 Drew Nassal
Drew Nassal Daniel Gratz
Daniel Gratz Thomas J. Hund
Thomas J. Hund- 1The Frick Center for Heart Failure and Arrhythmia and Dorothy M. Davis Heart and Lung Research Institute, The Ohio State University Wexner Medical Center, Columbus, OH, United States
- 2Department of Biomedical Engineering, College of Engineering, The Ohio State University, Columbus, OH, United States
- 3Department of Internal Medicine, The Ohio State University Wexner Medical Center, Columbus, OH, United States
Heart failure remains a major health burden around the world. Despite great progress in delineation of molecular mechanisms underlying development of disease, standard therapy has not advanced at the same pace. The multifunctional signaling molecule Ca2+/calmodulin-dependent protein kinase II (CaMKII) has received considerable attention over recent years for its central role in maladaptive remodeling and arrhythmias in the setting of chronic disease. However, these basic science discoveries have yet to translate into new therapies for human patients. This review addresses both the promise and barriers to developing translational therapies that target CaMKII signaling to abrogate pathologic remodeling in the setting of chronic disease. Efforts in small molecule design are discussed, as well as alternative targeting approaches that exploit novel avenues for compound delivery and/or genetic approaches to affect cardiac CaMKII signaling. These alternative strategies provide hope for overcoming some of the challenges that have limited the development of new therapies.
Introduction
For the past 50 years, therapy based on antagonism of beta adrenergic receptors, angiotensin converting enzyme (ACE), and/or AT2 receptors has been the staple for heart failure (HF) treatment (Bers, 2005). However, the incidence of HF and associated mortality rates continue to grow at an alarming pace, highlighting the need for novel therapies to target pathways that drive HF progression. While the field has shown tremendous strides in understanding mechanisms associated with pathologic remodeling leading to HF, translation of this information into new and effective therapies has been less successful. This review discusses the multifunctional signaling molecule Ca2+/calmodulin-dependent protein kinase II (CaMKII) as an example of both the promise and challenges of developing translational therapies based on our evolving understanding of pathologic remodeling in the setting of chronic disease. Despite the wealth of basic research supporting CaMKII as a master regulator of pathways important for cardiac remodeling and arrhythmia, the field has yet to witness translation of these findings to the clinic. The gap between advances at the bench and new treatments in human patients has not gone unnoticed, and efforts are underway that will hopefully fix the pipeline for development of effective therapy. In this review, we address a brief history of progress in small molecule drug design, followed by consideration of alternative approaches that take advantage of recent advances in novel delivery mechanisms as well as genetic approaches for manipulating CaMKII signaling in the heart. These alternative strategies address the ability to circumvent the challenges and limitations of small molecule drug design while highlighting next-generation therapeutic paradigms.
CaMKII Expression, Structure, and Function in Heart
CaMKII is a serine/threonine kinase important for translating changes in the levels of intracellular Ca2+/calmodulin (and other critical second messengers) into adaptations in cell function through direct phosphorylation of a large number of target proteins (Swaminathan et al., 2012; Westenbrink et al., 2013). Diversity in metazoan CaMKII expression arises from existence of four isoforms, α, β, γ, and δ, each encoded by a distinct gene. While α and β isoforms are primarily expressed in the brain where they regulate indispensable mechanisms of long-term potentiation/depression necessary for learning and memory (Silva et al., 1992a; Silva et al., 1992b; Stevens et al., 1994; Coultrap et al., 2014), the δ and γ isoforms are broadly expressed in multiple tissues, including the heart (Bennett et al., 1983; Tobimatsu and Fujisawa, 1989). Notably, CaMKIIδ, the predominant cardiac isoform, undergoes alternative splicing to give rise to CaMKIIδb and CaMKIIδc variants (Edman and Schulman, 1994). This splicing event confers CaMKIIδb with a nuclear localization signal not expressed in CaMKIIδc leading to differential subcellular distribution (Srinivasan et al., 1994; Ramirez et al., 1997). Despite important differences, there is high degree of homology across CaMKII isoforms, posing significant challenges for therapy around CaMKII inhibition. Given the ubiquitous nature of CaMKII with expression throughout the body, it is no small task to effectively target isoforms/splice variants involved in cardiac pathophysiology while avoiding those important for normal physiology in heart and other organ systems (e.g., brain) (Little et al., 2009; Peng et al., 2010; Lu et al., 2011b; Beckendorf et al., 2018).
Before considering strategies for targeting CaMKII, it is important to briefly discuss the kinase structure/function relationship. A single molecule of CaMKII is composed of three domains: (1) the N-terminal catalytic domain responsible for ATP binding and kinase function; (2) the regulatory domain responsible for binding Ca2+/calmodulin and subsequent kinase activation; and (3) the C-terminal association domain responsible for the oligomerization of individual CaMKII molecules to create a mature dodecameric-holoenzyme (Figure 1A). As will be discussed, the N-terminal catalytic domain has received the most attention for translatable drug design. Inactive CaMKII is folded in a closed conformation where the regulatory domain of each CaMKII monomer acts as a substrate binding the catalytic domain. Additionally, adjacent regulatory domains within the dodecameric structure block the binding of target substrates and ATP, maintaining a self-inhibited state. Activation of CaMKII occurs when Ca2+/calmodulin binds to a defined region in the regulatory domain resulting in a conformation shift that releases the catalytic domain, exposing the kinase substrate and ATP binding sites. Importantly, the regulatory domains of neighboring CaMKII monomers within the holoenzyme are themselves substrates for active CaMKII kinase activity, specifically at Thr287. Phosphorylation of this residue dramatically increases affinity for Ca2+/calmodulin and also prevents reassociation of the regulatory and catalytic domains, creating sustained CaMKII activity through beat-to-beat fluctuations in Ca2+ cycling.
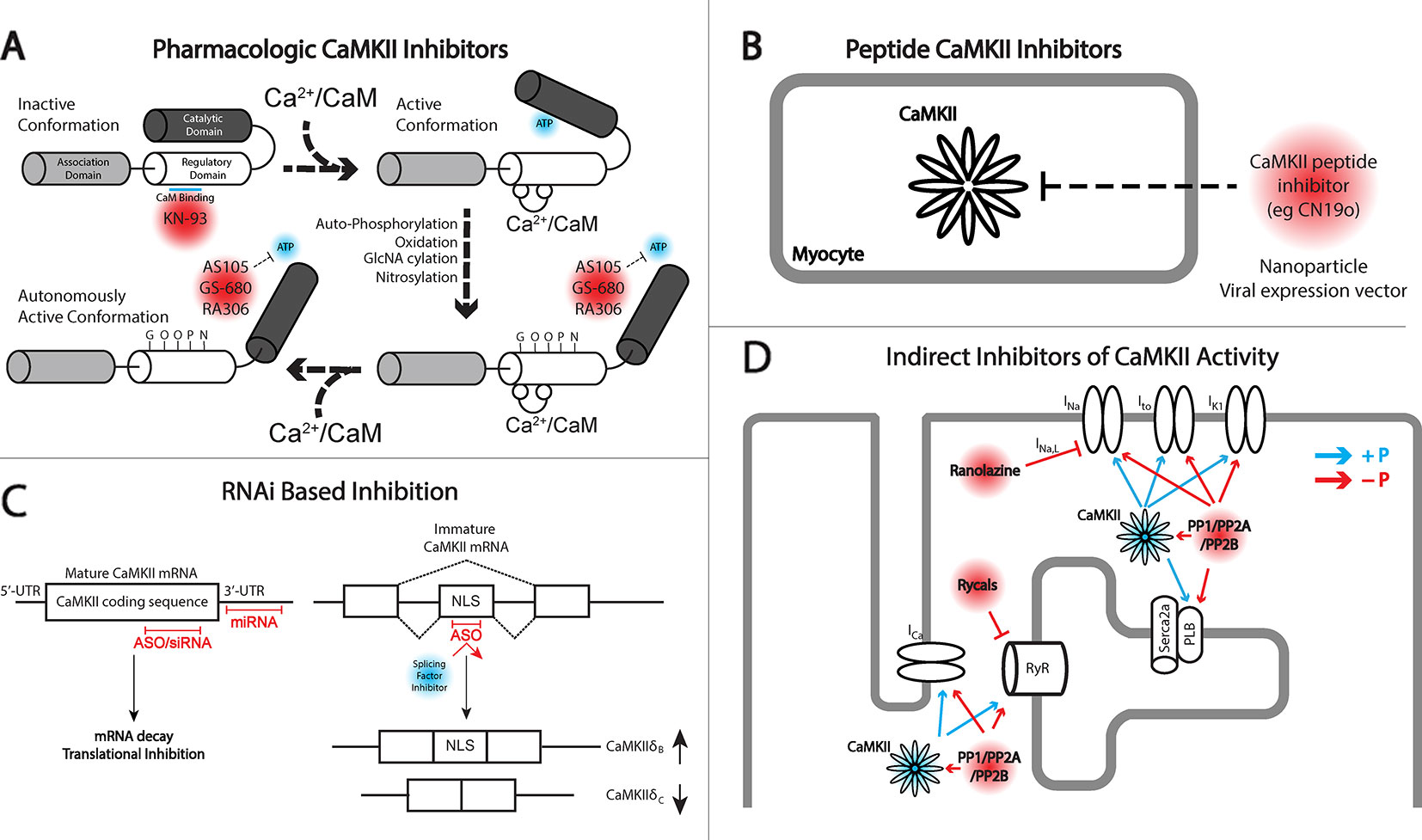
Figure 1 Different approaches for targeting Ca2+/calmodulin-dependent protein kinase II (CaMKII) signaling. (A) Depiction of inactive and active monomers of CaMKII showing the association, regulatory, and catalytic domains. The association domain is responsible for interaction with other CaMKII monomers and is necessary for forming the holoenzyme structure, consisting of 12 monomers. In its inactive state, the catalytic domain is obscured by interaction with the regulatory domain. This interaction is disrupted upon the binding of Ca2+/CaM leading to autophosphorylation by neighboring CaMKII monomers, in addition to other posttranslational modifications including oxidation, GlcNAcylation, and nitrosylation, maintaining CaMKII activation even upon release of Ca2+/CaM. KN-93 is a known allosteric inhibitor of CaM binding and therefore preferentially targets CaMKII in the inactive state. CaMKII inhibitors AS105, GS-680, and RA306 are novel pyrimidine–based, ATP-competitive inhibitors that inhibit the activated catalytic domain of CaMKII and represent potential therapeutic agents for translational CaMKII inhibition. (B) Peptide inhibitors of CaMKII (e.g., CN19o, refined from CaMKIItide) show favorable selectivity and potency for CaMKII inhibition but face challenges in delivery and bioavailability. Both viral gene delivery and novel advances in nanoparticles offer opportunities for delivery of these agents to the heart. (C) RNA interference (RNAi) is a novel approach for inhibiting CaMKII activity at the transcript level. Antisense oligonucleotides (ASOs), small interfering RNA (siRNA), and miRNAs provide opportunity for degrading CaMKII transcripts and/or inhibiting protein translation. ASOs can also be used to interfere with recruitment of splicing factors to enhance concentrations of CaMKIIδB which has been shown to have cardioprotective effects. (D) Indirect inhibition of CaMKII can be achieved by regulating downstream targets of CaMKII kinase activity. An example comes from the late Na+ current (INa,L) inhibitor ranolazine, or the RyR stabilizing agents, rycals. Alternatively, protein phosphatases may be targeted to antagonize kinase activity in cardiomyocytes. The development of phosphatase activators for cancer therapeutics may offer opportunity for drug applications in cardiovascular disease.
In addition to the autophosphorylation at Thr287 maintaining the active conformation, other posttranslational modifications have been identified on the regulatory domain, which maintain an active state and have been associated with cardiac disease states. These include the oxidation of Met281/282 in response to elevated reactive oxygen species (Erickson et al., 2008; He et al., 2011; Purohit et al., 2013; Anderson, 2015), O-linked N-acetylglucosamine (GlcNAcylation) targeting Ser279 as a result of elevated glucose levels (Erickson et al., 2008; Erickson et al., 2013), and reports of S-nitrosylation predicted on Cys290 as a result of increased NO production upon β-AR stimulation (Gutierrez et al., 2013; Erickson et al., 2015). Together, these posttranslational modifications establish a diverse set of pathways that contribute to sustained activation of CaMKII, reinforcing the strong association between CaMKII activity and the development of cardiac disease.
The physiologic targets and consequences of cardiac CaMKII signaling have been thoroughly addressed in previous review articles (Anderson et al., 2011; Westenbrink et al., 2013; Mattiazzi et al., 2015; Mustroph et al., 2017). Therefore, we will only briefly discuss CaMKII targets in the context of cardiac remodeling and disease to lay the foundation for more detailed treatment of efforts to inhibit CaMKII. Ample experimental evidence causally links chronic CaMKII activity to development of cardiac disease and arrhythmias (Maier and Bers, 2002; Swaminathan et al., 2012; Westenbrink et al., 2013). Animal models have shown proof-of-concept studies that transgenic overexpression of CaMKII is sufficient to induce structural and electrical remodeling in the heart, leading to compromised contractility and increased risk for sudden cardiac death (Zhang et al., 2002; Zhang et al., 2003; Wagner et al., 2011). Likewise, genetic and chemical inhibition of CaMKII has been shown to confer protection from the development of dilated cardiomyopathy and sustained contractile performance, following both pressure overload and ischemic stress (Zhang et al., 2005; Backs et al., 2009; Ling et al., 2009). Important when considering translation, human HF has also been associated with an increased expression/activity of CaMKII (Hoch et al., 1999; Kirchhefer et al., 1999). The central role for CaMKII in development of disease stems from its regulation of proteins involved in critical cell functions from Ca2+ cycling to mitochondrial function. CaMKII has been implicated in pathologic phosphorylation of a number of Ca2+ handling proteins including phospholamban, leading to activation of the sarcoplasmic reticulum (SR) ATP-driven Ca2+ pump SERCA2a (Mattiazzi and Kranias, 2014); the ryanodine receptor SR Ca2+ release channel (RyR2) (Witcher et al., 1991; Lokuta et al., 1995; Wehrens, 2011), promoting increased channel open probability and SR Ca2+ leak; and the L-type Ca2+ channel Cav1.2 and associated β-subunits, potentiating current amplitude and slowing inactivation (Hudmon et al., 2005; Grueter et al., 2006). Collectively, these events not only promote activation of hypertrophic remodeling cascades but also heighten the risk for inappropriate membrane potential depolarizations (afterdepolarizations) that serve as arrhythmia triggers (Wu et al., 2002). CaMKII-dependent phosphorylation of other ion channels like the primary cardiac voltage-gated Na+ channel Nav1.5 enhances late depolarizing current, leading to prolonged action potentials, further disrupting Ca2+ handling and providing additional substrates for the formation of arrhythmogenic afterdepolarizations (Koval et al., 2012; Glynn et al., 2015; Howard et al., 2018). Mitochondrial Ca2+ entry has also been identified as a target for CaMKII with consequences for mitochondrial function and apoptosis (Joiner et al., 2012; Erickson et al., 2013).
Beyond ion channels, CaMKII has been found to target chromatin remodeling protein class 2 histone deacetylases 4 and 5 (HDAC4/5). The phosphorylation of these proteins induces their nuclear export, freeing the transcription factor MEF2 from its repressed state, contributing to hypertrophic remodeling (Backs et al., 2006; Wu et al., 2006; Backs et al., 2008). Relevant to pathology, CaMKII signaling is notable for the presence of unique dynamical properties including 1) feedback where CaMKII affects downstream targets that in turn alter an input signal for CaMKII activation (Yao et al., 2011; Morotti et al., 2014; Onal et al., 2017); and 2) bistability where CaMKII is capable of toggling between resting and activated equilibrium states (Michalski, 2013). Together, these findings point to CaMKII as a prime candidate for next-generation therapies in managing heart disease in human patients. Despite our thorough understanding of CaMKII structure and function, the development of therapeutics based on this knowledge is in its infancy. Our goal for the remainder of the review will be to provide a comprehensive look at the progressing therapeutic opportunities for inhibiting CaMKII for HF management. Recent reviews have excellently covered the history and progress of CaMKII inhibition (Pellicena and Schulman, 2014; Mustroph et al., 2017). Here, we will provide an update of recent efforts toward the development of translational pharmacologic agents while considering more novel and unconsidered opportunities in drug delivery and genetic approaches (summarized in Table 1).
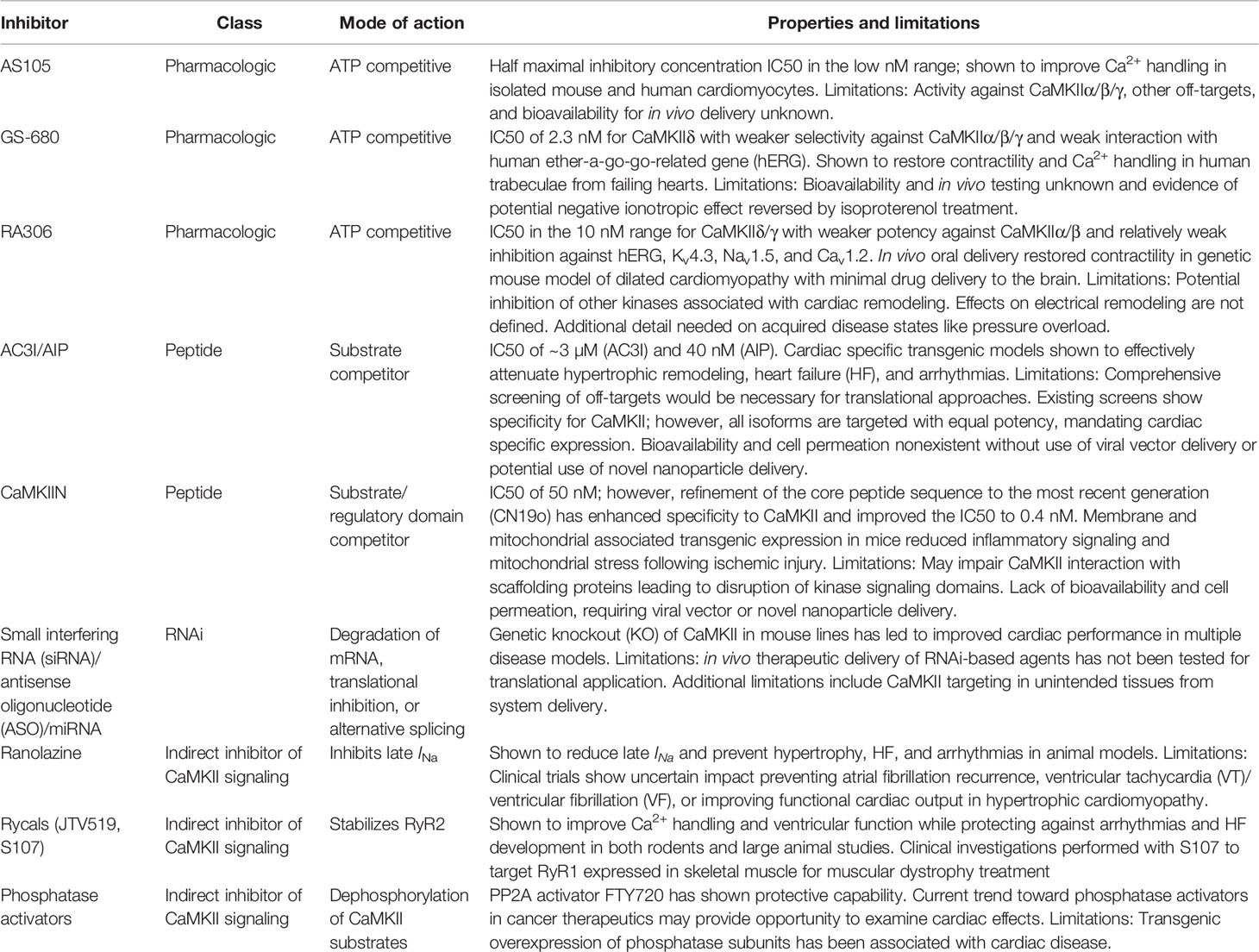
Table 1 Properties and limitations of Ca2+/calmodulin-dependent protein kinase II (CaMKII) inhibitory agents.
Pharmacologic Inhibitors of CaMKII
The approach of using pharmacologic inhibitors to target CaMKII activity has been used extensively in basic research with less progress in translational medicine, which is somewhat surprising given the widespread use of protein kinase inhibitors in cancer therapeutics for targeting tumor proliferation and cell survival (Bhullar et al., 2018). In fact, protein kinases are the second most targeted group of proteins, currently with 37 kinase inhibitors having received Food and Drug Administration (FDA) approval for cancer treatment, with another 150 in clinical trials (Bhullar et al., 2018). However, similar compounds have not been successfully developed for therapeutic purposes in the cardiac field due in part to a higher threshold for safety requirements, historical investment being more directed at ion channel blockers for anti-arrhythmics, and a larger burden of clinical trial costs and more uncertain return on investment compared to other therapeutics (Fordyce et al., 2015). Given the growing awareness of the slowdown in cardiovascular drug development (Fordyce et al., 2015) and the dominant academic literature on the contribution of CaMKII in pathologic cardiac remodeling, it seems the community is open and motivated for new pharmacologic drugs for CaMKII inhibition (Figure 1A).
Among the first small molecule inhibitors developed for CaMKII was KN-93 (Sumi et al., 1991), which has become one of the most widely used inhibitors in basic research. While its design was not intended for translational use, its prolific use over the years and unique mode of CaMKII inhibition make it important to discuss. While most kinase inhibitors target the ATP-binding domain, KN-93 is unique in that it allosterically disrupts CaM binding and stabilizes the interaction between the enzymatic and regulatory domains as evidenced by measured FRET signals associated with inactive versus activated structures of CaMKII (Erickson et al., 2011; Erickson et al., 2013). However, this mode of inhibition is thought to be less effective at inhibiting already active CaMKII, including autonomously activated CaMKII as a result of Thr287 autophosphorylation (Vest et al., 2010), which could also extend to additional autonomous modifications of oxidation and GlcNacylation. Another important limitation of KN-93 is that it is not particularly specific or potent against CaMKIIδ, with a half maximal inhibitory concentration (IC50) in a range of 1–4 µM. While KN-93 is reasonably selective for CaMKII over other protein kinases (Gao et al., 2013), it has been found to have a range of off-target effects, including voltage-gated potassium channels (Ledoux et al., 1999; Rezazadeh et al., 2006; Hegyi et al., 2015), L-type Ca2+ channels (Anderson et al., 1998), inositol triphosphate receptor Ca2+ release (Smyth et al., 2002), and even calmodulin (Johnson et al., 2019).
While KN-93 has proven a valuable research tool, it has not been until recently that concerted efforts have been made to move pharmacological CaMKII inhibition to the clinic. An early effort from the now defunct Scios sought to characterize CaMKII/activity relationships to identify new strategies for inhibiting CaMKII (Mavunkel et al., 2008). From this work, they developed the compound Scios 15b, a cell-permeable, ATP-competitive, pyridimine-based molecule that inhibits CaMKII with an IC50 of 9 nM in vitro and 320 nM in situ. These initial cell-based assays evaluated CaMKII inhibition by the extent to which it impaired CaMKII-dependent phosphorylation of vimentin. While the compound was not directly investigated with respect to its impact on pathological cardiac remodeling, this work laid the groundwork for the next generation of CaMKII pharmacologics that are being developed, particularly the pyrimidine, ATP-competitive compounds.
Indeed, a more recent novel CaMKII inhibitor, AS105, was also designed as a pyrimidine-based, ATP-competitive molecule by Allosteros Therapeutics following computational optimization of pyrimidine-based CaMKIIδ inhibitors (Neef et al., 2018). Achieving an IC50 in the low nanomolar range for in vitro binding, this drug was shown to therapeutically inhibit CaMKII in isolated cardiomyocytes from CaMKIIδC-overexpressing mice with HF by reducing SR Ca2+ leak, improving SR Ca2+ loading and Ca2+ transient amplitude, and restoring myocyte fractional shortening. Importantly, it was shown to not negatively impact basal excitation–contraction coupling in cardiomyocytes. Moreover, it was able to reduce SR Ca2+ leak within isolated human atrial myocytes and suppress arrhythmogenic Ca2+ release events. These studies provide promise for the development of a novel CaMKII inhibitor; however, it remains to be seen how specific this drug is for CaMKIIδ compared to CaMKIIα/β/γ and other potential off-targets, as well as its bioavailability.
Another pyrimidine-based, ATP-competitive molecule, GS-680, has been developed by Gilead Sciences (Lebek et al., 2018). GS-680 has a reported CaMKIIδ biochemical IC50 of 2.3 nM, with values 3.1-, 8.7-, and 22.5-fold less potent for CaMKIIɣ, α, and β, suggesting a selective potency of CaMKII inhibition for isotypes expressed within the myocardium, minimizing the risk of neuronal side effects. Measurement of phosphorylation levels of phospholamban in neonatal rat ventricular myocytes and neurons treated with GS-680 yielded estimates of EC50 of 98.9 nM and 9,005 nM, respectively, for CaMKII inhibition, consistent with selectivity of the compound for cardiac CaMKII. Moreover, assessment of additional safety factors found the drug to have an EC50 of 3,000 nM for human ether-a-go-go-related gene (hERG) channel inhibition, minimizing the risk of QT prolongation. GS-680 was shown to effectively inhibit premature atrial contractions (PACs) in human atrial trabeculae by exposure to increased external Ca2+ (3.5 mM) and isoproterenol (100 nM) and did so in a dose-dependent manner, completely eliminating PACs at 100 and 300 nM concentrations, which occurred in 50% of nontreated samples. In parallel, GS-680 was found to reduce Ca2+ sparks and SR Ca2+ leak and attenuated triggered activity, as well as increased action potential amplitude and maximal upstroke velocity within isolated human atrial myocytes. However, the drug was found to impair systolic atrial contractions, which was negated after pretreating the tissue with isoproterenol, suggesting that CaMKII inhibition at baseline had a negative ionotropic impact that could be compensated by PKA stimulation. Consistent with this idea, human HF ventricular trabeculae preparations, which are characterized by increased PKA activity and negative force–frequency relationships, experienced improved contractility and increased Ca2+ transients at higher pacing frequencies when treated with GS-680, showing that this compound holds promise for the potential treatment of both atrial arrhythmias and HF ventricular remodeling.
Like the compounds just discussed, the most recently designed compound named RA306 developed by Sanofi R&D was derived from a chemical optimization program based on the pyrimidine ATP-competitive class of CaMKII inhibitors (Beauverger et al., 2019). The drug was found to be a potent inhibitor of human CaMKIIδ and ɣ, the two main cardiac isoforms, having an IC50 of 15 and 25 nM, respectively, in enzymatic caliper assays and below 3 nM for each isoform in a more sensitive P33 assay. CaMKIIβ was also inhibited with an IC50 of 61 nM, and CaMKIIα was weakly inhibited with an IC50 of 420 nM (assessed using caliper assays). There were a limited number of other protein kinases found to be inhibited by the drug including MLK1, SIK, and Pyk2, which have also been associated with adverse cardiac remodeling. RA306 also displayed a favorable ion channel selectivity profile, showing IC50 values for hERG, Kv4.3, and Nav1.5 over 30 and 14 µM for Cav1.2. The therapeutic potential of this compound was tested in a transgenic mouse model of dilated cardiomyopathy, but unique to these studies was the delivery of the drug as an orally bioavailable agent, giving it significant advantage for potential clinical investigation. The drug was found to improve ejection fraction in a genetic mouse model of dilated cardiomyopathy, which associated with reduced levels of phosphorylated phospholamban (Thr17), a direct target of CaMKIIδ. Critically, a five-fold lower exposure for the drug was found in the brain compared to the heart, limiting the potential for off-target impacts of CaMKII inhibition in the brain.
The last several years has seen a strong push in the development of translational pharmacologic agents to inhibit CaMKII for the treatment of pathologic heart conditions. While only one compound (RA306) has been shown to be orally bioavailable, several new agents are available with favorable selectivity and potency profiles. Therefore, based on these efforts, it seems reasonable to remain optimistic about the development of a translationally relevant and novel HF pharmacologic.
Peptide Inhibitors of CaMKII
Peptide inhibitors (Figure 1B) represent some of the most effective agents to modulate CaMKII for research purposes. The identification of the auto-inhibitory state of the regulatory domain fueled the development of a long inhibitory peptide lacking both the CaM binding domain (Payne et al., 1988; Malinow et al., 1989) and the autophosphorylation residue Thr287 (mutated to be nonphosphorylatable). The result was the generation of the peptide inhibitors autocamtide-2-related inhibitory peptide (AIP) (Ishida et al., 1995) and autocamtide-3 derived inhibitory peptide (AC3-I) (Braun and Schulman, 1995), able to bind the CaMKII catalytic domain but unable to be displaced by canonical CaMKII activation mechanisms. As such, these inhibitors have been delivered through pharmacologic and genetic means to robustly inhibit CaMKII pathologic signaling in vitro and in vivo, mitigating HF, increasing cardiac function, and preventing lethal cardiac arrhythmias (Zhang et al., 2005; Khoo et al., 2006; Luo et al., 2013; Purohit et al., 2013). Subsequent to the design of these inhibitor peptides, natively expressed inhibitory proteins known as CaMKIINs were discovered. These small proteins were identified through a yeast two-hybrid screen where the CaMKII catalytic domain was used as bait (Chang et al., 1998; Chang et al., 2001), and while they have only been natively detected in the brain, they have been used effectively to target CaMKII activity. Notably, this peptide inhibitor binds with the kinase domain in the active conformation. Indeed, the CaMKIIN inhibitor has been used to limit myocyte death in response to MI, catecholamine stress, and ischemia–reperfusion (Swaminathan et al., 2011; Joiner and Koval, 2014) and reduce the occurrence of arrhythmogenic substrates (Koval et al., 2010) and in particular has utilized localization motifs to inhibit CaMKII within mitochondria or near plasma membrane domains (Joiner et al., 2012). Analysis of the peptide led to refinement and development of a 28-amino acid peptide inhibitor called CaMKIItide (Chang et al., 2001), while further refinement has led to the development of a shortened version only 19 amino acids long called CN19o (Coultrap and Bayer, 2011), with an IC50 of 0.4 nM. This was a >100-fold improvement in the IC50 than the native CaMKIIN, which also led to a stark improvement in the selectivity for CaMKII over other kinases (Coultrap and Bayer, 2011).
While the development and characterization of these highly selective peptide inhibitors has advanced cardiac research, there remains the rather significant challenge of delivery for therapeutic use. The greatest of these challenges is perhaps the limited bioavailability of short peptide sequences in vivo. Oral administration would provide limited enteral resorption and intravenous injection for the treatment of a chronic condition would offer challenges with continued compliance, let alone the quick half-life of a short peptide sequence in circulation (Otvos and Wade, 2014). Alternatively, gene therapy could be used to deliver a viral vector which expresses the short peptide. In the last 30 years, there have been four randomized clinical trials employing viral gene delivery for the treatment of HF with reduced ejection fraction (CUPID, CUPID-2, STOP-HF, and AC6) (Penny and Hammond, 2017). While the viral delivery was well tolerated in patients, the greatest burden they faced was insufficient gene transduction, where the patient with the lowest ejection fraction or those receiving the highest viral dosage were the only to experience improved endpoints. While these results do not preclude the future use of gene therapy, they do establish a more rigorous hurdle to overcome to pursue CaMKII inhibition.
A more novel and potentially robust opportunity, however, may reside in recent advances in nanotechnology. A recent report showed the successful delivery of peptide cargo to the heart through the use of biocompatible and biodegradable calcium phosphate nanoparticles introduced by inhalation (Miragoli et al., 2018). This strategy took advantage of the principle that oxygenated blood from the lungs moves from the pulmonary circulation to the heart first. This principle was supported by observations that nanoparticles and particulates from air pollution have been implicated in cardiac dysfunction and arrhythmia (Mills et al., 2009). Not only does the design of these particles allow for delivery to the heart and cell permeation but also protects peptides from enzymatic digestion. This approach has been used for successful delivery of a peptide impacting L-type Ca2+ channel expression, resulting in restored cardiac function in a mouse model of diabetic cardiomyopathy (Miragoli et al., 2018). Moreover, peptides have been delivered in a porcine model as well, with minimal delivery to tissues other than the heart. This mode of delivery not only represents a noninvasive and practical means of delivering novel cardiac peptide therapeutics but can take advantage of an already customized potency and precision of CaMKII peptide inhibitors like CaMKIItide or its derivative CN19o.
Use of RNA Interference (RNAi) to Target CaMKII
When it comes to therapeutic treatment of human disease, including cardiac disease, therapeutic intervention falls within two major classes of FDA-approved drugs, small molecules and proteins (Verdine and Walensky, 2007). As has already been discussed, small molecules typically must overcome the burden of target specificity and efficacy and require significant investments of time and resources to make them available for use. Alternatively, protein-based drugs, which frequently take the form of antibodies, display higher specificity, but limitations on size, stability, and deliverability to the right cellular compartments are frequent barriers to their application (Verdine and Walensky, 2007). An alternative approach to the targeting of specific genes implicated in disease may be found in RNA therapeutics (Figure 1C). For close to 30 years, RNA drugs [short hairpin RNAs (shRNAs), small-interfering RNAs (siRNAs), microRNAs (miRNAs)] have been used at the bench to target protein expression to identify critical gene targets in disease and signaling pathways. However, their use in clinical settings has been a non-factor given that short single- and double-stranded RNA is extremely susceptible to nuclease digestion, may lead to immune system activation, and is too large and negatively charged to cross cell membranes (Crooke, 1999; Laina et al., 2018). However, significant advances in the design of RNA therapeutics have been able to overcome these challenges to the point that numerous clinical trials are currently underway employing RNAi-based drugs for both cardiac and noncardiac targets (Kaczmarek et al., 2017; Laina et al., 2018).
RNAi therapeutics fall into several different categories, including single-stranded antisense oligonucleotides (ASOs), double-stranded siRNAs, and miRNAs (Laina et al., 2018). ASOs are short (typically 20 base pairs in length) single-stranded synthetic molecules that take advantage of Watson–Crick base-pairing to bind a target mRNA to induce its degradation through endogenous RNAse activity (Crooke, 1999). Depending on the design of the ASO, it can also block ribosomal attachment to reduce target protein expression or even redirect splicing factors to lead to the inclusion or exclusion of targeted exons (Dominski and Kole, 1993; Havens and Hastings, 2016). The latter mode is the mechanism of action of the FDA-approved RNA therapeutic eteplirsen (Exondys 51, Sarepta Therapeutics) for treating Duchenne muscular dystrophy, which induces the skipping of exon 51 of the mutant dystrophin gene and restoring the proper translational reading frame for dystrophin expression (Dystrophy et al., 2013). Therapeutic siRNAs are also synthetic molecules used to silence target genes but are instead double-stranded molecules ranging in length from 19 to 25 base pairs (Siomi and Siomi, 2009). These molecules are recognized by Argonaute 2 and the RNA-induced silencing complex (RISC) and unwound into single-stranded components. The sense strand is degraded and the antisense binds to a target mRNA sequence to induce cleavage by Argonaute 2 and degradation by exonucleases (Sledz et al., 2003; Liu et al., 2004; Rand et al., 2005; Meister, 2013; Ozcan et al., 2015). Finally, miRNAs are endogenous small noncoding RNAs that target multiple mRNAs, silencing target gene expression either by mRNA degradation or translational inhibition. Typically, miRNAs target the 3'-untranslated region (UTR) of the target mRNA through a primary seed sequence eight nucleotides long, with the degree of repression being modulated by the complementarity and nucleotide composition of flanking sequences. These molecules are natively expressed within the cell either under regulation of their own discrete promoter or found within intronic sequences of coding genes. Therefore, therapeutic treatments have focused both on their inhibition through the delivery of antisense sequences acting as complementary decoys to the miRNA as well as direct overexpression of miRNA mimics.
In effect, each of these RNAi-based platforms could theoretically be applied to the inhibition of CaMKII in the heart. While an ASO could be designed with a particular target sequence in mind, there is also precedence for physiologic regulation of CaMKIIδ by a number of native miRNAs to abrogate cardiac remodeling (Wang et al., 2010; Aurora et al., 2012; Cha et al., 2013; He et al., 2013). A multitude of preclinical and clinical trials have employed the use of chemically modified RNAi backbones and encapsulation in nanoparticles to improve pharmacokinetics, escape nuclease activity, enhance target mRNA binding, and minimize toxicity (Lucas and Dimmeler, 2016). Importantly, a major potential advantage in using RNA technology is that it offers the potential for increased precision. Small molecule drugs frequently suffer from impacting unintended secondary targets due to either conservation of enzymatic binding sites (like ATP-competitive sites) or impacting unintended pathways. However, all protein expression is derived from coding mRNA containing nucleic acid sequences significantly more unique to each individual gene, thereby providing a layer of intended target specificity difficult to match through pharmacologic drug design. Even more notably, this precision can even extend to specific isoform targeting of CaMKII. Not only could an siRNA molecule be designed that would specifically target transcripts encoding the CaMKIIδ isoform, circumventing the burden of CaMKII inhibition in the brain where CaMKIIα and β predominate, but may even offer the advantage of specific splice variant targeting of CaMKIIδ by focusing on specific exon–exon junctions unique to each variant. As previous literature has been able to identify, there are splice variant-specific effects of CaMKIIδ in cardiac disease and remodeling, revealing a potential protective role of CaMKIIδB, in comparison to the other cardiac variant CaMKIIδC (Little et al., 2009; Peng et al., 2010; Lu et al., 2011a).
Moreover, as mentioned, ASO-based therapeutics are also capable of influencing the access of splicing factors to induce the inclusion or skipping of certain exons based on their design. Through such a process, it would be possible to induce a state of increased exon inclusion that favors the variant CaMKIIδB, leading to its increased expression relative to CaMKIIδC. The consequence of this shift in relative abundances can alter the stoichiometry within the holoenzyme structure, imparting the localization pattern of the isoform that dominates within the complex. Such manipulation of the relative composition has led to the increased nuclear localization of the CaMKIIδC isoform when the holoenzyme is composed of more CaMKIIδB (Srinivasan et al., 1994). Given that nuclear activity of CaMKIIδC has been shown to lead to prosurvival mechanisms within cardiomyocytes and reduced pathologic remodeling (Little et al., 2009; Peng et al., 2010; Lu et al., 2011b), the manipulation of CaMKIIδ at the splicing level through RNA-based mechanisms offers a theoretically plausible mechanism for improving HF treatment that might not even require targeting CaMKII inhibition.
One of the remaining challenges for applying RNA-based CaMKII therapy is selective targeting of heart. Current application of these RNA agents is typically through intravenous or subcutaneous injection, exposing the drugs to systemic circulation. While chemical modification to these drugs can prevent crossing of the blood–brain barrier, this is likely insufficient to prevent undesired consequences for the ubiquitously expressed CaMKIIδ. While this barrier is not unique to RNA therapeutics (also a consideration for small molecule inhibition), there are options that are unique to RNA therapeutics for potentially overcoming this limitation, including gene therapy which could utilize cardiac specific promoters (limited to miRNAs for polymerase compatibility) or local delivery/injection directly into cardiac tissue. There is also exciting opportunity to combine emerging nanoparticle therapy (discussed in previous section) to carry RNA-based drugs instead of peptide cargos through the same inhalation delivery to deliver directly to the heart.
Therapeutic Targeting of the CaMKII Signaling Pathway
When considering CaMKII as a therapeutic target, it is important to acknowledge opportunities for manipulating the CaMKII signaling pathway without directly targeting the kinase itself. As discussed previously, CaMKII regulates a large number of intracellular substrates (ion channels, transcription factors, Ca2+ handling proteins) that may serve as attractive targets for electrical and hypertrophic remodeling. Perhaps the best known example of therapy aimed at a downstream target in the CaMKII pathway comes from ranolazine, an approved antianginal agent that inhibits late Na+ current among other actions (Rayner-Hartley and Sedlak, 2015). Late Na+ current, through the actions of the sodium Ca2+ exchanger, may impact intracellular Ca2+, contributing to further activation of CaMKII as well as driving the incidence of arrhythmogenic DADs. Ranolazine has been used successfully in rodents and large animals to block these changes and prevent the development of hypertrophy, HF, and arrhythmias (Rastogi et al., 2008; Figueredo et al., 2011; Glynn et al., 2015; Liang et al., 2016; Ellermann et al., 2018; Nie et al., 2019). However, results from clinical trials have been mixed with data supporting that ranolazine is safe with questionable efficacy in preventing AF recurrence (RAFAELLO trial), ventricular tachycardia (VT) or ventricular fibrillation (VF) following implantation of cardioverter defibrillator (RAID trial), or improving functional capacity in hypertrophic cardiomyopathy (RESTYLE-HCM trial) (De Ferrari et al., 2015; Bengel et al., 2017; Olivotto et al., 2018; Zareba et al., 2018).
RyR2 is another potential therapeutic target downstream of CaMKII signaling (Wehrens et al., 2004; Guo et al., 2006; Yang et al., 2007; Wehrens, 2011). Compounds referred to as rycals are small molecules that aim to stabilize RyR2 in its closed conformation through enhanced association with its binding partner calstabin, without blocking the channel or impairing normal Ca2+ signaling. JTV519 (K201) is one such compound that has been shown to improve Ca2+ leak and left ventricular function in a canine model of HF (Yano et al., 2003). However, translatability of JTV519 is somewhat limited by several off-target effects on other cardiac channels (Kaneko et al., 2009). S107 is another more selective rycal (Bellinger et al., 2008) with demonstrated ability to protect against ventricular arrhythmias in a mouse model with a RyR2 gain-of-function mutation (Lehnart et al., 2008) in addition to reversing the development of HF in a mouse model of constitutively phosphorylated RyR2 (Shan et al., 2010). Molecules such as these are being prepared for clinical testing and represent promising avenues for attenuating the impact of CaMKII activation.
Besides searching downstream of CaMKII for druggable targets, it is important to consider that the functional consequences of any kinase is determined by a delicate balance between the kinase and antagonism by a phosphatase. The majority of phosphatase activity in the heart is due to type-1 phosphatases (PP1) and type-2 phosphatases [PP2A, PP2B (calcineurin)] (El-Armouche and Eschenhagen, 2009), which collectively have been implicated in either dephosphorylating proteins targeted by CaMKII kinase activity or targeting CaMKII directly, removing its autonomous activation (Chiang et al., 2016; Lubbers and Mohler, 2016) (Figure 1D). Therefore, it may be possible to balance increased CaMKII activity in disease by simultaneously activating associated phosphatases. For example, the PP2A activator FTY720 has been shown to attenuate hypertrophic remodeling and protect hearts from ischemic injury in the mouse (Egom et al., 2010; Liu et al., 2011; Liu et al., 2013). However, one possible complication with this approach is that increased phosphatase activity has been linked to development of HF (Parra and Rothermel, 2017). Transgenic activation of CaMKII has itself been shown to increase phosphatase activity (Zhang et al., 2002), but it is unclear if this is a compensatory change or a cofactor in driving disease. While maximally activated phosphatase activity is likely to amplify adverse remodeling as the transgenic models have shown, it may be possible to implement a more nuanced approach with the goal of maintaining proper balance between kinase and phosphatase activity. Given that the majority of phosphatase activity is supplied by just three genes, unlike kinases which have numerous members with highly specific functions, phosphatase targeting is regulated by a multitude of binding partners and anchoring proteins (Shi, 2009). Cav1.2 dephosphorylation for example is dependent on PP2A and its interaction with regulatory subunits B56α and PR59 (Hall et al., 2006; Xu et al., 2010), while spinophilin and regulatory subunit PR130 target PP1 and PP2A to RyR2 (Bers, 2002; Lanner et al., 2010). Future studies may reveal critical subunits which direct phosphatases to relevant CaMKII targets, or to CaMKII itself, through which drug targeting or gene regulation would provide opportunity to influence the extent of that precise interaction. This may provide opportunity to limit adverse outcomes observed in global and unchecked phosphatase induction (Gergs et al., 2004; Hoehn et al., 2015), but clearly a great deal of work is still required to even establish proof of principle.
A unique opportunity for further discerning the potential impact of phosphatase activation may lie in the field of cancer therapeutics. Currently, cancer treatment strategies are beginning to consider phosphatase activators in earnest for tumor disruption (McClinch et al., 2018; Allen-Petersen and Sears, 2019; Remmerie and Janssens, 2019). It is well established that cardiac toxicity is a significant side effect following the use of the common cancer drug, doxorubicin, which has even been associated with CaMKII signaling (Yeh and Bickford, 2009; Sag et al., 2011). Therefore, it would be interesting to observe potential differences in the development of cardiomyopathy with the inclusion of these phosphatase activators when used in conjunction with doxorubicin to provide therapeutic insight and predictive data to the prospect of similar phosphatase activators being utilized for cardioprotective therapies.
Finally, when searching for alternative targets in the CaMKII pathway, it is worth considering scaffolding, cytoskeletal and other interacting proteins important for subcellular localization of CaMKII to specific signaling nanodomains. For example, a role for the cytoskeletal protein βIV-spectrin has been identified in targeting CaMKII to the cardiomyocyte intercalated disc to facilitate regulation of Nav1.5 (Hund et al., 2010). Genetic disruption of βIV-spectrin/CaMKII interaction selectively disrupted the subpopulation of CaMKII at the intercalated disc and prevented stress-induced phosphorylation of Nav1.5. Subsequent studies found that disruption of βIV-spectrin/CaMKII interaction abrogated maladaptive cardiac remodeling in a mouse model of pressure overload (Unudurthi et al., 2018). Thus, it may prove beneficial to develop pharmacological or genetic ways to interfere with spectrin/CaMKII interaction allowing for selective inhibition of a pathological CaMKII subpopulation while leaving the rest of the signaling pathway intact.
Conclusion
Given the effectiveness of CaMKII inhibition in improving cardiac function and reducing arrhythmia burden in animal models of disease, it is reasonable to remain optimistic that targeting of CaMKII signaling has a future in cardiac translational therapy. While conventional delivery routes and strategies merit further investigation, it will be important going forward to be aggressive in the search for new delivery routes and targets that may accelerate translation of basic science discoveries to human patients. Recent advances in genetic techniques and/or particle deliveries provide entirely new paradigms for not just treating familial diseases but acquired forms as well. Therefore, the development of CaMKII therapeutics might not only represent a breakthrough for cardiac treatment but, depending on the routes taken, may open up an entirely new branch in the models we use to treat a broader range of disease.
Author Contributions
DN, DG, and TH drafted and revised the manuscript.
Funding
This work was supported by the National Institutes of Health (grant numbers R01-HL135096 and R01-HL134824 to TH) and the American Heart Association (postdoctoral fellowship to DN).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Allen-Petersen, B. L., Sears, R. C. (2019). The use of protein phosphatase 2A activators in combination therapies for pancreas cancer. Oncotarget 10, 2008–2009. doi: 10.18632/oncotarget.26772
Anderson, M. E., Braun, A. P., Wu, Y., Lu, T., Wu, Y., Schulman, H., et al. (1998). KN-93, an inhibitor of multifunctional Ca++/calmodulin-dependent protein kinase, decreases early afterdepolarizations in rabbit heart. J. Pharmacol. Exp. Ther. 287, 996–1006.
Anderson, M. E., Brown, J. H., Bers, D. M. (2011). CaMKII in myocardial hypertrophy and heart failure. J. Mol. Cell. Cardiol. 51, 468–473. doi: 10.1016/j.yjmcc.2011.01.012
Anderson, M. E. (2015). Oxidant stress promotes disease by activating CaMKII. J. Mol. Cell. Cardiol. 89, 160–167. doi: 10.1016/j.yjmcc.2015.10.014
Aurora, A. B., Mahmoud, A. I., Luo, X., Johnson, B. A., van Rooij, E., Matsuzaki, S., et al. (2012). MicroRNA-214 protects the mouse heart from ischemic injury by controlling Ca2+ overload and cell death. J. Clin. Invest. 122, 1222–1232. doi: 10.1172/JCI59327
Backs, J., Song, K., Bezprozvannaya, S., Chang, S., Olson, E. N. (2006). CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J. Clin. Invest. 116, 1853–1864. doi: 10.1172/JCI27438
Backs, J., Backs, T., Bezprozvannaya, S., McKinsey, T. A., Olson, E. N. (2008). Histone deacetylase 5 acquires calcium/calmodulin-dependent kinase II responsiveness by oligomerization with histone deacetylase 4. Mol. Cell. Biol. 28, 3437–3445. doi: 10.1128/MCB.01611-07
Backs, J., Backs, T., Neef, S., Kreusser, M. M., Lehmann, L. H., Patrick, D. M., et al. (2009). The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc. Natl. Acad. Sci. U. S. A. 106, 2342–2347. doi: 10.1073/pnas.0813013106
Beauverger, P., Ozoux, M.-L., Bégis, G., Glénat, V., Briand, V., Philippo, M.-C., et al. (2019). Reversion of cardiac dysfunction by a novel orally available calcium/calmodulin-dependent protein kinase II inhibitor, RA306, in a genetic model of dilated cardiomyopathy. Cardiovasc. Res. doi: 10.1093/cvr/cvz097
Beckendorf, J., van den Hoogenhof, M. M. G., Backs, J. (2018). Physiological and unappreciated roles of CaMKII in the heart. Basic Res. Cardiol. 113, 29. doi: 10.1007/s00395-018-0688-8
Bellinger, A. M., Reiken, S., Dura, M., Murphy, P. W., Deng, S.-X., Landry, D. W., et al. (2008). Remodeling of ryanodine receptor complex causes “leaky” channels: a molecular mechanism for decreased exercise capacity. Proc. Natl. Acad. Sci. U. S. A. 105, 2198–2202. doi: 10.1073/pnas.0711074105
Bengel, P., Ahmad, S., Sossalla, S. (2017). Inhibition of Late Sodium Current as an Innovative Antiarrhythmic Strategy. Curr. Heart Fail. Rep. 14, 179–186. doi: 10.1007/s11897-017-0333-0
Bennett, M. K., Erondu, N. E., Kennedy, M. B. (1983). Purification and characterization of a calmodulin-dependent protein kinase that is highly concentrated in brain. J. Biol. Chem. 258, 12735–12744.
Bers, D. M. (2002). Cardiac excitation-contraction coupling. Nature 415, 198–205. doi: 10.1038/415198a
Bhullar, K. S., Lagarón, N. O., McGowan, E. M., Parmar, I., Jha, A., Hubbard, B. P., et al. (2018). Kinase-targeted cancer therapies: progress, challenges and future directions. Mol. Cancer 17, 48. doi: 10.1186/s12943-018-0804-2
Braun, A. P., Schulman, H. (1995). A non-selective cation current activated via the multifunctional Ca(2+)-calmodulin-dependent protein kinase in human epithelial cells. J. Physiol. 488, 37–55. doi: 10.1113/jphysiol.1995.sp020944
Cha, M.-J., Jang, J.-K., Ham, O., Song, B.-W., Lee, S.-Y., Lee, C. Y., et al. (2013). MicroRNA-145 suppresses ROS-induced Ca2+ overload of cardiomyocytes by targeting CaMKIIδ. Biochem. Biophys. Res. Commun. 435, 720–726. doi: 10.1016/j.bbrc.2013.05.050
Chang, B. H., Mukherji, S., Soderling, T. R. (1998). Characterization of a calmodulin kinase II inhibitor protein in brain. Proc. Natl. Acad. Sci. U. S. A. 95, 10890–10895. doi: 10.1073/pnas.95.18.10890
Chang, B. H., Mukherji, S., Soderling, T. R. (2001). Calcium/calmodulin-dependent protein kinase II inhibitor protein: Localization of isoforms in rat brain. Neuroscience 102, 767–777. doi: 10.1016/S0306-4522(00)00520-0
Chiang, D. Y., Heck, A. J. R., Dobrev, D., Wehrens, X. H. T. (2016). Regulating the regulator: insights into the cardiac protein phosphatase 1 interactome. J. Mol. Cell. Cardiol. 101, 165–172. doi: 10.1016/j.yjmcc.2016.09.009
Coultrap, S. J., Bayer, K. U. (2011). Improving a natural CaMKII inhibitor by random and rational design. PloS One 6, e25245. doi: 10.1371/journal.pone.0025245
Coultrap, S. J., Freund, R. K., O'Leary, H., Sanderson, J. L., Roche, K. W., Dell'Acqua, M. L., et al. (2014). Autonomous CaMKII mediates both LTP and LTD using a mechanism for differential substrate site selection. Cell Rep. 6, 431–437. doi: 10.1016/j.celrep.2014.01.005
Crooke, S. T. (1999). Molecular mechanisms of action of antisense drugs. Biochim. Biophys. Acta - Gene Struct. Expr. 1489, 31–43. doi: 10.1016/S0167-4781(99)00148-7
De Ferrari, G. M., Maier, L. S., Mont, L., Schwartz, P. J., Simonis, G., Leschke, M., et al. (2015). Ranolazine in the treatment of atrial fibrillation: results of the dose-ranging RAFFAELLO (ranolazine in atrial fibrillation following an electrical cardioversion) study. Heart Rhythm. 12, 872–878. doi: 10.1016/j.hrthm.2015.01.021
Dominski, Z., Kole, R. (1993). Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proc. Natl. Acad. Sci. U. S. A. 90, 8673–8677. doi: 10.1073/pnas.90.18.8673
Dystrophy, M., Mendell, J. R., Rodino-klapac, L. R. (2013). Eteplirsen for the treatment of duchenne eteplirsen for the treatment of duchenne muscular dystrophy. Ann. Neurol. 74, 637–647. doi: 10.1002/ana.23982
Edman, C. F., Schulman, H. (1994). Identification and characterization of delta B-CaM kinase and delta C-CaM kinase from rat heart, two new multifunctional Ca2+/calmodulin-dependent protein kinase isoforms. Biochim. Biophys. Acta 1221, 89–101. doi: 10.1016/0167-4889(94)90221-6
Egom, E. E. A., Ke, Y., Musa, H., Mohamed, T. M. A., Wang, T., Cartwright, E., et al. (2010). FTY720 prevents ischemia/reperfusion injury-associated arrhythmias in an ex vivo rat heart model via activation of Pak1/Akt signaling. J. Mol. Cell. Cardiol. 48, 406–414. doi: 10.1016/j.yjmcc.2009.10.009
El-Armouche, A., Eschenhagen, T. (2009). Beta-adrenergic stimulation and myocardial function in the failing heart. Heart Fail. Rev. 14, 225–241. doi: 10.1007/s10741-008-9132-8
Ellermann, C., Kohnke, A., Dechering, D. G., Kochhäuser, S., Reinke, F., Fehr, M., et al. (2018). Ranolazine prevents levosimendan-induced atrial fibrillation. Pharmacology. 102, 3–4. doi: 10.1159/000490572
Erickson, J. R., Joiner, M. A., Guan, X., Kutschke, W., Yang, J., Oddis, C. V., et al. (2008). A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133, 462–474. doi: 10.1016/j.cell.2008.02.048
Erickson, J. R., Patel, R., Ferguson, A., Bossuyt, J., Bers, D. M. (2011). Fluorescence resonance energy transfer-based sensor camui provides new insight into mechanisms of calcium/calmodulin-dependent protein kinase II activation in intact cardiomyocytes. Circ. Res. 109, 729–738. doi: 10.1161/CIRCRESAHA.111.247148
Erickson, J. R., Pereira, L., Wang, L., Han, G., Ferguson, A., Dao, K., et al. (2013). Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 502, 372–376. doi: 10.1038/nature12537
Erickson, J. R., Nichols, C. B., Uchinoumi, H., Stein, M. L., Bossuyt, J., Bers, D. M. (2015). S-nitrosylation induces both autonomous activation and inhibition of calcium/calmodulin-dependent protein Kinase II δ. J. Biol. Chem. 290, 25646–25656. doi: 10.1074/jbc.M115.650234
Figueredo, V. M., Pressman, G. S., Romero-Corral, A., Murdock, E., Holderbach, P., Morris, D. L. (2011). Improvement in left ventricular systolic and diastolic performance during ranolazine treatment in patients with stable angina. J. Cardiovasc. Pharmacol. Ther. 16, 168–172. doi: 10.1177/1074248410382105
Fordyce, C. B., Roe, M. T., Ahmad, T., Libby, P., Borer, J. S., Hiatt, W. R., et al. (2015). Cardiovascular drug development: is it dead or just hibernating? J. Am. Coll. Cardiol. 65, 1567–1582. doi: 10.1016/j.jacc.2015.03.016
Gao, Y., Davies, S. P., Augustin, M., Woodward, A., Patel, U. A., Kovelman, R., et al. (2013). A broad activity screen in support of a chemogenomic map for kinase signalling research and drug discovery. Biochem. J. 451, 313–328. doi: 10.1042/BJ20121418
Gergs, U., Boknik, P., Buchwalow, I., Fabritz, L., Matus, M., Justus, I., et al. (2004). Overexpression of the catalytic subunit of protein phosphatase 2A impairs cardiac function. J. Biol. Chem. 279, 40827–40834. doi: 10.1074/jbc.M405770200
Glynn, P., Musa, H., Wu, X., Unudurthi, S. D., Little, S., Qian, L., et al. (2015). Voltageg-gated sodium channel phosphorylation at Ser571 regulates late current, arrhythmia, and cardiac function in vivo. Circulation. 132, 567–577. doi: 10.1161/CIRCULATIONAHA.114.015218
Grueter, C. E., Abiria, S. A., Dzhura, I., Wu, Y., Ham, A.-J. L., Mohler, P. J., et al. (2006). L-type Ca2+ channel facilitation mediated by phosphorylation of the beta subunit by CaMKII. Mol. Cell 23, 641–650. doi: 10.1016/j.molcel.2006.07.006
Guo, T., Zhang, T., Mestril, R., Bers, D. M. (2006). Ca2+/Calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ. Res. 99, 398–406. doi: 10.1161/01.RES.0000236756.06252.13
Gutierrez, D. A., Fernandez-Tenorio, M., Ogrodnik, J., Niggli, E. (2013). NO-dependent Ca MKII activation during β-adrenergic stimulation of cardiac muscle. Cardiovasc. Res. 100, 392–401. doi: 10.1093/cvr/cvt201
Hall, D. D., Feekes, J. A., Arachchige Don, A. S., Shi, M., Hamid, J., Chen, L., et al. (2006). Binding of protein phosphatase 2A to the L-type calcium channel Cav1.2 next to Ser1928, its main PKA site, is critical for Ser1928 dephosphorylation. Biochemistry 45, 3448–3459. doi: 10.1021/bi051593z
Havens, M. A., Hastings, M. L. (2016). Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 44, 6549–6563. doi: 10.1093/nar/gkw533
He, B. J., Joiner, M.-L. A., Singh, M. V., Luczak, E. D., Swaminathan, P. D., Koval, O. M., et al. (2011). Oxidation of CaMKII determines the cardiotoxic effects of aldosterone. Nat. Med. 17, 1610–1618. doi: 10.1038/nm.2506
He, J., Jiang, S., Li, F., Zhao, X., Chu, E., Sun, M., et al. (2013). MicroRNA-30b-5p is involved in the regulation of cardiac hypertrophy by targeting CaMKIIδ. J. Investig. Med. 61, 604–612. doi: 10.2310/JIM.0b013e3182819ac6
Hegyi, B., Chen-Izu, Y., Jian, Z., Shimkunas, R., Izu, L. T., Banyasz, T. (2015). KN-93 inhibits IKr in mammalian cardiomyocytes. J. Mol. Cell. Cardiol. 89, 173–176. doi: 10.1016/j.yjmcc.2015.10.012
Hoch, B., Meyer, R., Hetzer, R., Krause, E. G., Karczewski, P. (1999). Identification and expression of delta-isoforms of the multifunctional Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circ. Res. 84, 713–721. doi: 10.1161/01.RES.84.6.713
Hoehn, M., Zhang, Y., Xu, J., Gergs, U., Boknik, P., Werdan, K., et al. (2015). Overexpression of protein phosphatase 2A in a murine model of chronic myocardial infarction leads to increased adverse remodeling but restores the regulation of β-catenin by glycogen synthase kinase 3β. Int. J. Cardiol. 183, 39–46. doi: 10.1016/j.ijcard.2015.01.087
Howard, T., Greer-Short, A., Satroplus, T., Patel, N., Nassal, D., Mohler, P. J., et al. (2018). CaMKII-dependent late Na+ current increases electrical dispersion and arrhythmia in ischemia-reperfusion. Am. J. Physiol. - Hear. Circ. Physiol. 315, H794–H801. doi: 10.1152/ajpheart.00197.2018
Hudmon, A., Schulman, H., Kim, J., Maltez, J. M., Tsien, R. W., Pitt, G. S. (2005). CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J. Cell Biol. 171, 537–547. doi: 10.1083/jcb.200505155
Hund, T. J., Koval, O. M., Li, J., Wright, P. J., Qian, L., Snyder, J. S., et al. (2010). A βIV-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J. Clin. Invest. 120, 3508–3519. doi: 10.1172/JCI43621
Ishida, A., Kameshita, I., Okuno, S., Kitani, T., Fujisawa, H. (1995). A novel highly specific and potent inhibitor of calmodulin-dependent protein kinase II. Biochem. Biophys. Res. Commun. 212, 806–812. doi: 10.1006/bbrc.1995.2040
Johnson, C. N., Pattanayek, R., Potet, F., Rebbeck, R. T., Blackwell, D. J., Nikolaienko, R., et al. (2019). The CaMKII inhibitor KN93-calmodulin interaction and implications for calmodulin tuning of NaV1.5 and RyR2 function. Cell Calcium. 82, 102063. doi: 10.1016/j.ceca.2019.102063
Joiner, M. L. A., Koval, O. M. (2014). CaMKII and stress mix it up in mitochondria. Front. Pharmacol. 5, 67. doi: 10.3389/fphar.2014.00067
Joiner, M.-L. A., Koval, O. M., Li, J., He, B. J., Allamargot, C., Gao, Z., et al. (2012). CaMKII determines mitochondrial stress responses in heart. Nature 491, 269–273. doi: 10.1038/nature11444
Kaczmarek, J. C., Kowalski, P. S., Anderson, D. G. (2017). Advances in the delivery of RNA therapeutics: from concept to clinical reality. Genome Med. 9, 60. doi: 10.1186/s13073-017-0450-0
Kaneko, N., Matsuda, R., Hata, Y., Shimamoto, K. (2009). Pharmacological characteristics and clinical applications of K201. Curr. Clin. Pharmacol. 4, 126–131. doi: 10.2174/157488409788184972
Khoo, M. S. C., Li, J., Singh, M. V., Yang, Y., Kannankeril, P., Wu, Y., et al. (2006). Death, cardiac dysfunction, and arrhythmias are increased by calmodulin kinase II in calcineurin cardiomyopathy. Circulation 114, 1352–1359. doi: 10.1161/CIRCULATIONAHA.106.644583
Kirchhefer, U., Schmitz, W., Scholz, H., Neumann, J. (1999). Activity of cAMP-dependent protein kinase and Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human hearts. Cardiovasc. Res. 42, 254–261. doi: 10.1016/S0008-6363(98)00296-X
Koval, O. M., Guan, X., Wu, Y., Joiner, M. L., Gao, Z., Chen, B., et al. (2010). Ca V 1.2 β-subunit coordinates CaMKII-triggered cardiomyocyte death and afterdepolarizations. Proc. Natl. Acad. Sci. U. S. A. 107, 4996–5000. doi: 10.1073/pnas.0913760107
Koval, O. M., Snyder, J. S., Wolf, R. M., Pavlovicz, R. E., Glynn, P., Curran, J., et al. (2012). Ca2+/calmodulin-dependent protein kinase ii-based regulation of voltage-gated na+ channel in cardiac disease. Circulation. 126, 2084–2094. doi: 10.1161/CIRCULATIONAHA.112.105320
Laina, A., Gatsiou, A., Georgiopoulos, G., Stamatelopoulos, K., Stellos, K. (2018). RNA therapeutics in cardiovascular precision medicine. Front. Physiol. 9, 953. doi: 10.3389/fphys.2018.00953
Lanner, J. T., Georgiou, D. K., Joshi, A. D., Hamilton, S. L. (2010). Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2, a003996. doi: 10.1101/cshperspect.a003996
Lebek, S., Plößl, A., Baier, M., Mustroph, J., Tarnowski, D., Lücht, C. M., et al. (2018). The novel CaMKII inhibitor GS-680 reduces diastolic SR Ca leak and prevents CaMKII-dependent pro-arrhythmic activity. J. Mol. Cell. Cardiol. 118, 159–168. doi: 10.1016/j.yjmcc.2018.03.020
Ledoux, J., Chartier, D., Leblanc, N. (1999). Inhibitors of calmodulin-dependent protein kinase are nonspecific blockers of voltage-dependent K+ channels in vascular myocytes. J. Pharmacol. Exp. Ther. 290, 1165–1174.
Lehnart, S. E., Mongillo, M., Bellinger, A., Lindegger, N., Chen, B.-X., Hsueh, W., et al. (2008). Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J. Clin. Invest. 118, 2230–2245. doi: 10.1172/JCI35346
Liang, F., Fan, P., Jia, J., Yang, S., Jiang, Z., Karpinski, S., et al. (2016). Inhibitions of late INa and CaMKII act synergistically to prevent ATX-II-induced atrial fibrillation in isolated rat right atria. J. Mol. Cell. Cardiol. 94, 122–130. doi: 10.1016/j.yjmcc.2016.04.001
Ling, H., Zhang, T., Pereira, L., Means, C. K., Cheng, H., Gu, Y., et al. (2009). Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J. Clin. Invest. 119, 1230–1240. doi: 10.1172/JCI38022
Little, G. H., Saw, A., Bai, Y., Dow, J., Marjoram, P., Simkhovich, B., et al. (2009). Critical role of nuclear calcium/calmodulin-dependent protein kinase IIδB in cardiomyocyte survival in cardiomyopathy. J. Biol. Chem. 284, 24857–24868. doi: 10.1074/jbc.M109.003186
Liu, J., Carmell, M. A., Rivas, F. V., Marsden, C. G., Thomson, J. M., Song, J. J., et al. (2004). Argonaute2 is the catalytic engine of mammalian RNAi. Science (80-.), 1437–1441. doi: 10.1126/science.1102513
Liu, W., Zi, M., Naumann, R., Ulm, S., Jin, J., Taglieri, D. M., et al. (2011). Pak1 as a novel therapeutic target for antihypertrophic treatment in the heart. Circulation 124, 2702–2715. doi: 10.1161/CIRCULATIONAHA.111.048785
Liu, W., Zi, M., Tsui, H., Chowdhury, S. K., Zeef, L., Meng, Q.-J., et al. (2013). A novel immunomodulator, FTY-720 reverses existing cardiac hypertrophy and fibrosis from pressure overload by targeting NFAT (nuclear factor of activated T-cells) signaling and periostin. Circ. Heart Fail. 6, 833–844. doi: 10.1161/CIRCHEARTFAILURE.112.000123
Lokuta, A. J., Rogers, T. B., Lederer, W. J., Valdivia, H. H. (1995). Modulation of cardiac ryanodine receptors of swine and rabbit by a phosphorylation-dephosphorylation mechanism. J. Physiol. 487, 609–622. doi: 10.1113/jphysiol.1995.sp020904
Lu, Y.-M., Huang, J., Shioda, N., Fukunaga, K., Shirasaki, Y., Li, X.-M., et al. (2011a). CaMKIIδB mediates aberrant NCX1 expression and the imbalance of NCX1/SERCA in transverse aortic constriction-induced failing heart. PloS One 6, e24724. doi: 10.1371/journal.pone.0024724
Lu, Y. M., Huang, J., Shioda, N., Fukunaga, K., Shirasaki, Y., Li, ,. X. M., et al. (2011b). Camkiiδb mediates aberrant NCX1 expression and the imbalance of NCX1/SERCA in transverse aortic Constriction-Induced failing heart. PloS One 6, e24724. doi: 10.1371/journal.pone.0024724
Lubbers, E. R., Mohler, P. J. (2016). Roles and regulation of protein phosphatase 2A (PP2A) in the heart. J. Mol. Cell. Cardiol. 101, 127–133. doi: 10.1016/j.yjmcc.2016.11.003
Lucas, T., Dimmeler, S. (2016). RNA therapeutics for treatment of cardiovascular diseases. Circ. Res. 119, 794–797. doi: 10.1161/CIRCRESAHA.116.308730
Luo, M., Guan, X., Luczak, E. D., Lang, D., Kutschke, W., Gao, Z., et al. (2013). Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J. Clin. Invest. 123, 1262–1274. doi: 10.1172/JCI65268
Maier, L. S., Bers, D. M. (2002). Calcium, calmodulin, and calcium-calmodulin kinase II: heartbeat to heartbeat and beyond. J. Mol. Cell. Cardiol. 34, 919–939. doi: 10.1006/jmcc.2002.2038
Malinow, R., Schulman, H., Tsien, R. W. (1989). Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science (80-.), 862–866. doi: 10.1126/science.2549638
Mattiazzi, A., Kranias, E. G. (2014). The role of CaMKII regulation of phospholamban activity in heart disease. Front. Pharmacol. 5, 5. doi: 10.3389/fphar.2014.00005
Mattiazzi, A., Bassani, R. A., Escobar, A. L., Palomeque, J., Valverde, C. A., Vila Petroff, M., et al. (2015). Chasing cardiac physiology and pathology down the caMKII cascade. Am. J. Physiol. - Hear. Circ. Physiol. 308, H1177–H1191. doi: 10.1152/ajpheart.00007.2015
Mavunkel, B., Xu, ,. Y. J., Goyal, B., Lim, D., Lu, Q., Chen, Z., et al. (2008). Pyrimidine-based inhibitors of CaMKIIδ. Bioorganic Med. Chem. Lett. 18, 2404–2408. doi: 10.1016/j.bmcl.2008.02.056
McClinch, K., Avelar, R. A., Callejas, D., Izadmehr, S., Wiredja, D., Perl, A., et al. (2018). Small-molecule activators of protein phosphatas 2A for the treatment of castration-resistant prostate cancer. Cancer Res. 78, 2065–2080. doi: 10.1158/0008-5472.CAN-17-0123
Meister, G. (2013). Argonaute proteins: functional insights and emerging roles. Nat. Rev. Genet. 14, 447–459. doi: 10.1038/nrg3462
Michalski, P. J. (2013). The delicate bistability of CaMKII. Biophys. J. 105, 794–806. doi: 10.1016/j.bpj.2013.06.038
Mills, N. L., Donaldson, K., Hadoke, P. W., Boon, N. A., MacNee, W., Cassee, F. R., et al. (2009). Adverse cardiovascular effects of air pollution. Nat. Clin. Pract. Cardiovasc. Med. 6, 36–44. doi: 10.1038/ncpcardio1399
Miragoli, M., Ceriotti, P., Iafisco, M., Vacchiano, M., Salvarani, N., Alogna, A., et al. (2018). Inhalation of peptide-loaded nanoparticles improves heart failure. Sci. Transl. Med. 10, eaan6205. doi: 10.1126/scitranslmed.aan6205
Morotti, S., Edwards, A. G., Mcculloch, A. D., Bers, D. M., Grandi, E. (2014). A novel computational model of mouse myocyte electrophysiology to assess the synergy between Na+loading and CaMKII. J. Physiol. 592, 1181–1197. doi: 10.1113/jphysiol.2013.266676
Mustroph, J., Neef, S., Maier, L. S. (2017). CaMKII as a target for arrhythmia suppression. Pharmacol. Ther. 176, 22–31. doi: 10.1016/j.pharmthera.2016.10.006
Neef, S., Steffens, A., Pellicena, P., Mustroph, J., Lebek, S., Ort, K. R., et al. (2018). Improvement of cardiomyocyte function by a novel pyrimidine-based CaMKII-inhibitor. J. Mol. Cell. Cardiol. 115, 73–81. doi: 10.1016/j.yjmcc.2017.12.015
Nie, J., Duan, Q., He, M., Li, X., Wang, B., Zhou, C., et al. (2019). Ranolazine prevents pressure overload-induced cardiac hypertrophy and heart failure by restoring aberrant Na + and Ca 2+ handling. J. Cell. Physiol. 234, 11587–11601. doi: 10.1002/jcp.27791
Olivotto, I., Camici, P. G., Merlini, P. A., Rapezzi, C., Patten, M., Climent, V., et al. (2018). Efficacy of ranolazine in patients with symptomatic hypertrophic cardiomyopathy. Circ. Hear. Fail. 11, e004124. doi: 10.1161/CIRCHEARTFAILURE.117.004124
Onal, B., Gratz, D., Hund, T. J. (2017). Ca2+/calmodulin-dependent kinase II-dependent regulation of atrial myocyte late Na+ current, Ca2+ cycling, and excitability: a mathematical modeling study. Am. J. Physiol. Heart Circ. Physiol. 313, H1227–H1239. doi: 10.1152/ajpheart.00185.2017
Otvos, L., Wade, J. D. (2014). Current challenges in peptide-based drug discovery. Front. Chem. 2, 62. doi: 10.3389/fchem.2014.00062
Ozcan, G., Ozpolat, B., Coleman, R. L., Sood, A. K., Lopez-Berestein, G. (2015). Preclinical and clinical development of siRNA-based therapeutics. Adv. Drug Deliv. Rev. 87, 108–119. doi: 10.1016/j.addr.2015.01.007
Parra, V., Rothermel, B. A. (2017). Calcineurin signaling in the heart: The importance of time and place. J. Mol. Cell. Cardiol. 103, 121–136. doi: 10.1016/j.yjmcc.2016.12.006
Payne, M. E., Fong, Y. L., Ono, T., Colbran, R. J., Kemp, B. E., Soderling, T. R., et al. (1988). Calcium/calmodulin-dependent protein kinase II. Characterization of distinct calmodulin binding and inhibitory domains. J. Biol. Chem. 263, 7190–7195.
Pellicena, P., Schulman, H. (2014). CaMKII inhibitors: from research tools to therapeutic agents. Front. Pharmacol. 5, 21. doi: 10.3389/fphar.2014.00021
Peng, W., Zhang, Y., Zheng, M., Cheng, H., Zhu, W., Cao, C. M., et al. (2010). Cardioprotection by CaMKII-δB is mediated by phosphorylation of heat shock factor 1 and subsequent expression of inducible heat shock protein 70. Circ. Res. 106, 102–110. doi: 10.1161/CIRCRESAHA.109.210914
Penny, W. F., Hammond, H. K. (2017). Randomized clinical trials of gene transfer for heart failure with reduced ejection fraction. Hum. Gene Ther. 28, 378–384. doi: 10.1089/hum.2016.166
Purohit, A., Rokita, A. G., Guan, X., Chen, B., Koval, O. M., Voigt, N., et al. (2013). Oxidized Ca(2+)/calmodulin-dependent protein kinase II triggers atrial fibrillation. Circulation 128, 1748–1757. doi: 10.1161/CIRCULATIONAHA.113.003313
Ramirez, M. T., Zhao, X. L., Schulman, H., Brown, J. H. (1997). The nuclear deltaB isoform of Ca2+/calmodulin-dependent protein kinase II regulates atrial natriuretic factor gene expression in ventricular myocytes. J. Biol. Chem. 272, 31203–31208. doi: 10.1074/jbc.272.49.31203
Rand, T. A., Petersen, S., Du, F., Wang, X. (2005). Argonaute2 cleaves the anti-guide strand of siRNA during RISC activation. Cell 123, 621–629. doi: 10.1016/j.cell.2005.10.020
Rastogi, S., Sharov, V. G., Mishra, S., Gupta, R. C., Blackburn, B., Belardinelli, L., et al. (2008). Ranolazine combined with enalapril or metoprolol prevents progressive LV dysfunction and remodeling in dogs with moderate heart failure. Am. J. Physiol. - Hear. Circ. Physiol. 295, H2149–H2155. doi: 10.1152/ajpheart.00728.2008
Rayner-Hartley, E., Sedlak, T. (2015). Ranolazine: a contemporary review. J. Am. Heart Assoc. 5, e003196. doi: 10.1161/JAHA.116.003196
Remmerie, M., Janssens, V. (2019). PP2A: a promising biomarker and therapeutic target in endometrial cancer. Front. Oncol. 9, 462. doi: 10.3389/fonc.2019.00462
Rezazadeh, S., Claydon, T. W., Fedida, D. (2006). KN-93 (2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)]amino-N-(4-chlorocinnamyl)-N-methylbenzylamine), a calcium/calmodulin-dependent protein kinase II inhibitor, is a direct extracellular blocker of voltage-gated potassium channels. J. Pharmacol. Exp. Ther. 317, 292–299. doi: 10.1124/jpet.105.097618
Sag, C. M., Köhler, A. C., Anderson, M. E., Backs, J., Maier, L. S. (2011). CaMKII-dependent SR Ca leak contributes to doxorubicin-induced impaired Ca handling in isolated cardiac myocytes. J. Mol. Cell. Cardiol. 51, 749–759. doi: 10.1016/j.yjmcc.2011.07.016
Shan, J., Betzenhauser, M. J., Kushnir, A., Reiken, S., Meli, A. C., Wronska, A., et al. (2010). Role of chronic ryanodine receptor phosphorylation in heart failure and β-adrenergic receptor blockade in mice. J. Clin. Invest. 120, 4375–4387. doi: 10.1172/JCI37649
Shi, Y. (2009). Serine/threonine phosphatases: mechanism through structure. Cell 139, 468–484. doi: 10.1016/j.cell.2009.10.006
Silva, A. J., Paylor, R., Wehner, J. M., Tonegawa, S. (1992a). Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science 257, 206–211. doi: 10.1126/science.1321493
Silva, A. J., Stevens, C. F., Tonegawa, S., Wang, Y. (1992b). Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science 257, 201–206. doi: 10.1126/science.1378648
Siomi, H., Siomi, M. C. (2009). On the road to reading the RNA-interference code. Nature 457, 396–404. doi: 10.1038/nature07754
Sledz, C. A., Holko, M., De Veer, M. J., Silverman, R. H., Williams, B. R. G. (2003). Activation of the interferon system by short-interfering RNAs. Nat. Cell Biol. 5, 834–839. doi: 10.1038/ncb1038
Smyth, J. T., Abbott, A. L., Lee, B., Sienaert, I., Kasri, N. N., De Smedt, H., et al. (2002). Inhibition of the inositol trisphosphate receptor of mouse eggs and A7r5 cells by KN-93 via a mechanism unrelated to Ca 2+/calmodulin-dependent protein kinase II antagonism. J. Biol. Chem. 277, 35061–35070. doi: 10.1074/jbc.M202928200
Srinivasan, M., Edman, C. F., Schulman, H. (1994). Alternative splicing introduces a nuclear localization signal that targets multifunctional CaM kinase to the nucleus. J. Cell Biol. 126, 839–852. doi: 10.1083/jcb.126.4.839
Stevens, C. F., Tonegawa, S., Wang, Y. (1994). The role of calcium-calmodulin kinase II in three forms of synaptic plasticity. Curr. Biol. 4, 687–693. doi: 10.1016/S0960-9822(00)00153-6
Sumi, M., Kiuchi, K., Ishikawa, T., Ishii, A., Hagiwara, M., Nagatsu, T., et al. (1991). The newly synthesized selective Ca2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochem. Biophys. Res. Commun. 181, 968–975. doi: 10.1016/0006-291X(91)92031-E
Swaminathan, P. D., Purohit, A., Soni, S., Voigt, N., Singh, M. V., Glukhov, A. V., et al. (2011). Oxidized CaMKII causes cardiac sinus node dysfunction in mice. J. Clin. Invest. 121, 3277–3288. doi: 10.1172/JCI57833
Swaminathan, P. D., Purohit, A., Hund, T. J., Anderson, M. E. (2012). Calmodulin-dependent protein kinase II: linking heart failure and arrhythmias. Circ. Res. 110, 1661–1677. doi: 10.1161/CIRCRESAHA.111.243956
Tobimatsu, T., Fujisawa, H. (1989). Tissue-specific expression of four types of rat calmodulin-dependent protein kinase II mRNAs. J. Biol. Chem. 264, 17907–17912. doi: 10.1016/0921-8696(89)90769-X
Unudurthi, S. D., Nassal, D., Greer-Short, A., Patel, N., Howard, T., Xu, X., et al. (2018). βIV-Spectrin regulates STAT3 targeting to tune cardiac response to pressure overload. J. Clin. Invest. 128, 5561–5572. doi: 10.1172/JCI99245
Verdine, G. L., Walensky, L. D. (2007). The challenge of drugging undruggable targets in cancer: lessons learned from targeting BCL-2 family members. Clin. Cancer Res. 13, 7264–7270. doi: 10.1158/1078-0432.CCR-07-2184
Vest, R. S., O'Leary, H., Coultrap, S. J., Kindy, M. S., Bayer, K. U. (2010). Effective post-insult neuroprotection by a novel Ca2+/calmodulin-dependent protein kinase II (CaMKII) inhibitor. J. Biol. Chem. 285, 20675–20682. doi: 10.1074/jbc.M109.088617
Wagner, S., Ruff, H. M., Weber, S. L., Bellmann, S., Sowa, T., Schulte, T., et al. (2011). Reactive oxygen species-activated Ca/calmodulin kinase IIδ is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circ. Res. 108, 555–565. doi: 10.1161/CIRCRESAHA.110.221911
Wang, X., Zhang, X., Ren, X.-P., Chen, J., Liu, H., Yang, J., et al. (2010). MicroRNA-494 targeting both proapoptotic and antiapoptotic proteins protects against ischemia/reperfusion-induced cardiac injury. Circulation 122, 1308–1318. doi: 10.1161/CIRCULATIONAHA.110.964684
Wehrens, X. H. T., Lehnart, S. E., Reiken, S. R., Marks, A. R. (2004). Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ. Res. 94, e61–e70. doi: 10.1161/01.RES.0000125626.33738.E2
Wehrens, X. H. T. (2011). CaMKII regulation of the cardiac ryanodine receptor and sarcoplasmic reticulum calcium release. Hear. Rhythm. 8, 323–325. doi: 10.1016/j.hrthm.2010.09.079
Westenbrink, B. D., Edwards, A. G., McCulloch, A. D., Brown, J. H. (2013). The promise of CaMKII inhibition for heart disease: preventing heart failure and arrhythmias. Expert Opin. Ther. Targets 17, 889–903. doi: 10.1517/14728222.2013.809064
Witcher, D. R., Kovacs, R. J., Schulman, H., Cefali, D. C., Jones, L. R. (1991). Unique phosphorylation site on the cardiac ryanodine receptor regulates calcium channel activity. J. Biol. Chem. 266, 11144–11152.
Wu, Y., Temple, J., Zhang, R., Dzhura, I., Zhang, W., Trimble, R., et al. (2002). Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation 106, 1288–1293. doi: 10.1161/01.CIR.0000027583.73268.E7
Wu, X., Zhang, T., Bossuyt, J., Li, X., McKinsey, T. A., Dedman, J. R., et al. (2006). Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J. Clin. Invest. 116, 675–682. doi: 10.1172/JCI27374
Xu, H., Ginsburg, K. S., Hall, D. D., Zimmermann, M., Stein, I. S., Zhang, M., et al. (2010). Targeting of protein phosphatases PP2A and PP2B to the C-terminus of the L-type calcium channel Ca v1.2. Biochemistry 49, 10298–10307. doi: 10.1021/bi101018c
Yang, D., Zhu, W.-Z., Xiao, B., Brochet, D. X. P., Chen, S. R. W., Lakatta, E. G., et al. (2007). Ca2+/calmodulin kinase II-dependent phosphorylation of ryanodine receptors suppresses Ca2+ sparks and Ca2+ waves in cardiac myocytes. Circ. Res. 100, 399–407. doi: 10.1161/01.RES.0000258022.13090.55
Yano, M., Kobayashi, S., Kohno, M., Doi, M., Tokuhisa, T., Okuda, S., et al. (2003). FKBP12.6-Mediated stabilization of calcium-release channel (ryanodine receptor) as a novel therapeutic strategy against heart failure. Circulation 107, 477–484. doi: 10.1161/01.CIR.0000044917.74408.BE
Yao, L., Fan, P., Jiang, Z., Viatchenko-Karpinski, S., Wu, Y., Kornyeyev, D., et al. (2011). Na v 1.5-dependent persistent Na + influx activates CaMKII in rat ventricular myocytes and N 1325 S mice. Am. J. Physiol. - Cell Physiol. 301, C577–C586. doi: 10.1152/ajpcell.00125.2011
Yeh, E. T. H., Bickford, C. L. (2009). Cardiovascular complications of cancer therapy: incidence, pathogenesis, diagnosis, and management. J. Am. Coll. Cardiol. 53, 2231–2247. doi: 10.1016/j.jacc.2009.02.050
Zareba, W., Daubert, J. P., Beck, C. A., Huang, D. T., Alexis, J. D., Brown, M. W., et al. (2018). Ranolazine in high-risk patients with implanted cardioverter-defibrillators: The RAID trial. J. Am. Coll. Cardiol. 72, 636–645. doi: 10.1016/j.jacc.2018.04.086
Zhang, T., Johnson, E. N., Gu, Y., Morissette, M. R., Sah, V. P., Gigena, M. S., et al. (2002). The cardiac-specific nuclear δ B isoform of Ca 2+/Calmodulin-dependent protein kinase II induces hypertrophy and dilated cardiomyopathy associated with increased protein phosphatase 2A activity. J. Biol. Chem. 277, 1261–1267. doi: 10.1074/jbc.M108525200
Zhang, T., Maier, L. S., Dalton, N. D., Miyamoto, S., Ross, J., Bers, D. M., et al. (2003). The δc isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ. Res. 92, 912–919. doi: 10.1161/01.RES.0000069686.31472.C5
Keywords: calmodulin kinase II, arrhythmias, heart failure, cardiovascular pharmacology, cardiac remodeling
Citation: Nassal D, Gratz D and Hund TJ (2020) Challenges and Opportunities for Therapeutic Targeting of Calmodulin Kinase II in Heart. Front. Pharmacol. 11:35. doi: 10.3389/fphar.2020.00035
Received: 04 November 2019; Accepted: 14 January 2020;
Published: 05 February 2020.
Edited by:
László Virág, University of Szeged, HungaryReviewed by:
Laetitia Pereira, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceBin Liu, Mississippi State University, United States
Copyright © 2020 Nassal, Gratz and Hund. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas J. Hund, VGhvbWFzLkh1bmRAb3N1bWMuZWR1