 Mariaconcetta Durante1
Mariaconcetta Durante1 Silvia Sgambellone1Cecilia Lanzi2Patrizia Nardini3Alessandro Pini3Flavio Moroni1
Silvia Sgambellone1Cecilia Lanzi2Patrizia Nardini3Alessandro Pini3Flavio Moroni1 Emanuela Masini1
Emanuela Masini1 Laura Lucarini1*
Laura Lucarini1*- 1Section of Pharmacology, Department of Neurosciences, Psychology, Drug Research and Child Health (NEUROFARBA), University of Florence, Florence, Italy
- 2Medical Toxicology Unit, Careggi-University Hospital (AOUC), Florence, Italy
- 3Section of Histology, Department of Clinical and Experimental Medicine, University of Florence, Florence, Italy
Pulmonary fibrosis is the most frequent form of interstitial lung disease. Effective therapies are not yet available; novel therapeutic approaches are needed for counteracting fibrosis. Poly(ADP-ribose) polymerases are enzymes, involved in DNA repair and cell apoptosis. PARP-1 deficient mice exhibited reduced lung fibrosis in response to bleomycin treatment compared to wild-type controls. Histamine H4 receptors (H4Rs) have been recognized as a new target for inflammatory and immune diseases, and H4R ligands reduced inflammation and oxidative stress in lung tissue. The aim of the study was to evaluate the cross-talk between PARP-1 and H4R in a model of bleomycin-induced lung fibrosis in PARP-1−/− and WT mice. Animals were treated with bleomycin or saline by intra-tracheal injection. JNJ7777120, an H4R antagonist, or VUF8430, an H4R agonist, were administered i.p for 21 days. Airway resistance to inflation was evaluated, and lung tissues were processed for PARylated protein content, oxidative stress evaluation, and histology of small bronchi. The levels of pro-inflammatory (IL-1β and TNF-α), regulatory (IL-10), and pro-fibrotic (TGF-β) cytokines were evaluated. The deposition of αSMA was determined by immunofluorescence analysis. The results indicate that JNJ7777120 reduces PARylated protein production, decreases oxidative stress damage, and MPO, a marker for leukocyte tissue infiltration, in PARP-1−/− mice. A significant decrease in the production of both IL-1β and TNF-α and a significant increase in IL-10 levels are observed in mice treated with H4R antagonist, suggesting a crucial anti-inflammatory activity of JNJ7777120. The smooth muscle layer thickness, the goblet cell relative number, and collagen deposition decreased following JNJ7777120 administration. The H4R antagonist treatment also reduces TGF-β production and αSMA deposition, suggesting an important role of JNJ7777120 in airway remodeling. Our results show that PARylation is essential for the pathogenesis of pulmonary fibrosis and propose that PARP-1 and H4Rs are both involved in inflammatory and fibrotic responses. JNJ7777120 treatment, in a condition of PARP-1 inhibition, exerts anti-inflammatory and anti-fibrotic effects, reducing airway remodeling and bronchoconstriction. Therefore, selective inhibition of H4Rs together with non-toxic doses of selective PARP-1 inhibitors could have clinical relevance for the treatment of idiopathic pulmonary fibrosis.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a severe and progressive lung disease with approximately 3 years of median survival from the time of diagnosis and great associated morbidity, with wide-ranging negative effects on quality of life (Swigris et al., 2005). Despite recent advances in understanding the disease pathobiology, IPF remains a disease with a poor prognosis and an incompletely understood pathogenesis. In the last few years, the European Medicines Agency and the Food and Drug Administration (FDA) approved the use of nintedanib and pirfenidone as new safe drugs for IPF treatment. However, effective treatments are not available, and new therapeutic strategies are needed as alternative options when the standards of care are not sufficient or effective (Wynn, 2007; Paz and Shoenfeld, 2010).
Despite an unknown etiology, many studies identified a significant genetic risk associated with the development of IPF, such as SFTPC, MUC5B, and telomerase mutations; however, mechanisms by which genetic risk factors promoting IPF remain unclear (Allen et al., 2017). It has been proposed that pulmonary fibrosis is due to an augmented proliferation of fibroblasts with massive formation and accumulation of extracellular matrix (ECM) proteins (e.g., vimentin, collagen, and fibronectin) that eventually damage lung function (Wilson and Wynn, 2009). Deposition of “scar” tissue in the interstitium of the lung causes an alteration in the homeostatic cross-talk between epithelial and mesenchymal cells. Epithelial cells secrete anti-fibrotic mediators like prostaglandin E2 (PGE2) (Lama et al., 2002); thus, the loss of epithelial cells results in lower levels of PGE2, which in turn, can allow resident fibroblasts to proliferate and differentiate into alpha-smooth muscle actin (αSMA) positive myofibroblasts (Kolodsick et al., 2003). Additionally, the release of the transforming growth factor-β (TGF-β), the most potent pro-fibrotic growth factor, promotes apoptosis of epithelial cells while simultaneously prevents apoptosis in lung fibroblasts (Thannickal and Horowitz, 2006). The apoptosis paradox allows resident fibroblasts to accumulate and become myofibroblasts. Myofibroblasts, organized into agglomerations of cells known as fibroblastic foci, are highly secretory cells producing an excessive tissue matrix, especially collagen, and highly contractile cells causing distortion of the alveolar architecture. When the synthesis of new collagen by myofibroblasts overcomes its degradation rate, pulmonary fibrosis occurs leading to the accumulation of collagen (Wynn, 2008), the common pathological hallmark of fibrotic disorders. This process results in multiple alterations in the lung structure, with progressive thickening of the air-blood membrane and airway stiffening; these lesions impair both gas diffusion and ventilation/perfusion relationship, with reduction or loss of gas exchange capacity (Plantier et al., 2018).
Poly(ADP-ribose) polymerases (PARPs) are enzymes, involved in DNA repair and apoptosis. PARP-1 is the most abundant member of the PARP family and the most widely studied enzyme of this class. PARP-1 is activated upon binding to single- and double-strand DNA breaks via its N-terminal zinc finger domains (Ali et al., 2012; Langelier et al., 2012). Once activated by DNA damage, PARP-1 widely poly(ADP-ribosyl)ates itself and promotes the enrollment of DNA repair proteins that are required for lesion processing and repair. However, when DNA damage is severe, PARP-1 becomes over-activated leading to excessive consumption of NAD+ and consequently to depletion of ATP that results in cellular dysfunction and necrotic cell death.
It has been reported that PARP activation characterizes a key pathway in many pathophysiological conditions associated with inflammation and oxidative stress. Interestingly, genes targeting approaches and the use of non-selective inhibitors have shown that PARP-1 is involved in a number of fibrotic diseases affecting the heart (Pacher et al., 2002), liver (Mukhopadhyay et al., 2014), vessels (Abdallah et al., 2007), and lungs (Genovese et al., 2005). Moreover, recent studies demonstrated that genetic depletion and pharmacological inhibition of PARP-1 reduced pulmonary fibrosis in an animal model of bleomycin-induced lung injury (Hu et al., 2013; Lucarini et al., 2017), suggesting that PARylation is important for myofibroblast differentiation and for the pathogenesis of the disease.
Histamine H4 receptor (H4R) has been identified as a novel target for inflammatory and immune disorders (Gschwandtner et al., 2013). Recent studies demonstrated that H4Rs are involved in the prevention of fibronectin-induced lung fibroblast migration, suggesting these receptors as new targets for the treatment of lung fibrosis (Kohyama et al., 2010). Moreover, H4R antagonists significantly reduce goblet cell hyperplasia and collagen deposition in a model of allergic asthma (Masini et al., 2013) and decrease inflammation and oxidative stress in a bleomycin-induced lung injury model (Rosa et al., 2014; Lucarini et al., 2017).
However, the cellular mechanism underlying the beneficial effects of H4R antagonists in lung diseases are not completely understood, and this topic needs to be investigated in details.
The aim of the present study is to evaluate a possible involvement of PARP-1 in the H4R antagonists’ beneficial effects in reducing airway inflammation and remodeling. For this purpose, we evaluated the effects of the H4R-selective antagonist, JNJ7777120, and of the H4R agonist, VUF8430, in an in vivo model of bleomycin-induced lung fibrosis, in PARP-1 knock-out and in wild-type mice.
Materials and Methods
Drugs and Reagents
Compound JNJ7777120 (JNJ; 1-[(5-chloro-1H-indol-2-yl)carbonyl]-4-methylpiperazine), a potent and selective H4R antagonist, has been used at a concentration of 2.5 mg/kg of body weight (b.wt.). This compound was kindly provided by Dr. Robin Thurmond (Janssen Research & Development, USA). Compound VUF8430, (VUF; 2-[(aminoiminomethyl)amino] ethyl carbamodithioic acid ester, S-(2-guanidylethyl)-isothiourea), an H4R agonist, (Sigma-Aldrich, San Louis, MO, USA), was used at a concentration of 2 mg/kg b.wt. Bleomycin (Merck-Millipore, Burlington, MA, USA) was used at a concentration of 0.05 IU for each mouse and dissolved in 50 μl of saline, in order to obtain the bleomycin-induced model of lung fibrosis. The drug doses and frequency of administrations were selected according to previous papers (Rosa et al., 2014; Lucarini et al., 2016).
Animals
Male wild-type (WT) C57BL/6 mice, approximately weighing 25–30 g and 8–9 weeks old, purchased from a commercial dealer (Harlan, Udine, Italy), and male C57BL/6 PARP-1 deficient mice (PARP-1−/−) (Wang et al., 1995), comparable in mice strain, age, and weight with WT mice, were used for the experiments. All the animals were fed with standard diet and water at libitum and housed under a 12 h light/dark photoperiod at 22°C for at least 48 h before the experiments.
The study protocol complied with the recommendations of the European Economic Community (86/609/CEE) and the Declaration of Helsinki on animal experimentation and was approved by the animal Ethical and Care Committee of the University of Florence (Florence, Italy) and by the Italian Health Ministry (Authorization n 874/2017-PR). Experiments were performed at the Centre for Laboratory Animal Housing and Experimentation (CeSAL) at the University of Florence. The ARRIVE guidelines were considered (McGrath et al., 2010).
Surgery and Treatments
Seventy mice (30 WT and 40 PARP-1−/−) were anesthetized with zolazepam/tiletamine (Zoletil, 50/50 mg/ml, Virbac Srl, Milan, Italy; 50 μg/g, in 100 μl of saline, i.p.); 50 of them (20 WT and 30 PARP-1−/−) were intra-tracheally treated with bleomycin (0.05 IU in 50 μl of saline), and the other 20 (10 WT and 10 PARP-1−/−) were intra-tracheally treated with 50 μl of saline (referred to as non-fibrotic negative controls, Naive).
Ten bleomycin-treated WT and 10 bleomycin-treated PARP-1 deficient mice received two daily intra-peritoneal (i.p.) injections of 100 μl of JNJ solution (2.5 mg/kg; i.p.), after bleomycin administration and for the next 21 days. These are referred to as Bleomycin+JNJ treated groups. Ten bleomycin treated PARP-1−/− mice received two daily intra-peritoneal injection of 100 μl of VUF solution (2 mg/kg; i.p.), after bleomycin administration and for the next 21 days. These are referred to as Bleomycin+VUF treated group. Ten WT mice and 10 PARP-1−/− mice were treated only with vehicle (PBS) and referred to as fibrotic positive controls (Bleomycin+Vehicle).
Functional Assay of Fibrosis
At the end of the treatment period, all mice were subjected to the measurement of airway resistance to inflation and static lung compliance, functional parameters for fibrosis-induced lung stiffness. The measurement is performed using a constant volume mechanical ventilation method with constant number of breaths in a minute; the static compliance determination is performed applying a positive end-expiratory pressure of 3 cm H2O, to mimic spontaneous ventilation (Pini et al., 2010, 2012; Manni et al., 2016). Briefly, a 22-gauge cannula (Venflon 2; Viggo Spectramed, Windlesham, UK, 0.8 mm diameter) was inserted into the trachea of the anesthetized mouse. The animal was ventilated with a small-animal respirator (Ugo Basile, Comerio, Italy) adjusted to deliver a tidal volume of 0.8 ml at a rate of 20 strokes/min. A high-sensitivity pressure transducer (settings: gain 1, chart speed 25 mm/sec.; P75 type 379; Harvard Apparatus Inc., Holliston, MA, USA) connected to a polygraph (Harvard Apparatus Inc. Edenbridge, UK; settings: gain 1, chart speed 25 mm/s) was used to register the changes in lung resistance to inflation, defined as the pressure at airway opening (PAO). Changes in lung resistance to inflation, expressed as millimeters on the chart and registered for at least 3 min, were carried out not less than 40 consecutive tracings of respiratory strokes and then averaged. For static lung compliance determination, multiple linear regression was used to fit pressure and volume in each individual mouse to the linear model of the lung (Manni et al., 2016).
Lung Tissue Sampling
After PAO measurements, the animals were killed with lethal dose of anesthetic drugs. The whole left lungs were removed and fixed with 4% paraformaldehyde in PBS for histological analysis. The right lungs were weighed, quickly frozen, and stored at −80°C. For biochemical measurements, the samples were thawed at 4°C, homogenized on ice in 50 mM Tris-HCl buffer (180 mM KCl and 10 mM EDTA, pH 7.4), and then centrifuged for 30 min at 10,000 g at 4°C, unless otherwise reported. The homogenized supernatants were collected.
Histology and Assessment of Collagen Deposition, Goblet Cell Hyperplasia, and Smooth Muscle Layer Thickness
Six μm thick histological sections were cut from the paraffin-embedded lung samples. In order to minimize artefactual differences in the staining process, all sections were stained in a single session. A digital camera connected to a light microscope equipped with an ×20 objective was used to randomly take photomicrographs of the histological slides. Computer-aided densitometry was performed to obtain quantitative assessment of the stained sections. The free-share ImageJ 1.33 image analysis program1 was used to measure optical density (OD) and surface area. For each measured parameter, values are means ± SEM of the OD measurements (arbitrary units) of individual mouse (five images each) from every experimental groups (tested blind).
The assessment of lung collagen was obtained by staining the histological sections with a simplified Azan method for collagen fibers (Smolle et al., 1996), omitting azocarminium and orange G to reduce parenchymal tissue background. OD measurements of the aniline blue-stained collagen fibers were performed after selection of a correct threshold to eliminate aerial air spaces and bronchial/alveolar epithelium (Formigli et al., 2007). To confirm the assessment of lung collagen, the Picrosirius red staining was carried out; the sections were stained in 0.1% Direct Red 80/Sirius Red F3B (Sigma-Aldrich) in saturated picric acid at room temperature for 1 h, and then they were differentiated in 0.5% acetic acid, prior to dehydration, clearing, and mounting (Lattouf et al., 2014; Marcos-Garcés et al., 2017).
Moreover, lung tissue sections were stained with hematoxylin and eosin or with periodic acid-Schiff (PAS) staining for mucins in order to obtain morphometry of smooth muscle layer thickness and bronchial goblet cell number, respectively, both key markers of airway remodeling. Digital microphotographs of small-sized bronchi were taken randomly. The thickness of the bronchial smooth muscle layer was measured on the digitized images using the above-mentioned software. Total bronchial epithelial cells and PAS-stained goblet cells were counted on bronchial cross-section profiles, and the percentage of goblet cells was calculated.
Western Blot Analysis of PARylated Protein Content
For determination of PARylated protein content in the lung, tissues were homogenized in RIPA buffer plus a cocktail of protease inhibitors and centrifuged at 12,000 g for 5 min. Supernatants were collected, and total protein levels were measured using Micro BCA Protein Assay (ThermoFisher Scientific, Waltham, MA, USA). Thirty μg of proteins were used for Western blot analysis, with a mouse monoclonal anti-PAR(10H) antibody (Alexis Biochemicals, Florence, Italy) diluted 1:1,000 in PBS-T containing 5% non-fat dry milk. The PAR(10H) monoclonal antibody recognizes poly(ADP-ribose) synthesized by PARP enzymes. A suitable peroxidase-conjugated secondary antibody (diluted 1:5,000 in PBS-T containing 5% non-fat dry milk) was used to determine the binding of the primary antibody. The loading transfer of equal amounts of proteins was controlled by reblotting the membrane with an anti-β-actin antibody (diluted 1:20,000 in 5% non-fat dry milk-PBS-T, Sigma-Aldrich). Bands were visualized by enhanced chemiluminescence (Luminata Crescendo Western HRP substrate, Merck Millipore) and quantified by densitometric analysis with the ImageJ software.
Determination of Transforming Growth Factor-β (TGF-β), Interleukin-1β (IL-1β), Tumor Necrosis Factor-α (TNF-α), and Interleukin-10 (IL-10)
The levels of TGF-β, the main profibrotic cytokine involved in fibroblast activation, and two pro-inflammatory cytokines, IL-1β and TNF-α, were measured on aliquots (20 μl) of lung samples homogenated in PBS by using the FlowCytomix assay (Bender Medsystems GmbH, Vienna, Austria), following the protocol provided by the manufacturer. Briefly, suspensions of anti-TGF-β, IL-1β or TNF-α-coated beads were incubated with the supernatant samples and then with biotin-conjugated secondary antibodies and streptavidin-phycoerythrin. Fluorescence was read with a cytofluorimeter (CyFlow® Space, Partec, Carate Brianza, MB, Italy).
The levels of the anti-inflammatory cytokine IL-10 were measured on aliquots (100 μl) of lung homogenate supernatants by using the mouse IL-10 ELISA Ready-SET&Go!® assay (eBioscience, San Diego, USA), following the protocol provided by the manufacturer. Values are indicated as means ± SEM of 10 individual mice from each group and expressed as pg/μg of total proteins determined over an albumin standard curve.
Determination of αSMA Deposition
Immunofluorescence analysis was performed as previously described (Lucarini et al., 2014). Briefly, histological sections of 5 μm thick were deparaffinized and boiled for 10 min in sodium citrate buffer (10 mM, pH 6.0, Bio-Optica, Milan, Italy) for antigen retrieval. A pre-incubation in 1.5% bovine serum albumin (BSA) in PBS, pH 7.4 for 20 min at RT was necessary to minimize the unspecific binding; whereupon, the sections were incubated overnight at 4°C with rabbit monoclonal anti-αSMA antibody (1:200 ABCAM, USA) followed by goat anti-rabbit Alexa Fluor 488-conjugated IgG (1:300 Invitrogen, San Diego, CA, USA) for 2 h in the dark at RT. Negative controls were performed with non-immune rabbit serum substituted for the primary antibody. The counterstaining of nuclei was obtained with 4′,6-diamidino-2-phenylindole (DAPI). Representative images were acquired with an Olympus BX63 microscope coupled to CellSens Dimension Imaging Software version 1.6 (Olympus, Milan, Italy). αSMA expression was quantified by densitometric analysis of fluorescence signal intensity, measured on digitized images using ImageJ software2. Twenty regions of interest (ROI) were evaluated for each sample. Values are expressed as mean ± SEM of the OD measurements (arbitrary units) of individual mouse from the different experimental groups.
Determination of 8-Hydroxy-Deoxyguanosine (8-OHdG)
Lung DNA isolation was performed as previously described (Lodovici et al., 2000) with minor modifications. In brief, lung samples were homogenized in 1 ml of 10 mM PBS, pH 7.4, sonicated on ice for 1 min, added to 1 ml of 10 mM Tris-HCl buffer, pH 8, containing 10 mM EDTA, 10 mM NaCl, and 0.5% SDS, and incubated for 1 h at 37°C with 20 μg/ml RNase 1 (Sigma-Aldrich). Samples were incubated at 37°C overnight in the presence of 100 μg/ml proteinase K (Sigma-Aldrich). The mixture was extracted with chloroform/isoamyl alcohol (10:2, v/v). DNA was precipitated from the aqueous phase with 0.2 volumes of 10 M ammonium acetate, solubilized in 200 μl of 20 mM acetate buffer, pH 5.3, and denatured at 90°C for 3 min. The extract was supplemented with 10 IU of P1 nuclease (Sigma-Aldrich) in 10 μl and incubated for 1 h at 37°C with 5 IU of alkaline phosphatase (Sigma-Aldrich) in 0.4 M phosphate buffer, pH 8.8. All the procedures were performed in the dark. The mixture was filtered by an Amicon Micropure-EZ filter (Merck-Millipore), and 100 μl of each sample were used for 8-OHdG determination by using an ELISA kit (JalCA, Shizuoka, Japan), following the instructions provided by the manufacturer. The absorbance of the chromogenic product was measured at 450 nm. The results were calculated from a standard curve based on an 8-OHdG solution and expressed as ng of 8-OHdG/ng of total DNA.
Determination of Myeloperoxidase Activity (MPO)
Frozen lung samples were weighed and homogenized (10 μl/mg of tissue) in 0.2 M phosphate buffer solution (PBS), pH 6, supplemented with protease inhibitors (1 mM PMSF, 20 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mg/ml Pefabloc SC, and 2.5 μg/ml aprotinin, Sigma-Aldrich) and were centrifuged at 10,000 g at 4°C for 30 min. MPO was measured in the supernatants with a specific immunoassay kit (CardioMPO; PrognostiX, Cleveland, OH), according to the manufacturer’s instructions (Pini et al., 2010). Total protein concentration in the lung tissue samples was determined over an albumin standard curve. The results are expressed as picomoles/mg of protein. Values are means ± SEM of individual mice from different experimental groups.
Statistical Analysis
Data were reported as mean values (±SEM) of individual average measures of the different animals per group, for each assay. Significance of differences among the groups was evaluated by one-way ANOVA followed by Newman-Keuls post hoc test for multiple comparisons. Calculations were made with Prism 5 statistical software (GraphPad Software, Inc., USA). A probability value p < 0.05 was considered significant.
Results
Functional Assay of Fibrosis (PAO)
Intra-tracheal administration of bleomycin causes an increase in airway stiffness leading to a clear-cut elevation of the pressure in airway opening (PAO) (Masini et al., 2005). Intra-tracheal delivery of bleomycin to rodents results in an intense inflammatory reaction within the first week, followed by the development of fibrosis by day 14, with maximal responses at day 21. Therefore, bleomycin administration caused a significant increase in airway stiffness, as judged by the elevation of PAO in the fibrotic positive controls (Bleomycin+Vehicle) compared with the non-fibrotic negative ones (Naïve), both in WT and PARP-1−/− mice (Figure 1).
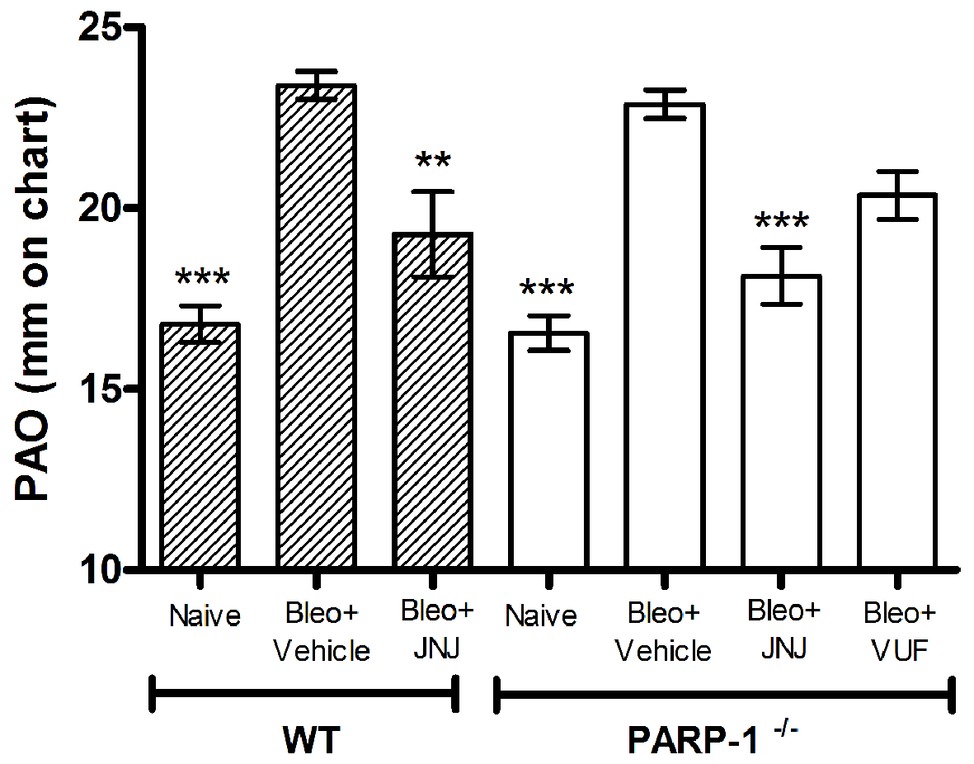
Figure 1. Spirometric evaluation. Bar graph and statistical analysis of differences in PAO values (means ± SEM) between different experimental groups (n = 10 animals per group). **p < 0.01 and ***p < 0.001 vs. Bleo+Vehicle of each related group WT or PARP−/− (Bleo = Bleomycin).
After 21 days, the treatment with JNJ significantly reduces PAO both in WT and in PARP-1−/− mice, in comparison to the relative group of animals treated with Vehicle; while VUF treatment seems to be not significantly effective in reducing the bleomycin-induced airway stiffness (Figure 1). The selected doses were based on previous papers (Pini et al., 2012; Lucarini et al., 2016). These results confirm our previous observation that H4R ligands could have beneficial effects in decreasing lung fibrosis in WT (Lucarini et al., 2016) and in PARP-1−/− animals, indicating that the treatment with H4R antagonists exerts its beneficial effects also in situation of PARP-1 deletion.
Changes in PARP-1 Activity
It has been demonstrated that bleomycin administration increases PARP activity in mouse lung tissues (Genovese et al., 2005). Moreover, previous studies provided evidence that PARylated protein levels, the major products of PARP activity, are significantly increased in lung homogenates of WT mice treated with bleomycin, while the treatment with HYDAMTIQ, a potent PARP-1/2 inhibitor, dose-dependently, prevents this PARylation (Lucarini et al., 2017).
To evaluate the effects of intra-tracheal administration of bleomycin on PARP activity, we investigated the formation of polyADP ribose (PAR) polymers in mouse lung tissues in WT and PARP-1−/− mice. As shown in Figure 2 and in Supplementary Figure 1, Western blot analysis with anti-PAR antibodies report an increase in PARylated protein content in lung homogenates of WT and in PARP-1−/− mice treated with bleomycin (Vehicle), compared to non-fibrotic controls (Naїve). Treatment with the H4R antagonist reduces the PARylated protein content in WT animals. Interestingly, our results indicate a reduction of PARylated protein levels in the lung tissues of PARP-1−/− mice treated with JNJ, compared to those treated with VUF, suggesting a positive role of H4R antagonism in suppressing catalytic activity of PARP enzymes different from PARP-1.
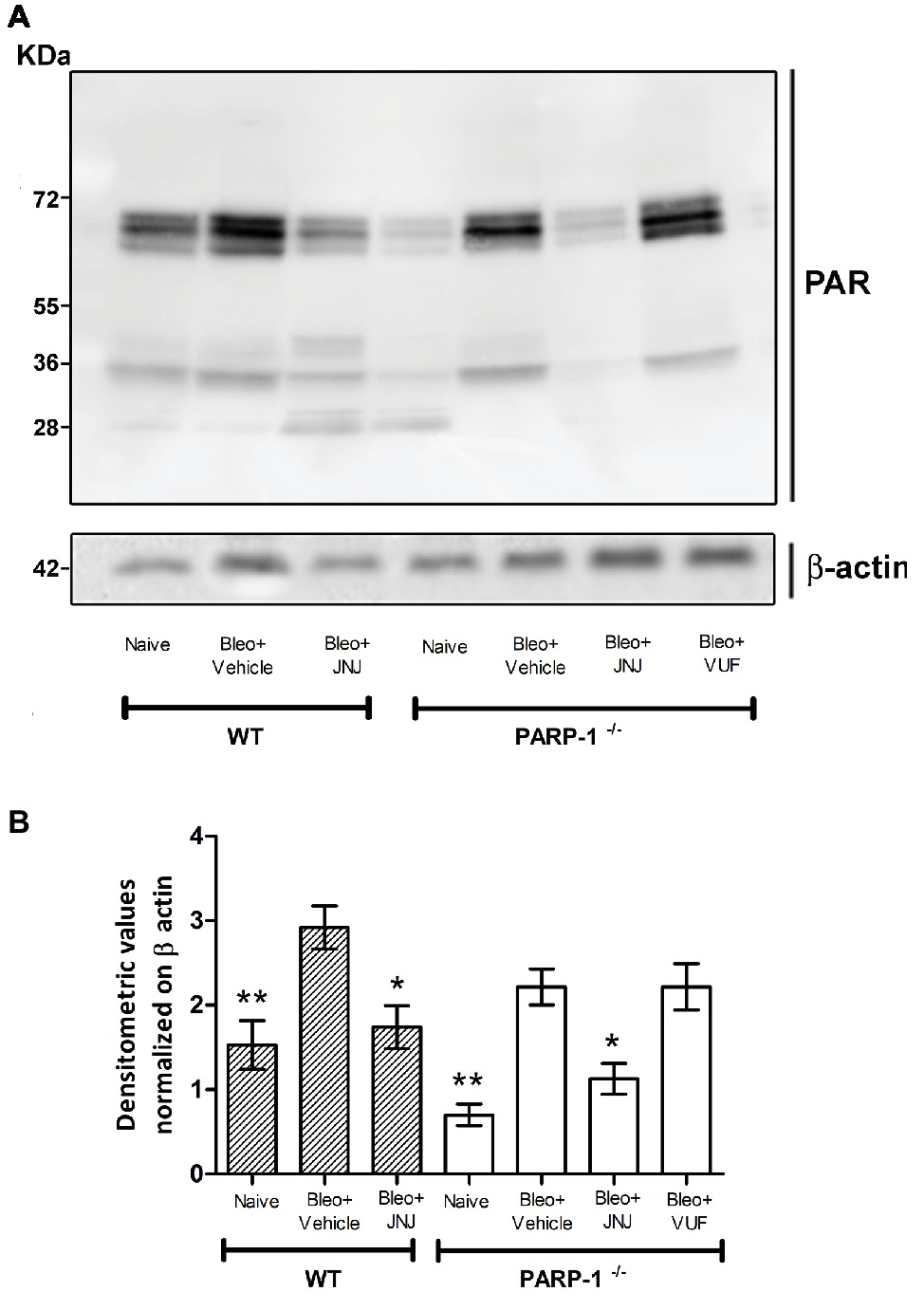
Figure 2. PARP activity. (A) Western blot analysis of PARylated protein content in lung samples from each experimental group. (B) Densitometric analysis was normalized on β-actin (n = 10 animals per group). *p < 0.05 and **p < 0.01 vs. Bleo+Vehicle of each related group WT or PARP−/− (Bleo = Bleomycin).
Histological and Morphometric Analysis
Acute inflammatory events and deposition of collagen fibers cause a pathological remodeling of the parenchyma of the lungs. Morphological observation and computer-aided densitometry on Azan-stained sections (Figure 3A) reveal a significant increase in collagen deposition in the lungs of all bleomycin-treated animals (Vehicle) compared to the non-fibrotic negative controls (Naïve). As already published, in WT animals, H4R antagonists reduce the deposition of collagen fibers in lungs compared to Vehicle (Rosa et al., 2014; Lucarini et al., 2016). The treatment with JNJ causes an important and significant reduction of the amount of lung collagen fibers in PARP-1−/− mice compared to Vehicle group. A slight effect is reported in VUF treated mice. To confirm the assessment of lung collagen, the Picrosirius red was used as second staining method; this specific staining for collagen (Lattouf et al., 2014; Marcos-Garcés et al., 2017) confirms the results obtained with Azan staining (Figure 3B).
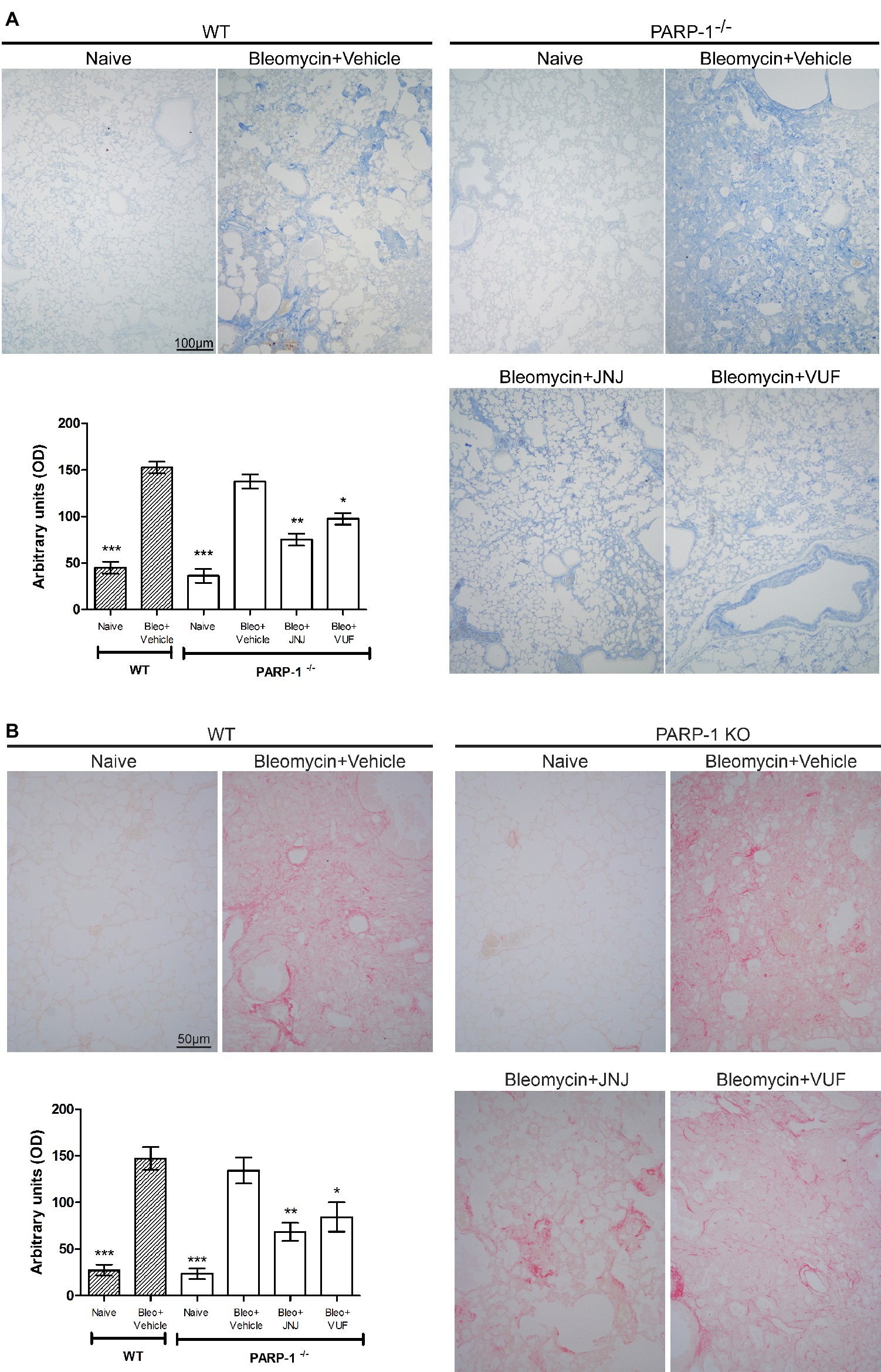
Figure 3. Evaluation of lung fibrosis. (A) Representative micrographs of Azan-stained sections from mice of the different experimental groups. Collagen fibers are stained deep blue. The lung from fibrotic controls treated with vehicle show marked fibrosis in peribronchial stroma, which is absent in non-fibrotic negative control lungs (Naïve) and reduced in JNJ treated animals. Bar graph showing the optical density (OD) (means ± SEM) of Azan-stained collagen fibers of the different experimental groups (n = 10 animals per group). *p < 0.05, **p < 0.01, and ***p < 0.001 vs. Bleomycin+Vehicle of each related group. (B) Representative micrographs of Picrosirius red-stained sections from mice of the different experimental groups. Collagen fibers are stained in red. Bar graph showing the optical density (OD) (means ± SEM) of Picrosirius red-stained collagen fibers of the different experimental groups (n = 10 animals per group). *p < 0.05, **p < 0.01, and ***p < 0.001 vs. Bleomycin+Vehicle of each related group.
We also evaluated the bronchial remodeling by measuring the number of goblet cells and the thickness of the smooth muscle layer, key histological parameters of inflammation-induced adverse airway remodeling (Bai and Knight, 2005). These two parameters are increased after intra-tracheal injection of bleomycin, both in WT and PARP-1 deficient mice. The treatment with JNJ significantly reduces the percentage of PAS-positive goblet cells over bronchial epithelial cells (Figure 4A), as well as the thickness of the airway smooth muscle layer (Figure 4B). On the contrary, VUF administration has no effect on both histological parameters, confirming our previous published results that on the effect of H4R antagonists on goblet cells and thickness of the smooth muscle layer in lungs, (Rosa et al., 2014; Lucarini et al., 2016).
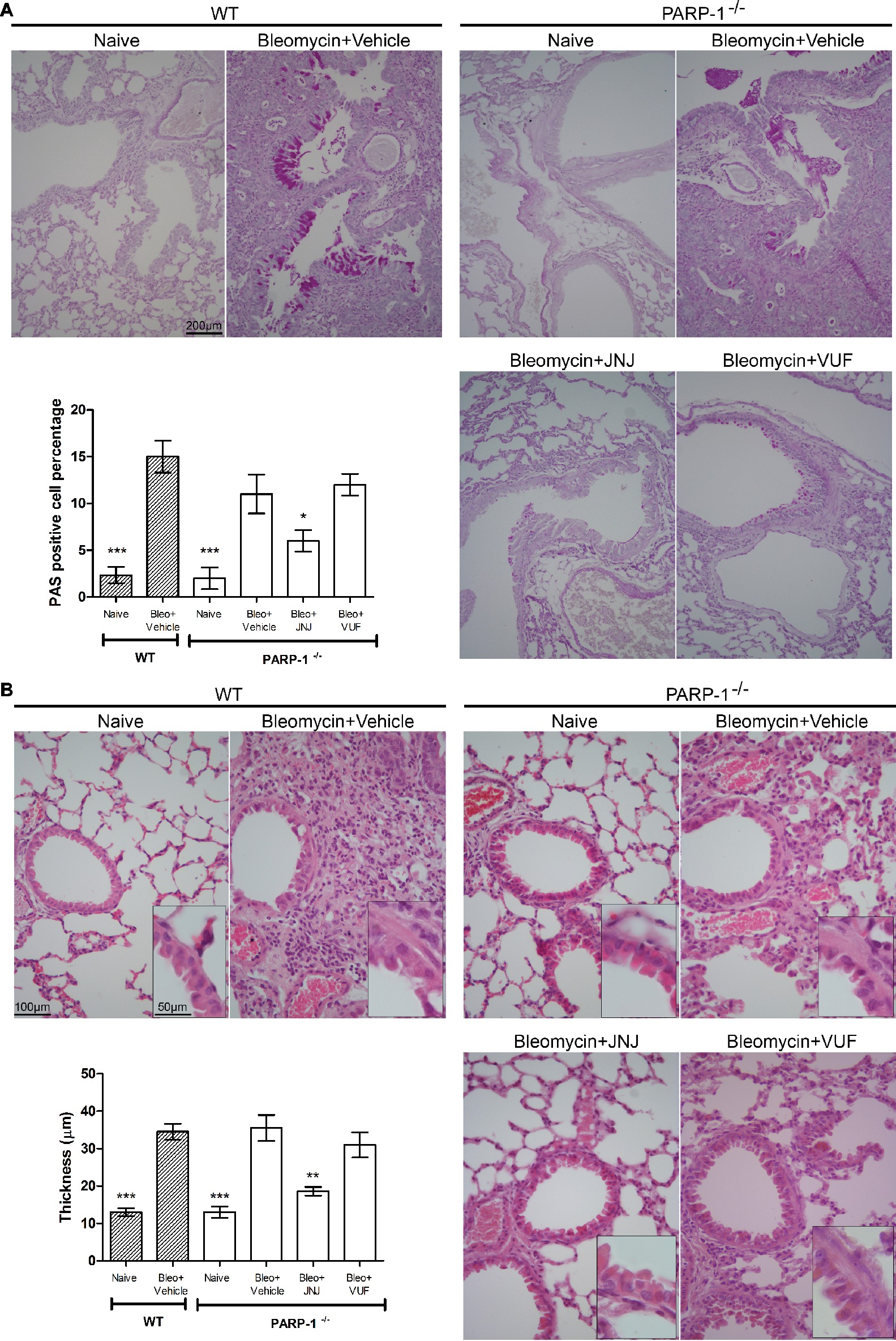
Figure 4. (A) Goblet cell hyperplasia. Representative micrographs of PAS-stained sections. Bar graph showing fraction of goblet cells (% means ± SEM) in the different experimental groups (n = 10 animals per group). *p < 0.05 and ***p < 0.001 of vs. Bleo+Vehicle of each related group. (B) Evaluation of muscular remodeling. Smooth muscle thickness was assessed by computer aided morphometry on H&E-stained lung sections. Representative micrographs of the sections. Bar graph showing the thickness of the muscular fiber (means ± SEM) in the different experimental groups (n = 10 animals per group). **p < 0.01 and ***p < 0.001 vs. Bleo+Vehicle of each related group.
TGF-β Signaling Pathway
An increased TGF-β expression and an amplified TGF-β signaling through Smad pathways contribute to the establishment and development of pulmonary fibrosis (Leask and Abraham, 2004; Chen et al., 2013; Chitra et al., 2015). Our results clearly indicate that TGF-β, a major pro-fibrotic cytokine, is increased in the vehicle groups, both in WT and PARP-1−/− mice. Systemic administration of JNJ causes a significant decrease of the levels of TGF-β in WT as well as PARP-1−/− animals, while the treatment with the H4R agonist has no effect on the production of this cytokine (Figure 5A). These results demonstrate the anti-fibrotic activity of JNJ both in WT and in PARP-1−/− mice.
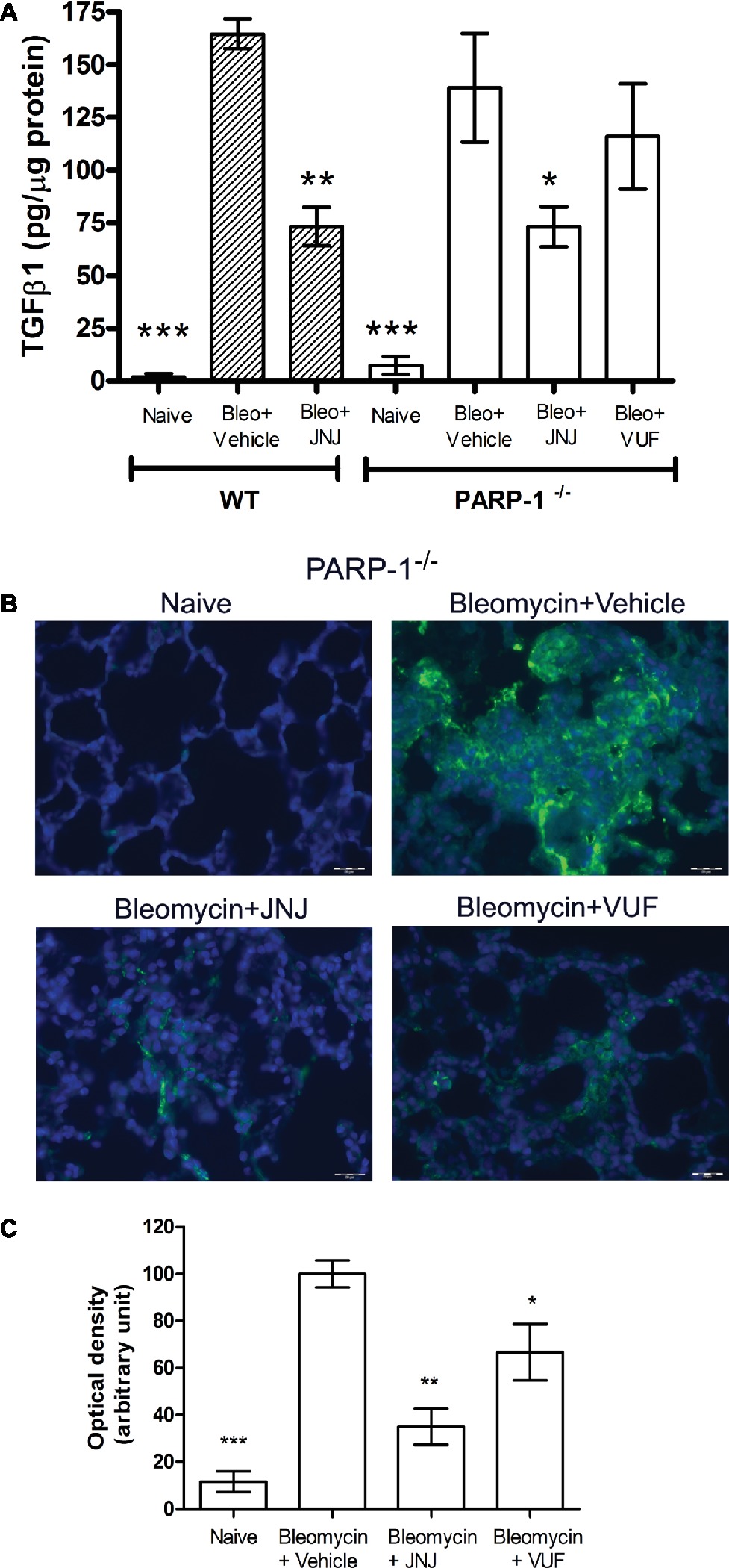
Figure 5. (A) Evaluation of TGF-β1 production. Bar graph showing lung tissue levels of the profibrotic cytokine TGFβ1 (means ± SEM) of the different experimental groups (n = 10 animals per group). *p < 0.05, **p < 0.01, and ***p < 0.001 vs. Bleo+Vehicle of each related group. (B) Evaluation of fibroblast activation. Immunofluorescence staining of lung tissue sections labeled with alpha smooth muscle actin (αSMA) (green) and nuclei (blue) counterstained with DAPI (20×). Images in the panels show the inhibition of αSMA expression, a marker of the transformation of fibroblasts into myofibroblasts (n = 10 animals/group). (C) Bar graph shows the optical density (means ± SEM) in the different experimental groups (n = 10 animals per group). *p < 0.05 and **p < 0.01 vs. Bleo+Vehicle. (Bleo = Bleomycin).
Fibroblast Activation
Transforming growth factor-β signaling has been reported to regulate the expression of αSMA, a marker of fibroblast activation, and myofibroblasts differentiation (Martin et al., 2007; Conte et al., 2014). To investigate the expression of αSMA during the fibrotic process, we performed immunofluorescence analysis in mouse lung tissues. Our results show a significant increase of αSMA levels in bleomycin-exposed PARP-1−/− animals (Figures 5B,C). Treatment with JNJ significantly reduces αSMA levels in lung tissue of PARP-1−/− mice compared to the vehicle group. Systemic administration of the H4R agonist provides no changes in αSMA lung levels. These results indicate that the use of an H4R antagonist, in the absence of PARP-1 enzyme, reduces the activation of fibroblasts and myofibroblasts differentiation and consequently the development of progressive fibrotic disease in bleomycin-exposed animals.
Effects of H4R Ligands on Pro-Inflammatory Cytokine Production
Chronic inflammation is determined by pathological wound-healing response, resulting in the accumulation of permanent scar tissue at the site of injury and consequently in the development of progressive fibrotic disease (Wynn and Ramalingam, 2012). Based on this evidence, we evaluated the late (21 days after challenge) inflammatory response to bleomycin by measuring the pro-inflammatory cytokines IL-1β and TNF-α, in lung homogenates of WT and PARP-1−/− mice. Bleomycin treatment increases the levels of IL-1β and TNF-α both in WT and PARP-1−/− mice; of note, in PARP-1−/− mice, this increase is less pronounced in comparison to WT mice. The systemic administration of JNJ significantly reduces the increase in both groups of animals, but in PARP-1−/− mice, this reduction is more evident. The treatment with VUF is not effective in the reduction of inflammatory cytokine production (Figures 6A,B).
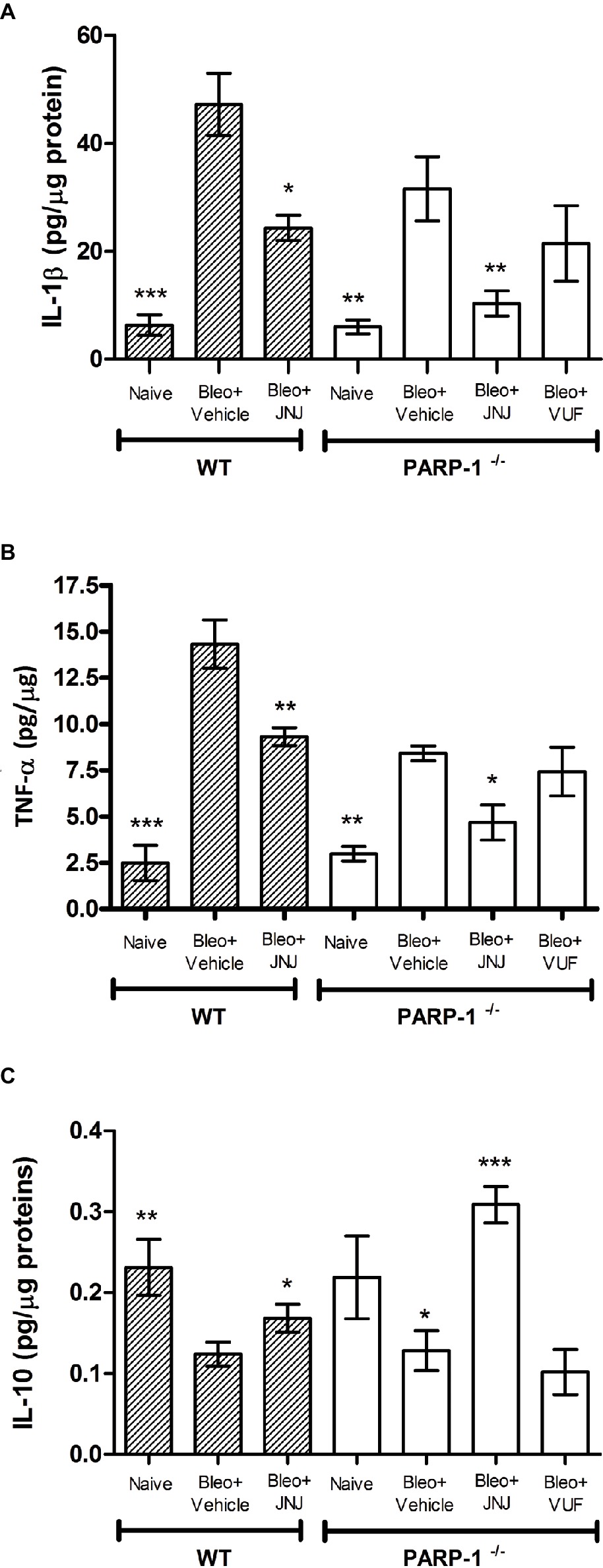
Figure 6. Determination of IL-1β (A), TNF-α (B), and IL-10 (C) levels in the supernatant of lung tissue homogenates. The values are expressed as pg/μg of total proteins (n = 10 animals per group). Values are mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 vs. Bleo+Vehicle of each related group (Bleo = Bleomycin).
In order to confirm the anti-inflammatory activity of the H4R antagonist, we evaluated the levels of interleukin-10 (IL-10), the most potent anti-inflammatory cytokine involved in resolution of different acute and chronic inflammatory diseases (Ajuebor et al., 1999). Our results report a significant increase in the production of IL-10 in WT and PARP-1−/− mice treated with JNJ in comparison to PARP-1−/− mice treated with vehicle or VUF (Figure 6C).
Our findings support the hypothesis that H4R antagonism exerts anti-inflammatory and anti-fibrotic effects in a model of bleomycin-induced lung fibrosis.
Determination of Leukocyte Lung Infiltration and of Oxidative Stress Marker
Lung MPO is a peroxidase enzyme abundantly expressed in neutrophils and monocytes/macrophages granules, and it is considered a reliable marker for leukocyte accumulation in inflamed tissues (Mullane et al., 1985). Levels of MPO were very low in the Naïve groups of both WT and PARP−/− mice; MPO levels increased significantly in the bleomycin-treated mice (Vehicle groups of WT and PARP−/− mice). A significant decrease in MPO was demonstrated after treatment with JNJ in WT and PARP−/− groups of animals in comparison to Vehicle; treatment with VUF has some non-significant effects (Figure 7A).
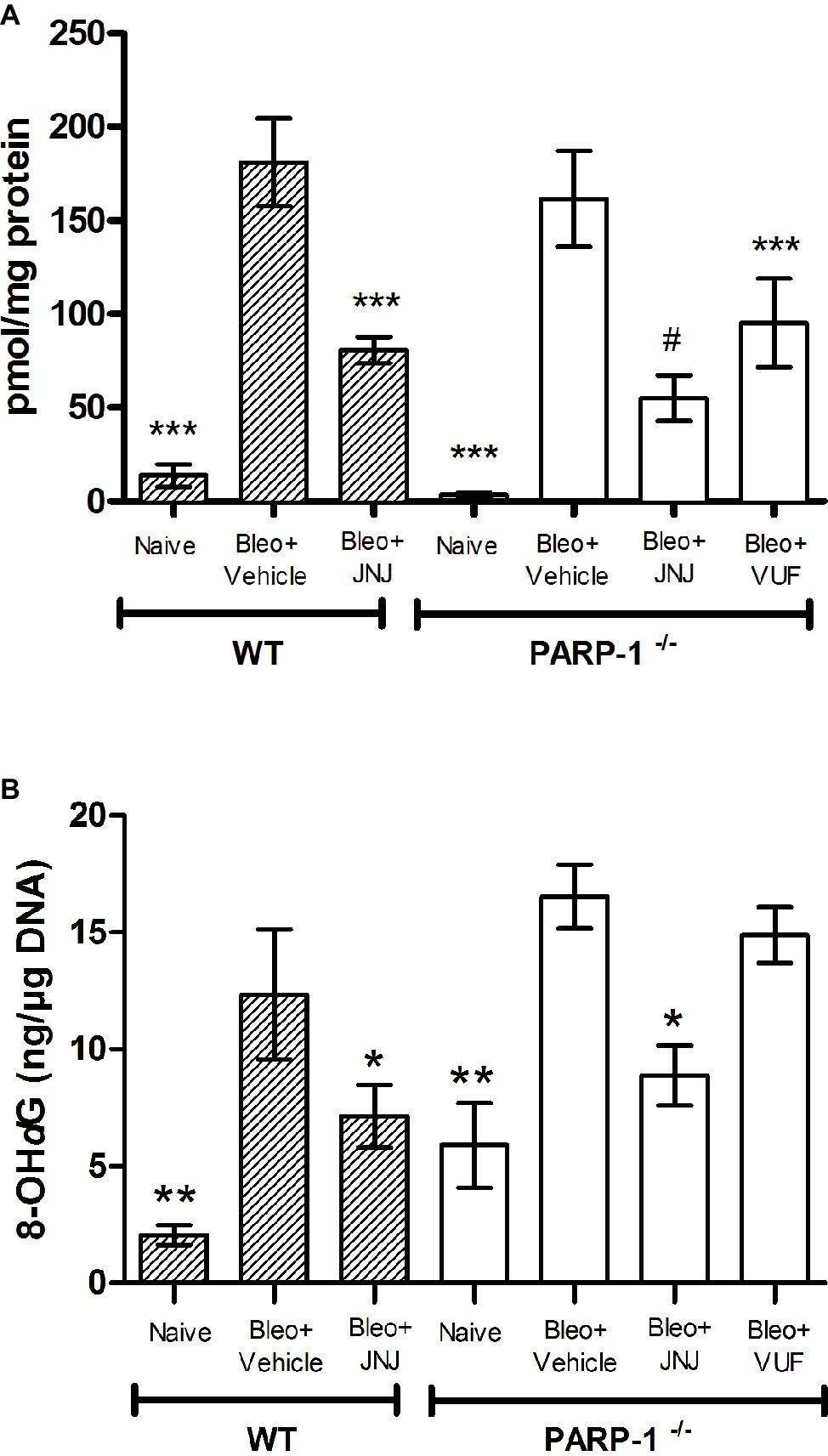
Figure 7. (A) MPO levels, a marker for leukocyte infiltration, are shown in the histogram. Values are expressed as mean ± SEM, expressed as picomoles per milligram of lung tissue protein (n = 10 animals per group) ***p < 0.001 vs. Bleo+Vehicle of each related group (Bleo = Bleomycin); #p < 0.05 vs. Bleo+VUF. (B) Evaluation of oxidative stress parameter in lung tissue. Bar graph shows the levels of 8-OHdG, a marker of free radicals-induced by DNA damage, (mean ± SEM) in the different experimental groups (n = 10 animals per group). *p < 0.05, **p < 0.01 vs. Bleo+Vehicle of each related group (Bleo = Bleomycin).
The determination of 8-OHdG, a biological marker of DNA damage under oxidative stress, demonstrates that it is significantly increased in bleomycin-exposed animals (Vehicle), compared with non-fibrotic negative ones (Naïve). Interestingly, our results show a significant reduction of 8-OHdG levels in WT and PARP-1−/− animals treated with JNJ (Figure 7B), but not with VUF.
Discussion
Pulmonary fibrosis is an unmet medical need with a median survival of ~3 years since diagnosis. It is often severe and difficult to manage, resulting in a chronic condition that negatively affects the quality of life. The pharmacological therapy of pulmonary fibrosis is challenging, and for many patients, effective treatment is lacking. Therefore, novel therapeutic strategies are required (Wynn, 2007; Paz and Shoenfeld, 2010).
Recent studies demonstrated the involvement of PARP enzymes in modulating airway inflammation and fibrosis. Particularly, PARP-1 inhibition improves functional, biochemical, and morphometric parameters in an in vivo allergen-induced asthma-like reaction model (Lucarini et al., 2014) and in a bleomycin-induced pulmonary fibrosis model (Lucarini et al., 2017).
Histamine H4 receptor (H4R), the last discovered histamine receptor subtype, is functionally expressed and distributed in white blood cells, mast cells, eosinophils, dendritic cells, and T cells (Gutzmer et al., 2009). Recent evidence strongly suggests that H4R ligands might be exploited as potential therapeutics in modulating allergy, inflammation, autoimmune disorders, and possibly cancer. In an animal model of allergic airway inflammation, H4R-knockout mice present lower inflammation, reduced pulmonary infiltrate of lymphocytes and eosinophils, and an attenuated Th2 response (Dunford et al., 2006). Moreover, blocking H4R in a model of pulmonary fibrosis alleviates the inflammatory response, reducing COX-2 expression and activity, leukocyte infiltration, TGF-β production, and collagen deposition (Lucarini et al., 2016).
Although these findings suggest a promising therapeutic use for H4R antagonists in the modulation of chronic lung diseases, the biochemical basis underlying the protective and beneficial effects of these drugs are unknown and need to be further elucidated.
Here, we tested the possible role that PARP-1 may have in the mechanism of action of H4R antagonists. Briefly, we demonstrate that JNJ, a selective antagonist of the histamine H4R, exerts its anti-inflammatory and anti-fibrotic properties independently of PARP-1 signaling pathway, in an in vivo mouse model of bleomycin-induced lung fibrosis.
Chronic inflammation is determined by pathological wound-healing response, resulting in the accumulation of scar tissue and consequently in the development of progressive fibrotic disease (Wynn and Ramalingam, 2012). Our model of pulmonary fibrosis is well described and widely accepted and includes an acute inflammatory phase (7–9 days), followed by a chronic inflammatory infiltrate and fibrotic process in the next 2 weeks, with maximal responses at day 21 (Moore and Hogaboam, 2008).
Therefore, we evaluated the inflammatory response to bleomycin in lung homogenates of WT and PARP-1−/− mice. The results show that the treatment with JNJ significantly reduces the production of IL-1β and TNF-α pro-inflammatory cytokines and the activity of MPO, as well as the levels of 8-OHdG, a reliable marker of oxidative stress, in WT and in PARP-1−/− mice. Collectively, our results demonstrate that H4R antagonist’s anti-inflammatory and anti-fibrotic effects are not dependent on PARP-1 expression, thus adding further evidence on the role of H4R in controlling leukocyte trafficking and pro-inflammatory responses (Zampeli and Tiligada, 2009).
Previous data demonstrated a role for histamine and H4R in the production of TGF-β, the major pro-fibrotic cytokine, and in fibroblast activation (Cowden et al., 2010). The cytokine TGF-β has been proposed to play a key role in lung fibrosis (Tatler and Jenkins, 2012), and drugs able to control TGF-β expression and/or signaling seem to be active in reducing fibroblast activation and clinical progression of the disease (Bonniaud et al., 2005). The TGF-β overexpression has been repeatedly associated to lung fibrosis (Sime et al., 1997), and the administration of TGF-β neutralizing antibodies was able to prevent the disease.
Here, we report the positive effects of the H4R antagonist on the lung TGF-β pathway, showing that the treatment with JNJ significantly reduces TGF-β levels also in PARP-1−/− mice. Thus, our results indicate that H4R antagonists can ameliorate lung fibrosis independently of PARP-1 expression. To further confirm the effects of the H4R antagonist JNJ, we performed experiments also with VUF8430, introduced as a selective H4R agonist. Surprisingly, in our model, VUF has some unexpected effects in reducing features of lung injury, in functional assay as well as α-SMA and collagen deposition. This discrepancy could be explained because this H4R agonist with minor modification on imidazole moiety has affinity also for H3R (Lim et al., 2009), thus reducing the endogenous release of histamine and H4R activation.
PARP-1 activity contributes to lung fibroblast activation and induces their proliferation with increased expression of αSMA, which plays a pivotal role in lung fibrosis (Hu et al., 2013). In order to confirm the above results, we demonstrate a significant reduction of αSMA levels in PARP-1−/− mice treated with JNJ, suggesting that the use of an H4R antagonist, in the absence of PARP-1 enzyme, strongly reduces the activation of fibroblasts and the differentiation of myofibroblasts, consequently blocking the development of progressive fibrotic disease.
Overall, these findings suggest that PARylation is a key factor for the pathogenesis of pulmonary fibrosis and provide evidence that PARP-1 and H4R are independently involved in the signaling pathways activated during inflammatory and fibrotic processes. The association of PARP-1 deficiency and H4R antagonist treatment exerts a cross-talk response with anti-inflammatory and anti-fibrotic effects, decreasing bronchoconstriction and airway inflammation, as also shown by the reduction of the percentage number of goblet cells and the thickness of the smooth muscle layer, key parameters of inflammation-induced adverse airway remodeling.
Actually, the treatment of idiopathic pulmonary fibrosis is based on the use of either pirfenidone, a drug able to reduce the production of fibrogenic mediators, such as TGF-β, and inflammatory mediators, such as TNFα and IL-1β (Inomata et al., 2015), or nintedanib, a tyrosine kinase inhibitor able to reduce the transduction pathway of growth factor receptors, such as platelet-derived growth factor receptor, fibroblast growth factor receptor, and vascular endothelial growth factor receptor, and to reduce the transduction pathways leading to cell activation and proliferation (Nanthakumar et al., 2015). Both agents have recently been introduced into clinical practice showing their ability to ameliorate lung fibrotic processes; however, it is still not clear whether these agents have a clinically meaningful efficacy in long-term patient survival (Karimi-Shah and Chowdhury, 2015). Effective therapies to contrast airway inflammation and remodeling are not available, and novel therapeutic strategies are needed as alternative options when the standards of care are not enough.
In this study, we showed that the beneficial effects of H4R antagonists in reducing progressive pulmonary fibrosis are not dependent upon PARP-1.
In conclusion, the therapeutic potential of the combination of H4R antagonists with non-toxic doses of selective PARP-1 inhibitors could significantly reduce the development of pulmonary fibrosis. Although JNJ itself is emerging as a promising therapeutic agent in lung inflammation, the combination with PARP inhibitors could have an advantage over the single drug for the potentiating effect on the inhibition of inflammatory and pro-fibrotic parameters.
Ethics Statement
The study protocol complied with the Declaration of Helsinki and the recommendations of the European Economic Community (86/609/CEE) on animal experimentation and was approved by the animal Ethical and Care Committee of the University of Florence (Florence, Italy) and by the Health Ministry (Authorization n 874/2017-PR). Experiments were carried out at the Centre for Laboratory Animal Housing and Experimentation (CeSAL), University of Florence. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (McGrath et al., 2010).
Author Contributions
LL, MD, CL, and EM designed the research study. LL, MD, CL, SS, PN, and AP performed the research. AP and LL analyzed the data. LL, MD, and EM wrote the paper. FM revised the scientific content of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Prof. Alberto Chiarugi, University of Florence, for providing us with PARP-1-/- mice, and Dr. Robin Thurmond (Janssen Research & Development, USA) for supplying the compound JNJ7777120.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/article/10.3389/fphar.2019.00525/full#supplementary-material
Abbreviations
αSMA, Alpha-smooth muscle actin; ECM, Extracellular matrix; 8-OHdG, 8-Hydroxy-deoxy-guanosine; H4R, Histamine H4 receptor; IL-10, Interleukin-10; IL-1β, Interleukin-1β; i.p., Intra-peritoneal; IPF, Idiopathic pulmonary fibrosis; JNJ, JNJ7777120; MPO, Myeloperoxidase; OD, Optical density; PARP, Poly(ADP-ribose) polymerase; PAO, Pressure at the airway opening; PAS, Periodic acid-Schiff; PGE2, Prostaglandin E2; TGF-β, Transforming growth factor-β; TNF-α, Tumor necrosis factor-α; WT, Wild-type; VUF, VUF8430.
Footnotes
References
Abdallah, Y., Gligorievski, D., Kasseckert, S. A., Dieterich, L., Schäfer, M., Kuhlmann, C. R., et al. (2007). The role of poly(ADP-ribose) polymerase (PARP) in the autonomous proliferative response of endothelial cells to hypoxia. Cardiovasc. Res. 73, 568–574. doi: 10.1016/j.cardiores.2006.11.027
Ajuebor, M. N., Das, A. M., Virág, L., Flower, R. J., Szabó, C., and Perretti, M. (1999). Role of resident peritoneal macrophages and mast cells in chemokine production and neutrophil migration in acute inflammation: evidence for an inhibitory loop involving endogenous IL-10. J. Immunol. 162, 1685–1691. Available at: http://www.ncbi.nlm.nih.gov/pubmed/9973430 [Accessed January 14, 2019]
Ali, A. A. E., Timinszky, G., Arribas-Bosacoma, R., Kozlowski, M., Hassa, P. O., Hassler, M., et al. (2012). The zinc-finger domains of PARP1 cooperate to recognize DNA strand breaks. Nat. Struct. Mol. Biol. 19, 685–692. doi: 10.1038/nsmb.2335
Allen, R. J., Porte, J., Braybrooke, R., Flores, C., Fingerlin, T. E., Oldham, J. M., et al. (2017). Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: a genome-wide association study. Lancet Respir. Med. 5, 869–880. doi: 10.1016/S2213-2600(17)30387-9
Bai, T. R., and Knight, D. A. (2005). Structural changes in the airways in asthma: observations and consequences. Clin. Sci. 108, 463–477. doi: 10.1042/CS20040342
Bonniaud, P., Margetts, P. J., Ask, K., Flanders, K., Gauldie, J., and Kolb, M. (2005). TGF-beta and Smad3 signaling link inflammation to chronic fibrogenesis. J. Immunol. 175, 5390–5395. doi: 10.4049/jimmunol.175.8.5390
Chen, Y.-L., Zhang, X., Bai, J., Gai, L., Ye, X.-L., Zhang, L., et al. (2013). Sorafenib ameliorates bleomycin-induced pulmonary fibrosis: potential roles in the inhibition of epithelial–mesenchymal transition and fibroblast activation. Cell Death Dis. 4:e665. doi: 10.1038/cddis.2013.154
Chitra, P., Saiprasad, G., Manikandan, R., and Sudhandiran, G. (2015). Berberine inhibits Smad and non-Smad signaling cascades and enhances autophagy against pulmonary fibrosis. J. Mol. Med. 93, 1015–1031. doi: 10.1007/s00109-015-1283-1
Conte, E., Gili, E., Fagone, E., Fruciano, M., Iemmolo, M., and Vancheri, C. (2014). Effect of pirfenidone on proliferation, TGF-b-induced myofibroblast differentiation and fibrogenic activity of primary human lung fibroblasts. Eur. J. Pharm. Sci. 58, 13–19. doi: 10.1016/j.ejps.2014.02.014
Cowden, J. M., Riley, J. P., Ma, J. Y., Thurmond, R. L., and Dunford, P. J. (2010). Histamine H4 receptor antagonism diminishes existing airway inflammation and dysfunction via modulation of Th2 cytokines. Respir. Res. 11:86. doi: 10.1186/1465-9921-11-86
Dunford, P. J., O’Donnell, N., Riley, J. P., Williams, K. N., Karlsson, L., and Thurmond, R. L. (2006). The histamine H4 receptor mediates allergic airway inflammation by regulating the activation of CD4+ T cells. J. Immunol. 176, 7062–7070. doi: 10.4049/jimmunol.176.11.7062
Formigli, L., Perna, A. M., Meacci, E., Cinci, L., Margheri, M., Nistri, S., et al. (2007). Paracrine effects of transplanted myoblasts and relaxin on post-infarction heart remodelling: In Focus. J. Cell. Mol. Med. 11, 1087–1100. doi: 10.1111/j.1582-4934.2007.00111.x
Genovese, T., Mazzon, E., Di Paola, R., Muià, C., Threadgill, M. D., Caputi, A. P., et al. (2005). Inhibitors of poly(ADP-ribose) polymerase modulate signal transduction pathways and the development of bleomycin-induced lung injury. J. Pharmacol. Exp. Ther. 313, 529–538. doi: 10.1124/jpet.104.080705
Gschwandtner, M., Koether, B., Werfel, T., Stark, H., and Gutzmer, R. (2013). Profiling of histamine H4 receptor agonists in native human monocytes. Br. J. Pharmacol. 170, 136–143. doi: 10.1111/bph.12237
Gutzmer, R., Mommert, S., Gschwandtner, M., Zwingmann, K., Stark, H., and Werfel, T. (2009). The histamine H4 receptor is functionally expressed on TH2 cells. J. Allergy Clin. Immunol. 123, 619–625. doi: 10.1016/j.jaci.2008.12.1110
Hu, B., Wu, Z., Hergert, P., Henke, C. A., Bitterman, P. B., and Phan, S. H. (2013). Regulation of myofibroblast differentiation by poly(ADP-ribose) polymerase 1. Am. J. Pathol. 182, 71–83. doi: 10.1016/j.ajpath.2012.09.004
Inomata, M., Nishioka, Y., and Azuma, A. (2015). Nintedanib: evidence for its therapeutic potential in idiopathic pulmonary fibrosis. Core Evid. 10, 89–98. doi: 10.2147/CE.S82905
Karimi-Shah, B. A., and Chowdhury, B. A. (2015). Forced vital capacity in idiopathic pulmonary fibrosis—FDA review of pirfenidone and nintedanib. N. Engl. J. Med. 372, 1187–1189. doi: 10.1056/NEJMp1414949
Kohyama, T., Yamauchi, Y., Takizawa, H., Kamitani, S., Kawasaki, S., and Nagase, T. (2010). Histamine stimulates human lung fibroblast migration. Mol. Cell. Biochem. 337, 77–81. doi: 10.1007/s11010-009-0287-y
Kolodsick, J. E., Peters-Golden, M., Larios, J., Toews, G. B., Thannickal, V. J., and Moore, B. B. (2003). Prostaglandin E2 inhibits fibroblast to myofibroblast transition via E. prostanoid receptor 2 signaling and cyclic adenosine monophosphate elevation. Am. J. Respir. Cell Mol. Biol. 29, 537–544. doi: 10.1165/rcmb.2002-0243OC
Lama, V., Moore, B. B., Christensen, P., Toews, G. B., and Peters-Golden, M. (2002). Prostaglandin E2 synthesis and suppression of fibroblast proliferation by alveolar epithelial cells is cyclooxygenase-2–dependent. Am. J. Respir. Cell Mol. Biol. 27, 752–758. doi: 10.1165/rcmb.4857
Langelier, M.-F., Planck, J. L., Roy, S., and Pascal, J. M. (2012). Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science 336, 728–732. doi: 10.1126/science.1216338
Lattouf, R., Younes, R., Lutomski, D., Naaman, N., Godeau, G., Senni, K., et al. (2014). Picrosirius red staining: a useful tool to appraise collagen networks in normal and pathological tissues. J. Histochem. Cytochem. 62, 751–758. doi: 10.1369/0022155414545787
Leask, A., and Abraham, D. J. (2004). TGF-beta signaling and the fibrotic response. FASEB J. 18, 816–827. doi: 10.1096/fj.03-1273rev
Lim, H. D., Adami, M., Guaita, E., Werfel, T., Smits, R. A., de Esch, I. J. P., et al. (2009). Pharmacological characterization of the new histamine H4 receptor agonist VUF 8430. Br. J. Pharmacol. 157, 34–43. doi: 10.1111/j.1476-5381.2009.00200.x
Lodovici, M., Casalini, C., Cariaggi, R., Michelucci, L., and Dolara, P. (2000). Levels of 8-hydroxydeoxyguanosine as a marker of DNA damage in human leukocytes. Free Radic. Biol. Med. 28, 13–17. doi: 10.1016/S0891-5849(99)00194-X
Lucarini, L., Durante, M., Lanzi, C., Pini, A., Boccalini, G., Calosi, L., et al. (2017). HYDAMTIQ, a selective PARP-1 inhibitor, improves bleomycin-induced lung fibrosis by dampening the TGF-β/SMAD signalling pathway. J. Cell. Mol. Med. 21, 324–335. doi: 10.1111/jcmm.12967
Lucarini, L., Pini, A., Gerace, E., Pellicciari, R., Masini, E., and Moroni, F. (2014). Poly(ADP-ribose) polymerase inhibition with HYDAMTIQ reduces allergen-induced asthma-like reaction, bronchial hyper-reactivity and airway remodelling. J. Cell. Mol. Med. 18, 468–479. doi: 10.1111/jcmm.12197
Lucarini, L., Pini, A., Rosa, A. C., Lanzi, C., Durante, M., Chazot, P. L., et al. (2016). Role of histamine H4 receptor ligands in bleomycin-induced pulmonary fibrosis. Pharmacol. Res. 111, 740–748. doi: 10.1016/j.phrs.2016.07.037
Manni, M. L., Mandalapu, S., McHugh, K. J., Elloso, M. M., Dudas, P. L., and Alcorn, J. F. (2016). Molecular mechanisms of airway hyperresponsiveness in a murine model of steroid-resistant airway inflammation. J. Immunol. 196, 963–977. doi: 10.4049/jimmunol.1501531
Marcos-Garcés, V., Harvat, M., Molina Aguilar, P., Ferrandez Izquierdo, A., and Ruiz-Saurì, A. (2017). Comparative measurement of collagen bundle orientation by Fourier analysis and semiquantitative evaluation: reliability and agreement in Masson’s trichrome, Picrosirius red and confocal microscopy techniques. J. Microsc. 267, 130–142. doi: 10.1111/jmi.12553
Martin, M. M., Buckenberger, J. A., Jiang, J., Malana, G. E., Knoell, D. L., Feldman, D. S., et al. (2007). TGF-beta1 stimulates human AT1 receptor expression in lung fibroblasts by cross talk between the Smad, p 38 MAPK, JNK, and PI3K signaling pathways. Am. J. Physiol. Lung Cell. Mol. Physiol. 293, L790–L799. doi: 10.1152/ajplung.00099.2007
Masini, E., Bani, D., Vannacci, A., Pierpaoli, S., Mannaioni, P. F., Comhair, S. A. A., et al. (2005). Reduction of antigen-induced respiratory abnormalities and airway inflammation in sensitized guinea pigs by a superoxide dismutase mimetic. Free Radic. Biol. Med. 39, 520–531. doi: 10.1016/j.freeradbiomed.2005.04.006
Masini, E., Lucarini, L., Sydbom, A., Dahlén, B., and Dahlén, S.-E. (2013). “Histamine in asthmatic and fibrotic lung disorders” in Histamine H4 Receptor: A Novel Drug Target in Immunoregulation and Inflammation. ed. H. Stark (London, Great Britain: Versita), 145–171.
McGrath, J. C., Drummond, G. B., McLachlan, E. M. E., Kilkenny, C., and Wainwright, C. L. (2010). Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br. J. Pharmacol. 160, 1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x
Moore, B. B., and Hogaboam, C. M. (2008). Murine models of pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 294, 152–160. doi: 10.1152/ajplung.00313.2007
Mukhopadhyay, P., Rajesh, M., Cao, Z., Horvth, B., Park, O., Wang, H., et al. (2014). Poly (ADP-ribose) polymerase-1 is a key mediator of liver inflammation and fibrosis. Hepatology 59, 1998–2009. doi: 10.1002/hep.26763
Mullane, K. M., Kraemer, R., and Smith, B. (1985). Myeloperoxidase activity as a quantitative assessment of neutrophil infiltration into ischemic myocardium. J. Pharmacol. Methods 14, 157–167. doi: 10.1016/0160-5402(85)90029-4
Nanthakumar, C. B., Hatley, R. J. D., Lemma, S., Gauldie, J., Marshall, R. P., and Macdonald, S. J. F. (2015). Dissecting fibrosis: therapeutic insights from the small-molecule toolbox. Nat. Rev. Drug Discov. 14, 693–720. doi: 10.1038/nrd4592
Pacher, P., Liaudet, L., Bai, P., Virag, L., Mabley, J. G., Haskó, G., et al. (2002). Activation of poly(ADP-ribose) polymerase contributes to development of doxorubicin-induced heart failure. J. Pharmacol. Exp. Ther. 300, 862–867. doi: 10.1124/jpet.300.3.862
Paz, Z., and Shoenfeld, Y. (2010). Antifibrosis: to reverse the irreversible. Clin. Rev. Allergy Immunol. 38, 276–286. doi: 10.1007/s12016-009-8157-7
Pini, A., Shemesh, R., Samuel, C. S., Bathgate, R. A. D., Zauberman, A., Hermesh, C., et al. (2010). Prevention of bleomycin-induced pulmonary fibrosis by a novel antifibrotic peptide with relaxin-like activity. J. Pharmacol. Exp. Ther. 335, 589–599. doi: 10.1124/jpet.110.170977
Pini, A., Viappiani, S., Bolla, M., Masini, E., and Bani, D. (2012). Prevention of bleomycin-induced lung fibrosis in mice by a novel approach of parallel inhibition of cyclooxygenase and nitric-oxide donation using NCX 466, a prototype cyclooxygenase inhibitor and nitric-oxide donor. J. Pharmacol. Exp. Ther. 341, 493–499. doi: 10.1124/jpet.111.190660
Plantier, L., Cazes, A., Dinh-Xuan, A.-T., Bancal, C., Marchand-Adam, S., and Crestani, B. (2018). Physiology of the lung in idiopathic pulmonary fibrosis. Eur. Respir. Rev. 27:170062. doi: 10.1183/16000617.0062-2017
Rosa, A. C., Pini, A., Lucarini, L., Lanzi, C., Veglia, E., Thurmond, R. L., et al. (2014). Prevention of bleomycin-induced lung inflammation and fibrosis in mice by naproxen and JNJ7777120 treatment. J. Pharmacol. Exp. Ther. 351, 308–316. doi: 10.1124/jpet.114.215152
Sime, P. J., Xing, Z., Graham, F. L., Csaky, K. G., and Gauldie, J. (1997). Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J. Clin. Invest. 100, 768–776. doi: 10.1172/JCI119590
Smolle, J., Fiebiger, M., Hofmann-Wellenhof, R., and Kerl, H. (1996). Quantitative morphology of collagen fibers in cutaneous malignant melanoma and melanocytic nevus. Am. J. Dermatopathol. 18, 358–363. doi: 10.1097/00000372-199608000-00005
Swigris, J. J., Kuschner, W. G., Jacobs, S. S., Wilson, S. R., and Gould, M. K. (2005). Health-related quality of life in patients with idiopathic pulmonary fibrosis: a systematic review. Thorax 60, 588–594. doi: 10.1136/thx.2004.035220
Tatler, A. L., and Jenkins, G. (2012). TGF-β activation and lung fibrosis. Proc. Am. Thorac. Soc. 9, 130–136. doi: 10.1513/pats.201201-003AW
Thannickal, V. J., and Horowitz, J. C. (2006). Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc. Am. Thorac. Soc. 3, 350–356. doi: 10.1513/pats.200601-001TK
Wang, Z. Q., Auer, B., Stingl, L., Berghammer, H., Haidacher, D., Schweiger, M., et al. (1995). Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 9, 509–520. doi: 10.1101/gad.9.5.509
Wilson, M. S., and Wynn, T. A. (2009). Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal Immunol. 2, 103–121. doi: 10.1038/mi.2008.85
Wynn, T. A. (2007). Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Invest. 117, 524–529. doi: 10.1172/JCI31487
Wynn, T. (2008). Cellular and molecular mechanisms of fibrosis. J. Pathol. 214, 199–210. doi: 10.1002/path.2277
Wynn, T. A., and Ramalingam, T. R. (2012). Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat. Med. 18, 1028–1040. doi: 10.1038/nm.2807
Keywords: histamine H4 receptors, poly(ADP-ribose) polymerase 1, pulmonary fibrosis, inflammation, oxidative stress, cytokines
Citation: Durante M, Sgambellone S, Lanzi C, Nardini P, Pini A, Moroni F, Masini E and Lucarini L (2019) Effects of PARP-1 Deficiency and Histamine H4 Receptor Inhibition in an Inflammatory Model of Lung Fibrosis in Mice. Front. Pharmacol. 10:525. doi: 10.3389/fphar.2019.00525
Edited by:
Ioanna Andreadou, National and Kapodistrian University of Athens, GreeceReviewed by:
Manfred Kietzmann, University of Veterinary Medicine Hannover, GermanyLongshuang Huang, University of Illinois at Chicago, United States
Copyright © 2019 Durante, Sgambellone, Lanzi, Nardini, Pini, Moroni, Masini and Lucarini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura Lucarini, bGF1cmEubHVjYXJpbmlAdW5pZmkuaXQ=