 Carlo Patrono
Carlo Patrono Bianca Rocca
Bianca Rocca
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 30 October 2019
Sec. Inflammation Pharmacology
Volume 10 - 2019 | https://doi.org/10.3389/fphar.2019.01244
This article is part of the Research TopicResolution of Inflammation: Mechanisms, Mediators & BiomarkersView all 14 articles
Thromboxane (TX) A2 is a chemically unstable lipid mediator involved in several pathophysiologic processes, including primary hemostasis, atherothrombosis, inflammation, and cancer. In human platelets, TXA2 is the major arachidonic acid derivative via the cyclooxygenase (COX)-1 pathway. Assessment of platelet TXA2 biosynthesis can be performed ex vivo through measurement of serum TXB2, an index of platelet COX-1 activity, as well as in vivo through measurement of urinary enzymatic metabolites, a non-invasive index of platelet activation. This article reviews the main findings of four decades of clinical investigation based on these analytical approaches, focusing on the measurement of TXA2 metabolites to characterize the pathophysiologic role of transiently or persistently enhanced platelet activation and to describe the clinical pharmacology of COX-1 inhibition in health and disease.
The pictorial description in 1962 of the aggregation of blood platelets by ADP, using a novel device called the Born aggregometer (Born, 1962), paved the way to quantitative assessment of platelet inhibition in vitro and ex vivo (reviewed by Born and Patrono, 2006). However, the possibility of establishing a mechanistic link between the inhibition of platelet prostanoid formation by aspirin (Smith and Willis, 1971) and inhibition of platelet aggregation had to wait the discovery of a novel pro-aggregating and vasoconstrictor prostanoid, thromboxane (TX)A2, as the major arachidonic acid derivative in human platelets (Hamberg et al., 1975). This discovery allowed the development of appropriate analytical tools to investigate platelet TXA2 biosynthesis and its inhibition by aspirin in human health and disease (reviewed by Born and Patrono, 2006). TXA2 is a pro-thrombotic, chemically unstable prostanoid, mostly synthesized via cyclooxygenase (COX)-1 and released by activated platelets (reviewed by Davì and Patrono, 2007). Two different biomarkers were characterized independently to assess TXA2 biosynthesis ex vivo, i.e., whole-blood TXB2 production (Patrono et al., 1980), and in vivo, i.e., urinary TXB2 metabolite excretion (Roberts et al., 1981) (Figure 1). By raising a specific antibody against TXB2, the chemically stable and biologically inactive hydrolysis product of TXA2, we were able to measure the time-dependent synthesis and release of platelet TXA2 induced by endogenous thrombin generated during whole-blood clotting and to demonstrate its suppression by low doses of aspirin (Patrono et al., 1980). The pioneering work of John Oates and his associates at Vanderbilt University was responsible for the development of a non-invasive approach to investigating prostanoid biosynthesis in man, based on the measurements of urinary prostanoid metabolites. In 1981, they reported the conversion of systemically infused TXB2 into 20 enzymatic derivatives, which were identified in the urine of a single healthy volunteer by gas chromatography/mass spectrometry (GC/MS) (Roberts et al., 1981). The availability of these analytical tools paved the way for investigating TXA2 biosynthesis in health and disease and its selective, cumulative inhibition by low-dose aspirin, that eventually led to its development as an antiplatelet drug for the treatment and prevention of atherothrombosis (reviewed by Patrono, 1994).
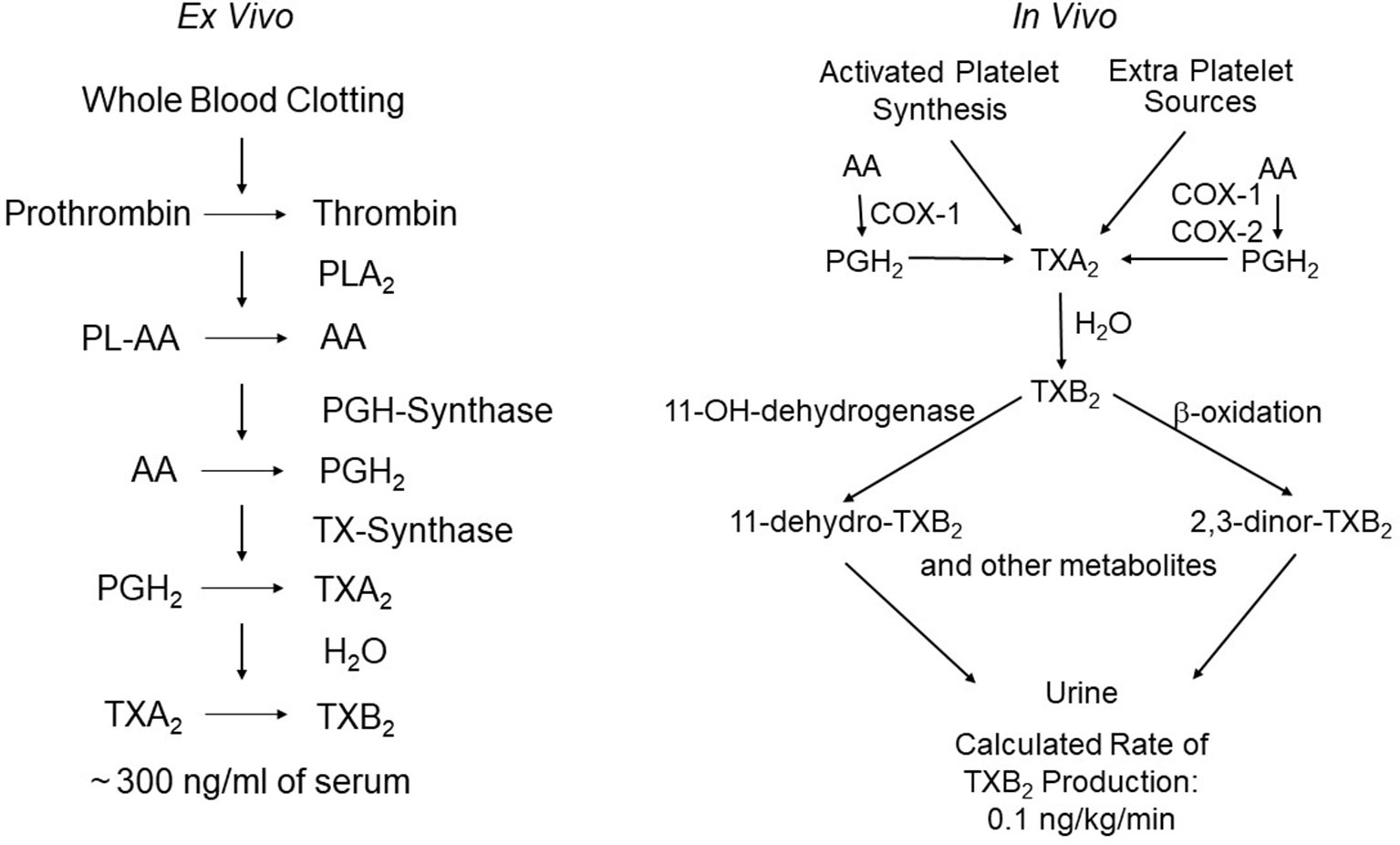
Figure 1 Assessment of platelet thromboxane (TX)A2 biosynthesis ex vivo and in vivo. The left panel depicts the chain of enzymatic reactions triggered by thrombin generation during whole-blood clotting, and the resulting formation of very large amounts of TXB2 (the stable hydration product of TXA2) that can be easily measured in serum as a sensitive and specific index of platelet COX-1 activity. The right panel depicts the metabolic fate of TXA2in vivo and the calculated rate of its production in healthy subjects on the basis of TXB2 infusions and measurement of its major urinary metabolites, 11-dehydro-TXB2 and 2,3-dinor-TXB2. The latter represent a non-invasive index of platelet activation in vivo. Modified and redrawn from Patrono et al. (2013).
This article reviews the main findings of four decades of clinical investigation based on these analytical approaches, focusing on the measurement of TXA2 metabolites in vivo and ex vivo as indexes of platelet activation and COX-1 activity, respectively, with emphasis on the authors’ contribution to the resulting pathophysiological and pharmacological developments.
In 1981, Roberts et al. reported the GC/MS characterization of 20 enzymatic metabolites of systemically infused [3H8]TXB2. Two major series of metabolites were identified based on a ring structure. One series retained the original TXB2 hemiacetal ring and included two metabolites, 2,3-dinor-TXB2 (the most abundant urinary metabolite) and 2,3,4,5-tetranor-TXB2, which were products of beta-oxidation (Roberts et al., 1981). The second group of derivatives was formed as a result of dehydrogenation of the hemiacetal alcohol group at C-11, and included 16 metabolites (Roberts et al., 1981). Among the compounds resulting from this single transformation, 11-dehydro-TXB2 was the most abundant urinary metabolite (Roberts et al., 1981). In their seminal paper in the Journal of Biological Chemistry, Roberts et al. accurately predicted the potential value of this analytical approach: “Since thromboxanes are released in substantial quantities from aggregating platelets, quantification of in vivo thromboxane production may provide a means to assess in vivo platelet aggregation and lead to a better understanding of the role of platelets in the pathophysiology of many cardiovascular diseases. It may also provide a means to assess the in vivo efficacy of anti-platelet drug therapy” (Roberts et al., 1981). Important limitations of this study were represented by a single high rate of TXB2 infusion and a single healthy subject being infused, precluding assessment of the linearity of conversion of TXB2 into its major enzymatic derivatives, as well as of the interindividual variability in the prevalence of the two main pathways of its metabolic transformation.
Together with Garret FitzGerald and Ian Blair, we reexamined the metabolic fate of TXB2 entering the systemic circulation, by measuring the urinary excretion of 2,3-dinor-TXB2 during the infusion of exogenous TXB2, in four aspirin-pretreated healthy volunteers randomized to receive 6-h i.v. infusions of vehicle alone and TXB2 at 0.1, 1.0, and 5.0 ng·kg−1·min−1 (Patrono et al., 1986). Plasma TXB2 and urinary 2,3-dinor-TXB2 were measured before, during, and up to 24 h after the infusions and in aspirin-free periods. Aspirin treatment suppressed baseline urinary 2,3-dinor-TXB2 excretion by 80%, consistent with a predominant platelet source of the parent compound. The fractional excretion of 2,3-dinor-TXB2 was independent of the rate of TXB2 infusion, over a 50-fold dose range, and averaged 5.3% ± 0.8% (Patrono et al., 1986). Insertion of 2,3-dinor-TXB2 excretion rates measured in aspirin-free periods into the linear relationship between the doses of infused TXB2 and the amounts of metabolite excreted in excess of control values permitted estimation of the rate of entry of endogenous TXB2 into the circulation as 0.11 ng·kg−1·min−1 (Patrono et al., 1986). Upon discontinuing TXB2 infusion, its rate of disappearance from the systemic circulation was linear over the first 10 min with an apparent half-life of 7 min. This resulted in a maximal estimate of the plasma concentration of endogenous TXB2 of 2.0 pg/ml, i.e., much lower than had been previously reported (Patrono et al., 1986). This finding argued for a local nature of TXA2 synthesis and action, as previously suggested for prostacyclin (PGI2) (FitzGerald et al., 1981). Similar to the endothelial synthesis of PGI2, the maximal TXA2 biosynthetic capacity of human platelets greatly exceeds its actual production in vivo. Thus, the platelets of 1 ml of whole blood clotted for 1 h in vitro can synthesize and release a similar amount of TXB2 as that secreted into the systemic circulation in vivo during the same time (Patrono et al., 1980; Patrono et al., 1986) (Figure 1), a finding that may help explain the unusual requirement for greater than 97% inhibition of TXA2 biosynthetic capacity to maximally inhibit TXA2-dependent platelet function (Reilly and FitzGerald, 1987; Santilli et al., 2009) (see below).
However, because of obvious safety concerns, it had not been possible to investigate the metabolic fate of TXA2 in humans, and it remained to be determined whether the enzymatic transformation of TXB2 to its major urinary metabolites accurately reflected TXA2 metabolism in vivo. Thus, together with Joe Rokach and his colleagues at Merck Frosst Research Laboratories, Patrignani et al. (1989) compared the metabolic handling of exogenously infused TXA2 and TXB2 in the cynomolgus monkey. The main finding of this study was that TXA2 and TXB2 are metabolized to 2,3-dinor-TXB2 and 11-dehydro-TXB2 with similar fractional conversion rates, thereby suggesting that TXA2 is hydrolyzed non-enzymatically to TXB2 prior to enzymatic degradation via the beta-oxidation and 11-OH-dehydrogenase pathways, and that the resulting urinary metabolites provide a quantitative index of TXA2 biosynthesis in vivo (Patrignani et al., 1989).
Because previous estimates of the rate of entry of TXB2 into the human systemic circulation had been based on monitoring the beta-oxidation pathway of TXB2 metabolism (Patrono et al., 1986), Ciabattoni et al. (1989) went on to measure the urinary excretion of immunoreactive 11-dehydro-TXB2 and 2,3-dinor-TXB2 (Ciabattoni et al., 1987) during the infusion of exogenous TXB2 over a 50-fold dose range in healthy volunteers, with the same protocol of the previous study (Patrono et al., 1986). The fractional elimination of both metabolites was independent of the rate of TXB2 infusion and averaged 6.0% to 7.0%, demonstrating that urinary 11-dehydro-TXB2 is at least as abundant a conversion product of exogenously infused TXB2 as 2,3-dinor-TXB2 (Ciabattoni et al., 1989) Furthermore, the study of Ciabattoni et al. (1987) showed that this analytical approach could detect changes in the urinary excretion of immunoreactive 11-dehydro-TXB2 associated with simulated short-term increases of TXB2 release into the human circulation (Ciabattoni et al., 1987).
Transient increases in the excretion of 2,3-dinor-TXB2 and 11-dehydro-TXB2 were described in patients with acute coronary syndromes and interpreted as reflecting repeated episodes of platelet activation (Fitzgerald et al., 1986; Vejar et al., 1990). Transient changes in TXA2 biosynthesis detected in patients with unstable angina were accompanied by concomitant increases in PGI2 biosynthesis, as reflected by urinary 2,3-dinor-6-keto-PGF1α excretion, suggesting a counter-regulatory endothelial activation in this setting (Fitzgerald et al., 1986). In contrast, patients with chronic stable angina did not display increased TXA2 biosynthesis, both under resting conditions and following exercise-induced myocardial ischemia (Fitzgerald et al., 1986). Biochemical evidence of episodic platelet activation in the setting of acute coronary syndromes was consistent with the post-mortem findings of Michael Davies and Erling Falk that suggested dynamic thrombotic events occurring over a disrupted plaque in the coronary vessels of patients dying after a diagnosis of unstable angina (reviewed by Falk et al., 1995; Davì and Patrono, 2007). These results provided a rationale for testing the efficacy and safety of low-dose aspirin in acute coronary syndromes, a clinical setting in which antiplatelet therapy reduced the risk of major atherothrombotic complications by approximately 50% (reviewed by Patrono et al., 2005). Episodic increases in TXB2 metabolite excretion were also characterized in patients with acute ischemic stroke (Koudstaal et al., 1993; van Kooten et al., 1997), though with a lower frequency and shorter duration than in acute coronary syndromes (Fitzgerald et al., 1986; Vejar et al., 1990), perhaps reflecting the heterogeneity of mechanisms responsible for ischemic stroke (Albers et al., 2004). Low-dose aspirin (50 mg daily) largely suppressed 11-dehydro-TXB2 excretion in this setting, reflecting the predominant platelet origin of TXA2 biosynthesis (Koudstaal et al., 1993). Enhanced platelet activation was independently associated with stroke severity on admission (van Kooten et al., 1997). Patients with a transient ischemic attack were characterized by infrequent episodes of platelet activation, suggesting that enhanced TXA2 biosynthesis was not secondary to cerebral ischemia (Koudstaal et al., 1993).
Persistent platelet activation, as reflected by persistently enhanced urinary excretion of TXB2 metabolites, was reported by different Groups in patients with a variety of cardiovascular risk factors that accelerate atherogenesis, including cigarette smoking (Nowak et al., 1987), type-2 diabetes mellitus (Davì et al., 1990), hypercholesterolemia (Davì et al., 1992), homozygous homocystinuria (Di Minno et al., 1993), and hypertension (Minuz et al., 2002). Because enhanced urinary excretion of 11-dehydro-TXB2 in diabetes mellitus might reflect either an abnormality in the biosynthesis of TXA2 or a shift in its metabolic fate through the two main enzymatic pathways of degradation, Davì et al. (1990) investigated the fractional conversion of infused TXB2 to urinary 11-dehydro-TXB2 in subjects with type-2 diabetes mellitus. Their finding of a linear conversion of infused TXB2 to urinary 11-dehydro-TXB2 over a 50-fold range of infusion rates, with a fractional elimination similar to that previously described in healthy subjects (Ciabattoni et al., 1989), was consistent with enhanced excretion of 11-dehydro-TXB2 reflecting a change in the biosynthesis of TXA2 rather than an alteration in its enzymatic transformations (Davì et al., 1990).
Urinary prostanoid metabolites, such as 11-dehydro-TXB2, do not reflect a specific site of prostanoid biosynthesis. To characterize the potential platelet versus non-platelet sources of TXA2 biosynthesis, Davì et al. (1990) used the unique property of aspirin to produce selective, cumulative acetylation of platelet cyclooxygenase (COX)-1 when it is given in low doses once daily (Patrignani et al., 1982). Renal COX-isozymes, which have been involved in enhanced TXA2 production in patients with systemic lupus erythematosus (Patrono et al., 1985), are not inhibited by low-dose aspirin to any detectable extent (Patrignani et al., 1982; Pierucci et al., 1989). The profound reduction in urinary 11-dehydro-TXB2 excretion that was found in subjects with type-2 diabetes mellitus after they received 50 mg of aspirin daily for 1 week and the platelet turnover-dependent return to a pre-treatment excretion rate over the next 10 days following aspirin discontinuation were consistent with a prevailing role for platelets as the source of TXA2 biosynthesis in this setting (Davì et al., 1990).
Persistently enhanced urinary excretion of TXB2 metabolites has been reported in the vast majority of patients with myeloproliferative neoplasms, such as essential thrombocythemia (ET) and polycythemia vera (PV) (reviewed by Patrono et al., 2013). As shown in Figure 2, the urinary excretion rates of 11-dehydro-TXB2 measured in untreated ET (Rocca et al., 1995) and PV patients (Landolfi et al., 1992) are comparable to the rate measured in patients with unstable angina (Fitzgerald et al., 1986; Vejar et al., 1990) and higher than values of metabolite excretion associated with a variety cardiovascular risk factors (Nowak et al., 1987; Davì et al., 1990; Davì et al., 1992; Di Minno et al., 1993; Minuz et al., 2002). The finding of persistently enhanced TXA2 biosynthesis in PV patients and its suppression by low-dose aspirin (Landolfi et al., 1992) provided a rationale for testing the efficacy and safety of this antiplatelet strategy in PV (Landolfi et al., 2004). Based on the positive results of the ECLAP (European Collaboration on Low-dose Aspirin in Polycythemia vera) trial (Landolfi et al., 2004), primary prophylaxis with low-dose aspirin (81–100 mg daily) is currently recommended for PV patients (Tefferi et al., 2018).
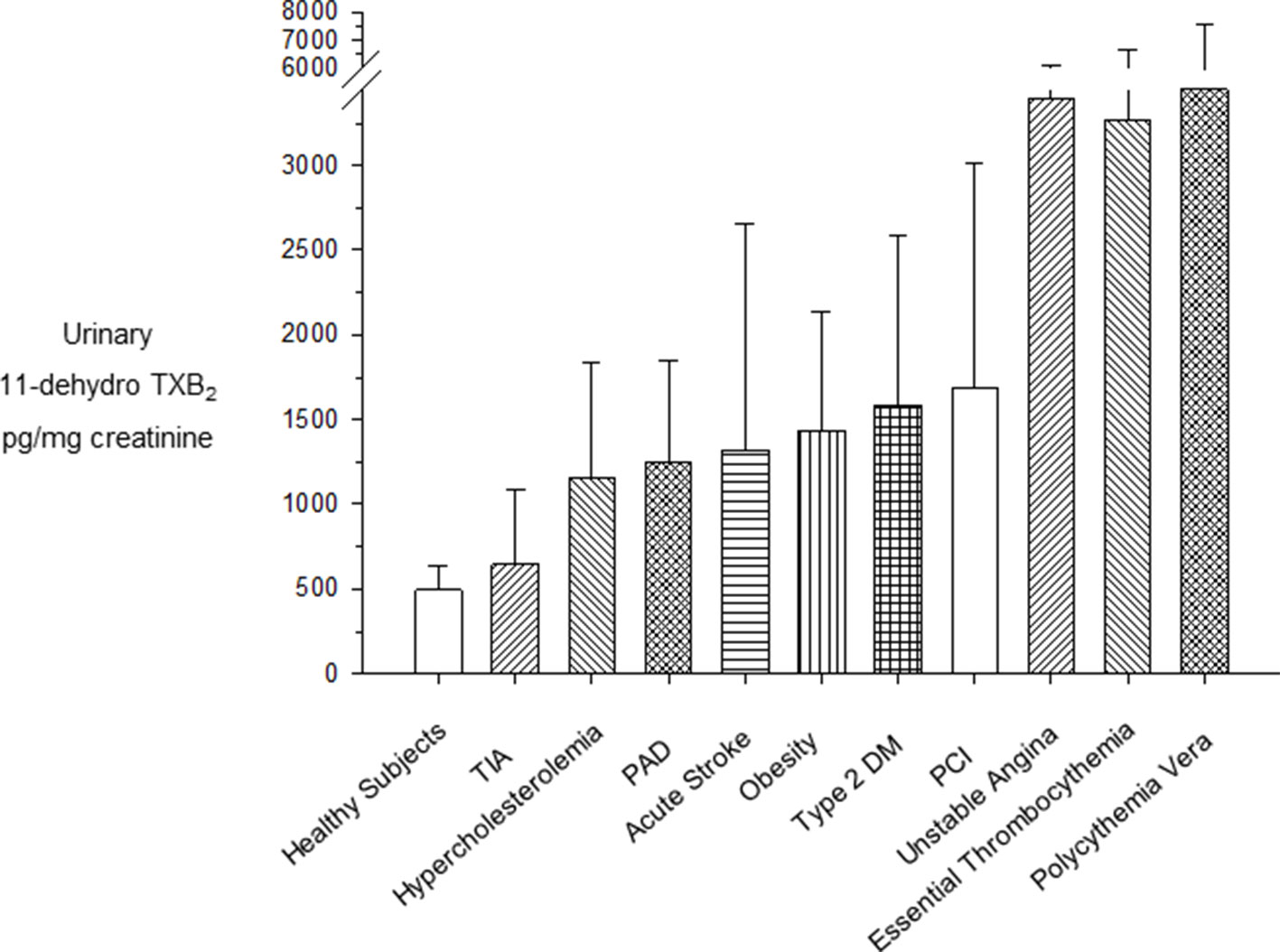
Figure 2 Urinary excretion rates of 11-dehydro-TXB2 in clinical settings at high cardiovascular risk. Mean (± standard deviation) or median (interquartile range) urinary excretion rates of 11-dehydro-TXB2 in clinical settings characterized by high cardiovascular risk. PAD, peripheral arterial disease; PCI, percutaneous coronary intervention; T2DM, type 2 diabetes mellitus; TIA, transient ischemic attack. Modified and redrawn from Patrono et al. (2013).
Urinary 11-dehydro-TXB2 excretion has been investigated as a potential biomarker of the future risk of major vascular events in aspirin-treated, high-risk patients enrolled in the Heart Outcomes Prevention Evaluation (HOPE) trial, by using a nested case-control design (Eikelboom et al., 2002). After adjustment for baseline differences, the odds for the composite end-point of myocardial infarction, stroke, or cardiovascular death increased with each increasing quartile of baseline urinary 11-dehydro-TXB2, with patients in the upper quartile having approximately 2-fold higher risk than those in the lower quartile (Eikelboom et al., 2002). This finding was largely confirmed by a similar substudy of the Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management and Avoidance (CHARISMA) trial (Eikelboom et al., 2008). However, because both HOPE and CHARISMA were mostly secondary prevention trials, an important limitation of these analyses is represented by the lack of a control group of subjects not treated with aspirin. Furthermore, the extent of biological variation in urinary 11-dehydro-TXB2 excretion rate, as well as the intrasubject variability in its reduction by low-dose aspirin are currently unknown, but could potentially limit the value of this biomarker to predict the risk of future cardiovascular events in an individual aspirin-treated patient. We are currently investigating the potential predictive value of urinary 11-dehydro-TXB2 excretion in a large sample of A Study of Cardiovascular Events in Diabetes (ASCEND) that randomized 15,480 adults with diabetes mellitus to long-term treatment with low-dose aspirin or placebo (ASCEND Study Collaborative Group, 2018).
Recently, the platelet origin of urinary thromboxane metabolite (TXM) excretion was challenged by the case report of a single patient with end-stage renal failure requiring dialysis who carried a rare genetic mutation in cPLA2α (cytosolic phospholipase A2), resulting in dramatically reduced urinary TXM and PGI2 metabolite (PGIM) excretion rates, with recovery of “normal” urinary levels of these prostanoid metabolites after kidney transplantation (Mitchell et al., 2018). The authors’ conclusion was that “urinary PGIM and TXM can be derived exclusively by the kidney without contribution from PGI2 made by endothelial cells or TXA2 by platelets in the systemic circulation” (Mitchell et al., 2018). However, the authors did not investigate the metabolic disposition of TXB2 and PGI2 in this patient, and no comparison was performed with renal failure and transplanted patients without the cPLA2α genetic defect (Grosser et al., 2018). Moreover, it should be emphasized that in patients with systemic lupus erythematosus, who displayed enhanced renal synthesis of TXA2, higher urinary TXB2 excretion was associated with unchanged urinary excretion of 2,3-dinor-TXB2 (Patrono et al., 1985), suggesting that TXA2 produced by the kidney is mostly excreted unchanged into the urine and does not undergo systemic metabolism to TXM (Remuzzi et al., 1992).
The effects of aspirin on the activity of platelet COX-1 have been investigated through measurements of serum TXB2 (Patrono et al., 1980; Patrignani et al., 1982) and urinary metabolites of TXB2 (FitzGerald et al., 1983). Serum TXB2 reflects the maximal biosynthetic capacity of blood platelets to generate TXA2 in response to endogenously formed thrombin during whole-blood clotting, and its measurement has been used extensively to assess the human pharmacology of platelet COX-1 inhibition in health and disease (reviewed by Patrono et al., 2008). Three important features of aspirin pharmacodynamics were characterized by measurements of serum TXB2 in healthy subjects (Patrignani et al., 1982): i) the cumulative nature of the inactivation of platelet COX-1 by repeated daily low doses (20–40 mg) of aspirin; ii) the saturability of this effect with single doses as low as 100 mg; and iii) the relative selectivity for platelet COX-1 inhibition at low doses. Our Research Group at the Catholic University School of Medicine in Rome described time-dependent, cumulative reduction in serum TXB2 (a product of platelet COX-1 activity), without statistically significant changes in urinary 6-keto-PGF1α excretion (mostly a product of renal COX-2 activity), a reverse paradigm of selective COX-isozyme inhibition by low-dose aspirin (30 mg daily) in man (Patrignani et al., 1982) (Figure 3). Measurements of serum TXB2 were also instrumental in characterizing the presystemic nature of platelet COX-1 acetylation by aspirin, an important feature of aspirin pharmacodynamics contributing to its biochemical selectivity (Pedersen and FitzGerald, 1984). Thus, serum TXB2 was reduced by about 40% five min after the oral administration of a 20-mg dose, before aspirin could be detected in the systemic circulation (Pedersen and FitzGerald, 1984).
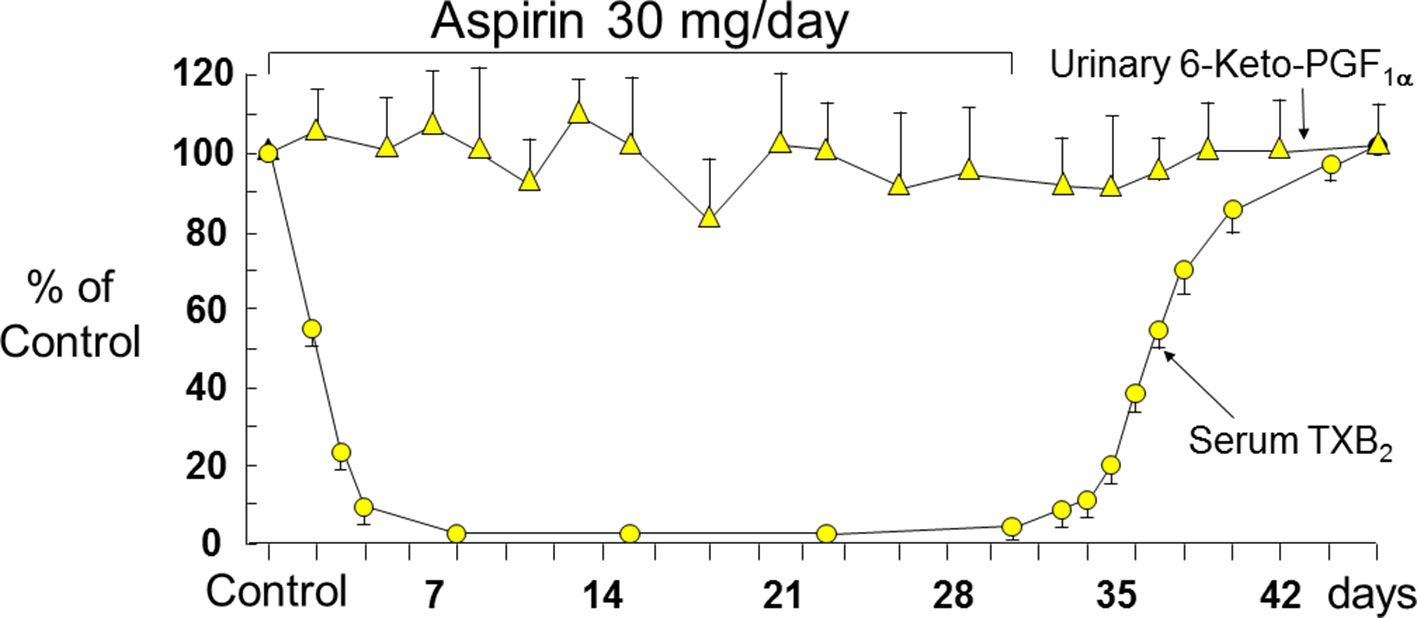
Figure 3 Long-term effects of low-dose (0.45 mg/kg per day) aspirin on platelet TXB2 and renal PGI2 synthesis. Serum TXB2 concentrations and urinary excretion of 6-keto-PGF1α were measured in three healthy subjects before, during, and after aspirin therapy. Mean values ± SEM are plotted as percentage of control measurements performed prior to aspirin administration. The arrows indicate duration of daily aspirin therapy. Redrawn from Patrignani et al. (1982).
It is important to realize that the serum TXB2 assay requires almost immediate whole-blood incubation at 37°C as a condition for optimal thrombin generation, arachidonic acid release and its sequential conversion by platelet COX-1 and TX-synthase to form PGH2 and TXA2, respectively. As a consequence of thrombin-induced platelet activation, the blood concentration of TXB2 increases over 60 min from 1 to 2 pg/ml (true circulating plasma level in vivo) to 300 to 400 ng/ml (maximally stimulated, COX-1-dependent production ex vivo) (Patrono et al., 1980). A variably delayed access to 37°C incubation, as well as different analytical methods to quantitate serum TXB2, may contribute to variable results in a multicenter setting (Frelinger et al., 2009; Reny et al., 2012). Petrucci et al. (2016) investigated whether a variable delay in 37°C incubation and/or analytical discrepancies may affect the assessment of aspirin pharmacodynamics based on serum TXB2 determinations. They found that a longer than 5-min delay in the 37°C incubation of whole-blood samples may variably influence the assessment of platelet COX-1 inhibition by low-dose aspirin and confound the analysis of aspirin responsiveness in the clinical setting (Petrucci et al., 2016; De Stefano et al., 2018). In contrast, a GC/MS-validated immunoassay and liquid chromatography-tandem mass-spectrometry yielded quite comparable TXB2 concentrations in the same serum samples (Petrucci et al., 2016).
The relationship between inhibition of platelet COX-1 activity, as reflected by serum TXB2, and arachidonic acid-dependent platelet function assays (i.e., arachidonate-induced optical aggregation), and urinary 11-dehydro-TXB2 excretion is strikingly non-linear (Figure 4) (Reilly and FitzGerald, 1987; Santilli et al., 2009). Thus, platelet COX-1 activity must be nearly completely (>97%) suppressed to fully inhibit in vivo platelet activation. Such stringent requirement may help explain the fact that the vast majority of traditional nonsteroidal antiinflammatory drugs (tNSAIDs), that are reversible inhibitors of platelet COX-1 with variable half-lives, are unable to achieve profound and persistent suppression of TXA2 biosynthesis, thereby unmasking their COX-2–dependent cardiovascular toxicity (reviewed by Patrono and Baigent, 2014). Although the variable COX-isozyme selectivity, half-life, daily dose, and duration of treatment of different COX-2 inhibitors could all influence the cardiovascular consequences of COX-2 inhibition, the dichotomous clinical read-outs of such inhibition are explained by the exponential relationship between inhibition of platelet COX-1 activity and suppression of TXA2-dependent platelet activation (Figure 4) (Patrono and Baigent, 2014). As depicted in Figure 4, NSAIDs inhibiting platelet COX-1 activity by 0% to 20% (e.g., highly selective COX-2 inhibitors, such as rofecoxib and etoricoxib), by 20% to 50% (e.g., COX-2 inhibitors with moderate COX-2 selectivity, such as diclofenac and celecoxib), or by 50% to 90% (most tNSAIDs, such as indomethacin and ibuprofen) will cause similarly modest suppression of TXA2-dependent platelet activation in vivo (Patrono and Baigent, 2014). Due to its longer half-life and modest COX-1 selectivity, naproxen 500 mg twice daily may suppress platelet COX-1 activity by >95% throughout the 12-h dosing interval and reduce TXA2-dependent platelet activation in vivo to a similar extent as aspirin 100 mg once daily (Capone et al., 2004; Capone et al., 2007).
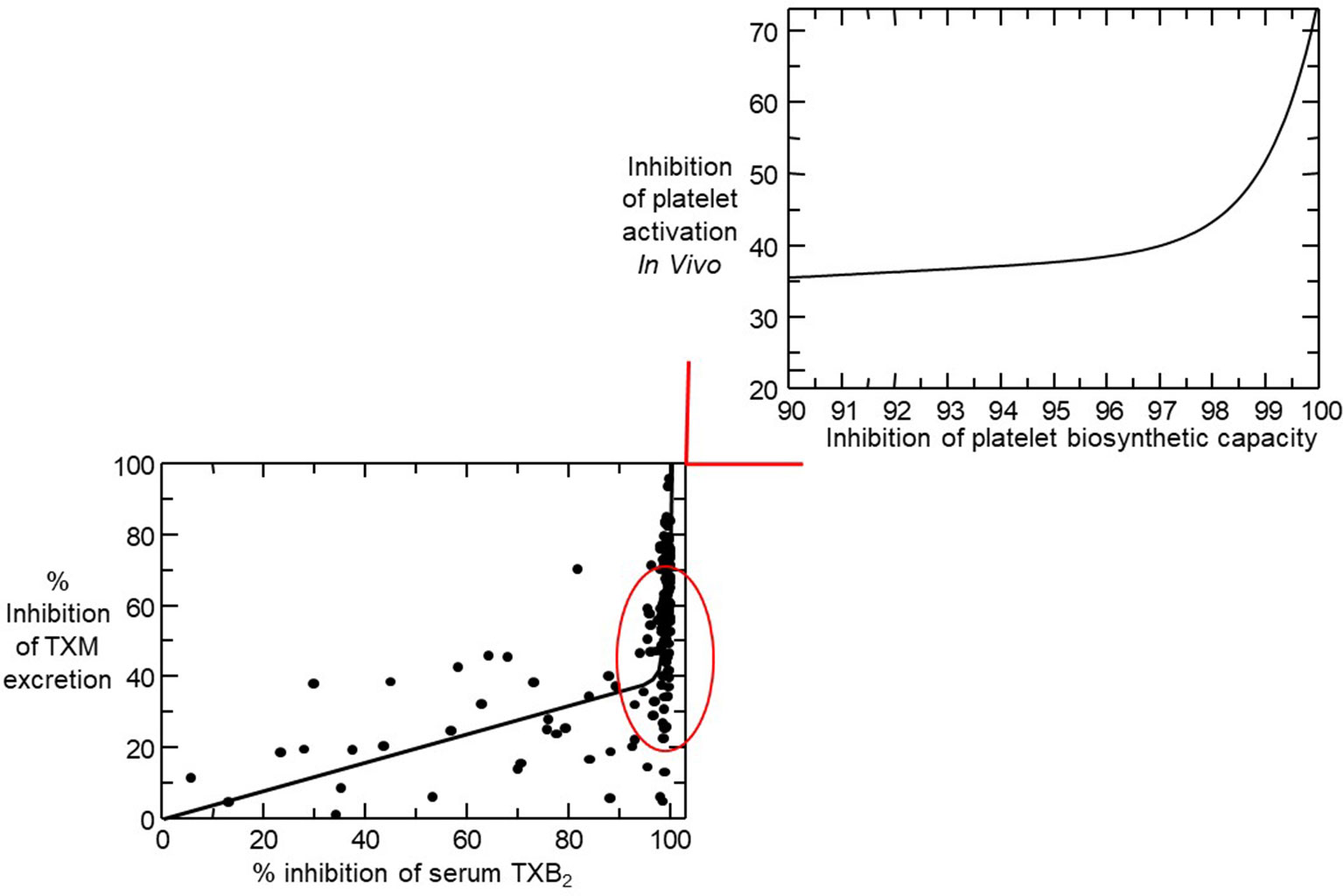
Figure 4 Nonlinear relationship between inhibition of serum thromboxane (TX) B2 and urinary 11-dehydro-TXB2 excretion. In the lower left panel, individual percentage inhibition values are depicted from all on-treatment and post-treatment measurements performed in 48 healthy subjects randomized to receive aspirin 100 mg daily for 1 to 8 weeks. The nonlinear relationship between percent inhibition of serum TXB2 and urinary 11-dehydro-TXB2 showed that for 0 to 97% of COX-1 inhibition, TXA2 biosynthesis in vivo was linearly inhibited by <40% and that >97% suppression of serum TXB2 was necessary to maximally reduce TX metabolite (TXM) excretion. The upper right panel represents a detail of the left panel and is based on mathematical modeling of the experimental data. Modified and redrawn from Santilli et al. (2009), with permission from the publisher.
Given the non-linear relationship depicted in Figure 4, platelet function assays, including urinary 11-dehydro-TXB2, do not accurately reflect the degree of platelet COX-1 inhibition (Pascale et al., 2012; Smith et al., 2012). Moreover, platelet function assays routinely used to measure aspirin response, are not necessarily related to its mechanism of action, display poor inter-assay agreement (Lordkipanidzé et al., 2007; Blais et al., 2009; Santilli et al., 2009; ), and give inconsistent results upon repeated measurements (Muir et al., 2009; Santilli et al., 2009). These methodological considerations may help explain the rise and fall of the aspirin “resistance” concept, typically defined as impaired platelet response to aspirin based on a single functional measurement performed at an often unspecified time point after dosing, usually without a reliable assessment of compliance (Michelson et al., 2005; Rocca and Patrono, 2005). In a study of 48 healthy volunteers, repeated measurements of platelet aggregation in response to different agonists demonstrated that occasionally “resistant” subjects could be classified as “responders” when examined previously or subsequently (Santilli et al., 2009). Not surprisingly, the incidence of “resistance” ranged from 1% up to 65% in different studies, was assay-dependent, fluctuated over time, and remains of unproven clinical significance (reviewed by Patrono and Rocca, 2008).
In contrast to the uniform effectiveness of low-dose aspirin in suppressing platelet COX-1 activity in healthy individuals, some clinical conditions are associated with transient or persistent suboptimal platelet inhibition by a conventional once daily regimen of low-dose aspirin (Rocca and Patrono, 2005). These include patients following on-pump coronary artery bypass surgery (CABG) (Cavalca et al., 2017), patients with ET (Dragani et al., 2010; Dillinger et al., 2012), patients with coronary artery disease and the metabolic syndrome (Smith et al., 2012), and some patients with type-2 diabetes mellitus (Spectre et al., 2011; Rocca et al., 2012; Bethel et al., 2016). Under these circumstances, most patients display biochemical evidence of TXA2-dependent platelet activation in vivo (Davì et al., 1990; Rocca et al., 1995), a finding that may help explain impaired aspirin pharmacodynamics. More specifically, less-than-optimal inactivation of platelet COX-1 could be a consequence of transiently (Cavalca et al., 2017) or persistently (Dragani et al., 2010) accelerated platelet renewal, or result from platelet activation-induced generation of hydroperoxides that may impair the acetylation of COX-1 by aspirin (Bala et al., 2008). The long-lasting duration of the antiplatelet effect of aspirin, despite its very short half-life, is explained by inactivation of COX-1 in bone-marrow platelet progenitors, as reflected by the 48-h delay between aspirin withdrawal and initial recovery of unacetylated COX-1 (Burch et al., 1978) and TXA2 biosynthetic capacity (Patrignani et al., 1982) in peripheral blood platelets. Thus, under conditions of normal thrombopoiesis, the efficacy of a short-lived drug given once daily reflects irreversible inactivation of a slowly renewable drug target (platelet COX-1) combined with an effect on bone-marrow platelet progenitors, leading to a new platelet progeny with largely non-functioning COX-1 for the vast majority of the 24-h dosing interval (Giaretta et al., 2017). However, reduced systemic bioavailability of aspirin, as may occur with some enteric-coated formulations (Maree et al., 2005) and in association with obesity (Petrucci et al., 2019), or faster renewal of platelet COX-1, as reported under conditions of altered megakaryopoiesis (Pascale et al., 2012), may shorten the duration of the antiplatelet effect of aspirin and dictate a more frequent dosing regimen (Patrono et al., 2013).
A standard once-daily regimen of low-dose aspirin administration cannot adequately suppress platelet TXA2 production, throughout the 24-h dosing interval, in the vast majority of ET patients (Dragani et al., 2010; Dillinger et al., 2012; Pascale et al., 2012). An accelerated turnover of platelet COX-1, reflecting abnormal megakaryopoiesis, has been hypothesized in ET (Pascale et al., 2012; Rocca and Patrono, 2015; Giaretta et al., 2017). A higher daily fraction of newly released platelets with unacetylated COX-isozymes would account for incomplete inhibition, as well as time-dependent recovery of TXA2-dependent platelet function during the standard 24-h dosing interval of low-dose aspirin administration (Dragani et al., 2010). Two relatively small studies in ET patients have shown that sub-optimal inhibition of platelet TXA2 production and TXA2-dependent platelet function can be largely rescued by a bid regimen of low-dose (100 mg) aspirin administration, but not by a higher dose (200–250 mg) given once daily (Dillinger et al., 2012; Pascale et al., 2012). The Aspirin Regimens in Essential Thrombocythemia Study (ARES) is a randomized, parallel-arm, dose-finding study recruiting 300 ET patients to address two main questions (De Stefano et al., 2018). First, whether a bid or tid 100-mg aspirin regimen is more effective than the standard once daily regimen in inhibiting platelet TXA2 production, without a major impact on vascular PGI2 biosynthesis. Second, whether superior biochemical efficacy of a multiple versus single dosing low-dose aspirin regimen can be safely maintained over long-term follow-up (De Stefano et al., 2018).
Similarly, several independent studies have consistently shown the superior biochemical efficacy of a strategy based on shortening the dosing interval versus a strategy of maintaining or increasing the once daily dose of aspirin in patients with type-2 diabetes mellitus (Spectre et al., 2011; Rocca et al., 2012; Bethel et al., 2016), undergoing CABG (Paikin et al., 2015; Cavalca et al., 2017), or presenting with an acute coronary syndrome (Parker et al., 2019).
Obesity is associated with biochemical evidence of persistently enhanced platelet activation (Davì et al., 2002; Petrucci et al., 2019) and high risk of atherothrombotic complications (reviewed by Rocca et al., 2018). Recently, Rothwell et al. (2018) analyzed individual data of 117,279 subjects recruited into 10 primary prevention trials and reported that low doses of aspirin (75–100 mg) were only effective in preventing major vascular events in subjects with body weight lower than 70 kg, and had no benefit in the vast majority of men and nearly 50% of all women weighing 70 kg or more. In contrast, higher doses (≥ 325 mg) of aspirin were only effective in subjects with body weight equal to or higher than 70 kg (Rothwell et al., 2018). Although the finding of effect modification by body weight has not been confirmed by some of the more recent aspirin trials (ASCEND Study Collaborative Group, 2018), these data appear consistent with the suggestion that the antiplatelet effect of aspirin, as reflected by serum TXB2 measurement, is influenced by body size (Maree et al., 2005). Increased body size, fat excess, and the associated changes in volume of distribution and liver function may all reduce the bioavailability of a lipophilic drug such as aspirin (Patrono and Rocca, 2017). Petrucci et al. (2019) recently reported measurements of serum TXB2 at the end of the 24-h dosing interval in 100 aspirin-treated subjects with a wide range of body mass index (BMI) and body weight values. A statistically significant exponential association was observed between body size, expressed as either BMI or body weight, and residual serum TXB2 values (Figure 5) (Petrucci et al., 2019). Thus, a standard, once‐daily 100-mg aspirin regimen appears to be inadequate to fully inhibit platelet COX‐1 activity throughout the 24-h dosing interval in moderately to severely obese subjects. However, based on the data depicted in Figure 5, one would not expect impaired aspirin pharmacodynamics in subjects with up to a body weight of ∼100 kg or a BMI of 35 kg/m2, corresponding to obesity of class 2 or higher (Petrucci et al., 2019). In silico modeling of the antiplatelet pharmacodynamics of aspirin in this setting suggested that either doubling the once‐daily dose or administering a lower dose (e.g., 85 mg) twice daily would be associated with adequate suppression of platelet TXA2 production(Petrucci et al., 2019). The consensus opinion that “it is reasonable to double the daily dose or shorten the dosing interval (twice-daily) for BMI ≥ 40 kg/m2” expressed by the Working Group on Thrombosis of the European Society of Cardiology (Rocca et al., 2018) is consistent with these experimental findings and in silico modeling (Petrucci et al., 2019).
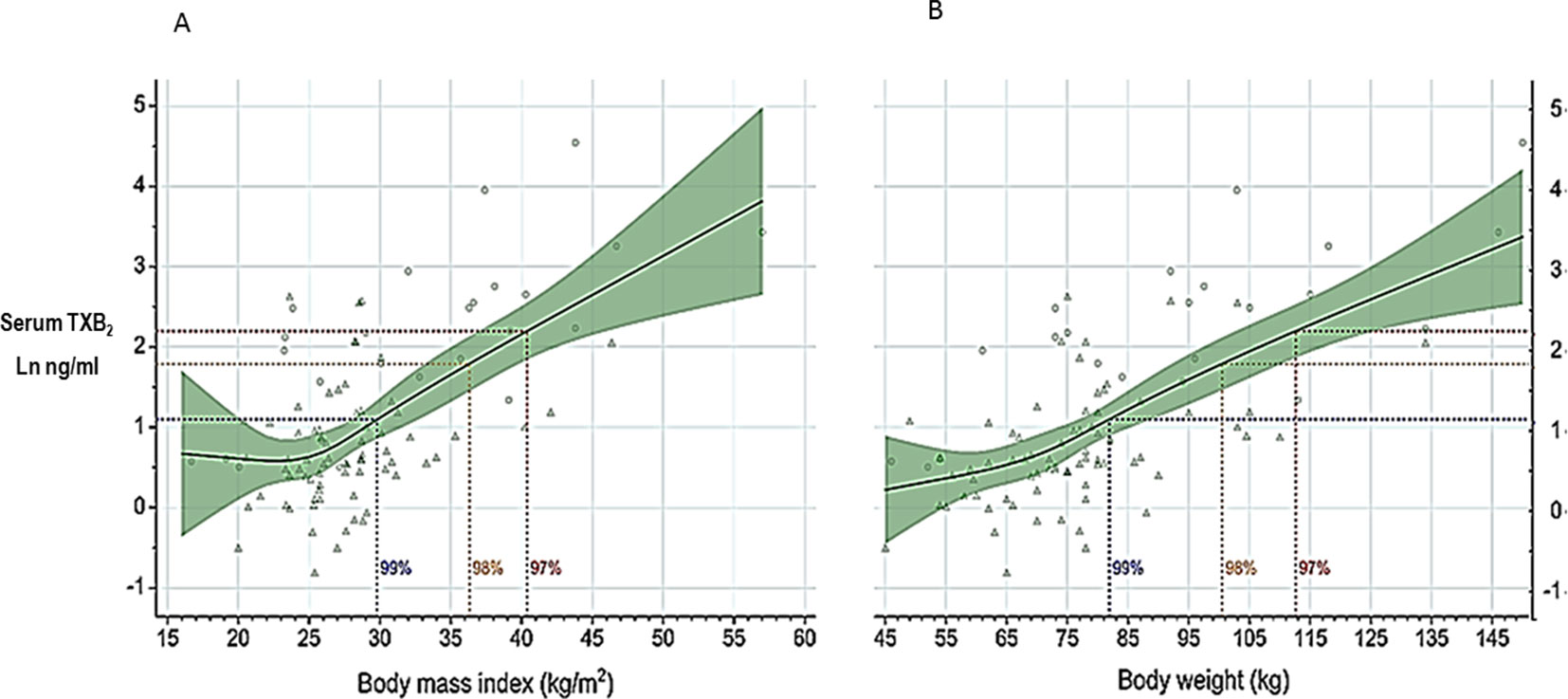
Figure 5 Serum thromboxane (TX)B2 levels at 24 h after aspirin intake (100 mg once daily) vs body mass index or body weight. Log-transformed serum TXB2 levels measured 24 h after a witnessed aspirin intake are represented in relation to body mass index (panel A) and body weight (panel B). Triangles represent individual patients receiving aspirin for primary or secondary cardiovascular prevention, n = 71; circles represent individual healthy subjects, n = 25. Solid lines and green areas: predicted serum TXB2 levels and 95% confidence intervals, respectively, as a function of increasing body mass index (panel A) and body weight (panel B) in the 96 study subjects. Horizontal dotted lines were drawn to indicate serum TXB2 levels of 3, 6, and 9 ng/mL, corresponding to 99%, 98%, and 97% inhibition of platelet COX-1 activity, respectively, measured in healthy subjects. The vertical dotted lines were drawn to indicate on the abscissa scales the highest calculated values of body mass index (panel A) and body weight (panel B) compatible with these different levels of platelet COX-1 inhibition. Modified and redrawn from Petrucci et al., 2019, with permission from the publisher.
If we look back at four decades of research on thromboxane biosynthesis and inhibition, the translational aspect of this research stands out in at least three areas. First, measurements of urinary TXM excretion, a non-invasive index of in vivo platelet activation, have been instrumental in identifying clinical conditions in which to assess the efficacy and safety of antiplatelet therapy. Notable examples are represented by acute coronary syndromes (Fitzgerald et al., 1986), acute ischemic stroke (Koudstaal et al., 1993), and polycythemia vera (Landolfi et al., 1992). The remarkable cardio-protective effects of low-dose aspirin in these settings (The RISC Group, 1990; Landolfi et al., 2004; Rothwell et al., 2016) are likely to reflect the important pathophysiologic role of transiently or persistently enhanced platelet activation unravelled by TXM measurements. Second, the development of serum TXB2 as a mechanism-based biomarker of platelet COX-1 inhibition (Patrono et al., 1980) has played a fundamental role in defining the human pharmacology of aspirin as an antiplatelet agent (Patrono, 1994). The results of a large series of randomized, placebo-controlled clinical trials of aspirin in the prevention of atherothrombosis in high-risk patients have confirmed the saturability of its antithrombotic effect at low doses (Antithrombotic Trialists’ Collaboration, 2002), consistent with saturability of platelet COX-1 inactivation by low-dose aspirin (Patrignani et al., 1982; FitzGerald et al., 1983). Third, a comparison of the extent and duration of serum TXB2 reduction in response to different NSAIDs has allowed predicting a similar cardiovascular hazard of tNSAIDs and coxibs (Baigent and Patrono, 2003): “If the vascular consequences of endothelial COX‐2 inhibition are modulated by profound and persistent blockade of platelet COX‐1 activity, as indirectly implied by genetic and pharmacologic manipulations in mice (Cheng et al., 2002), then the cardiovascular effects of most traditional NSAIDs, which only incompletely and transiently inhibit platelet COX‐1, may resemble those of selective COX‐2 inhibitors.” A prediction largely borne out by a tabular data meta-analysis published in 2006 (Kearney et al., 2006) and substantiated by an individual participant data meta-analysis of coxib and tNSAID trials in 2013 (CNT Collaboration et al., 2013).
Finally, as anticipated in the 1982 Journal of Clinical Investigation paper, “since the effect of low-dose aspirin is dependent upon platelet turnover as well as aspirin sensitivity of platelet and megakaryocyte cyclooxygenase, the adequacy of this therapeutic regimen might vary in different disease states” (Patrignani et al., 1982). More recent studies based on this methodological approach have largely confirmed this prediction both in specific clinical settings and by in silico modelling. Altogether, early and newer evidences have shown the desirability and practicality of adjusting the aspirin dosing interval based on two measurements of serum TXB2 at 12 and 24 h after dosing (Pascale et al., 2012; Rocca et al., 2012), a personalized approach that may be required under conditions of reduced systemic bioavailability of aspirin (Petrucci et al., 2019) or enhanced platelet turnover (Patrono et al., 2013).
CP has conceived and drafted the work. BR has contributed to the analysis and interpretation of the available literature in the field. CP and BR revised the paper critically for important intellectual content and provided approval for publication of the content.
CP reports consulting and lecture fees from Acticor Biotech, Amgen, Bayer, GlaxoSmithKline and Zambon, and institutional research grants from Bayer, Cancer Research UK (Catalyst Award—Aspirin for Cancer Prevention Collaboration), European Commission, and Italian Drug Agency (AIFA); he serves as the chairperson of the Scientific Advisory Board of the International Aspirin Foundation. BR reports consulting and lecture fees from Bayer, institutional research grants from Cancer Research UK (Catalyst Award—Aspirin for Cancer Prevention Collaboration), and from the Italian Drug Agency (AIFA).
Albers, G. W., Amarenco, P., Easton, J. D., Sacco, R. L., Teal, P. (2004). Antithrombotic and thrombolytic therapy for ischemic stroke: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 126 Suppl 3, 483S–512S. doi: 10.1378/chest.126.3_suppl.483S
Antithrombotic Trialists’ Collaboration. (2002). Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 324, 71–86. doi: 10.1136/bmj.324.7329.71
ASCEND Study Collaborative Group, Bowman, L., Mafham, M., Wallendszus, K., Stevens, W., Buck, G., et al. (2018). Effects of Aspirin for Primary Prevention in Persons with Diabetes Mellitus. N. Engl. J. Med. 379, 1529–1539. doi: 10.1056/NEJMoa1804988
Baigent, C., Patrono, C. (2003). Selective cyclooxygenase-2 inhibitors, aspirin and cardiovascular disease: a re-appraisal. Arthritis. Rheum. 48, 12–20. doi: 10.1002/art.10738
Bala, M., Chin, C. N., Logan, A. T., Amin, T., Marnett, L. J., Boutaud, O., et al. (2008). Acetylation of prostaglandin H2 synthases by aspirin is inhibited by redox cycling of the peroxidase. Biochem. Pharmacol. 75, 1472–1481. doi: 10.1016/j.bcp.2007.12.005
Bethel, M. A., Harrison, P., Sourij, H., Sun, Y., Tucker, L., Kennedy, I., et al. (2016). Randomized controlled trial comparing impact on platelet reactivity of twice-daily with once-daily aspirin in people with type 2 diabetes. Diabet. Med. 33, 224–230. doi: 10.1111/dme.12828
Blais, N., Pharand, C., Lordkipanidzé, M., Sia, Y. K., Merhi, Y., Diodati, J. G. (2009). Response to aspirin in healthy individuals. Cross comparison of light transmission aggregometry, VerifyNow system, platelet count drop, thromboelastography (TEG) and urinary 11-dehydrothromboxane B(2). Thromb Haemost. 102, 404–411. doi: 10.1160/TH09-02-0126
Born, G. V. (1962). Aggregation of blood platelets by adenosine diphosphate and its reversal. Nature 194, 927–929. doi: 10.1038/194927b0
Born, G., Patrono, C. (2006). Antiplatelet Drugs. Br. J. Pharmacol. 147, S241–S251. doi: 10.1038/sj.bjp.0706401
Burch, J. W., Stanford, N., Majerus, P. W. (1978). Inhibition of platelet prostaglandin synthetase by oral aspirin. J. Clin. Invest. 61, 314–319. doi: 10.1172/JCI108941
Capone, M. L., Tacconelli, S., Sciulli, M. G., Grana, M., Ricciotti, E., Minuz, P., et al. (2004). Clinical pharmacology of platelet, monocyte, and vascular cyclooxygenase inhibition by naproxen and low-dose aspirin in healthy subjects. Circulation 109, 1468–1471. doi: 10.1161/01.CIR.0000124715.27937.78
Capone, M. L., Tacconelli, S., Sciulli, M. G., Anzellotti, P., Di Francesco, L., Merciaro, G., et al. (2007). Human pharmacology of naproxen sodium. J. Pharmacol. Exp. Ther. 322, 453–460. doi: 10.1124/jpet.107.122283
Cavalca, V., Rocca, B., Veglia, F., Petrucci, G., Porro, B., Myasoedova, V., et al. (2017). On-pump cardiac surgery enhances platelet renewal and impairs aspirin pharmacodynamics: Effects of improved dosing regimens. Clin. Pharmacol. Ther. 102, 849–858. doi: 10.1002/cpt.702
Cheng, Y., Austin, S. C., Rocca, B., Koller, B. H., Coffman, T. M., Grosser, T., et al. (2002). Role of prostacyclin in the cardiovascular response to thromboxane A2. Science 296, 539–541. doi: 10.1126/science.1068711
Ciabattoni, G., Maclouf, J., Catella, F., FitzGerlad, G. A., Patrono, C. (1987). Radioimmunoassay of 11-dehydro-thromboxane B2 in human plasma and urine. Biochim. Biophys. Acta 918, 293–397. doi: 10.1016/0005-2760(87)90233-5
Ciabattoni, G., Pugliese, F., Davì, G., Pierucci, A., Simonetti, B. M., Patrono, C. (1989). Fractional conversion of thromboxane B2 to urinary 11-dehydro-thromboxane B2 in man. Biochim. Biophys. Acta 992, 66–70. doi: 10.1016/0304-4165(89)90051-2
CNT Collaboration, Bhala, N., Emberson, J., Merhi, A., Abramson, S., Arber, N., et al. (2013).Coxib and traditional NSAID Trialists’. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet 382, 769–779. doi: 10.1016/S0140-6736(13)60900-9
Davì, G., Catalano, I., Averna, M., Notarbartolo, A., Strano, A., Ciabattoni, G., et al. (1990). Thromboxane biosynthesis and platelet function in type II diabetes mellitus. N. Engl. J. Med. 322, 1769–1774. doi: 10.1056/NEJM199006213222503
Davì, G., Notarbartolo, A., Catalano, I., Averna, M., Barbagallo, C., Ciabattoni, G., et al. (1992). Increased thromboxane biosynthesis in type IIa hypercholesterolemia. Circulation 85, 1792–1798. doi: 10.1161/01.CIR.85.5.1792
Davì, G., Guagnano, M. T., Ciabattoni, G., Basili, S., Falco, A., Marinopiccoli, M., et al. (2002). Platelet activation in obese women: role of inflammation and oxidant stress. JAMA 288, 2008–2014. doi: 10.1001/jama.288.16.2008
Davì, G., Patrono, C. (2007). Platelet activation and atherothrombosis. N. Engl. J. Med. 357, 2482–2494. doi: 10.1056/NEJMra071014
De Stefano, V., Rocca, B., Tosetto, A., Soldati, D., Petrucci, G., Beggiato, E., et al. (2018). The Aspirin Regimens in Essential Thrombocythemia (ARES) phase II randomized trial design - implementation of the serum thromboxane B2 assay as an evaluation tool of different dosing regimens in the clinical setting. Blood Cancer J. 8, 49. doi: 10.1038/s41408-018-0078-3
Dillinger, J. G., Sideris, G., Henry, P., Bal dit Sollier, C., Ronez, E., Drouet, L. (2012). Twice daily aspirin to improve biological aspirin efficacy in patients with essential thrombocythemia. Thromb. Res. 129, 91–94. doi: 10.1016/j.thromres.2011.09.017
Dillinger, J. G., Drissa, A., Sideris, G., Bal dit Sollier, C., Voicu, S., Manzo Silberman, S., et al. (2012). Biological efficacy of twice daily aspirin in type 2 diabetic patients with coronary artery disease. Am. Heart J. 164, 600–606. doi: 10.1016/j.ahj.2012.06.008
Di Minno, G., Davì, G., Margaglione, M., Cirillo, F., Grandone, E., Ciabattoni, G., et al. (1993). Abnormally high thromboxane A2 biosynthesis in homozygous homocystinuria. Evidence for platelet involvement and probucol-sensitive mechanism. J. Clin. Invest. 92, 1400–1406. doi: 10.1172/JCI116715
Dragani, A., Pascale, S., Recchiuti, A., Mattoscio, D., Lattanzio, S., Petrucci, G., et al. (2010). The contribution of cyclooxygenase-1 and -2 to persistent thromboxane biosynthesis in aspirin-treated essential thrombocythemia: implications for antiplatelet therapy. Blood 115, 1054–1061. doi: 10.1182/blood-2009-08-236679
Eikelboom, J. W., Hirsh, J., Weitz, J. I., Johnston, M., Yi, Q., Yusuf, S. (2002). Aspirin-resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular events. Circulation 105, 1650–1655. doi: 10.1161/01.CIR.0000013777.21160.07
Eikelboom, J. W., Hankey, G. J., Thom, J., Bhatt, D. L., Steg, P. G., Montalescot, G., et al. (2008). Incomplete inhibition of thromboxane biosynthesis by acetylsalicylic acid: determinants and effect on cardiovascular risk. Circulation 118, 1705–1712. doi: 10.1161/CIRCULATIONAHA.108.768283
Falk, E., Shah, P. K., Fuster, V., Coronary plaque disruption. (1995). Circulation 92,657-671. doi: 10.1161/01.CIR.92.3.657
FitzGerald, G. A., Brash, A. R., Falardeau, P., Oates, J. A. (1981). Estimated rate of prostacyclin secretion into the circulation of normal man. J. Clin. Invest. 68, 1272–1276. doi: 10.1172/JCI110373
FitzGerald, G. A., Oates, J. A., Hawiger, J., Maas, R. L., Roberts, L. J., 2nd, Lawson, J. A., et al. (1983). Endogenous biosynthesis of prostacyclin and thromboxane and platelet function during chronic administration of aspirin in man. J. Clin. Invest. 71, 676–688. doi: 10.1172/JCI110814
Fitzgerald, D. J., Roy, L., Catella, F., FitzGerald, G. A. (1986). Platelet activation in unstable coronary disease. N. Engl. J. Med. 315, 983–989. doi: 10.1056/NEJM198610163151602
Frelinger, A. L., 3rd, Li, Y., Linden, M. D., Barnard, M. R., Fox, M. L., Christie, D. J., et al. (2009). Association of cyclooxygenase-1-dependent and -independent platelet function assays with adverse clinical outcomes in aspirin-treated patients presenting for cardiac catheterization. Circulation 120, 2586–2596. doi: 10.1161/CIRCULATIONAHA.109.900589
Giaretta, A., Rocca, B., Di Camillo, B., Toffolo, G. M., Patrono, C. (2017). In silico modeling of the antiplatelet pharmacodynamics of low-dose aspirin in health and disease. Clin. Pharmacol. Ther. 102, 823–831. doi: 10.1002/cpt.694
Grosser, T., Naji, A., FitzGerald, G. A. (2018). Urinary prostaglandin metabolites: An incomplete reckoning and a flush to judgment. Circ. Res. 122, 537–539. doi: 10.1161/CIRCRESAHA.118.312616
Hamberg, M., Svensson, J., Samuelsson, B. (1975). Thromboxanes: a new group of biologically active compounds derived from prostaglandin endoperoxides. Proc. Natl. Acad. Sci. U. S. A. 72, 2994–2998. doi: 10.1073/pnas.72.8.2994
Kearney, P. M., Baigent, C., Godwin, J., Halls, H., Emberson, J. R., Patrono, C. (2006). Do selective cyclooxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomized trials. BMJ 332, 1302–1308. doi: 10.1136/bmj.332.7553.1302
Koudstaal, P. J., Ciabattoni, G., Van Gijn, J., Nieuwenhuis, H. K., De Groot, P., Sixma, J. J., et al. (1993). Increased thromboxane biosynthesis in patients with acute cerebral ischemia. Stroke 24, 219–223. doi: 10.1161/01.STR.24.2.219
Landolfi, R., Ciabattoni, G., Patrignani, P., Castellana, M. A. L., Pogliani, E., Bizzi, B., et al. (1992). Increased thromboxane biosynthesis in patients with polycythemia vera. Evidence of aspirin-suppressible platelet activation in vivo. Blood 80, 1965–1971.
Landolfi, R., Marchioli, R., Kutti, J., Gisslinger, H., Tognoni, G., Patrono, C., et al. (2004). Efficacy and safety of low-dose aspirin in polycythemia vera. N. Engl. J. Med. 350, 114–124. doi: 10.1056/NEJMoa035572
Lordkipanidzé, M., Pharand, C., Schampaert, E., Turgeon, J., Palisaitis, D. A., Diodati, J. G. (2007). A comparison of six major platelet function tests to determine the prevalence of aspirin resistance in patients with stable coronary artery disease. Eur. Heart J. 28, 1702–1708. doi: 10.1093/eurheartj/ehm226
Maree, A. O., Curtin, R. J., Dooley, M., Conroy, R. M., Crean, P., Cox, D., et al. (2005). Platelet response to low-dose enteric-coated aspirin in patients with stable cardiovascular disease. J. Am. Coll. Cardiol. 46, 1258–1263. doi: 10.1016/j.jacc.2005.06.058
Michelson, A., Cattaneo, M., Eikelboom, J. W., Gurbel, P., Kottke-Marchant, K., Kunicki, T. J., et al. (2005). Aspirin resistance: position paper of the Working Group on Aspirin Resistance. J. Thromb. Haemost. 3, 1309–1311.
Minuz, P., Patrignani, P., Gaino, S., Degan, M., Menapace, L., Tommasoli, R., et al. (2002). Increased oxidative stress and platelet activation in patients with hypertension and renovascular disease. Circulation 106, 2800–2805. doi: 10.1161/01.CIR.0000039528.49161.E9
Mitchell, J. A., Knowles, R. B., Kirkby, N. S., Reed, D. M., Edin, M. L., White, W. E., et al. (2018). Kidney transplantation in a patient lacking cytosolic phospholipase A2 proves renal origins of urinary PGI-M and TX-M. Circ. Res. 122, 555–559. doi: 10.1161/CIRCRESAHA.117.312144
Muir, A. R., McMullin, M. F., Patterson, C., McKeown, P. P. (2009). Assessment of aspirin resistance varies on a temporal basis in patients with ischaemic heart disease. Heart 95, 1225–1229. doi: 10.1136/hrt.2008.150631
Nowak, J., Murray, J. J., Oates, J. A., FitzGerald, G. A. (1987). Biochemical evidence of a chronic abnormality in platelet and vascular function in healthy individuals who smoke cigarettes. Circulation 76, 6–14. doi: 10.1161/01.CIR.76.1.6
Paikin, J. S., Hirsh, J., Ginsberg, J. S., Weitz, J. I., Chan, N. C., Whitlock, R. P., et al. (2015). Multiple daily doses of acetyl-salicylic acid (ASA) overcome reduced platelet response to once-daily ASA after coronary artery bypass graft surgery: a pilot randomised controlled trial. J. Thromb. Haemost. 13, 448–456. doi: 10.1111/jth.12832
Parker, W. A. E., Orme, R. C., Hanson, J., Stokes, H. M., Bridge, C. M., Shaw, P. A., et al. (2019). Very-low-dose twice-daily aspirin maintains platelet inhibition and improves haemostasis during dual antiplatelet therapy for acute coronary syndrome. Platelets 30, 148–157. doi: 10.1080/09537104.2019.1572880
Pascale, S., Petrucci, G., Dragani, A., Habib, A., Zaccardi, F., Pocaterra, D., et al. (2012). Aspirin-insensitive thromboxane biosynthesis in essential thrombocythemia is explained by accelerated renewal of the drug target. Blood 119, 3595–3603. doi: 10.1182/blood-2011-06-359224
Patrignani, P., Filabozzi, P., Patrono, C. (1982). Selective cumulative inhibition of platelet thromboxane production by low-dose aspirin in healthy subjects. J. Clin. Invest. 69, 1366–1372. doi: 10.1172/JCI110576
Patrignani, P., Morton, H., Cirino, M., Lord, A., Charette, L., Gillard, J., et al. (1989). Fractional conversion of thromboxane A2 and B2 to urinary 2,3-dinor-thromboxane B2 and 11-dehydro-thromboxane B2 in the cynomolgus monkey. Biochim. Biophys. Acta 992, 71–77. doi: 10.1016/0304-4165(89)90052-4
Patrono, C. (1994). Aspirin as an antiplatelet drug. N. Engl. J. Med. 330, 1287–1294. doi: 10.1056/NEJM199405053301808
Patrono, C., Baigent, C. (2014). Nonsteroidal anti-inflammatory drugs and the heart. Circulation 129, 907–916. doi: 10.1161/CIRCULATIONAHA.113.004480
Patrono, C., Ciabattoni, G., Pinca, E., Pugliese, F., Castrucci, G., De Salvo, A., et al. (1980). Low-dose aspirin and inhibition of thromboxane B2 production in healthy subjects. Thromb. Res. 17, 317–327. doi: 10.1016/0049-3848(80)90066-3
Patrono, C., Ciabattoni, G., Pugliese, F., Pierucci, A., Blair, I. A., FitzGerald, G. A. (1986). Estimated rate of thromboxane secretion into the circulation of normal man. J. Clin. Invest. 77, 590–594. doi: 10.1172/JCI112341
Patrono, C., Ciabattoni, G., Remuzzi, G., Gotti, E., Bombardieri, S., Di Munno, O., et al. (1985). Functional significance of renal prostacyclin and thromboxane A2 production in patients with systemic lupus erythematosus. J. Clin. Invest. 76, 1011–1018. doi: 10.1172/JCI112053
Patrono, C., García Rodríguez, L. A., Landolfi, R., Baigent, C. (2005). Low-dose aspirin for the prevention of atherothrombosis. N. Engl. J. Med. 353, 2373–2383. doi: 10.1056/NEJMra052717
Patrono, C., Baigent, C., Hirsh, J., Roth, G. (2008). Antiplatelet drugs: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 133, 199S–233S. doi: 10.1378/chest.08-0672
Patrono, C., Rocca, B. (2008). Aspirin: promise and resistance in the new millennium. Arterioscler. Thromb. Vasc. Biol. 28, 25S–32S. doi: 10.1161/ATVBAHA.107.160481
Patrono, C., Rocca, B. (2017). Type 2 diabetes, obesity, and aspirin responsiveness. J. Am. Coll. Cardiol. 69, 613–615. doi: 10.1016/j.jacc.2016.11.049
Patrono, C., Rocca, B., De Stefano, V. (2013). Platelet activation and inhibition in polycythemia vera and essential thrombocythemia. Blood 121, 1701–1711. doi: 10.1182/blood-2012-10-429134
Pedersen, A. K., FitzGerald, G. A. (1984). Dose-related kinetics of aspirin. Presystemic acetylation of platelet cyclooxygenase. N. Engl. J. Med. 311, 1206–1211. doi: 10.1056/NEJM198411083111902
Petrucci, G., Rizzi, A., Cavalca, V., Habib, A., Pitocco, D., Veglia, F., et al. (2016). Patient-independent variables affecting the assessment of aspirin responsiveness by serum thromboxane measurement. Thromb. Haemost. 116, 891–896. doi: 10.1160/TH16-05-0349
Petrucci, G., Zaccardi, F., Giaretta, A., Cavalca, V., Capristo, E., Cardillo, C., et al. (2019). Obesity is associated with in vivo platelet activation and impaired responsiveness to once-daily, low-dose aspirin. J. Thromb. Haemost. 17, 885–895. doi: 10.1111/jth.14445
Pierucci, A., Simonetti, B. M., Pecci, G., Mavrikakis, G., Feriozzi, S., Cinotti, G. A., et al. (1989). Improvement of renal function with selective thromboxane antagonism in lupus nephritis. N. Engl. J. Med. 320, 421–425. doi: 10.1056/NEJM198902163200703
Reilly, I. A., FitzGerald, G. A. (1987). Inhibition of thromboxane formation in vivo and ex vivo: implications for therapy with platelet inhibitory drugs. Blood 69, 180–186.
Remuzzi, G., FitzGerald, G. A., Patrono, C. (1992). Thromboxane synthesis and action within the kidney. Kidney Int. 41, 1483–1493. doi: 10.1038/ki.1992.217
Reny, J. L., Berdagué, P., Poncet, A., Barazer, I., Nolli, S., Fabbro-Peray, P., et al. (2012). Antiplatelet drug response status does not predict recurrent ischemic events in stable cardiovascular patients: results of the Antiplatelet Drug Resistances and Ischemic Events study. Circulation 125, 3201–3210. doi: 10.1161/CIRCULATIONAHA.111.085464
Roberts, L. J., 2nd, Sweetman, B. J., Oates, J. A. (1981). Metabolism of thromboxane B2 in man. Identification of twenty urinary metabolites. J. Biol. Chem. 256, 8384–8393.
Rocca, B., Ciabattoni, G., Tartaglione, R., Cortellazzo, S., Barbui, T., Patrono, C., et al. (1995). Increased thromboxane biosynthesis in essential thrombocythemia. Thromb. Haemost. 74, 1225–1230. doi: 10.1055/s-0038-1649916
Rocca, B., Patrono, C. (2005). Determinants of the interindividual variability in response to antiplatelet drugs. J. Thromb. Haemost. 3, 1597–1602. doi: 10.1111/j.1538-7836.2005.01380.x
Rocca, B., Patrono, C. (2015). Platelet progenitors: the hidden drug target. Eur. Heart J. 36, 3211–3213. doi: 10.1093/eurheartj/ehv366
Rocca, B., Santilli, F., Pitocco, D., Mucci, L., Petrucci, G., Vitacolonna, E., et al. (2012). The recovery of platelet cyclooxygenase activity explains interindividual variability in responsiveness to low-dose aspirin in patients with and without diabetes. J. Thromb. Haemost. 10, 1220–1230. doi: 10.1111/j.1538-7836.2012.04723.x
Rocca, B., Fox, K. A. A., Ajjan, R. A., Andreotti, F., Baigent, C., Collet, J.-P., et al. (2018). Antithrombotic therapy and body mass: an expert position paper of the ESC Working Group on Thrombosis. Eur. Heart J. 39, 1672–1686f. doi: 10.1093/eurheartj/ehy066
Rothwell, P. M., Algra, A., Chen, Z., Diener, H. C., Norrving, B., Mehta, Z. (2016). Effects of aspirin on risk and severity of early recurrent stroke after transient ischaemic attack and ischaemic stroke: time-course analysis of randomised trials. Lancet 388, 365–375. doi: 10.1016/S0140-6736(16)30468-8
Rothwell, P. M., Cook, N. R., Gaziano, J. M., Price, J. F., Belch, J. F. F., Roncaglioni, M. C., et al. (2018). Effects of aspirin on risks of vascular events and cancer according to bodyweight and dose: analysis of individual patient data from randomised trials. Lancet 392, 387–399. doi: 10.1016/S0140-6736(18)31133-4
Santilli, F., Rocca, B., De Cristofaro, R., Lattanzio, S., Pietrangelo, L., Habib, A., et al. (2009). Platelet cyclooxygenase inhibition by low-dose aspirin is not reflected consistently by platelet function assays. Implications for aspirin “resistance”. J. Am. Coll. Cardiol. 53, 667–677. doi: 10.1016/j.jacc.2008.10.047
Smith, J. B., Willis, A. L. (1971). Aspirin selectively inhibits prostaglandin production in human platelets. Nat. New Biol. 231, 235–237. doi: 10.1038/newbio231235a0
Smith, J. P., Haddad, E. V., Taylor, M. B., Oram, D., Blakemore, D., Chen, Q., et al. (2012). Suboptimal inhibition of platelet cyclooxygenase-1 by aspirin in metabolic syndrome. Hypertension 59, 719–725. doi: 10.1161/HYPERTENSIONAHA.111.181404
Spectre, G., Arnetz, L., Ostenson, C. G., Brismar, K., Li, N., Hjemdahl, P. (2011). Twice daily dosing of aspirin improves platelet inhibition in whole blood in patients with type 2 diabetes mellitus and micro- or macrovascular complications. Thromb. Haemost. 106, 491–499. doi: 10.1160/TH11-04-0216
Tefferi, A., Vannucchi, A. M., Barbui, T. (2018). Polycythemia vera treatment algorithm 2018. Blood Cancer J. 8, 3. doi: 10.1038/s41408-017-0042-7
The RISC Group. (1990). Risk of myocardial infarction and death during treatment with low dose aspirin and intravenous heparin in men with unstable coronary artery disease. Lancet 336, 827–830. doi: 10.1016/0140-6736(90)92336-G
van Kooten, F., Ciabattoni, G., Patrono, C., Dippel, D. W. J., Koudstaal, P. J. (1997). Platelet activation and lipid peroxidation in patients with acute ischemic stroke. Stroke 28, 1557–1563. doi: 10.1161/01.STR.28.8.1557
Vejar, M., Fragasso, G., Hackett, D., Lipkin, P. D., Maseri, A., Born, G. V., et al. (1990). Dissociation of platelet activation and spontaneous myocardial ischemia in unstable angina. Thromb. Haemost. 63, 163–168. doi: 10.1055/s-0038-1645038
Keywords: thromboxane, prostanoids biosynthesis, aspirin, cardiovasular disease, platelet activation
Citation: Patrono C and Rocca B (2019) Measurement of Thromboxane Biosynthesis in Health and Disease. Front. Pharmacol. 10:1244. doi: 10.3389/fphar.2019.01244
Received: 03 June 2019; Accepted: 27 September 2019;
Published: 30 October 2019.
Edited by:
G.Enrico Rovati, University of Milan, ItalyReviewed by:
Anthony Ashton, University of Sydney, AustraliaCopyright © 2019 Patrono and Rocca. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlo Patrono, Y2FybG8ucGF0cm9ub0B1bmljYXR0Lml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.