, S., Sun, T. T., Leyfer, D., Xia, E., Sangrey, G. R., et al. (2012). Genome-wide histone acetylation is altered in a transgenic mouse model of Huntington's disease. PloS One 7 (7), e41423–e41423. doi: 10.1371/journal.pone.0041423
Michan, S., Sinclair, D. (2007). Sirtuins in mammals: insights into their biological function. Biochem. J. 404 (1), 1–13. doi: 10.1042/bj20070140
Minamiyama, M., Inukai, A., Sang, C., Tanaka, F., Sobue, G., Adachi, H., et al. (2004). Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Hum. Mol. Genet. 13 (11), 1183–1192. doi: 10.1093/hmg/ddh131
Mohseni, J., Zabidi-Hussin, Z. A. M. H., Sasongko, T. H. (2013). Histone deacetylase inhibitors as potential treatment for spinal muscular atrophy. Genet. Mol. Biol. 36 (3), 299–307. doi: 10.1590/S1415-47572013000300001
Nör, C., Sassi, F. A., de Farias, C. B., Schwartsmann, G., Abujamra, A. L., Lenz, G., et al. (2013). The Histone Deacetylase Inhibitor Sodium Butyrate Promotes Cell Death and Differentiation and Reduces Neurosphere Formation in Human Medulloblastoma Cells. Mol. Neurobiol. 48 (3), 533–543. doi: 10.1007/s12035-013-8441-7
Neuwald, A. (1997). GCN5-related histone N-acetyltransferases belong to a diverse superfamily that includes the yeast SPT10 protein. Trends Biochem. Sci. 22 (5), 154–155. doi: 10.1016/s0968-0004(97)01034-7
Noh, H., Seo, H. (2014). Age-dependent effects of valproic acid in Alzheimer's disease (AD) mice are associated with nerve growth factor (NGF) regulation. Neuroscience 266, 255–265. doi: 10.1016/j.neuroscience.2014.02.012
Obianyo, O., Ye, K. (2013). Novel small molecule activators of the Trk family of receptor tyrosine kinases. Biochim. Biophys. Acta 1834 (10), 2213–2218. doi: 10.1016/j.bbapap.2012.08.021
Pagliarini, V., Guerra, M., Di Rosa, V., Compagnucci, C., Sette, C. (2019). Combined treatment with the histone deacetylase inhibitor LBH589 and a splice-switch antisense oligonucleotide enhances SMN2 splicing and SMN expression in Spinal Muscular Atrophy cells. J. Neurochem., e14935. doi: 10.1111/jnc.14935
Pallos, J., Bodai, L., Lukacsovich, T., Purcell, J. M., Steffan, J. S., Thompson, L. M., et al. (2008). Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington’s disease. Hum. Mol. Genet. 17 (23), 3767–3775. doi: 10.1093/hmg/ddn273
Pandey, U. B., Nie, Z., Batlevi, Y., McCray, B. A., Ritson, G. P., Nedelsky, N. B., et al. (2007). HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447 (7146), 859–863. doi: 10.1038/nature05853
Parthun, M. R. (2007). Hat1: the emerging cellular roles of a type B histone acetyltransferase. Oncogene 26 (37), 5319–5328. doi: 10.1038/sj.onc.1210602
Peng, G.-S., Li, G., Tzeng, N.-S., Chen, P.-S., Chuang, D.-M., Hsu, Y.-D., et al. (2005). Valproate pretreatment protects dopaminergic neurons from LPS-induced neurotoxicity in rat primary midbrain cultures: role of microglia. Mol. Brain Res. 134 (1), 162–169. doi: 10.1016/j.molbrainres.2004.10.021
Pesavento, E., Capsoni, S., Domenici, L., Cattaneo, A. (2002). Acute cholinergic rescue of synaptic plasticity in the neurodegenerating cortex of anti-nerve-growth-factor mice. Eur. J. Neurosci. 15 (6), 1030–1036. doi: 10.1046/j.1460-9568.2002.01937.x
Petri, S., Kiaei, M., Kipiani, K., Chen, J., Calingasan, N. Y., Crow, J. P., et al. (2006). Additive neuroprotective effects of a histone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 22 (1), 40–49. doi: 10.1016/j.nbd.2005.09.013
Pigna, E., Simonazzi, E., Sanna, K., Bernadzki, K. M., Proszynski, T., Heil, C., et al. (2019). Histone deacetylase 4 protects from denervation and skeletal muscle atrophy in a murine model of amyotrophic lateral sclerosis. EBioMedicine 40, 717–732. doi: 10.1016/j.ebiom.2019.01.038
Pinho, B. R., Reis, S. D., Guedes-Dias, P., Leitão-Rocha, A., Quintas, C., Valentão, P., et al. (2016). Pharmacological modulation of HDAC1 and HDAC6 in vivo in a zebrafish model: Therapeutic implications for Parkinson's disease. Pharmacol. Res. 103, 328–339. doi: 10.1016/j.phrs.2015.11.024
Poplawski, S. G., Garbett, K. A., McMahan, R. L., Kordasiewicz, H. B., Zhao, H., Kennedy, A. J., et al. (2020). An Antisense Oligonucleotide Leads to Suppressed Transcription of Hdac2 and Long-Term Memory Enhancement. Mol. Ther. Nucleic Acids 19, 1399–1412. doi: 10.1016/j.omtn.2020.01.027
Qing, H., He, G., Ly, P. T. T., Fox, C. J., Staufenbiel, M., Cai, F., et al. (2008). Valproic acid inhibits Aβ production, neuritic plaque formation, and behavioral deficits in Alzheimer's disease mouse models. J. Exp. Med. 205 (12), 2781–2789. doi: 10.1084/jem.20081588
Rabal, O., Sánchez-Arias, J. A., Cuadrado-Tejedor, M., de Miguel, I., Pérez-González, M., García-Barroso, C., et al. (2016). Design, Synthesis, and Biological Evaluation of First-in-Class Dual Acting Histone Deacetylases (HDACs) and Phosphodiesterase 5 (PDE5) Inhibitors for the Treatment of Alzheimer's Disease. J. Med. Chem. 59 (19), 8967–9004. doi: 10.1021/acs.jmedchem.6b00908
Rabal, O., Sánchez-Arias, J. A., Cuadrado-Tejedor, M., de Miguel, I., Pérez-González, M., García-Barroso, C., et al. (2018). Discovery of in Vivo Chemical Probes for Treating Alzheimer's Disease: Dual Phosphodiesterase 5 (PDE5) and Class I Histone Deacetylase Selective Inhibitors. ACS Chem. Neurosci. 10, 1765–1782. doi: 10.1021/acschemneuro.8b00648
Rao, R., Fiskus, W., Ganguly, S., Kambhampati, S., Bhalla, K. N. (2012). HDAC inhibitors and chaperone function. Adv. Cancer Res. 116, 239–262. doi: 10.1016/b978-0-12-394387-3.00007-0
Ren, M., Leng, Y., Jeong, M., Leeds, P. R., Chuang, D. M. (2004). Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: potential roles of histone deacetylase inhibition and heat shock protein induction. J. Neurochem. 89 (6), 1358–1367. doi: 10.1111/j.1471-4159.2004.02406.x
Ricobaraza, A., Cuadrado-Tejedor, M., Pérez-Mediavilla, A., Frechilla, D., Del Río, J., García-Osta, A. (2009). Phenylbutyrate Ameliorates Cognitive Deficit and Reduces Tau Pathology in an Alzheimer's Disease Mouse Model. Neuropsychopharmacology 34 (7), 1721–1732. doi: 10.1038/npp.2008.229
Riessland, M., Brichta, L., Hahnen, E., Wirth, B. (2006). The benzamide M344, a novel histone deacetylase inhibitor, significantly increases SMN2 RNA/protein levels in spinal muscular atrophy cells. Hum. Genet. 120 (1), 101–110. doi: 10.1007/s00439-006-0186-1
Riessland, M., Ackermann, B., Forster, A., Jakubik, M., Hauke, J., Garbes, L., et al. (2010). SAHA ameliorates the SMA phenotype in two mouse models for spinal muscular atrophy. Hum. Mol. Genet. 19 (8), 1492–1506. doi: 10.1093/hmg/ddq023
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362 (6415), 59–62. doi: 10.1038/362059a0
Roth, S. Y., Denu, J. M., Allis, C. D. (2001). Histone Acetyltransferases. Annu. Rev. Biochem. 70 (1), 81–120. doi: 10.1146/annurev.biochem.70.1.81
Rouaux, C., Panteleeva, I., Rene, F., Gonzalez de Aguilar, J. L., Echaniz-Laguna, A., Dupuis, L., et al. (2007). Sodium Valproate Exerts Neuroprotective Effects In Vivo through CREB-Binding Protein-Dependent Mechanisms But Does Not Improve Survival in an Amyotrophic Lateral Sclerosis Mouse Model. J. Neurosci. 27 (21), 5535–5545. doi: 10.1523/jneurosci.1139-07.2007
Rousseaux, S., Khochbin, S. (2015). Histone Acylation beyond Acetylation: Terra Incognita in Chromatin Biology. Cell J. 17 (1), 1–6. doi: 10.22074/cellj.2015.506
Rowland, L. P., Shneider, N. A. (2001). Amyotrophic Lateral Sclerosis. New Engl. J. Med. 344 (22), 1688–1700. doi: 10.1056/nejm200105313442207
Ryu, H., Smith, K., Camelo, S. I., Carreras, I., Lee, J., Iglesias, A. H., et al. (2005). Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J. Neurochem. 93 (5), 1087–1098. doi: 10.1111/j.1471-4159.2005.03077.x
Sánchez-Arias, J. A., Rabal, O., Cuadrado-Tejedor, M., de Miguel, I., Pérez-González, M., Ugarte, A., et al. (2017). Impact of Scaffold Exploration on Novel Dual-Acting Histone Deacetylases and Phosphodiesterase 5 Inhibitors for the Treatment of Alzheimer's Disease. ACS Chem. Neurosci. 8 (3), 638–661. doi: 10.1021/acschemneuro.6b00370
Sadoul, K., Wang, J., Diagouraga, B., Khochbin, S. (2011). The Tale of Protein Lysine Acetylation in the Cytoplasm. J. Biomed. Biotechnol. 2011, 1–15. doi: 10.1155/2011/970382
Sadri-Vakili, G., Jang-Ho, J. C. (2006). Histone Deacetylase Inhibitors: A Novel Therapeutic Approach to Huntingtons Disease (Complex Mechanism of Neuronal Death). Curr. Alzheimer Res. 3 (4), 403–408. doi: 10.2174/156720506778249407
Sadri-Vakili, G., Bouzou, B., Benn, C. L., Kim, M. O., Chawla, P., Overland, R. P., et al. (2007). Histones associated with downregulated genes are hypo-acetylated in Huntington's disease models. Hum. Mol. Genet. 16 (11), 1293–1306. doi: 10.1093/hmg/ddm078
Saha, R. N., Pahan, K. (2005). HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ 13 (4), 539–550. doi: 10.1038/sj.cdd.4401769
Schroeder, F. A., Lin, C. L., Crusio, W. E., Akbarian, S. (2007). Antidepressant-Like Effects of the Histone Deacetylase Inhibitor, Sodium Butyrate, in the Mouse. Biol. Psychiatry 62 (1), 55–64. doi: 10.1016/j.biopsych.2006.06.036
Schwechter, B. R., Millet, L. E., Levin, L. A. (2007a). Histone deacetylase inhibition-mediated differentiation of RGC-5 cells and interaction with survival. Invest. Ophthalmol. Visual Sci. 48 (6), 2845–2857. doi: 10.1167/iovs.06-1364
Schwechter, B. R., Millet, L. E., Levin, L. A. (2007b). Histone Deacetylase Inhibition–Mediated Differentiation of RGC-5 Cells and Interaction with Survival. Invest. Opthalmol. Visual Sci. 48 (6), 2845. doi: 10.1167/iovs.06-1364
Seigneurin-Berny, D., Verdel, A., Curtet, S., Lemercier, C., Garin, J., Rousseaux, S., et al. (2001). Identification of components of the murine histone deacetylase 6 complex: link between acetylation and ubiquitination signaling pathways. Mol. Cell Biol. 21 (23), 8035–8044. doi: 10.1128/mcb.21.23.8035-8044.2001
SG, G., Dangond, F. (2006). Rationale for the Use of Histone Deacetylase Inhibitors as a Dual Therapeutic Modality in Multiple Sclerosis. Epigenetics 1 (2), 67–75. doi: 10.4161/epi.1.2.2678
Sharma, S., Taliyan, R. (2015a). Targeting histone deacetylases: a novel approach in Parkinson's disease. Parkinson's Dis. 2015, 303294–303294. doi: 10.1155/2015/303294
Sharma, S., Taliyan, R. (2015b). Transcriptional dysregulation in Huntington's disease: The role of histone deacetylases. Pharmacol. Res. 100, 157–169. doi: 10.1016/j.phrs.2015.08.002
Shukla, S., Shariat-Madar, Z., Walker, L. A., Tekwani, B. L. (2016). Mechanism for neurotropic action of vorinostat, a pan histone deacetylase inhibitor. Mol. Cell. Neurosci. 77, 11–20. doi: 10.1016/j.mcn.2016.09.003
Siebzehnrübl, F. A., Raber, K. A., Urbach, Y. K., Schulze-Krebs, A., Canneva, F., Moceri, S., et al. (2018). Early postnatal behavioral, cellular, and molecular changes in models of Huntington disease are reversible by HDAC inhibition. Proc. Natl. Acad. Sci. U. S. A. 115 (37), E8765–E8774. doi: 10.1073/pnas.1807962115
Siebzehnrubl, F. A., Buslei, R., Eyupoglu, I. Y., Seufert, S., Hahnen, E., Blumcke, I. (2007). Histone deacetylase inhibitors increase neuronal differentiation in adult forebrain precursor cells. Exp. Brain Res. 176 (4), 672–678. doi: 10.1007/s00221-006-0831-x
Steffan, J. S., Bodai, L., Pallos, J., Poelman, M., McCampbell, A., Apostol, B. L., et al. (2001). Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 413 (6857), 739–743. doi: 10.1038/35099568
Sterner, D. E., Berger, S. L. (2000). Acetylation of Histones and Transcription-Related Factors. Microbiol. Mol. Biol. Rev. 64 (2), 435–459. doi: 10.1128/mmbr.64.2.435-459.2000
Stertz, L., Fries, G. R., de Aguiar, B. W., Pfaffenseller, B., Valvassori, S. S., Gubert, C., et al. (2014). Histone deacetylase activity and brain-derived neurotrophic factor (BDNF) levels in a pharmacological model of mania. Rev. Bras. Psiquiatr. 36 (1), 39–46. doi: 10.1590/1516-4446-2013-1094
Su, M., Shi, J. J., Yang, Y. P., Li, J., Zhang, Y. L., Chen, J., et al. (2011). HDAC6 regulates aggresome-autophagy degradation pathway of alpha-synuclein in response to MPP+-induced stress. J. Neurochem. 117 (1), 112–120. doi: 10.1111/j.1471-4159.2011.07180.x
Sugai, F., Yamamoto, Y., Miyaguchi, K., Zhou, Z., Sumi, H., Hamasaki, T., et al. (2004). Benefit of valproic acid in suppressing disease progression of ALS model mice. Eur. J. Neurosci. 20 (11), 3179–3183. doi: 10.1111/j.1460-9568.2004.03765.x
Sumner, C. J., Huynh, T. N., Markowitz, J. A., Perhac, J. S., Hill, B., Coovert, D. D., et al. (2011). Valproic acid increases SMN levels in spinal muscular atrophy patient cells. Ann. Neurol. 54 (5), 647–654. doi: 10.1002/ana.10743
Sumner, C. J. (2006). Therapeutics development for spinal muscular atrophy. J. Am. Soc. Exp. Neurother. 3 (2), 235–245. doi: 10.1016/j.nurx.2006.01.010
Sun, G., Yu, R. T., Evans, R. M., Shi, Y. (2007). Orphan nuclear receptor TLX recruits histone deacetylases to repress transcription and regulate neural stem cell proliferation. Proc. Natl. Acad. Sci. 104 (39), 15282–15287. doi: 10.1073/pnas.0704089104
Sun, G., Fu, C., Shen, C., Shi, Y. (2011). Histone Deacetylases in Neural Stem Cells and Induced Pluripotent Stem Cells. J. Biomed. Biotechnol. 2011, 1–6. doi: 10.1155/2011/835968
Sun, L., Telles, E., Karl, M., Cheng, F., Luetteke, N., Sotomayor, E. M., et al. (2018). Loss of HDAC11 ameliorates clinical symptoms in a multiple sclerosis mouse model. Life Sci. Alliance 1 (5), e201800039–e201800039. doi: 10.26508/lsa.201800039
Suo, H., Wang, P., Tong, J., Cai, L., Liu, J., Huang, D., et al. (2015). NRSF is an essential mediator for the neuroprotection of trichostatin A in the MPTP mouse model of Parkinson's disease. Neuropharmacology 99, 67–78. doi: 10.1016/j.neuropharm.2015.07.015
Tagawa, K., Marubuchi, S., Qi, M. L., Enokido, Y., Tamura, T., Inagaki, R., et al. (2007). The induction levels of heat shock protein 70 differentiate the vulnerabilities to mutant huntingtin among neuronal subtypes. J. Neurosci. 27 (4), 868–880. doi: 10.1523/jneurosci.4522-06.2007
Tan, Y., Delvaux, E., Nolz, J., Coleman, P. D., Chen, S., Mastroeni, D. (2018). Upregulation of histone deacetylase 2 in laser capture nigral microglia in Parkinson's disease. Neurobiol. Aging 68, 134–141. doi: 10.1016/j.neurobiolaging.2018.02.018
Temple, S. (2001). Stem cell plasticity — building the brain of our dreams. Nat. Rev. Neurosci. 2 (7), 513–520. doi: 10.1038/35081577
Thomas, E. A., Coppola, G., Desplats, P. A., Tang, B., Soragni, E., Burnett, R., et al. (2008). The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington's disease transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 105 (40), 15564–15569. doi: 10.1073/pnas.0804249105
Thomson, J. A. (1998). Embryonic Stem Cell Lines Derived from Human Blastocysts. Science 282 (5391), 1145–1147. doi: 10.1126/science.282.5391.1145
Tomioka, T., Maruoka, H., Kawa, H., Yamazoe, R., Fujiki, D., Shimoke, K., et al. (2014). The histone deacetylase inhibitor trichostatin A induces neurite outgrowth in PC12 cells via the epigenetically regulated expression of the nur77 gene. Neurosci. Res. 88, 39–48. doi: 10.1016/j.neures.2014.07.009
Trapp, J., Meier, R., Hongwiset, D., Kassack, M. U., Sippl, W., Jung, M. (2007). Structure–Activity Studies on Suramin Analogues as Inhibitors of NAD+-Dependent Histone Deacetylases (Sirtuins). ChemMedChem 2 (10), 1419–1431. doi: 10.1002/cmdc.200700003
Tseng, H.-J., Lin, M.-H., Shiao, Y.-J., Yang, Y.-C., Chu, J.-C., Chen, C.-Y., et al. (2020). Synthesis and biological evaluation of acridine-based histone deacetylase inhibitors as multitarget agents against Alzheimer's disease. Eur. J. Med. Chem. 192, 112193. doi: 10.1016/j.ejmech.2020.112193
Tsilimigras, D. I., Ntanasis-Stathopoulos, I., Moris, D., Spartalis, E., Pawlik, T. M. (2018). Histone deacetylase inhibitors in hepatocellular carcinoma: A therapeutic perspective. Surg. Oncol. 27 (4), 611–618. doi: 10.1016/j.suronc.2018.07.015
Valenzuela-Fernandez, A., Cabrero, J. R., Serrador, J. M., Sanchez-Madrid, F. (2008). HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 18 (6), 291–297. doi: 10.1016/j.tcb.2008.04.003
Vaughan, E. E., Geiger, R. C., Miller, A. M., Loh-Marley, P. L., Suzuki, T., Miyata, N., et al. (2008). Microtubule Acetylation Through HDAC6 Inhibition Results in Increased Transfection Efficiency. Mol. Ther. 16 (11), 1841–1847. doi: 10.1038/mt.2008.190
Vecsey, C. G., Hawk, J. D., Lattal, K. M., Stein, J. M., Fabian, S. A., Attner, M. A., et al. (2007). Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J. Neurosci. 27 (23), 6128–6140. doi: 10.1523/jneurosci.0296-07.2007
Wang, Y., Wang, X., Liu, L., Wang, X. (2009). HDAC inhibitor trichostatin A-inhibited survival of dopaminergic neuronal cells. Neurosci. Lett. 467 (3), 212–216. doi: 10.1016/j.neulet.2009.10.037
Wang, Y., Wu, X., Zhong, Y., Shen, J., Wu, X., Ju, S., et al. (2014). Effects of histone deacetylase inhibition on the survival, proliferation and migration of Schwann cells, as well as on the expression of neurotrophic factors and genes associated with myelination. Int. J. Mol. Med. 34, 599–605. doi: 10.3892/ijmm.2014.1792
Wapenaar, H., Dekker, F. J. (2016). Histone acetyltransferases: challenges in targeting bi-substrate enzymes. Clin. Epigenet. 8, 59–59. doi: 10.1186/s13148-016-0225-2
Wiech, N., Fisher, J., Helquist, P., Wiest, O. (2009). Inhibition of Histone Deacetylases: A Pharmacological Approach to the Treatment of Non-Cancer Disorders. CTMC 9 (3), 257–271. doi: 10.2174/156802609788085241
Wright, D. J., Renoir, T., Gray, L. J., Hannan, A. J. (2017). ““Huntington's Disease: Pathogenic Mechanisms and Therapeutic Targets,”,” in Neurodegenerative Diseases: Pathology, Mechanisms, and Potential Therapeutic Targets. Eds. Beart, P., Robinson, M., Rattray, M., Maragakis, N. J. ((Cham: Springer International Publishing)), 93–128.
Wu, J. I., Lessard, J., Olave, I. A., Qiu, Z., Ghosh, A., Graef, I. A., et al. (2007). Regulation of Dendritic Development by Neuron-Specific Chromatin Remodeling Complexes. Neuron 56 (1), 94–108. doi: 10.1016/j.neuron.2007.08.021
Wu, X., Li, S., Wu, Q., Peng, Y., Yu, D., Wang, H., et al. (2013). Histone deacetylase inhibition leads to neuroprotection through regulation on glial function. Mol. Neurodegeneration 8 (Suppl. 1), P49. doi: 10.1186/1750-1326-8-s1-p49
Xu, K., Dai, X.-L., Huang, H.-C., Jiang, Z.-F. (2011). Targeting HDACs: A Promising Therapy for Alzheimer's Disease. Oxid. Med. Cell. Longevity 2011, 1–5. doi: 10.1155/2011/143269
Xu, A., He, F., Zhang, X., Li, X., Ran, Y., Wei, C., et al. (2020). Tacrine-hydroxamate derivatives as multitarget-directed ligands for the treatment of Alzheimer's disease: Design, synthesis, and biological evaluation. Bioorg. Chem. 98, 103721. doi: 10.1016/j.bioorg.2020.103721
Yakhine-Diop, S. M. S., Niso-Santano, M., Rodríguez-Arribas, M., Gómez-Sánchez, R., Martínez-Chacón, G., Uribe-Carretero, E., et al. (2018). Impaired Mitophagy and Protein Acetylation Levels in Fibroblasts from Parkinson's Disease Patients. Mol. Neurobiol. 56, 2466–2481. doi: 10.1007/s12035-018-1206-6
Yamaguchi, M. (2005). Histone deacetylase 1 regulates retinal neurogenesis in zebrafish by suppressing Wnt and Notch signaling pathways. Development 132 (13), 3027–3043. doi: 10.1242/dev.01881
Yang, X.-J., Seto, E. (2008). The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 9 (3), 206–218. doi: 10.1038/nrm2346
Yang, S.-S., Zhang, R., Wang, G., Zhang, Y.-F. (2017). The development prospection of HDAC inhibitors as a potential therapeutic direction in Alzheimer's disease. Trans. Neurodegeneration 6, 19–19. doi: 10.1186/s40035-017-0089-1
Yoo, Y.-E., Ko, C.-P. (2011). Treatment with trichostatin A initiated after disease onset delays disease progression and increases survival in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 231 (1), 147–159. doi: 10.1016/j.expneurol.2011.06.003
Yu, I. T., Park, J.-Y., Kim, S. H., Lee, J.-s., Kim, Y.-S., Son, H. (2009). Valproic acid promotes neuronal differentiation by induction of proneural factors in association with H4 acetylation. Neuropharmacology 56 (2), 473–480. doi: 10.1016/j.neuropharm.2008.09.019
Yuan, P. X. (2001). The Mood Stabilizer Valproic Acid Activates Mitogen-activated Protein Kinases and Promotes Neurite Growth. J. Biol. Chem. 276 (34), 31674–31683. doi: 10.1074/jbc.m104309200
Zhang, Z. Z., Gong, Y. Y., Shi, Y. H., Zhang, W., Qin, X. H., Wu, X. W. (2012). Valproate promotes survival of retinal ganglion cells in a rat model of optic nerve crush. Neuroscience 224, 282–293. doi: 10.1016/j.neuroscience.2012.07.056
Zhang, L., Liu, C., Wu, J., Tao, J.-J., Sui, X.-L., Yao, Z.-G., et al. (2014). Tubastatin A/ACY-1215 Improves Cognition in Alzheimer's Disease Transgenic Mice. J. Alzheimer's Dis. 41 (4), 1193–1205. doi: 10.3233/JAD-140066
Zhao, S., Xu, W., Jiang, W., Yu, W., Lin, Y., Zhang, T., et al. (2010). Regulation of Cellular Metabolism by Protein Lysine Acetylation. Science 327 (5968), 1000–1004. doi: 10.1126/science.1179689
Zheng, Z., Kim, J. Y., Ma, H., Lee, J. E., Yenari, M. A. (2008). Anti-Inflammatory Effects of the 70 kDa Heat Shock Protein in Experimental Stroke. J. Cereb. Blood Flow Metab. 28 (1), 53–63. doi: 10.1038/sj.jcbfm.9600502
Zhou, Q., Dalgard, C. L., Wynder, C., Doughty, M. L. (2011). Histone deacetylase inhibitors SAHA and sodium butyrate block G1-to-S cell cycle progression in neurosphere formation by adult subventricular cells. BMC Neurosci. 12 (1), 50. doi: 10.1186/1471-2202-12-50
Zhu, X., Wang, S., Yu, L., Jin, J., Ye, X., Liu, Y., et al. (2017). HDAC3 negatively regulates spatial memory in a mouse model of Alzheimer's disease. Aging Cell 16 (5), 1073–1082. doi: 10.1111/acel.12642
Ziemka-Nalecz, M., Jaworska, J., Sypecka, J., Zalewska, T. (2018). Histone Deacetylase Inhibitors: A Therapeutic Key in Neurological Disorders? J. Neuropathol. Exp. Neurol. 77 (10), 855–870. doi: 10.1093/jnen/nly073
Keywords: histone deacetylase, neurodegenerative disease, histone deacetylases inhibitors, neuritogenesis, neuronal differentiation, neuroprotection
Citation: Shukla S and Tekwani BL (2020) Histone Deacetylases Inhibitors in Neurodegenerative Diseases, Neuroprotection and Neuronal Differentiation. Front. Pharmacol. 11:537. doi: 10.3389/fphar.2020.00537
Received: 27 March 2019; Accepted: 06 April 2020;
Published: 24 April 2020.
Edited by:
Luis F. Callado, University of the Basque Country, SpainReviewed by:
Paul Dent, Virginia Commonwealth University, United StatesCopyright © 2020 Shukla and Tekwani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Surabhi Shukla, c3NodWtsYUB1bGFya2luLm9yZw==; Babu L. Tekwani, YnRla3dhbmlAc291dGhlcm5yZXNlYXJjaC5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.