
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pharmacol. , 24 September 2019
Sec. Translational Pharmacology
Volume 10 - 2019 | https://doi.org/10.3389/fphar.2019.01057
This article is part of the Research Topic New Horizons in Health-Promoting Technologies: From Development to Rational Use View all 31 articles
 Renata Carvalho Feitosa1,2
Renata Carvalho Feitosa1,2 Danilo Costa Geraldes1,2
Danilo Costa Geraldes1,2 Viviane Lucia Beraldo-de-Araújo1
Viviane Lucia Beraldo-de-Araújo1 Juliana Souza Ribeiro Costa1,2
Juliana Souza Ribeiro Costa1,2 Laura Oliveira-Nascimento1*
Laura Oliveira-Nascimento1*Oral route maintains its predominance among the ones used for drug delivery, especially when medicines are self-administered. If the dosage form is solid, therapy gains in dose precision and drug stability. Yet, some active pharmaceutical substances do not present the required solubility, permeability, or release profile for incorporation into traditional matrices. The combination of nanostructured drugs (nanoparticle [NP]) with these matrices is a new and little-explored alternative, which could bring several benefits. Therefore, this review focused on combined delivery systems based on nanostructures to administer drugs by the oral cavity, intended for buccal, sublingual, gastric, or intestinal absorption. We analyzed published NP-in-matrix systems and compared main formulation characteristics, pharmacokinetics, release profiles, and physicochemical stability improvements. The reported formulations are mainly semisolid or solid polymers, with polymeric or lipid NPs and one active pharmaceutical ingredient. Regarding drug specifics, most of them are poorly permeable or greatly metabolized. The few studies with pharmacokinetics showed increased drug bioavailability and, sometimes, a controlled release rate. From our knowledge, the gathered data make up the first focused review of these trendy systems, which we believe will help to gain scientific deepness and future advancements in the field.
Medicines administered by the oral cavity have different fates, according to their varied processes: (i) immediate delivery and absorption, (ii) slow delivery from an adherent drug delivery system (DDS), followed by absorption or local action; and (iii) transport to the gastrointestinal region for absorption or local action. The latter is the gold standard for medicines and the most common process used for self-administered drug intake. Recently, buccal permeation (items i and ii) strategies increased in number of developments; this route bypasses hepatic and gastrointestinal effects, which is advantageous to sensitive drugs (Barua et al., 2016).
Regardless of the process, drug solubility can impair efficient release from DDS and subsequent mucosal crossing. Therefore, several solubilization strategies are available, such as salt forms of active molecules, pH modifiers, cosolvents, amorphization, solid dispersions, inclusion complexes, microemulsions, and nanotechnology (Kalepu and Nekkanti, 2015). The latter represents a large portion of published research but a few marketed products and no buccal-based options.
In addition to the solubility enhancement, nanoparticles (NPs) aid drug efficacy in at least four different ways (Figure 1). The simpler way relates to transport throughout the gastrointestinal lumen and local release (mode 1). Another possibility consists of carrier adherence to the mucosal surface/mucus to enhance drug absorption, but without carrier crossing (mode 2). Nanoparticles may also be absorbed and transport the drug systemically, enhancing its plasma half-life (mode 3). Absorbed NPs can then perform passive targeting due to charge, size, morphology, and constituents. For a detailed review on mechanisms of NP/absorption and uptake, see Griffin et al. (2016). Mode 4 happens when specific surface molecules or pH-responsive mechanisms allow the carrier to perform an active targeting after absorption. Modes 3 and 4 can still avoid first-pass metabolism if absorption happens in the buccal mucosa or lymphatic vessels. However, like other delivery strategies, NPs can fail to liberate drugs at an expected rate or target, as described in the following paragraphs.
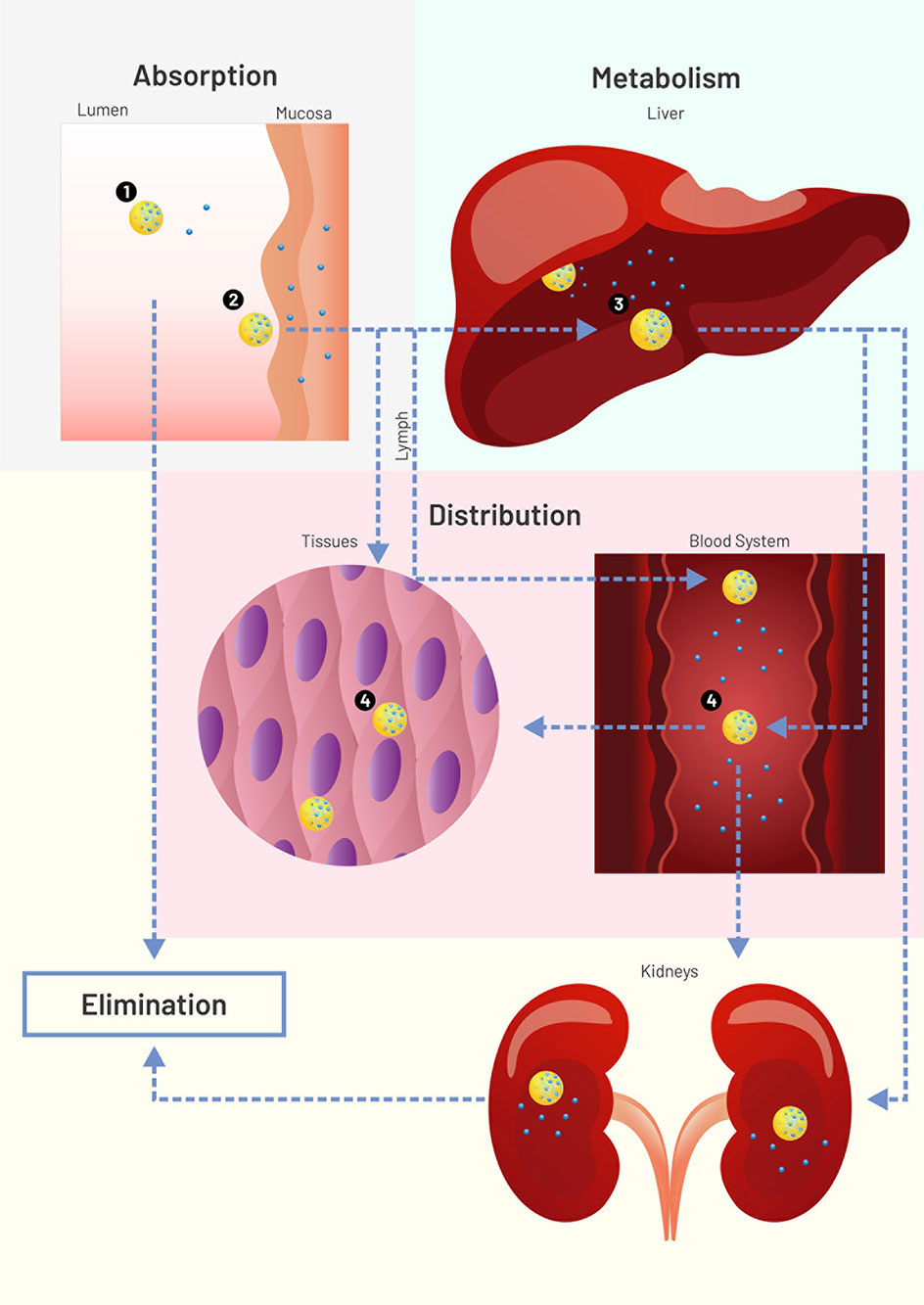
Figure 1 Scheme (not to scale) of drug (blue circles) loaded nanoparticles (NPs, yellow circles) interacting with physiological environments. Nanoparticle modes to enhance drug systemic delivery through the oral or buccal routes. 1: Lumen release (solubility enhancement), followed by NP excretion or degradation. 2: Adherence to mucus or mucosal surface for enhanced drug absorption (no NP permeation), followed by NP excretion or degradation. 3: NP absorption for enhanced plasma half-life, followed by NP liver degradation or lymphatic drainage. 4: Tissue or cell targeting. Figure created for this review in CorelDRAW Graphics Suite X7.
For drug uptake enhancement, NPs need to interact with the respective mucosal surface/mucus during an adequate amount of time. Some particles do stick to mucosa under continuous rinsing, such as thiolated chitosan NPs. But, in this case, NPs were freeze dried and applied on top of the mucosa in an ex vivo test (Bernkop-Schnürch et al., 2006). Since powder formulations require specific devices or encapsulation for a precise dosage, they generally require an additional delivery strategy to be administered.
A noteworthy NP disadvantage is the burst effect: a rapid initial drug release followed by a sustained or controlled release. Rapid plasma peaks or local drug concentration are desirable in some therapies; however, burst behavior is frequently uncontrollable and irreproducible. The phenomenon occurs due to several reasons, including weakly bound drugs, molecule migration to the particle surface, and nanomatrix heterogeneity (Kamaly et al., 2016). Strategies to prevent it include coating or nanomatrix reformulation (Kamaly et al., 2016), not always straightforward because both processes change NP physicochemical characteristics.
The majority of oral DDSs are biodegradable, so they will erode or degrade at some point. In fact, some systems rely on both properties as a release mechanism. If the carrier liberates as desired, it may not be resistant to pH/enzymes during traffic or at the action site. An earlier instability may interfere with the desired drug liberation profile, such as for regular gelatin NPs: their degradation in the stomach requires coating, chemical modification or an outermatrix embedment to allow intestinal release (Imperiale and Sosnik, 2013).
Apart from degradation, NPs can change their fate, uptake, or release rate due to in vitro or in vivo aggregation. The in vitro facet shows that cell media for cell culture is quite different from lumen or saliva environments, which makes it hard to predict aggregation during in vivo absorption (Moore et al., 2015).
All failure modes described above can be solved or attenuated with NP dispersion in micromatrices and macromatrices (Figure 2), reasons why this technique became important for buccal and oral delivery. As opposed to coating and core modifications, matrix embedment brings an extra added value: it can be applied to several kinds of NPs, with a better general prediction of the release behavior.
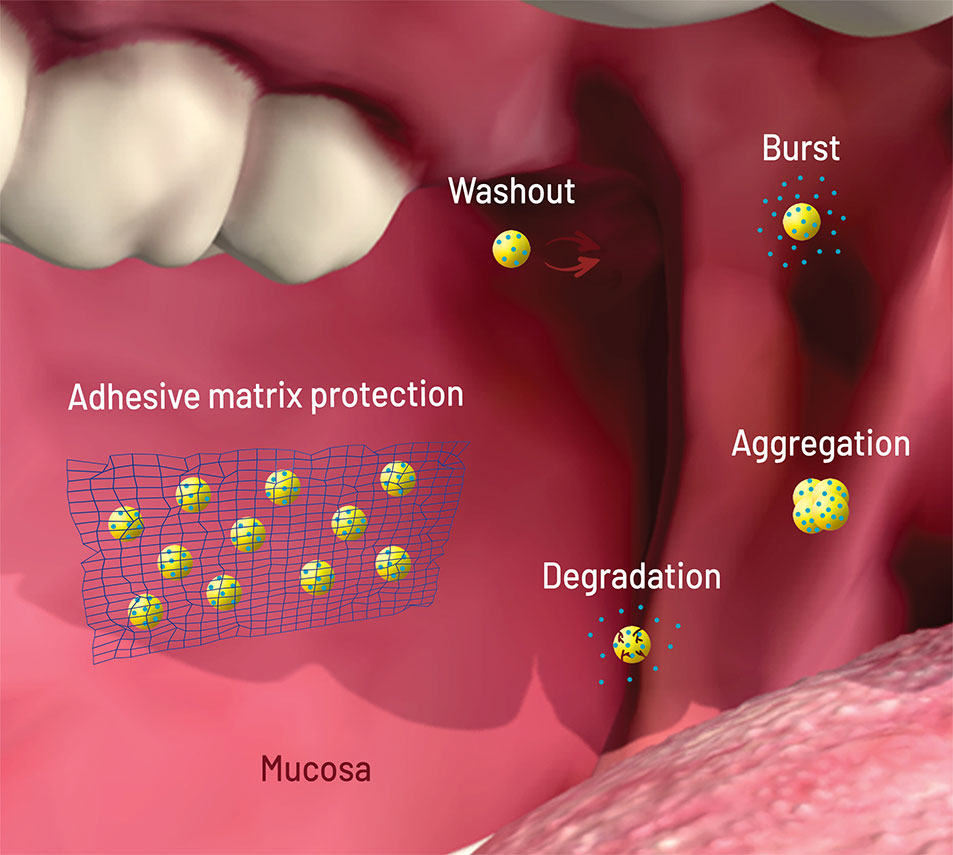
Figure 2 Scheme (not to scale) of the failure modes of nanoparticles (NPs, yellow circles) loaded with drug (blue circles) after oral/buccal administration: early washout in mouth or intestinal cavities; early degradation due to pH or enzymes; burst release upon contact with aqueous media (saliva, stomach acid); aggregation due to pH, osmotic environment, and protein binding. The network on the left shows matrix protection through adhesion (wash out elimination), shielding (degradation and burst elimination/decrease), and physical separation (aggregation elimination). Figure created for this review in CorelDRAW Graphics Suite X7 and Adobe Photoshop CS6.
Considering all points above, this review intends to analyze pharmacokinetic improvements with the use of NPs incorporated in matrices for drug delivery (NP-in-matrix). Therefore, we focused here on pharmacokinetic data from animal and humans (when available). Several NP-in-matrix systems were tested only with in vitro assays. However, correlation of in vivo behavior is not guaranteed, and an extensive array of protocols impairs model comparisons (discussed in the following topic). Combined systems resulting in nanosized carriers (NP-in-NP) were not included in this work. Based on the theoretical analysis, we intend to verify and clarify matrix role on permeation and bioavailability of nanocarriers.
Pharmacokinetics describes absorption, distribution, metabolism, and excretion processes after drug intake. This knowledge remains essential for safety and efficacy assessments of the therapy in the search for the optimal DDS. However, in vivo tests must be a final step toward product development. It should succeed an extensive physicochemical characterization, together with promising in vitro data assessing biorelevant properties. Table 1 lists the most common assays used to evaluate and predict in vivo parameters related to pharmacokinetics, together with drug and NP predictors. Because matrix-based formulations aid mainly in absorption, this process will be discussed in more detail below.
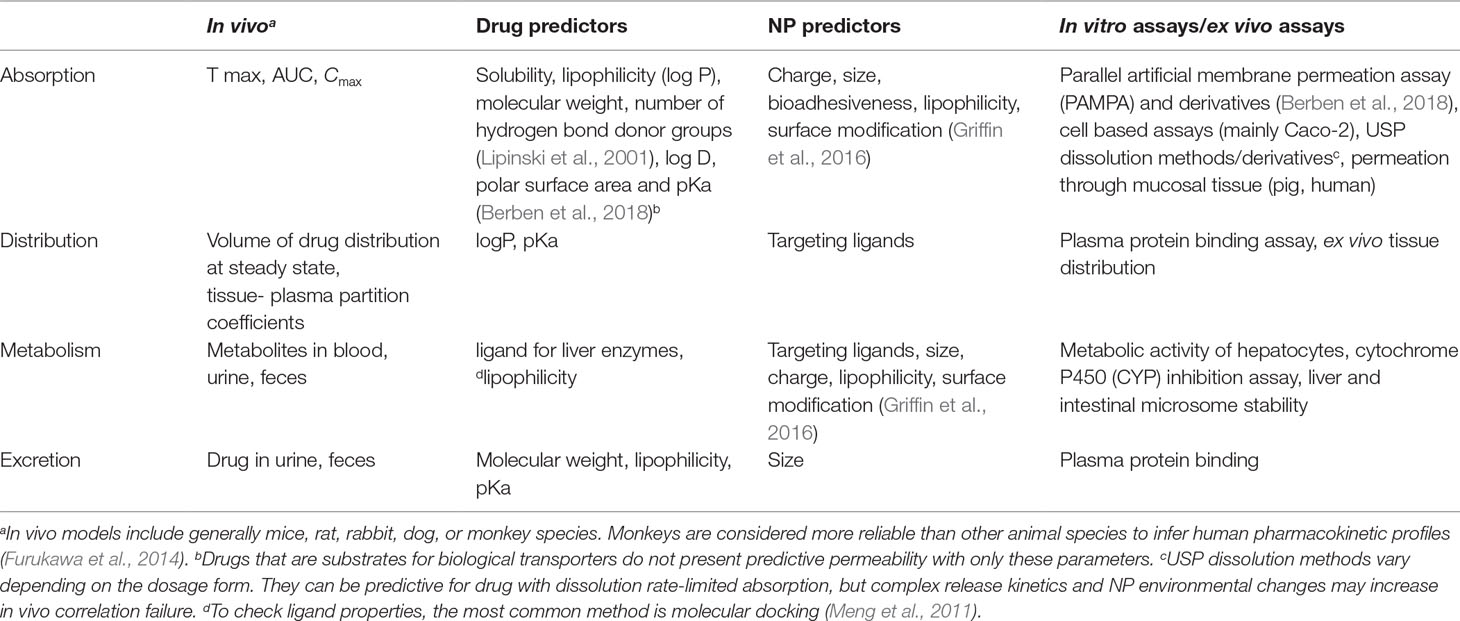
Table 1 Pharmacokinetic parameters and evaluation strategies for oral/buccal medicines.
Nanoparticle permeation happens only after mucus barrier crossing (gastrointestinal) and subsequent cellular uptake for transcytosis (M cells, enterocytes) or paracellular transport. In vivo absorption is generally better predicted by ex vivo mucosal permeation than in vitro tests with synthetic membranes; the extracted tissue offers a biochemical, anatomical, and structural resemblance to its in vivo counterpart that is difficult to replicate (Berben et al., 2018).
When it comes to gastrointestinal absorption, Caco-2 cell model stands out among the options, joining cell layer with synthetic membrane permeation. This system is frequently used to classify drugs in a permeation rank to direct dosage form development. However, a review covering Caco-2 correlation with in vivo intestinal permeability stressed the test fragility. Based on several studies, the authors concluded that the model is applicable to hydrophobic drugs but fails to predict hydrophilic molecule absorption. Even with hydrophobic drugs, variability is high yet minimized with internal standards. The paracellular route and active transport seem to correlate with higher variability and in vivo–in vitro lack of correlation (Larregieu and Benet, 2013).
Regardless of the in vitro–ex vivo model, most of them do not promote predigestion of samples. One cannot estimate the effect of gastrointestinal fluids upon NPs without this assessment. Even nondegradable polystyrene NPs (unmodified, aminated, or carboxylated) promoted different ex vivo permeation profiles with predigestion, showing its relevance for in vivo prediction (Westerhout et al., 2017).
Drug/NP in vivo absorption can be estimated by plasma detection if metabolism does not influence their integrity. In vivo pharmacokinetics focuses on blood sampling and drug quantification at several time points, generally with rats, rabbits, or humans. The most common parameters include the amount of drug detected over time (area under the curve [AUC]), the highest drug level detected (Cmax), and the time the latter happened (tmax). On the other hand, NPs are seldom classified based on plasma levels. Nanoparticle tracking relies mainly on marked particles with fluorescent dyes to observe biodistribution in tissues. An important reminder is that plasma levels do not discriminate buccal from gastric or intestinal absorption.
The following items present a discussion of all articles we could find with in vivo pharmacokinetic data of NP-in-matrix DDSs. The NP type directed subdivisions based on their main constituent. Exact values and formulation details are presented in Table 2.
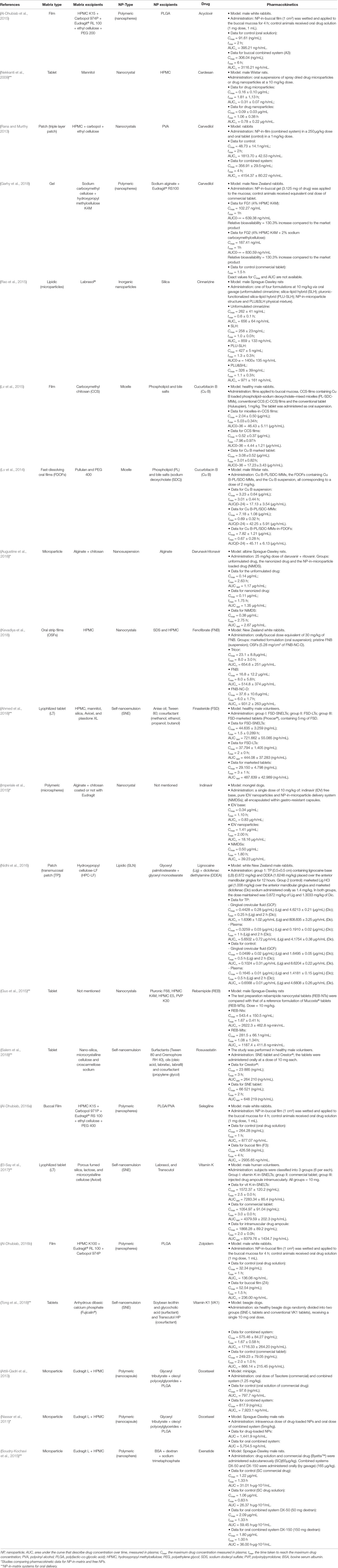
Table 2 Pharmacokinetic data for NP-in-matrix systems.
Polymers vary on their degradation, polarity, source, and chain size properties (Hallan et al., 2016). Polymeric based NPs present scalable manufacturing methods and capability to load a wide range of drug types (Crucho and Barros, 2017). The most common natural polymers for NPs are chitosan, sodium alginate, dextran, gelatin, and albumin. These hydrophilic proteins and carbohydrates degrade in physiological conditions, besides their biocompatibility, which helps to avoid side effects (Boateng and Areago, 2014; Madkhali et al., 2019; Bronze-Uhle et al., 2017; Kim et al., 2001). Alginate and chitosan still exhibit bioadhesive nature, which increases NP efficacy for mucosal delivery (Boateng and Areago, 2014). In turn, albumin discharges drugs via desorption without significant burst effects (Jiang and Stenzel, 2016). We have not found any pharmacokinetics data for NP-in-matrix systems based on gelatin or chitosan NPs; however, several studies discussed in this article address these excipients as matrix components.
Concerning alginate NPs, Garhy et al. loaded carvedilol in these carriers and further incorporated this system in buccoadhesive gels. The in vitro release assay showed burst behavior for all 12 different NP formulations. However, NP-in-gel formulations (FG1 and FG2) indicated that the gelling agent delayed carvedilol release, which diminished burst. FG1 gel contained hydroxypropyl methylcellulose (HPMC), whereas le FG2 presented the same HPMC concentration and sodium carboxymethylcellulose. Rabbits treated with FG2 formulation showed a two-fold increase in relative bioavailability compared to the market product. The increase in bioavailability occurred probably due to NP enhancement in drug solubility and the buccal bypass of the first-pass effect (Garhy et al., 2018).
Albumin properties were tested in exenatide-loaded bovine serum albumin/dextran NPs; the peptide was adsorbed to the carrier protein and released in a sustained manner due to dextran crosslinkings. This loaded NP was incorporated in gastroresistant microparticles of Eudragit L/HPMC. The incorporation led to macromolecule protection and decreased release rates of the peptide cargo. Consequently, pharmacokinetic data showed that NP-in-matrix resulted in a high relative oral bioavailability of 77% compared to a subcutaneous injection of the commercial equivalent medicine (Soudry-Kochavi et al., 2015).
Among the synthetic polymers, the aliphatic polyesters and their copolymers are the most used for drug delivery because of their biodegradability and biocompatibility. One of the most popular is the poly(lactic acid), often combined with glycolide to form the hydrophobic copolymer poly(lactic-co-glycolic acid) (PLGA) (Washington et al., 2017); PLGA exhibit high stability in biological fluids and long clinical experience; it was the only synthetic polymer to compose a nanostructure part of an NP-in-matrix dosage form with pharmacokinetic data. All the drugs entrapped into these studied systems are considered poorly water-soluble drugs.
Three different studies reported buccal films as matrices for PLGA NPs. The incorporated drugs were acyclovir (Al-Dhubiab et al., 2015), selegiline (Al-Dhubiab, 2016a) and zolpidem (Al-Dhubiab, 2016b). In vitro studies showed that film formulations prolonged drug liberation in a composition-dependent mode. The results of ex vivo studies with rabbit buccal mucosa showed that these nanospheres can permeate the tissue. In vivo results confirmed the predictions, whereas male rabbits demonstrated an increased drug bioavailability with the NP-in-film strategy. After incorporating NPs in polymeric films, the bioadhesive properties increased the residence time in oral cavity. It was also observed that Cmax, AUC, and tmax improved with the use of combined systems when compared to controls (oral drug solutions, results in Table 2).
The PLGA NP improvements may come as well from a micro matrix strategy. Nassar et al. (Nassar et al., 2011) used docetaxel-loaded PLGA nanocapsules in entero-coated microparticles. The microcarrier released NPs that penetrated the enterocytes of rats, bypassed permeability-glycoprotein pump, and apparently circumvented gut metabolism of the drug. An oral administration of NP-in-matrix resulted in higher bioavailability than intravenous solution of the free drug (commercial formulation, 276%) and its NP formulation (400%). A subsequent study with minipigs confirmed the pattern obtained with rats; the superiority of NP-in-matrix over NPs was attributed to lymphatic transportation that changed drug biodistribution (Attili-Qadri et al., 2013).
Lipid NPs are well-established DDSs due to their high biocompatibility, biodegradability, low toxicity, and applicability to various administration routes (Chime and Onyishi, 2013; Wissing et al., 2004; Shastri, 2017; Teixeira et al., 2017; Puri et al., 2009). The main lipid NPs reported are liposomes, solid lipid nanoparticles (SLNs), nanostructured lipid carriers, and nanoemulsions. The hydrophobic cores enhance drug solubility and protect it from the environment; the surfactant layer separates particles by steric or electrical hindrance. The exception is the liposome vesicle form, which allows loading of hydrophobic and hydrophilic drug molecules into its outer layer or aqueous core, respectively (Karamanidou et al., 2016). Among the cited particles, only NLC did not present in vivo data, probably because it is the most recent development among this group.
Lipidic NPs may deform under mechanical stress, such as tableting; they can also undergo lipid phase transitions upon heating, such as for film casting. Therefore, Hazzah et al. incorporated curcumin SLN in freeze-dried polymeric sponges to avoid the cited issues. Human studies showed that curcumin SLN-in-polycarbophil sponge has higher Cmax, tmax, and AUC than the SLN-in-HPMC sponge (Table 2). In accordance, polycarbophil formulation adhered to the mucosa for a longer time (15 h, compared to 4 h) and presented higher matrix porosity and homogeneous distribution of SLNs. The decreased porosity of HPMC sponges diminished swelling and consequent interaction with mucin. Also, SLNs remained onto the surface of HPMC sponges, lowering its adhesion property to the oral mucosa and releasing the NPs faster than polycarbophil. Although pure NPs were not tested in vivo, in vitro release showed that NP-in-sponge eliminated the burst effect of the nanodispersions, which improves prediction of therapy outcomes and avoids possible plasma peaks (Hazzah et al., 2015).
Ahmed et al. also used freeze-dried polymeric matrix (gelatin “tablets,” no compression step) to load lipidic self-nanoemulsions (SNEs) of finasteride. Different from the mucoadhesive sponges, the freeze-dried tablets melt on the mouth upon contact with saliva. These macrocarriers aim as fast release as possible for buccal and gastrointestinal absorption. Both formulated tablets (with or without SNE) presented in human higher Cmax and shorter tmax of the drug than the marketed tablets. Likewise, AUC and Mean Residence Time (MRT) indicated superiority of NP-in-tablet system. The improved drug bioavailability may enhance the therapeutic effects (Ahmed et al., 2018).
A variety of this technique decreased the low bioavailability of oral vitamin K. The SNE was loaded on porous silica carriers and later incorporated in lyophilized tablets. SNE-in-lyophilized tablets increased absorption rate and extent of vitamin K in humans compared to marketed tablets. Even better, the NP-in-matrix system presented AUC similar to the commercial intramuscular injection (El-Say et al., 2017). Powdered SNE loaded with vitamin K1 was also incorporated into regular tablets, but with different vitamin loadings. Because SNEs form nanoparticles upon contact with gastrointestinal fluid, NP deformation was not a concern. Beagle dogs administered with SNE-in-tablet showed a 2.3-fold increase in vitamin K1Cmax and 1.98-fold in AUC (Tong et al., 2018). The increment in bioavailability probably happened due to higher surface area and consequent higher drug dissolution rate in the gastrointestinal tract (Gong et al., 2016).
SNE-in-tablet was also applied to rosuvastatin delivery, increasing solubility and bypassing hepatic first-pass metabolism. Male humans taking SNE-in-tablet had rosuvastatin AUC increased 2.45 times and Cmax increased 2.78 times compared with the intake of commercial rosuvastatin tablets. Similarly, tmax decreased with drug administration in SNE tablet, highlighting its benefits (Salem et al., 2018).
The latter lipid NP-in-matrix with published in vivo pharmacokinetics is the phospholipid-bile salts-mixed micelles. Cucurbitacin B was loaded in these micelles to address its water insolubility, toxicity, and gastrointestinal side effects. The use of fast-dissolving oral films of pullulan as a matrix maintained the in vivo absorption properties like micelles and promoted a significant increase in the oral bioavailability of cucurbitacin B in Wistar rats (highest values of Cmax and AUC, lowest tmax), when compared to the free suspension drug. Also, the matrix did not interfere with Cu B-micelles original structure (Lv et al., 2014). These micelles in carboxymethyl chitosan buccal film rendered a mucoadhesive formulation, which released the drug for a longer period. It resulted in 2.69-fold increase in bioavailability in rabbits when compared to marketed tablets and 10.46 times the film formulation without NP. The team reported that buccal mucosa barrier probably explains higher Cmax and lower tmax of oral tablets; high AUC from film formulations may be owed to the presence of permeation enhancers in it and the nanosized drug, ensuring the increase in the amount of drug penetration into the blood (Lv et al., 2015). It would be interesting to compare the influence of the different matrices in cucurbitacin B kinetics, but the animal models differ in species, drug dose, and period of evaluation.
Inorganic nanoparticles are flexible carriers that allow surface modification, drug targeting, and modified drug release. This group includes silica, clay, and metals as excipients arranged in nanoparticles, nanotubes, or nanorods/nanoparticles, respectively (Kerdsakundee et al., 2017; Rao et al., 2011; Zhang et al., 2018). The silica-based ones were the only inorganic NP-in-matrix compositions tested in vivo for oral administration. Although biocompatible and with a well-defined/modifiable structure, they do not adhere to mucosa and are good candidates for matrix incorporation (Slowing et al., 2008).
To improve the oral delivery of the poorly soluble drug cinnarizine, an antihistamine and calcium-channel blocker, Rao S. et al. developed a pluronic functionalized silica–lipid hybrid microparticle. Pluronic acts as a polymeric precipitation inhibitor, avoiding recrystallization of cinnarizine dissolution; the silica–lipid hybrid microparticle improved drug partition by producing a hydrophobic microenvironment. Bioavailability was compared between the cinnarizine loaded functionalized silica-lipid hybrid system, unformulated cinnarizine and a cinnarizine loaded nonfunctionalized silica-lipid system; the in vivo design included a single dose of 10 mg/kg of each formulation via oral gavage. The study resulted in more than 2.1-fold improvement in the AUC and 1.6-fold improvement in Cmax of cinnarizine of the functionalized NP-in-microparticle structure in comparison to the unformulated one and a 1.6-fold improvement in both AUC and Cmax in comparison with the nonfunctionalized formulation, resulting in an overall improved bioavailability of cinnarizine (Rao et al., 2015).
Poor solubility, low bioavailability, and short stability in vivo are limiting problems in the development and delivery of new active ingredients (Liu et al., 2012). To overcome these issues, one can entrap the drug in a nanocarrier or reduce its particles to obtain a nanosized range, such as nanocrystals. Compared to NPs, drug-based particles offer higher drug loading (nearly 100%) with generally less excipients and uniform/stable physical nature (Liu et al., 2012).
Accordingly, indinavir nanonization increased absorption (from 0.83 to 18.16 µg·h/mL) and tmax (from 1.10 to 2.50 h) of the free base drug after administration of a single oral dose in mongrel dogs; however, NP-in-microparticle alginate/chitosan particles performed much better than pure nanocrystals (AUC of 39.23 µg·h/mL and t½ value of 76.3 h). This represented an increase of the oral bioavailability and the apparent t½ of 47 and 95 times, compared to the free drug. The increment in release time may improve HIV treatment, which demands long-term therapy and frequent dosing (Imperiale et al., 2015). Likewise, darunavir and ritonavir benefited from an NP-in-microparticle oral delivery system for nanocrystals. The difference was that NP-in-microparticle increased the oral bioavailability of the combined drugs by 2.3-fold compared with the NP only and the free drug. In this case, nanonization alone was not able to increase absorption (Augustine et al., 2018).
Nanocrystals vehicled in tablets and buccal films allow easier self-administration, increased dosage precision, and superior performance. Rebamipide presents only 10% of oral bioavailability in humans due to poor solubility, which is the reason Guo and coworkers formulated an NP-in-tablet version of the medicine. Drug nanocrystals were stabilized with HPMC and polyvinylpyrrolidone before tablet incorporation. The relative oral bioavailability of REB nanocrystal tablets was 256.8% in rats (reference Mucosta® tablets) (Guo et al., 2015). Nekkanti’s group also used HPMC-stabilized nanocrystals for candesartan cilexetil delivery, further incorporated in mannitol-based tablets. This prodrug belongs to the low solubility/long-term therapy group, such as rebamipide. In vivo studies confirmed dosage form benefits: Wistar rats presented 2.51-fold increase in AUC, a 1.77-fold increase in Cmax, and a decreased tmax (1.81–1.06 h) compared to the free prodrug (Nekkanti et al., 2009).
Rana and Murthy developed a three-layer buccal film: a mucoadhesive layer, a layer containing nanosuspension of carvedilol nanocrystals and a backing membrane. The structure aimed to prevent the first-pass metabolism, raising drug bioavailability. Cmax of the buccal patch was 7.3 times higher than that of the oral tablet, and tmax exhibited by the patch was 4 h in comparison to 2 h for oral tablet. The NP-in-microparticle structured buccal patch has been designed as a novel platform for potential buccal delivery of drugs having high first-pass metabolism (Rana and Murthy, 2013).
Another application of nanosized drugs is the development of better suitable dosage forms of existing drugs to attend the patient needs. Kevadiya et al. developed an oral strip-film containing nanocrystals of the cholesterol-reducing agent fenofibrate, a very low solubility prodrug, aiming to create a fast disintegrating solid dosage form ideal for emergency administration and for patients with swallowing difficulties. New Zealand white rabbits were divided in three groups: the first received the oral commercial formulation Tricor in suspension form; the second received the suspension of fenofibrate nanocrystals (pristine FNB) and the third received the oral striped-films containing fenofibrate nanocrystals (OSF). Pharmacokinetics data showed a higher Cmax and lower tmax of OSF when compared to the marketed Tricor formulation and pristine FNB. The AUC was also higher for OSF formulation (931.26 µg·h/mL) compared to Tricor (654.6 ± 251 µg·h/mL) and pristine FNB (514.8 ± 374 µg·h/mL); and the tmax (h) of OSFs was found to be 2 h earlier than the tmax of Tricor and pristine FNB (Kevadiya et al., 2018).
Because most of the articles we gathered date from this decade, NP-in-matrix approach still has a long way for exploitation. In our findings, most matrices belong to the polymeric group, based on classical excipients in the market, like HPMC. However, they vary from buccal to oral delivery, with rapid or slow degradation to offer an immediate or modified release. The use of an external matrix to incorporate nanoparticles brought several advantages to formulations. This type of system reduced burst effect, avoided NP degradation in gastrointestinal tract, increased residence time in the mouth, modulated tmax, and bypassed first-pass metabolism (buccal forms and some oral forms). Thus, the systemic bioavailability of the tested drugs was successfully enhanced.
Buccal release belongs to the trending strategies for drugs, so we expect an increase in pharmacokinetic studies concerning NP-in-matrix buccal delivery. Nanoparticle types will probably expand too, as the actual group is mainly PLGA, nanocrystals, and some lipid particles. In summary, we believe this article compiled several evidences and possible pitfalls of this strategy, which will help future developments on the field.
RC: synthetic polymeric NP-in-matrix research and writing. DG— inorganic NP-in-matrix research and writing. VB-d-A: lipidic NP-in-matrix research and writing. JC: tables, figures and conclusion. LO-N: review design, introduction and pharmacokinetics writing, scientific revision.
FAPESP 14/14457-5 - article charges funding CAPES 001 - scholarships for graduate students (RF, DG, VB-d-A, JC). This study was financed by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) - Finance Code 001 and FAPESP project 2019/08281-5 and 2019/19696-1.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ahmed, T. A., El-Say, K. M., Hosny, K. M., Aljaeid, B. M. (2018). Development of optimized self-nanoemulsifying lyophilized tablets (SNELTs) to improve finasteride clinical pharmacokinetic behavior. Drug Dev. Ind. Pharm. 44 (4), 652–615. doi: 10.1080/03639045.2017.1405977
Al-Dhubiab, B. E., Nair, A. B., Kumria, R., Attimarad, M., Harsha, S. (2015). Formulation and evaluation of nano based drug delivery system for the buccal delivery of acyclovir. Colloids and Surfaces. B. Biointerfaces 136, 878–884. doi: 10.1016/j.colsurfb.2015.10.045
Al-Dhubiab, B. E. (2016a). Development and evaluation of buccal films impregnated with selegiline-loaded nanospheres. Drug Delivery 23 (7), 2154–2162. doi: 10.3109/10717544.2014.948644
Al-Dhubiab, B. E. (2016b). In vitro and in vivo evaluation of nano-based films for buccal delivery of zolpidem. Braz. Oral Res. 30 (1), e126. doi: 10.1590/1807-3107bor-2016.vol30.0126
Attili-Qadri, S., Karra, N., Nemirovski, A., Schwob, O., Talmon, Y., Nassar, T., et al. (2013). Oral delivery system prolongs blood circulation of docetaxel nanocapsules via lymphatic absorption. Proc.Nat. Acad. Sci. 110 (43), 17498–17503. doi: 10.1073/pnas.1313839110
Augustine, R., Ashkenazi, D. L., Arzi, R. S., Zlobin, V., Shofti, R., Sosnik, A. (2018). Nanoparticle-in-microparticle oral drug delivery system of a clinically relevant darunavir/ritonavir antiretroviral combination. Acta Biomater. 74, 344–359. doi: 10.1016/j.actbio.2018.04.045
Barua, S., Kim, H., Jo, K., Seo, C. W., Park, T. J., Lee, K. B., et al. (2016). Drug delivery techniques for buccal route: formulation strategies and recent advances in dosage form design. J. Pharm. Invest. 46 (7), 593–6135. doi: 10.1007/s40005-016-0281-9
Berben, P., Bauer-Brandl, A., Brandl, M., Faller, B., Flaten, G. E., Jacobsen, A. C., et al. (2018). Drug permeability profiling using cell-free permeation tools: overview and applications. Eur. J. Pharmaceut. Sci.: Off. J. Eur. Fed. Pharmaceut. Sci. 119, 219–233. doi: 10.1016/j.ejps.2018.04.016
Bernkop-Schnürch, A., Weithaler, A., Albrecht, K., Greimel, A. (2006). Thiomers: preparation and in vitro evaluation of a mucoadhesive nanoparticulate drug delivery system. Int. J. Pharm. 317 (1), 76–81. doi: 10.1016/j.ijpharm.2006.02.044
Boateng, J. S., Areago, D. (2014). Composite sodium alginate and chitosan based wafers for buccal delivery of macromolecules. Austin J. Anal. Pharmaceut. Chem. 1, 1–7.
Bronze-Uhle, E. S., Costa, B. C., Ximenes, V. F., Lisboa-Filho, P. N. (2017). Synthetic nanoparticles of bovine serum albumin with entrapped salicylic acid. Nanotechnol. Sci. Appl. 10, 11–21. doi: 10.2147/NSA.S117018
Chime, S. A., Onyishi, I. V. (2013). Lipid-based drug delivery systems (LDDS): recent advances and applications of lipids in drug delivery. Afr. J. Pharm. Pharmacol. 7 (48), 3034–3595. doi: 10.5897/AJPPX2013.0004
Crucho, C. I. C., Barros, M. T. (2017). Polymeric nanoparticles: a study on the preparation variables and characterization methods. Mater. Sci. Eng. C. Mater. Biol. Appl. 80, 771–784. doi: 10.1016/j.msec.2017.06.004
El-Say, K. M., Ahmed, T. A., Ahmed, O. A. A., Hosny, K. M., Abd-Allah, F. I. (2017). Self-nanoemulsifying lyophilized tablets for flash oral transmucosal delivery of vitamin K: development and clinical evaluation. J. Pharm. Sci. 106 (9), 2447–2565. doi: 10.1016/j.xphs.2017.01.001
Furukawa, T., Naritomi, Y., Tetsuka, K., Nakamori, F., Moriguchi, H., Yamano, K., et al. (2014). Species differences in intestinal glucuronidation activities between humans, rats, dogs and monkeys. Xenobiotica 44 (3), 205–165. doi: 10.3109/00498254.2013.828362
Garhy, D. M. A., Ismail, S., Ibrahim, H. K., Ghorab, M. M. (2018). Buccoadhesive gel of carvedilol nanoparticles for enhanced dissolution and bioavailability. J. Drug Delivery Sci. Technol. 47, 151–158. doi: 10.1016/j.jddst.2018.07.009
Gong, W., Wang, Y., Sun, L., Yang, J., Shan, L., Yang, M., et al. (2016). Development of itraconazole liquisolid compact: effect of polyvinylpyrrolidone on the dissolution properties. Curr. Drug Delivery 13 (3), 452–615. doi: 10.2174/1567201813666160216144323
Griffin, B. T., Guo, J., Presas, E., Donovan, M. D., Alonso, M. J., O’Driscoll, C. M. (2016). Pharmacokinetic, pharmacodynamic and biodistribution following oral administration of nanocarriers containing peptide and protein drugs. Adv. Drug Delivery Rev. Oral Delivery Pept. 106, 367–380. doi: 10.1016/j.addr.2016.06.006
Guo, Y., Wang, Y., Xu, L. (2015). Enhanced bioavailability of rebamipide nanocrystal tablets: formulation and in vitro/in vivo evaluation. Asian J. Pharmaceut. Sci. 10 (3), 223–229. doi: 10.1016/j.ajps.2014.09.006
Hallan, S. S., Kaur, P., Kaur, V., Mishra, N., Vaidya, B. (2016). Lipid polymer hybrid as emerging tool in nanocarriers for oral drug delivery. Artif. Cells Nanomed. Biotechnol. 44 (1), 334–495. doi: 10.3109/21691401.2014.951721
Hazzah, H. A., Farid, R. M., Nasra, M. M. A., El-Massik, M. A., Abdallah, O. Y. (2015). Lyophilized sponges loaded with curcumin solid lipid nanoparticles for buccal delivery: development and characterization. Int. J. Pharm. 492 (1–2), 248–257. doi: 10.1016/j.ijpharm.2015.06.022
Imperiale, J. C., Nejamkin, P., del Sole, M. J., Lanusse, C. E., Sosnik, A. (2015). Novel protease inhibitor-loaded nanoparticle-in-microparticle delivery system leads to a dramatic improvement of the oral pharmacokinetics in dogs. Biomaterials 37, 383–394. doi: 10.1016/j.biomaterials.2014.10.026
Imperiale, J. C., Sosnik, A. (2013). Nanoparticle-in-microparticle delivery systems (NiMDS): production, administration routes and clinical potential. Text. 3 (1), 22–38. doi: 10.1166/jbt.2013.1064
Jiang, Y., Stenzel, M. (2016). Drug delivery vehicles based on albumin-polymer conjugates. Macromol. Biosci. 16 (6), 791–8025. doi: 10.1002/mabi.201500453
Kalepu, S., Nekkanti, V. (2015). Insoluble drug delivery strategies: review of recent advances and business prospects. Acta Pharm. Sin. B. 5 (5), 442–535. doi: 10.1016/j.apsb.2015.07.003
Kamaly, N., Yameen, B., Wu, J., Farokhzad, O. C. (2016). Degradable controlled-release polymers and polymeric nanoparticles: mechanisms of controlling drug release. Chem. Rev. 116 (4), 2602–2635. doi: 10.1021/acs.chemrev.5b00346
Karamanidou, T., Bourganis, V., Kammona, O., Kiparissides, C. (2016). Lipid-based nanocarriers for the oral administration of biopharmaceutics. Nanomed. (London, Engl.) 11 (22), 3009–3255. doi: 10.2217/nnm-2016-0265
Kerdsakundee, N., Li, W., Martins, J. P., Liu, Z., Zhang, F., Kemell, M., et al. (2017). Multifunctional nanotube-mucoadhesive poly(methyl vinyl ether-co-maleic acid) @hydroxypropyl methylcellulose acetate succinate composite for site-specific oral drug delivery. Adv. Healthcare Mater. 6 (20), 1700629. doi: 10.1002/adhm.201700629.
Kevadiya, B. D., Barvaliya, M., Zhang, L., Anovadiya, A., Brahmbhatt, H., Paul, P., et al. (2018). Fenofibrate nanocrystals embedded in oral strip-films for bioavailability enhancement. Bioeng. (Basel, Switzerland) 5 (1), 16. doi: 10.3390/bioengineering5010016
Kim, I. S., Jeong, Y. I., Kim, D. H., Lee, Y. H., Kim, S. H. (2001). Albumin release from biodegradable hydrogels composed of dextran and poly(ethylene glycol) macromer. Arch. Pharmacal Res. 24 (1), 69–735. doi: 10.1007/BF02976496
Larregieu, C. A., Benet, L. Z. (2013). Drug discovery and regulatory considerations for improving in silico and in vitro predictions that use Caco-2 as a surrogate for human intestinal permeability measurements. AAPS J. 15 (2), 483–497. doi: 10.1208/s12248-013-9456-8
Lipinski, C. A., Lombardo, F., Dominy, B. W., e, and Feeney, P. J. (2001). “Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings1PII of original article: S0169-409X(96)00423-1. The article was originally published in Advanced Drug Delivery Reviews 23 (1997) 3–25.1”. Adv. Drug Delivery Rev., Special issue dedicated to Dr. Eric Tomlinson, Advanced drug delivery reviews, a selection of the most highly cited articles, 1991–1998, 46 (1): 3–26. doi: 10.1016/S0169-409X(00)00129-0
Liu, Y., Xie, P., Zhang, D., Zhang, Q. (2012). A mini review of nanosuspensions development. J. Drug Targeting 20 (3), 209–235. doi: 10.3109/1061186X.2011.645161
Meng, X. Y., Zhang, H. X., Mezei, M., Cui, M. (2011). Molecular docking: a powerful approach for structure-based drug discovery. Curr. comput. Aided Drug Des. 7(2), 146–157.
Moore, T. L., Rodriguez-Lorenzo, L., Hirsch, V., Balog, S., Urban, D., Jud, C., et al. (2015). Nanoparticle colloidal stability in cell culture media and impact on cellular interactions. Chem. Soc. Rev. 44 (17), 6287–6305. doi: 10.1039/C4CS00487F
Lv, Q., Li, X., Shen, B., Dai, L., Xu, H., Shen, C., et al. (2014). A solid phospholipid-bile salts-mixed micelles based on the fast dissolving oral films to improve the oral bioavailability of poorly water-soluble drugs. J. Nanopart. Res. 16 (6), 24555. doi: 10.1007/s11051-014-2455-6
Lv, Q., Shen, C., Li, X., Shen, B., Yu, C., Xu, P., et al. (2015). Mucoadhesive buccal films containing phospholipid-bile salts-mixed micelles as an effective carrier for cucurbitacin B delivery. Drug Delivery 22 (3), 351–585. doi: 10.3109/10717544.2013.876459
Madkhali, O., Mekhail, G., Wettig, S. D. (2019). Modified gelatin nanoparticles for gene delivery. Int. J. Pharm. 554, 224–234. doi: 10.1016/j.ijpharm.2018.11.001
Nassar, T., Attili-Qadri, S., Harush-Frenkel, O., Farber, S., Lecht, S., Lazarovici, P., et al. (2011). High plasma levels and effective lymphatic uptake of docetaxel in an orally available nanotransporter formulation. Cancer Res. 71 (8), 3018–3285. doi: 10.1158/0008-5472.CAN-10-3118
Nekkanti, V., Pillai, R., Venkateshwarlu, V., Harisudhan, T. (2009). Development and characterization of solid oral dosage form incorporating candesartan nanoparticles. Pharm. Dev. Technol. 14 (3), 290–985. doi: 10.1080/10837450802585278
Nidhi, M., Patro, M. N., Kusumvalli, S., Kusumdevi, V. (2016). Development of transmucosal patch loaded with anesthetic and analgesic for dental procedures and in vivo evaluation. Int. J. Nanomed. 11, 2901–2920. doi: 10.2147/IJN.S94658
Puri, A., Loomis, K., Smith, B., Lee, J.-H., Yavlovich, A., Heldman, E., et al. (2009). Lipid-based nanoparticles as pharmaceutical drug carriers: from concepts to clinic. Critical Rev. Ther. Drug Carrier Syst. 26 (6), 523–805. doi: 10.1615/CritRevTherDrugCarrierSyst.v26.i6.10
Rana, P., Murthy, R. S. R. (2013). Formulation and evaluation of mucoadhesive buccal films impregnated with carvedilol nanosuspension: a potential approach for delivery of drugs having high first-pass metabolism”. Drug Delivery 20 (5), 224–355. doi: 10.3109/10717544.2013.779331
Rao, S., Richter, K., Nguyen, T.-H., Boyd, B. J., Porter, C. J. H., Tan, A., et al. (2015). Pluronic-functionalized silica–lipid hybrid microparticles: improving the oral delivery of poorly water-soluble weak bases. Mol. Pharm. 12 (12), 4424–4335. doi: 10.1021/acs.molpharmaceut.5b00622
Rao, S., Song, Y., Peddie, F., Evans, A. M. (2011). Particle size reduction to the nanometer range: a promising approach to improve buccal absorption of poorly water-soluble drugs. Int. J. Nanomed. 6, 1245–1251. doi: 10.2147/IJN.S19151
Salem, H. F., Kharshoum, R. M., Halawa, A. K. A., Naguib, D. M. (2018). Preparation and optimization of tablets containing a self-nano-emulsifying drug delivery system loaded with rosuvastatin. J. Liposome Res. 28 (2), 149–605. doi: 10.1080/08982104.2017.1295990
Shastri, D. H. (2017). Effective delivery routes and strategies for solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC). Curr. Pharm. Des. 23 (43), 6592–6601. doi: 10.2174/1381612823666171122111132
Slowing, I. I., Vivero-Escoto, J. L., Wu, C.-W., Lin, V. S. Y. (2008). Mesoporous silica nanoparticles as controlled release drug delivery and gene transfection carriers. Adv. Drug Delivery Rev. Inorg. Nanopart. Drug Delivery 60 (11), 1278–1288. doi: 10.1016/j.addr.2008.03.012
Soudry-Kochavi, L., Naraykin, N., Nassar, T., Benita, S. (2015). Improved oral absorption of exenatide using an original nanoencapsulation and microencapsulation approach. J. Controlled Release: Off. J. Controlled Release Soc. 217, 202–210. doi: 10.1016/j.jconrel.2015.09.012
Teixeira, M. C., Carbone, C., Souto, E. B. (2017). Beyond liposomes: recent advances on lipid based nanostructures for poorly soluble/poorly permeable drug delivery. Prog. Lipid Res. 68, 1–11. doi: 10.1016/j.plipres.2017.07.001
Tong, Y., Wang, Y., Yang, M., Yang, J., Chen, L., Chu, X., et al. (2018). Systematic development of self-nanoemulsifying liquisolid tablets to improve the dissolution and oral bioavailability of an oily drug, vitamin K1. Pharmaceutics 10 (3), 96. doi: 10.3390/pharmaceutics10030096
Washington, K. E., Kularatne, R. N., Karmegam, V., Biewer, M. C., Stefan, M. C. (2017). Recent advances in aliphatic polyesters for drug delivery applications. Rev. Nanomed. Nanobiotechnol. 9 (4), e1446. doi: 10.1002/wnan.1446.
Westerhout, J., Bellmann, S., van Ee, R., Havenaar, R., Leeman, W., et al. (2017). Prediction of Oral Absorption of Nanoparticles from Biorelevant Matrices Using a Combination of Physiologically Relevant In Vitro and Ex Vivo Models. J. Food Chem. Nanotechnol. 3 (4), 111–119. doi: 10.17756/jfcn.2017-046.
Wissing, S. A., Kayser, O., Müller, R. H. (2004). Solid lipid nanoparticles for parenteral drug delivery. Adv. Drug Delivery Rev. Adv. Lipid-Based Drug Solubilization Targeting 56 (9), 1257–1272. doi: 10.1016/j.addr.2003.12.002
Keywords: nanoparticle, oral delivery, buccal delivery, matrix delivery, drug absorption
Citation: Feitosa RC, Geraldes DC, Beraldo-de-Araújo VL, Costa JSR and Oliveira-Nascimento L (2019) Pharmacokinetic Aspects of Nanoparticle-in-Matrix Drug Delivery Systems for Oral/Buccal Delivery. Front. Pharmacol. 10:1057. doi: 10.3389/fphar.2019.01057
Received: 26 February 2019; Accepted: 20 August 2019;
Published: 24 September 2019.
Edited by:
Silvio Barberato-Filho, Universidade de Sorocaba, BrazilReviewed by:
Han Qiao, Shanghai Jiao-Tong University, School of Medicine, ChinaCopyright © 2019 Feitosa, Geraldes, Beraldo-de-Araújo, Costa and Oliveira-Nascimento. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura Oliveira-Nascimento, bGF1cmFvbkB1bmljYW1wLmJy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.