 Kendall L. Mores
Kendall L. Mores Benjamin R. Cummins2†
Benjamin R. Cummins2† Richard M. van Rijn
Richard M. van Rijn
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 17 April 2019
Sec. Neuropharmacology
Volume 10 - 2019 | https://doi.org/10.3389/fphar.2019.00407
Between 2000 and 2005 several studies revealed that morphine is more potent and exhibits fewer side effects in beta-arrestin 2 knockout mice. These findings spurred efforts to develop opioids that signal primarily via G protein activation and do not, or only very weakly, recruit beta-arrestin. Development of such molecules targeting the mu opioid receptor initially outpaced those targeting the kappa, delta and nociceptin opioid receptors, with the G protein-biased mu opioid agonist oliceridine/TRV130 having completed phase III clinical trials with improved therapeutic window to treat moderate-to-severe acute pain. Recently however, there has been a sharp increase in the development of novel G protein-biased kappa agonists. It is hypothesized that G protein-biased kappa agonists can reduce pain and itch, but exhibit fewer side effects, such as anhedonia and psychosis, that have thus far limited the clinical development of unbiased kappa opioid agonists. Here we summarize recently discovered G protein-biased kappa agonists, comparing structures, degree of signal bias and preclinical effects. We specifically reviewed nalfurafine, 22-thiocyanatosalvinorin A (RB-64), mesyl-salvinorin B, 2-(4-(furan-2-ylmethyl)-5-((4-methyl-3-(trifluoromethyl)benzyl)thio)-4H-1,2,4-triazol-3-yl)pyridine (triazole 1.1), 3-(2-((cyclopropylmethyl)(phenethyl)amino)ethyl)phenol (HS666), N-n-butyl-N-phenylethyl-N-3-hydroxyphenylethyl-amine (compound 5/BPHA), 6-guanidinonaltrindole (6′GNTI), and collybolide. These agonists encompass a variety of chemical scaffolds and range in both their potency and efficacy in terms of G protein signaling and beta-arrestin recruitment. Thus unsurprisingly, the behavioral responses reported for these agonists are not uniform. Yet, it is our conclusion that the kappa opioid field will benefit tremendously from future studies that compare several biased agonists and correlate the degree of signaling bias to a particular pharmacological response.
The majority of clinically used opioids selectively target the μ opioid receptor (μOR). Their use however, particularly in patients with chronic pain disorders, is complicated by side effects including opioid dependence, tolerance, constipation, itch and respiratory depression (Chou et al., 2009). The beginning of the 21st century saw the emergence of the hypothesis that the side effect profile of μOR based drugs may be attributed to β-arrestin 2 signaling, as preclinical studies showed that mice lacking this protein displayed reduced morphine tolerance and respiratory depression (Bohn et al., 1999, 2000; Raehal et al., 2005). Despite morphine being already a relatively low efficacious β-arrestin 2 recruiter (Whistler and von Zastrow, 1998), the β-arrestin 2 KO mice studies were the driving factor for the development of so-called G protein-biased μOR agonists that preferentially signaled via the canonical G protein pathway, while further minimizing β-arrestin 2 recruitment and signaling. Such signal-biased opioids like TRV130 (Chen et al., 2013) and PZM21 indeed appeared to have improved therapeutic windows (Soergel et al., 2014; Manglik et al., 2016), and TRV130 advanced through all three clinical trial phases under the brand-name Olinvo®(oliceridine) for the treatment of moderate-to-severe pain via intravenous injection for example following abdominoplasty (Singla et al., 2017). However, recent preclinical studies have sowed doubt regarding the potential for these G protein-biased μOR agonists to reduce side effects like constipation, respiratory depression and dependence (Altarifi et al., 2017; Austin Zamarripa et al., 2018; Hill et al., 2018; Kliewer et al., 2019). Moreover, in October of 2018, the Food and Drug Administration (FDA) decided on a 8–7 vote not to approve Olinvo®, as the committee still had doubts as to whether the benefits associated with the drug outweighed the risks.
The μOR is not the only opioid receptor modulating nociceptive transmission; the κ-opioid receptor (κOR) is a ubiquitously expressed G protein-coupled receptor (GPCR) whose signaling is involved in a wide range of biological processes, including nociception, stress, anxiety, depression, and substance use disorder (Al-Hasani and Bruchas, 2011; Chavkin, 2011; Bruchas and Roth, 2016). Whereas, μOR agonists like morphine are known to induce itch, κOR/dynorphin system has been linked with reducing pruritis as potential therapeutic action (Kardon et al., 2014; Cowan et al., 2015). However, compared to μOR and δ opioid receptor (δOR), the κOR/dynorphin system is more heavily associated with negative affect and stress responses of drug use (Chavkin and Koob, 2016). Therefore from a drug development point of view antagonism of κOR has received most attention, with κOR antagonists relieving depression-like and anxiety-like behaviors, attenuate stress responses and alcohol and cocaine use (Butelman et al., 2012; Walker et al., 2012; Karkhanis et al., 2017). Negative affect is an important factor in chronic pain management and the amygdala plays an important role in the circuitry associated with negative affect (Corder et al., 2019). Like μOR, activation of κOR produces analgesia, however the κOR/dynorphin system is heavily present in the amygdala (Land et al., 2008; Knoll et al., 2011; Kissler et al., 2014; Crowley et al., 2016). Thus there is a therapeutic promise for utilizing κORs in chronic pain settings, yet this requires producing κOR agonists with optimized pharmacological properties to ensure the drug produces analgesia, but are capable of mitigating the negative affect. Currently, the therapeutic potential of κOR agonists is limited by negative side effects they can produce, which include sedation, motor incoordination and dysphoria (or aversion in rodents) and psychotomimesis, the latter two effects being specific to κOR (Pfeiffer et al., 1986; Dykstra et al., 1987; Roth et al., 2002; Land et al., 2009) (Figure 1). The FDA has approved several non-selective opioids that target both the μOR and the κOR. However, these drugs act either as partial agonists (nalbuphine, nalmefene, pentazocine, butorphanol) or antagonists (buprenorphine) at the κOR, thus largely avoiding the side effects associated with strong κOR activation. Yet beyond partial agonism, an additional strategy may include biasing the κOR agonists signaling to a specific downstream pathway.
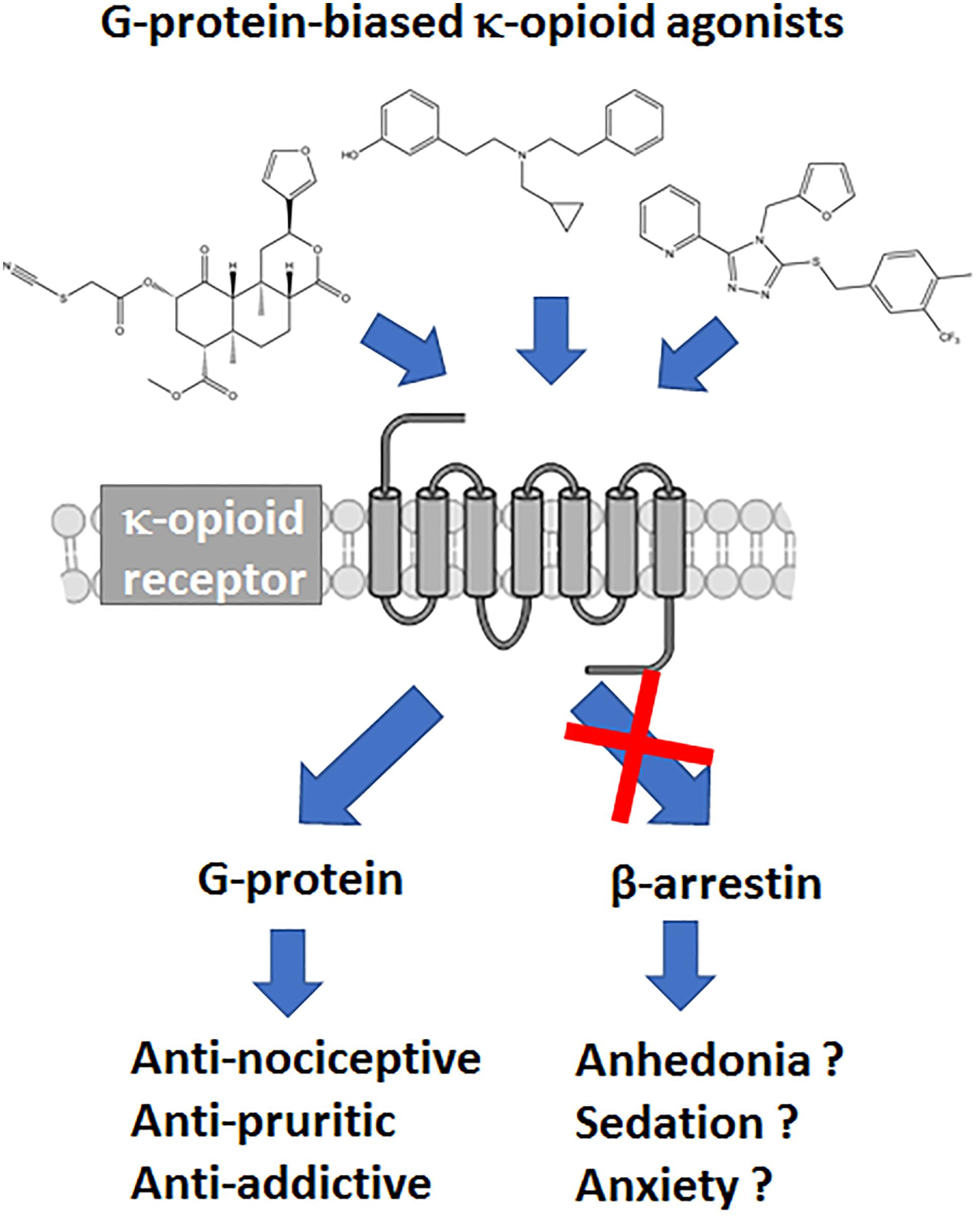
Figure 1. Hypothesized benefits of G protein-biased κ opioid receptor agonists.
Similar to studies of μOR signaling bias, studies investigating κOR signaling have indicated that some of the negative side effects, such as aversion, could be mediated by β-arrestin 2 (Bruchas and Chavkin, 2010). Specifically, Bruchas et al. (2007), first revealed that U50,488 induced aversion requires p38 activation, which largely depends on G protein receptor kinase 3, which has been linked to β-arrestin 2 recruitment (Bruchas et al., 2006). In a follow up study, mice virally expressing the S369A κOR mutant, which does not get phosphorylated by G protein receptor kinase 3, in the dorsal raphe nucleus neurons projecting to the nucleus accumbens did not show U50,488 conditioned place aversion (CPA) (Land et al., 2009). Importantly, both antinociceptive and anti-pruritic efficacy of κOR agonists are retained in β-arrestin 2 knockout mice (Morgenweck et al., 2015; White et al., 2015) suggesting that G protein signaling is key for those beneficial effects. To harness the therapeutic potential of κOR activation, there has been an escalated search for κOR agonists which favor G protein signaling over β-arrestin recruitment. In this review, we provide a summary of the cellular bias and behavioral profiles of a dozen recently discovered G protein-biased κOR agonists (Figure 2), with the goal of discovering patterns or correlations between bias and specific adverse effects. This is particularly important as a study in 2015 found that κOR agonists-mediated aversion did not depend on β-arrestin 2 (White et al., 2015). In contrast to the earlier studies which indirectly associated β-arrestin 2 to the aversive effects, this particular study utilized β-arrestin 2 knockout mice. This study thus introduces some counterweight to the hypothesis that G protein-biased κOR agonists will produce fewer adverse effects. We will describe this controversy in more detail in the discussion.
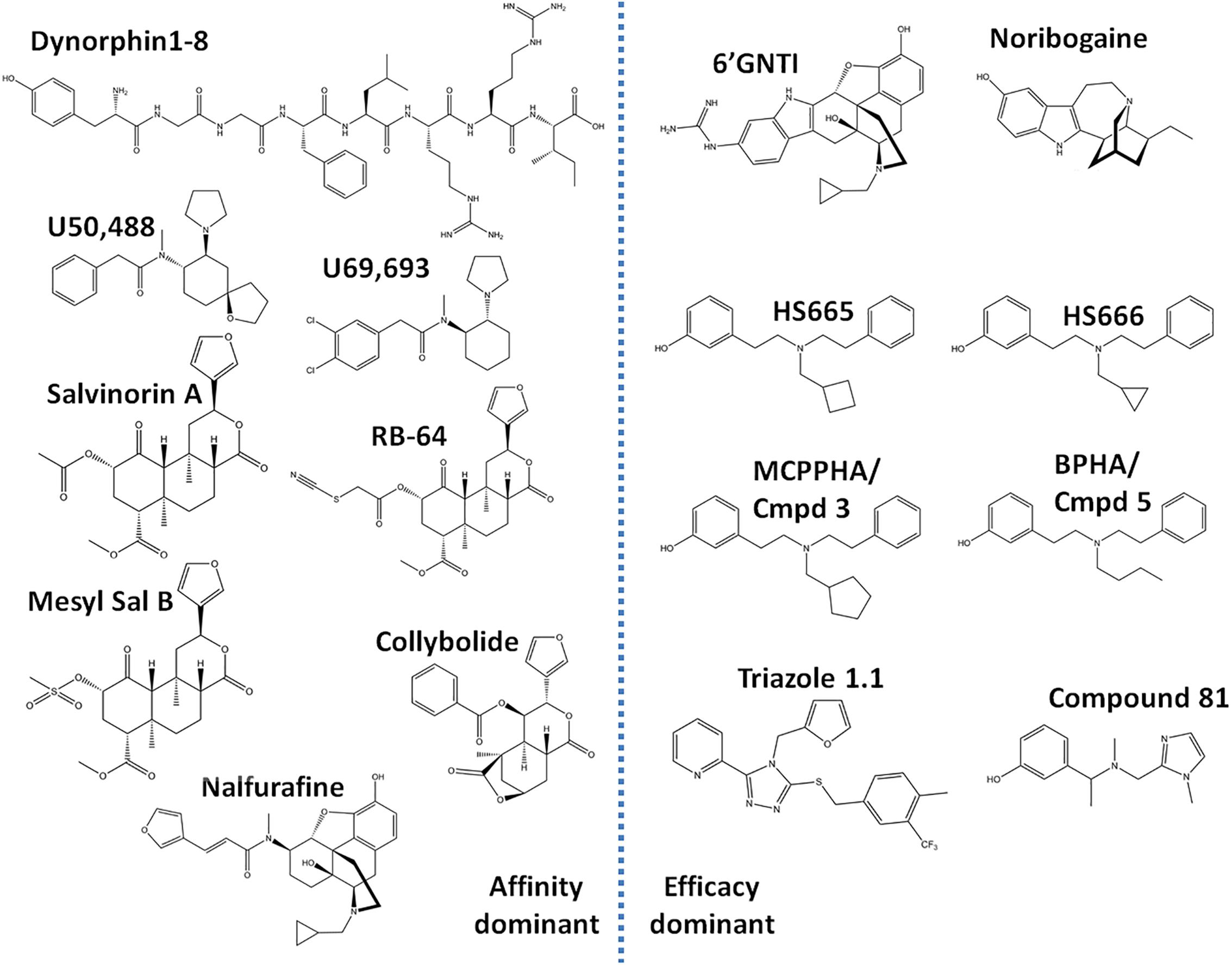
Figure 2. Chemical structures of ‘affinity-dominant’ and ‘efficacy-dominant’ G protein-biased κ opioid receptor agonists.
The cellular environment strongly impacts the efficacy with which an agonist can engage a signal transduction pathway. For example strong expression/activity of regulators of G protein signaling will dampen G protein efficacy, whereas strong expression of G protein receptor kinases will enhance potency and efficacy of β-arrestin recruitment (Miess et al., 2018). Similarly, G-protein signaling can be measured at multiple levels, e.g., at the level of GTP exchange (GTPγS), at the level of second messenger production (e.g., cAMP) or at the level of gene transcription, resulting in different levels of signal amplification. Given the influence of cell environment and choice of readout it is important to be aware of limitations of detecting G protein or β-arrestin signaling. Therefore a lack of signaling is not evidence of antagonism or an exclusive preference for a single transduction pathway (Kenakin, 2015). To provide a strong measure of ligand bias it is recommended that G protein signaling and β-arrestin recruitment are measured in the same cell (Luttrell et al., 2015).
For the κOR agonists discussed here, the predominant assay for G protein signaling utilized [35S]GTPγS, whereas β-arrestin recruitment was primarily assessed using the proprietary PathHunter cells from DiscoverX. Although G protein signaling has also been assessed by measuring inhibition of cAMP (using a cAMP biosensor) and β-arrestin recruitment has been assessed using a TANGO assay and using bioluminescence resonance energy transfer (BRET). A 2014 study showed similarities and differences in potency and efficacy for κOR agonists when assessed in the TANGO or BRET assay, with the primary difference that partial agonism was more apparent in the TANGO assay. This however is not a rule, as partial agonism of β-arrestin recruitment at dopamine D2 receptor was more apparent using the PathHunter and BRET assay than using the TANGO assay (Allen et al., 2011).
The ability of agonists to engage multiple independent signal transduction routes means that one cannot rely on potency rank-order to compare signaling preference. Instead new methodologies were introduced to calculate a bias factor for agonists, which is a score for an agonist to preferentially signal via one pathway over another, relative to a reference compound. The endogenous κOR opioid dynorphin, the natural occurring opioid salvinorin A, and the synthetic κOR selective agonists U50,488 and U69,593 (Figure 2) are relatively unbiased, acting as equipotent and fully efficacious agonists for both G protein signaling and β-arrestin recruitment. Because of their unbiased nature, these κOR opioids often serve as a standard reference compound to assess the signaling bias for novel κOR agonists. The choice of reference compound across the studies discussed here was not consistent as some groups chose U50,488, while others used U69,593, or salvinorin A. The two most commonly used methods to calculate bias are the operational model/transduction coefficient method, and the equiactive method (Black and Leff, 1983; Rajagopal et al., 2011; Kenakin et al., 2012). The equiactive method, requires agonist responses to exhibit a perfect hill-slope, but can be quickly calculated solely using the EC50 and Emax values, however it does not account for receptor reserve (Rajagopal et al., 2011). A study on dopamine D2 agonists found little differences between these methods in calculated bias factor (Brust et al., 2015). The preferred method for the discussed papers in this review is the operational model/transduction coefficient method. A downside of both methods is that they work best when comparing full agonists, but fare worse when the agonist is a weak partial agonist in one of the assays. To optimize calculations the more data points (e.g., half-log dilution steps vs. full log step dilutions) that are used to create the dose response curve the more accurate the bias factor, this is particularly important for weak partial agonists, with a small signal-to-background window. To overcome the limitation of the partial agonist, it is advisable to concurrently run the cellular assay in competition mode; here the partial agonist is tested in the presence of a non-saturating concentration of the reference compound (Stahl et al., 2015; Dunn et al., 2018).
17-Cyclopropylmethyl-3, 14 beta-dihydroxy-4,5 alpha-epoxy-6 beta-[N-methyl-trans-3-(3-furyl) acrylamido]morphinan hydrochloride (TRK-820) was discovered in 1998 in Japan in the lab of Dr. Hiroshi Nagase and found to produce antinociception without aversion (Nagase et al., 1998) and act as a selective κOR agonist (Seki et al., 1999). In 2002, TRK-820 was reported to reduce pruritis (Togashi et al., 2002), and is currently marketed as nalfurafine hydrochloride (Remitch®) in Japan as an antipruritic. Nalfurafine is the first, and currently only, κOR-selective agonist to have been approved for clinical use (Kumagai et al., 2010). Using early stage ERK phosphorylation as a proxy for G protein signaling and p38 phosphorylation as a measure for β-arrestin mediated signaling, nalfurafine was found to act as a G protein-biased agonist (Table 1) with a bias factor (Kenakin and Christopoulos, 2013; van der Westhuizen et al., 2014) of 7 at the rat κOR and 300 at the human κOR (Schattauer et al., 2017) relative to U50,488. It should be noted that MAPK activation is not a great endpoint to assess signal bias (Lovell et al., 2015). In a later study nalfurafine β-arrestin 2 recruitment was assessed in HEK293 cells using a galactosidase-based assay, which is similar to the PathHunter assay with a potency of 1.4 nM and efficacy of 129% relative to U50,488 (Liu et al., 2019). In CHO cells, G protein signaling as measured by [35S]GTPγS for nalfurafine is 0.025 nM (Fujij et al., 2012), while in mouse neuro2A cells the potency of nalfurafine, measured by GTPγS is lower at 0.11 nM (Liu et al., 2019) which would suggest nalfurafine is only slightly G protein-biased, with the caveat that the GTPγS was not measured in the HEK 293 cells used to measure β-arrestin recruitment. Preclinical behavioral studies in male C57BL/6 and CD-1 mice found that nalfurafine attenuated 5′-GNTI-induced scratching and produced antinociception in the warm water tail withdrawal assay. These behavioral effects were mediated by the κOR as they were blocked by the administration of the κOR antagonist norbinaltorphimine, and absent in κOR knockout mice (Inan et al., 2009; Schattauer et al., 2017; Liu et al., 2019). Additionally, in CD-1 mice, nalfurafine was not aversive up to a dose of 20 μg/kg, as measured in the CPA test. The same tested doses of nalfurafine did not cause anhedonia as evident from a lack in change in baseline intracranial self-stimulation (ICSS) threshold. Nalfurafine produced mild locomotor incoordination in the rotarod assay compared to U50,488 (Liu et al., 2019). Reportedly, nalfurafine only produced sedation at doses much higher (ED50 = 27 μg/kg) than needed to produce antinociception (ED50 = 3.3 μg/kg) (Endoh et al., 1999). In contrast to nalfurafine, U50,488 (0.5 mg/kg and higher) caused anhedonia and CPA (Liu et al., 2019). In Fisher 344 rats, a low dose of nalfurafine did not induce CPA, but reduced cocaine conditioned placed preference (CPP) (Mori et al., 2002) as well as morphine CPP (Tsuji et al., 2001).
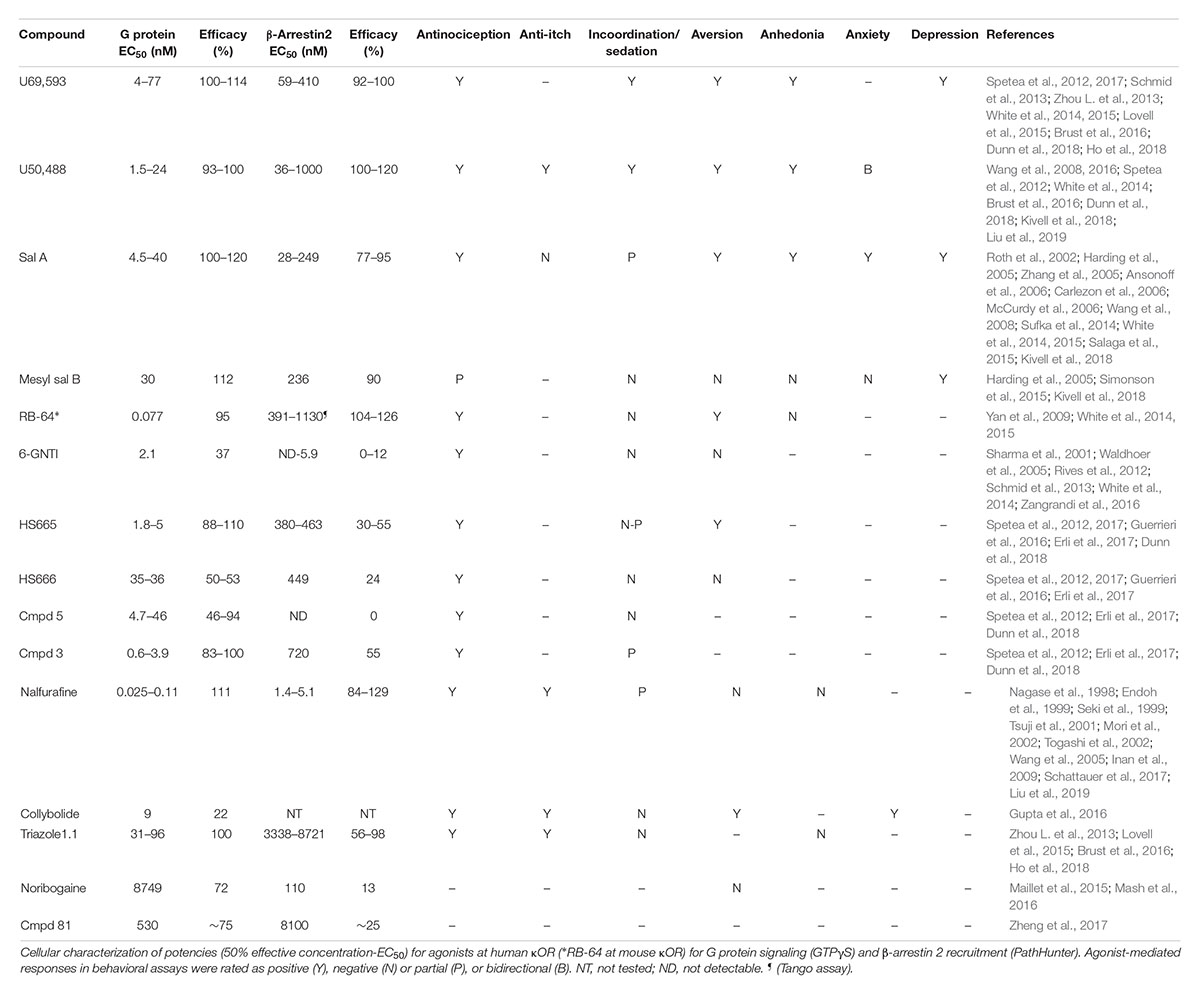
Table 1. Overview of potency and efficacy of unbiased and G protein biased κOR agonists at human κOR for G protein and β-arrestin coupling, and of behavioral responses induced by the agonists.
Salvia divinorum is a psychedelic plant that contains a non-nitrogenous diterpene, salvinorin A, with high κOR affinity (Roth et al., 2002). Depending on the technique and endpoint used, salvinorin A can be relatively unbiased (White et al., 2015) or G protein-biased (Kivell et al., 2018) (Figure 3). In male mice, salvinorin A is antinociceptive (Ansonoff et al., 2006; McCurdy et al., 2006) and was shown to non-significantly reduce scratching (Salaga et al., 2015), and induce aversion (Zhang et al., 2005; Sufka et al., 2014) and sedation (Zhang et al., 2005). The high affinity and selectivity of salvinorin A makes it an intriguing start point for the development G protein-biased κOR agonists. One such endeavor by the lab of Dr. Bryan Roth led to the development of RB-64 (22-thiocyanatosalvinorin A), a semi-synthetic structural derivative of salvinorin A (Yan et al., 2009). RB-64 was identified as a G protein-biased κOR agonist (Table 1) with a measured bias factor of 35–96 for G protein signaling relative to salvinorin A (White et al., 2014, 2015). However, this bias is purely driven by a 70–210 fold lower potency for RB-64 to recruit β-arrestin 2, as measured using the TANGO assay (Kroeze et al., 2015), compared to the potency to activate G protein signaling (cAMP GloSensor assay); in both assays RB-64 is a full agonist or even superagonist (Table 1). In both male and female C57BL/6 mice, RB-64 was shown to produce antinociception in the hot-plate assay to a similar degree as U69,593 and salvinorin A. However, in this study RB-64 was the only κOR agonist that did not produce locomotor incoordination in the rotarod assay (White et al., 2015). Surprisingly, based on the hypothesis that aversion/anhedonia is mediated by β-arrestin 2, RB-64, as well as salvinorin A and U69,593, produced CPA in both wild-type and β-arrestin 2 knockout mice (White et al., 2015), suggesting aversion is not mediated by β-arrestin 2. Given that G protein-biased μOR agonist have not consistently reproduced the observed phenotype of μOR agonists in β-arrestin 2 knockout mice, we should still be cautious in interpreting results from global knockout mice. The recently developed conditional β-arrestin 2 knockout mice (Urs et al., 2016) should be a more precise tool to study the role of β-arrestin 2 in CPA. RB-64 caused a weak right-ward shift in ICSS response rate, compared to a moderate shift by salvinorin A and a strong shift by U69,593, suggesting RB-64 did not induce pronounced anhedonia. It should be noted that sedation is a possible confound in interpreting the anhedonic effects of salvinorin A and U69,593 in this assay. The correlation between ICSS reduction and bias factor may indicate a role for β-arrestin 2 signaling in producing anhedonia (White et al., 2015).
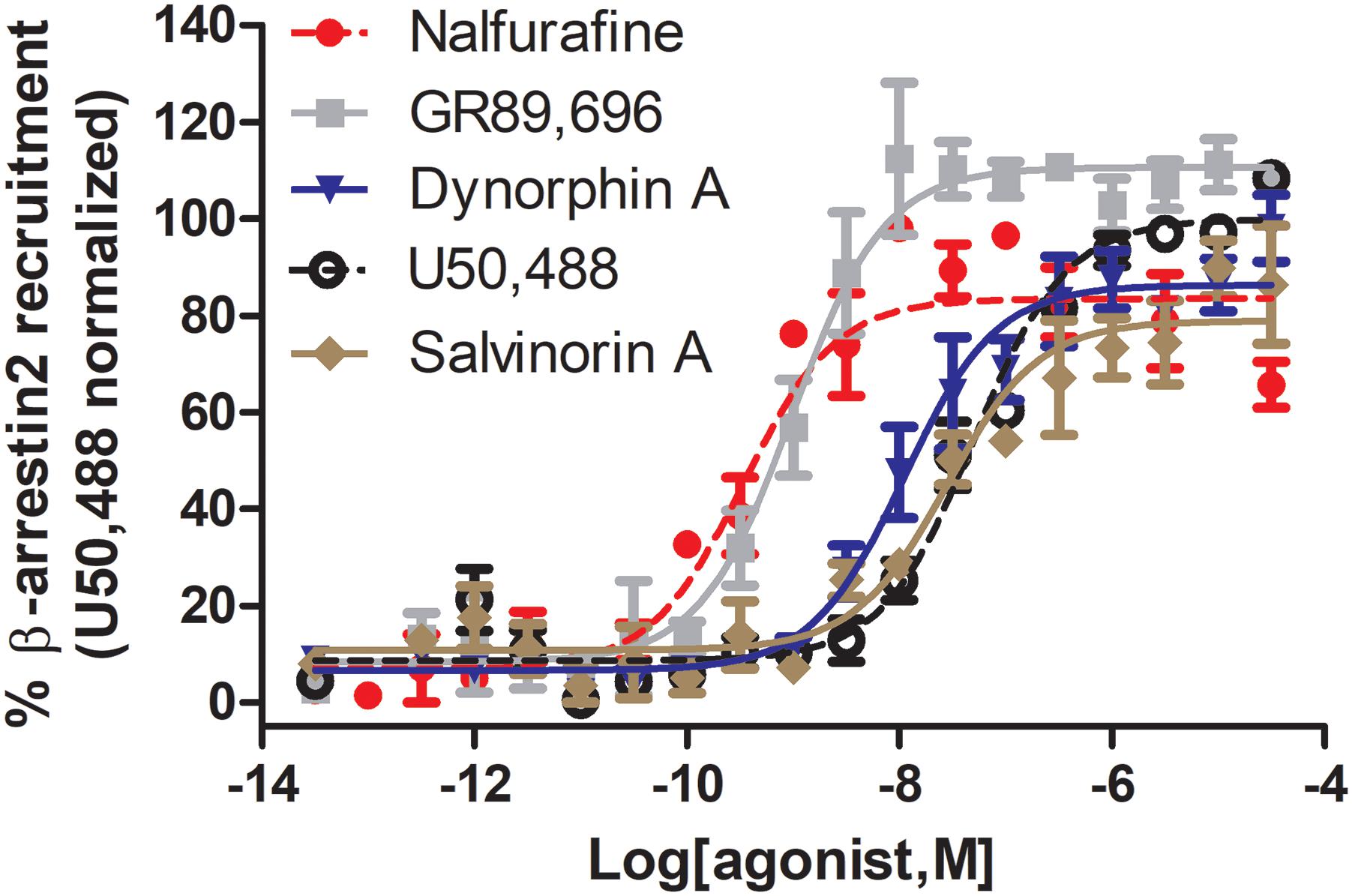
Figure 3. Nalfurafine and GR89,696, respectively potently and efficaciously recruit β-arrestin 2 recruitment following κOR activation. Dose-dependent recruitment of β-arrestin 2 to κOR following activation with nalfurafine (n = 3, red  ), GR89,696 (n = 4,
), GR89,696 (n = 4,  ), dynorphin A (n = 4, blue
), dynorphin A (n = 4, blue  ), U50,488 (n = 4, ) and salvinorin A (n = 4, brown
), U50,488 (n = 4, ) and salvinorin A (n = 4, brown  ) in PathHunter U2OS hOPRK1/β-arrestin-2 cells (DiscoverX, Fremont, CA, United States). Each concentration was tested in duplicate, and three independent dose-response curves were produced for each agonist as previously described (Chiang et al., 2016). Data is normalized to U50,488. Nalfurafine was purchased from AdooQ (Irvine, CA, United States) all other compounds were from Tocris (Minneapolis, MN, United States).
) in PathHunter U2OS hOPRK1/β-arrestin-2 cells (DiscoverX, Fremont, CA, United States). Each concentration was tested in duplicate, and three independent dose-response curves were produced for each agonist as previously described (Chiang et al., 2016). Data is normalized to U50,488. Nalfurafine was purchased from AdooQ (Irvine, CA, United States) all other compounds were from Tocris (Minneapolis, MN, United States).
((2S,4aR,6aR,7R,9S,10aS,10bR)-9-(methanesulfonyloxy)-2-(3-furanyl)dodecahydro-6a, 10b-dimethyl-4,10-dioxy-2H-naptho [2,1-c]pyran-7-carboxylic acid methyl ester) is a semi-synthetic derivative of salvinorin A (Harding et al., 2005), originating from the lab of Dr. Thomas Prisinzano, that has a G protein bias similar to that of RB-64, in that it is a full agonist for both G protein and β-arrestin 2 recruitment (Table 1), but >1000 fold more potent in the G protein activation assay (Kivell et al., 2018). Mesyl sal B was only weakly analgesic in the warm-water tail withdrawal test, but did reduce cocaine-induced hyperlocomotor activity and cocaine seeking behavior (Simonson et al., 2015; Kivell et al., 2018). In male Sprague-Dawley male rats mesyl sal B did not produce CPA or anxiety-like behavior nor did it reduce sucrose-reinforced responding, a measure of anhedonia, although it did increase depression-like behavior in the forced swim test (Kivell et al., 2018). In B6.SJL male mice, Mesyl sal B did not impact rotarod performance (Kivell et al., 2018).
In 2012, a team of researchers from Duke University, the Scripps Research Institute, Sanford-Burnham Medical Research Institute and Kansas University employed a high-throughput strategy utilizing κOR β-arrestin 2 PathHunter cells to screen nearly 300,000 compounds to identify novel κOR agonists and antagonists (Frankowski et al., 2012). The screen identified novel antagonist and agonist chemotypes including triazole 1.1 (Frankowski et al., 2012), which was shown to be biased toward G protein coupling relative to U69,593 as determined by [35S]GTPγS and PathHunter β-arrestin 2 recruitment assays (Table 1) and produce antinociception in male C57BL/6 mice in the warm-water tail immersion test (Zhou L. et al., 2013; Lovell et al., 2015). A more detailed analysis of the behavioral effects of triazole 1.1 revealed that the biased κOR agonist also reduced chloroquine-mediated scratching, but did not reduce ambulatory locomotion at analgesic doses (Brust et al., 2016). Unlike the unbiased κOR agonist U50,488, triazole 1.1 did not strongly reduce dopamine release in the nucleus accumbens, which could indicate that triazole 1.1 will not produce dysphoria/aversion. In line with these findings, U50,488, but not triazole 1.1, produced a rightward shift in ICSS in rats, suggesting unlike U50,488 triazole 1.1 did not produce anhedonia (Brust et al., 2016). It should be noted that in this study the positive control U50,488 decreased the maximum response rate, which may imply sedation and confound data interpretation.
In 2012, first diphenethylamine derivatives with κOR activity and antinociceptive efficacy created by the lab of Drs. Mariana Spetea/Helmut Schmidhammer were reported (Spetea et al., 2012). In a follow up study, Spetea et al. (2017) used [35S]GTPγS and the PathHunter β-arrestin 2 recruitment assays to determine if HS665 and HS666 exhibit bias toward the activation of G protein over β−arrestin 2−mediated signaling (Table 1), when compared with U69,593; the bias factors for HS665 and HS666 were determined to be 389 and 62, respectively. In a warm-water tail withdrawal assay HS665 and HS666, administered intracerebroventricularly, dose−dependently produced antinociception in male C57BL/6 wild-type mice, but not in κOR knockout mice. Neither HS665 nor HS666 produced locomotor coordination issues as measured in the rotarod assay, however only HS665, but not HS666 induced CPA (Spetea et al., 2017). When injected systemically (intraperitoneally) HS665 (although named MCBPHA in this paper) produced modest motor incoordination in male C57BL/6 mice in the rotarod assay, which was similar to MCPPHA (which was compound 3 in Spetea et al., 2012), but less than U50,488 (Dunn et al., 2018). A library of derivatives of HS665 and HS665 were synthesized with several compounds showing subnanomolar affinity and exceptional κOR selectivity, as well as high G-protein potency acting as either full or partial agonists (Erli et al., 2017). While those new derivatives were not assessed for β-arrestin recruitment, the new derivatives displayed increased antinociceptive potencies compared with U50,488, HS665 and HS666 in the acetic acid-induced writhing test (Erli et al., 2017). Another derivative of the trialkylamine scaffold, BPHA (compound 5 in Spetea et al., 2012), did not measurably recruit β-arrestin 2 as determined using the PathHunter cell assay using U50,488 as reference compound (Table 1) and did not cause locomotor incoordination (Dunn et al., 2018). These results appear to indicate a correlation between bias factor or β-arrestin 2 recruitment efficacy and κOR agonist-induced rotarod incoordination.
After its initial synthesis in 2001 in the lab of Dr. Philip Portoghese (Sharma et al., 2001), the κOR agonist 6′-guanidinonaltrindole (6′-GNTI) was deemed to mediate antinociception through interacting specifically with heteromers of the κOR and δOR in a publication by Waldhoer et al. (2005). In 2012, using BRET assays, Rives et al. (2012) found that 6′-GNTI is a partial G protein agonist (Table 1), with no detectable β-arrestin 2 recruitment. The κOR agonist GR89,696 has been suggested to also interact with κOR-δOR heteromers (Brissett et al., 2012), however, in contrast to 6′-GNTI, GR89,696 reportedly displays β-arrestin2 bias (White et al., 2014), giving pause to a hypothesis that the κOR-δOR heteromer adopts a G protein biased conformation. Similar to HS666, 6′GNTI was not significantly aversive in male C57BL/6 mice in the CPA paradigm, nor was it sedative (Zangrandi et al., 2016). Activation of the κOR in striatal neurons with 6′-GNTI led to phosphorylation of Akt, but not ERK1/2, which is in contrast to the unbiased κOR agonist U69,593 which induced phosphorylation of both kinases (Schmid et al., 2013). In striatal neurons from β-arrestin 2 knockout mice, persistent ERK activation by 6′-GNTI was β-arrestin 2-dependent, whereas Akt phosphorylation was pertussis toxin sensitive, indicative of a Gαi-protein-mediated mechanism (Schmid et al., 2013). The chronology of the pharmacological characterization of 6′-GNTI mimics that of the δOR agonist TAN-67, which was suggested to act on δOR-μOR heteromers (van Rijn and Whistler, 2009), and was subsequently found to be G protein-biased (Chiang et al., 2016) in vitro and in mouse dorsal striatum (Robins et al., 2018).
Collybolide is a natural product first extracted from the fungus Collybia maculata by the research group of Dr. Pierre Potier (Bui et al., 1974). Collybolide shares structural similarity, particularly a familiar furyl-δ-lactone core, with salvinorin A (Gupta et al., 2016). In male C57BL/6 mice, systemic (intraperitoneal) injection of collybolide produced antinociception in a tail-flick assay and reduced chloroquine-induced itch. Collybolide was not sedative at a dose that produced antinociception, but did produce aversion in a CPA paradigm and also produced mild, norbinaltorphimine reversible anxiety (Gupta et al., 2016). In vitro, collybolide was a potent but partial agonist in the [35S]GTPγS assay, though β-arrestin 2 recruitment was not tested (Table 1). However, compared to U69,593, collybolide preferentially induced phosphorylation of Akt over ERK (Gupta et al., 2016), which is a pharmacological profile also seen for the G protein-biased agonist 6′-GNTI.
A team of researchers from the Universities of North Carolina and Southern California utilized the crystal structure of the κOR, which was resolved in 2012 bound to κOR antagonist JDTic (Wu et al., 2012), to virtually screen five million fragment-like and lead-like compounds resulting in the identification of 11 hits subcategorized into four chemotypes (Zheng et al., 2017). Compound 81 (3-(1-(methyl((1-methyl-1H-imidazol-2-yl)methyl)amino)ethyl)phenol), a derivative of one of the hits (the balanced agonist compound 23), was determined to act as a G protein-biased κOR agonist (Table 1), using the cAMP GloSensor assay, but was found to only weakly recruit β-arrestin 2 using the TANGO assay, with a bias factor of 6 against salvinorin A (Zheng et al., 2017). Thus far, compound 81 has not been tested in vivo, perhaps in part because of its relatively low potency (530 nM). Structurally, compound 81 resembles the G protein-biased κOR agonists, HS666 and BPHA/compound 5 (Figure 2) and thus perhaps derivatization of compound 81 may yield a more potent G protein-biased κOR agonist that can be assessed in animals. Another low potency G protein-biased κOR agonist is noribogaine, which is an active metabolite of the psychoactive alkaloid ibogaine found in plants belonging to the Apocynaceae family such as Tabernanthe iboga (Davis et al., 2017; Malcolm et al., 2018; Mash et al., 2018). In [35S]GTPγS binding assays, noribogaine displayed partial κOR agonist activity with an EMax of 72% relative to U69,593 (Table 1). In the DiscoverX PathHunter β-arrestin 2 recruitment assay, noribogaine was a very weak recruiter (EMax of 13% normalized to U69,593). No significant CPP or CPA was observed for 10, 30, and 100 mg/kg (oral) noribogaine (Mash et al., 2016), although given the low potency of noribogaine κORs may not play a major role in any observed behavior at these doses.
G protein bias may either be affinity/potency-dominant or efficacy-dominant (Table 1). A potential concern is that, despite using the same cellular assays, variations in agonist potency were determined by different, or even the same labs, potentially due to differences in expression levels of the receptor and signaling proteins that occurred during cell passaging. For example the reported arrestin recruitment potency for U69,593 has been reported to be as low as 67.7 nM (Spetea et al., 2017) and as high as 410 nM (Dunn et al., 2018), similarly for U50,488 potency has ranged from 36 to 1000 nM (Dunn et al., 2018; Liu et al., 2019). In our hands the values were slightly different as well, finding the following potencies and efficacies for U50,488 (EC50 = 51.5 ± 12, α = 100 ± 1), nalfurafine (EC50 (nM) = 5.19 ± 1.3, α = 84 ± 7), and salvinorin A (EC50 = 27.5 ± 10, α = 76 ± 9), whilst also confirming super recruitment by GR89,696 (EC50 = 9.90 ± 3.6, α = 112 ± 3), but partial recruitment of dynorphin A (EC50 = 21.0 ± 13, α = 87 ± 5) relative to U50,488 (Figure 3). These potency differences as well as which reference compound is used will impact the calculated bias factor, which is the reason why we did not include bias factors in Table 1. It would be helpful for the field if the endogenous agonist dynorphin A was used as reference compound to ensure easier comparison between studies.
As detailed in Section “Can Specifically Targeting G Protein-Biased Signaling Lead to the Development of Clinically Effective, κOR-Selective, Full Agonists?,” cellular context strongly impacts the calculation of signaling bias. The majority of the discussed papers utilized U20S human bone osterosarcoma cells stable overexpressing β-arrestin2 and κOR, which most likely do not resemble the cellular context of, for example, striatal neurons, a region selectively and strongly expressing regulator of G protein signaling 9-2 (Gold et al., 1997; Rahman et al., 1999). Moreover, cellular context is known to change during a chronic pain state (Xiao et al., 2002; Obara et al., 2009; Zhou Y. et al., 2013). Unfortunately, many of the studies investigating the analgesic potency of the G protein-biased κOR agonists discussed here were conducted in naïve mice using acute/reflexive pain models that are not as translatable to patients with chronic pain. Native receptor expression and G protein signaling can be assessed in cultured neurons or cryopreserved brain sections using radioligand binding. However, the biggest hurdle currently is the lack of a radiolabel or biosensor to assess β-arrestin signaling in such cultures or brain sections, at least without exogenously introducing a genetic β-arrestin construct. If a peptide could be produced that can reach and bind to the intracellular side of GPCRs and that can be competitively displaced upon β-arrestin binding to the GPCR, such a peptide could be turned into a radiolabel to assess bias factor in neurons under native conditions.
It is important to note the limitation of comparing bias factors of two G protein-biased agonists even if they are calculated using the same reference compound. Specifically two compounds may have the same bias factor, but have completely different β-arrestin recruitment efficacy [affinity-dominant vs. efficacy-dominant (Kenakin, 2015)]. The physiological difference would be that a weak/partial β-arrestin recruiter would serve as a functional antagonist for β-arrestin recruitment relative to the endogenous full agonist response (Kenakin, 2015) (Figure 4). For example, HS665 is a partial recruiter of β-arrestin 2 but has a higher calculated bias factor than HS666 and BPHA, which show minimal β-arrestin 2 recruitment. Yet, despite the lower bias factor it is HS666 and BPHA that do not produce locomotor incoordination. This is not too dissimilar to findings that β-arrestin 2 recruitment efficacy of δOR agonists was tightly correlated with modulation of alcohol use (Chiang et al., 2016).
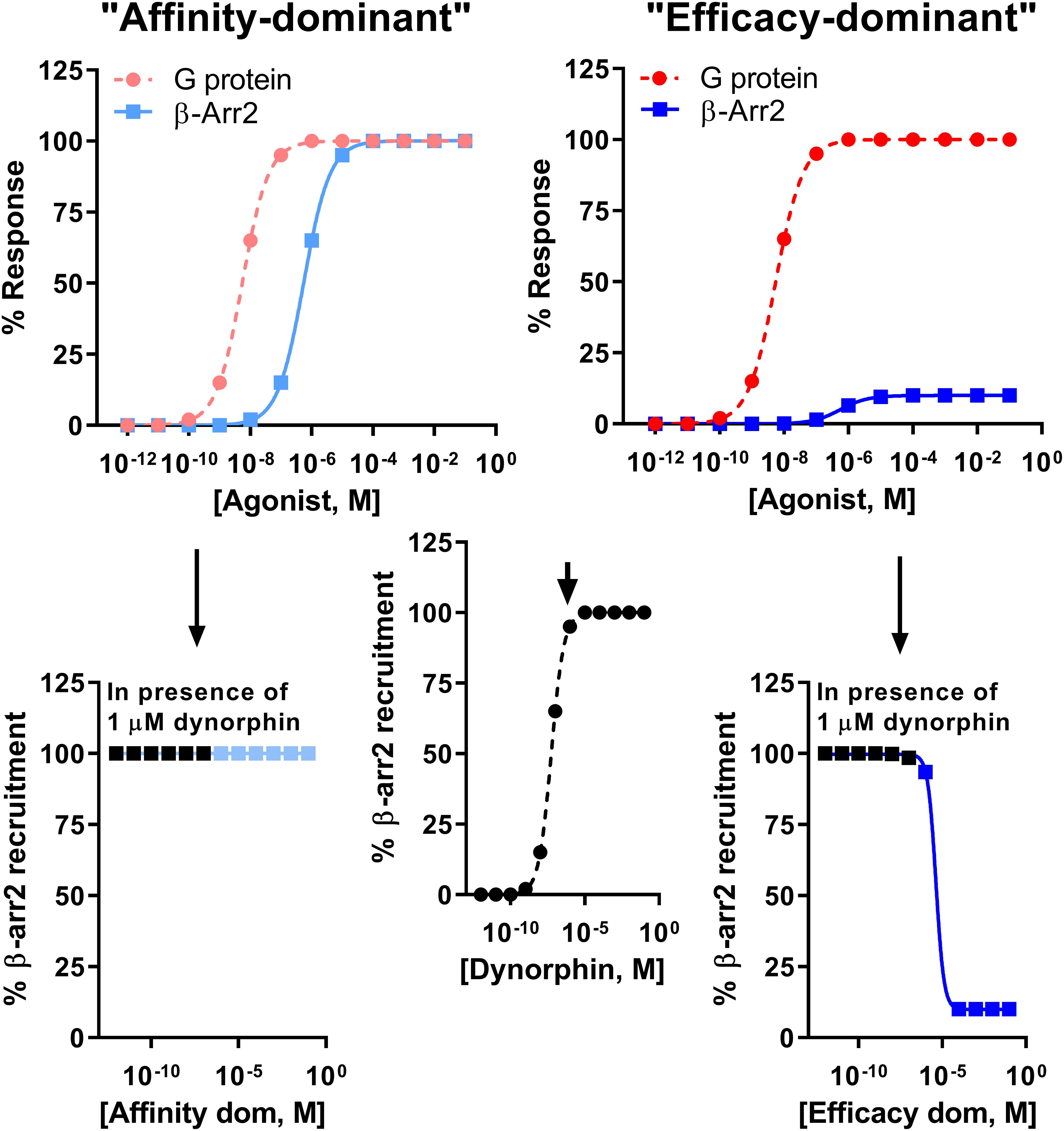
Figure 4. Difference between an “affinity-dominant” and “efficacy-dominant” G protein-biased agonist. An “affinity-dominant” κOR agonist has a higher affinity for the κOR conformation that activates G proteins than that recruits β-arrestin 2: see top left panel for an example of a κOR agonist that resembles RB-64, with a G protein-coupling EC50 potency of 5.5 nM (dotted line, pink  , G-protein) and EC50 potency for β-arrestin 2 recruitment of 550 nM and 100% efficacy (solid line, light blue
, G-protein) and EC50 potency for β-arrestin 2 recruitment of 550 nM and 100% efficacy (solid line, light blue  ). In contrast an ‘efficacy-dominant” κOR agonist, that resembles HS666 with an EC50 potency for β-arrestin 2 recruitment of 550 nM, but 10% efficacy (solid line, blue
). In contrast an ‘efficacy-dominant” κOR agonist, that resembles HS666 with an EC50 potency for β-arrestin 2 recruitment of 550 nM, but 10% efficacy (solid line, blue  ), only weakly recruits β-arrestin 2 even at high concentrations (top right panel). Consider the endogenous agonist dynorphin A, which recruits β-arrestin 2 at 100% efficacy (middle panel). At high concentrations the affinity-dominant agonist will displace dynorphin from the κOR, yet retain highly efficacious recruitment of β-arrestin 2 (bottom left panel). In contrast the efficacy-dominant agonist will reduce β-arrestin 2 recruitment efficacy once this type of agonists displaces dynorphin from the κOR (bottom right) panel.
), only weakly recruits β-arrestin 2 even at high concentrations (top right panel). Consider the endogenous agonist dynorphin A, which recruits β-arrestin 2 at 100% efficacy (middle panel). At high concentrations the affinity-dominant agonist will displace dynorphin from the κOR, yet retain highly efficacious recruitment of β-arrestin 2 (bottom left panel). In contrast the efficacy-dominant agonist will reduce β-arrestin 2 recruitment efficacy once this type of agonists displaces dynorphin from the κOR (bottom right) panel.
The finding that κOR agonists regardless of signaling bias were still producing CPA in β-arrestin 2 knockout mice is disconcerting for the therapeutic promise of G protein-biased κOR agonists. A recent study by Liu et al. (2018) compared the phosphoproteome of the striatum (amongst other regions) following exposure to U50,488, HS665, RB-64, 6′-GNTI and HS666. Notably, compared to the arrestin recruiting agonists U50,488, HS665 and RB64, the G protein-biased 6′GNTI and HS666 did not activate mechanistic target of rapamycin (mTOR) signaling. Inhibition of mTOR abolished CPA induced by U50,488 thus linking the mTOR transduction pathway to this important side effect. Close examination of their results revealed that the weak β-arrestin 2 recruiting κOR agonists 6′-GNTI and HS666 often displayed unique modulation of protein phosphorylation relative to the more efficacious β-arrestin 2 recruiters (Liu et al., 2018). Thus, even if β-arrestin 2 is not involved in all these phosphorylation events/signaling cascades, searching for efficacy-dominant G protein-biased κOR agonists may provide the desired therapeutic window to treat pain with reduced side effects. In a follow up study, nalfurafine surprisingly, as it still efficaciously recruits β-arrestin 2, also did not activate mTOR nor induce CPA (Liu et al., 2019). Activation of mTOR however could not explain for κOR agonist-induced locomotor inhibition, but importantly mTOR also did not play a role in the antipruritic and antinociceptive effects of U50,488 (Liu et al., 2019). Thus, currently it is unclear what precise role β-arrestin 2 plays, if any, in mediating κOR agonist-mediated aversion and in general there does not yet seem to be a consistent consensus on the therapeutic and side-effect profile of affinity-dominant and efficacy-dominant G protein-biased κOR agonists.
After the initial antagonist-bound crystal structure of the κOR (Wu et al., 2012), two agonist-bound κOR structures have been produced, one in which the κOR was bound to the endogenous agonist dynorphin 1–13 (O’Connor et al., 2015) and the other bound to the unbiased agonist MP1104 (Varadi et al., 2015; Che et al., 2018). It would be incredibly insightful for the opioid field to study an opioid receptor bound to a signal-biased ligand. However, crystallizing ligand-bound structures requires the use of ligands that have very strong affinity for the receptor. The currently available agonist-bound structures may aid in the rational design of high affinity G protein-biased κOR agonists, which can then be used to crystallize a G protein-biased conformation of the κOR. The recent discovery of numerous G protein-biased κOR agonists has substantially expanded the pharmacological toolbox and perhaps one of the recently developed efficacy-dominant G protein-biased opioids, like HS665, may already have the correct properties to accomplish this goal. Hopefully, additional κOR biased agonists will be developed to strengthen the diversity of the current crop and they will be used in tandem to investigate the role of biased signaling in human (patho)physiology in more detail, including by studying downstream phosphorylation events. Accordingly, they can aid in the search for better, more efficacious therapies for disorders, such as chronic pain, and pruritis.
RvR supervised KM, BC, and RC. RC carried out the β-arrestin 2 recruitment experiments. KM, BC, and RvR wrote the first draft of the manuscript. KM, BC, RC, and RvR edited and proofread the manuscript and approved the final draft.
RvR was supported by funds from NIH grants (AA025368, AA026949, AA026675, and DA045897), the Purdue Institute for Drug Discovery and the Department of Medicinal Chemistry and Molecular Pharmacology at Purdue University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Al-Hasani, R., and Bruchas, M. R. (2011). Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology 115, 1363–1381. doi: 10.1097/ALN.0b013e318238bba6
Allen, J. A., Yost, J. M., Setola, V., Chen, X., Sassano, M. F., Chen, M., et al. (2011). Discovery of beta-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc. Natl. Acad. Sci. U.S.A. 108, 18488–18493. doi: 10.1073/pnas.1104807108
Altarifi, A. A., David, B., Muchhala, K. H., Blough, B. E., Akbarali, H., and Negus, S. S. (2017). Effects of acute and repeated treatment with the biased mu opioid receptor agonist TRV130 (oliceridine) on measures of antinociception, gastrointestinal function, and abuse liability in rodents. J. Psychopharmacol. 31, 730–739. doi: 10.1177/0269881116689257
Ansonoff, M. A., Zhang, J., Czyzyk, T., Rothman, R. B., Stewart, J., Xu, H., et al. (2006). Antinociceptive and hypothermic effects of Salvinorin A are abolished in a novel strain of kappa-opioid receptor-1 knockout mice. J. Pharmacol. Exp. Ther. 318, 641–648. doi: 10.1124/jpet.106.101998
Austin Zamarripa, C., Edwards, S. R., Qureshi, H. N., Yi, J. N., Blough, B. E., and Freeman, K. B. (2018). The G-protein biased mu-opioid agonist, TRV130, produces reinforcing and antinociceptive effects that are comparable to oxycodone in rats. Drug Alcohol. Depend. 192, 158–162. doi: 10.1016/j.drugalcdep.2018.08.002
Black, J. W., and Leff, P. (1983). Operational models of pharmacological agonism. Proc. R. Soc. Lond. B. Biol. Sci. 220, 141–162. doi: 10.1098/rspb.1983.0093
Bohn, L. M., Gainetdinov, R. R., Lin, F. T., Lefkowitz, R. J., and Caron, M. G. (2000). [mu]-Opioid receptor desensitization by [beta]-arrestin-2 determines morphine tolerance but not dependence. Nature 408, 720–723. doi: 10.1038/35047086
Bohn, L. M., Lefkowitz, R. J., Gainetdinov, R. R., Peppel, K., Caron, M. G., and Lin, F. T. (1999). Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 286, 2495–2498. doi: 10.1126/science.286.5449.2495
Brissett, D. I., Whistler, J. L., and van Rijn, R. M. (2012). Contribution of mu and delta opioid receptors to the pharmacological profile of kappa opioid receptor subtypes. Eur. J. Pain 16, 327–337. doi: 10.1002/j.1532-2149.2011.00022.x
Bruchas, M. R., and Chavkin, C. (2010). Kinase cascades and ligand-directed signaling at the kappa opioid receptor. Psychopharmacology (Berl.) 210, 137–147. doi: 10.1007/s00213-010-1806-y
Bruchas, M. R., Land, B. B., Aita, M., Xu, M., Barot, S. K., Li, S., et al. (2007). Stress-induced p38 mitogen-activated protein kinase activation mediates kappa-opioid-dependent dysphoria. J. Neurosci. 27, 11614–11623. doi: 10.1523/jneurosci.3769-07.2007
Bruchas, M. R., Macey, T. A., Lowe, J. D., and Chavkin, C. (2006). Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J. Biol. Chem. 281, 18081–18089. doi: 10.1074/jbc.m513640200
Bruchas, M. R., and Roth, B. L. (2016). New technologies for elucidating opioid receptor function. Trends Pharmacol. Sci. 37, 279–289. doi: 10.1016/j.tips.2016.01.001
Brust, T. F., Hayes, M. P., Roman, D. L., Burris, K. D., and Watts, V. J. (2015). Bias analyses of preclinical and clinical D2 dopamine ligands: studies with immediate and complex signaling pathways. J. Pharmacol. Exp. Ther. 352, 480–493. doi: 10.1124/jpet.114.220293
Brust, T. F., Morgenweck, J., Kim, S. A., Rose, J. H., Locke, J. L., Schmid, C. L., et al. (2016). Biased agonists of the kappa opioid receptor suppress pain and itch without causing sedation or dysphoria. Sci. Signal. 9:ra117. doi: 10.1126/scisignal.aai8441
Bui, A. M., Cave, A., Janot, M. M., Parello, J., and Potier, P. (1974). Isolement et analyse structurale du collybolide, nouveau sesquiterpene extrait de collybia maculata alb. et sch. ex fries (basidomycetes). Tetrahedron 30, 1327–1336. doi: 10.1016/s0040-4020(01)97243-6
Butelman, E. R., Yuferov, V., and Kreek, M. J. (2012). kappa-opioid receptor/dynorphin system: genetic and pharmacotherapeutic implications for addiction. Trends Neurosci. 35, 587–596. doi: 10.1016/j.tins.2012.05.005
Carlezon, W. A. Jr., Béguin, C., DiNieri, J. A., Baumann, M. H., Richards, M. R., Todtenkopf, M. S., et al. (2006). Depressive-like effects of the kappa-opioid receptor agonist salvinorin A on behavior and neurochemistry in rats. J. Pharmacol. Exp. Ther. 316, 440–447. doi: 10.1124/jpet.105.092304
Chavkin, C. (2011). The therapeutic potential of kappa-opioids for treatment of pain and addiction. Neuropsychopharmacology 36, 369–370. doi: 10.1038/npp.2010.137
Chavkin, C., and Koob, G. F. (2016). Dynorphin, dysphoria, and dependence: the stress of addiction. Neuropsychopharmacology 41, 373–374. doi: 10.1038/npp.2015.258
Che, T., Majumdar, S., Zaidi, S. A., Ondachi, P., McCorvy, J. D., Wang, S., et al. (2018). Structure of the nanobody-stabilized active state of the kappa opioid receptor. Cell 172, 55.e15–67.e15.
Chen, X. T., Pitis, P., Liu, G., Yuan, C., Gotchev, D., Cowan, C. L., et al. (2013). Structure-activity relationships and discovery of a G protein biased mu opioid receptor ligand, [(3-methoxythiophen-2-yl)methyl]({2-[(9R)-9-(pyridin-2-yl)-6-oxaspiro-[4.5]decan- 9-yl]ethyl})amine (TRV130), for the treatment of acute severe pain. J. Med. Chem. 56, 8019–8031. doi: 10.1021/jm4010829
Chiang, T., Sansuk, K., and van Rijn, R. M. (2016). Beta-arrestin 2 dependence of delta opioid receptor agonists is correlated with alcohol intake. Br. J. Pharmacol. 173, 323–343. doi: 10.1111/bph.13374
Chou, R., Fanciullo, G. J., Fine, P. G., Adler, J. A., Ballantyne, J. C., Davies, P., et al. (2009). Clinical guidelines for the use of chronic opioid therapy in chronic noncancer pain. J. Pain 10, 113–130.
Corder, G., Ahanonu, B., Grewe, B. F., Wang, D., Schnitzer, M. J., and Scherrer, G. (2019). An amygdalar neural ensemble that encodes the unpleasantness of pain. Science 363, 276–281. doi: 10.1126/science.aap8586
Cowan, A., Kehner, G. B., and Inan, S. (2015). Targeting itch with ligands selective for kappa opioid receptors. Handb. Exp. Pharmacol. 226, 291–314. doi: 10.1007/978-3-662-44605-8_16
Crowley, N. A., Bloodgood, D. W., Hardaway, J. A., Kendra, A. M., McCall, J. G., Al-Hasani, R., et al. (2016). Dynorphin controls the gain of an amygdalar anxiety circuit. Cell Rep. 14, 2774–2783. doi: 10.1016/j.celrep.2016.02.069
Davis, A. K., Barsuglia, J. P., Windham-Herman, A. M., Lynch, M., and Polanco, M. (2017). Subjective effectiveness of ibogaine treatment for problematic opioid consumption: short- and long-term outcomes and current psychological functioning. J. Psychedelic Stud. 1, 65–73. doi: 10.1556/2054.01.2017.009
Dunn, A. D., Reed, B., Guariglia, C., Dunn, A. M., Hillman, J. M., and Kreek, M. J. (2018). Structurally related kappa opioid receptor agonists with substantial differential signaling bias: neuroendocrine and behavioral effects in C57BL6 mice. Int. J. Neuropsychopharmacol. 21, 847–857. doi: 10.1093/ijnp/pyy034
Dykstra, L. A., Gmerek, D. E., Winger, G., and Woods, J. H. (1987). Kappa opioids in rhesus monkeys. Diuresis, I., sedation, analgesia and discriminative stimulus effects. J. Pharmacol. Exp. Therap. 242, 413–420.
Endoh, T., Matsuura, H., Tajima, A., Izumimoto, N., Tajima, C., Suzuki, T., et al. (1999). Potent antinociceptive effects of TRK-820, a novel kappa-opioid receptor agonist. Life Sci. 65, 1685–1694. doi: 10.1016/s0024-3205(99)00417-8
Erli, F., Guerrieri, E., Ben Haddou, T., Lantero, A., Mairegger, M., Schmidhammer, H., et al. (2017). Highly potent and selective new diphenethylamines interacting with the kappa-opioid receptor: synthesis, pharmacology, and structure-activity relationships. J. Med. Chem. 60, 7579–7590. doi: 10.1021/acs.jmedchem.7b00981
Frankowski, K. J., Hedrick, M. P., Gosalia, P., Li, K., Shi, S., Whipple, D., et al. (2012). Discovery of small molecule kappa opioid receptor agonist and antagonist chemotypes through a HTS and hit refinement strategy. ACS Chem. Neurosci. 3, 221–236. doi: 10.1021/cn200128x
Fujij, H., Hirayama, S., and Nagase, H. (2012). “4 opioid kappa receptor selective agonist TRK-820 (Nalfurafine Hydrochloride ),” in Pharmacology, ed. G. Luca (London: InTech), 81–98. doi: 10.1021/cn200128x
Gold, S. J., Ni, Y. G., Dohlman, H. G., and Nestler, E. J. (1997). Regulators of G-protein signaling (RGS) proteins: region-specific expression of nine subtypes in rat brain. J. Neurosci. 17, 8024–8037. doi: 10.1523/jneurosci.17-20-08024.1997
Guerrieri, E., Bermudez, M., Wolber, G., Berzetei-Gurske, I. P., Schmidhammer, H., and Spetea, M. (2016). Structural determinants of diphenethylamines for interaction with the kappa opioid receptor: synthesis, pharmacology and molecular modeling studies. Bioorg. Med. Chem. Lett. 26, 4769–4774. doi: 10.1016/j.bmcl.2016.08.031
Gupta, A., Gomes, I., Bobeck, E. N., Fakira, A. K., Massaro, N. P., Sharma, I., et al. (2016). Collybolide is a novel biased agonist of kappa-opioid receptors with potent antipruritic activity. Proc. Natl. Acad. Sci. U.S.A. 113, 6041–6046. doi: 10.1073/pnas.1521825113
Harding, W. W., Tidgewell, K., Byrd, N., Cobb, H., Dersch, C. M., Butelman, E. R., et al. (2005). Neoclerodane diterpenes as a novel scaffold for mu opioid receptor ligands. J. Med. Chem. 48, 4765–4771. doi: 10.1021/jm048963m
Hill, R., Disney, A., Conibear, A., Sutcliffe, K., Dewey, W., Husbands, S., et al. (2018). The novel mu-opioid receptor agonist PZM21 depresses respiration and induces tolerance to antinociception. Br. J. Pharmacol. 175, 2653–2661. doi: 10.1111/bph.14224
Ho, J. H., Stahl, E. L., Schmid, C. L., Scarry, S. M., Aube, J., and Bohn, L. M. (2018). G protein signaling-biased agonism at the kappa-opioid receptor is maintained in striatal neurons. Sci. Signal. 11:eaar4309. doi: 10.1126/scisignal.aar4309
Inan, S., Dun, N. J., and Cowan, A. (2009). Nalfurafine prevents 5′-guanidinonaltrindole- and compound 48/80-induced spinal c-fos expression and attenuates 5′-guanidinonaltrindole-elicited scratching behavior in mice. Neuroscience 163, 23–33. doi: 10.1016/j.neuroscience.2009.06.016
Kardon, A. P., Polgar, E., Hachisuka, J., Snyder, L. M., Cameron, D., Savage, S., et al. (2014). Dynorphin acts as a neuromodulator to inhibit itch in the dorsal horn of the spinal cord. Neuron 82, 573–586. doi: 10.1016/j.neuron.2014.02.046
Karkhanis, A., Holleran, K. M., and Jones, S. R. (2017). Dynorphin/kappa opioid receptor signaling in preclinical models of alcohol, drug, and food addiction. Int. Rev. Neurobiol. 136, 53–88. doi: 10.1016/bs.irn.2017.08.001
Kenakin, T. (2015). The effective application of biased signaling to new drug discovery. Mol. Pharmacol. 88, 1055–1061. doi: 10.1124/mol.115.099770
Kenakin, T., and Christopoulos, A. (2013). Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat. Rev. Drug Discov. 12, 205–216. doi: 10.1038/nrd3954
Kenakin, T., Watson, C., Muniz-Medina, V., Christopoulos, A., and Novick, S. (2012). A simple method for quantifying functional selectivity and agonist bias. ACS Chem. Neurosci. 3, 193–203. doi: 10.1021/cn200111m
Kissler, J. L., Sirohi, S., Reis, D. J., Jansen, H. T., Quock, R. M., Smith, D. G., et al. (2014). The one-two punch of alcoholism: role of central amygdala dynorphins/kappa-opioid receptors. Biol. Psychiatry 75, 774–782. doi: 10.1016/j.biopsych.2013.03.014
Kivell, B. M., Paton, K. F., Kumar, N., Morani, A. S., Culverhouse, A., Shepherd, A., et al. (2018). Kappa opioid receptor agonist mesyl Sal B attenuates behavioral sensitization to cocaine with fewer aversive side-effects than salvinorin A in Rodents. Molecules 23:E2602. doi: 10.1016/j.biopsych.2013.03.014
Kliewer, A., Schmiedel, F., Sianati, S., Bailey, A., Bateman, J. T., Levitt, E. S., et al. (2019). Phosphorylation-deficient G-protein-biased mu-opioid receptors improve analgesia and diminish tolerance but worsen opioid side effects. Nat. Commun. 10:367. doi: 10.1038/s41467-018-08162-1
Knoll, A. T., Muschamp, J. W., Sillivan, S. E., Ferguson, D., Dietz, D. M., Meloni, E. G., et al. (2011). Kappa opioid receptor signaling in the basolateral amygdala regulates conditioned fear and anxiety in rats. Biol. Psychiatry 70, 425–433. doi: 10.1016/j.biopsych.2011.03.017
Kroeze, W. K., Sassano, M. F., Huang, X. P., Lansu, K., McCorvy, J. D., Giguere, P. M., et al. (2015). PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol. 22, 362–369. doi: 10.1038/nsmb.3014
Kumagai, H., Ebata, T., Takamori, K., Muramatsu, T., Nakamoto, H., and Suzuki, H. (2010). Effect of a novel kappa-receptor agonist, nalfurafine hydrochloride, on severe itch in 337 haemodialysis patients: a Phase III, randomized, double-blind, placebo-controlled study. Nephrol. Dial. Trans. 25, 1251–1257. doi: 10.1093/ndt/gfp588
Land, B. B., Bruchas, M. R., Lemos, J. C., Xu, M., Melief, E. J., and Chavkin, C. (2008). The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J. Neurosci. 28, 407–414. doi: 10.1523/JNEUROSCI.4458-07.2008
Land, B. B., Bruchas, M. R., Schattauer, S., Giardino, W. J., Aita, M., Messinger, D., et al. (2009). Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. Proc. Natl. Acad. Sci. U.S.A. 106, 19168–19173. doi: 10.1073/pnas.0910705106
Liu, J. J., Chiu, Y. T., DiMattio, K. M., Chen, C., Huang, P., Gentile, T. A., et al. (2019). Phosphoproteomic approach for agonist-specific signaling in mouse brains: mTOR pathway is involved in kappa opioid aversion. Neuropsychopharmacology 44, 939–949. doi: 10.1038/s41386-018-0155-0
Liu, J. J., Sharma, K., Zangrandi, L., Chen, C., Humphrey, S. J., Chiu, Y. T., et al. (2018). In vivo brain GPCR signaling elucidated by phosphoproteomics. Science 360:eaao4927. doi: 10.1126/science.aao4927
Lovell, K. M., Frankowski, K. J., Stahl, E. L., Slauson, S. R., Yoo, E., Prisinzano, T. E., et al. (2015). Structure-activity relationship studies of functionally selective kappa opioid receptor agonists that modulate ERK 1/2 phosphorylation while preserving G protein over betaarrestin2 signaling bias. ACS Chem. Neurosci. 6, 1411–1419. doi: 10.1021/acschemneuro.5b00092
Luttrell, L. M., Maudsley, S., and Bohn, L. M. (2015). Fulfilling the promise of “biased” G protein-coupled receptor agonism. Mol. Pharmacol. 88, 579–588. doi: 10.1124/mol.115.099630
Maillet, E. L., Milon, N., Heghinian, M. D., Fishback, J., Schurer, S. C., Garamszegi, N., et al. (2015). Noribogaine is a G-protein biased kappa-opioid receptor agonist. Neuropharmacology 99, 675–688. doi: 10.1016/j.neuropharm.2015.08.032
Malcolm, B. J., Polanco, M., and Barsuglia, J. P. (2018). Changes in withdrawal and craving scores in participants undergoing opioid detoxification utilizing ibogaine. J. Psychoact. Drugs 50, 256–265. doi: 10.1080/02791072.2018.1447175
Manglik, A., Lin, H., Aryal, D. K., McCorvy, J. D., Dengler, D., Corder, G., et al. (2016). Structure-based discovery of opioid analgesics with reduced side effects. Nature 537, 185–190. doi: 10.1038/nature19112
Mash, D. C., Ameer, B., Prou, D., Howes, J. F., and Maillet, E. L. (2016). Oral noribogaine shows high brain uptake and anti-withdrawal effects not associated with place preference in rodents. J. Psychopharmacol. 30, 688–697. doi: 10.1177/0269881116641331
Mash, D. C., Duque, L., Page, B., and Allen-Ferdinand, K. (2018). Ibogaine detoxification transitions opioid and cocaine abusers between dependence and abstinence: clinical observations and treatment outcomes. Front. Pharmacol. 9:529. doi: 10.3389/fphar.2018.00529
McCurdy, C. R., Sufka, K. J., Smith, G. H., Warnick, J. E., and Nieto, M. J. (2006). Antinociceptive profile of salvinorin A, a structurally unique kappa opioid receptor agonist. Pharmacol. Biochem. Behav. 83, 109–113. doi: 10.1016/j.pbb.2005.12.011
Miess, E., Gondin, A. B., Yousuf, A., Steinborn, R., Mosslein, N., Yang, Y., et al. (2018). Multisite phosphorylation is required for sustained interaction with GRKs and arrestins during rapid mu-opioid receptor desensitization. Sci. Signal. 11:eaas9609. doi: 10.1016/j.pbb.2005.12.011
Morgenweck, J., Frankowski, K. J., Prisinzano, T. E., Aube, J., and Bohn, L. M. (2015). Investigation of the role of betaarrestin2 in kappa opioid receptor modulation in a mouse model of pruritus. Neuropharmacology 99, 600–609. doi: 10.1016/j.neuropharm.2015.08.027
Mori, T., Nomura, M., Nagase, H., Narita, M., and Suzuki, T. (2002). Effects of a newly synthesized kappa-opioid receptor agonist, TRK-820, on the discriminative stimulus and rewarding effects of cocaine in rats. Psychopharmacology 161, 17–22. doi: 10.1007/s00213-002-1028-z
Nagase, H., Hayakawa, J., Kawamura, K., Kawai, K., Takezawa, Y., Matsuura, H., et al. (1998). Discovery of a structurally novel opioid kappa-agonist derived from 4,5-epoxymorphinan. Chem. Pharm. Bull. 46, 366–369. doi: 10.1248/cpb.46.366
Obara, I., Parkitna, J. R., Korostynski, M., Makuch, W., Kaminska, D., Przewlocka, B., et al. (2009). Local peripheral opioid effects and expression of opioid genes in the spinal cord and dorsal root ganglia in neuropathic and inflammatory pain. Pain 141, 283–291. doi: 10.1016/j.pain.2008.12.006
O’Connor, C., White, K. L., Doncescu, N., Didenko, T., Roth, B. L., Czaplicki, G., et al. (2015). NMR structure and dynamics of the agonist dynorphin peptide bound to the human kappa opioid receptor. Proc. Natl. Acad. Sci. U.S.A. 112, 11852–11857. doi: 10.1073/pnas.1510117112
Pfeiffer, A., Brantl, V., Herz, A., and Emrich, H. M. (1986). Psychotomimesis mediated by kappa opiate receptors. Science 233, 774–776. doi: 10.1126/science.3016896
Raehal, K. M., Walker, J. K., and Bohn, L. M. (2005). Morphine side effects in beta-arrestin 2 knockout mice. J. Pharmacol. Exp. Ther. 314, 1195–1201. doi: 10.1124/jpet.105.087254
Rahman, Z., Gold, S. J., Potenza, M. N., Cowan, C. W., Ni, Y. G., He, W., et al. (1999). Cloning and characterization of RGS9-2: a striatal-enriched alternatively spliced product of the RGS9 gene. J. Neurosci. 19, 2016–2026. doi: 10.1523/jneurosci.19-06-02016.1999
Rajagopal, S., Ahn, S., Rominger, D. H., Gowen-MacDonald, W., Lam, C. M., Dewire, S. M., et al. (2011). Quantifying ligand bias at seven-transmembrane receptors. Mol. Pharmacol. 80, 367–377. doi: 10.1124/mol.111.072801
Rives, M. L., Rossillo, M., Liu-Chen, L. Y., and Javitch, J. A. (2012). 6′-Guanidinonaltrindole (6′-GNTI) is a G protein-biased kappa-opioid receptor agonist that inhibits arrestin recruitment. J. Biol. Chem. 287, 27050–27054. doi: 10.1074/jbc.C112.387332
Robins, M. T., Chiang, T., Mores, K. L., Alongkronrusmee, D., and van Rijn, R. M. (2018). Critical ROLE for Gi/o-protein activity in the dorsal striatum in the reduction of voluntary alcohol intake in C57Bl/6 Mice. Front. Psychiatry 9:112. doi: 10.3389/fpsyt.2018.00112
Roth, B. L., Baner, K., Westkaemper, R., Siebert, D., Rice, K. C., Steinberg, S., et al. (2002). Salvinorin A: a potent naturally occurring nonnitrogenous kappa opioid selective agonist. Proc. Natl. Acad. Sci. U.S.A. 99, 11934–11939. doi: 10.1073/pnas.182234399
Salaga, M., Polepally, P. R., Zielinska, M., Marynowski, M., Fabisiak, A., and Murawska, N. (2015). Salvinorin A analogues PR-37 and PR-38 attenuate compound 48/80-induced itch responses in mice. Br. J. Pharmacol. 172, 4331–4341. doi: 10.1111/bph.13212
Schattauer, S. S., Kuhar, J. R., Song, A., and Chavkin, C. (2017). Nalfurafine is a G-protein biased agonist having significantly greater bias at the human than rodent form of the kappa opioid receptor. Cell. Signal. 32, 59–65. doi: 10.1016/j.cellsig.2017.01.016
Schmid, C. L., Streicher, J. M., Groer, C. E., Munro, T. A., Zhou, L., and Bohn, L. M. (2013). Functional selectivity of 6′-guanidinonaltrindole (6′-GNTI) at kappa-opioid receptors in striatal neurons. J. Biol. Chem. 288, 22387–22398. doi: 10.1074/jbc.M113.476234
Seki, T., Awamura, S., Kimura, C., Ide, S., Sakano, K., Minami, M., et al. (1999). Pharmacological properties of TRK-820 on cloned mu-, delta- and kappa-opioid receptors and nociceptin receptor. Eur. J. Pharmacol. 376, 159–167. doi: 10.1016/s0014-2999(99)00369-6
Sharma, S. K., Jones, R. M., Metzger, T. G., Ferguson, D. M., and Portoghese, P. S. (2001). Transformation of a kappa-opioid receptor antagonist to a kappa-agonist by transfer of a guanidinium group from the 5′- to 6′-position of naltrindole. J. Med. Chem. 44, 2073–2079. doi: 10.1021/jm010095v
Simonson, B., Morani, A. S., Ewald, A. W., Walker, L., Kumar, N., Simpson, D., et al. (2015). Pharmacology and anti-addiction effects of the novel kappa opioid receptor agonist Mesyl Sal B, a potent and long-acting analogue of salvinorin A. Br. J. Pharmacol. 172, 515–531. doi: 10.1111/bph.12692
Singla, N., Minkowitz, H. S., Soergel, D. G., Burt, D. A., Subach, R. A., Salamea, M. Y., et al. (2017). A randomized, phase IIb study investigating oliceridine (TRV130), a novel micro-receptor G-protein pathway selective (mu-GPS) modulator, for the management of moderate to severe acute pain following abdominoplasty. J. Pain Res. 10, 2413–2424. doi: 10.2147/JPR.S137952
Soergel, D. G., Subach, R. A., Burnham, N., Lark, M. W., James, I. E., Sadler, B. M., et al. (2014). Biased agonism of the mu-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: a randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain 155, 1829–1835. doi: 10.1016/j.pain.2014.06.011
Spetea, M., Berzetei-Gurske, I. P., Guerrieri, E., and Schmidhammer, H. (2012). Discovery and pharmacological evaluation of a diphenethylamine derivative (HS665), a highly potent and selective kappa opioid receptor agonist. J. Med. Chem. 55, 10302–10306. doi: 10.1021/jm301258w
Spetea, M., Eans, S. O., Ganno, M. L., Lantero, A., Mairegger, M., Toll, L., et al. (2017). Selective kappa receptor partial agonist HS666 produces potent antinociception without inducing aversion after i.c.v. administration in mice. Br. J. Pharmacol. 174, 2444–2456. doi: 10.1111/bph.13854
Stahl, E. L., Zhou, L., Ehlert, F. J., and Bohn, L. M. (2015). A novel method for analyzing extremely biased agonism at G protein-coupled receptors. Mol. Pharmacol. 87, 866–877. doi: 10.1124/mol.114.096503
Sufka, K. J., Loria, M. J., Lewellyn, K., Zjawiony, J. K., Ali, Z., Abe, N., et al. (2014). The effect of Salvia divinorum and Mitragyna speciosa extracts, fraction and major constituents on place aversion and place preference in rats. J. Ethnopharmacol. 151, 361–364. doi: 10.1016/j.jep.2013.10.059
Togashi, Y., Umeuchi, H., Okano, K., Ando, N., Yoshizawa, Y., Honda, T., et al. (2002). Antipruritic activity of the kappa-opioid receptor agonist, TRK-820. Eur. J. Pharmacol. 435, 259–264.
Tsuji, M., Takeda, H., Matsumiya, T., Nagase, H., Narita, M., and Suzuki, T. (2001). The novel kappa-opioid receptor agonist TRK-820 suppresses the rewarding and locomotor-enhancing effects of morphine in mice. Life Sci. 68, 1717–1725. doi: 10.1016/s0024-3205(01)00957-2
Urs, N. M., Gee, S. M., Pack, T. F., McCorvy, J. D., Evron, T., Snyder, J. C., et al. (2016). Distinct cortical and striatal actions of a beta-arrestin-biased dopamine D2 receptor ligand reveal unique antipsychotic-like properties. Proc. Natl. Acad. Sci. U.S.A. 113, E8178–E8186.
van der Westhuizen, E. T., Breton, B., Christopoulos, A., and Bouvier, M. (2014). Quantification of ligand bias for clinically relevant beta2-adrenergic receptor ligands: implications for drug taxonomy. Mol. Pharmacol. 85, 492–509. doi: 10.1124/mol.113.088880
van Rijn, R. M., and Whistler, J. L. (2009). The delta(1) opioid receptor is a heterodimer that opposes the actions of the delta(2) receptor on alcohol intake. Biol. Psychiatry 66, 777–784. doi: 10.1016/j.biopsych.2009.05.019
Varadi, A., Marrone, G. F., Eans, S. O., Ganno, M. L., Subrath, J. J., Le Rouzic, V., et al. (2015). Synthesis and characterization of a dual kappa-delta opioid receptor agonist analgesic blocking cocaine reward behavior. ACS Chem. Neurosci. 6, 1813–1824. doi: 10.1021/acschemneuro.5b00153
Waldhoer, M., Fong, J., Jones, R. M., Lunzer, M. M., Sharma, S. K., Kostenis, E., et al. (2005). A heterodimer-selective agonist shows in vivo relevance of G protein-coupled receptor dimers. Proc. Natl. Acad. Sci. U.S.A. 102, 9050–9055. doi: 10.1073/pnas.0501112102
Walker, B. M., Valdez, G. R., McLaughlin, J. P., and Bakalkin, G. (2012). Targeting dynorphin/kappa opioid receptor systems to treat alcohol abuse and dependence. Alcohol 46, 359–370. doi: 10.1016/j.alcohol.2011.10.006
Wang, Y., Chen, Y., Xu, W., Lee, D. Y., Ma, Z., Rawls, S. M., et al. (2008). 2-Methoxymethyl-salvinorin B is a potent kappa opioid receptor agonist with longer lasting action in vivo than salvinorin A. J. Pharmacol. Exp. Ther. 324, 1073–1083. doi: 10.1124/jpet.107.132142
Wang, Y., Tang, K., Inan, S., Siebert, D., Holzgrabe, U., Lee, D. Y., et al. (2005). Comparison of pharmacological activities of three distinct kappa ligands (Salvinorin A, TRK-820 and 3FLB) on kappa opioid receptors in vitro and their antipruritic and antinociceptive activities in vivo. J. Pharmacol. Exp. Ther. 312, 220–230. doi: 10.1124/jpet.104.073668
Wang, Y. J., Hang, A., Lu, Y. C., Long, Y., Zan, G. Y., Li, X. P., et al. (2016). kappa Opioid receptor activation in different brain regions differentially modulates anxiety-related behaviors in mice. Neuropharmacology 110, 92–101. doi: 10.1016/j.neuropharm.2016.04.022
Whistler, J. L., and von Zastrow, M. (1998). Morphine-activated opioid receptors elude desensitization by beta-arrestin. Proc. Natl. Acad. Sci. U.S.A. 95, 9914–9919. doi: 10.1073/pnas.95.17.9914
White, K. L., Robinson, J. E., Zhu, H., DiBerto, J. F., Polepally, P. R., Zjawiony, J. K., et al. (2015). The G protein-biased kappa-opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo. J. Pharmacol. Exp. Ther. 352, 98–109. doi: 10.1124/jpet.114.216820
White, K. L., Scopton, A. P., Rives, M. L., Bikbulatov, R. V., Polepally, P. R., Brown, P. J., et al. (2014). Identification of novel functionally selective kappa-opioid receptor scaffolds. Mol. Pharmacol. 85, 83–90. doi: 10.1124/mol.113.089649
Wu, H., Wacker, D., Mileni, M., Katritch, V., Han, G. W., Vardy, E., et al. (2012). Structure of the human kappa-opioid receptor in complex with JDTic. Nature 485, 327–332. doi: 10.1038/nature10939
Xiao, H. S., Huang, Q. H., Zhang, F. X., Bao, L., Lu, Y. J., Guo, C., et al. (2002). Identification of gene expression profile of dorsal root ganglion in the rat peripheral axotomy model of neuropathic pain. Proc. Natl. Acad. Sci. U.S.A. 99, 8360–8365. doi: 10.1073/pnas.122231899
Yan, F., Bikbulatov, R. V., Mocanu, V., Dicheva, N., Parker, C. E., Wetsel, W. C., et al. (2009). Structure-based design, synthesis, and biochemical and pharmacological characterization of novel salvinorin A analogues as active state probes of the kappa-opioid receptor. Biochemistry 48, 6898–6908. doi: 10.1021/bi900605n
Zangrandi, L., Burtscher, J., MacKay, J. P., Colmers, W. F., and Schwarzer, C. (2016). The G-protein biased partial kappa opioid receptor agonist 6′-GNTI blocks hippocampal paroxysmal discharges without inducing aversion. Br. J. Pharmacol. 173, 1756–1767. doi: 10.1111/bph.13474
Zhang, Y., Butelman, E. R., Schlussman, S. D., Ho, A., and Kreek, M. J. (2005). Effects of the plant-derived hallucinogen salvinorin A on basal dopamine levels in the caudate putamen and in a conditioned place aversion assay in mice: agonist actions at kappa opioid receptors. Psychopharmacology 179, 551–558. doi: 10.1007/s00213-004-2087-0
Zheng, Z., Huang, X. P., Mangano, T. J., Zou, R., Chen, X., Zaidi, S. A., et al. (2017). Structure-based discovery of new antagonist and biased agonist chemotypes for the kappa opioid receptor. J. Med. Chem. 60, 3070–3081. doi: 10.1021/acs.jmedchem.7b00109
Zhou, L., Lovell, K. M., Frankowski, K. J., Slauson, S. R., Phillips, A. M., Streicher, J. M., et al. (2013). Development of functionally selective, small molecule agonists at kappa opioid receptors. J. Biol. Chem. 288, 36703–36716. doi: 10.1074/jbc.M113.504381
Keywords: kappa opioid receptor, beta-arrestin, G protein, signaling bias, nalfurafine, diphenethylamine, antinociception, side effects
Citation: Mores KL, Cummins BR, Cassell RJ and van Rijn RM (2019) A Review of the Therapeutic Potential of Recently Developed G Protein-Biased Kappa Agonists. Front. Pharmacol. 10:407. doi: 10.3389/fphar.2019.00407
Received: 10 January 2019; Accepted: 01 April 2019;
Published: 17 April 2019.
Edited by:
Dominique Massotte, UPR3212 Institut des Neurosciences Cellulaires et Intégratives (INCI), FranceReviewed by:
Helmut Schmidhammer, University of Innsbruck, AustriaCopyright © 2019 Mores, Cummins, Cassell and van Rijn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard M. van Rijn, cnZhbnJpam5AcHVyZHVlLmVkdQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.