 Saveg Yadav
Saveg Yadav Shrish Kumar Pandey
Shrish Kumar Pandey Yugal Goel
Yugal Goel Mithlesh Kumar Temre
Mithlesh Kumar Temre Sukh Mahendra Singh
Sukh Mahendra Singh- School of Biotechnology, Institute of Science, Banaras Hindu University, Varanasi, India
Malignant cells possess a unique metabolic machinery to endure unobstructed cell survival. It comprises several levels of metabolic networking consisting of 1) upregulated expression of membrane-associated transporter proteins, facilitating unhindered uptake of substrates; 2) upregulated metabolic pathways for efficient substrate utilization; 3) pH and redox homeostasis, conducive for driving metabolism; 4) tumor metabolism-dependent reconstitution of tumor growth promoting the external environment; 5) upregulated expression of receptors and signaling mediators; and 6) distinctive genetic and regulatory makeup to generate and sustain rearranged metabolism. This feat is achieved by a “battery of molecular patrons,” which acts in a highly cohesive and mutually coordinated manner to bestow immortality to neoplastic cells. Consequently, it is necessary to develop a multitargeted therapeutic approach to achieve a formidable inhibition of the diverse arrays of tumor metabolism. Among the emerging agents capable of such multifaceted targeting of tumor metabolism, an alkylating agent designated as 3-bromopyruvate (3-BP) has gained immense research focus because of its broad spectrum and specific antineoplastic action. Inhibitory effects of 3-BP are imparted on a variety of metabolic target molecules, including transporters, metabolic enzymes, and several other crucial stakeholders of tumor metabolism. Moreover, 3-BP ushers a reconstitution of the tumor microenvironment, a reversal of tumor acidosis, and recuperative action on vital organs and systems of the tumor-bearing host. Studies have been conducted to identify targets of 3-BP and its derivatives and characterization of target binding for further optimization. This review presents a brief and comprehensive discussion about the current state of knowledge concerning various aspects of tumor metabolism and explores the prospects of 3-BP as a safe and effective antineoplastic agent.
Introduction
Oncogenic transformation is associated with a massive metabolic reprogramming in neoplastic cells, which bestows unmatched self-sufficiency of biosynthetic, bioenergetic, and redox homeostasis (Tarrado-Castellarnau et al., 2016; Costa and Frezza, 2017). The reorganized metabolism leads to the generation of unique intrinsic and extrinsic environments conducive for an unhindered neoplastic transformation, accelerated tumor progression, the evolution of chemoresistance, invasion, and metastasis (Sánchez-García, 2009; Tao et al., 2014). Thus, tumor metabolism is recognized as an emerging hallmark of cancer (Hanahan and Weinberg, 2011; Pavlova and Thompson, 2016). The importance of unique metabolic characteristics of cancer cells was aptly recognized by Nobel laureate Sir Otto Heinrich Warburg, who described that neoplastic cells predominantly acquire energy through the fermentation of glucose to lactate irrespective of the presence or absence of oxygen and functional mitochondrial machinery (Warburg, 1956). This phenomenon of “aerobic glycolysis” is also denoted as the “Warburg effect” (Hanahan and Weinberg, 2011). Despite the ongoing debate regarding the universal applicability of the Warburg hypothesis on neoplastic cells (Hsu and Sabatini, 2008; Xu et al., 2015), the last decade has witnessed a vast resurgence of research to recognize tumor metabolism as the central driving force underlying the manifestation of the oncogenic phenotype of neoplastic cells (Kroemer and Pouyssegur, 2008; Liberti and Locasale, 2016). Interestingly, tumor cells display a tremendous heterogeneity concerning the execution of the metabolic hallmarks, depending on a variety of internal and external regulatory factors (Marusyk and Polyak, 2010; Polyak, 2011; Dagogo-Jack and Shaw, 2018). Moreover, metabolic reprogramming of neoplastic cells is accompanied by an equally matching utilization and dissemination machinery, mainly composed of the rewired metabolic pathways (Cantor and Sabatini, 2012; Quail and Joyce, 2013; Wang et al., 2017).
As depicted in Figure 1, the crucial aspects of tumor metabolism can be broadly categorized into the following major groups of functionalities: 1) quantitative and qualitative alterations of transporters’ repertoire for optimization of nutrient uptake; 2) acceleration of metabolic pathways caused by upregulated expression of the catalyzing enzymes and upstream signaling events; 3) altered pH and redox homeostasis, which facilitate the progression of metabolic pathways; and 4) metabolism-dependent alterations in the soluble, biophysical, and cellular components of the tumor microenvironment (TME), imparting diverse consequences on tumor progression. In the following sections of the review, we will discuss the above-mentioned crucial aspects of tumor metabolism and associated modulation of the TME. Furthermore, the review focuses on the promising ability of alkylating agent 3-bromopyruvate (3-BP) to circumvent these hallmarks of cancer metabolism, accompanying a discussion on issues related to its safety in antineoplastic therapeutic applications.
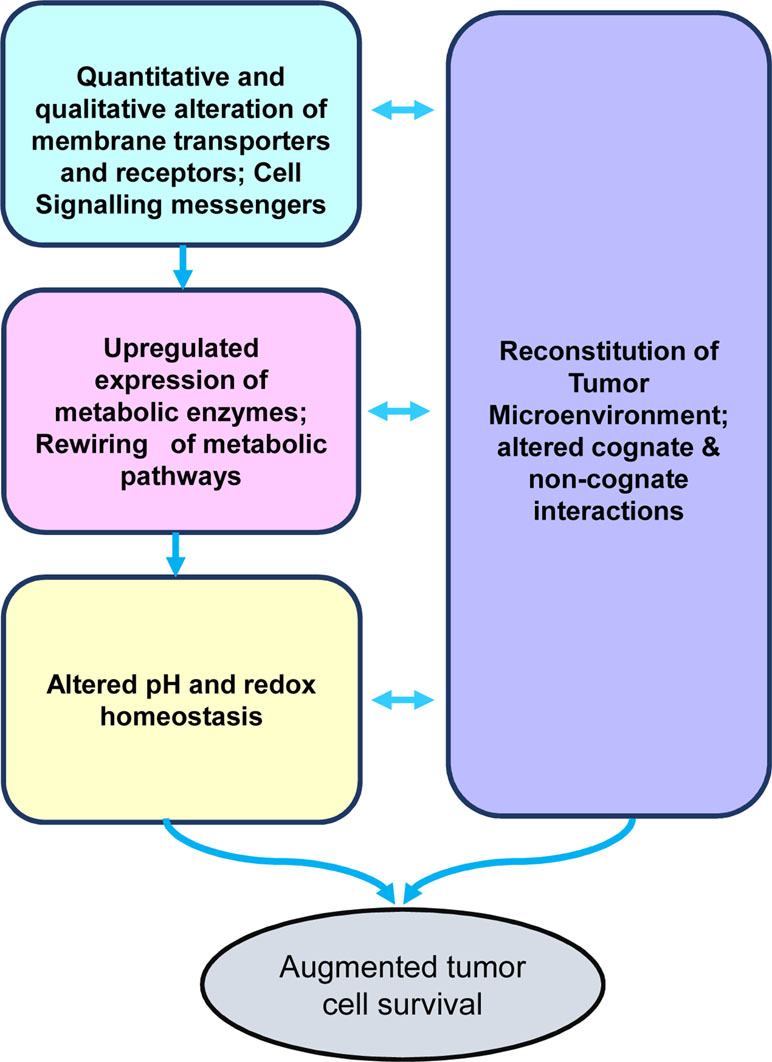
Figure 1 Stakeholders of tumor metabolism. The diagram depicts the primary stakeholders of the tumor metabolism, namely, nutrient uptake mechanisms, rewired metabolic pathways, altered pH homeostasis, and reconstituted tumor microenvironment, which collectively lead to the creation of a protumor survival scenario conducive for rapid tumor progression.
Although elegant reviews are addressing various aspects of the antineoplastic potential of 3-BP (Shoshan, 2012; Lis et al., 2016; Baghdadi, 2017), the novelty of the present review is a collation, on a single platform, of the updated and comprehensive information of multiple stakeholders of tumor metabolism. The particular focus of this review is on the repertoire of transporters involved in cancer metabolism vis-à-vis the ability of 3-BP to target most of the metabolic stakeholders. Furthermore, this review analyzes the available literature on the molecular characterization of the docking ability of 3-BP with some critical metabolic targets. Additionally, the review discusses the current literature addressing the recently reported effects of 3-BP on components of the TME and hematological homeostasis. This review also incorporates evidence addressing issues related to the safety of 3-BP on vital organs/systems, which was feebly discussed earlier.
Optimization of Nutrient Uptake
Glucose Metabolism
Neoplastic cells display an elaborate and upregulated expression of a plethora of transporters on their cell surface mainly meant for the uptake of nutrients required to fuel the accelerated metabolic pathways (Figure 2). Sugar transporters are particularly crucial, considering their role in fueling glycolysis. Neoplastic cells display an upregulated expression of glucose transporters (GLUT) like GLUT-1 and GLUT-3, facilitating the uptake of a huge quantity of glucose (Douard and Ferraris, 2008; Hamanaka and Chandel, 2012). Nevertheless, fructose transporter, GLUT-5, is also reported to be highly upregulated in several types of neoplastic cells (Douard and Ferraris, 2008). Moreover, studies have demonstrated an upregulated expression of additional sugar transports belonging to the sodium-dependent glucose cotransporter (SGLT) family, such as SGLT-1 and SLC5A/SGLT (Scafoglio et al., 2015). The role of other glucose transporters, however, remains debatable concerning their ability to fuel the metabolic pathways in neoplastic cells. Sporadic studies have implicated transporters such as GLUT-8 and GLUT-12 in specific types of neoplastic cells (Barron et al., 2012; Mueckler and Thorens, 2013). However, GLUT-1 and GLUT-3 remain as the unequivocally recognized universal glucose transporters responsible for the uptake of large quantities of glucose required by the highly glycolytic malignant cells (Mueckler and Thorens, 2013; Yu et al., 2017b). Thus, therapies aimed to target the implicated GLUTs are envisaged as novel antineoplastic strategies to interfere with neoplastic cells’ bioenergetic and biosynthetic homeostasis (Hamanaka and Chandel, 2012; Labak et al., 2016). Hence, many inhibitors of sugar transporters are being explored for therapeutic efficacy. As depicted in Table 1, drugs like cytochalasin B, resveratrol, naringenin, phloretin, WZB117, and thiazolidinedione have been demonstrated to display an inhibitory action on glucose transporters through direct (competitive) or indirect (noncompetitive) mechanisms (Kapoor et al., 2016; Siebeneicher et al., 2016). Furthermore, SGLT inhibitors such as dapagliflozin, canagliflozin, and empagliflozin have been explored for antineoplastic effectiveness (Lin and Tseng, 2014; Scafoglio et al., 2015). Approaches using short hairpin RNA (shRNA) and small interfering RNA (siRNA) to target the expression of various GLUTs are demonstrated to hold promising antineoplastic potential (Shimanishi et al., 2013; Jian et al., 2015). However, tumor cell specificity of these inhibitors is debatable because many healthy cells also express high levels of GLUT-1 and GLUT-3 under certain physiological conditions (Krzeslak et al., 2012). Additional indirect strategies to interfere with the “sugar tooth” of cancer cells include the targeting of signaling pathways, which regulate GLUT expression, implicating regulators such as phosphoinositide 3-kinase (PI3K), mechanistic target of rapamycin (mTOR), c-Myc, and hypoxia inducible factor (HIF)-1α (Choi, 2017). Inhibitors of such signaling mediator systems have been identified (Table 1) and are under active evaluation for therapeutic potential (Logue and Morrison, 2012). Similarly, vascular endothelial growth factor (VEGF)-dependent regulation of GLUT expression carries potential as a targetable entity of carbohydrate metabolism (Choi, 2017). Additionally, strategies involving lowering the availability of dietary sugars to minimize their uptake by the neoplastic cells have also been explored (Hamanaka and Chandel, 2012; Vishvakarma et al., 2013). Optimization of such approaches will be of potential benefit in circumventing the glucose dependence of cancer metabolism.
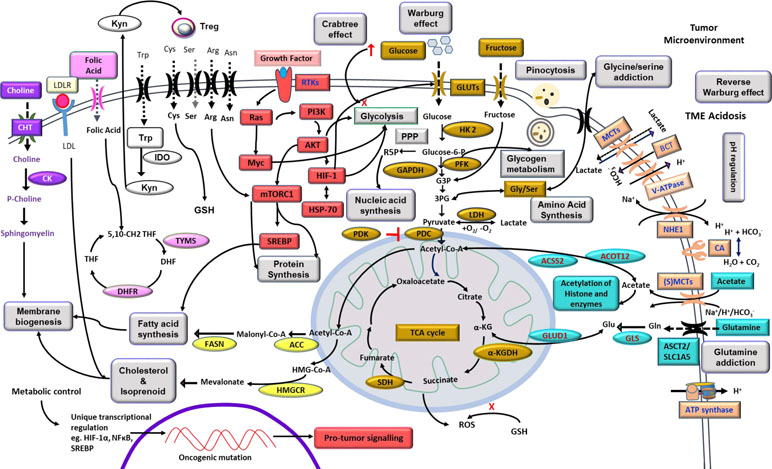
Figure 2 Highlights of tumor metabolism. The diagram depicts metabolic reprogramming in tumor cells with overexpression of transporters meant for nutrient uptake, pH regulation, and receptors for cytokines, hormones, and other ligands. Internal components include prominent metabolic pathways of bioenergetics and biosynthetic machinery, including carbohydrate and fatty acid metabolism, integration of metabolic networking for efficient utilization of substrates like glucose, fructose, lactate, acetate, amino acids, and precursor of membrane components. Signal transduction events indicate a crucial role of PI3K, HIF-1α, AKT, Ras, Myc, and mTOR downstream to receptor–ligand ligation with promoting consequences on metabolic pathways. The exterior of the membrane depicts a manifestation of the Warburg effect and modulation of the TME. Altered mitochondrial functions and its correlation to lipid metabolism, ROS generation, and glutamine assimilation are also depicted. The overall effect of such cross-talk of metabolic pathways resulted in the promotion of neoplastic cell survival. The diagram also indicates the metabolism-dependent regulation of gene expression. Carbohydrate metabolism is indicated by the golden color; cell signaling is indicated by the red color; alternative fuels and their cytoplasmic fates are indicated by the teal color; pH regulators are depicted by the orange color; choline metabolism is shown in purple color; enzymes of fatty acid and cholesterol synthesis are indicated by the yellow color; pink color represents folic acid metabolism; kynurenine and tryptophan pathways are depicted by the black and white boxes; amino acid transporters are shown in black; and major phenomena are indicated in boxes of gray color. Abbreviations: αKG, α-ketoglutarate; 3PG, 3phosphoglycerate; ACC, acetyl-CoA carboxylase; ACSS2, acyl-coenzyme A synthetase short-chain family member 2; ACOT12, acyl-CoA thioesterase 12; SLC1A5, neutral amino acid transporter B(0)/solute carrier family 1 member 5; CK, choline kinase; CHT, choline transporter; FASN, fatty acid synthase; GAPDH, glyceraldehyde-3phosphate dehydrogenase; DHF, dihydrofolic acid; DHFR, dihydrofolate reductase; GLUD1, glutamate dehydrogenase 1; GLS, glutaminase; GLUT, glucose transporter; G3P, glucose-3-phosphate; HIF, hypoxia inducible factor; HK2, hexokinase 2; HMG-CoA, 3-hydroxy-3–methyl-glutaryl coenzyme A; HMGCR, HMG-CoA reductase; HSP-70, 70-kilodalton heat shock protein; IDO, indoleamine 2,3-dioxygenase; Kyn, kynurenine; LDH, lactate dehydrogenase; MCT, monocarboxylate transporter; mTOR, mechanistic target of rapamycin; PDC, pyruvate dehydrogenase complex; NHE1, Na+/H+ exchanger 1; PDK, pyruvate dehydrogenase kinase; PI3K, phosphoinositide 3-kinase; PFK, phosphofructokinase; PPP, pentose phosphate pathway; R5P, ribose-5phosphate; ROS, reactive oxygen species; Treg, regulatory T cells; RTK, receptor tyrosine kinase; THF, tetrahydrofolic acid; TYMS, thymidylate synthetase; (S)MCT, sodium-coupled monocarboxylate transporter; SDH, succinate dehydrogenase; SREBP, sterol regulatory element-binding protein.

Table 1 Inhibitors of tumor metabolism.
Table 1 highlights the stage of preclinical and clinical trials of the indicated metabolic inhibitors. Some of these inhibitors yielded limited therapeutic success. The inhibitor of monocarboxylate transporter (MCT) lonidamine did not pass phase III clinical trial (Berruti et al., 2002). Likewise, failure is reported for oxidative stress inducer STA-4783 (Sborov et al., 2015). Similarly, mTOR inhibitors fetched limited success in clinical trials (Faes et al., 2017). Moreover, VEGF inhibitor bevacizumab and other such agents fetched limited success in clinical trials (Zirlik and Duyster, 2018). The possible reasons underlying the observed failure of clinical trials could be as follows: 1) lack of adequate basic and preclinical research foundation before translational application in cancer patients; 2) obtaining approval for the clinical trials is a time-consuming process, requiring liaison of basic researchers, clinicians, pharmacologists, and financial sponsors; 3) limitations regarding the bioavailability of inhibitors within the tumor milieu in adequate cytotoxic concentration; 4) toxicity and other side effects of inhibitors in clinical applications; and 5) limitations of knowledge regarding the metabolomics of human cancers to determine their susceptibility in an inhibitor-specific manner.
Lipid Metabolism
Lipid metabolism is crucial for tumor cell survival, particularly concerning membrane biogenesis and cell signaling to sustain rapid cellular proliferation (Beloribi-Djefaflia et al., 2016). Moreover, levels of lipids like cholesterol, high-density lipoprotein, and low-density lipoprotein (LDL), and their metabolic by-products are significantly elevated in tumor-bearing hosts (Beloribi-Djefaflia et al., 2016). Lipid uptake of neoplastic cells is mediated via various modes. Passive diffusion of lipids is considered as one of the main routes through which fatty acids gain entry in neoplastic cells (Harjes et al., 2016). Accumulating experimental evidence has demonstrated that the neoplastic cells mostly produce their lipids by fatty acid synthase (FASN) catalyzed de novo fatty acid synthesis (Santos and Schulze, 2012). Orlistat, an inhibitor of FASN, has been demonstrated to circumvent tumor cell survival effectively (Kant et al., 2012, Kant et al., 2014b; Schcolnik-Cabrera et al., 2018). Nevertheless, many studies have also indicated lipolysis as an additional source of fatty acids (Zaidi et al., 2013; Zhao et al., 2017). Moreover, lipophagy is yet another alternative source of lipids, which is associated with oncogenic transformation and metastasis (Maan et al., 2018). Thus, neoplastic cells display altered “lipid metabolic network” to sustain their bioenergetic and biosynthetic processes (Maan et al., 2018). Neoplastic cells also display upregulated expression of a transmembrane fatty acid translocase (CD36), a scavenger receptor, which is responsible for fatty acid and protein uptake (Enciu et al., 2018). Hence, approaches to inhibit the transporter functions of CD36 (Table 1) can cause inhibition of both protein and lipid supply to cancer cells (Watt et al., 2019). Moreover, the expression of LDL receptor is highly upregulated in neoplastic cells of diverse etiologies, which are internalized after the ligation to the LDL (Furuya et al., 2016). The expression of LDL receptor is mainly regulated by a membrane-bound transcription factor designated as sterol regulatory element-binding protein (SREBP1) (Streicher et al., 1996). Additionally, SREBP has been shown to regulate several key processes of lipid metabolism, including the uptake of cholesterol, fatty acids, triglycerides, phospholipid, and Nicotinamide adenine dinucleotide phosphate (NADPH) (Guo et al., 2014). Furthermore, the upregulated lipid metabolism of cancer cells is dependent on various factors, including hypoxia, tumor acidosis, and upregulated SREBP1c via signaling of Ras, extracellular signal-regulated kinase1/2, Phosphatase and tensin homolog, PI3K, and Protein kinase B (PKB or AKT) (Santos and Schulze, 2012; Beloribi-Djefaflia et al., 2016). SREBP also activates adenosine triphosphate citrate lyase (ACLY), acetyl-CoA carboxylase (ACC), and FASN in neoplastic cells (Baenke et al., 2013; Guo et al., 2014). ACLY, in turn, catalyzes the conversion of citrate to acetyl-CoA, which is then converted to malonyl CoA by the action of ACC. The malonyl CoA serves as a substrate for FASN to produce fatty acids. The fatty acids thus generated serve as a major source of signaling proteins, membrane phospholipids, and production of acyl-CoA to be channelized into the tricarboxylic acid cycle (TCA) cycle (Baenke et al., 2013). The electrons released from the β-oxidation of lipids are utilized for the production of NADPH and flavin adenine dinucleotide (FADH2) for redox balancing and adenosine triphosphate (ATP) production (Santos and Schulze, 2012). Moreover, lipids are stored as lipid droplets in cancer cells, which render protective action in cellular adaptations during the stressful condition of nutrient depletion (Baenke et al., 2013). Reports also indicate a major contribution of lipid metabolism in the metastasis of neoplastic cells (Luo et al., 2017). Consequently, lipid metabolism appears to be a multifaceted “metabolic lifeline” of neoplastic cells, indicating a strong potential for one or more of these pathways as targets for therapeutic maneuvering. Furthermore, the therapeutic utility of targeting LDL receptor in pancreatic cancer cells has been demonstrated to hold a promising antineoplastic potential (Vasseur and Guillaumond, 2016). The approach of shRNA-mediated interference of LDL receptor expression was also shown to chemosensitize neoplastic cells (Gallagher et al., 2017) (Table 1).
Alternative Nutrients
Neoplastic cells gain an advantage in their battle for resources by the modulation of their sole metabolic dependence on sugars (Cantor and Sabatini, 2012; DeBerardinis and Chandel, 2016) by utilizing other nutrients, which serve as substrates for driving various metabolic pathways. Such nutrients are collectively designated as “alternative fuels” (Keenan and Chi, 2015; Cairns and Mak, 2016). The alternative fuels of neoplastic cells include a variety of biomolecules such as lactate, acetate, glutamine, cysteine, alanine, and several proteins, which can be channeled into the metabolic pathways (Keenan and Chi, 2015; Sousa et al., 2016). In addition to the utility of alternative fuels in carbohydrate catabolism, they are also used in lipid, protein, and histone metabolism (Keenan and Chi, 2015). Moreover, glutamine contributes to the redox homeostasis of tumor cells (Vučetić et al., 2017; Choi and Park, 2018). Many neoplastic cells overexpress transporters for glutamine and other amino acids (Hensley et al., 2013; Zhao et al., 2015a; Lukey et al., 2017; Choi and Park, 2018). Additionally, transporters including MCT-1 (SLC16A1) and MCT-4 (SLC16A3) mediate the uptake of lactate and branched-chain keto acids (Kennedy and Dewhirst, 2010; Silva et al., 2017), SLC7A11 for cysteine (Huang et al., 2005), and ASCT2 for glutamine (Cormerais et al., 2018), facilitating the uptake of alternative fuels by cancer cells. Moreover, neoplastic cells display an upregulated expression of MCTs for lactate and acetate uptake (Birsoy et al., 2013). Acetate is also imported via the Na+/H+/HCO3 − transporter (Pandey et al., 2018). Additionally, acetate is endogenously generated in neoplastic cells (Pandey et al., 2018). The uptake of acetate by cancer cells is dependent on facilitated diffusion via aquaporins and through transporters like MCT-1/2 and Na+/monocarboxylate transport 1 (SMCT1) (Ferro et al., 2016; Pandey et al., 2018). Accumulating pieces of evidence demonstrate that neoplastic cells use acetate for the synthesis of acetyl-CoA and thus feed the TCA cycle and fatty acid synthesis (Lyssiotis and Cantley, 2014). Furthermore, tumor cells display upregulated expression of acetyl-CoA synthetase II (Lyssiotis and Cantley, 2014; Research, 2015). Acetate is also utilized for acetylation of histone of several metabolic enzymes. Moreover, acetate uptake by the tumor cells is linked to the uptake of Na+ and HCO3 −, which can be utilized in the metabolic processes via the upregulated expression of SMCTs (Sterling and Casey, 2002). Lactate can be converted to pyruvate in neoplastic cells by the action of lactate dehydrogenase (LDH), showing upregulated expression in neoplastic cells (Miao et al., 2013). Additionally, lactate is implicated in the modulation of signaling events leading to activation of HIF-1α (Jiang, 2017). Nevertheless, lactate is also a source of carbon for cancer cells under normoxic conditions (Jiang, 2017). Furthermore, neoplastic cells display upregulated expression of the transporters of several non-glutamine and cysteine amino acids including serine, methionine, arginine, valine, leucine, asparagine, and glycine, collectively contributing to a higher uptake of amino acids by neoplastic cells (Schulze and Harris, 2012; Keenan and Chi, 2015; Hayase et al., 2017). Amino acid transporter LAT-1 (SLC7A5) and its chaperone CD98 also mediate uptake of neutral amino acids in cancer cells (Haase et al., 2007). Interestingly, whole proteins can be taken up via scavenger receptor CD36 and other processes including macropinocytosis, which facilitates the uptake of the lipids (Bonen et al., 2004; Ha et al., 2016). Signaling via Ras and Src facilitates the process of macropinocytosis by increasing vesicular transport (Commisso et al., 2013; Recouvreux and Commisso, 2017). Macropinocytosis is followed by a breakdown of engulfed molecules by the action of lysosomal enzymes (Recouvreux and Commisso, 2017) to be utilized in metabolic processes. Strategies are thus being designed to utilize the weakness of tumor cells for nutrient uptake for the import of anticancer drugs (Ha et al., 2016).
Approaches to inhibiting the transporters of alternative fuels have been experimented for designing antineoplastic strategies. MCT inhibitors α-CHC and AZD3965 have been demonstrated to inhibit tumor cell survival in a variety of neoplastic cells (Doherty and Cleveland, 2013; Kumar et al., 2013a, Kumar et al., 2013b; Polański et al., 2014; Curtis et al., 2017). Similarly, SMCT inhibitor ibuprofen and its derivatives show promising anticancer activity (Ganapathy et al., 2008). Moreover, inhibitors of other transporters like those of various amino acids and carbonic anhydrase display antineoplastic potential (Bhutia et al., 2015) (Table 1).
Rewiring of Metabolic Pathways in Cancer
Commensurate to the repertoire of nutrient import mechanisms, neoplastic cells display highly upregulated metabolic pathways, particularly those implicating carbohydrates, amino acids, other alternative fuels, and lipids as substrates (Kroemer and Pouyssegur, 2008). These geared-up metabolic pathways are effectively maneuvered by an upregulated repertoire of metabolic enzymes (Lincet and Icard, 2015) and their regulatory elements (Mossmann et al., 2018). As tumor cells mainly depend on glycolysis for their ATP production, they display an augmented expression of mitochondrial membrane-associated hexokinase (HK), which catalyzes the conversion of glucose to glucose-6-phosphate, the first crucial step of glycolysis (Wilson, 2003; Mathupala et al., 2006). Additionally, the neoplastic cells display massive upregulation in the expression of other glycolytic enzymes, including phosphofructokinase, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and isoforms of pyruvate kinase (Ganapathy-Kanniappan and Geschwind, 2013). Interestingly, using a natural GAPDH inhibitor, koninginic acid, the group of Locasale (Liberti et al., 2017; Liberti et al., 2019) demonstrated that aerobic glycolysis (Warburg effect) and glucose metabolism are functionally distinct phenomena, a promising rationale for therapeutic targeting of the Warburg effect (Liberti et al., 2019). Besides the upregulated expression of transporters and enzymes mentioned above, HIF-1α contributes in the suppression of the mitochondrial OxPhos metabolism of glucose by inhibiting pyruvate dehydrogenase (PDH) via the upregulated expression of pyruvate dehydrogenase I (Kim et al., 2006; Singh et al., 2017). As stated earlier, neoplastic cells display an upregulated expression of LDH (Miao et al., 2013). LDH is also under the regulatory control of HIF-1α-associated signal transduction events (Luo et al., 2017). Additionally, glucose metabolism is regulated by other upstream signaling mediators, including p53, Ras, c-Myc, AKT, and mTOR (Hay, 2016). It is important to note that the accelerated metabolic machinery is regulated by oncogene activation in the normoxic condition itself, rendering tumor cells to manifest the Warburg effect. Thus, the HIF-1α-dependent upregulation of glycolysis in neoplastic cells is ancillary and manifested only under hypoxic conditions (Yu et al., 2017a). Nevertheless, the upregulated metabolism of glucose through glycolysis caters to the anabolic machinery for the synthesis of other biomolecules (Ganapathy-Kanniappan and Geschwind, 2013). Moreover, neoplastic cells are reported to display elevated channelization of glucose to the pentose phosphate pathway, which has a significant contribution in the biosynthesis and bioenergetic machinery (Patra and Hay, 2014). Additionally, the pentose phosphate pathway contributes to the generation of protons and, hence, in the maintenance of tumor acidosis (Zhang et al., 2017).
Although many earlier studies have suggested a truncated role of TCA cycle in carbohydrate metabolism, recent experimental evidence strongly indicates that even Krebs’ cycle is functionally operative in neoplastic cells and has a crucial role in the bioenergetics of carbohydrate, lipid, and aspartate metabolism, accompanying maintenance of redox homeostasis of cancer cells (Martínez-Reyes et al., 2016; Anderson et al., 2018; Sullivan et al., 2018). Moreover, it is overwhelmingly suggested that the TCA cycle facilitates neoplastic cells to utilize alternative fuels, such as glutamine and acetate (Keenan and Chi, 2015). Additionally, the TCA cycle plays a crucial role in cancer cells for anaplerotic reactions to support their biosynthetic machinery (Griss et al., 2015; Anderson et al., 2018). The metabolic signaling apparatus composed of Myc, HIF-1α, p53, and Ras plays a crucial regulatory role to reinforce the TCA cycle of neoplastic cells by triggering alterations in the expression of constituent enzymes such as succinate dehydrogenase (SDH), fumarase, and isocitrate dehydrogenase (IDH) (Raimundo et al., 2011). Moreover, the de novo synthesis of lipids utilizes citrate generated through the TCA cycle by the catalytic action of ACLY, ACC, and FASN, all of which are regulated by membrane-bound transcription factor SREBP (Madison, 2016). Furthermore, the electron transport chain (ETC) functions, in particular, respiratory complex I, are crucial for redox homeostasis in addition to its role in oxygen sensing and lipid and aspartate metabolism (Galkin et al., 2009; Vatrinet et al., 2015; Gui et al., 2016; Martínez-Reyes et al., 2016; Kurelac et al., 2019).
One carbon metabolism is also considered crucial for supporting processes such as nucleotide synthesis, methylation, and nicotinamide adenine dinucleotide (NAD+/NADH) generation (Newman and Maddocks, 2017; Rosenzweig et al., 2018). Neoplastic cells utilize molecules like folic acid, serine, and glycine to fuel the one-carbon metabolism (Figure 2) (Rosenzweig et al., 2018). Hence, antifolate agents have been considered for antineoplastic activity (Newman and Maddocks, 2017). It is also proposed that targeting one-carbon metabolism may render a promising contribution to the designing of novel anticancer therapeutic strategies (Newman and Maddocks, 2017).
Unique pH Homeostasis of Neoplastic Cells: Generation of Tumor Acidosis
Tumor cells generate a massive amount of protons, which necessitates a tight regulation of the intracellular pH to prevent cytosolic acidification. Consequent pumping of these protons to the exterior causes the pH of the external milieu to reach the range of 6.5 to 6.9, designated as tumor acidosis (Damaghi et al., 2013). Interestingly, tumor acidosis also contributes to the manifestation of metabolic reprogramming of cancer cells (Peppicelli et al., 2017). Besides the significant contribution of accelerated metabolism in lowering pH, many other factors also contribute to the modulation of pH in cancer cells, which include, but is not limited to, hypoxia, hypercapnia, and a reduced diffusion of gases in the TME (Damaghi et al., 2013; Swietach et al., 2014; Damaghi et al., 2015). pH regulation results in a relative alkalinization of cytosol, accompanied by acidification of the external milieu (Chiche et al., 2010; Robey, 2012; Persi et al., 2018), having an up-regulatory action on tumor progression, metastasis, chemo-, and immuno-resistance (Riemann et al., 2016; Corbet and Feron, 2017; Huber et al., 2017; Persi et al., 2018). Accelerated glycolysis of neoplastic cells generates an enormous quantity of lactate, which is pumped to the exterior by MCTs (Swietach et al., 2014). Furthermore, recent reports highlight abnormal Golgi pH homeostasis in cancer cells, which is implicated in targeting carcinoembryonic antigen (Kokkonen et al., 2018). Moreover, glutamine metabolism is involved in the manifestation of tumor acidosis (Romero-Garcia et al., 2016). A hypoxic TME has also been demonstrated to be a critical trigger in regulating the expression of pH regulators, which play a crucial role in pH homeostasis and the manifestation of tumor acidosis (Miranda-Gonçalves et al., 2016). Nevertheless, the amount of glucose in the TME and transporters of nutritive molecules regulate the expression of various pH regulators (De Saedeleer et al., 2014; McGuire et al., 2016). Moreover, hypoxic conditions of the TME induce HIF-dependent cell signaling, which creates a glycolytic bias of glucose metabolism, leading to the high uptake of glucose, accelerated glycolysis, and production of lactate and H+ manifesting tumor acidosis (Petrova et al., 2018). HIF also promotes the expression of GLUTs and other nutrient transporters, fueling the upregulated glycolysis (Petrova et al., 2018). Furthermore, HIF supports pH homeostasis by promoting carbonic anhydrase (CA) IX expression (Iwasaki et al., 2015).
Besides MCTs, other membrane-associated pH regulators (Figure 2) include Na+/H+ exchanger (NHE), vacuolar ATPase (V-ATPase), CA, bicarbonate transporter (BCT), and ATP synthase (Damaghi et al., 2013; Swietach et al., 2014). Collectively, these pH regulators generate the characteristic “reverse pH gradient” across the plasma membrane of neoplastic cells, which is also recognized as a hallmark of the oncogenic transformation (Hanahan and Weinberg, 2011; Corbet and Feron, 2017). In addition to pH regulation, NHE, which belongs to the family of SLC cation/proton antiporters (CPAs), plays a crucial role in oncogenesis, tumor progression, and metastasis (Loo et al., 2012). NHE is responsible for exporting H+ with associated exchange of Na+ (Loo et al., 2012). Similar to other pH regulators, NHE expression is also dependent on signaling via PI3K, ras-related C3 botulinum toxin substrate 1 (Rac1), ERK1/2, and mitogen-activated protein kinase (MAPK) (Orlowski and Grinstein, 1997; Sartori et al., 1999; Vallés et al., 2015). In an interesting study using MCF-7 cells, it was demonstrated that malignant cells adapting to acidosis upregulate the expression of lysosomal protein LAMP2, which is translocated to the plasma membrane, rendering protection against acid-induced lysis (Damaghi et al., 2015). V-ATPases are yet another family of prominent pH regulators of neoplastic cells, which are known for a variety of normal cellular functions (Stransky et al., 2016). A wide spectrum of neoplastic cells is demonstrated to overexpress V-ATPase on their membrane (Stransky et al., 2016). Moreover, V-ATPase plays an indispensable role in pH homeostasis of neoplastic cells (Cotter et al., 2015). The expression of V-ATPase is under the regulatory control of signaling via Wnt/Notch and mTOR (Cotter et al., 2015; Stransky et al., 2016). In addition to its role in mediating export of H+ ion at the expense of the breakdown of ATP, V-ATPase facilitates autophagy, which is crucial in the biology of neoplastic cells (Stransky et al., 2016). Interestingly, recent studies have demonstrated the interaction between V-ATPase and microdomains of cholesterol in the manifestation of tumor metastasis (Stransky et al., 2016; Whitton et al., 2018). Thus, V-ATPase inhibitors are being explored for anticancer potential (Table 1). Furthermore, several reports emphasize the crucial role played by BCT in the regulation of pH in neoplastic cells (Gorbatenko et al., 2014). BCT belong to Na+/HCO3 − (SLC4 family) cotransporters and Cl−/HCO3 −(SLC 26 family) exchangers. They display a modulated expression in neoplastic cells and mediate the process of pH regulation and several other functions of neoplastic cells (Gorbatenko et al., 2014). However, despite the promising potential of BCTs, only a few studies have been carried out to understand and evaluate their relative contribution in the maintenance of pH homeostasis of neoplastic cells (Kant et al., 2014a). CA II, CA IX, and CA XII subtypes are reported to play a crucial role in manifesting tumor acidosis (Mboge et al., 2018). CA IX is demonstrated to play a vital role in pH homeostasis of neoplastic cells (Benej et al., 2014). In addition to its role as a pH regulator, CA IX plays various other contributing roles in the biology of cancer cells, including epithelial–mesenchymal transition, reshaping other cognate cellular interactions in the TME, and altered chemosensitivity (Benej et al., 2014; Mboge et al., 2018). Among the anionic exchangers, anionic exchanger 2 has been well demonstrated for its role in pH regulation of neoplastic cells via its function to exchange chloride with HCO3 − (Shiozaki et al., 2018). It also plays a crucial role in tumor metabolism (Xu et al., 2009). Additionally, ectopic localization of F1/F0 ATP synthase from the mitochondrial membrane to cell surface plasma membrane in neoplastic cells is envisaged to play a role in tumor acidosis because it serves as a proton channel in addition to its participation in energy generation (Mowery and Pizzo, 2008). Nevertheless, the membrane of tumor cells displays an upregulated expression of numerous pH-sensing proteins, which include ovarian cancer G-protein-coupled receptor 1, G-protein-coupled receptor 4, T-cell death-associated gene 8, acid-sensitive ion channel, and transient receptor potential of channel vanilloid subfamily, which cooperate with pH regulators (Damaghi et al., 2013; Justus et al., 2013; Huber et al., 2017). Other pH-sensing proteins such as actin-depolymerizing factor/cofilin, talin, and guanine nucleotide exchange factors collectively cooperate to regulate microfilament remodeling, vital for epithelial-mesenchymal transition, tumor cell invasion, and metastasis (Damaghi et al., 2013). Thus, all of these pH regulators and pH-sensing proteins collectively act in a concerted manner to regulate prosurvival signaling, tumor progression, and metastasis (Kato et al., 2013; Riemann et al., 2016). Furthermore, acid-sensing ion channels, particularly acid-sensing ion channel 2, which are voltage-independent have been associated with tumor invasion and metastasis (Zhou et al., 2017). The transient receptor potential channel of vanilloid subfamily I (TRPVI) is a proton-sensitive channel associated with the regulation of the process of tumorigenesis (Bode et al., 2009). However, more studies will be required to decipher its role in pH regulation in cancer cells. In the view of the crucial role of pH-dependent metabolic reprogramming in cancer cells, various components of the pH regulatory machinery have been explored for therapeutic targeting. These include approaches such as alkalinization of the TME (Kato et al., 2013; Pilon-Thomas et al., 2016) and use of specific inhibitors of various pH regulators (Vishvakarma and Singh, 2010; Vishvakarma and Singh, 2011; Kuchuk et al., 2018). These approaches indicate that targeting pH homeostasis can result in a cytostatic action on tumor cell survival, proliferation, metastasis, and invasion (Swietach et al., 2014; Huber et al., 2017). Reversal of tumor acidosis also ushers augmented chemosensitivity, elimination of acidosis-induced immunosuppression, and retardation of angiogenesis (Justus et al., 2013; Thews et al., 2014; Huber et al., 2017; Lacroix et al., 2018).
Metabolic Linking in the Tumor Microenvironment: A Platform for Redefining Cellular Relations
Although optimization of self-sufficiency is the main “motto” of neoplastic cells, external conditions become ultimately harsher, along with the progression of tumor, leading to a depleted supply of nutrients (DeBerardinis and Chandel, 2016). Moreover, tumor-infiltrating cells of the immune system compete for the available nutrients in the TME (Chang et al., 2015). Nutrient competition between tumor cells and tumor-infiltrating T lymphocytes has been elegantly demonstrated (Chang et al., 2015). However, being blessed with the Warburg phenomenon, neoplastic cells win the competition by comparatively higher uptake of glucose, leading to its depletion in the TME, thereby depriving the tumor-infiltrating cells of the immune system of essential glucose required for sustaining metabolism (Chang et al., 2015). Neoplastic cells evolve into a unique relationship with components of the TME, which could be symbiotic, parasitic, or competitive (Gatenby and Gillies, 2008; Lyssiotis and Kimmelman, 2017). Gradients of nutrients and gases in the TME create pockets of oxygenated, nutrient-rich, and depleted microniches (Lyssiotis and Kimmelman, 2017). Accordingly, both neoplastic and normal cells differentially adapt to these niches. Tumor cells optimize nutrient uptake by entering into a unique “metabolic symbiosis” operated between tumor cells themselves and with other normal cells in their vicinity (Nakajima and Van Houten, 2013). Thus, neoplastic cells can create a network of cognate and noncognate cellular interactions, among themselves, of “metabolic cross-feeding” to support nutritional uptake as an additional avenue. Lactate symbiosis is one of such well worked out examples, operational between aerobic and anaerobic tumor cells (Nakajima and Van Houten, 2013). Lactate produced by tumor cells is used as a nutrient not only by the OxPhos cancer cells but also by other cells of the TME, including mesenchymal stem cells, cancer-associated fibroblasts (CAFs), tumor-associated macrophages, and T lymphocytes (Allen et al., 2016; Romero-Garcia et al., 2016; Lyssiotis and Kimmelman, 2017). Furthermore, neoplastic cell-derived lactate has multiple effects on the metabolism of these stromal cells of the TME (Lyssiotis and Kimmelman, 2017), including M2 polarization of tumor-associated macrophages, which are protumorigenic, and inhibition of T-cell functions (Romero-Garcia et al., 2016; Yang and Zhang, 2017; Mu et al., 2018). Lactate is also reported to modulate redox and nitrogen balance in tumor cells (San-Millán and Brooks, 2017).
Availability of metabolites produced by normal cells in the TME also mediates the modulated metabolism of neoplastic cells (Lyssiotis and Kimmelman, 2017). Moreover, studies demonstrate that CAF produces lactate upon uptake of glucose, which in turn can be utilized by tumor cells (Lyssiotis and Kimmelman, 2017). Metabolic reprogramming of CAF leads to increased synthesis of glutamine (Lyssiotis and Kimmelman, 2017). Furthermore, increased uptake of glucose and tryptophan by tumor cells can deprive T cells of these nutrients, leading to an inhibition of their antitumor functions (Sukumar et al., 2015). Moreover, tryptophan metabolism of tumor cells produces kynurenine, which is reported to promote regulatory T cells to inhibit the functions of T helper (TH) cells and contribute to tumor growth promotion (Lyssiotis and Kimmelman, 2017). Moreover, a recent study strongly indicates the role of CD4+ TH cells in gearing antitumor immune responses (Morales Del Valle et al., 2019). Hence, TH cells are envisaged for anticancer therapeutic applications. Besides immune cells, adipocytes of the TME contribute to lipid homeostasis of neoplastic cells (Lyssiotis and Kimmelman, 2017). TME adipocytes produce fatty acids that are taken up by cancer cells to support their metabolism (Lyssiotis and Kimmelman, 2017). Tumor cells can also import mitochondria and exosomes containing metabolites (Lyssiotis and Kimmelman, 2017). Furthermore, the immune cells of the TME get suppressed because of nutrient deprivation caused by not only tumor cells but also other triggers derived from neoplastic cells and normal cells (Lyssiotis and Kimmelman, 2017). Indeed, arginine deprivation in the TME by cells of myeloid lineage is reported to be the cause of T-cell inhibition (Hanahan and Weinberg, 2011). Additionally, recent reports indicate the role of tumor acidosis in suppressing the activity of T effector cells and macrophages (Choi et al., 2013). Tumor acidosis is also conducive for tumor infiltration of immune cells, which, however, get suppressed or polarized to promote tumor progression (Choi et al., 2013). Furthermore, metabolites of the TME are capable of epigenetic modulations, such as histone acetylation, with consequences of modified genetic regulation of tumor cell metabolic plasticity (Etchegaray and Mostoslavsky, 2016). However, much still needs to be understood regarding the precise definition of cellular interactions at tumor–tumor and tumor–immune cells’ synapse, which has the potential for being explored for therapeutic reconstitution of the TME based on the interference of metabolic coupling between the constituent cells. Studies on 3D multicellular spheroids can be an important tool in understanding such dimensions concerning “metabolic coupling” operating in the TME (Nath and Devi, 2016).
3-Bromopyruvate Is Capable of Multifaceted Targeting of Tumor Metabolism and Constituents of the Tumor Microenvironment
Given the diverse stakeholders of tumor metabolism, it is essential to evolve a multifaceted targeting strategy for effective control of neoplastic cells’ survival, invasion, and metastasis. Our survey of literature for drugs fulfilling the objective of targeting multiple aspects of tumor metabolism has fetched a promising hope from an agent, which is a brominated derivative of pyruvate, designated as 3-BP. It has shown tremendous antineoplastic potential with several merits over other metabolic inhibitors (Ko et al., 2001; Azevedo-Silva et al., 2016). The following section highlights the broad spectrum of the antineoplastic actions of 3-BP, along with the possible underlying mechanisms. Future possibilities for its applications in anticancer regimens are also discussed. Interestingly, 3-BP is capable of inhibiting several aspects of tumor metabolism related to nutrient uptake, rewired metabolic pathways, pH homeostasis, and metabolism-dependent cellular interactions in the TME. Being an alkylating agent, 3-BP targets a plethora of biomolecules of neoplastic cells (Azevedo-Silva et al., 2016; Lis et al., 2016). Moreover, 3-BP shows a high degree of specificity for its anticancer activity (Buijs et al., 2013; Ganapathy-Kanniappan et al., 2013). The tumor cell-specific selectivity of 3-BP mainly depends on the similarity of this molecule with lactate and pyruvate, and hence it utilizes common transporters to gain cellular entry. On the contrary, other alkylating agents mainly enter by diffusion across the plasma membrane and, therefore, lack specificity (Ganapathy-Kanniappan et al., 2013). MCT-1 and MCT-4, which are specifically upregulated in neoplastic cells, mediate transport of 3-BP (Queirós et al., 2012; Baltazar, 2014; Counillon et al., 2016). Nevertheless, the acidic environment of the tumor milieu proves thermodynamically favorable for the uptake of 3-BP by cancer cells because of the pH difference across the plasma membrane (Azevedo-Silva et al., 2015). Another important reason for its selectivity against neoplastic cells is caused by the unmatched ability of 3-BP to alkylate and inactivate metabolic enzymes, which are selectively upregulated in malignant cells (Chen et al., 2009; Ganapathy-Kanniappan et al., 2009; Gandham et al., 2015; Azevedo-Silva et al., 2016; Jardim-Messeder and Moreira-Pacheco, 2016). Being an electrophile, 3-BP covalently and irreversibly modifies its targets’ nucleophilic moieties via SN2 mechanism of alkylation (Azevedo-Silva et al., 2016; Oronsky et al., 2012) (Figure 3). Interestingly, it is also reported that 3-BP is a prodrug, which gets activated in the vicinity of target nucleophiles only (Oronsky et al., 2012). As shown in Figure 4, 3-BP is capable of alkylating and modifying several target enzymes of glycolysis and the TCA cycle and consequently is highly capable of reversing the Warburg effect in neoplastic cells, leading to induction of cell death (Lis et al., 2016). Critical metabolic enzymes reported to be inhibited by 3-BP include hexokinase 2 (HK2) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in the glycolytic pathway; LDH and PDH in the linker pathway to TCA cycle; and SDH, isocitrate dehydrogenase, and α-ketoglutarate dehydrogenase in the TCA cycle (Chen et al., 2009; Ganapathy-Kanniappan et al., 2009; Ganapathy-Kanniappan et al., 2013; Azevedo-Silva et al., 2015; Jardim-Messeder and Moreira-Pacheco, 2016; Yadav et al., 2017b). Moreover, many other targets have been identified, including V-ATPase, pyruvate kinase, ribonuclease A, and glutathione (Dell’Antone, 2006; Ganapathy-Kanniappan et al., 2009; Ehrke et al., 2015). Nevertheless, 3-BP can alkylate several amino acids, particularly the cysteine moieties in several proteins (Hanau et al., 1995; Ganapathy-Kanniappan et al., 2009). Additionally, 3-BP also reported inhibiting glyoxalase and serine hydroxyl ethyl transferase (Valenti et al., 2015; Paiardini et al., 2016). Given the broad spectrum of the inhibitory action of 3-BP on metabolic enzymes, it is capable of ushering a “metabolic catastrophe” in cancer cells, leading to the depletion of ATP generation, causing declined neoplastic cell survival (Davidescu et al., 2015; Sun et al., 2015). Because of the massive antimetabolic potential, an ever-increasing list of cancer targets is building up, which display susceptibility to the antineoplastic action of 3-BP. Cytotoxic action of 3-BP is exerted against neoplastic cells and animal tumor models of diverse origins such as breast, prostate, pancreas, cervix, kidney, colon, hematological, and pulmonary (Xu et al., 2005; Cao et al., 2008; Hulleman et al., 2009; Schaefer et al., 2012; Attia et al., 2015; Nilsson et al., 2015; Sun et al., 2015; Valenti et al., 2015; Azevedo-Silva et al., 2016). Recently, our group reported a strong antitumor action of 3-BP against tumor cells of thymic origin, which are one of the rarest cancers and hence difficult for therapeutic exploration (Yadav et al., 2017a; Yadav et al., 2017b). In addition to its ability to inhibit metabolic enzymes, 3-BP also causes overexpression of the reactive oxygen species (ROS), along with depletion of ROS scavenger glutathione (GSH), in neoplastic cells, which in turn can induce cell death by induction of apoptosis and necrosis (Kim et al., 2008; Valenti et al., 2015). Nevertheless, ROS increases cellular and endoplasmic reticular (ER) stress (Ganapathy-Kanniappan et al., 2010). Moreover, reports suggest that, in addition to ER stress, 3-BP can contribute to the inhibition of protein synthesis (Kwiatkowska et al., 2016). Such perturbations are also associated with unfavorable modulation of redox homeostasis, accompanying mitochondrial damage (Kwiatkowska et al., 2016; Lis et al., 2016). Additionally, 3-BP treatment leads to the release of VADC-associated HK2, diminished mitochondrial potential, the release of cytochrome c, downregulation of antiapoptotic Bcl-2 and Mcl-1, and activation of caspase-3, indicating the mitochondrial mode of apoptosis (Chen et al., 2009; Yadav et al., 2017a; Yadav et al., 2017b).
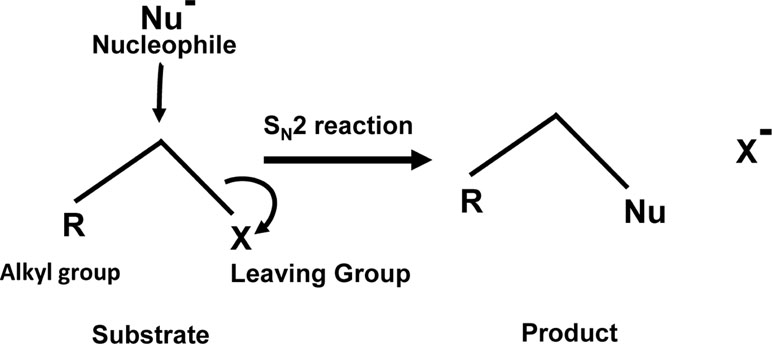
Figure 3 SN2 alkylation: Alkylation of the target by 3-BP follows the SN2 mechanism.
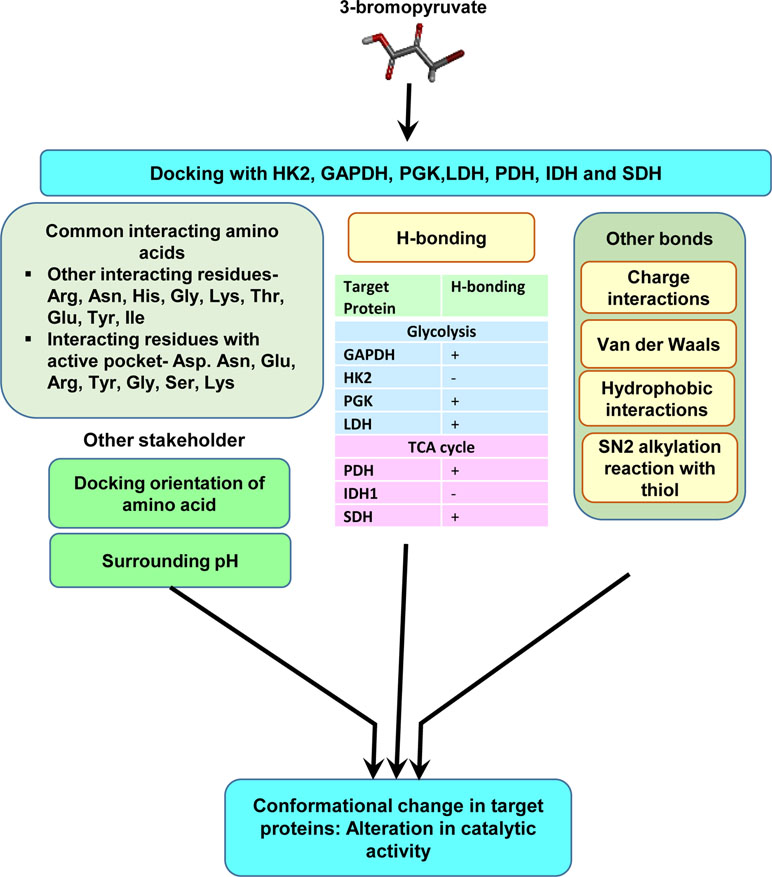
Figure 4 Molecular characterization of the interaction of 3-BP with multiple target molecules. Molecular docking studies indicate that, in addition to alkylation by SN2 reaction, 3-BP may impart modulatory actions on targets via multiple interactions including H-bonds, charged, and hydrophobic interactions. The diagram depicts interacting amino acids of the target molecules involved in docking. The nature of interactions could depend on docking orientations, the composition of active sites, and other environmental parameters, such as pH.
Furthermore, it is demonstrated that 3-BP can render tumor cells susceptible to the induction of cell death by additional mechanisms, including modulated expression of HIF-1α and by modulating pH homeostasis associated with altered glycolysis and TCA cycle (Marín-Hernández et al., 2009; Semenza, 2010). As already elaborated, HIF-1α can alter carbohydrate metabolism, oxidative stress, expression of cell survival-modulating cytokines, and mediators of drug resistance, enhancing chemosensitivity of tumor cells (Semenza, 2010; Masoud and Li, 2015). Nevertheless, 3-BP-dependent alterations in HIF-1α expression can lead to a declined expression of GLUT-1 and consequently glucose uptake by neoplastic cells, interfering with the lifeline of the nutrition supply of neoplastic cells (Orue et al., 2016; Yadav et al., 2017b). 3-BP-dependent inhibition of MCT-1 expression and, hence, lactate transport can cause a rise in intracellular pH accompanied by altered tumor acidosis (Sun et al., 2015; Yadav et al., 2017b). In vivo study in the murine tumor-bearing model has demonstrated 3-BP-dependent reconstitution of the cellular and soluble component of the TME. The TME of 3-BP-administered tumor-bearing hosts displayed repolarization of macrophages to tumoricidal M1 phenotype, accompanied by an increase in CD4, CD8, CD49, CD25 (IL-2R), and CD62L, CD11c, and TLR-4 expressing cells, indicating an altered repertoire of NK cells and T lymphocytes in the TME and alleviation of tumor-associated immunosuppression (Yadav et al., 2017a). Interestingly, it was demonstrated that 3-BP administration could inhibit the expression of V-ATPase in tumor cells, further contributing to the deregulation of pH homeostasis (Dell’Antone, 2006). The 3-BP-dependent altered internal milieu in tumor cells could also be linked to the decline of FASN expression, which is suggestive of inhibited de novo fatty acid synthesis, necessary for membrane biogenesis (Yadav et al., 2017a). Additionally, 3-BP can lead to a decline in HSP70 expression, suggesting the declined ability of 3-BP-exposed tumor cells to cope with stressful conditions, rendering them susceptible to induction of cell death (Yadav et al., 2017a). Indeed, other workers have indicated an increase in stress markers in 3-BP-treated cancer cells (Ganapathy-Kanniappan et al., 2010; Chiasserini et al., 2017). A decline of the stress-bearing capacity can augment cell death and the expression of VEGF, which triggers a diminished vasculature and blood flow in the TME (Attia et al., 2015; Yadav et al., 2017a). The 3-BP-dependent cell cycle arrest in tumor cells can be yet another trigger leading to induction of cell death (Chong et al., 2017; Yadav et al., 2017a). Additionally, 3-BP has been shown to interfere with oxidative phosphorylation (Lis et al., 2016). Furthermore, 3-BP targets complexes I and II of ETC, which also contributes to ATP depletion (Jardim-Messeder and Moreira-Pacheco, 2016).
Given the fact that most anticancer drugs inflict cytotoxicity to normal cells, tissues, and organs in cancer patients (Cheok, 2012), safety concerns are of primary focus while designing and developing chemotherapeutic agents. It has been demonstrated that the antitumor action of 3-BP is accompanied by protective and recuperative effects on immunological, hepatic, and renal homeostasis, with normalization of liver and kidney functions, reduction of tumor growth-associated splenomegaly, restored thymic homeostasis, normalization of blood lymphocytes, and upregulated myelopoiesis (Yadav et al., 2018). Additionally, other studies showed that 3-BP was safe to various tissues (Kunjithapatham et al., 2013; Pan et al., 2016), displaying minimal hepatic and nephrotoxicity (Pan et al., 2016). In Figure 5, a summary of novel antitumor mechanisms of 3-BP is depicted, showing its ability of multifaceted antitumor action, encompassing aspects such as membrane transport, inhibiting metabolic pathways, pH homeostasis, reconstitution of the TME, declined lipid biosynthesis, mitochondrial stress, restored organ homeostasis, and chemosensitivity.
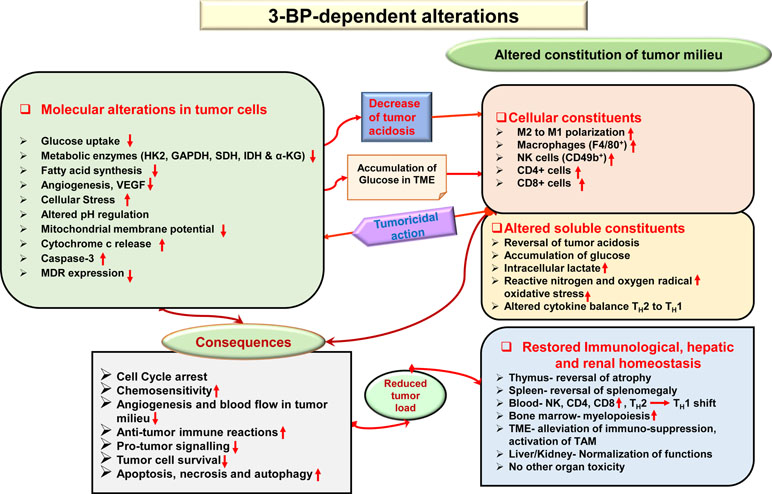
Figure 5 Multifaceted antineoplastic actions of 3-BP. Molecular mechanisms of the antineoplastic action of 3-BP involve multifaceted targeting of critical molecules involved in tumor metabolism leading to a metabolic catastrophe in neoplastic cells. A broad spectrum of antitumor actions can be manifested by 3-BP, which included the generation of ER and mitochondrial stress, inhibition in the expression of multidrug resistance (MDR) molecules, altered pH and redox homeostasis, depleted glucose uptake, and reconstitution of cellular, soluble, and biophysical components of the TME. Many of these actions are direct, whereas others could be manifested indirectly via other molecules such as altered cytokine balance and ROS. The antitumor action of 3-BP implicates cell cycle arrest, altered chemosensitivity, a decline of blood supply in the TME, inhibition of immune evasion, immune augmentation, and a decline of protumor signaling. Consequently, because of multiple effects and depletion of energy generation, tumor cells undergo cell death via induction of apoptosis, necrosis, and autophagy. Recuperative effect of 3-BP is imparted on the function of vital organs, such as the liver and kidney. Abbreviations: Cyc c, cytochrome c; pHi, intercellular pH; V-ATPase, vascular-ATPase; VEGF, vascular endothelial growth factor.
Most of the antineoplastic actions of 3-BP are mainly attributed to its ability to alkylate a variety of target molecules in neoplastic cells. However, because of the lacuna concerning the biochemical mechanism(s) of its binding to various heterogeneous target molecules, we carried out an extensive in silico-based investigation to precisely understand the molecular nature of the binding of 3-BP to its target proteins of glycolysis and TCA cycle (Yadav et al., 2017c). Docking analysis of 3-BP was carried out against the most vulnerable targets, namely, HK2, GAPDH, LDH, SDH, PDH, phosphoglycerate kinase (PGK), and IDH1 enzymes of carbohydrate metabolism (Figure 4) (Yadav et al., 2017c). Interestingly, this study demonstrated the implication of H-bonding between 3-BP and its targets, except for HK2 and IDH1. Moreover, Arg, Asn, Gly, His, Ser, and Thr were suggested to determine the binding strength between 3-BP and its target enzymes involving active sites (Yadav et al., 2017c). Another study has demonstrated the binding of 3-BP with one or more amino acids at the active site of the target enzymes (Silverman and Eoyang, 1987). Based on the calculation of geometric shape complementarity score, approximate interface, binding energy, and dissociation constant of the docking of 3-BP with target enzymes, it was demonstrated that 3-BP shows a stable binding to its targets (Yadav et al., 2017c). Furthermore, it was indicated that HK2, PDH, and SDH were the most preferred targets over the other enzymes. In addition to the H-bonds, other prominent biochemical interactions included hydrophobic interaction and Van der Waals forces, which vary by the amino acids of the respective docking sites (Pan et al., 2013; Yadav et al., 2017c).
Based on the ability to inhibit target enzymes, 3-BP derivatives have been tested for their antineoplastic activity. A derivative of 3-BP, named the 3-bromo-2-oxopropionate-1-propyl ester (3-BrOP), acts similarly as the 3-BP prodrug but was reported to be more stable than 3-BP and possessed a superior ability to deplete ATP in neoplastic cells (Lis et al., 2016). Similarly, we compared the docking ability of 3-BP derivatives dibromopyruvate (DBPA) and propionic acid (PA) with 3-BP target enzymes. Interestingly, DBPA was found to display a better docking ability than 3-BP and PA to various target enzymes (Yadav et al., 2017c), indicating strong antineoplastic potential, which needs to be explored further. These studies will also aid in optimizing the therapeutic efficacy of 3-BP by achieving a better understanding of the inhibition of target enzymes by modification of the catalytic site. In addition to the protective and recuperative actions of 3-BP in a tumor-bearing host, it is noteworthy that 3-BP and its derivatives DBPA and PA have been predicted for drug-likeness criteria and found to satisfactorily pass the parameters of drug-likeness on Lipinski filter and FAFDrugs 3 analysis tools (Yadav et al., 2017c).
Limitations and Prospects
Despite approval for phase I trial by the FDA, the clinical trials with 3-BP have not yet been realized because of 1) limitation of financial resources for executing the trials and 2) death of three patients being attributed to 3-BP. However, it was later on reported that these deaths were not likely caused by 3-BP (Lis et al., 2016). Moreover, these controversies associated with the lethality of 3-BP when used in inappropriate dose regimens (Feldwisch-Drentrup, 2016) need to be addressed adequately under proper scientifically validated and controlled settings before its applications for human use as an anticancer drug. It is essential to consider physiological and physical parameters capable of influencing the antitumor efficacy of 3-BP. Despite this, isolated sporadic clinical applications of 3-BP in humans and xenograft models of human cancers have raised positive optimism for its use as a potent cancer therapeutic agent (Lee et al., 2017a). A study using a volunteer cancer patient demonstrated fruitful outcomes for the use of 3-BP as an anticancer drug (El Sayed et al., 2014). Moreover, these limited clinical trials have shown minimal side effects, except minor concerns regarding burning sensation and phlebitis (El Sayed, 2018). Most preclinical and clinical trials report about its safety concerning hepatic functions (Ko et al., 2012; El Sayed et al., 2014; Pan et al., 2016; Yadav et al., 2018). The main recommendations for overcoming the limitations regarding the use of 3-BP include strict implementation of only formulated preparations in human applications (El Sayed, 2018; Fan et al., 2019b), use of GSH scavengers accompanying 3-BP administration because GSH can inactivate 3-BP (El Sayed, 2018), and strict monitoring of the dose regimens (El Sayed, 2018). Furthermore, approaches of liposome-encapsulated 3-BP formulations are suggested to improve its adequate concentrations in the tumor milieu (Gandham et al., 2015). Use of 3-BP as an adjunct agent along with other conventional approaches is considered to hold therapeutic potential (Ganapathy-Kanniappan et al., 2013; Zhang et al., 2015; Chong et al., 2017).
There is a promising potential for 3-BP and its derivatives for being assessed further in preclinical and clinical settings to predict about anticancer efficacy. Critical checkpoints of tumor metabolism right from the level of import of nutrients through their metabolic channelization and generation of ATP are affected by 3-BP. Further, cellular signaling and pH homeostasis are also influenced by 3-BP and hence it is a highly capable agent for modulating metabolic linking in the TME. Thus, 3-BP displays multifaceted antineoplastic activity via its direct inhibitory action on metabolic targets of neoplastic cells, by harnessing the antitumor potential of the immune system, and by rendering the TME unfavorable for tumor progression. Hence, 3-BP ushers a promising hope in the combat against cancer because of its low cost, a broad spectrum of antineoplastic potential, desirable drugability characteristics, and a track record of safety, necessitating initiation of further clinical optimization.
Author Contributions
SY: conceiving of the idea; a survey of the literature, writing, and preparation of the manuscript; SS: conceiving of the idea and manuscript preparation; SP, YG, and MT: writing manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thankfully acknowledge fellowship support to SY [Award No. 09/013(0577)/2015-EMR-I] from CSIR, New Delhi; SP [Award No. 3/1/3/JRF-2015(2)HRD], YG [Award No.3/1/3/JRF-2016/LS/HRD-10(80666)] from ICMR, New Delhi; and MT [Award No. DBT/JRF/BET-18/I/2018/AL/154] from DBT, New Delhi. The work contained in this manuscript is a component of the PhD dissertation of SY.
References
Advani, A. S., McDonough, S., Copelan, E., Willman, C., Mulford, D. A., List, A. F., et al. (2014). SWOG0919: a Phase 2 study of idarubicin and cytarabine in combination with pravastatin for relapsed acute myeloid leukaemia. Br. J. Haematol. 167, 233–237. doi: 10.1111/bjh.13035
Allen, E., Miéville, P., Warren, C. M., Saghafinia, S., Li, L., Peng, M.-W., et al. (2016). Metabolic symbiosis enables adaptive resistance to anti-angiogenic therapy that is dependent on mTOR signaling. Cell Rep. 15, 1144–1160. doi: 10.1016/j.celrep.2016.04.029
Amith, S. R., Wilkinson, J. M., Fliegel, L. (2016). KR-33028, a potent inhibitor of the Na+/H+exchanger NHE1, suppresses metastatic potential of triple-negative breast cancer cells. Biochem. Pharmacol. 118, 31–39. doi: 10.1016/j.bcp.2016.08.010
Anderson, N. M., Mucka, P., Kern, J. G., Feng, H. (2018). The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 9, 216–237. doi: 10.1007/s13238-017-0451-1
Attia, Y. M., El-Abhar, H. S., Al Marzabani, M. M., Shouman, S. A. (2015). Targeting glycolysis by 3-bromopyruvate improves tamoxifen cytotoxicity of breast cancer cell lines. BMC Cancer 15, 838. doi: 10.1186/s12885-015-1850-4
Azevedo-Silva, J., Queirós, O., Baltazar, F., Ułaszewski, S., Goffeau, A., Ko, Y. H., et al. (2016). The anticancer agent 3-bromopyruvate: a simple but powerful molecule taken from the lab to the bedside. J. Bioenerg. Biomembr. 48, 349–362. doi: 10.1007/s10863-016-9670-z
Azevedo-Silva, J., Queirós, O., Ribeiro, A., Baltazar, F., Young, K. H., Pedersen, P. L., et al. (2015). The cytotoxicity of 3-bromopyruvate in breast cancer cells depends on extracellular pH. Biochem. J. 467, 247–258. doi: 10.1042/BJ20140921
Baenke, F., Peck, B., Miess, H., Schulze, A. (2013). Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Dis. Model. Mech. 6, 1353–1363. doi: 10.1242/dmm.011338
Baggstrom, M. Q., Qi, Y., Koczywas, M., Argiris, A., Johnson, E. A., Millward, M. J., et al. (2011). A phase II study of AT-101 (Gossypol) in chemotherapy-sensitive recurrent extensive-stage small cell lung cancer. J. Thorac. Oncol. 6, 1757–1760. doi: 10.1097/JTO.0b013e31822e2941
Baghdadi, H. H. (2017). Targeting cancer cells using 3-bromopyruvate for selective cancer treatment. Saudi J. Med. Med. Sci. 5, 9–19. doi: 10.4103/1658-631X.194253
Baltazar, F. (2014). Significance of monocarboxylate transporter (MCT) expression in human tumors. Front. Pharmacol. doi: 10.3389/conf.fphar.2014.61.00004
Barr, P. M., Miller, T. P., Friedberg, J. W., Peterson, D. R., Baran, A. M., Herr, M., et al. (2014). Phase 2 study of imexon, a prooxidant molecule, in relapsed and refractory B-cell non-Hodgkin lymphoma. Blood 124, 1259–1265. doi: 10.1182/blood-2014-04-570044
Barron, C., Tsiani, E., Tsakiridis, T. (2012). Expression of the glucose transporters GLUT1, GLUT3, GLUT4 and GLUT12 in human cancer cells. BMC Proc. 6, P4. doi: 10.1186/1753-6561-6-S3-P4
Bayat Mokhtari, R., Baluch, N., Hon Tsui, M., Kumar, S., Homayouni, T., Aitken, K., et al. (2017). Acetazolamide potentiates the anti-tumor potential of HDACi, MS-275, in neuroblastoma. BMC Cancer 17, 156. doi: 10.1186/s12885-017-3126-7
Beckers, A., Organe, S., Timmermans, L., Scheys, K., Peeters, A., Brusselmans, K., et al. (2007). Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res. 67, 8180–8187. doi: 10.1158/0008-5472.CAN-07-0389
Beloribi-Djefaflia, S., Vasseur, S., Guillaumond, F. (2016). Lipid metabolic reprogramming in cancer cells. Oncogenesis 5, e189. doi: 10.1038/oncsis.2015.49
Benej, M., Pastorekova, S., Pastorek, J. (2014). Carbonic anhydrase IX: regulation and role in cancer. Subcell. Biochem. 75, 199–219. doi: 10.1007/978-94-007-7359-2_11
Beppu, T., Sato, Y., Sasaki, T., Terasaki, K., Yamashita, F., Sasaki, M., et al. (2019). Comparisons between PET with 11C-methyl-L-methionine and arterial spin labeling perfusion imaging in recurrent glioblastomas treated with bevacizumab. Clin. Nucl. Med. 44, 186–193. doi: 10.1097/RLU.0000000000002417
Berruti, A., Bitossi, R., Gorzegno, G., Bottini, A., Alquati, P., De Matteis, A., et al. (2002). Time to progression in metastatic breast cancer patients treated with epirubicin is not improved by the addition of either cisplatin or lonidamine: final results of a phase III study with a factorial design. J. Clin. Oncol. 20, 4150–4159. doi: 10.1200/JCO.2002.08.012
Bhutia, Y. D., Babu, E., Ramachandran, S., Ganapathy, V. (2015). Amino acid transporters in cancer and their relevance to “Glutamine Addiction”: novel targets for the design of a new class of anticancer drugs. Cancer Res. 75, 1782–1788. doi: 10.1158/0008-5472.CAN-14-3745
Birsoy, K., Wang, T., Possemato, R., Yilmaz, O. H., Koch, C. E., Chen, W. W., et al. (2013). MCT1-mediated transport of a toxic molecule is an effective strategy for targeting glycolytic tumors. Nat. Genet. 45, 104–108. doi: 10.1038/ng.2471
Boddu, P., Borthakur, G. (2017). Therapeutic targeting of isocitrate dehydrogenase mutant AML. Expert Opin. Investig. Drugs 26, 525–530. doi: 10.1080/13543784.2017.1317745
Bode, A. M., Cho, Y.-Y., Zheng, D., Zhu, F., Ericson, M. E., Ma, W.-Y., et al. (2009). The transient receptor potential type vanilloid 1 suppresses skin carcinogenesis. Cancer Res. 69, 905–913. doi: 10.1158/0008-5472.CAN-08-3263
Bonen, A., Campbell, S. E., Benton, C. R., Chabowski, A., Coort, S. L. M., Han, X.-X., et al. (2004). Regulation of fatty acid transport by fatty acid translocase/CD36. Proc. Nutr. Soc. 63, 245–249. doi: 10.1079/PNS2004331
Brana, I., Ocana, A., Chen, E. X., Razak, A. R. A., Haines, C., Lee, C., et al. (2014). A phase I trial of pantoprazole in combination with doxorubicin in patients with advanced solid tumors: evaluation of pharmacokinetics of both drugs and tissue penetration of doxorubicin. Invest. New Drugs 32, 1269–1277. doi: 10.1007/s10637-014-0159-5
Brochez, L., Chevolet, I., Kruse, V. (2017). The rationale of indoleamine 2, 3-dioxygenase inhibition for cancer therapy. Eur. J. Cancer 76, 167–182. doi: 10.1016/j.ejca.2017.01.011
Buijs, M., Wijlemans, J. W., Kwak, B. K., Ota, S., Geschwind, J.-F. H. (2013). Antiglycolytic therapy combined with an image-guided minimally invasive delivery strategy for the treatment of breast cancer. J. Vasc. Interv. Radiol. 24, 737–743. doi: 10.1016/j.jvir.2013.01.013
Burstein, H. J., Demetri, G. D., Mueller, E., Sarraf, P., Spiegelman, B. M., Winer, E. P. (2003). Use of the peroxisome proliferator-activated receptor (PPAR) gamma ligand troglitazone as treatment for refractory breast cancer: a phase II study. Breast Cancer Res. Treat. 79, 391–397. doi: 10.1023/A:1024038127156
Cairns, R. A., Mak, T. W. (2016). An alternative sugar fuels AML. Cancer Cell 30, 660–662. doi: 10.1016/j.ccell.2016.10.015
Cantor, J. R., Sabatini, D. M. (2012). Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2, 881–898. doi: 10.1158/2159-8290.CD-12-0345
Cao, X., Bloomston, M., Zhang, T., Frankel, W. L., Jia, G., Wang, B., et al. (2008). Synergistic antipancreatic tumor effect by simultaneously targeting hypoxic cancer cells with HSP90 inhibitor and glycolysis inhibitor. Clin. Cancer Res. 14, 1831–1839. doi: 10.1158/1078-0432.CCR-07-1607
Chan, J. K., Brady, W., Monk, B. J., Brown, J., Shahin, M. S., Rose, P. G., et al. (2018). A phase II evaluation of sunitinib in the treatment of persistent or recurrent clear cell ovarian carcinoma: an NRG Oncology/Gynecologic Oncology Group Study (GOG-254). Gynecol. Oncol. 150, 247–252. doi: 10.1016/j.ygyno.2018.05.029
Chang, C.-H., Qiu, J., O’Sullivan, D., Buck, M. D., Noguchi, T., Curtis, J. D., et al. (2015). Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 162, 1229–1241. doi: 10.1016/j.cell.2015.08.016
Chen, Z., Zhang, H., Lu, W., Huang, P. (2009). Role of mitochondria-associated hexokinase II in cancer cell death induced by 3-bromopyruvate. Biochim. Biophys. Acta 1787, 553–560. doi: 10.1016/j.bbabio.2009.03.003
Cheok, C. F. (2012). Protecting normal cells from the cytotoxicity of chemotherapy. Cell Cycle 11, 2227. doi: 10.4161/cc.20961
Chiasserini, D., Davidescu, M., Orvietani, P. L., Susta, F., Macchioni, L., Petricciuolo, M., et al. (2017). 3-Bromopyruvate treatment induces alterations of metabolic and stress-related pathways in glioblastoma cells. J. Proteomics 152, 329–338. doi: 10.1016/j.jprot.2016.11.013
Chiche, J., Brahimi-Horn, M. C., Pouysségur, J. (2010). Tumour hypoxia induces a metabolic shift causing acidosis: a common feature in cancer. J. Cell. Mol. Med. 14, 771–794. doi: 10.1111/j.1582-4934.2009.00994.x
Choi, S. Y. C., Collins, C. C., Gout, P. W., Wang, Y. (2013). Cancer-generated lactic acid: a regulatory, immunosuppressive metabolite? J. Pathol. 230, 350–355. doi: 10.1002/path.4218
Choi, Y. K. (2017). A positive circuit of VEGF increases Glut-1 expression by increasing HIF-1α gene expression in human retinal endothelial cells. Arch. Pharm. Res. 40, 1433–1442. doi: 10.1007/s12272-017-0971-5
Choi, Y.-K., Park, K.-G. (2018). Targeting glutamine metabolism for cancer treatment. Biomol. Ther. 26, 19–28. doi: 10.4062/biomolther.2017.178
Chong, D., Ma, L., Liu, F., Zhang, Z., Zhao, S., Huo, Q., et al. (2017). Synergistic antitumor effect of 3-bromopyruvate and 5-fluorouracil against human colorectal cancer through cell cycle arrest and induction of apoptosis. Anticancer Drugs 28, 831–840. doi: 10.1097/CAD.0000000000000517
Chu, Q. S.-C., Sangha, R., Spratlin, J., Vos, L. J., Mackey, J. R., McEwan, A. J. B., et al. (2015). A phase I open-labeled, single-arm, dose-escalation, study of dichloroacetate (DCA) in patients with advanced solid tumors. Invest. New Drugs 33, 603–610. doi: 10.1007/s10637-015-0221-y
Commisso, C., Davidson, S. M., Soydaner-Azeloglu, R. G., Parker, S. J., Kamphorst, J. J., Hackett, S., et al. (2013). Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637. doi: 10.1038/nature12138
Corbet, C., Feron, O. (2017). Tumour acidosis: from the passenger to the driver’s seat. Nat. Rev. Cancer 17, 577–593. doi: 10.1038/nrc.2017.77
Cormerais, Y., Massard, P. A., Vucetic, M., Giuliano, S., Tambutté, E., Durivault, J., et al. (2018). The glutamine transporter ASCT2 (SLC1A5) promotes tumor growth independently of the amino acid transporter LAT1 (SLC7A5). J. Biol. Chem. 293, 2877–2887. doi: 10.1074/jbc.RA117.001342
Costa, A. S. H., Frezza, C. (2017). Metabolic reprogramming and oncogenesis: one hallmark, many organelles. Int. Rev. Cell Mol. Biol. 332, 213–231. doi: 10.1016/bs.ircmb.2017.01.001
Cotter, K., Stransky, L., McGuire, C., Forgac, M. (2015). Recent insights into the structure, regulation, and function of the V-ATPases. Trends Biochem. Sci. 40, 611–622. doi: 10.1016/j.tibs.2015.08.005
Counillon, L., Bouret, Y., Marchiq, I., Pouysségur, J. (2016). Na+/H+ antiporter (NHE1) and lactate/H+ symporters (MCTs) in pH homeostasis and cancer metabolism. Biochim. Biophys. Acta BBA - Mol. Cell Res. 1863, 2465–2480. doi: 10.1016/j.bbamcr.2016.02.018
Curtis, N. J., Mooney, L., Hopcroft, L., Michopoulos, F., Whalley, N., Zhong, H., et al. (2017). Pre-clinical pharmacology of AZD3965, a selective inhibitor of MCT1: DLBCL, NHL and Burkitt’s lymphoma anti-tumor activity. Oncotarget 8, 69219–69236. doi: 10.18632/oncotarget.18215
Dagogo-Jack, I., Shaw, A. T. (2018). Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 15, 81–94. doi: 10.1038/nrclinonc.2017.166
Damaghi, M., Tafreshi, N. K., Lloyd, M. C., Sprung, R., Estrella, V., Wojtkowiak, J. W., et al. (2015). Chronic acidosis in the tumour microenvironment selects for overexpression of LAMP2 in the plasma membrane. Nat. Commun. 6, 8752. doi: 10.1038/ncomms9752
Damaghi, M., Wojtkowiak, J. W., Gillies, R. J. (2013). pH sensing and regulation in cancer. Front. Physiol. 4, 370. doi: 10.3389/fphys.2013.00370
Davidescu, M., Macchioni, L., Scaramozzino, G., Cristina Marchetti, M., Migliorati, G., Vitale, R., et al. (2015). The energy blockers bromopyruvate and lonidamine lead GL15 glioblastoma cells to death by different p53-dependent routes. Sci. Rep. 5, 14343. doi: 10.1038/srep14343
De Saedeleer, C. J., Porporato, P. E., Copetti, T., Pérez-Escuredo, J., Payen, V. L., Brisson, L., et al. (2014). Glucose deprivation increases monocarboxylate transporter 1 (MCT1) expression and MCT1-dependent tumor cell migration. Oncogene 33, 4060–4068. doi: 10.1038/onc.2013.454
DeBerardinis, R. J., Chandel, N. S. (2016). Fundamentals of cancer metabolism. Sci. Adv. 2, e1600200. doi: 10.1126/sciadv.1600200
Dell’Antone, P. (2006). Inactivation of H+-vacuolar ATPase by the energy blocker 3-bromopyruvate, a new antitumour agent. Life Sci. 79, 2049–2055. doi: 10.1016/j.lfs.2006.06.043
Doherty, J. R., Cleveland, J. L. (2013). Targeting lactate metabolism for cancer therapeutics. J. Clin. Invest. 123, 3685–3692. doi: 10.1172/JCI69741
Douard, V., Ferraris, R. P. (2008). Regulation of the fructose transporter GLUT5 in health and disease. Am. J. Physiol. Endocrinol. Metab. 295, E227–237. doi: 10.1152/ajpendo.90245.2008
Ehrke, E., Arend, C., Dringen, R. (2015). 3-bromopyruvate inhibits glycolysis, depletes cellular glutathione, and compromises the viability of cultured primary rat astrocytes. J. Neurosci. Res. 93, 1138–1146. doi: 10.1002/jnr.23474
El Sayed, S. M. (2018). Enhancing anticancer effects, decreasing risks and solving practical problems facing 3-bromopyruvate in clinical oncology: 10 years of research experience. Int. J. Nanomedicine 13, 4699–4709. doi: 10.2147/IJN.S170564
El Sayed, S. M., Mohamed, W. G., Seddik, M.-A. H., Ahmed, A.-S. A., Mahmoud, A. G., Amer, W. H., et al. (2014). Safety and outcome of treatment of metastatic melanoma using 3-bromopyruvate: a concise literature review and case study. Chin. J. Cancer 33, 356–364. doi: 10.5732/cjc.013.10111
Enciu, A.-M., Radu, E., Popescu, I. D., Hinescu, M. E., Ceafalan, L. C. (2018). Targeting CD36 as biomarker for metastasis prognostic: how far from translation into clinical practice? BioMed Res. Int. 2018, 7801202. doi: 10.1155/2018/7801202
Esen, A., Khan, A. A., Chan, J., Mahmud, N., Quigley, J. G. (2016). Targeted depletion of amino acids as a novel therapy for acute leukemia and other cancers: mechanisms and countermechanisms. Blood 128, 4716–4716.
Etchegaray, J.-P., Mostoslavsky, R. (2016). Interplay between metabolism and epigenetics: a nuclear adaptation to environmental changes. Mol. Cell 62, 695–711. doi: 10.1016/j.molcel.2016.05.029
Faes, S., Demartines, N., Dormond, O. (2017). Resistance to mTORC1 inhibitors in cancer therapy: from kinase mutations to intratumoral heterogeneity of kinase activity. Oxid. Med. Cell. Longev. 2017, 1726078. doi: 10.1155/2017/1726078
Fan, T., Sun, G., Sun, X., Zhao, L., Zhong, R., Peng, Y. (2019b). Tumor energy metabolism and potential of 3-bromopyruvate as an inhibitor of aerobic glycolysis: implications in tumor treatment. Cancers 11, 317. doi: 10.3390/cancers11030317
Feldwisch-Drentrup (2016). Aug. 12, H., 2016, and Pm, 2:00. Sci. AAAS. Available at: https://www.sciencemag.org/news/2016/08/candidate-cancer-drug-suspected-after-death-three-patients-alternative-medicine-clinic [Accessed May 19, 2019]. doi: 10.1126/science.aah7192
Fenner, M., Oing, C., Dieing, A., Gauler, T., Oechsle, K., Lorch, A., et al. (2019). Everolimus in patients with multiply relapsed or cisplatin refractory germ cell tumors: results of a phase II, single-arm, open-label multicenter trial (RADIT) of the german testicular cancer study group. J. Cancer Res. Clin. Oncol. 145, 717–723. doi: 10.1007/s00432-018-2752-z
Ferro, S., Azevedo-Silva, J., Casal, M., Côrte-Real, M., Baltazar, F., Preto, A. (2016). Characterization of acetate transport in colorectal cancer cells and potential therapeutic implications. Oncotarget 7, 70639–70653. doi: 10.18632/oncotarget.12156
Furuya, Y., Sekine, Y., Kato, H., Miyazawa, Y., Koike, H., Suzuki, K. (2016). Low-density lipoprotein receptors play an important role in the inhibition of prostate cancer cell proliferation by statins. Prostate Int. 4, 56–60. doi: 10.1016/j.prnil.2016.02.003
Galkin, A., Abramov, A. Y., Frakich, N., Duchen, M. R., Moncada, S. (2009). Lack of oxygen deactivates mitochondrial complex I: implications for ischemic injury? J. Biol. Chem. 284, 36055–36061. doi: 10.1074/jbc.M109.054346
Gallagher, E. J., Zelenko, Z., Neel, B. A., Antoniou, I. M., Rajan, L., Kase, N., et al. (2017). Elevated tumor LDLR expression accelerates LDL cholesterol-mediated breast cancer growth in mouse models of hyperlipidemia. Oncogene 36, 6462–6471. doi: 10.1038/onc.2017.247
Ganapathy, V., Thangaraju, M., Gopal, E., Martin, P. M., Itagaki, S., Miyauchi, S., et al. (2008). Sodium-coupled monocarboxylate transporters in normal tissues and in cancer. AAPS J. 10, 193–199. doi: 10.1208/s12248-008-9022-y
Ganapathy-Kanniappan, S., Geschwind, J.-F. H. (2013). Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol. Cancer 12, 152. doi: 10.1186/1476-4598-12-152
Ganapathy-Kanniappan, S., Geschwind, J.-F. H., Kunjithapatham, R., Buijs, M., Syed, L. H., Rao, P. P., et al. (2010). 3-Bromopyruvate induces endoplasmic reticulum stress, overcomes autophagy and causes apoptosis in human HCC cell lines. Anticancer Res. 30, 923–935.
Ganapathy-Kanniappan, S., Geschwind, J.-F. H., Kunjithapatham, R., Buijs, M., Vossen, J. A., Tchernyshyov, I., et al. (2009). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is pyruvylated during 3-bromopyruvate mediated cancer cell death. Anticancer Res. 29, 4909–4918.
Ganapathy-Kanniappan, S., Kunjithapatham, R., Geschwind, J.-F. (2012). glyceraldehyde-3-phosphate dehydrogenase: a promising target for molecular therapy in hepatocellular carcinoma. Oncotarget 3, 940–953. doi: 10.18632/oncotarget.623
Ganapathy-Kanniappan, S., Kunjithapatham, R., Geschwind, J.-F. (2013). Anticancer efficacy of the metabolic blocker 3-bromopyruvate: specific molecular targeting. Anticancer Res. 33, 13–20.
Gandham, S. K., Talekar, M., Singh, A., Amiji, M. M. (2015). Inhibition of hexokinase-2 with targeted liposomal 3-bromopyruvate in an ovarian tumor spheroid model of aerobic glycolysis. Int. J. Nanomedicine 10, 4405–4423. doi: 10.2147/IJN.S82818
Gatenby, R. A., Gillies, R. J. (2008). A microenvironmental model of carcinogenesis. Nat. Rev. Cancer 8, 56–61. doi: 10.1038/nrc2255
Gorbatenko, A., Olesen, C. W., Boedtkjer, E., Pedersen, S. F. (2014). Regulation and roles of bicarbonate transporters in cancer. Front. Physiol. 5, 130. doi: 10.3389/fphys.2014.00130
Granchi, C., Minutolo, F. (2012). Anti-cancer agents counteracting tumor glycolysis. ChemMedChem 7, 1318–1350. doi: 10.1002/cmdc.201200176
Griss, T., Vincent, E. E., Egnatchik, R., Chen, J., Ma, E. H., Faubert, B., et al. (2015). Metformin antagonizes cancer cell proliferation by suppressing mitochondrial-dependent biosynthesis. PLoS Biol. 13, e1002309. doi: 10.1371/journal.pbio.1002309
Gui, D. Y., Sullivan, L. B., Luengo, A., Hosios, A. M., Bush, L. N., Gitego, N., et al. (2016). Environment dictates dependence on mitochondrial complex I for NAD+ and aspartate production and determines cancer cell sensitivity to metformin. Cell Metab. 24, 716–727. doi: 10.1016/j.cmet.2016.09.006
Guo, D., Bell, E. H., Mischel, P., Chakravarti, A. (2014). Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr. Pharm. Des. 20, 2619–2626. doi: 10.2174/13816128113199990486
Ha, K. D., Bidlingmaier, S. M., Liu, B. (2016). Macropinocytosis exploitation by cancers and cancer therapeutics. Front. Physiol. 7, 381. doi: 10.3389/fphys.2016.00381
Haase, C., Bergmann, R., Fuechtner, F., Hoepping, A., Pietzsch, J. (2007). L-type amino acid transporters LAT1 and LAT4 in cancer: uptake of 3-O-methyl-6- 18F-fluoro-l-dopa in human adenocarcinoma and squamous cell carcinoma in vitro and in vivo. J. Nucl. Med. 48, 2063–2071. doi: 10.2967/jnumed.107.043620
Hamanaka, R. B., Chandel, N. S. (2012). Targeting glucose metabolism for cancer therapy. J. Exp. Med. 209, 211–215. doi: 10.1084/jem.20120162
Hanahan, D., Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Hanau, S., Bertelli, M., Dallocchio, F., Rippa, M. (1995). Bromopyruvate for the affinity labelling of a single cysteine residue near the carboxylate binding site of lamb liver 6-phosphogluconate dehydrogenase. Biochem. Mol. Biol. Int. 37, 785–793.
Harjes, U., Kalucka, J., Carmeliet, P. (2016). Targeting fatty acid metabolism in cancer and endothelial cells. Crit. Rev. Oncol. Hematol. 97, 15–21. doi: 10.1016/j.critrevonc.2015.10.011
Harriman, G., Greenwood, J., Bhat, S., Huang, X., Wang, R., Paul, D., et al. (2016). Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Proc. Natl. Acad. Sci. U.S.A. 113, E1796–E1805. doi: 10.1073/pnas.1520686113
Hawkins, J. L., Robbins, M. D., Warren, L. C., Xia, D., Petras, S. F., Valentine, J. J., et al. (2008). Pharmacologic inhibition of site 1 protease activity inhibits sterol regulatory element-binding protein processing and reduces lipogenic enzyme gene expression and lipid synthesis in cultured cells and experimental animals. J. Pharmacol. Exp. Ther. 326, 801–808. doi: 10.1124/jpet.108.139626
Hay, N. (2016). Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat. Rev. Cancer 16, 635–649. doi: 10.1038/nrc.2016.77
Hayase, S., Kumamoto, K., Saito, K., Kofunato, Y., Sato, Y., Okayama, H., et al. (2017). L-type amino acid transporter 1 expression is upregulated and associated with cellular proliferation in colorectal cancer. Oncol. Lett. 14, 7410–7416. doi: 10.3892/ol.2017.7148
Hensley, C. T., Wasti, A. T., DeBerardinis, R. J. (2013). Glutamine and cancer: cell biology, physiology, and clinical opportunities. J. Clin. Invest. 123, 3678–3684. doi: 10.1172/JCI69600
Hsu, P. P., Sabatini, D. M. (2008). Cancer cell metabolism: warburg and beyond. Cell 134, 703–707. doi: 10.1016/j.cell.2008.08.021
Huang, Y., Dai, Z., Barbacioru, C., Sadée, W. (2005). Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res. 65, 7446–7454. doi: 10.1158/0008-5472.CAN-04-4267
Huber, V., Camisaschi, C., Berzi, A., Ferro, S., Lugini, L., Triulzi, T., et al. (2017). Cancer acidity: an ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin. Cancer Biol. 43, 74–89. doi: 10.1016/j.semcancer.2017.03.001
Hulleman, E., Kazemier, K. M., Holleman, A., VanderWeele, D. J., Rudin, C. M., Broekhuis, M. J. C., et al. (2009). Inhibition of glycolysis modulates prednisolone resistance in acute lymphoblastic leukemia cells. Blood 113, 2014–2021. doi: 10.1182/blood-2008-05-157842
Huttunen, K. M., Gynther, M., Huttunen, J., Puris, E., Spicer, J. A., Denny, W. A. (2016). A selective and slowly reversible inhibitor of l-type amino acid transporter 1 (LAT1) potentiates antiproliferative drug efficacy in cancer cells. J. Med. Chem. 59, 5740–5751. doi: 10.1021/acs.jmedchem.6b00190
Imai, H., Kaira, K., Oriuchi, N., Shimizu, K., Tominaga, H., Yanagitani, N., et al. (2010). Inhibition of L-type amino acid transporter 1 has antitumor activity in non-small cell lung cancer. Anticancer Res. 30, 4819–4828.
Iwasaki, K., Yabushita, H., Ueno, T., Wakatsuki, A. (2015). Role of hypoxia-inducible factor-1α, carbonic anhydrase-IX, glucose transporter-1 and vascular endothelial growth factor associated with lymph node metastasis and recurrence in patients with locally advanced cervical cancer. Oncol. Lett. 10, 1970–1978. doi: 10.3892/ol.2015.3524
Jardim-Messeder, D., Moreira-Pacheco, F. (2016). 3-Bromopyruvic acid inhibits tricarboxylic acid cycle and glutaminolysis in HepG2 Cells. Anticancer Res. 36, 2233–2241.
Jian, F., Yuan, F., Jiong, M., Zhu, X., Yu, G., Lu, D. (2015). Silencing of glucose transporter protein-1 by RNA interference inhibits human osteosarcoma Mg63 cells growth in vivo. Technol. Cancer Res. Treat. 14, 243–248. doi: 10.7785/tcrt.2012.500412
Jiang, B. (2017). Aerobic glycolysis and high level of lactate in cancer metabolism and microenvironment. Genes Dis. 4, 25–27. doi: 10.1016/j.gendis.2017.02.003
Justus, C. R., Dong, L., Yang, L. V. (2013). Acidic tumor microenvironment and pH-sensing G protein-coupled receptors. Front. Physiol. 4, 354. doi: 10.3389/fphys.2013.00354
Kamisuki, S., Shirakawa, T., Kugimiya, A., Abu-Elheiga, L., Choo, H.-Y. P., Yamada, K., et al. (2011). Synthesis and evaluation of diarylthiazole derivatives that inhibit activation of sterol regulatory element-binding proteins. J. Med. Chem. 54, 4923–4927. doi: 10.1021/jm200304y
Kant, S., Kumar, A., Singh, S. M. (2012). Fatty acid synthase inhibitor orlistat induces apoptosis in T cell lymphoma: role of cell survival regulatory molecules. Biochim. Biophys. Acta 1820, 1764–1773. doi: 10.1016/j.bbagen.2012.07.010
Kant, S., Kumar, A., Singh, S. M. (2014a). Bicarbonate transport inhibitor SITS modulates pH homeostasis triggering apoptosis of Dalton’s lymphoma: implication of novel molecular mechanisms. Mol. Cell. Biochem. 397, 167–178. doi: 10.1007/s11010-014-2184-2
Kant, S., Kumar, A., Singh, S. M. (2014b). Tumor growth retardation and chemosensitizing action of fatty acid synthase inhibitor orlistat on T cell lymphoma: implication of reconstituted tumor microenvironment and multidrug resistance phenotype. Biochim. Biophys. Acta 1840, 294–302. doi: 10.1016/j.bbagen.2013.09.020
Kapoor, K., Finer-Moore, J. S., Pedersen, B. P., Caboni, L., Waight, A., Hillig, R. C., et al. (2016). Mechanism of inhibition of human glucose transporter GLUT1 is conserved between cytochalasin B and phenylalanine amides. Proc. Natl. Acad. Sci. U.S.A. 113, 4711–4716. doi: 10.1073/pnas.1603735113
Kato, Y., Ozawa, S., Miyamoto, C., Maehata, Y., Suzuki, A., Maeda, T., et al. (2013). Acidic extracellular microenvironment and cancer. Cancer Cell Int. 13, 89. doi: 10.1186/1475-2867-13-89
Keenan, M., Chi, J.-T. (2015). Alternative fuels for cancer cells. Cancer J. 21, 49–55. doi: 10.1097/PPO.0000000000000104
Kennedy, K. M., Dewhirst, M. W. (2010). Tumor metabolism of lactate: the influence and therapeutic potential for MCT and CD147 regulation. Future Oncol. 6, 127–148. doi: 10.2217/fon.09.145
Kim, C.-W., Addy, C., Kusunoki, J., Anderson, N. N., Deja, S., Fu, X., et al. (2017). Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: a bedside to bench investigation. Cell Metab. 26, 394–406.e6. doi: 10.1016/j.cmet.2017.07.009
Kim, J. S., Ahn, K. J., Kim, J.-A., Kim, H. M., Lee, J. D., Lee, J. M., et al. (2008). Role of reactive oxygen species-mediated mitochondrial dysregulation in 3-bromopyruvate induced cell death in hepatoma cells: ROS-mediated cell death by 3-BrPA. J. Bioenerg. Biomembr. 40, 607–618. doi: 10.1007/s10863-008-9188-0
Kim, J., Tchernyshyov, I., Semenza, G. L., Dang, C. V. (2006). HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3, 177–185. doi: 10.1016/j.cmet.2006.02.002
Kluckova, K., Bezawork-Geleta, A., Rohlena, J., Dong, L., Neuzil, J. (2013). Mitochondrial complex II, a novel target for anti-cancer agents. Biochim. Biophys. Acta BBA - Bioenerg. 1827, 552–564. doi: 10.1016/j.bbabio.2012.10.015
Ko, Y. H., Pedersen, P. L., Geschwind, J. F. (2001). Glucose catabolism in the rabbit VX2 tumor model for liver cancer: characterization and targeting hexokinase. Cancer Lett. 173, 83–91. doi: 10.1016/S0304-3835(01)00667-X
Ko, Y. H., Verhoeven, H. A., Lee, M. J., Corbin, D. J., Vogl, T. J., Pedersen, P. L. (2012). A translational study “case report” on the small molecule “energy blocker” 3-bromopyruvate (3BP) as a potent anticancer agent: from bench side to bedside. J. Bioenerg. Biomembr. 44, 163–170. doi: 10.1007/s10863-012-9417-4
Kokkonen, N., Khosrowabadi, E., Hassinen, A., Harrus, D., Glumoff, T., Kietzmann, T., et al. (2018). Abnormal golgi pH homeostasis in cancer cells impairs apical targeting of carcinoembryonic antigen by inhibiting its glycosyl-phosphatidylinositol anchor-mediated association with lipid rafts. Antioxid. Redox Signal. doi: 10.1089/ars.2017.7389
Koselny, K., Green, J., Favazzo, L., Glazier, V. E., DiDone, L., Ransford, S., et al. (2016). Antitumor/antifungal celecoxib derivative AR-12 is a non-nucleoside inhibitor of the ANL-family adenylating enzyme acetyl CoA synthetase. ACS Infect. Dis. 2, 268–280. doi: 10.1021/acsinfecdis.5b00134
Kristeleit, R., Davidenko, I., Shirinkin, V., El-Khouly, F., Bondarenko, I., Goodheart, M. J., et al. (2017). A randomised, open-label, phase 2 study of the IDO1 inhibitor epacadostat (INCB024360) versus tamoxifen as therapy for biochemically recurrent (CA-125 relapse)-only epithelial ovarian cancer, primary peritoneal carcinoma, or fallopian tube cancer. Gynecol. Oncol. 146, 484–490. doi: 10.1016/j.ygyno.2017.07.005
Kroemer, G., Pouyssegur, J. (2008). Tumor cell metabolism: cancer’s achilles’ heel. Cancer Cell 13, 472–482. doi: 10.1016/j.ccr.2008.05.005
Krzeslak, A., Wojcik-Krowiranda, K., Forma, E., Jozwiak, P., Romanowicz, H., Bienkiewicz, A., et al. (2012). Expression of GLUT1 and GLUT3 glucose transporters in endometrial and breast cancers. Pathol. Oncol. Res. 18, 721–728. doi: 10.1007/s12253-012-9500-5
Kuchuk, O., Tuccitto, A., Citterio, D., Huber, V., Camisaschi, C., Milione, M., et al. (2018). pH regulators to target the tumor immune microenvironment in human hepatocellular carcinoma. OncoImmunology 0, 1–60. doi: 10.1080/2162402X.2018.1445452
Kumar, A., Kant, S., Singh, S. M. (2013a). Targeting monocarboxylate transporter by α-cyano-4-hydroxycinnamate modulates apoptosis and cisplatin resistance of Colo205 cells: implication of altered cell survival regulation. Apoptosis 18, 1574–1585. doi: 10.1007/s10495-013-0894-7
Kumar, A., Kant, S., Singh, S. M. (2013b). α-Cyano-4-hydroxycinnamate induces apoptosis in Dalton’s lymphoma cells: role of altered cell survival-regulatory mechanisms. Anticancer Drugs 24, 158–171. doi: 10.1097/CAD.0b013e3283586743
Kunjithapatham, R., Geschwind, J.-F. H., Rao, P. P., Boronina, T. N., Cole, R. N., Ganapathy-Kanniappan, S. (2013). Systemic administration of 3-bromopyruvate reveals its interaction with serum proteins in a rat model. BMC Res. Notes 6, 277. doi: 10.1186/1756-0500-6-277
Kurelac, I., Iommarini, L., Vatrinet, R., Amato, L. B., De Luise, M., Leone, G., et al. (2019). Inducing cancer indolence by targeting mitochondrial Complex I is potentiated by blocking macrophage-mediated adaptive responses. Nat. Commun. 10, 903. doi: 10.1038/s41467-019-08839-1
Kwiatkowska, E., Wojtala, M., Gajewska, A., Soszyński, M., Bartosz, G., Sadowska-Bartosz, I. (2016). Effect of 3-bromopyruvate acid on the redox equilibrium in non-invasive MCF-7 and invasive MDA-MB-231 breast cancer cells. J. Bioenerg. Biomembr. 48, 23–32. doi: 10.1007/s10863-015-9637-5
Labak, C. M., Wang, P. Y., Arora, R., Guda, M. R., Asuthkar, S., Tsung, A. J., et al. (2016). Glucose transport: meeting the metabolic demands of cancer, and applications in glioblastoma treatment. Am. J. Cancer Res. 6, 1599–1608.
Lacroix, R., Rozeman, E. A., Kreutz, M., Renner, K., Blank, C. U. (2018). Targeting tumor-associated acidity in cancer immunotherapy. Cancer Immunol. Immunother. CII 67, 1331–1348. doi: 10.1007/s00262-018-2195-z
Lane, H. A., Wood, J. M., McSheehy, P. M. J., Allegrini, P. R., Boulay, A., Brueggen, J., et al. (2009). mTOR inhibitor RAD001 (everolimus) has antiangiogenic/vascular properties distinct from a VEGFR tyrosine kinase inhibitor. Clin. Cancer Res. 15, 1612–1622. doi: 10.1158/1078-0432.CCR-08-2057
Lee, M., Jo, A., Lee, S., Kim, J. B., Chang, Y., Nam, J. Y., et al. (2017a). 3-bromopyruvate and buthionine sulfoximine effectively kill anoikis-resistant hepatocellular carcinoma cells. PloS One 12, e0174271. doi: 10.1371/journal.pone.0174271
Lee, Y., Lee, K. H., Lee, G. K., Lee, S.-H., Lim, K. Y., Joo, J., et al. (2017b). Randomized phase II study of afatinib plus simvastatin versus afatinib alone in previously treated patients with advanced nonadenocarcinomatous non-small cell lung cancer. Cancer Res. Treat. 49, 1001–1011. doi: 10.4143/crt.2016.546
Li, H., Wozniak, A., Sciot, R., Cornillie, J., Wellens, J., Van Looy, T., et al. (2014a). Pazopanib, a Receptor Tyrosine Kinase Inhibitor, Suppresses Tumor Growth through Angiogenesis in Dedifferentiated Liposarcoma Xenograft Models. Transl. Oncol. 7, 665–671. doi: 10.1016/j.tranon.2014.09.007
Li, X., Chen, Y.-T., Hu, P., Huang, W.-C. (2014b). Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol. Cancer Ther. 13, 855–866. doi: 10.1158/1535-7163.MCT-13-0797
Li, Y., Liu, L., Gomez, D., Chen, J., Tong, Z., Palmisano, M., et al. (2018). Pharmacokinetics and safety of Enasidenib following single oral doses in Japanese and Caucasian subjects. Pharmacol. Res. Perspect. 6, e00436. doi: 10.1002/prp2.436
Liberti, M. V., Allen, A. E., Ramesh, V., Dai, Z., Singleton, K. R., Guo, Z., et al. (2019). Evolved resistance to GAPDH inhibition results in loss of the Warburg Effect but retains a different state of glycolysis. bioRxiv 602557. doi: 10.1101/602557
Liberti, M. V., Dai, Z., Wardell, S. E., Baccile, J. A., Liu, X., Gao, X., et al. (2017). A predictive model for selective targeting of the Warburg Effect through GAPDH inhibition with a natural product. Cell Metab. 26, 648–659.e8. doi: 10.1016/j.cmet.2017.08.017
Liberti, M. V., Locasale, J. W. (2016). The Warburg Effect: how does it benefit cancer cells? Trends Biochem. Sci. 41, 211–218. doi: 10.1016/j.tibs.2015.12.001
Lim, S. H., Kim, T. W., Hong, Y. S., Han, S.-W., Lee, K.-H., Kang, H. J., et al. (2015). A randomised, double-blind, placebo-controlled multi-centre phase III trial of XELIRI/FOLFIRI plus simvastatin for patients with metastatic colorectal cancer. Br. J. Cancer 113, 1421–1426. doi: 10.1038/bjc.2015.371
Lin, H.-W., Tseng, C.-H. (2014). A review on the relationship between SGLT2 inhibitors and cancer. Int. J. Endocrinol. 2014, 719578. doi: 10.1155/2014/719578
Lincet, H., Icard, P. (2015). How do glycolytic enzymes favour cancer cell proliferation by nonmetabolic functions? Oncogene 34, 3751–3759. doi: 10.1038/onc.2014.320
Lis, P., Dyląg, M., Niedźwiecka, K., Ko, Y. H., Pedersen, P. L., Goffeau, A., et al. (2016). The HK2 dependent “Warburg Effect” and mitochondrial oxidative phosphorylation in cancer: targets for effective therapy with 3-Bromopyruvate. Molecules 21, 1730. doi: 10.3390/molecules21121730
Logue, J. S., Morrison, D. K. (2012). Complexity in the signaling network: insights from the use of targeted inhibitors in cancer therapy. Genes Dev. 26, 641–650. doi: 10.1101/gad.186965.112
Loo, S. Y., Chang, M. K. X., Chua, C. S. H., Kumar, A. P., Pervaiz, S., Clement, M. V. (2012). NHE-1: a promising target for novel anti-cancer therapeutics. Curr. Pharm. Des. 18, 1372–1382. doi: 10.2174/138161212799504885
Lukey, M. J., Katt, W. P., Cerione, R. A. (2017). Targeting amino acid metabolism for cancer therapy. Drug Discov. Today 22, 796–804. doi: 10.1016/j.drudis.2016.12.003
Luo, X., Cheng, C., Tan, Z., Li, N., Tang, M., Yang, L., et al. (2017). Emerging roles of lipid metabolism in cancer metastasis. Mol. Cancer 16, 76. doi: 10.1186/s12943-017-0646-3
Lyssiotis, C. A., Cantley, L. C. (2014). Acetate fuels the cancer engine. Cell 159, 1492–1494. doi: 10.1016/j.cell.2014.12.009
Lyssiotis, C. A., Kimmelman, A. C. (2017). Metabolic interactions in the tumor microenvironment. Trends Cell Biol. 27, 863–875. doi: 10.1016/j.tcb.2017.06.003
Maan, M., Peters, J. M., Dutta, M., Patterson, A. D. (2018). Lipid metabolism and lipophagy in cancer. Biochem. Biophys. Res. Commun. 504, 582–589. doi: 10.1016/j.bbrc.2018.02.097
Madison, B. B. (2016). Srebp2: a master regulator of sterol and fatty acid synthesis1. J. Lipid Res. 57, 333–335. doi: 10.1194/jlr.C066712
Marín-Hernández, A., Gallardo-Pérez, J. C., Ralph, S. J., Rodríguez-Enríquez, S., Moreno-Sánchez, R. (2009). HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev. Med. Chem. 9, 1084–1101. doi: 10.2174/138955709788922610
Martínez-Reyes, I., Diebold, L. P., Kong, H., Schieber, M., Huang, H., Hensley, C. T., et al. (2016). TCA cycle and mitochondrial membrane potential are necessary for diverse biological functions. Mol. Cell 61, 199–209. doi: 10.1016/j.molcel.2015.12.002
Marusyk, A., Polyak, K. (2010). Tumor heterogeneity: causes and consequences. Biochim. Biophys. Acta 1805, 105. doi: 10.1016/j.bbcan.2009.11.002
Masoud, G. N., Li, W. (2015). HIF-1α pathway: role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 5, 378–389. doi: 10.1016/j.apsb.2015.05.007
Mathupala, S. P., Ko, Y. H., Pedersen, P. L. (2006). Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 25, 4777–4786. doi: 10.1038/sj.onc.1209603
Mboge, M. Y., Mahon, B. P., McKenna, R., Frost, S. C. (2018). Carbonic anhydrases: role in pH control and cancer. Metabolites 8, 19. doi: 10.3390/metabo8010019
McGuire, C., Cotter, K., Stransky, L., Forgac, M. (2016). Regulation of V-ATPase assembly and function of V-ATPases in tumor cell invasiveness. Biochim. Biophys. Acta 1857, 1213–1218. doi: 10.1016/j.bbabio.2016.02.010
McKeown, M. R., Bradner, J. E. (2014). Therapeutic strategies to inhibit MYC. Cold Spring Harb. Perspect. Med. 4, 014266. doi: 10.1101/cshperspect.a014266
Miao, P., Sheng, S., Sun, X., Liu, J., Huang, G. (2013). Lactate dehydrogenase A in cancer: a promising target for diagnosis and therapy. IUBMB Life 65, 904–910. doi: 10.1002/iub.1216
Miranda-Gonçalves, V., Granja, S., Martinho, O., Honavar, M., Pojo, M., Costa, B. M., et al. (2016). Hypoxia-mediated upregulation of MCT1 expression supports the glycolytic phenotype of glioblastomas. Oncotarget 7, 46335–46353. doi: 10.18632/oncotarget.10114
Morales Del Valle, C., Maxwell, J. R., Xu, M. M., Menoret, A., Mittal, P., Tsurutani, N., et al. (2019). Costimulation induces CD4 T cell antitumor immunity via an innate-like mechanism. Cell Rep. 27, 1434–1445.e3. doi: 10.1016/j.celrep.2019.04.016
Moreira, I. S., Fernandes, P. A., Ramos, M. J. (2007). Vascular endothelial growth factor (VEGF) inhibition–a critical review. Anticancer Agents Med. Chem. 7, 223–245. doi: 10.2174/187152007780058687
Morsy, S. M. I., Badawi, A. M., Cecchi, A., Scozzafava, A., Supuran, C. T. (2009). Carbonic anhydrase inhibitors. J. Enzyme Inhib. Med. Chem. 24, 499–505. doi: 10.1080/14756360802218441
Mossmann, D., Park, S., Hall, M. N. (2018). mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 18, 744–757. doi: 10.1038/s41568-018-0074-8
Mowery, Y. M., Pizzo, S. V. (2008). Targeting cell surface F1F0 ATP synthase in cancer therapy. Cancer Biol. Ther. 7, 1836–1838. doi: 10.4161/cbt.7.11.7155
Mu, X., Shi, W., Xu, Y., Xu, C., Zhao, T., Geng, B., et al. (2018). Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle 17, 428–438. doi: 10.1080/15384101.2018.1444305
Mueckler, M., Thorens, B. (2013). The SLC2 (GLUT) family of membrane transporters. Mol. Aspects Med. 34, 121–138. doi: 10.1016/j.mam.2012.07.001
Nakajima, E. C., Van Houten, B. (2013). Metabolic symbiosis in cancer: refocusing the Warburg lens. Mol. Carcinog. 52, 329–337. doi: 10.1002/mc.21863
Nath, S., Devi, G. R. (2016). Three-dimensional culture systems in cancer research: focus on tumor spheroid model. Pharmacol. Ther. 163, 94–108. doi: 10.1016/j.pharmthera.2016.03.013
Newman, A. C., Maddocks, O. D. K. (2017). One-carbon metabolism in cancer. Br. J. Cancer 116, 1499–1504. doi: 10.1038/bjc.2017.118
Nilsson, H., Lindgren, D., Mandahl Forsberg, A., Mulder, H., Axelson, H., Johansson, M. E. (2015). Primary clear cell renal carcinoma cells display minimal mitochondrial respiratory capacity resulting in pronounced sensitivity to glycolytic inhibition by 3-Bromopyruvate. Cell Death Dis. 6, e1585. doi: 10.1038/cddis.2014.545
O’Day, S. J., Eggermont, A. M. M., Chiarion-Sileni, V., Kefford, R., Grob, J. J., Mortier, L., et al. (2013). Final results of phase III SYMMETRY study: randomized, double-blind trial of elesclomol plus paclitaxel versus paclitaxel alone as treatment for chemotherapy-naive patients with advanced melanoma. J. Clin. Oncol. 31, 1211–1218. doi: 10.1200/JCO.2012.44.5585
Orlowski, J., Grinstein, S. (1997). Na+/H+ exchangers of mammalian cells. J. Biol. Chem. 272, 22373–22376. doi: 10.1074/jbc.272.36.22373
Oronsky, B. T., Reid, T., Knox, S. J., Scicinski, J. J. (2012). The scarlet letter of alkylation: a mini review of selective alkylating agents. Transl. Oncol. 5, 226–229. doi: 10.1593/tlo.12187
Orue, A., Chavez, V., Strasberg-Rieber, M., Rieber, M. (2016). Hypoxic resistance of KRAS mutant tumor cells to 3-Bromopyruvate is counteracted by Prima-1 and reversed by N-acetylcysteine. BMC Cancer 16, 902. doi: 10.1186/s12885-016-2930-9
Oudard, S., Carpentier, A., Banu, E., Fauchon, F., Celerier, D., Poupon, M. F., et al. (2003). Phase II study of lonidamine and diazepam in the treatment of recurrent glioblastoma multiforme. J. Neurooncol. 63, 81–86. doi: 10.1023/A:1023756707900
Paiardini, A., Tramonti, A., Schirch, D., Guiducci, G., di Salvo, M. L., Fiascarelli, A., et al. (2016). Differential 3-bromopyruvate inhibition of cytosolic and mitochondrial human serine hydroxymethyltransferase isoforms, key enzymes in cancer metabolic reprogramming. Biochim. Biophys. Acta 1864, 1506–1517. doi: 10.1016/j.bbapap.2016.08.010
Pan, A. C., Borhani, D. W., Dror, R. O., Shaw, D. E. (2013). Molecular determinants of drug–receptor binding kinetics. Drug Discov. Today 18, 667–673. doi: 10.1016/j.drudis.2013.02.007
Pan, Q., Sun, Y., Jin, Q., Li, Q., Wang, Q., Liu, H., et al. (2016). Hepatotoxicity and nephrotoxicity of 3-bromopyruvate in mice. Acta Cir. Bras. 31, 724–729. doi: 10.1590/s0102-865020160110000004
Pandey, S. K., Yadav, S., Temre, M. K., Singh, S. M. (2018). Tracking acetate through a journey of living world: evolution as alternative cellular fuel with potential for application in cancer therapeutic. Life Sci. 215, 86–95 doi: 10.1016/j.lfs.2018.11.004
Patil, M. D., Bhaumik, J., Babykutty, S., Banerjee, U. C., Fukumura, D. (2016). Arginine dependence of tumor cells: targeting a chink in cancer’s armor. Oncogene 35, 4957–4972. doi: 10.1038/onc.2016.37
Patra, K. C., Hay, N. (2014). The pentose phosphate pathway and cancer. Trends Biochem. Sci. 39, 347–354. doi: 10.1016/j.tibs.2014.06.005
Pavlova, N. N., Thompson, C. B. (2016). The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 23, 27–47. doi: 10.1016/j.cmet.2015.12.006
Peppicelli, S., Andreucci, E., Ruzzolini, J., Margheri, F., Laurenzana, A., Bianchini, F., et al. (2017). Acidity of microenvironment as a further driver of tumor metabolic reprogramming. J. Clin. Cell. Immunol. 8, 1–5. doi: 10.4172/2155-9899.1000485
Pérez-Sayáns, M., Somoza-Martín, J. M., Barros-Angueira, F., Rey, J. M. G., García-García, A. (2009). V-ATPase inhibitors and implication in cancer treatment. Cancer Treat. Rev. 35, 707–713. doi: 10.1016/j.ctrv.2009.08.003
Persi, E., Duran-Frigola, M., Damaghi, M., Roush, W. R., Aloy, P., Cleveland, J. L., et al. (2018). Systems analysis of intracellular pH vulnerabilities for cancer therapy. Nat. Commun. 9, 2997. doi: 10.1038/s41467-018-05261-x
Petrova, V., Annicchiarico-Petruzzelli, M., Melino, G., Amelio, I. (2018). The hypoxic tumour microenvironment. Oncogenesis 7, 10. doi: 10.1038/s41389-017-0011-9
Pilon-Thomas, S., Kodumudi, K. N., El-Kenawi, A. E., Russell, S., Weber, A. M., Luddy, K., et al. (2016). Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res. 76, 1381–1390. doi: 10.1158/0008-5472.CAN-15-1743
Pinard, M. A., Boone, C. D., Rife, B. D., Supuran, C. T., McKenna, R. (2013). Structural study of interaction between brinzolamide and dorzolamide inhibition of human carbonic anhydrases. Bioorg. Med. Chem. 21, 7210–7215. doi: 10.1016/j.bmc.2013.08.033
Polański, R., Hodgkinson, C. L., Fusi, A., Nonaka, D., Priest, L., Kelly, P., et al. (2014). Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin. Cancer Res. 20, 926–937. doi: 10.1158/1078-0432.CCR-13-2270
Polyak, K. (2011). Heterogeneity in breast cancer. J. Clin. Invest. 121, 3786–3788. doi: 10.1172/JCI60534
Popat, R., Plesner, T., Davies, F., Cook, G., Cook, M., Elliott, P., et al. (2013). A phase 2 study of SRT501 (resveratrol) with bortezomib for patients with relapsed and or refractory multiple myeloma. Br. J. Haematol. 160, 714–717. doi: 10.1111/bjh.12154
Porta, C., Paglino, C., Mosca, A. (2014). Targeting PI3K/Akt/mTOR signaling in cancer. Front. Oncol. 4, 64. doi: 10.3389/fonc.2014.00064
Quail, D. F., Joyce, J. A. (2013). Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19, 1423–1437. doi: 10.1038/nm.3394
Queirós, O., Preto, A., Pacheco, A., Pinheiro, C., Azevedo-Silva, J., Moreira, R., et al. (2012). Butyrate activates the monocarboxylate transporter MCT4 expression in breast cancer cells and enhances the antitumor activity of 3-bromopyruvate. J. Bioenerg. Biomembr. 44, 141–153. doi: 10.1007/s10863-012-9418-3
Raimundo, N., Baysal, B. E., Shadel, G. S. (2011). Revisiting the TCA cycle: signaling to tumor formation. Trends Mol. Med. 17, 641–649. doi: 10.1016/j.molmed.2011.06.001
Recouvreux, M. V., Commisso, C. (2017). Macropinocytosis: a Metabolic adaptation to nutrient stress in cancer. Front. Endocrinol. 8, 261. doi: 10.3389/fendo.2017.00261
Research, A. A. (2015). Acetyl-CoA synthetase 2 confers a growth advantage under metabolic stress. Cancer Discov. 5, OF10–OF10. doi: 10.1158/2159-8290.CD-RW2015-011
Riemann, A., Schneider, B., Gündel, D., Stock, C., Gekle, M., Thews, O. (2016). Acidosis promotes metastasis formation by enhancing tumor cell motility. Adv. Exp. Med. Biol. 876, 215–220. doi: 10.1007/978-1-4939-3023-4_27
Riess, C., Shokraie, F., Classen, C. F., Kreikemeyer, B., Fiedler, T., Junghanss, C., et al. (2018). Arginine-depleting enzymes - An increasingly recognized treatment strategy for therapy-refractory malignancies. Cell. Physiol. Biochem. 51, 854–870. doi: 10.1159/000495382
Roberts, D. J., Miyamoto, S. (2015). Hexokinase II integrates energy metabolism and cellular protection: akting on mitochondria and TORCing to autophagy. Cell Death Differ. 22, 248–257. doi: 10.1038/cdd.2014.173
Robey, I. F. (2012). Examining the relationship between diet-induced acidosis and cancer. Nutr. Metab. 9, 72. doi: 10.1186/1743-7075-9-72
Romero-Garcia, S., Moreno-Altamirano, M. M. B., Prado-Garcia, H., Sánchez-García, F. J. (2016). Lactate contribution to the tumor microenvironment: mechanisms, effects on immune cells and therapeutic relevance. Front. Immunol. 7, 52. doi: 10.3389/fimmu.2016.00052
Rosenzweig, A., Blenis, J., Gomes, A. P. (2018). Beyond the Warburg Effect: how do cancer cells regulate one-carbon metabolism? Front. Cell Dev. Biol. 6, 90. doi: 10.3389/fcell.2018.00090
Sánchez-García, I. (2009). The crossroads of oncogenesis and metastasis. N. Engl. J. Med. 360, 297–299. doi: 10.1056/NEJMcibr0808031
San-Millán, I., Brooks, G. A. (2017). Reexamining cancer metabolism: lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis 38, 119–133. doi: 10.1093/carcin/bgw127
Santos, C. R., Schulze, A. (2012). Lipid metabolism in cancer. FEBS J. 279, 2610–2623. doi: 10.1111/j.1742-4658.2012.08644.x
Sartori, M., Ceolotto, G., Semplicini, A. (1999). MAPKinase and regulation of the sodium-proton exchanger in human red blood cell. Biochim. Biophys. Acta 1421, 140–148. doi: 10.1016/S0005-2736(99)00121-2
Sato, Y., Nishiofuku, H., Yasumoto, T., Nakatsuka, A., Matsuo, K., Kodama, Y., et al. (2018). Multicenter Phase II clinical trial of sorafenib combined with transarterial chemoembolization for advanced stage hepatocellular carcinomas (Barcelona clinic liver cancer stage C): STAB Study. J. Vasc. Interv. Radiol. 29, 1061–1067. doi: 10.1016/j.jvir.2018.03.020
Sborov, D. W., Haverkos, B. M., Harris, P. J. (2015). Investigational cancer drugs targeting cell metabolism in clinical development. Expert Opin. Investig. Drugs 24, 79–94. doi: 10.1517/13543784.2015.960077
Scafoglio, C., Hirayama, B. A., Kepe, V., Liu, J., Ghezzi, C., Satyamurthy, N., et al. (2015). Functional expression of sodium-glucose transporters in cancer. Proc. Natl. Acad. Sci. U.S.A. 112, E4111–4119. doi: 10.1073/pnas.1511698112
Schaefer, N. G., Geschwind, J. F., Engles, J., Buchanan, J. W., Wahl, R. L. (2012). Systemic administration of 3-bromopyruvate in treating disseminated aggressive lymphoma. Transl. Res. J. Lab. Clin. Med. 159, 51–57. doi: 10.1016/j.trsl.2011.08.008
Schcolnik-Cabrera, A., Chávez-Blanco, A., Domínguez-Gómez, G., Taja-Chayeb, L., Morales-Barcenas, R., Trejo-Becerril, C., et al. (2018). Orlistat as a FASN inhibitor and multitargeted agent for cancer therapy. Expert Opin. Investig. Drugs 27, 475–489. doi: 10.1080/13543784.2018.1471132
Schulze, A., Harris, A. L. (2012). How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 491, 364–373. doi: 10.1038/nature11706
Semenza, G. L. (2010). HIF-1: upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 20, 51–56. doi: 10.1016/j.gde.2009.10.009
Shimanishi, M., Ogi, K., Sogabe, Y., Kaneko, T., Dehari, H., Miyazaki, A., et al. (2013). Silencing of GLUT-1 inhibits sensitization of oral cancer cells to cisplatin during hypoxia. J. Oral Pathol. Med. 42, 382–388. doi: 10.1111/jop.12028
Shiozaki, A., Hikami, S., Ichikawa, D., Kosuga, T., Shimizu, H., Kudou, M., et al. (2018). Anion exchanger 2 suppresses cellular movement and has prognostic significance in esophageal squamous cell carcinoma. Oncotarget 9, 25993–26006. doi: 10.18632/oncotarget.25417
Shoshan, M. C. (2012). 3-Bromopyruvate: targets and outcomes. J. Bioenerg. Biomembr. 44, 7–15. doi: 10.1007/s10863-012-9419-2
Siebels, M., Rohrmann, K., Oberneder, R., Stahler, M., Haseke, N., Beck, J., et al. (2011). A clinical phase I/II trial with the monoclonal antibody cG250 (RENCAREX®) and interferon-alpha-2a in metastatic renal cell carcinoma patients. World J. Urol. 29, 121–126. doi: 10.1007/s00345-010-0570-2
Siebeneicher, H., Bauser, M., Buchmann, B., Heisler, I., Müller, T., Neuhaus, R., et al. (2016). Identification of novel GLUT inhibitors. Bioorg. Med. Chem. Lett. 26, 1732–1737. doi: 10.1016/j.bmcl.2016.02.050
Siegel, A. B., Narayan, R., Rodriguez, R., Goyal, A., Jacobson, J. S., Kelly, K., et al. (2014). A phase I dose-finding study of silybin phosphatidylcholine (milk thistle) in patients with advanced hepatocellular carcinoma. Integr. Cancer Ther. 13, 46–53. doi: 10.1177/1534735413490798
Silva, L. S., Poschet, G., Nonnenmacher, Y., Becker, H. M., Sapcariu, S., Gaupel, A.-C., et al. (2017). Branched-chain ketoacids secreted by glioblastoma cells via MCT1 modulate macrophage phenotype. EMBO Rep. 18, 2172–2185. doi: 10.15252/embr.201744154
Silverman, P. M., Eoyang, L. (1987). Alkylation of acetohydroxyacid synthase I from Escherichia coli K-12 by 3-bromopyruvate: evidence for a single active site catalyzing acetolactate and acetohydroxybutyrate synthesis. J. Bacteriol. 169, 2494–2499. doi: 10.1128/jb.169.6.2494-2499.1987
Singh, D., Arora, R., Kaur, P., Singh, B., Mannan, R., Arora, S. (2017). Overexpression of hypoxia-inducible factor and metabolic pathways: possible targets of cancer. Cell Biosci. 7, 62. doi: 10.1186/s13578-017-0190-2
Sousa, C. M., Biancur, D. E., Wang, X., Halbrook, C. J., Sherman, M. H., Zhang, L., et al. (2016). Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536, 479–483. doi: 10.1038/nature19084
Stein, E., Tallman, M., Pollyea, D. A., Flinn, I. W., Fathi, A. T., Stone, R. M., et al. (2014). Abstract CT103: clinical safety and activity in a phase I trial of AG-221, a first in class, potent inhibitor of the IDH2-mutant protein, in patients with IDH2 mutant positive advanced hematologic malignancies. Cancer Res. 74, CT103–CT103. doi: 10.1158/1538-7445.AM2014-CT103
Sterling, D., Casey, J. R. (2002). Bicarbonate transport proteins. Biochem. Cell Biol. Biochim. Biol. Cell. 80, 483–497. doi: 10.1139/o02-152
Stransky, L., Cotter, K., Forgac, M. (2016). The Function of V-ATPases in Cancer. Physiol. Rev. 96, 1071–1091. doi: 10.1152/physrev.00035.2015
Streicher, R., Kotzka, J., Müller-Wieland, D., Siemeister, G., Munck, M., Avci, H., et al. (1996). SREBP-1 mediates activation of the low density lipoprotein receptor promoter by insulin and insulin-like growth factor-I. J. Biol. Chem. 271, 7128–7133. doi: 10.1074/jbc.271.12.7128
Sukumar, M., Roychoudhuri, R., Restifo, N. P. (2015). Nutrient competition: a new axis of tumor immunosuppression. Cell 162, 1206–1208. doi: 10.1016/j.cell.2015.08.064
Sullivan, L. B., Luengo, A., Danai, L. V., Bush, L. N., Diehl, F. F., Hosios, A. M., et al. (2018). Aspartate is an endogenous metabolic limitation for tumour growth. Nat. Cell Biol. 20, 782–788. doi: 10.1038/s41556-018-0125-0
Sun, J.-M., Lee, K. H., Kim, B.-S., Kim, H.-G., Min, Y. J., Yi, S. Y., et al. (2018). Pazopanib maintenance after first-line etoposide and platinum chemotherapy in patients with extensive disease small-cell lung cancer: a multicentre, randomised, placebo-controlled Phase II study (KCSG-LU12-07). Br. J. Cancer 118, 648–653. doi: 10.1038/bjc.2017.465
Sun, Y., Liu, Z., Zou, X., Lan, Y., Sun, X., Wang, X., et al. (2015). Mechanisms underlying 3-bromopyruvate-induced cell death in colon cancer. J. Bioenerg. Biomembr. 47, 319–329. doi: 10.1007/s10863-015-9612-1
Swietach, P., Vaughan-Jones, R. D., Harris, A. L., Hulikova, A. (2014). The chemistry, physiology and pathology of pH in cancer. Philos. Trans. R. Soc. B Biol. Sci. 369, 20130099. doi: 10.1098/rstb.2013.0099
Takeuchi, T., Schumacker, P. T., Kozmin, S. A. (2015). Identification of fumarate hydratase inhibitors with nutrient-dependent cytotoxicity. J. Am. Chem. Soc. 137, 564–567. doi: 10.1021/ja5101257
Tang, J.-J., Li, J.-G., Qi, W., Qiu, W.-W., Li, P.-S., Li, B.-L., et al. (2011). Inhibition of SREBP by a Small Molecule, Betulin, Improves Hyperlipidemia and Insulin Resistance and Reduces Atherosclerotic Plaques. Cell Metab. 13, 44–56. doi: 10.1016/j.cmet.2010.12.004
Tao, S., Wang, S., Moghaddam, S. J., Ooi, A., Chapman, E., Wong, P. K., et al. (2014). Oncogenic KRAS Confers Chemoresistance by Upregulating NRF2. Cancer Res. 74, 7430–7441. doi: 10.1158/0008-5472.CAN-14-1439
Tarrado-Castellarnau, M., de Atauri, P., Cascante, M. (2016). Oncogenic regulation of tumor metabolic reprogramming. Oncotarget 7, 62726–62753. doi: 10.18632/oncotarget.10911
Thews, O., Riemann, A., Nowak, M., Gekle, M. (2014). Impact of hypoxia-related tumor acidosis on cytotoxicity of different chemotherapeutic drugs in vitro and in vivo. Adv. Exp. Med. Biol. 812, 51–58. doi: 10.1007/978-1-4939-0620-8_7
Valenti, D., Vacca, R. A., de Bari, L. (2015). 3-Bromopyruvate induces rapid human prostate cancer cell death by affecting cell energy metabolism, GSH pool and the glyoxalase system. J. Bioenerg. Biomembr. 47, 493–506. doi: 10.1007/s10863-015-9631-y
Vallés, P. G., Bocanegra, V., Gil Lorenzo, A., Costantino, V. V. (2015). Physiological Functions and Regulation of the Na+/H+ Exchanger [NHE1] in Renal Tubule Epithelial Cells. Kidney Blood Press. Res. 40, 452–466. doi: 10.1159/000368521
Vasseur, S., Guillaumond, F. (2016). LDL Receptor: an open route to feed pancreatic tumor cells. Mol. Cell. Oncol. 3, e1033586. doi: 10.1080/23723556.2015.1033586
Vatrinet, R., Iommarini, L., Kurelac, I., De Luise, M., Gasparre, G., Porcelli, A. M. (2015). Targeting respiratory complex I to prevent the Warburg effect. Int. J. Biochem. Cell Biol. 63, 41–45. doi: 10.1016/j.biocel.2015.01.017
Vishvakarma, N. K., Kumar, A., Singh, V., Singh, S. M. (2013). Hyperglycemia of tumor microenvironment modulates stage-dependent tumor progression and multidrug resistance: implication of cell survival regulatory molecules and altered glucose transport. Mol. Carcinog. 52, 932–945. doi: 10.1002/mc.21922
Vishvakarma, N. K., Singh, S. M. (2010). Immunopotentiating effect of proton pump inhibitor pantoprazole in a lymphoma-bearing murine host: implication in antitumor activation of tumor-associated macrophages. Immunol. Lett. 134, 83–92. doi: 10.1016/j.imlet.2010.09.002
Vishvakarma, N. K., Singh, S. M. (2011). Mechanisms of tumor growth retardation by modulation of pH regulation in the tumor-microenvironment of a murine T cell lymphoma. Biomed. Pharmacother. Biomedecine Pharmacother. 65, 27–39. doi: 10.1016/j.biopha.2010.06.012
Vučetić, M., Cormerais, Y., Parks, S. K., Pouysségur, J. (2017). The Central Role of Amino Acids in Cancer Redox Homeostasis: vulnerability Points of the Cancer Redox Code. Front. Oncol. 7, 319. doi: 10.3389/fonc.2017.00319
Wang, M., Zhao, J., Zhang, L., Wei, F., Lian, Y., Wu, Y., et al. (2017). Role of tumor microenvironment in tumorigenesis. J. Cancer 8, 761–773. doi: 10.7150/jca.17648
Warburg, O. (1956). On the origin of cancer cells. Science 123, 309–314. doi: 10.1126/science.123.3191.309
Watt, M. J., Clark, A. K., Selth, L. A., Haynes, V. R., Lister, N., Rebello, R., et al. (2019). Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci. Transl. Med. 11, eaau5758. doi: 10.1126/scitranslmed.aau5758
Whitton, B., Okamoto, H., Packham, G., Crabb, S. J. (2018). Vacuolar ATPase as a potential therapeutic target and mediator of treatment resistance in cancer. Cancer Med. 7, 3800–3811. doi: 10.1002/cam4.1594
Wilson, J. E. (2003). Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J. Exp. Biol. 206, 2049–2057. doi: 10.1242/jeb.00241
Xie, J., Wang, X., Proud, C. G. (2016). mTOR inhibitors in cancer therapy. F1000Research 5, 2078. doi: 10.12688/f1000research.9207.1
Xu, R.-H., Pelicano, H., Zhou, Y., Carew, J. S., Feng, L., Bhalla, K. N., et al. (2005). Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 65, 613–621.
Xu, W.-Q., Song, L.-J., Liu, Q., Zhao, L., Zheng, L., Yan, Z.-W., et al. (2009). Expression of anion exchanger 1 is associated with tumor progress in human gastric cancer. J. Cancer Res. Clin. Oncol. 135, 1323–1330. doi: 10.1007/s00432-009-0573-9
Xu, X. D., Shao, S. X., Jiang, H. P., Cao, Y. W., Wang, Y. H., Yang, X. C., et al. (2015). Warburg effect or reverse Warburg effect? A review of cancer metabolism. Oncol. Res. Treat. 38, 117–122. doi: 10.1159/000375435
Yadav, S., Kujur, P. K., Pandey, S. K., Goel, Y., Maurya, B. N., Verma, A., et al. (2017a). Antitumor action of 3-bromopyruvate implicates reorganized tumor growth regulatory components of tumor milieu, cell cycle arrest and induction of mitochondria-dependent tumor cell death. Toxicol. Appl. Pharmacol. 339, 52–64. doi: 10.1016/j.taap.2017.12.004
Yadav, S., Pandey, S. K., Goel, Y., Kujur, P. K., Maurya, B. N., Verma, A., et al. (2018). Protective and recuperative effects of 3-bromopyruvate on immunological, hepatic and renal homeostasis in a murine host bearing ascitic lymphoma: implication of niche dependent differential roles of macrophages. Biomed. Pharmacother. 99, 970–985. doi: 10.1016/j.biopha.2018.01.149
Yadav, S., Pandey, S. K., Kumar, A., Kujur, P. K., Singh, R. P., Singh, S. M. (2017b). Antitumor and chemosensitizing action of 3-bromopyruvate: implication of deregulated metabolism. Chem. Biol. Interact. 270, 73–89. doi: 10.1016/j.cbi.2017.04.015
Yadav, S., Pandey, S. K., Singh, V. K., Goel, Y., Kumar, A., Singh, S. M. (2017c). Molecular docking studies of 3-bromopyruvate and its derivatives to metabolic regulatory enzymes: implication in designing of novel anticancer therapeutic strategies. PloS One 12, e0176403. doi: 10.1371/journal.pone.0176403
Yang, L., Zhang, Y. (2017). Tumor-associated macrophages: from basic research to clinical application. J. Hematol. Oncol. 10, 58. doi: 10.1186/s13045-017-0430-2
Yu, L., Chen, X., Sun, X., Wang, L., Chen, S. (2017a). The glycolytic switch in tumors: how many players are involved? J. Cancer 8, 3430–3440. doi: 10.7150/jca.21125
Yu, M., Yongzhi, H., Chen, S., Luo, X., Lin, Y., Zhou, Y., et al. (2017b). The prognostic value of GLUT1 in cancers: a systematic review and meta-analysis. Oncotarget 8, 43356–43367. doi: 10.18632/oncotarget.17445
Yuan, J.-M., Stepanov, I., Murphy, S. E., Wang, R., Allen, S., Jensen, J., et al. (2016). Clinical trial of 2-Phenethyl isothiocyanate as an inhibitor of metabolic activation of a tobacco-specific lung carcinogen in cigarette smokers. Cancer Prev. Res. (Phila.) 9, 396–405. doi: 10.1158/1940-6207.CAPR-15-0380
Zaidi, N., Lupien, L., Kuemmerle, N. B., Kinlaw, W. B., Swinnen, J. V., Smans, K. (2013). Lipogenesis and lipolysis: the pathways exploited by the cancer cells to acquire fatty acids. Prog. Lipid Res. 52, 585–589. doi: 10.1016/j.plipres.2013.08.005
Zhang, Q., Pan, J., Lubet, R. A., Komas, S. M., Kalyanaraman, B., Wang, Y., et al. (2015). Enhanced antitumor activity of 3-bromopyruvate in combination with rapamycin in vivo and in vitro. Cancer Prev. Res. (Phila.) 8, 318–326. doi: 10.1158/1940-6207.CAPR-14-0142
Zhang, W., Guo, C., Jiang, K., Ying, M., Hu, X. (2017). Quantification of lactate from various metabolic pathways and quantification issues of lactate isotopologues and isotopmers. Sci. Rep. 7, 8489. doi: 10.1038/s41598-017-08277-3
Zhang, Y., Fu, R., Liu, Y., Li, J., Zhang, H., Hu, X., et al. (2016a). Dephosphorylation and mitochondrial translocation of cofilin sensitizes human leukemia cells to cerulenin-induced apoptosis via the ROCK1/Akt/JNK signaling pathway. Oncotarget 7, 20655–20668. doi: 10.18632/oncotarget.7994
Zhang, Z., Garzotto, M., Beer, T. M., Thuillier, P., Lieberman, S., Mori, M., et al. (2016b). Effects of ω-3 fatty acids and catechins on fatty acid synthase in the prostate: a randomized controlled trial. Nutr. Cancer 68, 1309–1319. doi: 10.1080/01635581.2016.1224365
Zhao, H., Zhu, W., Xie, P., Li, H., Zhang, X., Sun, X., et al. (2014). A phase I study of concurrent chemotherapy and thoracic radiotherapy with oral epigallocatechin-3-gallate protection in patients with locally advanced stage III non-small-cell lung cancer. Radiother. Oncol. 110, 132–136. doi: 10.1016/j.radonc.2013.10.014
Zhao, J., Zhi, Z., Wang, C., Xing, H., Song, G., Yu, X., et al. (2017). Exogenous lipids promote the growth of breast cancer cells via CD36. Oncol. Rep. 38, 2105–2115. doi: 10.3892/or.2017.5864
Zhao, Y., Wang, L., Pan, J. (2015a). The role of L-type amino acid transporter 1 in human tumors. Intractable Rare Dis. Res. 4, 165–169. doi: 10.5582/irdr.2015.01024
Zhao, Z., Han, F., Yang, S., Wu, J., Zhan, W. (2015b). Oxamate-mediated inhibition of lactate dehydrogenase induces protective autophagy in gastric cancer cells: involvement of the Akt-mTOR signaling pathway. Cancer Lett. 358, 17–26. doi: 10.1016/j.canlet.2014.11.046
Zhou, Z.-H., Song, J.-W., Li, W., Liu, X., Cao, L., Wan, L.-M., et al. (2017). The acid-sensing ion channel, ASIC2, promotes invasion and metastasis of colorectal cancer under acidosis by activating the calcineurin/NFAT1 axis. J. Exp. Clin. Cancer Res. CR 36, 130. doi: 10.1186/s13046-017-0599-9
Zirlik, K., Duyster, J. (2018). Anti-angiogenics: current situation and future perspectives. Oncol. Res. Treat. 41, 166–171. doi: 10.1159/000488087
Keywords: tumor metabolism, tumor microenvironment, metabolic inhibitors, 3-bromopyruvate, organ homeostasis
Citation: Yadav S, Pandey SK, Goel Y, Temre MK and Singh SM (2019) Diverse Stakeholders of Tumor Metabolism: An Appraisal of the Emerging Approach of Multifaceted Metabolic Targeting by 3-Bromopyruvate. Front. Pharmacol. 10:728. doi: 10.3389/fphar.2019.00728
Received: 08 March 2019; Accepted: 05 June 2019;
Published: 04 July 2019.
Edited by:
Simona Rapposelli, University of Pisa, ItalyReviewed by:
Mehdi Damaghi, Moffitt Cancer Center,United StatesIvana Kurelac, University of Bologna, Italy
Copyright © 2019 Yadav, Pandey, Goel, Temre and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sukh Mahendra Singh, smsinghbiotech@bhu.ac.in