 Matthew L. M. Jones1,2,3
Matthew L. M. Jones1,2,3 Gulcan Sarila1
Gulcan Sarila1 Pierre Chapuis2
Pierre Chapuis2 John M. Hutson1,4,5Sebastian K. King1,2,3,4
John M. Hutson1,4,5Sebastian K. King1,2,3,4 Warwick J. Teague1,2,3,4*
Warwick J. Teague1,2,3,4*- 1F. Douglas Stephens Surgical Research Laboratory, Murdoch Children’s Research Institute, Melbourne, VIC, Australia
- 2Discipline of Surgery, Sydney Medical School, The University of Sydney, Sydney, NSW, Australia
- 3Department of Paediatric Surgery, The Royal Children’s Hospital, Melbourne, VIC, Australia
- 4Department of Paediatrics, The University of Melbourne, Melbourne, VIC, Australia
- 5Department of Urology, The Royal Children’s Hospital Melbourne, Melbourne, VIC, Australia
Introduction: Duodenal atresia (DA) is a congenital bowel obstruction requiring major surgery in the first week of life. Three morphological phenotypes are described, reflecting increasing degrees of obstruction and discontinuity of the duodenum. The cause of DA is not known. Tandler’s original “solid cord” hypothesis conflicts with recent biological evidence, and is unable to account for differing DA types. In humans, a genetic etiology is supported by the association between Trisomy 21 and DA, and reports of familial inheritance patterns. Interruption of FGF10/FGFR2b signaling is the best demonstrated genetic link to DA in mice, with 35–75% of homozygous knockout embryos developing DA.
Purpose: This review examines the current evidence surrounding the etiology of DA. We focus on research regarding FGF10/FGFR2b signaling and its role in duodenal and other intestinal atresia. Further, we outline planned future research in this area, that we consider necessary to validate and better understand this murine model in order to successfully translate this research into clinical practice.
Conclusion: Determining the etiology of DA in humans is a clinical and scientific imperative. Fgf10/Fgfr2b murine models represent current science’s best key to unlocking this mystery. However, further research is required to understand the complex role of FGF10/FGFR2b signaling in DA development. Such complexity is expected, given the lethality of their associated defects makes ubiquitous interruption of either Fgf10 or Fgfr2b genes an unlikely cause of DA in humans. Rather, local or tissue-specific mutation in Fgf10, Fgfr2b, or their downstream targets, is the hypothesized basis of DA etiology.
Introduction
Duodenal atresia (DA) is a congenital foregut malformation affecting 1 in 5,000–10,000 live births (Escobar et al., 2004). Three morphological types of DA are described, reflecting increasing degrees of obstruction and discontinuity; type 1: bowel continuity but luminal obstruction or stenosis, type 2: bowel discontinuity with a connecting “bridge” of tissue, and, type 3: bowel discontinuity with complete separation (Skandalakis and Gray, 1994; Figures 1A–D). Duodenal atresia was first described in humans by Calder (1733), but it was not until the early twentieth century that Vidal and Ernst performed the first reported successful surgical repair (Vidal, 1905; Ernst, 1916). Nowadays, DA is routinely surgically repaired in the first week of life, with re-establishment of duodenal continuity and minimal postoperative morbidity or mortality (Zani et al., 2017).
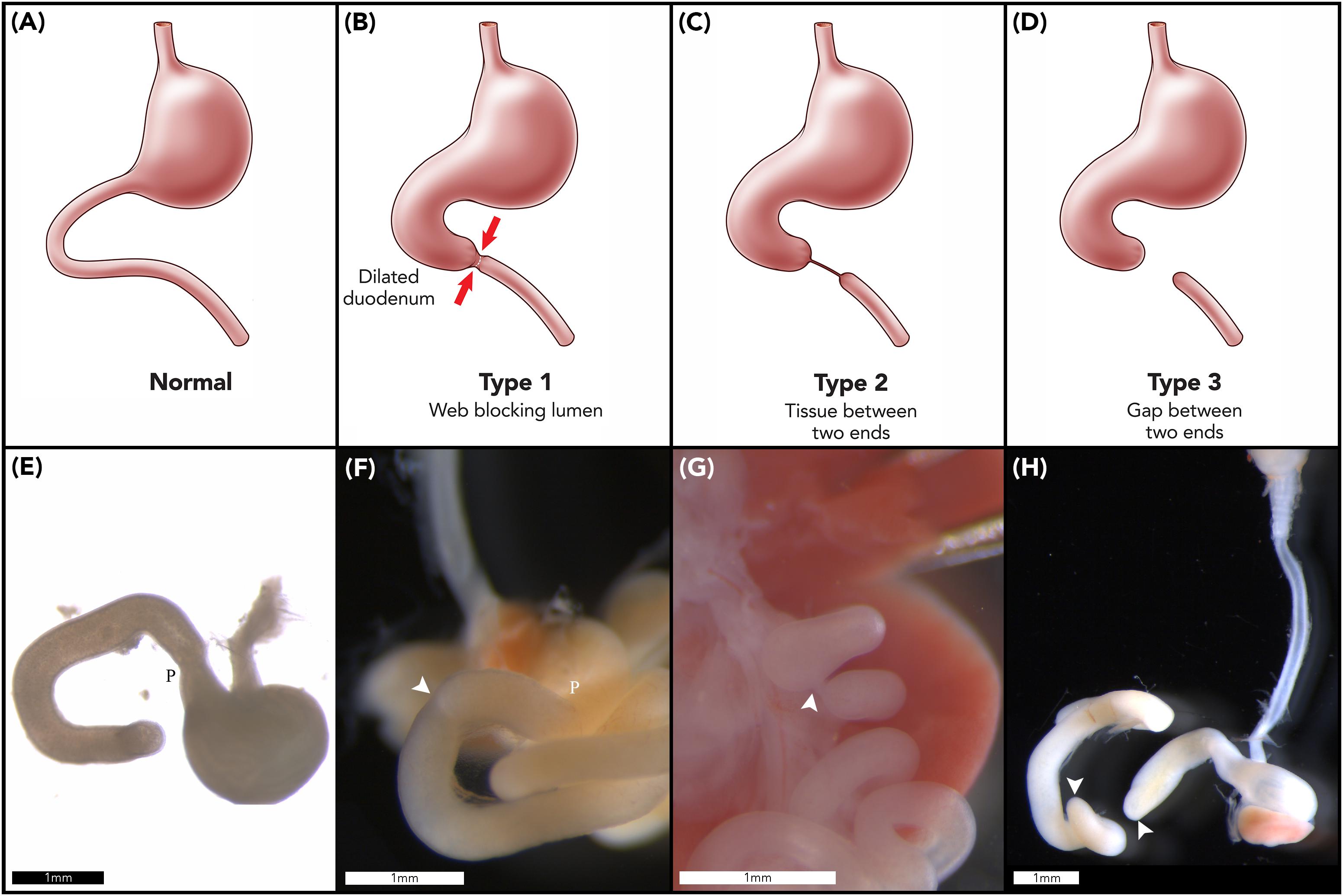
Figure 1. Duodenal atresia phenotype characterization. Normal gastric, pyloric, and duodenal morphology demonstrated pictorially (A) and as seen in wildtype murine embryos (E). Type 1 DA is characterized by bowel continuity with luminal obstruction or stenosis (B). Type 2 DA demonstrates bowel discontinuity with a connecting “bridge” of tissue (C). Type 3 DA shows bowel discontinuity with complete separation (D). Our CRISPR-derived Fgf10 knockout embryos provided examples of type 1 DA (F); type 2 DA (G); and type 3 DA (H) including demonstration of an intact esophagus, in the presence of tracheal atresia (H). Annotations denote scale bars, and location of the pylorus, P. Arrows indicate location of an atresia. Figures (E–H) reused from Teague et al. (2018), held under the CC-BY 4.0 license.
The cause of DA is not known. In 1900, Julius Tandler published a hypothesis on the origins of DA, based on his studies of normal duodenal development (Tandler, 1900). Tandler meticulously dissected 11 human embryos and theorized, on the basis of his macroscopic observations, that normal duodenal development includes a “solid cord” phase due to exuberant endodermal growth. As such, he proposed that failure of this “solid cord” to re-canalize may result in DA in humans. Even then, Tandler cautioned readers, stating: “It is clear to me that the opinion represented here does not exceed the status of a new hypothesis, and it is not meant to exceed this.” (Nichol et al., 2011). Notwithstanding, Tandler’s “solid cord” hypothesis is generally accepted worldwide by the majority of pediatric surgeons. This is despite conflicting developmental biology findings (Cheng and Tam, 1998; Botham et al., 2012), and failure of Tandler’s theory to account for the morphological variations of DA (Merrot et al., 2006).
As will be discussed in detail in this review, murine studies have shown that interruption of the Fgf10-Fgfr2b signaling axis may result in DA in 35–75% of knockout mice (Fairbanks et al., 2004b; Kanard et al., 2005; Botham et al., 2012; Reeder et al., 2012b; Teague et al., 2018). This also supports a genetic etiology for DA in humans, however, no specific genetic cause has been demonstrated to date. In humans, the association between DA and Trisomy 21 is well recognized, with DA occurring in 20% of cases (Khan et al., 2017). Yet, animal models of Trisomy 21 fail to demonstrate DA or other associated gastrointestinal malformations (Delabar et al., 2006). Again in humans, an autosomal recessive inheritance pattern for familial cases has been proposed, albeit based on a limited number of isolated and historical case reports (Fonkalsrud et al., 1969; Berant and Kahana, 1970; Best et al., 1989; Gross et al., 1996; Lambrecht and Kluth, 1998). Finally, whilst associations between DA and specific chromosomal anomalies have been reported (Mora et al., 2014; Zamfir et al., 2016), none relate to Chromosome 21, or to Chromosome 5 which houses the FGF10 gene in humans.
FGF Signaling in Gastrointestinal Development
The mammalian fibroblast growth factors (FGF) family contains 22 genes, 18 of which have been shown to bind and activate tyrosine kinase FGF receptors (FGFR), using either heparin-like molecules or klotho cofactors receptor binding (Ornitz and Itoh, 2015). These 18 FGFs have been grouped into six sub-families based on biochemical function, sequence homology and evolutionary traits (Ornitz and Itoh, 2001). The FGF7 sub-family is comprised of FGF3, FGF7, FGF10, and FGF22, and preferentially binds and activates FGFR2b (the IIIb splice variant of the FGFR2 receptor). This receptor is expressed in the epithelial layer and can be activated by FGF7 and FGF10 ligands (Ibrahimi et al., 2001). These ligands show no binding activity with the mesenchyme-expressed FGFR2c variant (Igarashi et al., 1998; Zhang et al., 2006). Conversely, FGF8 ligand preferentially binds and activates FGFR2c, with no binding activity toward FGFR2b (Macarthur et al., 1995; Ornitz et al., 1996).
The activation of FGFR2b by FGF10 is associated with mesenchymal/epithelial interactions which instruct branching and budding morphogenesis. This is a key signaling pathway for normal development of the gastrointestinal tract [e.g., stomach (Spencer-Dene et al., 2006), duodenum (Kanard et al., 2005), and large intestine (Burns et al., 2004)] and its derivatives [e.g., lung (Park et al., 1998)]. Indeed, gastrointestinal development involves multiple signaling pathways and their associated transcription factors, including FGF10-FGFR2b, which are summarized in Figure 2A and reviewed elsewhere (Kim and Shivdasani, 2016; Danopoulos et al., 2017).
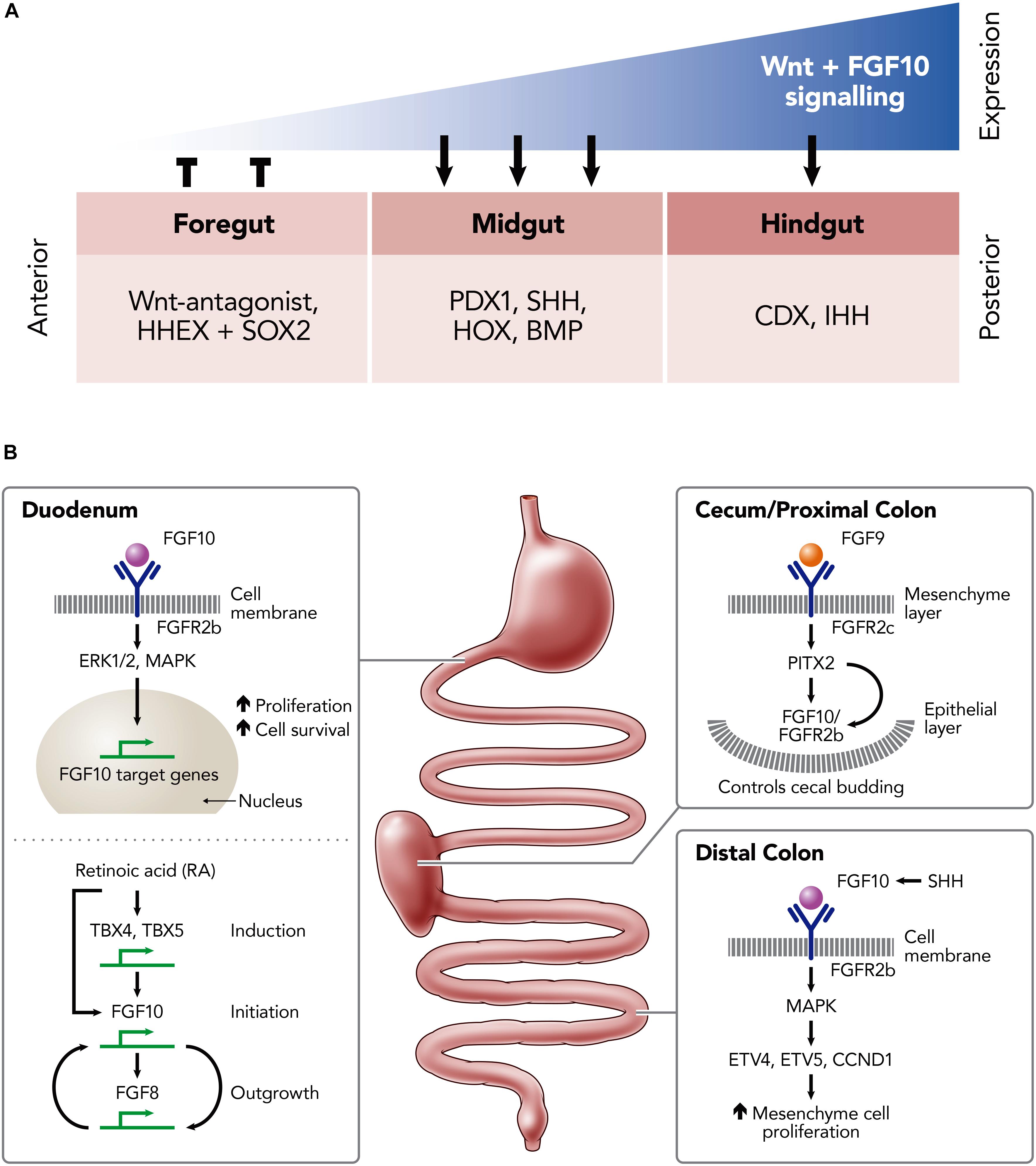
Figure 2. Schematic illustrations of the role of FGF10 in the developing and developed murine intestine. (A) The expression-dependent changes of key transcription factors in the foregut, midgut and hindgut mediated by differing levels of FGF10 and Wnt-signaling expression; hematopoietically expressed homeobox protein (HHEX), SRY (sex determining region Y)-box 2 (SOX2) in the foregut; pancreatic and duodenal homeobox 1 (PDX1), Sonic Hedgehog (SHH), homeobox genes (HOX), Bone morphogenetic proteins (BMP) in the midgut; Caudal-Type Homeobox Protein (CDX), Indian hedgehog (IHH) in the hindgut. (B) Key FGF10-pathways involved the development of the duodenum, cecum/proximal colon and distal colon; Fibroblast growth factor (FGF) 8, 9, and 10; Tyrosine kinase fibroblast growth factor receptors (FGFR) 2b, 2c; extracellular signal-regulated kinases (ERK1/2); mitogen-activated protein kinase (MAPK); Thromboxanes B4 (TXB4); Thromboxanes B5 (TBX5); ETS translocation variant 4 (ETV4); ETS translocation variant 5 (ETV5); Cyclin D1 (CCND1); Sonic Hedgehog (SHH).
FGF10-FGFR2b binding is implicated in the activation of multiple intracellular signaling pathways, including the ERK1/2, MAP kinase (MAPK) pathway, to induce downstream activation of proliferation and cell survival genes (Luo et al., 2008; Yang et al., 2011; Figure 2B). TBX4/TBX5 may induce Fgf10 expression to promote region-specific stimulation of the mesenchymal proliferation at the initial sites of foregut growth. This growth depends on the regulation of the HOX and TBX genes, which are mediated by retinoic acid (RA) (Wang et al., 2006). Potentially, RA acts on TBX genes, such as TBX4 and TBX5, as a co-factor to activate FGF10 transcription and, as a result, mediates the FGF10-FGF8 positive feedback loop which is required for outgrowth (Nishimoto et al., 2015). Embryos that are RA-deficient fail to outgrow due to defective FGF10 signaling, resulting in a lack of FGF-target genes in the epithelium. Interestingly, the development of the foregut may be partly restored in the defective embryos by introducing RA supplementation early on in development (between E7.5 and E8.5), suggesting that RA regulation is upstream of FGF10 (Figure 2B; Wang et al., 2006).
Within the cecum and proximal colon FGF9 binds to the mesenchyme-expressed FGFR2c receptor to direct cecal mesenchyme expansion via the expression of PITX2. In conjunction with this, FGF10 acts to trigger the budding of the epithelium into the cecal mesenchyme, again through mediation of PITX2 expression. Thus, the interaction between FGF10 and PITX2 is critical for the maintenance and the proliferation of the epithelial progenitor cells of the colon (Figure 2B). During embryonic development, FGF10 is also expressed in the mesenchyme of the distal colon, where it binds and activates FGFR1b and FGFR2b to promote proliferation, differentiation and survival of the epithelium. This activation maintains progenitor cells within the embryonic small intestine by targeting E26 transformation-specific (ETS)-Family genes such as ETV4, ETV5, and CCND1 through the MAPK pathway (Nyeng et al., 2011; Figure 2B).
The homozygous deletion of the Fgf10 or Fgfr2b gene results in mice with multiple organ defects, including lung, limb and mammary glands (with failure to form buds 1, 2, 3, and 5). The resultant mice are non-viable after birth due to lung agenesis (De Moerlooze et al., 2000; Mailleux et al., 2002).
Fgf10/Fgfr2b Signaling in Duodenal Atresia
Fairbanks et al. (2004b) were the first to identify a link between the Fgf10/Fgfr2b signaling pathway and intestinal atresia in mice. Using Fgfr2b knockout mice, they observed DA in 35% of null embryos when examined at gestational stage E18.5. These authors reported examples of both type 1 DA (57%) and type 3 DA (43%), but no type 2 DA (Fairbanks et al., 2004b). The same group then turned their attention to interruption of the ligand instead of receptor, knocking out Fgf10 (Kanard et al., 2005). Interestingly, Fgf10 knockout embryos were found to have a similar penetrance of DA (38%) compared to Fgfr2b knockouts, but with notably different relative frequency of DA types; type 1 DA (8%), type 3 DA (92%), and still no type 2 DA (Kanard et al., 2005). Further emphasizing the importance of the Fgf10/Fgfr2b pathway in intestinal development, deletion of either Fgf10 or Fgfr2b also resulted in a more distally located intestinal atresia, i.e., colonic atresia (Fairbanks et al., 2004a). In contrast with the incomplete penetrance of DA, colonic atresia was present in 100% of Fgf10 and Fgfr2b knockout mice embryos (Fairbanks et al., 2004a).
Importantly, the morphology of the duodenum is normal in Fgf10 hypomorphic mice, with no examples of DA in this setting of Fgf10 under-expression (Teague et al., 2018). Thus, even reduced Fgf10 signaling is sufficient for morphologically normal duodenal development. Similarly, Sala et al. (2006) found normal colonic development in Fgf10 hypomorphic mice, whereas complete loss of Fgf10 gene function resulted in colonic atresia. In the colon, Fgf10 expression was shown to be expressed first distally before extending proximally, and was found to be critical for the maintenance and proliferation of the epithelial progenitor cells in the colon. Furthermore, crypt formation seen within the proximal colon in the absence of Fgf10 suggested other signaling pathways were active in the development of this region, reinforcing the early anatomical and functional regionalization of the GI tract early during development. Another important finding of Sala et al. (2006) was that atresia occured without and associated loss of mesenchyme. This finding makes vascular compromise an unlikely mechanism for colonic atresia in Fgf10 knockouts, as a vascular event would be expected to cause loss of both mesenchyme and epithelium.
The impact of Fgf10 over-expression has also been investigated in both ex vivo and in vivo models. Torashima et al. (2016) showed that the addition of Fgf10 to tissue-engineered small intestine (TESI) resulted in increased size and weight of the sample, and increased villi height and crypt depth. In this setting, epithelial differentiation was not inhibited by over-expression of Fgf10, a finding attributed to the lack of normal feedback inhibitory mechanisms in the ex vivo TESI samples. Nyeng et al. (2011) studied in vivo over-expression of Fgf10 within the developing duodenum. In these embryos, Fgf10 over-expression caused attenuation of cell differentiation and expansion of the progenitor niche along with upregulation of FGFR signaling targets known to exert feedback inhibition on the FGF pathway (e.g., Sprouty2, Etv4, and Etv5) (Minowada et al., 1999; Nyeng et al., 2011). Conversely, complete loss of Fgf10 led to the premature differentiation of all enteric cell lineages in the duodenum (Nyeng et al., 2011). These findings support the inference that Fgf10 is necessary to prevent premature differentiation of enteric cell lineages, and so maintain the epithelial progenitor cell population during development. In extension of this conclusion, these authors hypothesized that premature differentiation and resultant depletion of the progenitor cell population is responsible for the epithelial deficiencies of DA in Fgf10 knockout mice (Nyeng et al., 2011).
An alternative to the premature differentiation hypothesis, is that proposed by Botham et al. (2012). These investigators concluded that exaggerated endodermal apoptosis, rather than terminal differentiation of progenitor cells was the root cause for DA in Fgfr2b knockout mice. Apoptosis was seen in the endoderm at E10.5, disappearance of the endoderm in the atretic region by E11.5, followed by involution of the mesoderm in the absence of further apoptosis at E13.5. This timeline corresponded with first recognition of DA in these Fgfr2b knockout mice, also at E13.5. Comparisons were also made between the duodenum of Fgfr2b knockout mice and otherwise normal, human embryo samples. In mice, loss of endoderm is complete at an earlier stage of development (E11.5, Carnegie Stage 16). In humans, however, “epithelial crowding” with corresponding apparent loss of duodenal lumen is not seen until Carnegie Stage 17 with “recanalization” only at Carnegie Stage 18. Thus, there is a temporal dislocation between luminal occlusion in atretic murine and normal human duodenal development (Matsumoto et al., 2002; Botham et al., 2012). The timeline for murine DA is supported by the findings of Nichol et al. (2012), who showed murine duodenal narrowing evident from E11.5 and DA from E13.5.
Yet another hypothesis is that of Merei (2004), who proposed that intestinal atresia may be the result of aberrant notochord signaling. Reeder et al. (2012a) used Fgfr2b knockout mice, reporting 42% penetrance of DA and 100% penetrance of colonic atresia. These investigators refuted the role of aberrant notochord signaling as the basis for murine DA, showing neither disruption of sonic hedgehog (Shh) expression nor notochordal discontinuity in Fgfr2b knockouts. Furthermore, organ culture of explanted wildtype murine gut in the presence of supra-physiological concentrations of Shh failed to induce atresia in either the duodenum or colon.
One striking feature of the DA reported in the aforementioned, seminal studies is the morphological types of DA reported in Fgf10 and Fgfr2b knockout mice (Fairbanks et al., 2004b; Kanard et al., 2005; Botham et al., 2012; Reeder et al., 2012b). In humans, the distribution of DA types shows similar proportions of type 1 and type 3 DA, and few cases of type 2 DA; e.g., 54% type 1, 4% type 2, and 42% type 3 DA (Khan et al., 2017). In contrast to this, when collated together, 93% of all murine DA cases reported in these studies were examples of type 3 DA, i.e., the most severe type of DA with complete discontinuity of the duodenum (Figure 1D; Fairbanks et al., 2004b; Kanard et al., 2005; Botham et al., 2012; Reeder et al., 2012b). Also, none of these studies reported any examples of type 2 DA in mice.
Of relevance to DA type and the severity of malformation is the work by Reeder et al. (2012b). Reeder et al. (2012b) investigated a role for retinaldehyde dehydrogenase 2 (Raldh2), a gene which encodes the enzyme responsible for the final step of vitamin A conversion to RA. Raldh2 is critical for normal posterior foregut development (Wang et al., 2006), and is under-expressed in the duodenum of Fgfr2b knockout mice (Botham et al., 2012). Therefore, Reeder et al. combined heterozygous Raldh2 deletion with homozygous deletion of Fgfr2b with the hypothesis of generating a more severe DA phenotype than with homozygous deletion of Fgfr2b alone. Contrary to this hypothesis, addition of Raldh2 haploinsufficiency resulted in a reduced rather than increased penetrance of atresia (21 vs 45%), and a shift toward less severe atresia types: Fgfr2IIIb−/−; Raldh2± (80% type 1 DA, 20% type 2 DA, and no type 3) versus Fgfr2IIIb-/-; Raldh2+/+ (7% type 1 DA, 0% type 2 DA, and 93% type 3 DA). These authors suggested that signaling downstream of Fgf10/Fgfr2b, i.e., Raldh2, was responsible for the phenotypic variations in DA type morphology.
We have recently reported a more human-like distribution of DA types, including type 2 DA, in two strains of CRISPR-derived Fgf10 knockout mouse (Teague et al., 2018). Of note, these newly reported strains; tm1 (B6-Fgf10<c.[464_470dup;506_645del]APNMu>); and tm2 (B6-Fgf10<c.495_507delAPNMu>) (Eppig et al., 2006) each demonstrate all three DA types (Figures 1F–H), and a significantly higher penetrance of DA than previous animal models [74% (Teague et al., 2018) versus 35–45% (Fairbanks et al., 2004b; Kanard et al., 2005; Botham et al., 2012; Reeder et al., 2012b). The basis for such differences in type-distribution and penetrance when compared with previously reported Fgf10 knockout mouse strains remains unclear. The various strains are of consistent background, namely C57/Bl6 (Fairbanks et al., 2004b; Kanard et al., 2005), and whilst the genetics differ, each represents a nonsense mutation (Eppig et al., 2006). We hypothesize that signaling molecules and pathways downstream of Fgf10-Fgfr2b are moderating the morphology and penetrance of the DA phenotype in these various strains.
Our hypothesis is supported by the findings of Al Alam et al. (2012). While an FGF9/Pitx2/FGF10 signaling axis has been proposed within the lung (De Langhe et al., 2008), Al Alam et al. demonstrated that the same signaling axis is active in developing gut (Al Alam et al., 2012). Using tissue specific Fgf9 and Pitx2 conditional knockout they demonstrated that, within the cecum, FGF9 controls mesenchymal Pitx2 expression and, in turn, Fgf10 expression. They also found that Pitx2 induced the expression of Fgf10, even in the absence of Fgf9, suggesting that Pitx2 is downstream of Fgf9. In further attempts to define pathways upstream of Fgf10 involved in atresia, Reeder et al. investigated the role that Shh plays with Fgfr2b (Reeder et al., 2014). Using a Matrigel culture system and focusing on the colon of Fgfr2b knockout mice, they found a downregulation of Shh at E11.5 throughout the entire colon, and then loss of Foxf1 expression 12 h later in the mid-to-distal colon prior to its involution. They also found that the exogenous addition of Shh to the Fgfr2b deficient colon failed to prevent the formation of atresia, suggesting that the loss of Shh is not a critical event in atresia formation.
Forth-Coming Research Into Duodenal Atresia Etiology
Based on the currently available literature regarding Fgf10-Fgfr2b signaling in duodenal atresia, we hypothesize that the etiology of DA in humans is genetically driven, and that the causative genetic changes are downstream of Fgf10-Fgfr2b. This downstream locus may account for both the incomplete penetrance of DA in the murine model, and the different morphological types (i.e., severity) of DA. It may also explain why human DA patients lack the non-survivable associations of Fgf10 deletion such as pulmonary agenesis. Therefore, we plan to test this hypothesis by further investigating the up- and down-regulation of genes and pathways in atretic and morphologically normal duodenum. Coordinated RNA sequencing, validation, and protein interaction analyses are planned, focusing on the duodenum of CRISPR-derived Fgf10 knockout mice with and without duodenal atresia, as well as additional wildtype controls. We consider that all three of these mice populations are necessary to discern candidate genes and pathways responsible for DA within the murine model. Also, given the complete penetrance of colonic atresia in the setting of Fgf10-Fgfr2b signaling interruption, we plan additional experiments to delineate the up- and down-regulation of genes and pathways in atretic and morphologically normal colon. We consider the results of colonic analyses may aid discernment of the genetic changes which hold true relevance for atresia formation, amongst the high volume of genetic changes we expect to observe in this model. Furthermore, we plan to test the aforementioned dose-dependence hypothesis, by scrutinizing gene expression differences between the three morphological types of DA in our CRISPR-derived Fgf10 knockout mice.
In order to determine the translational relevance of the genetic analysis within our murine DA model, we plan to study the genetic profile of a thought-provoking subset of human DA patients with known associated anomalies akin to Fgf10-Fgfr2b signaling-related defects, e.g., craniofacial, limb, and lung anomalies. We consider this human DA subset to be at particular “risk” of demonstrating an underlying genetic basis for their DA related to the FGF10-FGFR2b signaling pathway. For this analysis, we will screen our institutional DA patient database for case with potential Fgf10-Fgfr2b signaling-related defects, and perform detailed clinical genetic phenotyping and exon sequencing of phenotypically homogenous patient groups. During the subsequent bioinformatic and genetic analyses, we will pay particular attention those genes and pathways evidenced to be associated with DA in our murine model.
Ultimate translation of these further studies will be to determine the cause of DA in humans, which will have immediate and therapeutic application in the clinical context of antenatal counseling. Also, these may provide the bases for future hypothesis-driven clinical targets and trials to ameliorate or prevent the development of DA in humans.
Summary
Interruption of FGF10-FGFR2b signaling is the best demonstrated genetic link to DA in mice. This corresponds in importance, and hypothesis-formulating relevance, to the association between DA and Trisomy 21 in humans. FGF10 haploinsufficiency and FGF10 mutations have been linked to human disease, principally to craniofacial syndromes (Entesarian et al., 2005; Milunsky et al., 2006; Rohmann et al., 2006), chronic respiratory disease (Klar et al., 2011), but no human gastrointestinal tract atresia cases to date (Tatekawa et al., 2007). In contrast to this narrative of haploinsufficiency, homozygous Fgf10 invalidation appears necessary for DA in our animal models (Teague et al., 2018), albeit accompanied by non-survivable anomalies such as lung agenesis. Therefore, we hypothesize that DA is caused by conditional tissue-specific deletion in Fgf10, Fgfr2b or their downstream targets (e.g., spatially limited to the duodenum or foregut). Constitutional deletion of this nature is less likely to explain DA in humans for reasons of non-survivable anomalies as detailed previously.
We consider determining the etiology of DA in humans to be a clinical and scientific imperative. Therefore, further study is required to understand the exact role of FGF10-FGFR2b signaling in this. Murine models of DA based on interruption of this signaling will be key to unlocking the mystery of DA in humans. Parallel genetic studies of human DA patients will enable translation of model-derived lessons from bench to bedside.
Author Contributions
MJ conceived the project, conducted the review of literature, analyzed the evidence, and wrote and finalized the manuscript. GS provided specific scientific input into the analysis of evidence, and assisted in writing and finalizing the manuscript. PC mentored MJ and assisted in finalization of the manuscript. JH mentored MJ, and assisted in the analysis of evidence and finalization of the manuscript. SK mentored MJ, and assisted in the review of literature, analysis of evidence, and finalization of the manuscript. WT mentored MJ, conceived the project, and assisted in the review of literature, analysis of evidence, and writing and finalization of the manuscript.
Funding
Our laboratory’s past and future work included in this review is supported by a Royal Australasian College of Surgeons (RACS) Foundation in Surgery Small Project Grant, a Colorectal Surgical Society of Australia and New Zealand (CSSANZ) Foundation Research Grant, and an Australian New Zealand Association of Paediatric Surgeons (ANZAPS) Douglas Stephens Research Grant. MJ is supported in his higher degree research through the John Brooke Moore Scholarship at the University of Sydney. In addition, WT and SK are both generously supported by The Royal Children’s Hospital Foundation, Melbourne.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge Mr. Bill Reid, The Royal Children’s Hospital Creative Studio, for his assistance in the art production of the figures for this review.
References
Al Alam, D., Sala, F. G., Baptista, S., Galzote, R., Danopoulos, S., Tiozzo, C., et al. (2012). FGF9-Pitx2-FGF10 signaling controls cecal formation in mice. Dev. Biol. 369, 340–348. doi: 10.1016/j.ydbio.2012.07.008
Berant, M., and Kahana, D. (1970). Familial duodenal atresia. Arch. Dis. Child. 45, 281–282. doi: 10.1136/adc.45.240.281
Best, L. G., Wiseman, N. E., and Chudley, A. E. (1989). Familial duodenal atresia: a report of two families and review. Am. J. Med. Genet. 34, 442–444. doi: 10.1002/ajmg.1320340322
Botham, R. A., Franco, M., Reeder, A. L., Lopukhin, A., Shiota, K., Yamada, S., et al. (2012). Formation of duodenal atresias in fibroblast growth factor receptor 2IIIb-/- mouse embryos occurs in the absence of an endodermal plug. J. Pediatr. Surg. 47, 1369–1379. doi: 10.1016/j.jpedsurg.2012.02.001
Burns, R. C., Fairbanks, T. J., Sala, F., De Langhe, S., Mailleux, A., Thiery, J. P., et al. (2004). Requirement for fibroblast growth factor 10 or fibroblast growth factor receptor 2-IIIb signaling for cecal development in mouse. Dev. Biol. 265, 61–74. doi: 10.1016/j.ydbio.2003.09.021
Calder, E. (1733). Two examples of children born with preternatural conformation of the guts. Med. Essays 1:203.
Cheng, W., and Tam, P. K. (1998). Murine duodenum does not go through a “solid core” stage in its embryological development. Eur. J. Pediatr. Surg. 8, 212–215. doi: 10.1055/s-2008-1071156
Danopoulos, S., Schlieve, C. R., Grikscheit, T. C., and Al Alam, D. (2017). Fibroblast growth factors in the gastrointestinal tract: twists and turns. Dev. Dyn. 246, 344–352. doi: 10.1002/dvdy.24491
De Langhe, S. P., Carraro, G., Tefft, D., Li, C., Xu, X., Chai, Y., et al. (2008). Formation and differentiation of multiple mesenchymal lineages during lung development is regulated by beta-catenin signaling. PLoS One 3:e1516. doi: 10.1371/journal.pone.0001516
De Moerlooze, L., Spencer-Dene, B., Revest, J. M., Hajihosseini, M., Rosewell, I., and Dickson, C. (2000). An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development 127, 483–492.
Delabar, J. M., Aflalo-Rattenbac, R., and Créau, N. (2006). Developmental defects in trisomy 21, and mouse models. Sci. World J. 6, 1945–1964. doi: 10.1100/tsw.2006.322
Entesarian, M., Matsson, H., Klar, J., Bergendal, B., Olson, L., Arakaki, R., et al. (2005). Mutations in the gene encoding fibroblast growth factor 10 are associated with aplasia of lacrimal and salivary glands. Nat. Genet. 37, 125–127.
Eppig, J. J., Fox, J., Barthold, S., Davvison, M., Newcomber, C., Quimby, F., et al. (2006). Mouse strain and genetic nomenclature: an abbreviated guide. Mouse Biomed. Res. 1, 79–98. doi: 10.1016/b978-012369454-6/50017-0
Ernst, N. P. (1916). A case of congenital atresia of the duodenum treated successfully by operation. Br. Med. J. 1, 644–645. doi: 10.1136/bmj.1.2888.644
Escobar, M. A., Ladd, A. P., Grosfeld, J. L., West, K. W., Rescorla, F. J., Scherer, L. R., et al. (2004). Duodenal atresia and stenosis: long-term follow-up over 30 years. J. Pediatr. Surg. 39, 867–871. discussion 867–871, doi: 10.1016/j.jpedsurg.2004.02.025
Fairbanks, T. J., Kanard, R., Del Moral, P. M., Sala, F. G., De Langhe, S., Warburton, D., et al. (2004b). Fibroblast growth factor receptor 2 IIIb invalidation–a potential cause of familial duodenal atresia. J. Pediatr. Surg. 39, 872–874. doi: 10.1016/j.jpedsurg.2004.02.026
Fairbanks, T. J., Kanard, R. C., De Langhe, S. P., Sala, F. G., Del Moral, P. M., Warburton, D., et al. (2004a). A genetic mechanism for cecal atresia: the role of the Fgf10 signaling pathway. J. Surg. Res. 120, 201–209. doi: 10.1016/j.jss.2003.12.017
Fonkalsrud, E. W., Delorimier, A. A., and Hays, D. M. (1969). Congenital atresia and stenosis of the duodenum. A review compiled from the members of the Surgical Section of the American Academy of Pediatrics. Pediatrics 43, 79–83.
Gross, E., Armon, Y., Abu-Dalu, K., Gale, R., and Schiller, M. (1996). Familial combined duodenal and jejunal atresia. J. Pediatr. Surg. 31:1573. doi: 10.1016/s0022-3468(96)90182-7
Ibrahimi, O. A., Eliseenkova, A. V., Plotnikov, A. N., Yu, K., Ornitz, D. M., and Mohammadi, M. (2001). Structural basis for fibroblast growth factor receptor 2 activation in Apert syndrome. Proc. Natl. Acad. Sci. U.S.A. 98, 7182–7187. doi: 10.1073/pnas.121183798
Igarashi, M., Finch, P. W., and Aaronson, S. A. (1998). Characterization of recombinant human fibroblast growth factor (FGF)-10 reveals functional similarities with keratinocyte growth factor (FGF-7). J. Biol. Chem. 273, 13230–13235. doi: 10.1074/jbc.273.21.13230
Kanard, R. C., Fairbanks, T. J., De Langhe, S. P., Sala, F. G., Del Moral, P. M., Lopez, C. A., et al. (2005). Fibroblast growth factor-10 serves a regulatory role in duodenal development. J. Pediatr. Surg. 40, 313–316. doi: 10.1016/j.jpedsurg.2004.10.057
Khan, A., Tanny, S. T., Perkins, E. J., Hunt, R. W., Hutson, J. M., King, S. K., et al. (2017). Is selective echocardiography in duodenal atresia the future standard of care? J. Pediatr. Surg. 52, 1952–1955. doi: 10.1016/j.jpedsurg.2017.08.046
Kim, T. H., and Shivdasani, R. A. (2016). Stomach development, stem cells and disease. Development 143, 554–565. doi: 10.1242/dev.124891
Klar, J., Blomstrand, P., Brunmark, C., Badhai, J., Hakansson, H. F., Brange, C. S., et al. (2011). Fibroblast growth factor 10 haploinsufficiency causes chronic obstructive pulmonary disease. J. Med. Genet. 48, 705–709. doi: 10.1136/jmedgenet-2011-100166
Lambrecht, W., and Kluth, D. (1998). Hereditary multiple atresias of the gastrointestinal tract: report of a case and review of the literature. J. Pediatr. Surg. 33, 794–797. doi: 10.1016/s0022-3468(98)90225-1
Luo, B., Cheung, H. W., Subramanian, A., Sharifnia, T., Okamoto, M., Yang, X., et al. (2008). Highly parallel identification of essential genes in cancer cells. Proc. Natl. Acad. Sci. U.S.A. 105, 20380–20385. doi: 10.1073/pnas.0810485105
Macarthur, C. A., Lawshe, A., Xu, J., Santos-Ocampo, S., Heikinheimo, M., Chellaiah, A. T., et al. (1995). FGF-8 isoforms activate receptor splice forms that are expressed in mesenchymal regions of mouse development. Development 121, 3603–3613.
Mailleux, A. A., Spencer-Dene, B., Dillon, C., Ndiaye, D., Savona-Baron, C., Itoh, N., et al. (2002). Role of FGF10/FGFR2b signaling during mammary gland development in the mouse embryo. Development 129, 53–60.
Matsumoto, A., Hashimoto, K., Yoshioka, T., and Otani, H. (2002). Occlusion and subsequent re-canalization in early duodenal development of human embryos: integrated organogenesis and histogenesis through a possible epithelial-mesenchymal interaction. Anat Embryol. 205, 53–65. doi: 10.1007/s00429-001-0226-5
Merei, J. M. (2004). Notochord-gut failure of detachment and intestinal atresia. Pediatr. Surg. Int. 20, 439–443.
Merrot, T., Anastasescu, R., Pankevych, T., Tercier, S., Garcia, S., Alessandrini, P., et al. (2006). Duodenal duplications. Clinical characteristics, embryological hypotheses, histological findings, treatment. Eur. J. Pediatr. Surg. 16, 18–23. doi: 10.1055/s-2006-923798
Milunsky, J. M., Zhao, G., Maher, T. A., Colby, R., and Everman, D. B. (2006). LADD syndrome is caused by FGF10 mutations. Clin. Genet. 69, 349–354. doi: 10.1111/j.1399-0004.2006.00597.x
Minowada, G., Jarvis, L. A., Chi, C. L., Neubuser, A., Sun, X., Hacohen, N., et al. (1999). Vertebrate Sprouty genes are induced by FGF signaling and can cause chondrodysplasia when overexpressed. Development 126, 4465–4475.
Mora, M. C., Volk, J., Cuevas-Ocampo, A. K., Wong, K. E., Rockwell, G., Tirabassi, M. V., et al. (2014). Martinez-Frias syndrome: evidence of linkage to RFX6 mutation. J. Pediatr. Surg. Case Rep. 2, 492–494. doi: 10.1016/j.epsc.2014.10.003
Nichol, P. F., Reeder, A., and Botham, R. (2011). Humans, mice, and mechanisms of intestinal atresias: a window into understanding early intestinal development. J. Gastrointest. Surg. 15, 694–700. doi: 10.1007/s11605-010-1400-y
Nichol, P. F., Tyrrell, J. D., and Saijoh, Y. (2012). Retinaldehyde dehydrogenase 2 is down-regulated during duodenal atresia formation in Fgfr2IIIb-/- mice. J. Surg. Res. 175, 82–87. doi: 10.1016/j.jss.2011.02.040
Nishimoto, S., Wilde, S. M., Wood, S., and Logan, M. P. (2015). RA Acts in a Coherent Feed-Forward Mechanism with Tbx5 to Control Limb Bud Induction and Initiation. Cell Rep. 12, 879–891. doi: 10.1016/j.celrep.2015.06.068
Nyeng, P., Bjerke, M. A., Norgaard, G. A., Qu, X., Kobberup, S., and Jensen, J. (2011). Fibroblast growth factor 10 represses premature cell differentiation during establishment of the intestinal progenitor niche. Dev. Biol. 349, 20–34. doi: 10.1016/j.ydbio.2010.09.010
Ornitz, D. M., and Itoh, N. (2015). the fibroblast growth factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 4, 215–266. doi: 10.1002/wdev.176
Ornitz, D. M., Xu, J., Colvin, J. S., Mcewen, D. G., Macarthur, C. A., Coulier, F., et al. (1996). Receptor specificity of the fibroblast growth factor family. J. Biol. Chem. 271, 15292–15297.
Ornitz, D. M., and Itoh, N. (2001). Fibroblast growth factors. Genome Biol. 2, reviews3005.1. doi: 10.1186/gb-2001-2-3-reviews3005
Park, W. Y., Miranda, B., Lebeche, D., Hashimoto, G., and Cardoso, W. V. (1998). FGF-10 is a chemotactic factor for distal epithelial buds during lung development. Dev. Biol. 201, 125–134. doi: 10.1006/dbio.1998.8994
Reeder, A. L., Botham, R. A., Franco, M., Zaremba, K. M., and Nichol, P. F. (2012a). Formation of intestinal atresias in the Fgfr2IIIb-/- mice is not associated with defects in notochord development or alterations in Shh expression. J. Surg. Res. 177, 139–145. doi: 10.1016/j.jss.2012.04.024
Reeder, A. L., Botham, R. A., Zaremba, K. M., and Nichol, P. F. (2012b). Haploinsufficiency of retinaldehyde dehydrogenase 2 decreases the severity and incidence of duodenal atresia in the fibroblast growth factor receptor 2IIIb-/- mouse model. Surgery 152, 768–775. doi: 10.1016/j.surg.2012.07.022
CrossRef Full Text discussion 775–776.,
Reeder, A. L., Zaremba, K. M., Liebl, R. M., Kowalkowski, A., and Nichol, P. F. (2014). Exogenous Sonic hedgehog protein does not rescue cultured intestine from atresia formation. J. Surg. Res. 187, 14–18. doi: 10.1016/j.jss.2013.11.1114
Rohmann, E., Brunner, H. G., Kayserili, H., Uyguner, O., Nurnberg, G., Lew, E. D., et al. (2006). Mutations in different components of FGF signaling in LADD syndrome. Nat. Genet. 38, 414–417. doi: 10.1038/ng1757
Sala, F. G., Curtis, J. L., Veltmaat, J. M., Del Moral, P. M., Le, L. T., Fairbanks, T. J., et al. (2006). Fibroblast growth factor 10 is required for survival and proliferation but not differentiation of intestinal epithelial progenitor cells during murine colon development. Dev. Biol. 299, 373–385. doi: 10.1016/j.ydbio.2006.08.001
Skandalakis, J. E., and Gray, S. W. (1994). Embryology for Surgeons: the Embryological Basis for the Treatment of Congenital Anomalies. Baltimore: Williams & Wilkins.
Spencer-Dene, B., Sala, F. G., Bellusci, S., Gschmeissner, S., Stamp, G., and Dickson, C. (2006). Stomach development is dependent on fibroblast growth factor 10/fibroblast growth factor receptor 2b-mediated signaling. Gastroenterology 130, 1233–1244. doi: 10.1053/j.gastro.2006.02.018
Tandler, J. (1900). Zur Entwicklingsgeschichte des menschlichen duodenums im frühen embryonalstadium. Morphol. Jahrb. 29, 187–216.
Tatekawa, Y., Kanehiro, H., and Nakajima, Y. (2007). Duodenal atresia associated with “apple peel” small bowel without deletion of fibroblast growth factor-10 or fibroblast growth factor receptor 2IIIb: report of a case. Surg. Today 37, 430–433. doi: 10.1007/s00595-006-3415-2
Teague, W. J., Jones, M. L. M., Hawkey, L., Smyth, I. M., Catubig, A., King, S. K., et al. (2018). FGF10 and the mystery of duodenal atresia in humans. Front. Genet. 9:530. doi: 10.3389/fgene.2018.00530
Torashima, Y., Levin, D. E., Barthel, E. R., Speer, A. L., Sala, F. G., Hou, X., et al. (2016). Fgf10 overexpression enhances the formation of tissue-engineered small intestine. J. Tissue Eng. Regen. Med. 10, 132–139. doi: 10.1002/term.1720
Vidal, E. (1905). 18e Congrès de Chirurgie. Paris: Procès verbaux mémoires et discussion, Ass Fr Chir.
Wang, Z., Dolle, P., Cardoso, W. V., and Niederreither, K. (2006). Retinoic acid regulates morphogenesis and patterning of posterior foregut derivatives. Dev. Biol. 297, 433–445. doi: 10.1016/j.ydbio.2006.05.019
Yang, Q. E., Giassetti, M. I., and Ealy, A. D. (2011). Fibroblast growth factors activate mitogen-activated protein kinase pathways to promote migration in ovine trophoblast cells. Reproduction 141, 707–714. doi: 10.1530/REP-10-0541
Zamfir, C., Dassonville, M., Rodesch, G., and Steyaert, H. (2016). A rare malformation: double duodenal atresia associated with malrotation in a patient with “Cri du Chat” syndrome. J. Pediatr. Surg. Case Rep. 12, 3–5. doi: 10.1016/j.epsc.2016.06.003
Zani, A., Yeh, J. B., King, S. K., Chiu, P. P., and Wales, P. W. (2017). Duodeno-duodenostomy or duodeno-jejunostomy for duodenal atresia: is one repair better than the other? Pediat. r Surg. Int. 33, 245–248. doi: 10.1007/s00383-016-4016-9
Keywords: duodenal obstruction, congenital intestinal atresia, fibroblast growth factor 10, fibroblast growth factor receptor 2b, morphogenesis
Citation: Jones MLM, Sarila G, Chapuis P, Hutson JM, King SK and Teague WJ (2020) The Role of Fibroblast Growth Factor 10 Signaling in Duodenal Atresia. Front. Pharmacol. 11:250. doi: 10.3389/fphar.2020.00250
Received: 22 November 2019; Accepted: 24 February 2020;
Published: 10 March 2020.
Edited by:
Saverio Bellusci, University of Giessen, GermanyReviewed by:
Stefano Rivetti, University of Giessen, GermanyDenise Al Alam, University of Southern California, United States
Copyright © 2020 Jones, Sarila, Chapuis, Hutson, King and Teague. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Warwick J. Teague, d2Fyd2ljay50ZWFndWVAcmNoLm9yZy5hdQ==