 Markus Herkt
Markus Herkt Ariana Foinquinos1
Ariana Foinquinos1 Sandor Batkai
Sandor Batkai Andreas Pich
Andreas Pich
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 25 March 2020
Sec. Experimental Pharmacology and Drug Discovery
Volume 11 - 2020 | https://doi.org/10.3389/fphar.2020.00220
Cardiac diseases are the most frequent causes of death in industrialized countries. Pathological remodeling of the heart muscle is caused by several etiologies such as prolonged hypertension or injuries that can lead to myocardial infarction and in serious cases also the death of the patient. The micro-RNA miR-132 has been identified as a master-switch in the development of cardiac hypertrophy and adverse remodeling. In this study, MALDI-TOF mass spectrometry (MS) was utilized to establish a robust and fast method to sensitively detect and accurately quantify anti-microRNA (antimiR) oligonucleotides in blood plasma. An antimiR oligonucleotide isolation protocol containing an ethanol precipitation step with glycogen as oligonucleotide carrier as well as a robust and reproducible MS-analysis procedure has been established. Proteinase K treatment was crucial for releasing antimiR oligonucleotides from plasma- as well as cellular proteins and reducing background derived from biological matrices. AntimiR oligonucleotide detection was achieved from samples of studies in different animal models such as mouse and pig where locked nucleic acids-(LNA)-modified antimiR oligonucleotides have been used to generate pharmacokinetic data.
MALDI-TOF mass spectrometry (MS) has been widely used for the analysis of many different molecules including oligonucleotides. Its advantages lie in efficient and fast sample preparation and precise analysis using highly resolving reflector TOF systems. Due to its negatively charged backbone, DNA is difficult to stabilize. Cobalt (III) hexamine and ammonium citrate dibasic have been utilized to stabilize DNA during desorption (Distler and Allison, 2002; Sudha and Zenobi, 2002). Natural and modified oligonucleotides were characterized by MALDI-MS and the use of 2,4,6-trihydroxy acetophenone (THAP) as ionization matrix. Di- and triammonium salts of organic and inorganic acids were utilized to remove various ion adducts and prevent peak widening (Pieles et al., 1993). An ionization matrix of 3,4-diaminoparabenzophenone (DABP) has been proposed to yield lower detection limits, less fragmentation and less ion adducts especially at low oligonucleotide concentrations. In addition, samples were highly homogenous resulting in excellent shot-to-shot reproducibility, resolution, and low background noise (Fu et al., 2006).
Extraction of analytes from an organic matrix is mandatory for analysis of oligonucleotides. One of the least complex methodologies is reducing the background noise by diluting samples. Several approaches use a proteinase K digestion or methanol precipitation to remove proteins from the biological samples with a recovery of oligonucleotides reaching 98% (Bellon et al., 2000; Waters et al., 2000; Arora et al., 2002). In addition, many approaches include a phenol/chloroform extraction (Griffey et al., 1997; Beverly et al., 2006). For oligonucleotide extraction, chemical extraction using denaturing agents followed by organic solvent precipitation and solid-phase extraction by immobilizing the RNAs on a glass support are two classical approaches (Beverly et al., 2006).
As for oligonucleotides, molecular masses can indicate modifications and composition. However, sequence informations are not available. This analysis can be performed by digestion of oligonucleotides with subsequent analysis of fragmentation products by MS analysis (Pomerantz and McCloskey, 2005). MS-based fragmentation techniques as for instance in-source decay (ICD) of digested oligonucleotides yield ions that are typical for base and sugar fragments of the nucleoside and allow further localization of the modification in the structure of the parent ion. With this nuclease digestion sequencing and partial sequencing of larger oligonucleotides can be accomplished. For this approach, aliquots from the digestion reaction are measured at specific time intervals. By using either a 3′ or a 5′ specific exonuclease, approximately 3–4 bases from each end can be determined (Hossain and Limbach, 2007).
The fragments arise from cleavages along the phosphodiester backbone resulting in a series of ions that can be used to determine the order within an oligonucleotide. Secondary fragmentation of large oligonucleotides resulting in more complex MS/MS-spectra or a different fragmentation behavior due to backbone modifications, complicates further analysis (Pomerantz and McCloskey, 1990; Bartlett et al., 1996). Derivatization is one way to enhance fragmentation behavior that occurs due to the intrinsic character of these sugar or nucleobase modifications (Emmerechts et al., 2005). The detection limit is still the most challenging issue for analyzing oligonucleotides. Detection limit for single stranded DNA with a phosphorothioate modification was described at 4 ng/mL (Zhang et al., 2007). A method for relative quantification of tRNAs was described where tRNAs were isotopically labeled resulting in quantification in the femtomole range (Castleberry and Limbach, 2010).
Sufficiently low detection limits are still the most challenging issues in oligonucleotide analysis besides efficient separation and ionization efficiency. Finding the fine balance between separations and obtaining high signal intensity requires further research. The analytical detection of oligonucleotides will be a major point to facilitate effective research and clinical applications of oligonucleotides.
Cardiac disorders are amongst the leading causes of morbidity and mortality worldwide. Adverse remodeling causes contractile dysfunction and the failing of the heart that will lead ultimately to the patient’s death (Hill and Olson, 2008). One of the central mechanism in the remodeling process is cardiomyocyte hypertrophy. At the molecular level, the FoxO transcription factor family suppresses calcineurin/NFAT signaling and thus functions as an anti-hypertrophic agent due to induction of autophagy and apoptosis in cardiomyocytes (Ni et al., 2006; Glas, 2010; Ronnebaum and Patterson, 2010). The miR-212/132 family was identified as a master-switch in the development of cardiac hypertrophy and adverse remodeling. MiR-132 is upregulated due to myocardial stress resulting in cardiac hypertrophy in vivo and pathological hypertrophy of cardiomyocytes in vitro (Ucar et al., 2012). Administration of miR-132 inhibitors (antimiRs) inhibited the development of cardiac hypertrophy mimicking the effects seen in miR212/132 KO mice (Ucar et al., 2012). Thus, miR-132 seems to be as a promising target in heart failure drug development.
The combination of MALDI and a time-of-flight (TOF) mass analyzer (MALDI-TOF) is widely used to analyze many different analytes, especially oligonucleotides. Therefore, the aim of this study was the development of a novel, fast, and reliable MALDI-MS-based technique for quantification of therapeutic oligonucleotides to enable pharmacokinetic studies of therapeutic oligonucleotides in mouse and pig.
All chemicals and reagents are listed and described below in alphabetical order (Table 1). For all substances the purity is at least p.a. or higher. Purity of LNA-modified oligonucleotides containing a phosphorothioate backbone was determined by EXIQON with HPLC-ESI-MS analysis (Table 2).

Table 1. Chemicals and reagents used in this study.

Table 2. Oligonucleotides used in this study.
In this study murine, as well as porcine EDTA-plasma was used. As an internal standard 1.5 μL of a 200 pmol/μL antimiR oligonucleotide Scr, or 132 solution were added to 28.5 μL of plasma to achieve a final concentration of 10 pmol/μL. For Proteinase K digestion 30 μL of plasma containing antimiR oligonucleotides were mixed with 40 μL proteinase K solution and 330 μL Proteinase K digestion buffer. The mixture was incubated at 55°C with 450 rpm on an Eppendorf Thermomixer comfort for 16 h overnight. Subsequently, antimiR oligonucleotides were precipitated by adding 100 μg of glycogen and 2 volumes ethanol followed by incubation for 4 h at −20°C. Then the samples were centrifuged at 21100 x g for 30 min and 4°C with a HERAEUS FRESCO 21 centrifuge. Supernatant was removed and pellets were dissolved in 30 μL ammonium citrate dibasic (AHC) solution at RT.
For MALDI-mass spectrometry (MALDI-MS) analysis a 5800 MALDI-TOF/TOF mass spectrometer (Sciex) was used. A special metal polish paste provided by Sciex successfully removed even the slightest impurities on the MALDI target plate so that a homogenous crystallization behavior of the analyte/matrix mix and thus a higher reproducibility of the analytical results was ensured. 5 μL of an antisense oligonucleotide (ASO) solution were mixed thoroughly with 5 μL of THAP matrix-solution in a 0.5 mL tube by vortexing with subsequent spotting of 0.5 μL of this solution per spot onto an MALDI target plate. This added a dilution factor of four on spot in terms of molarity. The Acquisition method was set up to the following parameters: Fixed Laser Intensity: 6100; Mass Range (Da): 4.000 to 6.000; Focus Mass: 5.000; Delay Times (ns): 600; X2 Deflector: 0.055; Shots/sub-spectrum: 200; Total Shots/Spectrum: 3000; Stage Velocity (μm/s): 600; Bin Size (ns): 1.0; Vertical Offset (% full scale): −0.5; Detector Voltage Multiplier: 0.67; Laser Pulse Rate (Hz): 400. The other parameters were set to standard. For analysis the TOF/TOFTM Series ExplorerTM Software Version 4.1.0 (build 12), Oracle Database Schema Version 4.0.0, Data Version 4.0.4 was used.
For quantification of oligonucleotides, peak areas were utilized. For each analysis, three technical replicates per sample were generated and the values were averaged with subsequent determination of standard deviation (SD) as well as the coefficient of variation (CV).
The limit of detection (LOD) and lower limit of quantification (LLOQ) of antimiR oligonucleotides in mice as well as pig plasma were determined by setting up several dilutions of antimiR132. In addition, antimiRScr was added to each sample at a final concentration of 10 pmol/μL as an internal standard. Afterward 5 μL of antimiR oligonucleotide solution were mixed thoroughly with 5 μL of THAP matrix-solution (Table 1) in a 0.5 mL tube by vortexing with subsequent spotting of 0.5 μL of this solution per spot on an MALDI target plate. MS analysis was performed as described above. Ratios obtained from antimiR132 and antimiRScr were inserted into the calibration curve equation (Figure 3), which was then resolved to x to obtain the amount of substance. Taking into account all dilution factors, the concentration of the substance was then calculated.
All experimental procedures involving mice were performed in accordance with the local and national regulations and approved by local animal welfare bodies of the Hannover Medical School, Germany or University of Kaposvar, Hungary. Male C57BL/6N Crl mice were administered once at the beginning of the experiment with 20 mg/kg antimiR132 intravenous (i.v.) and sacrificed after various time points to remove plasma and organs, namely heart and liver. The respective substance and dose combination was applied intravenously by administration into the lateral tail veins. For blood collection, the mouse was placed under anesthesia using isoflurane. The study plan is depicted in Table 3.

Table 3. Mouse PK study plan.
Additionally, heart and liver were taken 24 h, 72 h, and 1 weeks post application of antimiR132.
The tissue and plasma samples for this assay development study were derived from an animal study and used in all described experiments in different quantities and specification.
The study was conducted in female domestic pigs (Danish Landrace bread, 29 – 31 kg body weight), using three experimental groups. There was a treated group of three animals, which were injected with antimiR132. A placebo group (Placebo) of three animals was injected with 0.9% physiological saline. A control group of three animals received no treatment.
AntimiR132 and placebo was injected as a 20 mL solution via cardiac, intracoronary (i.c.) perfusion (80 mL/h for 15 ± 5 min) in three concentrations: 0.5 mg/kg (low dose), 1.17 mg/kg (medium dose), and 5 mg/kg (high dose). The treatment procedure was carried out on day 0 under general anesthesia following a coronary angiography. Blood samples were taken from both treated as well as untreated group. On day 1 organs and blood samples were taken from both groups before i.c. application (baseline) and at 15 min, 30 min, 1 h, 6 h, 12 h, and 24 h post treatment. After last blood sampling was completed, animals were euthanized and organs and blood samples were taken. Control animals were also euthanized and tissue and blood (only one) samples collected. Blood samples were collected in EDTA and serum tubes and processed within 1 h post collection. The tissue samples were collected at study end-point, cut into pieces and snap frozen on dry ice. Tissue and aliquots of blood samples were stored at −80°C until analysis. The study plan is depicted in Table 4.

Table 4. Pig PK study plan.
The protein concentration of mouse plasma was determined by Bradford protein assay. Plasma or tissue extracts were diluted with Millipore water if necessary and 5 μL were mixed with 200 μL of Bradford reagent (Table 1) and 800 μL of Millipore water with subsequent incubation for 5 min at RT. Afterward absorbance levels were determined at λ = 595 nm in triplicates. Protein concentration was then calculated for the obtained absorbance levels based on the calibration curve done with bovine serum albumin (Supplementary Figure 1).
Protein samples (50 – 100 μg) were mixed with 0.25 volumes of Laemmli buffer (Table 1) and incubated for 10 min at 95°C. If necessary, proteins were alkylated by addition of 1 μL of a 40% acrylamide solution and incubation at RT for 30 min. Proteins were applied to a 12% Acryl-/Bisacrylamid SDS-gel (Table 1) and separation was performed with a SDS-running buffer (Table 1) and a 5% stacking gel (Table 1) for 15 min at 100 V and 1h at 180 V. The Page RulerTM Prestained Protein Ladder (Table 1) was used. After separation gels were washed 3 times with Millipore water for 10 min with subsequent treatment with a fixation solution (Table 1). Staining was performed with 20 mL PageBlue Protein Staining Solution (Table 1). Destaining was done 3 times with 200 mL Millipore water each for 15 min.
An example for the quantification of antimiR132 in porcine Plasma 1 h post high dose (5 mg/kg) treatment is shown below:
Converted to x this results in equation (2)
With y = 0.194 the result is equation (3)
Multiplied by the dilution factor of 4 (derived from 1:1 mixture with the THAP matrix-solution and that only 0.5 microL were applied on spot) results in equation (4):
Six matrices were prepared: 9-aminoacridine (AA), α-cyano-4-hydroxycinnamic acid (CHCA), 2,5-dihydroxybenzoic acid (DHB), 6-Aza-2-thiothymine (ATT), 3-hydroxypicolinic acid, 2,4,6-Trihydroxyacetophenone (THAP), and 2,5-diaminobenzophenone (DABP) (Table 1). In addition, a final concentration of 5 mg/mL L-Fucose was added to every matrix solution. Every matrix was then mixed with 30 pmol/μL antimiR oligonucleotide 24 and spotted on a MALDI target plate and analyzed. Analysis was performed in linear negative- as well as in linear positive detection mode for all matrix/antimiR oligonucleotide solutions. Reflector detection mode and linear positive detection mode did not yield any evaluable results. Only HPA and THAP could show evaluable results in linear detection mode. Best signal intensities were obtained using THAP in linear negative detection mode (Supplementary Figure 2). These results indicated best ionization efficiency for THAP as ionization matrix and thus it was decided to use THAP for further analysis.
Full extraction of an analyte from a biological sample is crucial for quantification. Since phosphorothioate modified oligonucleotides were reported to strongly bind to plasma proteins as well as cell surface proteins, a proteinase K digestion was used to digest those proteins and thus releasing oligonucleotides and decreasing the background noise originated from complex matrix components (Beltinger et al., 1995; Geary et al., 2001). The workflow consisted of two main steps: a) protein digestion at 55°C using proteinase K and b) glycogen-facilitated precipitation of the oligonucleotide. Subsequently, oligonucleotides were measured by MALDI-MS (Figure 1). Effectiveness of protein removal is shown in Supplementary Figures 3, 4.

Figure 1. Workflow of antimiR oligonucleotide detection using MALDI-MS.
For extraction of antimiR oligonucleotides from complex matrices four steps were necessary. With being the most important step, Proteinase K digestion was followed by precipitation of antimiR oligonucleotides with subsequent resuspension and MS analysis.
For assay validation purposes, the LOD, LLOQ and area of quantification (AOQ) values for detection of antimiR132 were determined. Validation of the isolation protocol was performed by adding antimiR132 at various concentrations (10, 5, 2, 1, 0.5, 0.25, and 0.1 pmol) together with 10 pmol antimiRScr as internal standard to murine plasma from untreated mice and measured with the developed assay. Analysis was performed with 10 replicates for determination of the LOD being at 0.25 pmol and three replicates for determination of the LLOQ and the AOQ. The LLOQ was determined to be at 0.25 pmol. AOQ was determined to be linear in the range from 0.25 up to 25 pmol (Figures 2, 3).
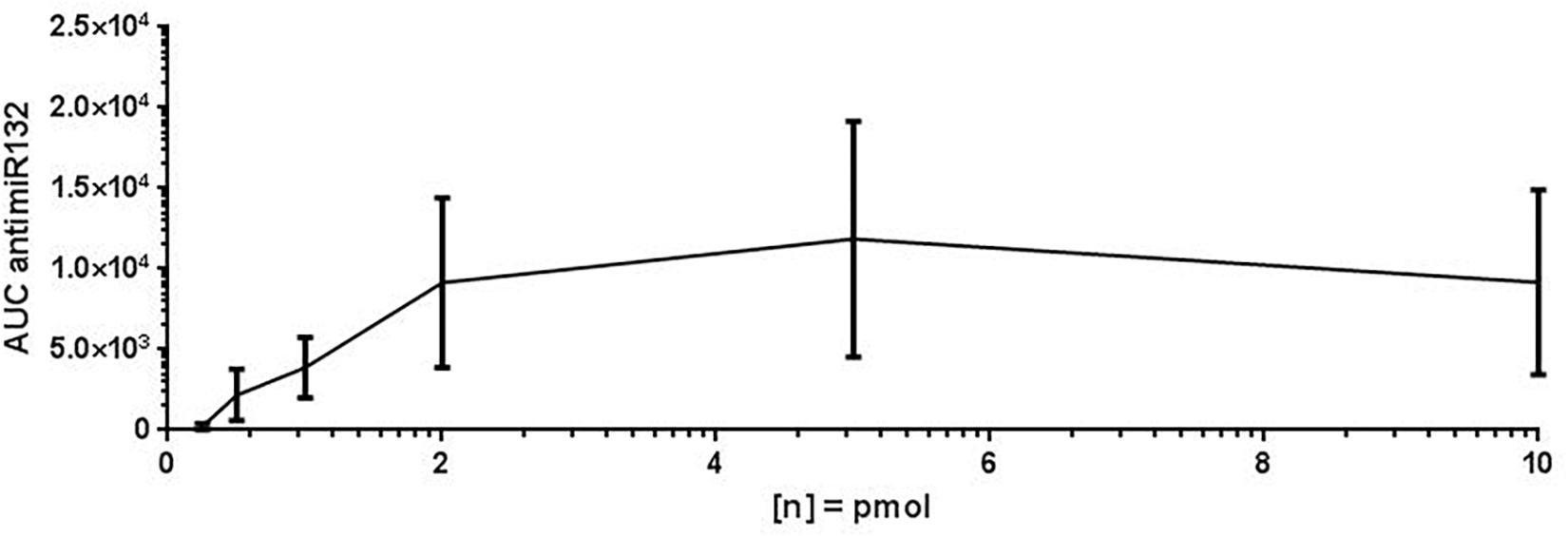
Figure 2. Limit of detection (LOD) of antimiR132. For determination of the LOD, antimiR oligonucleotides were spotted on a MALDI target plate after isolation from murine plasma. Analyses were done in 10 replicates and the peak area (=signal intensity) were plotted against concentration. The LOD is 0.5 pmol.
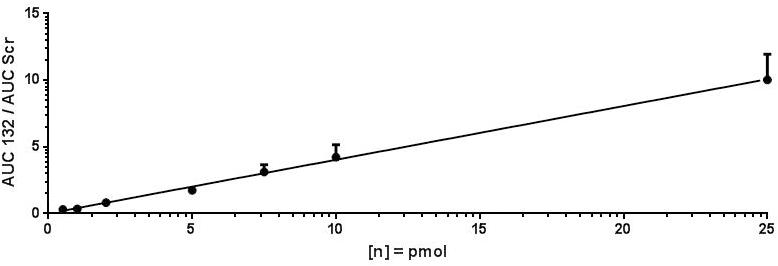
Figure 3. LLOQ and area of quantification of antimiR132. For determination of the LLOQ and AOQ with MALDI-MS, antimiR oligonucleotides were spotted into murine plasma, extracted, and analyzed by MALDI-MS in three replicates per oligonucleotide concentration with the LLOQ being at 0.25. The equation of the calibration curve was y = 0.4013x + 0.0331 with a coefficient of determination of 99.77%. The LLOQ is indicated by a red box.
For the quantification of antimiR132 in murine plasma, 20 mg/kg antimiR132 was delivered i.v. to 21 mice. At seven timepoints (3 min; 30 min; 1 h; 9 h, 1 days; 3 days; 1 week) blood samples were taken from which 100 – 200 μL plasma were obtained. AntimiRScr was added as an internal standard in a final concentration of 10 pmol/μL. By increasing the NaCl concentration from 100 mM to 250 mM in the Proteinase K buffer, a higher efficiency of the enzymatic reaction was achieved. The quantification of antimiR oligonucleotides was carried out as described previously. The plasma concentrations determined for each time were averaged over all three biological replicates with the standard deviation and the coefficient of variation (CV) being determined (Figure 4).
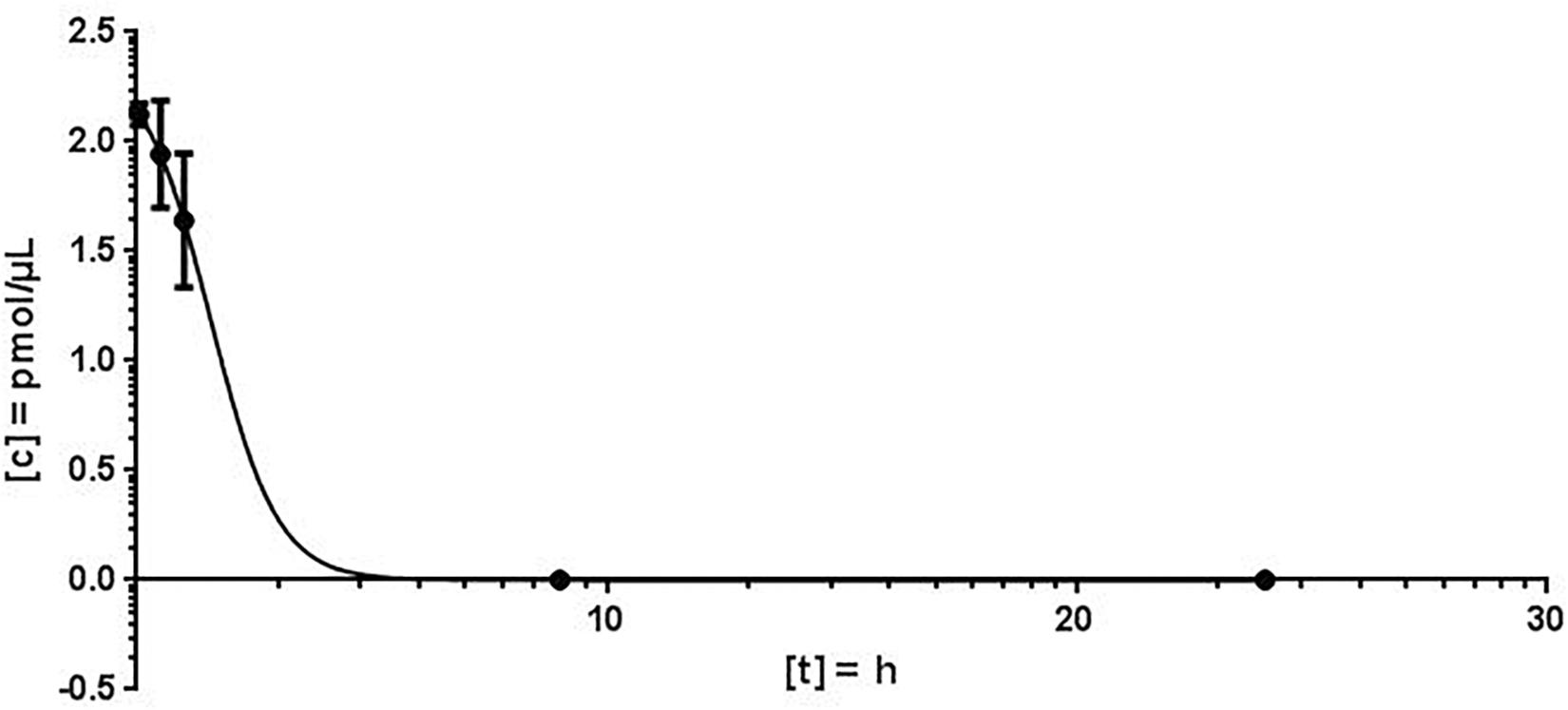
Figure 4. Murine plasma concentration of antimiR132. After application of 20 mg/kg antimiR132 i.v., mouse plasma levels were assessed utilizing MALDI-MS.
Using the MALDI-MS analysis, a plausible kinetic could be determined. The coefficient of variation (CV) varied from 2.4 to 18.7% and the peak concentration was calculated to 2.124 pmol/μL plasma. The plasma concentration decreased rapidly and plasma half-life of antimiR132 could be determined to be approximately 2 h. However, at least 9 h post treatment no antimiR132 was detected (Figure 5).
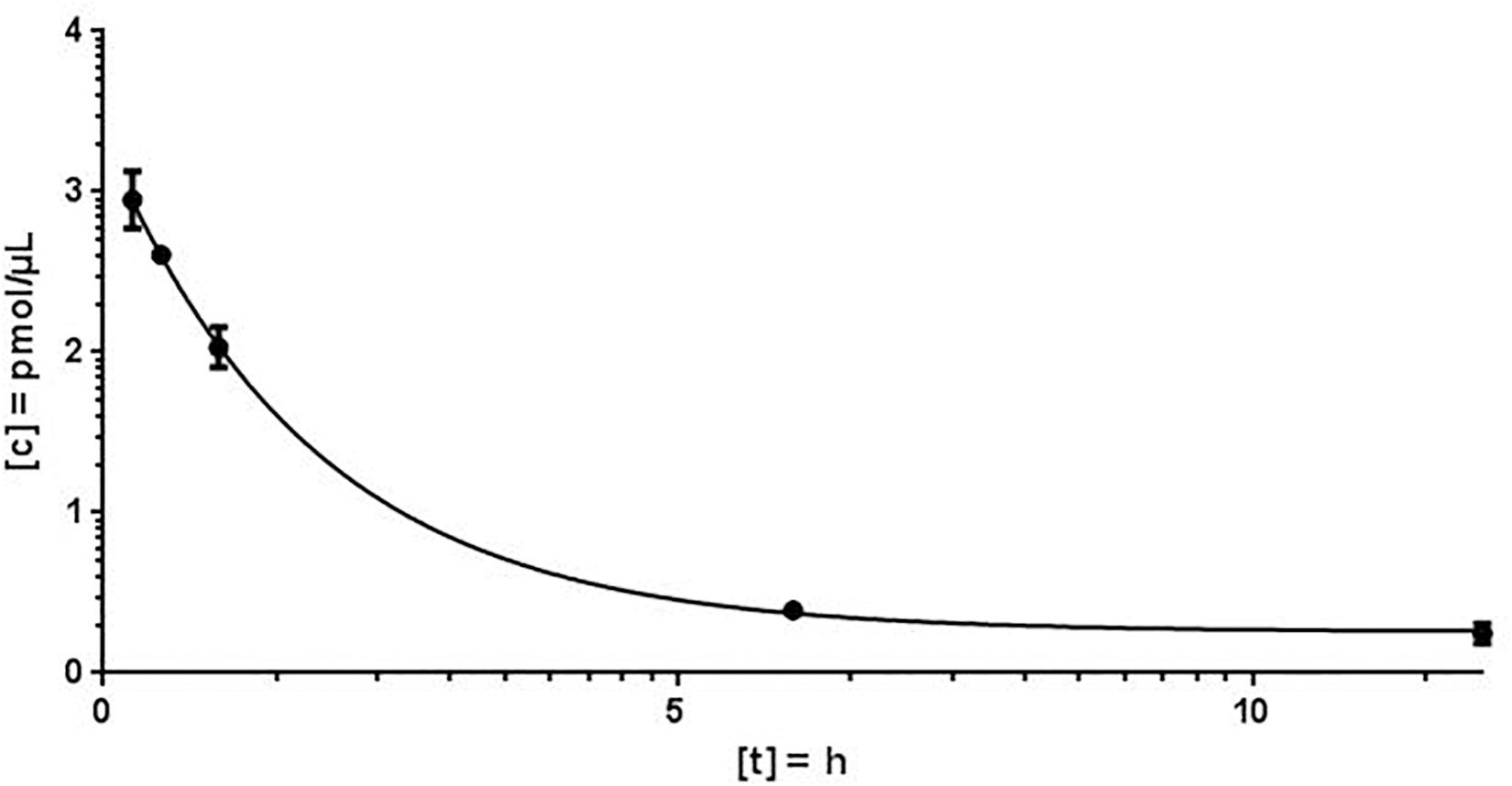
Figure 5. Porcine plasma concentration of antimiR132. After application of 5 mg/kg antimiR132 i.c., pig plasma levels were assessed using a mass spectrometry-based method for quantification. CV = 3.6 – 24.2%.
To evaluate the pharmacokinetic properties of antimiR132, further blood plasma samples from pig taken 15 and 30 min; 1, 6, 12, and 24 h after application were examined for the presence of antimiR132 and subsequently quantified (Figure 5).
Using MALDI-MS, a plausible kinetic could be determined. The peak concentration could be calculated at 2.124 pmol/μL. As expected, the plasma concentration decreased rapidly with the plasma half-life of antimiR132 being after approximately 2 h with no longer detection after 9 h. The concentration fell below the LOD and LLOQ after 12 h (Figure 4).
An antimiR oligonucleotide isolation protocol as well as a robust and reproducible MALDI-MS analysis procedure has been established. Proteinase K digestion was the important releasing antimiR oligonucleotides bound to plasma and cellular proteins. MALDI-MS analysis in linear negative detection mode with THAP as ionization matrix was proven to be best suited for oligonucleotide analysis in MALDI-MS and highly sensitive and reproducible values for antimiR132 could be determined. The high salt conditions might have reduced the binding affinity of LNA-based antimiR132 to plasma proteins. With this experimental setup the LOD and LLOQ for antimiR132 were determined and showed high sensitivity of this approach. The LOD as well as the LLOQ were at 0.25 pmol (1 pmol/μL) while the AOQ ranged to 25 pmol (100 pmol/μL, allowing a wide range for quantification (Figures 2, 3). These results show that MALDI-MS is a very sensitive and highly specific method for derivatized oligonucleotides of interest. That is less than is achieved with LC-MS approaches where reported sensitivity was 10 fmol/μL assuming for an oligonucleotide 5000 g/mol (Gallo-Cantafio et al., 2016). However, the LOD and LLOQ values are at an acceptable low range to quantify effective concentrations that are used in model systems to test efficacy of oligonucleotides as pharmacological agent (Tuntland et al., 2014). The advantage of MALDI-MS is its easy handling and very fast analysis times that allow measurements of e.g., 100 samples in about 15 min.
With MALDI-MS, a mouse PK study with antimiR132 was performed in which mice were treated i.v. with 20 mg/kg of this substance and sacrificed afterward with blood and organs taken. After reaching the peak concentration 3 min after application the plasma levels decreased strongly and antimiR132 could not be detected in the plasma after 9 h (Figure 4). In addition to the qualitative detection of antimiR132 in pig plasma, pharmacokinetic parameters were also obtained (Figure 5). After 12 h antimiR132 was no longer quantitatively detectable. Accordingly, the latter was either renally excreted or absorbed by the tissue. This relatively short plasma half-life of antimiR132 was to be expected since earlier studies with similar antimiR oligonucleotides also showed a similar trend even across species and support the assumption that plasma clearance of phosphorothioate-modified oligonucleotides might be species independent since plasma half-lifes were comparable (Yu et al., 2009).
MALDI-MS is a very precise and quick method for quantification of modified antisense oligonucleotides. Despite being mostly used for qualitative analysis, MALDI-MS can also be utilized for quantitative purposes, if sample size is adequate. It is more straightforward compared to other approaches such as ESI-MS, since sample preparation time is noticeable reduced and a liquid chromatography system is not needed, which further reduces preparative effort. However, its sensitivity is high enough to detect lower amounts of antimiR oligonucleotides in blood samples or maybe tissues.
All datasets generated for this study are included in the article/Supplementary Material.
The animal study was reviewed and approved by the Local Animal Welfare Bodies of the Hannover Medical School, Hannover, Germany and the University of Kaposvar, Kaposvár, Hungary.
MH, AF, SB, TT, and AP conceived and performed the experiments, and wrote the manuscript. AP and TT secured the funding. SB, AP, and TT provided the expertise and feedback.
The authors acknowledge funding by the EU Consolidator Grant Longheart, the ERANET network EXPERT, and the Deutsche Forschungsgemeinschaft (Th903/22-1 to TT).
TT is founder and shareholder of Cardior Pharmaceutical. He has filed and licensed patents about noncoding RNAs. SB is an employee of Cardior Pharmaceuticals.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Special thanks goes to Karsten Heidrich and Ulrike Schrammeck for the maintenance of the MALDI-TOF MS.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2020.00220/full#supplementary-material
Arora, V., Knapp, D. C., Reddy, M. T., Weller, D. D., and Iversen, P. L. (2002). Bioavailability and efficacy of antisense morpholino oligomers targeted to c-myc and cytochrome P-450 3A2 following oral administration in rats. J. Pharm. Sci. 91, 1009–1018. doi: 10.1002/jps.10088
Bartlett, M., McCloskey, J., Manalili, S., and Giffey, R. H. (1996). The effect of backbone charge on the collision-induced dissociation of oligonucleotides. J. Mass Spectrom. 31, 1277–1283. doi: 10.1002/(sici)1096-9888(199611)31:11<1277::aid-jms418>3.0.co;2-f
Bellon, L., Maloney, L., Zinnen, S. P., Sandberg, J. A., and Johnson, K. E. (2000). Quantitative determination of a chemically modified hammerhead ribozyme in blood plasma using 96-well solid-phase extraction coupled with high-performance liquid chromatography or capillary gel electrophoresis. Anal. Biochem. 283, 228–240. doi: 10.1006/abio.2000.4638
Beltinger, C., Saragovi, H. U., Smith, R. M., LeSauteur, L., Shah, N., DeDionisio, L., et al. (1995). Binding, uptake, and intracellular trafficking of phosphorothioate-modified oligodeoxynucleotides. J. Clin. Invest. 95, 1814–1823. doi: 10.1172/JCI117860
Beverly, M., Hartsough, K., Machemer, L., Pavco, P., and Ockridge, J. (2006). Liquid chromatography electrospray ionization mass spectrometry analysis of the ocular metabolites from a short interfering RNA duplex. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 835, 62–70. doi: 10.1016/j.jchromb.2006.03.008
Castleberry, C., and Limbach, P. (2010). Relative quantitation of transfer RNAs using liquid chromatography mass spectrometry and signature digestion products. Nucleic Acids Res. 38, e162. doi: 10.1093/nar/gkq578
Distler, A., and Allison, J. (2002). Additives for the stabilization of double-stranded DNA in UV-MALDI MS. J. Am. Soc. Spectrom. 13, 1129–1137. doi: 10.1016/S1044-0305(02)00430-0
Emmerechts, G., Herdewijn, P., and Rozenski, J. (2005). Pseudouridine detection improvement by derivatization with methyl vinyl sulfone and capillary HPLC-mass spectrometry. J. Chromatogr. B Technol. Biomed. Life Sci. 825, 233–238. doi: 10.1016/j.jchromb.2005.06.041
Fu, Y., Songyun, X., Chensong Mingliang, Y., Hanfa, Z., and Baochuan, G. (2006). A matrix of 3,4-diaminobenzophenone for the analysis of oligonucleotides by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Nucleic Acids Res. 34:13. doi: 10.1093/nar/gkl509
Gallo-Cantafio, M. E., Schnack-Nielsen, B., Mignogna, C., Arbitrio, M., Botta, C., Frandsen, N. M., et al. (2016). Pharmacokinetics and pharmacodynamics of a 13-mer LNA-inhibitor-miR-221 in mice and non-human primates. Mol. Thera. Nucleic. Acids 5:e336. doi: 10.1038/mtna.2016.36
Geary, R. S., Watanabe, T. A., Truong, L., Freier, S., Lesnik, E. A., Sioufi, N. B., et al. (2001). Pharmacokinetic properties of 2’-O-(2-Methoxyethyl)-modified oligonucleotide analogs in rats. J. Pharmacol. Exp. Ther. 296, 890–897.
Glas, D. J. (2010). PI3 kinase regulation of skeletal muscle hypertrophy and atrophy. Curr. Top. Microbiol. Immunol. 346, 267–278. doi: 10.1007/82_2010_78
Griffey, R., Greig, M., Gaus, H., Liu, K., Monteith, D., Winniman, M., et al. (1997). Characterization of oligonucleotide metabolism in vivo via liquid chromatography/electrospray tandem mass spectrometry with a quadrupole ion trap mass spectrometer. J Mass Spectrom. 32, 305–313. doi: 10.1002/(sici)1096-9888(199703)32:3<305::aid-jms482>3.0.co;2-r
Hill, J. A., and Olson, E. N. (2008). Cardiac plasticity. N. Engl. J. Med. 358, 1370–1380. doi: 10.1056/NEJMra072139
Hossain, M., and Limbach, P. (2007). Mass spectrometry-based detection of transfer RNAs by their signature endonuclease digestion products. RNA 13:295. doi: 10.1261/rna.272507
Ni, Y. G., Berenji, K., Wang, N., Oh, M., Sachan, N., Dey, A., et al. (2006). FoxO transcription factors blunt cardiac hypertrophy by inhibiting calcineurin signaling. Circulation 114, 1159–1168. doi: 10.1161/CIRCULATIONAHA.106.637124
Pieles, U., Zürcher, W., and Schär Moser, H. E. (1993). Matrix-Assisted Laser Desorption Ionization Time-of-Flight Mass Spectrometry: A Powerful tool for the Mass and Sequence Analysis of Natural and Modified Oligonucleotides. Oxford: Oxford University Press.
Pomerantz, S., and McCloskey, J. (1990). Analysis of RNA hydrolysates by liquid chromatography-mass spectrometry. Methods Enzymol. 193:796. doi: 10.1016/0076-6879(90)93452-q
Pomerantz, S., and McCloskey, J. (2005). Detection of the common RNA nucleoside pseudouridine in mixtures of oligonucleotides by mass spectrometry. Anal Chem. 77, 4687–4697. doi: 10.1021/ac058023p
Ronnebaum, S. M., and Patterson, C. (2010). The FoxO family in cardiac function and dysfunction. Annu. Rev. Physiol. 72, 81–94. doi: 10.1146/annurev-physiol-021909-135931
Sudha, R., and Zenobi, R. (2002). The detection and stability of DNA duplexes probed by MALDI mass spectrometry. Helvetica Chim. Acta 85, 3136–3143. doi: 10.1002/1522-2675(200210)85:10<3136::aid-hlca3136>3.0.co;2-f
Tuntland, T., Ethell, B., Kosaka, T., Blasco, F., Zang, R. X., Jain, M., et al. (2014). Implementation of pharmacokinetik and pharmacodynamic strategies in early research phases of drug discovery and development at Novartis Institute of Biomedical Research. Front. Phamracol. 5:174. doi: 10.3389/fphar.2014.00174
Ucar, A., Gupta, S. K., Fiedler, J., Erikci, E., Kardasinski, M., Batkai, S., et al. (2012). The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat. Commun. 3:1078. doi: 10.1038/ncomms2090
Waters, J., Webb, A., Cunningham, D., Clarke, P. A., Raynaud, F., DiStefano, F., et al. (2000). Phase I clinical and pharmacokinetic study of bcl-2 antisense oligonucleotide therapy in patients with non-Hodgkin’s lymphoma. J. Clin. Oncol. 18:1812. doi: 10.1200/JCO.2000.18.9.1812
Yu, R. Z., Lemonidis, K. M., Graham, M. J., Matson, J. E., Crooke, R. M., Tribble, D. L., et al. (2009). Cross-species comparison of in vivo PK/PD relationships for second-generation antisense oligonucleotides targeting apolipoprotein B-100. Biochem. Pharmacol. 77, 910–919. doi: 10.1016/j.bcp.2008.11.005
Keywords: microRNAs, therapeutic oligonucleotides, mass spectrometry, absolute quantification, pharmacokinctics
Citation: Herkt M, Foinquinos A, Batkai S, Thum T and Pich A (2020) Pharmacokinetic Studies of Antisense Oligonucleotides Using MALDI-TOF Mass Spectrometry. Front. Pharmacol. 11:220. doi: 10.3389/fphar.2020.00220
Received: 25 November 2019; Accepted: 18 February 2020;
Published: 25 March 2020.
Edited by:
Salvatore Salomone, University of Catania, ItalyReviewed by:
Sylwia Studzińska, Nicolaus Copernicus University in Toruń, PolandCopyright © 2020 Herkt, Foinquinos, Batkai, Thum and Pich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Markus Herkt, aGVya3QubWFya3VzQG1oLWhhbm5vdmVyLmRl
†These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.