 Zhiru Wang1,2,3*
Zhiru Wang1,2,3* Wenting Kang1,2
Wenting Kang1,2 Yinghua You1,2
Yinghua You1,2 Jingru Pang1,2
Jingru Pang1,2 Hongmei Ren1,2
Hongmei Ren1,2 Zhenhe Suo3*Hongmin Liu1,2*
Zhenhe Suo3*Hongmin Liu1,2* Yichao Zheng1,2*
Yichao Zheng1,2*- 1School of Pharmaceutical Sciences, Zhenghzou University, Zhengzhou, China
- 2Collaborative Innovation Centre of New Drug Research and Safety Evaluation, Henan Province, and Key Laboratory of Advanced Drug Preparation Technologies, Zhengzhou University, and Key Laboratory of Henan Province for Drug Quality and Evaluation, Ministry of Education of China, Zhengzhou, China
- 3Pathology, Institute of Clinical Medicine, Faculty of Medicine, University of Oslo, Oslo, Norway
Ubiquitin specific protease 7 (USP7) is one of the deubiquitinating enzymes (DUB) that erases ubiquitin and protects substrate protein from degradation. Full activity of USP7 requires the C-terminal Ub-like domains fold back onto the catalytic domain, allowing the remodeling of the active site to a catalytically competent state by the C-terminal peptide. Until now, numerous proteins have been identified as substrates of USP7, which play a key role in cell cycle, DNA repair, chromatin remodeling, and epigenetic regulation. Aberrant activation or overexpression of USP7 may promote oncogenesis and viral disease, making it a target for therapeutic intervention. Currently, several synthetic small molecules have been identified as inhibitors of USP7, and applied in the treatment of diverse diseases. Hence, USP7 may be a promising therapeutic target for the treatment of cancer.
Introduction
Post-translational modification (PTM) is generally enzymatic modification of proteins following protein biosynthesis. Examples of PTM include methylation, acetylation, phosphorylation, glycosylation, ubiquitination, S-nitrosylation, and so on (Chatterjee and Thakur, 2018). As one of the most studied PTMs, ubiquitination involves in the intracellular proteolytic machinery and regulates numerous physical activities in the cell (Dybas et al., 2018). The process of the addition of ubiquitin to a substrate protein is named ubiquitination, which may contribute to the protein degradation. Ubiquitination of target protein can be catalyzed by a cascade reaction comprising the ubiquitin-activating enzymes (E1), the ubiquitin conjugation enzymes (E2) and the ubiquitin ligases (E3). First, ubiquitin is activated by E1 with the participation of ATP and transferred to E2 through a trans-thiolation reaction, and then conjugated to a lysine or α-amino group of the substrate protein in the presence of E3 (Cheon and Baek, 2006). Eventually, proteins labels with more than four ubiquitin molecules can be recognized and subjected to the 26S proteasome at which they are degraded, generating small polypeptides (Figure 1).
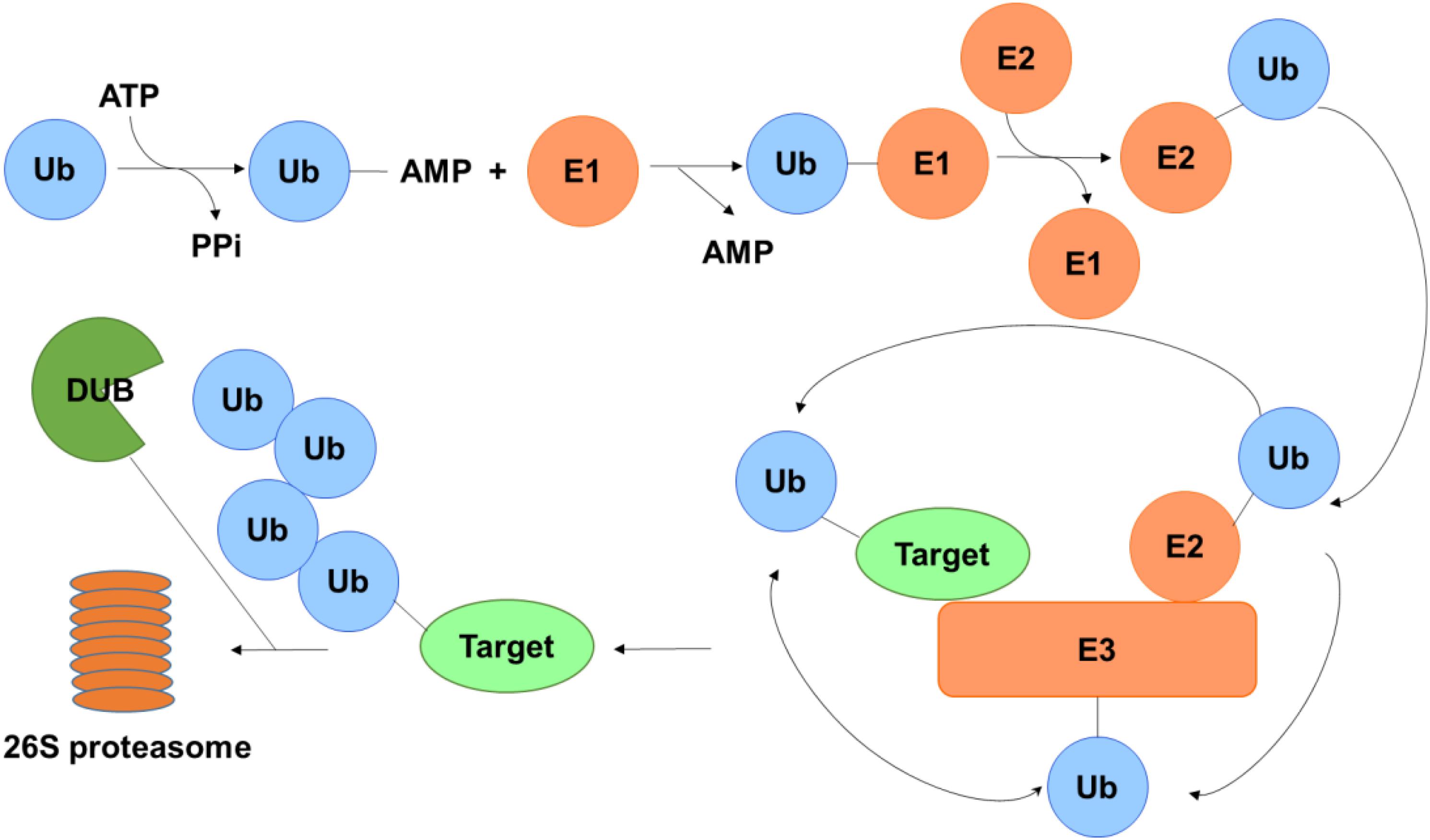
Figure 1. Schematic of the ubiquitin-proteasome system. Ubiquitin is activated by E1 in the presence of ATP and transferred to E2 and then conjugated to a lysine or α-amino group of the substrate protein with the aid of E3. Polyubiquinated targets are recognized and degraded by the 26S proteasome, while the ubiquitin on the substrate can be erased by DUBs to protect it from degradation.
Deubiquitinating enzymes (DUBs) are responsible for the removal of ubiquitin and keeping the stability of the substrate by rescuing them from degradation (Nijman et al., 2005; Clague et al., 2013). Until now, approximately 100 DUBs have been identified and can be classified into five subclasses based on their Ub-protease domains: ubiquitin-specific proteases (USPs), ubiquitin C-terminal hydrolases (UCHs), ovarian tumor proteases (OTUs), Machado-Joseph disease proteases (MJDs) belonging to cysteine-dependent proteases, and JAB1/MPN/Mov34 (JAMMs) belonging to zinc metalloproteases (Zhou et al., 2018). With approximately 50 members, the USPs family is the largest one among all the DUB subfamilies. All these members include conserved domains, i.e., three primary functional domains of Cys, His and Asp/Asn boxes which are in charge of the reorganization of ubiquitin conjugated molecules.
Among the members of USP family, ubiquitin specific protease USP7, also known as herpes-associated ubiquitin-specific protease (HAUSP), is a unique deubiquitinating enzyme which was identified in 1997, and it characterized as a novel member of the ubiquitin-specific protease family to interact with herpes simplex virus type 1 immediate-early protein (Vmw110) of the herpes simplex virus type 1 (HSV-1) regulatory protein (Everett et al., 1997). Later, USP7 was found to interact with other viral proteins such as the Epstein-Barr nuclear antigen 1 (EBNA1) of Epstein-Barr virus (EBV) and the vIRF1 (viral interferon regulatory factor 1) protein of Kaposi’s sarcoma associated herpesvirus (KSHV) (Holowaty et al., 2003), therefore indicating it as a general target of herpes viruses and giving it the name herpes-associated ubiquitin specific protease. Up to now, USP7 is the most widely studied deubiquitinating enzymes, and is considered as an oncogene by promoting tumor growth and negatively affecting the patient immune response to tumors (Everett, 2014; Lu et al., 2016).
Structure of USP7
The full length USP7 includes 1102 amino acids. There are four domains: an N-terminal poly-glutamine stretch (poly Q), the tumor necrosis factor receptor- associated factors (TRAF) domain (amino acids 62–205), the catalytic domain (amino acids 208–560), and the C-terminal tandem ubiquitin-like (Ubl) domain (amino acids 560–1102) (Kim and Sixma, 2017) (Figure 2A).
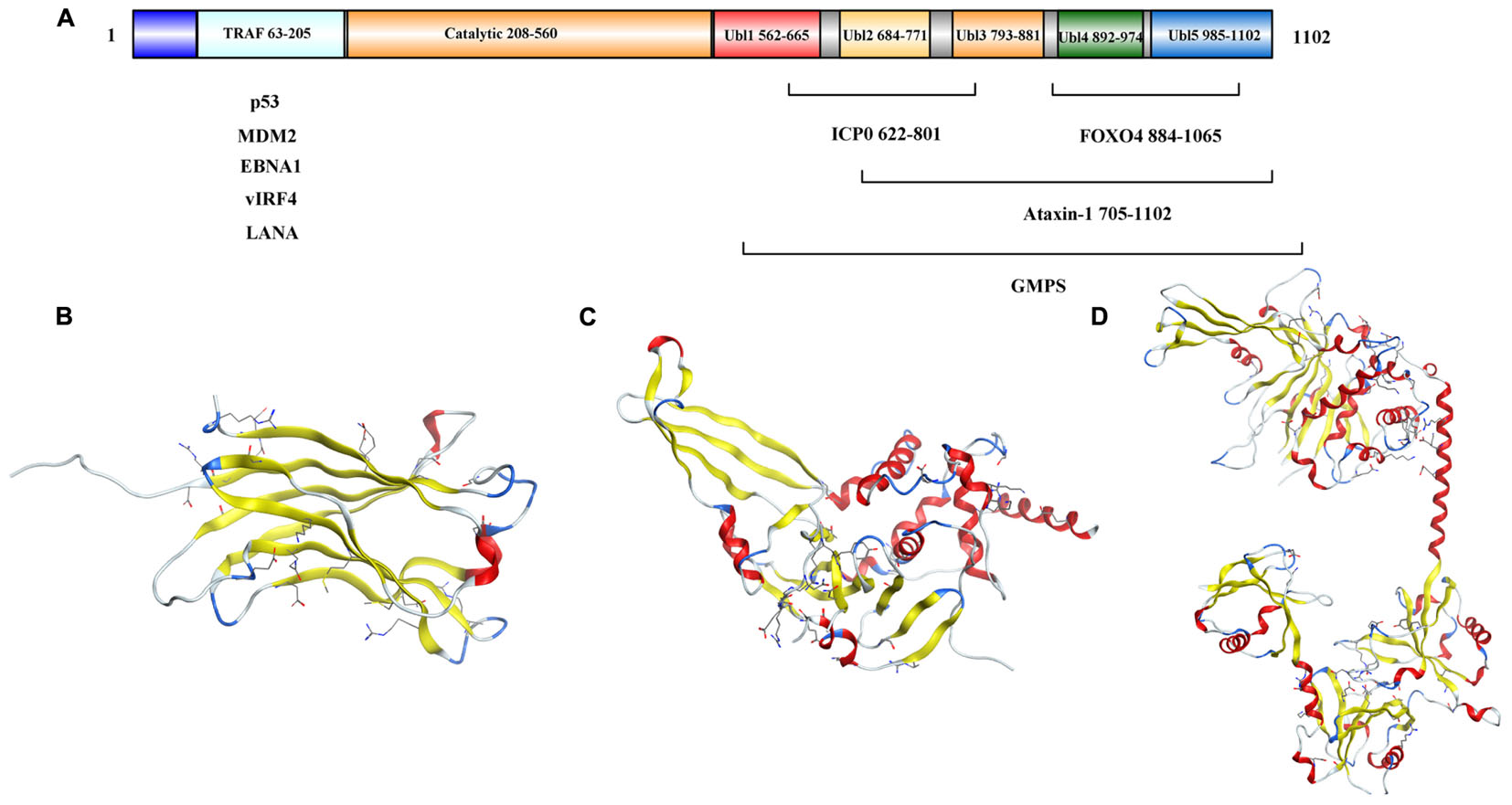
Figure 2. Structure analysis of USP7. (A) USP7 primary sequence map. (B) Structure of the USP7 N-terminal domain (PDB 2F1W). (C) Structure of USP7 catalytic domain and five UBl-domains (PDB 1NB8). (D) Structure of the inactive state of USP7 catalytic domain (PDB 5FWI).
As reported, the amino acids 62–205 of USP7 (Figure 2B) bind to EBNA1 (Holowaty et al., 2003), mouse double minute 2 homolog (MDM2) and p53 (Hu et al., 2006; Sheng et al., 2006) through a PA-x-x-S motif (Saridakis et al., 2005), and the TRAF (amino acids 62–205) domain contributes to the nuclear localization of the USP7 (Fernandez-Montalvan et al., 2007). Besides, the USP7 truncation (amino acids 208–1102) performed similar activity as the full length protein (Ma et al., 2010).
Hu et al. (2002) identified a 40 kDa fragment of USP7 as the catalytic domain (amino acids 208–560), which mediates ubiquitin binding and deubiquitination of the substrate. The structure of the catalytic core domain reveals novel three-domain architecture, including Fingers, Palm, and Thumb domains (Figure 2C). This catalytic core domain binds to ubiquitin aldehyde, which reveals a conformational change in the active site (Hu et al., 2002). With the aid of molecular dynamics simulations, it is found that the transition of USP7 from the inactive to the active can only be captured when H294 was neutralized with a deprotonated C223 and charged H464. In the inactive apo state, positively charged H294 stabilizes an electrostatic network with W285, E298, and Y224. However, neutral H294 in the active state cannot make charge interactions, so the electrostatic network is disrupted. That would results in the C223 unfavorable backbone angles improved by helical refolding, thus, the active site is formed (Ozen et al., 2018).
Ubl shares the ubiquitin β-grasp fold, however, it lacks the C-terminal Gly–Gly residues that are required for conjugation to a target and is located outside the boundaries of the catalytic core domain (Faesen et al., 2011). There are five Ubl domains that are detected in the C-terminal and are organized in a 2-1-2 manner as Ubl-12, Ubl-3, and Ubl-45 (Figure 2D) (Zhu et al., 2007). Among them, Ubl-45 is sufficient to reconstitute the USP7 activation in vitro and in vivo. In the C-terminal, the 19 residues of USP7 (amino acids 1084–1102) are conserved across species (Faesen et al., 2011). Rouge et al. (2016) revealed how the C terminal 19 amino acids of the USP7 contribute to the enhancement of USP7 activity by stabilizing the ubiquitin binding conformation of the catalytic domain. And the individual point mutations at residues I1100 or I1098 are able to abolish the deubiquitinase activity of USP7 (Rouge et al., 2016).
USP7: One Protein, Multiple Roles
Many proteins have been identified as potential substrates and binding partners of USP7, such as viral proteins, transcription factors, and epigenetic modulators (Figure 8), and most of these substrates play important roles in viral replication, immune response, tumor suppression, epigenetic control, and DNA repair. Here, functions of USP7 on these substrate are as detailed below (Table 1).
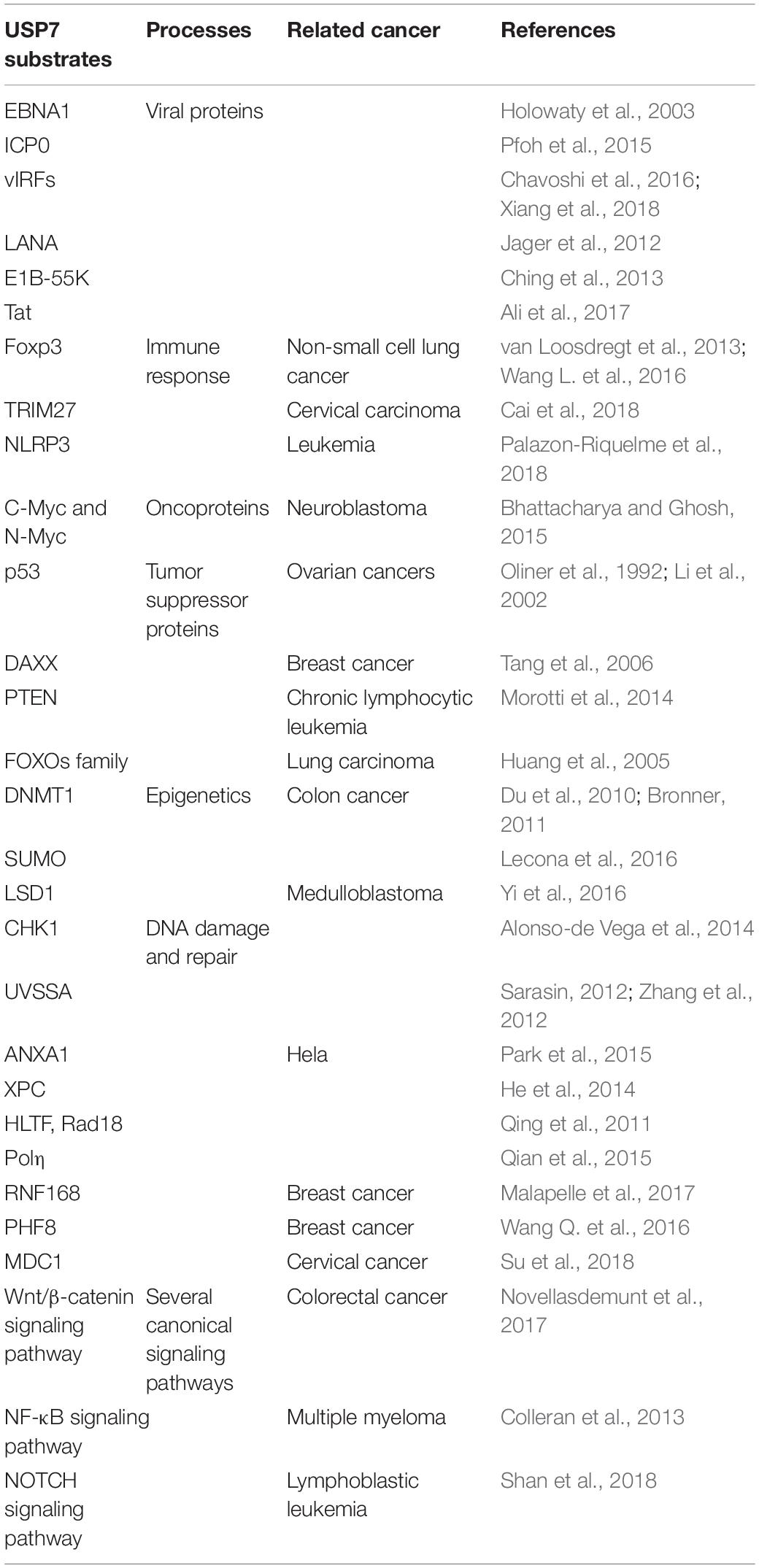
Table 1. Proteins regulated by USP7.
Viral Proteins
EBNA1
EBNA1 of EBV is important for the replication, segregation, and transcriptional activation of latent EBV genomes, it has been implicated in host cell immortalization, and avoids proteasome processing and cell-surface presentation. The amino acids 395–450 of EBNA1 bind to the USP7 N-terminal domain with a dissociation constant of 0.9–2 μM. The △395–450 mutant that selectively disrupted the binding to USP7 was found to increase fourfold EBNA1 replication activity than wild-type, but performed no impact on EBNA1 turnover and cell-surface presentation (Holowaty et al., 2003). As p53 and EBNA1 share similar binding sites with USP7, EBNA1 peptide efficiently competes with p53 peptide for USP7 binding, which results the decreasing stability of p53, and protects cells from apoptosis (Saridakis et al., 2005).
ICP0
Infected cell protein 0 (ICP0) of HSV is a multifunctional protein containing 775 amino acids that acts as a promiscuous trans-activator linked to the degradation of several proteins. The 618PRKCARKT625 of ICP0 binds to a negatively charged region on Ubl2, where the residues K620 and K624 of ICP0 form direct contacts with residues D762 and D764 in Ubl2 of USP7 (Pfoh et al., 2015). Overexpression of USP7 had no effect on the mRNA level of ICP0, but could accelerate the mRNA accumulation of thymidine kinase (TK) and gI, which are important for HSV infection of non-replicating cells. The mutations at residues 620 to 626 of ICP0 (named as R6702) can abolish the interaction between USP7 and ICP0, and the replication of R6702 in cells cannot be impaired (Kalamvoki et al., 2012). Hence, inhibition of USP7 and/or its interaction with ICP0 using small molecule inhibitors may decrease the virulence of HSV.
vIRFs
Among the vIRFs, vIRF1 could interact with the TRAF domain of USP7 via EGPS motif. The vIRF1 interaction with USP7 can decrease p53 levels by blocking the deubiquitination and stabilization of USP7 on p53. Thus the KSHV could have a lifelong infection when p53 is destabilized by USP7 coupled with vIRF1 (Chavoshi et al., 2016). Besides, vIRF3 is expressed in human herpes virus 8 (HHV-8) – infected primary effusion lymphoma (PEL) cells. The vIRF3 has two copies of EGPS, and both support the vIRF3 – USP7 interaction. This interaction plays important roles in PEL cell growth and viability and contributes to the suppression of productive virus replication (Xiang et al., 2018). For another vIRF family member, amino residues 210–216 of KSHV vIRF4 bind to the same surface groove of the USP7 TRAF domain as that can be recognized by MDM2 and p53. Moreover, the amino residues 202–208 of vIRF4 interact with the β-sheet in TRAF domain. The vIRF4-derived vif1 and vif2 peptides can restore p53 dependent apoptosis in wild-type p53 cancer cells by suppressing the USP7 activity. Thus the two peptides may be considered as potential backbones for peptide mimic small molecule inhibitors development for anti-cancer therapies (Lee et al., 2011).
LANA
The viral latency-associated nuclear antigen 1 (LANA) is expressed in all latency KSHV-infected cells and involves in viral latent replication and maintenance of the viral genome. The amino residues 971–986 of LANA interact with TRAF domain of USP7 with similar binding sites as EBNA1, while the △971–986 mutant shows an enhanced ability to replicate latent viral DNA. These results indicate that USP7 may influence accessibility of the viral DNA for latent replication or LANA-mediated viral persistence (Jager et al., 2012). Because of the role of USP7 in EBNA1 – dependent latent replication of EBV, USP7 may play the same role in the replication of latent viral DNA among gamma-1 and gamma-2 herpesviridae.
E1B-55K
Adenovirus E1B protein refers to one or two proteins transcribed from the E1B gene of the adenovirus: a 55 kDa protein and a 19 kDa protein. The N-terminal 79 amino acids of E1B-55K interact with the TRAF domain of USP7. Abrogation of USP7 decreases the protein level of E1B-55K and reduces progeny viral production. Therefore, the small inhibitors of USP7 may be used to treat adenovirus infections (Ching et al., 2013).
Tat
Human immunodeficiency virus (HIV) Tat is synthesized early after infection and mainly responsible for enhancing viral production. USP7 deubiquitinates and stabilizes Tat and enhances HIV-1 production. In turn, HIV-1 infection leads to the overexpression of USP7. These results show that the small inhibitors of USP7 can be used as a novel anti-HIV approach (Ali et al., 2017).
In sum, these results show that USP7 is recruited by these viruses to promote their survival in the host. So we speculate that USP7 may be an attractive target for controlling infection and other malignancies caused by these viruses.
Immune Response
Foxp3
Recent years, more and more reports have identified the importance of USP7 on keeping T regulatory cells (Treg) functions. As the major factor that restrains autoimmune responses, Treg cell expresses the forkhead transcription factor Foxp3, which is necessary for Treg cell development (Bettelli et al., 2005; Laurence et al., 2013). According to a report in 2016, five distinct lysine residues (K249, K251, K263, K267, and K393) in Foxp3 were identified to be ubiquitinated, and Foxp3 can be stabilized by USP7 mediated deubiquitination, resulting in the maintenance of Treg cell number and function (Wang L. et al., 2016). In fact, a study in 2013 showed that aberrant USP7 overexpression decreases Foxp3 polyubiquitination and protects it from proteasome degradation, resulting in Treg-cell-mediated suppression and tumor growth. On the contrary, USP7 knockdown decreases Foxp3 level and abrogates Treg cell-induced suppression of autoimmune responses in vitro and in vivo (van Loosdregt et al., 2013).
Later studies gave the mechanism how the level of Foxp3 is regulated. Foxp3 could be ubiquitinated and degraded by the E3 ubiquitin ligase stress inducible protein 1 homology and U-Box containing protein 1 (STUB1). In addition, Foxp3, Heat Shock Protein 70 (Hsp70) and STUB1 associate together as a complex, indicating that these proteins bind and promote Foxp3 ubiquitination (Figure 3) (van Loosdregt and Coffer, 2014). Moreover, it is found that mesenchymal stem cells (MSCs) – induced Treg cells express high level of USP7 and low level of STUB1. Besides, Foxp3 mRNA expression was positively associated with USP7 and negatively associated with STUB1 (Khosravi et al., 2018). So, it provides us an opportunity to find a new way to study the unique role of USP7 in Treg cells and makes USP7 as a target in immunology.
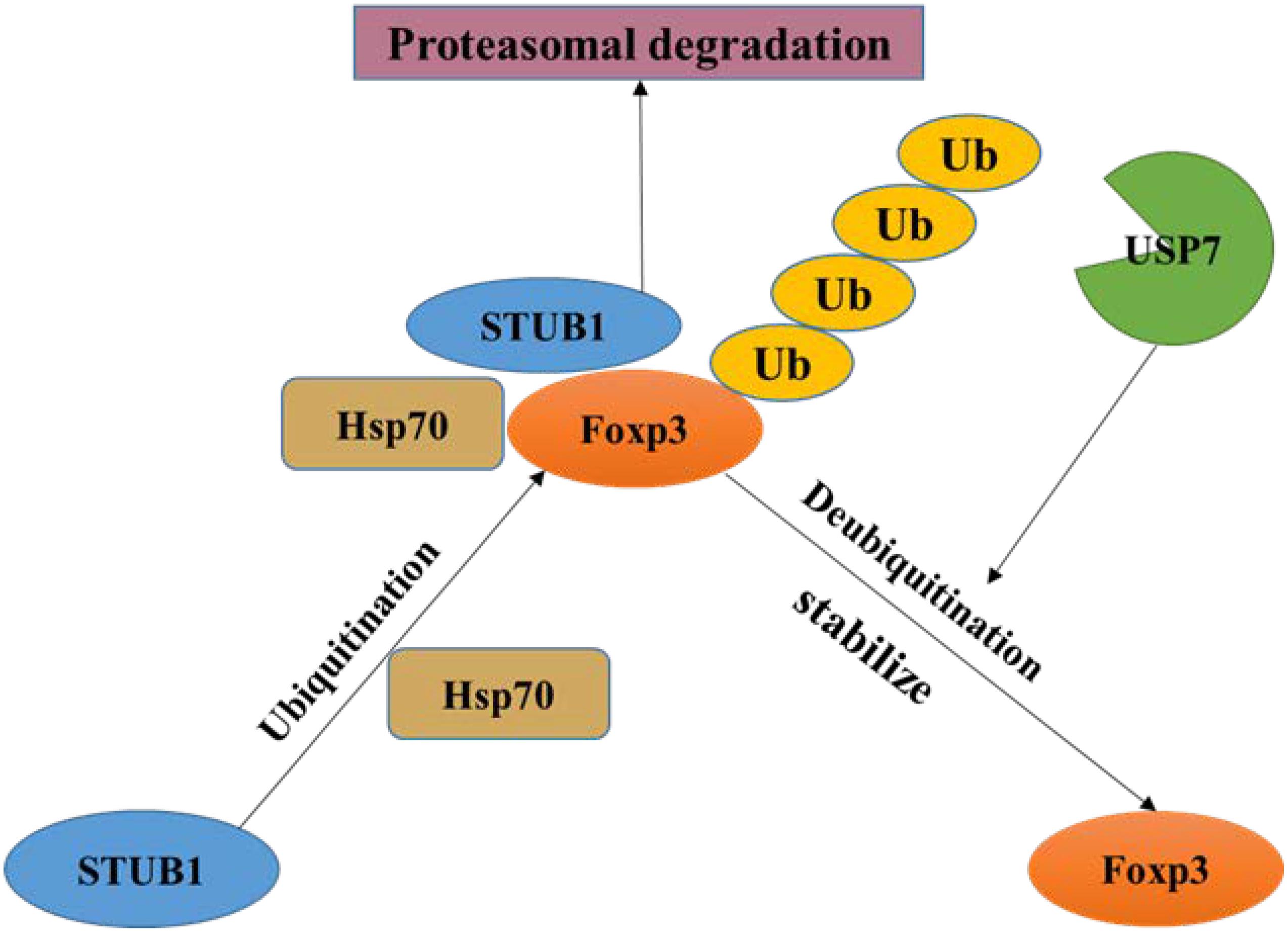
Figure 3. Regulation of Foxp3 by USP7. Foxp3 is ubiquitinated by STUB1 and then produces a complex containing Foxp3, Hsp70 and STUB1, which leading to proteasome degradation of Foxp3. USP7 can remove the ubiquitin on Foxp3 and stabilize it.
TRIM27
Among the binding partners of USP7, tripartite motif 27 (TRIM27) is an ubiquitin E3 ligase that negatively regulates antiviral signaling by promoting the ubiquitination and degradation of TRAF family member-associated NF-κ-B activator – binding kinase 1 (TBK1). USP7 interacts with TRIM27 and forms the USP7-TRIM27-TBK1 complex, and the interaction between USP7 and TRIM27 can be enhanced after Sendai virus (SeV) infection. When USP7 was overexpressed, TRIM27 can be protected from degradation, which contributed to the ubiquitination and degradation of TBK1, resulting in decreased type I interferons (IFNs) signaling (Cai et al., 2018). As IFNs are a series of signaling proteins which are produced and released by host cells to cope with the presence of pathogens, USP7 can enhance the effects of TRIM27 on TBK1-induced IFN – stimulated response element (ISRE) and IFN-β activation (Zaman et al., 2013). Therefore, USP7 may act as a significant host protein to bridge the viral proteins with the antiviral immune response. Therapeutic methods against the USP7-TRIM27 complex may overcome the immune escape mediated by various viruses.
NLRP3
USP7 may also impact on regulating NLR family pyrin domain containing 3 (NLRP3) inflammasome activation. NLRP3 is expressed primarily in macrophages as a component of the inflammasome to monitor products of damaged cells such as extracellular ATP and crystalline uric acid. The ubiquitination status of NLRP3 itself can be altered by USP7 and USP47. Furthermore, researchers discovered that the activity of USP7 and USP47 were augmented once the inflammasome was activated. In the meantime, they discovered that abrogation of both USP7 and USP47 resulted in reduction of inflammasome activation (Palazon-Riquelme et al., 2018).
To sum up, there is a remarkable connection between USP7 and immune-associated proteins, and so many studies have shown that the important roles of USP7 on regulating these proteins. It’s worth thinking about USP7 inhibitors in combination with immunotherapy will be applied to cancer therapy so that the antitumor effect can be promoted. We hope to see their potential dual antitumor activity will be applied to clinical trials on day.
Oncoproteins
C-Myc and N-Myc
There are three members in Myc family: C-Myc, l-Myc, and N-Myc. Myc family is the most frequent amplified oncogene in human, which contributing to the formation of cancer. Among them, C-Myc and N-Myc are the substrates of USP7. USP7 overexpression can promote C-Myc stability by deubiquitination as well as transformation/transcription domain-associated protein (TRRAP), which is an adaptor protein known as a regulator of C-Myc. On the other hand, C-Myc mRNA can be accumulated by TRRAP indirectly (Bhattacharya and Ghosh, 2015).
N-Myc is another transcription factor that can be stabilized by USP7 via deubiquitination (Tavana et al., 2016). Hence, USP7 inhibitor p5091 was applied to decrease N-Myc expression in a dose dependent manner in neuroblastoma (Tavana et al., 2016). As a consequence, USP7 can be considered as a drug target to modulate C-Myc and N-Myc amount in order to block tumor development.
Tumor Suppressor Proteins
p53
p53 participates in cell cycle arrest, DNA repair, apoptosis, senescence and plays a key role in maintaining normal cell growth (Levine, 1997). USP7 plays a paradoxical role in regulating p53 functions through a variety of mechanisms. On one hand, p53 binds to TRAF domain and C-terminal (amino acids 880–1050) of USP7, and then USP7 ubiquitinates p53 directly and prevents it from degradation. On the other hand, TRAF domain and C-terminal (amino acids 801–1050) of USP7 can interact with MDM2 to increase its stability by erasing the ubiquitin on MDM2, an E3 ligase of p53 (Oliner et al., 1992), and protect it from proteasome degradation. Subsequently, MDM2 ubiquitinates p53 and causes its proteasome degradation, resulting in low expression of p53 in cancer cells (Figure 4) (Li et al., 2002, 2004). In addition, MDM2 can also inhibit the transcription of p53 (Wade et al., 2013). Therefore, inhibition of the interaction between MDM2 and p53 can stabilize p53 (Vassilev et al., 2004). It is noteworthy that, crystal structures analysis and binding studies suggest that the MDM2 peptide and p53 peptide bind to the same surface groove in USP7, but MDM2 performs more extensive interaction and stronger affinity (Hu et al., 2006). Taken together, the activation of USP7-MDM2-p53 interaction can promote the occurrence and development of tumors. The design of small molecules that disrupt or prevent the interaction may be an important target for cancer therapy by regulating p53 pathway.
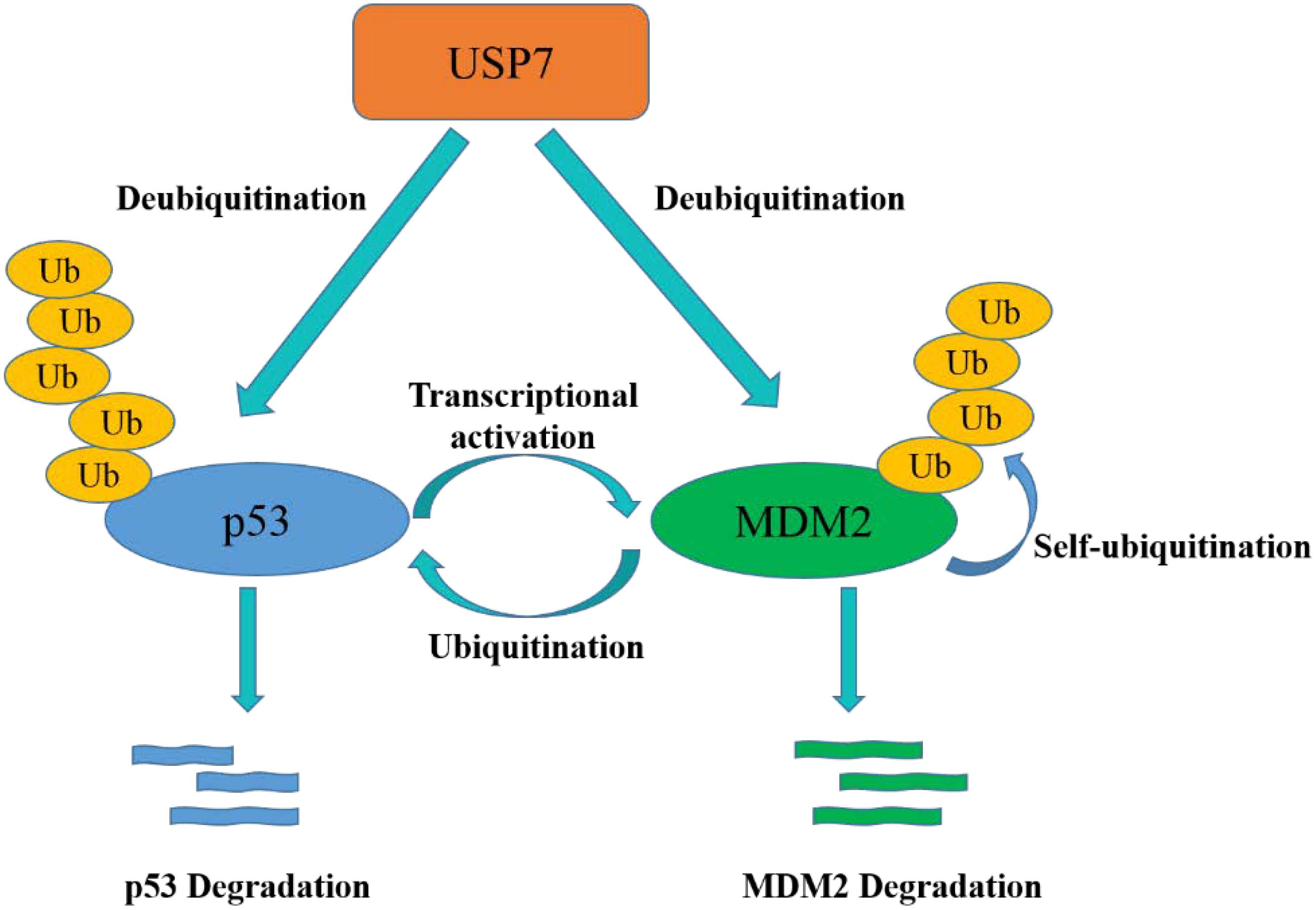
Figure 4. USP7-p53-MDM2 axis interactions control the stability of p53 and MDM2. USP7 can stabilize p53 by deubiquitination, meanwhile, USP7 can also remove the ubiquitin of MDM2, which promotes to p53 degradation.
DAXX
Death-domain-associated protein (DAXX) is a highly conserved and developmentally essential nuclear protein, which participates in many cellular processes (Lindsay et al., 2008). The N-terminal 160 amino acids and amino acids 347–570 of DAXX associate with USP7, which are far from the binding sites of MDM2 on DAXX. In unstressed cells, DAXX interacts with USP7 and MDM2, and mediates the stabilization of USP7 on MDM2, thus blocking p53 activation. In response to DNA damage, self-ubiquitination of MDM2 is accelerated when MDM2 is stripped from DAXX and USP7. That is to say, DAXX directs the ligase activity of MDM2 through regulating USP7 (Tang et al., 2006). Recent reports also show that USP7 and DAXX are critical in regulating the correct execution of mitosis by forming a tertiary complex as MDM2/DAXX/USP7 (Zhang et al., 2010). DAXX binding increases USP7 activity toward MDM2. Disassemble the MDM2-DAXX-USP7 complex can increase MDM2 self-ubiquitination and degradation, which leads to the stabilization and accumulation of p53 (Kumar et al., 2018).
PTEN
Phosphatase and tensin homolog (PTEN) is a tumor suppressor gene that displays dual specific phosphatase activity. PTEN inhibits the proliferation and migration of tumor cells (Blanco-Aparicio et al., 2007). It is reported that nuclear PTEN import is promoted by its mono-ubiquitination (Trotman et al., 2007). However, USP7 can remove the mono-ubiquitination of PTEN, triggering its nuclear exclusion and PTEN inactivation (Morotti et al., 2014). Likewise, USP7 inhibitor, P5091, regains PTEN nuclear pool and restores its tumor suppressive functions in chronic lymphocytic leukemia (CLL) (Carra et al., 2017). In addition, PTEN deletion leads to accumulation of activated AKT, and subsequent phosphorylation of MDM2 by AKT (Blanco-Aparicio et al., 2007), which results in the ubiquitination and degradation of p53 (Freeman et al., 2003). Therefore, PTEN deficiency causes p53-dependent cancer-promoting processes. This suggests how important it is to inhibit USP7 to ensure PTEN protein localization and activity.
FOXOs Family
The Forkhead box O (FOXO) family members, including FOXO1, FOXO3, FOXO4 and FOXO6, are transcription factors that take part in regulating several cellular responses, including cell cycle progression and apoptosis and so on (van der Horst and Burgering, 2007). It is reported that USP7 can remove ubiquitin from FOXO1, which is written by Skp2 as an E3 ligase (Huang et al., 2005). Besides, mono-ubiquitination FOXO4 localizes in the nucleus and exhibits stronger transcriptional promotion activity (Brenkman et al., 2008). USP7 can suppress FOXO4 activity due to its deubiquitination and re-localization (van der Horst et al., 2006). In a word, USP7 affects tumor progression by interacting with FOXOs and affecting their activity and localization.
Epigenetics
DNMT1
DNMT1 (DNA methyltransferase 1) contributes to the maintenance of DNA methylation. As reported, USP7 can deubiquitinate and stabilize DNMT1 when its acetylation is erased by histone deacetylase 1 (HDAC1), which protects DNMT1 from proteasome degradation (Bronner, 2011). When the KG linker of DNMT1 is acetylated by Tip60, USP7 breaks away from DNMT1 and results in the degradation of DNMT1 mediated by proteasome system (Figure 5) (Du et al., 2010). Thus, HDAC and USP7 inhibitors can be applied in combination for cancer treatment (Cheng et al., 2015).
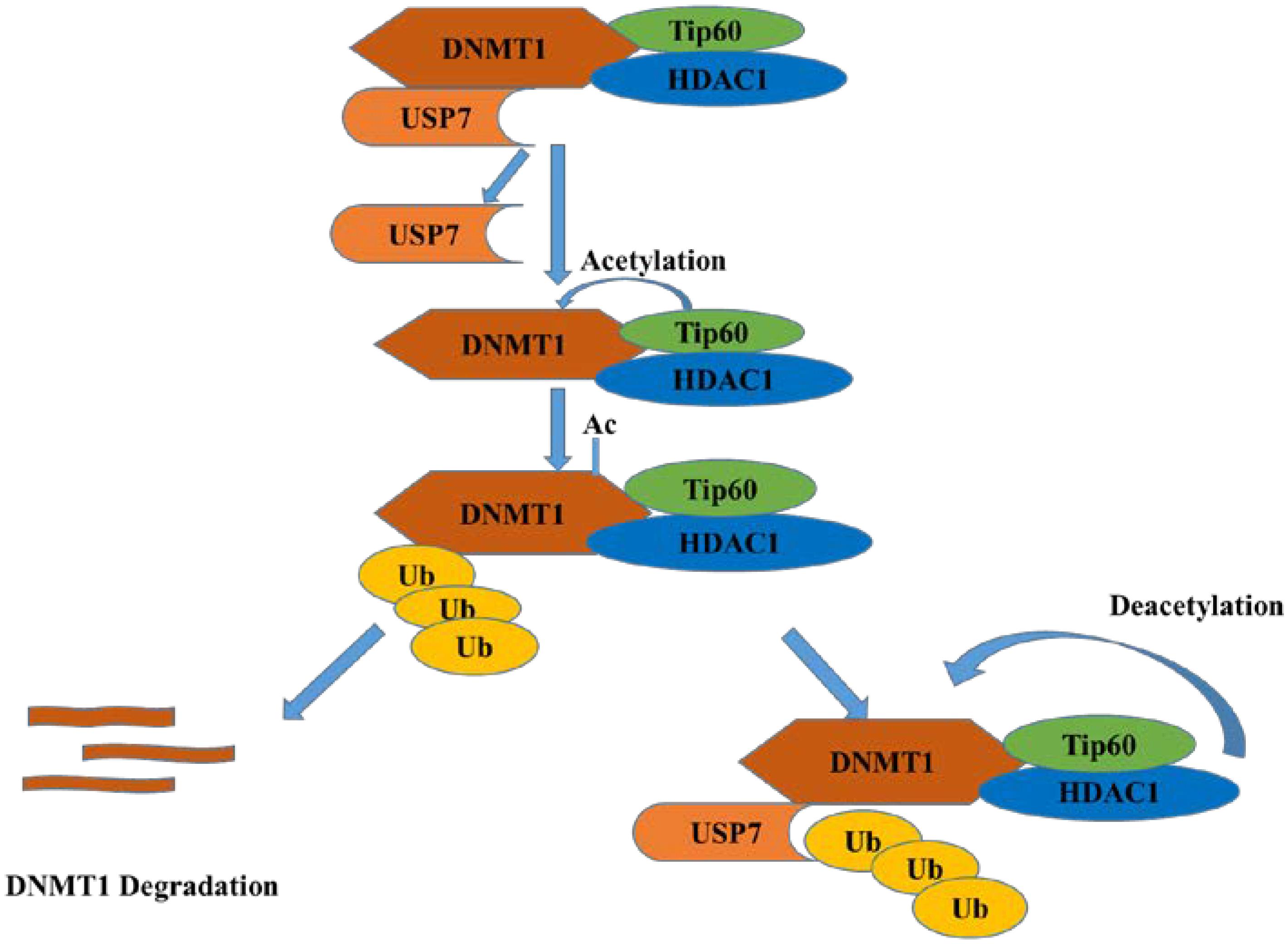
Figure 5. USP7 and HDAC1 protect DNMT1 from degradation while Tip60 acetylates DNMT1 and promotes its degradation.
SUMO
Small ubiquitin related modifier (SUMO) is a ubiquitin-like molecule, which binds to its substrate by E3 SUMO ligase in a similar way as ubiquitination (Geoffroy and Hay, 2009). Like ubiquitin, proteins can be SUMOylated as mono-SUMOylation or poly-SUMOylation, but differently, poly-SUMOylation cannot lead to target degradation directly (Smits and Freire, 2016). Recent research shows that USP7 is associated with DNA synthesis (Smits and Freire, 2016). USP7 associates with an active DNA replication fork and inhibition of USP7 can reduce DNA replication. Moreover, Lecona et al. (2016) identified SUMO2 as a new USP7 substrate and demonstrated that USP7 can deubiquitinate SUMO2 in vitro and in vivo. However, the fate of SUMO2 after deubiquitination and its biological function are still unclear (Lecona et al., 2016).
LSD1
Histone lysine specific demethylase 1 (LSD1) is the first histone demethylase identified in 2004 and can remove methyl groups of histone H3K4, H3K9 (Shi et al., 2004). As reported, LSD1 can be ubiquitinated by E3 ligase JADE2 (Han et al., 2014). Since ubiquitination of LSD1 is considered as reversible process as ubiquitination and deubiquitination always exit in pair, LSD1 was identified to be deubiquitinated by USP7 and protected it from proteasome degradation (Yi et al., 2016). Besides, patients with high expression of USP7, REST, and LSD1 performed poorer outcomes in medulloblastoma (Callegari et al., 2018). And they found that p53 was a vital downstream transcription factor in the action of USP7 and LSD1.
DNA Damage and Repair
CHK1
USP7 can regulate CHK1 in three manners. The first one is the indirect regulation, USP7 deubiquitinated and prolonged the half-life of Claspin, which leaded to the sustaining phosphorylation of checkpoint kinase 1 (Chk1) in response to genotoxic stress (Faustrup et al., 2009). For the rest two manners, in DNA damage, USP7 deubiquitinates and stabilizes Chk1 via direct deubiquitination in the presence of zinc finger E-box binding homeobox 1 (ZEB1) (Zhang et al., 2014) or not (Alonso-de Vega et al., 2014), while ZEB1 binds to USP7 may result in promoting homologous recombinant-dependent DNA repair and resistant to radiation. In addition, USP7 can also directly regulate the stability of CDC25A, a Cdk-activating phosphatase as the substrate of CHK1, with the aid of brain and reproductive organ expressed protein (BRE). These results show that USP7 is an important modulator of Chk1.
CHFR
Checkpoint with Forkhead and Ring domains (CHFR), a RING family Ub-ligase, is a mitotic checkpoint that delays the transition to metaphase in response to mitotic stress. USP7 binds with CHFR in vivo and regulates its stability (Figure 6). These results indicate that USP7 may play a role in the cell cycle progression via the deubiquitination of CHFR (Oh et al., 2007).
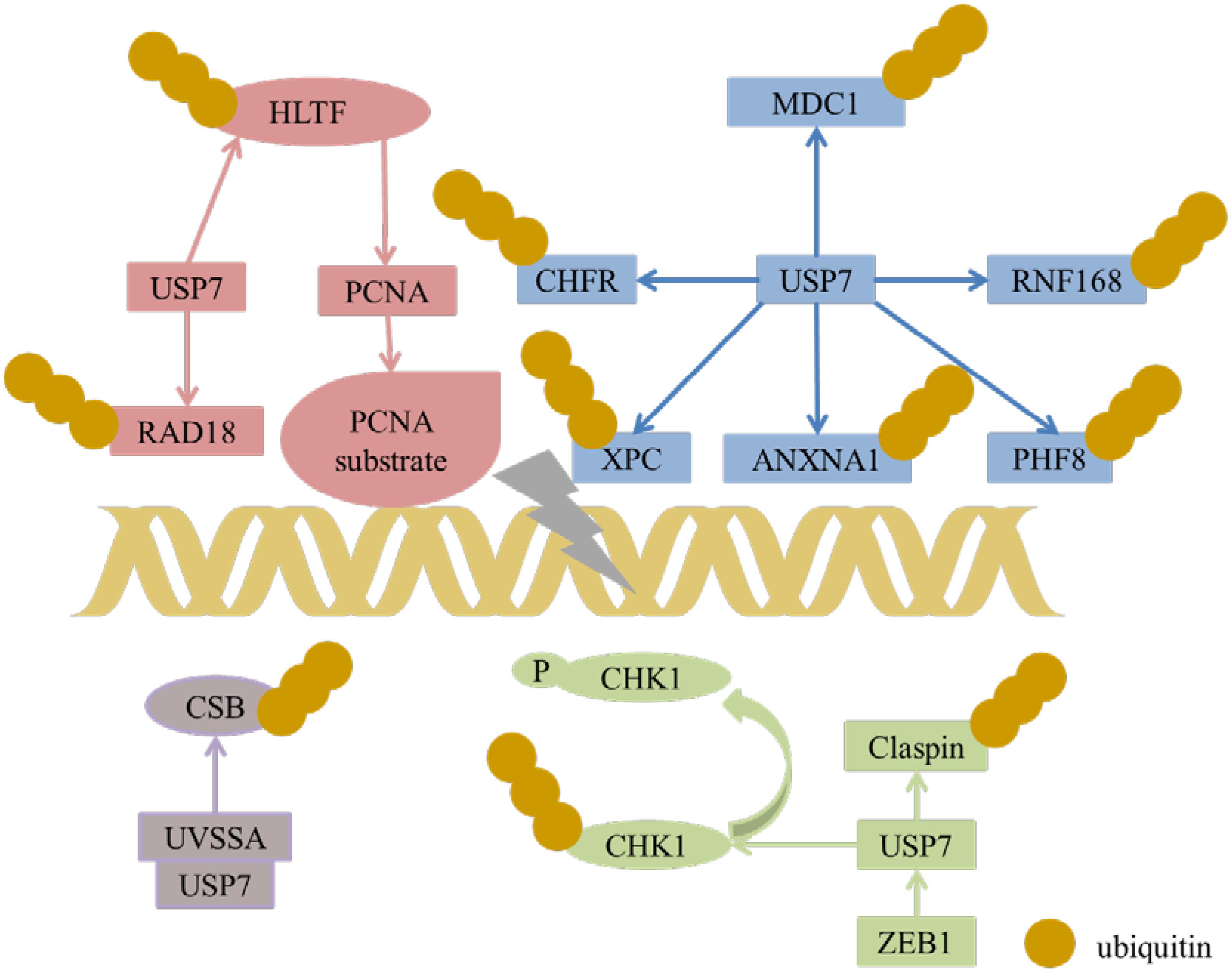
Figure 6. USP7 interacts with a number of substrates in DNA damage response.
UVSSA
Transcription-coupled nucleotide excision repair (TC-NER) removes DNA damage of actively transcribed genes. Defect in TC-NER is associated with cockayne syndrome (CS) and ultraviolet – sensitive syndrome (UVSS). Cockayne syndrome B (CSB/ERCC6) and UVSS protein are two important proteins in TC-NER. UVSSA binds with USP7 to stabilize CSB and restores the hypophosphorylated form of RNA polymerase II after UV irradiation (Figure 6) (Zhang et al., 2012). UVSSA and USP7 play roles in controlling the fate of stalled RNA polymerase II, the steady-state level of CSB, the efficiency of TC-NER and cell survival following DNA damage (Sarasin, 2012).
ANXA1
ANXA1 is a 37-kDa protein identified as the first member of the annexin superfamily. In response to DNA damage, ANXA1 is cleaved and generates the N-terminal fragment (Ac2-26) and the cleaved form of ANXA1. Both the full length of ANXA1 and Ac2-26 can be translocated to the cell membrane and induce apoptotic cell clearance through recruiting monocytes. The N-terminal of ANXA1 shares the USP7-binding motif sequences (AMVS and ALLS) and interacts with USP7. Hence, USP7 can deubiquitinate and stabilize ANXA1 (Figure 6). USP7 may participate in the DDR after UV-induced DNA damage in certain types via ANXA1 (Park et al., 2015).
XPC
Xeroderma pigmentosum complementation group C (XPC) is a critical damage recognition factor which binds to helix-distorting DNA lesions and initiates nucleotide excision repair (NER). During the early stage of NER of UV light-induced DNA lesions, XPC is ubiquitinated. Ubl1 domain (amino acids 560–644) of USP7 can bind and erase the ubiquitination on XPC and prevents XPC from proteolysis (Figure 6). Taken together, USP7 plays a vital role in regulating NER through deubiqitinating XPC (He et al., 2014).
HLTF, Rad18, and Polη
Helicase-like transcription factor (HLTF) is a double-stranded DNA translocase that can promote the polyubiquitination of proliferating cell nuclear antigen (PCNA), while Rad6–Rad18 monoubiquitinates PCNA, both of which make PCNA work as a molecular switch between various DNA damage bypass processes. On one hand, USP7 stabilizes HLTF after genotoxic stress, resulting in prolonging the half-life of HLTF, thus in turn increases polyubiquitination of PCNA (Figure 6) (Qing et al., 2011). Besides, USP7 and DNA polymerase eta (Polη), a key player in several DNA damage-tolerance pathways, interact with each other, and USP7 increases UV-induced PCNA ubiquitination through stabilizing Polη and in turn facilitates the recruitment of DNA translesion synthesis (TLS) polymerases to bypass DNA lesions. Therefore USP7 promotes monoUb-PCNA mediated stress-tolerance pathways via the stabilization of Polη. These results provide new mechanistic for USP7-related tumorigenesis and therapeutic strategy (Qian et al., 2015). On the other hand, the amino acids 110–251 of Rad18 interact with USP7 and contain two USP7-binding motifs. Loss of USP7 destabilizes Rad18 and compromises UV-induced PCNA monoubiquitylation and Polη recruitment to stalled replication forks (Zlatanou et al., 2016).
RNF168
During DDR, histone ubiquitination by RNF168 orchestrates the recruitment of downstream DDR factors, e.g., breast cancer type 1 susceptibility protein (BRCA1) and p53 binding protein 1 (53BP1). The Ubl1 domain of USP7 binds to RNF168 (Figure 6). USP7 regulates H2A monoubiquitination and H2A/X polyubiquitination via its regulation on RNF168. In summary, USP7 plays a vital role in regulation of Ub-dependent signaling in DDR via monitoring RNF168 (Malapelle et al., 2017).
PHF8
Plant homeodomain finger-containing protein 8 (PHF8) consists of an N-terminal plant homeodomain and recognizes and binds tri-methyl histone 3 lysine 4 at transcription start sites. The C-terminal region of PHF8 binds with the TRAF domain of USP7, and USP7 promotes the stability of PHF8 via deubiquitinase activity and contributes to the maintenance of genome integrity, which is implemented in DDR (Figure 6). The USP7/PHF8 is involved in breast carcinogenesis, indicating these molecules may be as potential targets for breast cancer intervention (Wang Q. et al., 2016).
MDC1
DNA damage checkpoint protein 1 (MDC1) is important for the initiation and amplification of the DDR. USP7 deubiquitinates and stabilizes of MDC1, resulting in sustaining the DDR, while depletion of USP7 influences the engagement of MRE11-RAD50-NBS1(MRN)-MDC1 complex and the recruitment of the downstream factors 53BP1 and BRCA1 at DNA lesions. USP7 promotes cervical cancer cell survival and confrere cellular resistance to genotoxic insults via the stabilization of MDC1 (Su et al., 2018).
In a nutshell, USP7 plays a vital role in the DNA damage response (Figure 6), and it can be targeted for the treatment of malignancies with DDR defects. Besides, USP7 inhibitors can be combined with genotoxic agents as a novel therapeutic strategy for the treatment of cancer.
USP7 stabilizes HLTF to result in polyubiquitination of PCNA and induces monoubiquitination of PCNA through regulating Rad18. USP7 plays a role in DDR through regulating MDC1, CHFR, XPC, ANXNA1, RNF168, and PHF8. USP7 and UVSSA interact with each other to control steady state of CSB following DNA damage. USP7 can regulate stability of CDC25A via deubiquitination CDC25A directly and through regulating Claspin and CHK1 expression.
Several Canonical Signaling Pathways
Wnt/β-Catenin Signaling Pathway
Wnt signaling was initially found for its function in cancer and embryonic development and then was found responsible for tissue regeneration in adult bone marrow, skin and intestine. β-Catenin, a key element in Wnt signaling pathway, is regulated by diverse PTMs, including ubiquitination (Ma et al., 2014). According to a research in 2017, β-catenin can be deubiquitinated and stabilized by USP7 in adenomatous polyposis coli (APC) truncating mutated colorectal cancer (CRC) but not APC wide type CRC, which resulting in the activation of Wnt pathway (Novellasdemunt et al., 2017). Mechanism study suggested that APC β-catenin inhibitory domain (CID) protects β-catenin from USP7-mediated deubiquitination, while APC lacking CID exposes β-catenin to USP7 for deubiquitination. Hence, abrogation of USP7 in APC-mutated CRC suppresses Wnt activation by regaining β-catenin ubiquitination, which leads to the cell differentiation, and inhibits tumor growth (Novellasdemunt et al., 2017). With the aid of USP7 inhibitor P5091, Wnt pathway can be inactivated by improving ubiquitination and degradation of β-catenin, which provides evidence for the rationality for developing USP7 inhibitors as anti-CRC agent (Figure 7A) (An et al., 2017). In a nutshell, USP7 can be considered as a Wnt activator for tumor-specific therapeutic target for most CRCs.
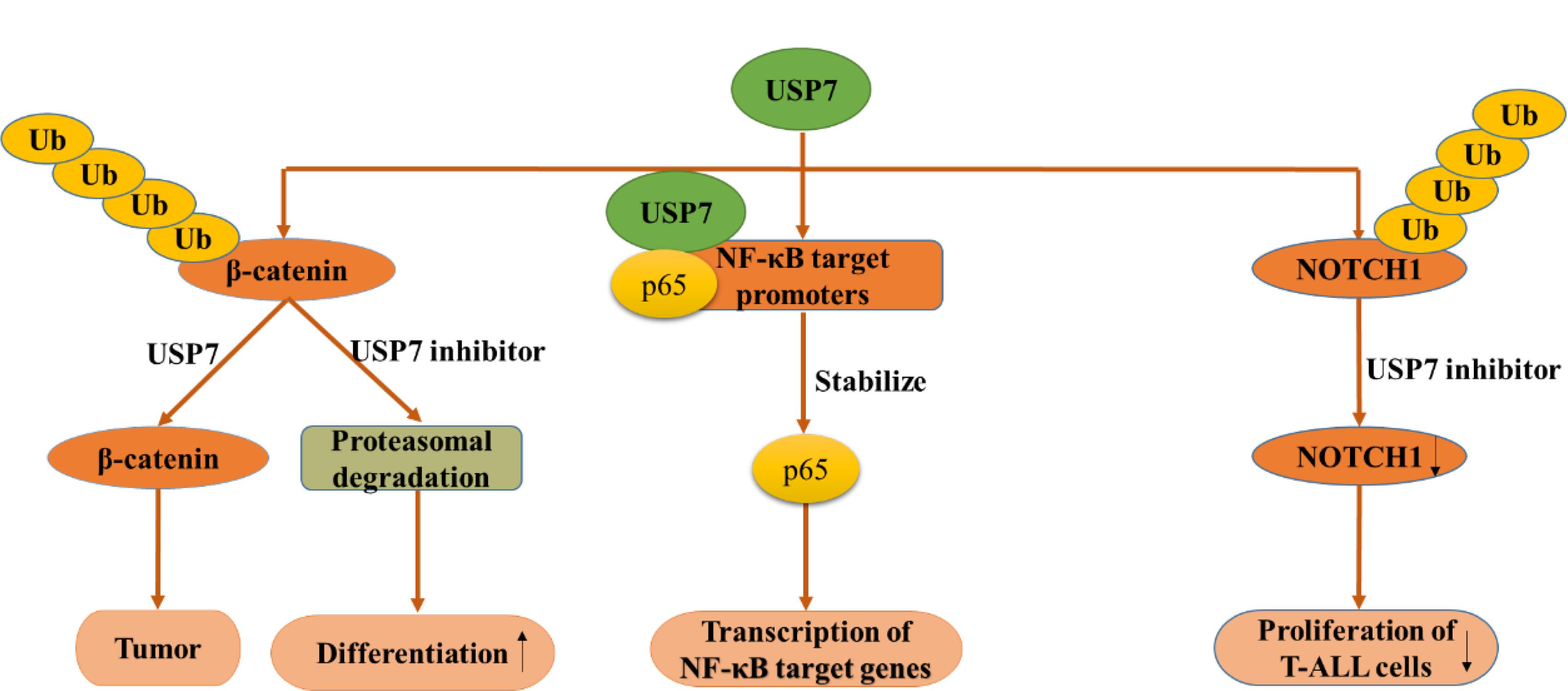
Figure 7. USP7 functions in several canonical signaling pathways. USP7 functions in Wnt/β-catenin signaling pathway (left), NF-κB signaling pathway (middle), NOTCH signaling pathway (right).
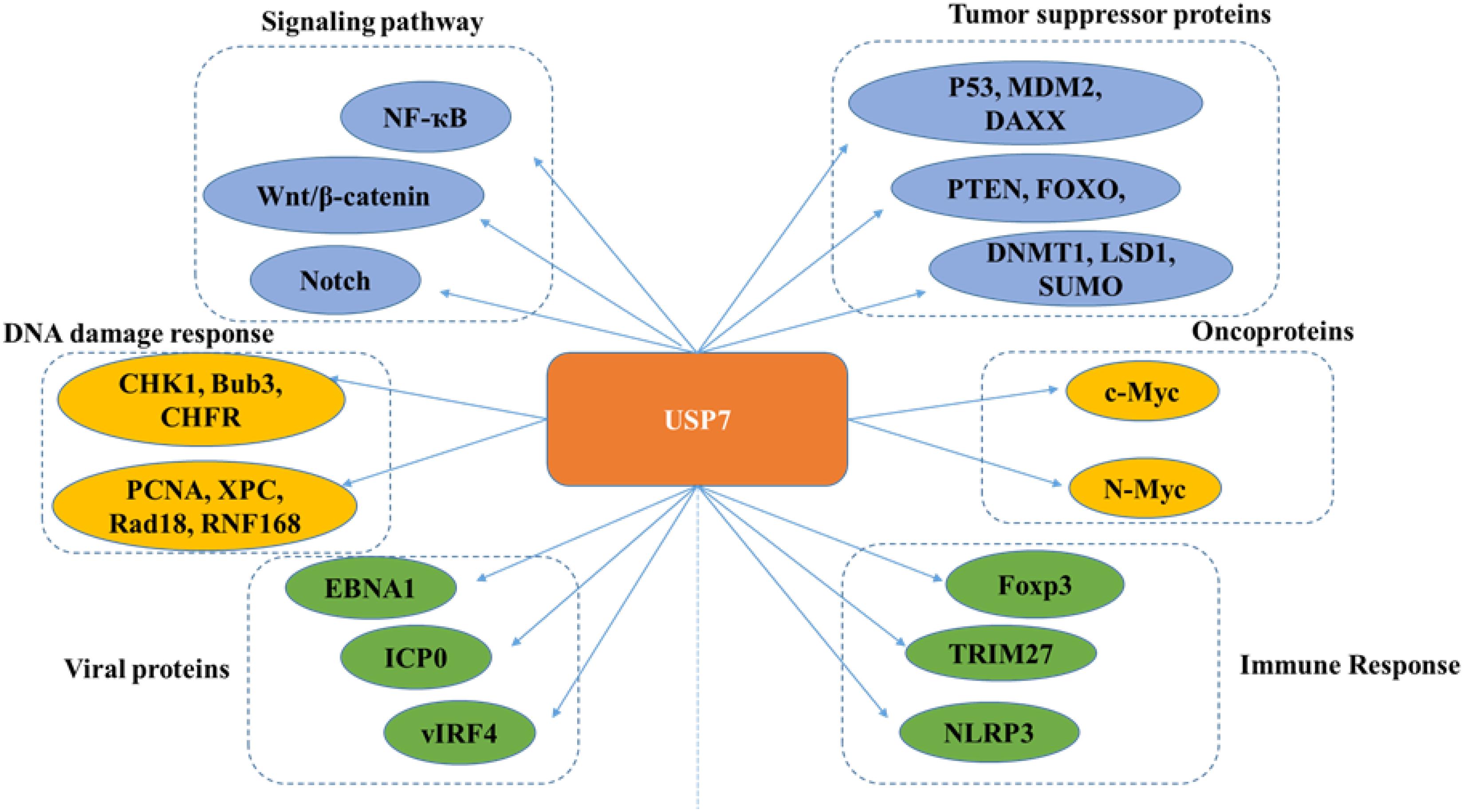
Figure 8. Overview of USP7 functions.
NF-κB Signaling Pathway
Nuclear factor kappa B (NF-κB) signaling pathway is responsible for the transcription of a series of genes that controlling inflammation and immunity. As an essential regulator of Toll-like-receptor (TLR) and tumor necrosis factor receptor (TNFR)-inducible inflammatory gene expression, NF-κB is regulated by USP7 in a research in 2013. Different from other USP7 partners and substrates, NF-κB p65 and USP7 interact together after USP7 is recruited to NF-κB target promoters. Besides, the inhibition of USP7 lead to decreased TLR and TNFR-induced expression of Interleukin (IL-6), TNFα (NF-κB reporter) indicates that the deubiquitination of NF-κB by USP7 may have therapeutic potential (Figure 7B) (Colleran et al., 2013).
In 2018, some researchers found that knockout of USP7 dramatically increased the sensitivity of multiple myeloma (MM) cells to bortezomib (BTZ) which led to myeloma cell death and inhibited NF-κB activation by stabilizing IκBα. As expected, usage of USP7 inhibitors also inhibited the activation of NF-κB and the combination of USP7 inhibitor with BTZ triggered the synergistic antitumor activity in bortezomib-resistant MM cells. Taken together, this study provides a new application for USP7 inhibitors alone or in combination with BTZ to overcome BTZ resistance and improve the patient prognosis in MM (Yao et al., 2018).
In all, several reports have illustrated the mechanism that how USP7 and its related proteins regulate NF-κB signaling pathway. However, more deep studies should be conducted to make the mechanism more clearly and logically and there are still great challenges for researchers to face.
NOTCH Signaling Pathway
Notch signaling pathway is highly conserved and presents in most multicellular organisms. This intercellular signaling cascade is involved in cell differentiation, proliferation, and contributes to the fate of cells and occurs in multiple organisms and tissues, containing early T cell development in the thymus and peripheral T cell differentiation (Auderset et al., 2012; Bailis et al., 2013; Amsen et al., 2015). There are four notch receptors in mammals possessing NOTCH1-4 in which NOTCH1 can be stabilized through USP7-mediated deubiquitination. Previous studies have revealed that ubiquitination regulates the stability, activity, and localization of NOTCH1. However, the specific deubiquitinase that affects NOTCH1 protein stability was clarified recently. Researchers reported that USP7 can deubiquitinate and stabilize NOTCH1 in vivo and in vitro, on the other hand, knockdown of USP7 increased the ubiquitination of NOTCH1. Used up of USP7 significantly restrained the proliferation of T-cell acute lymphoblastic leukemia (T-ALL) cells in vitro and in vivo, accompanied by downregulation of the NOTCH1 protein level, suggesting that targeting the USP7/NOTCH1 axis is a novel strategy to combat T-ALL and other NOTCH1-related malignancies (Figure 7C) (Shan et al., 2018). Almost at the same time, researchers found USP7 can bind several oncogenes by interacting and stabilizing NOTCH1 and JmjC Domain-Containing Protein 3 (JMJD3) in order to control leukemia growth. What’s more, USP7 and NOTCH1 bind T-ALL superenhancers, and inhibition of USP7 leads to a decrease of the transcriptional levels of NOTCH1 targets and T-ALL cell growth in vitro and in vivo. Therefore, USP7 cooperating with NOTCH1 can improve the oncogenic transcriptional program in T-ALL (Jin et al., 2018).
The functions of USP7 on different signaling pathways indicate the brand new role of USP7 as a great target. To be sure, other classical signaling pathways which may be regulated by USP7 is yet to be found. It provides us a great challenge to find the new mechanisms between USP7 and other classical signaling pathways.
USP7 in Cancer
USP7 is highly expressed in a wide variety of cancers and affects the progression of cancer diseases. Moreover, USP7 assumes different roles in different tumors. In prostate cancer, high expression of USP7 is directly related to tumor invasion (Song et al., 2008). USP7 plays a key role in carcinogenesis via p53-dependent pathways in non-small cell lung carcinoma (NSCLCs) (Masuya et al., 2006). Studies have shown that changes of USP7 regulate colon carcinoma growth and apoptotic sensitivity in vivo (Becker et al., 2008). USP7 maintains DNA damage response and promotes cervical cancer, and is positively correlated with poor survival rate in patients with cervical cancer (Su et al., 2018). USP7 regulates human terminal erythroid differentiation by stabilizing GATA1, providing a certain treatment for leukemia (Liang et al., 2019). In short, USP7 plays an important role in a variety of pathologies and is a good target from a therapeutic point of view.
USP7 Inhibitors
USP7 is a promising target not only for its roles in cellular pathways including regulators of viral proteins, immune response, oncogenes, and DNA damage but also because of its aberrant expression in various cancers. Due to lack of co-crystal structures between USP7 and small molecule inhibitors, there is no potent and selective USP7 inhibitor for a long time (Colland et al., 2009; Altun et al., 2011; Chauhan et al., 2012; Reverdy et al., 2012) (Figure 9). However, several groups reported the structures of USP7 in complex with small molecule inhibitors last year (Kategaya et al., 2017; Turnbull et al., 2017) (Figure 10) and these structures gives guidance to obtain structure-based small molecule inhibitors.
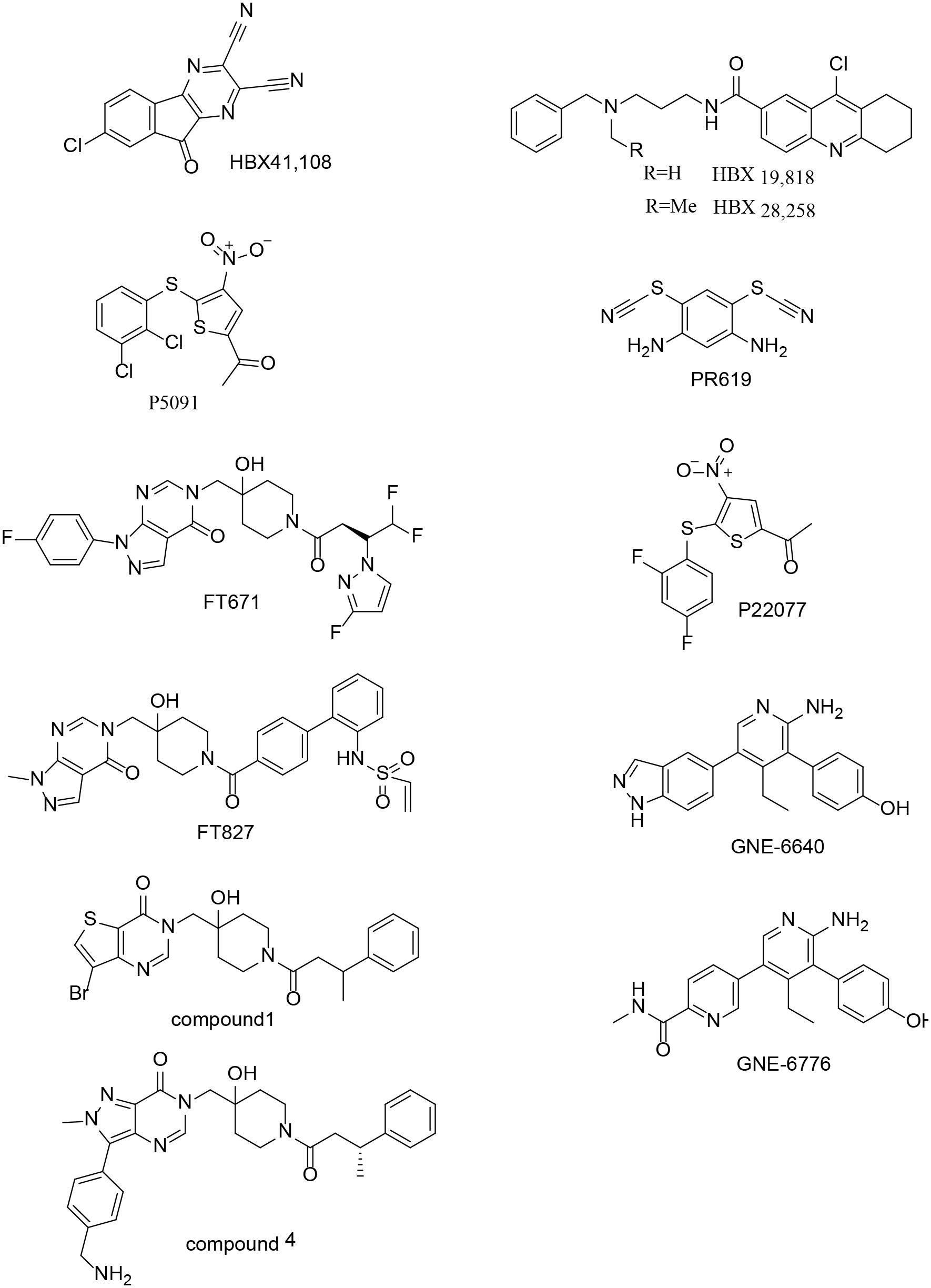
Figure 9. Chemical structures of USP7 inhibitors.
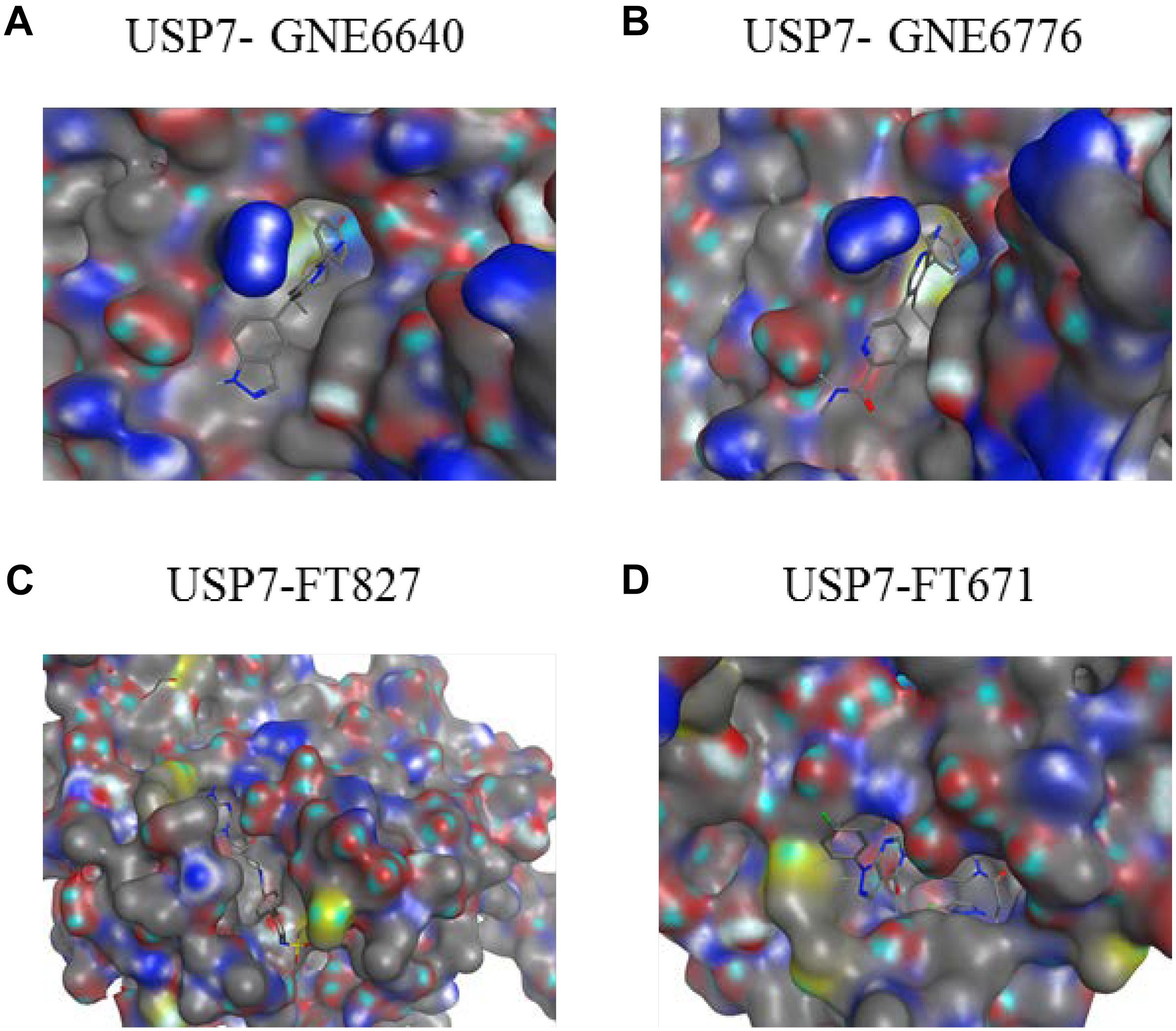
Figure 10. Co-crystal structures of USP7 in complex with inhibitors. The electrostatic surface representation of the CD of USP7 is shown along with compounds (A) USP7-GNE6640 (PDB code 5UQV). (B) USP7-GNE6776 (PDB code 5UQX). (C) USP7-FT827 (PDB code 5NGF). (D) USP7-FT671 (PDB code 5NGE). The images were generated with molecular operating environment (MOE).
Conclusion and Future Perspectives
This review illustrates our current knowledge of USP7, including its source and characterization, structure, binding partners and substrates in various biological processes. Besides, how USP7 regulates various aspects of a cell under both normal and pathological states are elaborated in detail. As the processes of ubiquitination and deubiquitination are extremely dynamic and context-specific, a series of studies have linked USP7 to different cancers. The biology, particularly the immune oncology mechanisms, reveal that USP7 inhibitors would be useful drugs, thus it is vital to develop highly selective and specific inhibitors of USP7. The association of USP7 with several canonical signaling pathways still needs characterized in order to search new targets and regulatory mechanisms. Last but not least, USP7 may be a promising target for cancer therapy and it therefore merits further studies.
Author Contributions
ZW discusses the structure of USP7, the viral protein targets of USP7, the DNA damage substrates of USP7 and conclusion. WK talks about the introduction, immune function of USP7, Wnt/β-catenin signaling pathway, and NF-κB signaling pathway. YY illustrates oncoproteins and tumor suppressor proteins. JP states about the epigenetics substrates of USP7. HR talks about the NOTCH signaling pathway. ZS and YZ provide expertise and feedback. HL provides funding.
Funding
This work was supported by National Natural Science Foundation of China (Project No. 81602961 for YZ; Project Nos. 81430085 and 81773562 for HL); Outstanding Young Talent Research Fund of Zhengzhou University (Project No. 1521331002 for YZ); National Key Research Program of Proteins (Project No. 2016YFA0501800 for HL; Project No. 2017YFD0501401 for YZ); and Key Research Program of Henan Province (Project No. 161100310100 for HL).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ali, A., Raja, R., Farooqui, S. R., Ahmad, S., and Banerjea, A. C. (2017). USP7 deubiquitinase controls HIV-1 production by stabilizing Tat protein. Biochem. J. 474, 1653–1668. doi: 10.1042/bcj20160304
Alonso-de Vega, I., Martin, Y., and Smits, V. A. (2014). USP7 controls Chk1 protein stability by direct deubiquitination. Cell Cycle 13, 3921–3926. doi: 10.4161/15384101.2014.973324
Altun, M., Kramer, H. B., Willems, L. I., McDermott, J. L., Leach, C. A., Goldenberg, S. J., et al. (2011). Activity-based chemical proteomics accelerates inhibitor development for deubiquitylating enzymes. Chem. Biol. 18, 1401–1412. doi: 10.1016/j.chembiol.2011.08.018
Amsen, D., Helbig, C., and Backer, R. A. (2015). Notch in T Cell differentiation: all things considered. Trends Immunol. 36, 802–814. doi: 10.1016/j.it.2015.10.007
An, T., Gong, Y., Li, X., Kong, L., Ma, P., Gong, L., et al. (2017). USP7 inhibitor P5091 inhibits Wnt signaling and colorectal tumor growth. Biochem. Pharmacol. 131, 29–39. doi: 10.1016/j.bcp.2017.02.011
Auderset, F., Coutaz, M., and Tacchini-Cottier, F. (2012). The role of notch in the differentiation of CD4(+) T helper cells. Curr. Top. Microbiol. Immunol. 360, 115–134. doi: 10.1007/82_2012_227
Bailis, W., Yashiro-Ohtani, Y., Fang, T. C., Hatton, R. D., Weaver, C. T., Artis, D., et al. (2013). Notch simultaneously orchestrates multiple helper T cell programs independently of cytokine signals. Immunity 39, 148–159. doi: 10.1016/j.immuni.2013.07.006
Becker, K., Marchenko, N. D., Palacios, G., and Moll, U. M. (2008). A role of HAUSP in tumor suppression in a human colon carcinoma xenograft model. Cell Cycle 7, 1205–1213. doi: 10.4161/cc.7.9.5756
Bettelli, E., Dastrange, M., and Oukka, M. (2005). Foxp3 interacts with nuclear factor of activated T cells and NF-kappa B to repress cytokine gene expression and effector functions of T helper cells. Proc. Natl. Acad. Sci. U.S.A. 102, 5138–5143. doi: 10.1073/pnas.0501675102
Bhattacharya, S., and Ghosh, M. K. (2015). HAUSP regulates c-MYC expression via de-ubiquitination of TRRAP. Cell Oncol. 38, 265–277. doi: 10.1007/s13402-015-0228-6
Blanco-Aparicio, C., Renner, O., Leal, J. F., and Carnero, A. (2007). PTEN, more than the AKT pathway. Carcinogenesis 28, 1379–1386. doi: 10.1093/carcin/bgm052
Brenkman, A. B., de Keizer, P. L., van den Broek, N. J., Jochemsen, A. G., and Burgering, B. M. (2008). Mdm2 induces mono-ubiquitination of FOXO4. PLoS One 3:e2819. doi: 10.1371/journal.pone.0002819
Bronner, C. (2011). Control of DNMT1 abundance in epigenetic inheritance by acetylation, ubiquitylation, and the histone code. Sci. Signal. 4:pe3. doi: 10.1126/scisignal.2001764
Cai, J., Chen, H. Y., Peng, S. J., Meng, J. L., Wang, Y., Zhou, Y., et al. (2018). USP7-TRIM27 axis negatively modulates antiviral type I IFN signaling. FASEB J. 32, 5238–5249. doi: 10.1096/fj.201700473RR
Callegari, K., Maegawa, S., Bravo-Alegria, J., and Gopalakrishnan, V. (2018). Pharmacological inhibition of LSD1 activity blocks REST-dependent medulloblastoma cell migration. Cell Commun. Signal. 16:60. doi: 10.1186/s12964-018-0275-5
Carra, G., Panuzzo, C., Torti, D., Parvis, G., Crivellaro, S., Familiari, U., et al. (2017). Therapeutic inhibition of USP7-PTEN network in chronic lymphocytic leukemia: a strategy to overcome TP53 mutated/deleted clones. Oncotarget 8, 35508–35522. doi: 10.18632/oncotarget.16348
Chatterjee, B., and Thakur, S. S. (2018). Investigation of post-translational modifications in type 2 diabetes. Clin. Proteomics 15:32. doi: 10.1186/s12014-018-9208-y
Chauhan, D., Tian, Z., Nicholson, B., Kumar, K. G., Zhou, B., Carrasco, R., et al. (2012). A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell 22, 345–358. doi: 10.1016/j.ccr.2012.08.007
Chavoshi, S., Egorova, O., Lacdao, I. K., Farhadi, S., Sheng, Y., and Saridakis, V. (2016). Identification of kaposi sarcoma herpesvirus (KSHV) vIRF1 protein as a novel interaction partner of human deubiquitinase USP7. J. Biol. Chem. 291, 6281–6291. doi: 10.1074/jbc.M115.710632
Cheng, J., Yang, H., Fang, J., Ma, L., Gong, R., Wang, P., et al. (2015). Molecular mechanism for USP7-mediated DNMT1 stabilization by acetylation. Nat. Commun. 11:7023. doi: 10.1038/ncomms8023
Cheon, K. W., and Baek, K. H. (2006). HAUSP as a therapeutic target for hematopoietic tumors (review). Int. J. Oncol. 28, 1209–1215.
Ching, W., Koyuncu, E., Singh, S., Arbelo-Roman, C., Hartl, B., Kremmer, E., et al. (2013). A ubiquitin-specific protease possesses a decisive role for adenovirus replication and oncogene-mediated. PLoS Pathog. 9:e1003273. doi: 10.1371/journal.ppat.1003273
Clague, M. J., Barsukov, I., Coulson, J. M., Liu, H., Rigden, D. J., and Urbe, S. (2013). Deubiquitylases from genes to organism. Physiol. Rev. 93, 1289–1315. doi: 10.1152/physrev.00002.2013
Colland, F., Formstecher, E., Jacq, X., Reverdy, C., Planquette, C., Conrath, S., et al. (2009). Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol. Cancer Ther. 8, 2286–2295. doi: 10.1158/1535-7163.mct-09-0097
Colleran, A., Collins, P. E., O’Carroll, C., Ahmed, A., Mao, X., McManus, B., et al. (2013). Deubiquitination of NF-kappaB by ubiquitin-specific protease-7 promotes transcription. Proc. Natl. Acad. Sci. U.S.A. 110, 618–623. doi: 10.1073/pnas.1208446110
Du, Z., Song, J., Wang, Y., Zhao, Y., Guda, K., Yang, S., et al. (2010). DNMT1 stability is regulated by proteins coordinating deubiquitination and acetylation-driven ubiquitination. Sci. Signal. 3:ra80. doi: 10.1126/scisignal.2001462
Dybas, J. M., Herrmann, C., and Weitzman, M. D. (2018). Ubiquitination at the interface of tumor viruses and DNA damage responses. Curr. Opin. Virol. 32, 40–47. doi: 10.1016/j.coviro.2018.08.017
Everett, R. D. (2014). HSV-1 biology and life cycle. Methods Mol. Biol. 1444, 1–17. doi: 10.1007/978-1-4939-0428-0_1
Everett, R. D., Meredith, M., Orr, A., Cross, A., Kathoria, M., and Parkinson, J. (1997). A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. EMBO J. 16, 1519–1530. doi: 10.1093/emboj/16.7.1519
Faesen, A. C., Dirac, A. M., Shanmugham, A., Ovaa, H., Perrakis, A., and Sixma, T. K. (2011). Mechanism of USP7/HAUSP activation by its C-terminal ubiquitin-like domain and allosteric regulation by GMP-synthetase. Mol. Cell 44, 147–159. doi: 10.1016/j.molcel.2011.06.034
Faustrup, H., Bekker-Jensen, S., Bartek, J., Lukas, J., and Mailand, N. (2009). USP7 counteracts SCFbetaTrCP- but not APCCdh1-mediated proteolysis of Claspin. J. Cell Biol. 184, 13–19. doi: 10.1083/jcb.200807137
Fernandez-Montalvan, A., Bouwmeester, T., Joberty, G., Mader, R., Mahnke, M., Pierrat, B., et al. (2007). Biochemical characterization of USP7 reveals post-translational modification sites and structural requirements for substrate processing and subcellular localization. FEBS J. 274, 4256–4270. doi: 10.1111/j.1742-4658.2007.05952.x
Freeman, D. J., Li, A. G., Wei, G., Li, H. H., Kertesz, N., Lesche, R., et al. (2003). PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell 3, 117–130. doi: 10.1016/s1535-6108(03)00021-7
Geoffroy, M. C., and Hay, R. T. (2009). An additional role for SUMO in ubiquitin-mediated proteolysis. Nat. Rev. Mol. Cell Biol. 10, 564–568. doi: 10.1038/nrm2707
Han, X., Gui, B., Xiong, C., Zhao, L., Liang, J., Sun, L., et al. (2014). Destabilizing LSD1 by Jade-2 promotes neurogenesis: an antibraking system in neural development. Mol. Cell 55, 482–494. doi: 10.1016/j.molcel.2014.06.006
He, J., Zhu, Q., Wani, G., Sharma, N., Han, C., Qian, J., et al. (2014). Ubiquitin-specific protease 7 regulates nucleotide excision repair through deubiquitinating XPC protein and preventing XPC protein from undergoing ultraviolet light-induced and VCP/p97 protein-regulated proteolysis. J. Biol. Chem. 289, 27278–27289. doi: 10.1074/jbc.M114.589812
Holowaty, M. N., Zeghouf, M., Wu, H., Tellam, J., Athanasopoulos, V., Greenblatt, J., et al. (2003). Protein profiling with epstein-barr nuclear antigen-1 reveals an interaction with the herpesvirus-associated ubiquitin-specific protease HAUSP/USP7. J. Biol. Chem. 278, 29987–29994. doi: 10.1074/jbc.M303977200
Hu, M., Gu, L., Li, M., Jeffrey, P. D., Gu, W., and Shi, Y. (2006). Structural basis of competitive recognition of p53 and MDM2 by HAUSP/USP7: implications for the regulation of the p53-MDM2 pathway. PLoS Biol. 4:e27. doi: 10.1371/journal.pbio.0040027
Hu, M., Li, P., Li, M., Li, W., Yao, T., Wu, J. W., et al. (2002). Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell 111, 1041–1054. doi: 10.1016/s0092-8674(02)01199-6
Huang, H., Regan, K. M., Wang, F., Wang, D., Smith, D. I., van Deursen, J. M., et al. (2005). Skp2 inhibits FOXO1 in tumor suppression through ubiquitin-mediated degradation. Proc. Natl. Acad. Sci. U.S.A. 102, 1649–1654. doi: 10.1073/pnas.0406789102
Jager, W., Santag, S., Weidner-Glunde, M., Gellermann, E., Kati, S., Pietrek, M., et al. (2012). The ubiquitin-specific protease USP7 modulates the replication of Kaposi’s sarcoma-associated herpesvirus latent episomal DNA. J. Virol. 86, 6745–6757. doi: 10.1128/jvi.06840-11
Jin, Q., Martinez, C. A., Arcipowski, K. M., Zhu, Y., Gutierrez-Diaz, B. T., Wang, K. K., et al. (2018). USP7 cooperates with NOTCH1 to drive the oncogenic transcriptional program in T cell leukemia. Clin. Cancer Res. 25, 222–239. doi: 10.1158/1078-0432.CCR-18-1740
Kalamvoki, M., Gu, H., and Roizman, B. (2012). Overexpression of the ubiquitin-specific protease 7 resulting from transfection or mutations in the ICP0 binding site accelerates rather than depresses herpes simplex virus 1 gene expression. J. Virol. 86, 12871–12878. doi: 10.1128/jvi.01981-12
Kategaya, L., Di Lello, P., Rouge, L., Pastor, R., Clark, K. R., Drummond, J., et al. (2017). USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature 550, 534–538. doi: 10.1038/nature24006
Khosravi, M., Bidmeshkipour, A., Cohen, J. L., Moravej, A., Hojjat-Assari, S., Naserian, S., et al. (2018). Induction of CD4(+)CD25(+)FOXP3(+) regulatory T cells by mesenchymal stem cells is associated with modulation of ubiquitination factors and TSDR demethylation. Stem Cell Res. Ther. 9:273. doi: 10.1186/s13287-018-0991-1
Kim, R. Q., and Sixma, T. K. (2017). Regulation of USP7: a high incidence of E3 complexes. J. Mol. Biol. 429, 3395–3408. doi: 10.1016/j.jmb.2017.05.028
Kumar, S., Brown, A., and Tchounwou, P. B. (2018). Trisenox disrupts MDM2-DAXX-HAUSP complex and activates p53, cell cycle regulation and apoptosis in acute leukemia cells. Oncotarget 9, 33138–33148. doi: 10.18632/oncotarget.26025
Laurence, A., Belkaid, Y., and O’Shea, J. J. (2013). A degrading view of regulatory T cells. Immunity 39, 201–203. doi: 10.1016/j.immuni.2013.08.017
Lecona, E., Rodriguez-Acebes, S., Specks, J., Lopez-Contreras, A. J., Ruppen, I., Murga, M., et al. (2016). USP7 is a SUMO deubiquitinase essential for DNA replication. Nat. Struct. Mol. Biol. 23, 270–277. doi: 10.1038/nsmb.3185
Lee, H. R., Choi, W. C., Lee, S., Hwang, J., Hwang, E., Guchhait, K., et al. (2011). Bilateral inhibition of HAUSP deubiquitinase by a viral interferon regulatory factor protein. Nat. Struct. Mol. Biol. 18, 1336–1344. doi: 10.1038/nsmb.2142
Levine, A. J. (1997). p53, the cellular gatekeeper for growth and division. Cell 88, 323–331. doi: 10.1016/s0092-8674(00)81871-1
Li, M., Brooks, C. L., Kon, N., and Gu, W. (2004). A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol. Cell 13, 879–886. doi: 10.1016/s1097-2765(04)00157-1
Li, M., Chen, D., Shiloh, A., Luo, J., Nikolaev, A. Y., Qin, J., et al. (2002). Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 416, 648–653. doi: 10.1038/nature737
Liang, L., Peng, Y., Zhang, J., Zhang, Y., Roy, M., Han, X., et al. (2019). Deubiquitylase USP7 regulates human terminal erythroid differentiation by stabilizing GATA1. Haematologica
Lindsay, C. R., Morozov, V. M., and Ishov, A. M. (2008). PML NBs (ND10) and Daxx: from nuclear structure to protein function. Front. Biosci. 13, 7132–7142. doi: 10.2741/3216
Lu, Y., Orr, A., and Everett, R. D. (2016). Stimulation of the replication of ICP0-null mutant herpes simplex virus 1 and pp71-deficient human cytomegalovirus by epstein-barr virus tegument protein BNRF1. J. Virol. 90, 9664–9673. doi: 10.1128/jvi.01224-16
Ma, J., Martin, J. D., Xue, Y., Lor, L. A., Kennedy-Wilson, K. M., Sinnamon, R. H., et al. (2010). C-terminal region of USP7/HAUSP is critical for deubiquitination activity and contains a second mdm2/p53 binding site. Arch. Biochem. Biophys. 503, 207–212. doi: 10.1016/j.abb.2010.08.020
Ma, P., Yang, X., Kong, Q., Li, C., Yang, S., Li, Y., et al. (2014). The ubiquitin ligase RNF220 enhances canonical Wnt signaling through USP7-mediated deubiquitination of beta-catenin. Mol. Cell Biol. 34, 4355–4366. doi: 10.1128/mcb.00731-14
Malapelle, U., Morra, F., Ilardi, G., Visconti, R., Merolla, F., Cerrato, A., et al. (2017). USP7 inhibitors, downregulating CCDC6, sensitize lung neuroendocrine cancer cells to PARP-inhibitor drugs. Lung Cancer 107, 41–49. doi: 10.1016/j.lungcan.2016.06.015
Masuya, D., Huang, C., Liu, D., Nakashima, T., Yokomise, H., Ueno, M., et al. (2006). The HAUSP gene plays an important role in non-small cell lung carcinogenesis through p53-dependent pathways. J. Pathol. 208, 724–732. doi: 10.1002/path.1931
Morotti, A., Panuzzo, C., Crivellaro, S., Pergolizzi, B., Familiari, U., Berger, A. H., et al. (2014). BCR-ABL disrupts PTEN nuclear-cytoplasmic shuttling through phosphorylation-dependent activation of HAUSP. Leukemia 28, 1326–1333. doi: 10.1038/leu.2013.370
Nijman, S. M., Luna-Vargas, M. P., Velds, A., Brummelkamp, T. R., Dirac, A. M., Sixma, T. K., et al. (2005). genomic and functional inventory of deubiquitinating enzymes. Cell 123, 773–786. doi: 10.1016/j.cell.2005.11.007
Novellasdemunt, L., Foglizzo, V., Cuadrado, L., Antas, P., Kucharska, A., Encheva, V., et al. (2017). USP7 Is a tumor-specific WNT activator for APC-mutated colorectal cancer by mediating beta-catenin deubiquitination. Cell Rep. 21, 612–627. doi: 10.1016/j.celrep.2017.09.072
Oh, Y. M., Yoo, S. J., and Seol, J. H. (2007). Deubiquitination of Chfr, a checkpoint protein, by USP7/HAUSP regulates its stability and activity. Biochem. Biophys. Res. Commun. 357, 615–619. doi: 10.1016/j.bbrc.2007.03.193
Oliner, J. D., Kinzler, K. W., Meltzer, P. S., George, D. L., and Vogelstein, B. (1992). Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 358, 80–83. doi: 10.1038/358080a0
Ozen, A., Rouge, L., Bashore, C., Hearn, B. R., Skelton, N. J., and Dueber, E. C. (2018). Selectively modulating conformational states of USP7 catalytic domain for activation. Structure 26, 72–84.e7. doi: 10.1016/j.str.2017.11.010
Palazon-Riquelme, P., Worboys, J. D., Green, J., Valera, A., Martin-Sanchez, F., Pellegrini, C., et al. (2018). USP7 and USP47 deubiquitinases regulate NLRP3 inflammasome activation. EMBO Rep. 19:e44766. doi: 10.15252/embr.201744766
Park, J. J., Lim, K. H., and Baek, K. H. (2015). Annexin-1 regulated by HAUSP is essential for UV-induced damage response. Cell Death Dis. 2015:e1654. doi: 10.1038/cddis.2015.32
Pfoh, R., Lacdao, I. K., Georges, A. A., Capar, A., Zheng, H., Frappier, L., et al. (2015). Crystal structure of USP7 ubiquitin-like domains with an ICP0 peptide reveals a novel mechanism used by viral and cellular proteins to target. PLoS Pathog. 11:e1004950. doi: 10.1371/journal.ppat.1004950
Qian, J., Pentz, K., Zhu, Q., Wang, Q., He, J., Srivastava, A. K., et al. (2015). USP7 modulates UV-induced PCNA monoubiquitination by regulating DNA polymerase eta stability. Oncogene 34, 4791–4796. doi: 10.1038/onc.2014.394
Qing, P., Han, L., Bin, L., Yan, L., and Ping, W. X. (2011). USP7 regulates the stability and function of HLTF through deubiquitination. J. Cell Biochem. 112, 3856–3862. doi: 10.1002/jcb.23317
Reverdy, C., Conrath, S., Lopez, R., Planquette, C., Atmanene, C., Collura, V., et al. (2012). Discovery of specific inhibitors of human USP7/HAUSP deubiquitinating enzyme. Chem. Biol. 19, 467–477. doi: 10.1016/j.chembiol.2012.02.007
Rouge, L., Bainbridge, T. W., Kwok, M., Tong, R., Di Lello, P., Wertz, I. E., et al. (2016). Molecular understanding of USP7 substrate recognition and C-terminal activation. Structure 24, 1335–1345. doi: 10.1016/j.str.2016.05.020
Sarasin, A. (2012). UVSSA and USP7: new players regulating transcription-coupled nucleotide excision repair in human cells. Genome Med. 4:44. doi: 10.1186/gm343
Saridakis, V., Sheng, Y., Sarkari, F., Holowaty, M. N., Shire, K., Nguyen, T., et al. (2005). Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein-Barr nuclear antigen 1 implications for EBV-mediated immortalization. ESE Mol. Cell 18, 25–36. doi: 10.1016/j.molcel.2005.02.029
Shan, H., Li, X., Xiao, X., Dai, Y., Huang, J., Song, J., et al. (2018). USP7 deubiquitinates and stabilizes NOTCH1 in T-cell acute lymphoblastic leukemia. Signal. Transduct. Target. Ther. 3:29. doi: 10.1038/s41392-018-0028-3
Sheng, Y., Saridakis, V., Sarkari, F., Duan, S., Wu, T., Arrowsmith, C. H., et al. (2006). Molecular recognition of p53 and MDM2 by USP7/HAUSP. Nat. Struct. Mol. Biol. 13, 285–291. doi: 10.1038/nsmb1067
Shi, Y., Lan, F., Matson, C., Mulligan, P., Whetstine, J. R., Cole, P. A., et al. (2004). Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953. doi: 10.1016/j.cell.2004.12.012
Smits, V. A., and Freire, R. (2016). USP7/HAUSP: a SUMO deubiquitinase at the heart of DNA replication. Bioessays 38, 863–868. doi: 10.1002/bies.201600096
Song, M. S., Salmena, L., Carracedo, A., Egia, A., Lo-Coco, F., Teruya-Feldstein, J., et al. (2008). The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature 455, 813–817. doi: 10.1038/nature07290
Su, D., Ma, S., Shan, L., Wang, Y., Wang, Y., Cao, C., et al. (2018). Ubiquitin-specific protease 7 sustains DNA damage response and promotes cervical carcinogenesis. J. Clin. Invest. 128, 4280–4296. doi: 10.1172/jci120518
Tang, J., Qu, L. K., Zhang, J., Wang, W., Michaelson, J. S., Degenhardt, Y. Y., et al. (2006). Critical role for Daxx in regulating Mdm2. Nat. Cell Biol. 8, 855–862. doi: 10.1038/ncb1442
Tavana, O., Li, D., Dai, C., Lopez, G., Banerjee, D., Kon, N., et al. (2016). HAUSP deubiquitinates and stabilizes N-Myc in neuroblastoma. Nat. Med. 22, 1180–1186. doi: 10.1038/nm.4180
Trotman, L. C., Wang, X., Alimonti, A., Chen, Z., Teruya-Feldstein, J., Yang, H., et al. (2007). Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 128, 141–156. doi: 10.1016/j.cell.2006.11.040
Turnbull, A. P., Ioannidis, S., Krajewski, W. W., Pinto-Fernandez, A., Heride, C., Martin, A. C. L., et al. (2017). Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature 550, 481–486. doi: 10.1038/nature24451
van der Horst, A., and Burgering, B. M. (2007). Stressing the role of FoxO proteins in lifespan and disease. Nat. Rev. Mol. Cell Biol. 8, 440–450. doi: 10.1038/nrm2190
van der Horst, A., de Vries-Smits, A. M., Brenkman, A. B., van Triest, M. H., van den Broek, N., Colland, F., et al. (2006). FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat. Cell Biol. 8, 1064–1073. doi: 10.1038/ncb1469
van Loosdregt, J., and Coffer, P. J. (2014). Post-translational modification networks regulating FOXP3 function. Trends Immunol. 35, 368–378. doi: 10.1016/j.it.2014.06.005
van Loosdregt, J., Fleskens, V., Fu, J., Brenkman, A. B., Bekker, C. P., Pals, C. E., et al. (2013). Stabilization of the transcription factor Foxp3 by the deubiquitinase USP7 increases Treg-cell-suppressive capacity. Immunity 39, 259–271. doi: 10.1016/j.immuni.2013.05.018
Vassilev, L. T., Vu, B. T., Graves, B., Carvajal, D., Podlaski, F., Filipovic, Z., et al. (2004). In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848. doi: 10.1126/science.1092472
Wade, M., Li, Y. C., and Wahl, G. M. (2013). MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 13, 83–96. doi: 10.1038/nrc3430
Wang, L., Kumar, S., Dahiya, S., Wang, F., Wu, J., Newick, K., et al. (2016). Ubiquitin-specific protease-7 inhibition impairs Tip60-dependent Foxp3+ T-regulatory Cell function and promotes antitumor immunity. EBioMedicine 13, 99–112. doi: 10.1016/j.ebiom.2016.10.018
Wang, Q., Ma, S., Song, N., Li, X., Liu, L., Yang, S., et al. (2016). Stabilization of histone demethylase PHF8 by USP7 promotes breast carcinogenesis. J. Clin. Invest. 126, 2205–2220. doi: 10.1172/jci85747
Xiang, Q., Ju, H., Li, Q., Mei, S. C., Chen, D., and Choi, Y. B. (2018). Human herpesvirus 8 interferon regulatory factors 1 and 3 mediate replication and latency activities via interactions with USP7 Deubiquitinase. J. Virol. 92:e2003–17. doi: 10.1128/jvi.02003-17
Yao, Y., Zhang, Y., Shi, M., Sun, Y., Chen, C., Niu, M., et al. (2018). Blockade of deubiquitinase USP7 overcomes bortezomib resistance by suppressing NF-kappaB signaling pathway in multiple myeloma. J. Leukoc. Biol. 104, 1105–1115. doi: 10.1002/JLB.2A1017-420RR
Yi, L., Cui, Y., Xu, Q., and Jiang, Y. (2016). Stabilization of LSD1 by deubiquitinating enzyme USP7 promotes glioblastoma cell tumorigenesis and metastasis through suppression of the p53 signaling pathway. Oncol. Rep. 36, 2935–2945. doi: 10.3892/or.2016.5099
Zaman, M. M., Nomura, T., Takagi, T., Okamura, T., Jin, W., Shinagawa, T., et al. (2013). Ubiquitination-deubiquitination by the TRIM27-USP7 complex regulates tumor necrosis factor alpha-induced apoptosis. Mol. Cell Biol. 33, 4971–4984. doi: 10.1128/mcb.00465-13
Zhang, P., Wei, Y., Wang, L., Debeb, B. G., Yuan, Y., Zhang, J., et al. (2014). ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat. Cell Biol. 16, 864–875. doi: 10.1038/ncb3013
Zhang, X., Gu, L., Li, J., Shah, N., He, J., Yang, L., et al. (2010). Degradation of MDM2 by the interaction between berberine and DAXX leads to potent apoptosis in MDM2-overexpressing cancer cells. Cancer Res. 70, 9895–9904. doi: 10.1158/0008-5472.can-10-1546
Zhang, X., Horibata, K., Saijo, M., Ishigami, C., Ukai, A., Kanno, S., et al. (2012). Mutations in UVSSA cause UV-sensitive syndrome and destabilize ERCC6 in transcription-coupled DNA repair. Nat. Genet. 44, 593–597. doi: 10.1038/ng.2228
Zhou, J., Wang, J., Chen, C., Yuan, H., Wen, X., and Sun, H. (2018). USP7: target validation and drug discovery for cancer therapy. Med. Chem. 14, 3–18. doi: 10.2174/1573406413666171020115539
Zhu, X., Menard, R., and Sulea, T. (2007). High incidence of ubiquitin-like domains in human ubiquitin-specific proteases. Proteins 69, 1–7. doi: 10.1002/prot.21546
Keywords: deubiquitination, USP7, structure, immune, DNA damage
Citation: Wang Z, Kang W, You Y, Pang J, Ren H, Suo Z, Liu H and Zheng Y (2019) USP7: Novel Drug Target in Cancer Therapy. Front. Pharmacol. 10:427. doi: 10.3389/fphar.2019.00427
Received: 28 January 2019; Accepted: 04 April 2019;
Published: 30 April 2019.
Edited by:
Zhe-Sheng Chen, St. John’s University, United StatesReviewed by:
Hong-Yu Hu, Shanghai Institutes for Biological Sciences (CAS), ChinaXiaoming Zha, China Pharmaceutical University, China
Copyright © 2019 Wang, Kang, You, Pang, Ren, Suo, Liu and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiru Wang, c2ltcGxlYXJ1QDE2My5jb20= Zhenhe Suo, emhlbmhlLnN1b0BtZWRpc2luLnVpby5ubw== Hongmin Liu, bGl1aG1Aenp1LmVkdS5jbg== Yichao Zheng, eWljaGFvemhlbmdAenp1LmVkdS5jbg==