 Sophie J. Hopkin
Sophie J. Hopkin Jonathan W. Lewis
Jonathan W. Lewis Franziska Krautter
Franziska Krautter Myriam Chimen
Myriam Chimen Helen M. McGettrick
Helen M. McGettrick- 1Institute of Cardiovascular Sciences, University of Birmingham, Birmingham, United Kingdom
- 2Rheumatology Research Group, Arthritis Research UK Centre of Excellence in the Pathogenesis of Rheumatoid Arthritis, Institute of Inflammation and Ageing, University of Birmingham, Birmingham, United Kingdom
Leukocyte recruitment is a pivotal process in the regulation and resolution of an inflammatory episode. It is vital for the protective responses to microbial infection and tissue damage, but is the unwanted reaction contributing to pathology in many immune mediated inflammatory diseases (IMIDs). Indeed, it is now recognized that patients with IMIDs have defects in at least one, if not multiple, check-points regulating the entry and exit of leukocytes from the inflamed site. In this review, we will explore our understanding of the imbalance in recruitment that permits the accumulation and persistence of leukocytes in IMIDs. We will highlight old and novel pharmacological tools targeting these processes in an attempt to trigger resolution of the inflammatory response. In this context, we will focus on cytokines, chemokines, known pro-resolving lipid mediators and potential novel lipids (e.g., sphingosine-1-phosphate), along with the actions of glucocorticoids mediated by 11-beta hydroxysteroid dehydrogenase 1 and 2.
Introduction
Acute inflammation is a self-limiting, resolving response in which leukocyte entry and exit is tightly controlled. An imbalance in these processes permits accumulation and persistence of leukocytes within inflamed tissue, leading to damaging chronic non-resolving inflammation that underpins immune-mediated inflammatory diseases (IMIDs). Although significant advances have been made, we still do not fully understand the physiological processes regulating resolution of inflammation, and whether tissue-specific, or stimuli-specific processes exist. Current therapeutic strategies target leukocytes directly or their cytokine products, and hence the activation process of inflammation, rather than promoting resolution. Identifying the immune components that actively induce resolution of inflammation may be the key to novel therapeutic strategies for the treatment of IMIDs (Fullerton and Gilroy, 2016). In this review, we will focus on the pharmacological tools that influence the migration of leukocytes during an inflammatory response and whether such agents can trigger resolution (Figure 1).
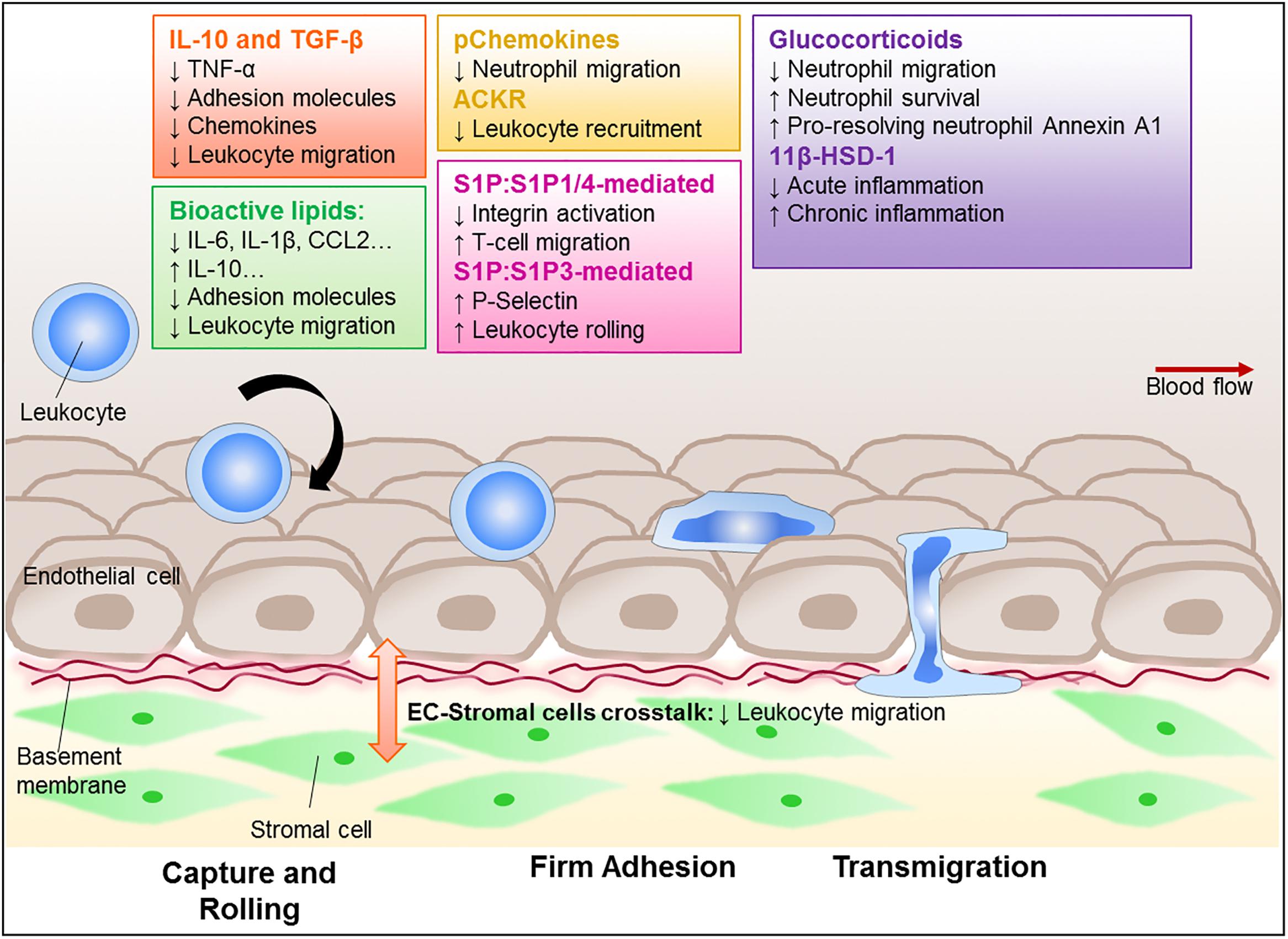
Figure 1. Regulation of leukocyte trafficking as a way to enhance resolution. Leukocytes, such as neutrophils, monocytes, and lymphocytes, are recruited to and migrate through the vessel wall to reach the site of inflammation. This process is tightly regulated and involves adhesion molecules and chemokine-chemokine receptor signal transduction, as well as interaction with the stromal compartment. Critically, these processes become dysregulated in chronic inflammatory diseases leading to aberrant recruitment that contributes to the diseases. There is a growing interest in finding ways to control leukocyte trafficking as a means to reduce the numbers of leukocyte in inflammatory sites, and therefore potentially allow resolution. Amongst those potential targets are cytokines, such as IL-10 and TGF-β; lipid mediators (e.g., lipoxins, resolvins, maresin-1, and S1P); annexin A1, glucocorticoids and its regulating enzyme 11β-HSD-1, as well as the recently characterized pChemokines and ACKRs. These potential targets are all capable of modulating leukocyte migration and further investigation is required to establish their potential pro-resolution roles.
Leukocyte Trafficking in Health
Upon inflammation, blood vascular endothelial cells (EC) up-regulate adhesion molecules and chemokines necessary to support dynamic EC-leukocyte interactions and allow leukocytes to cross the EC barrier (Nourshargh and Alon, 2014; Mellado et al., 2015). The leukocytes themselves receive a series of sequential signals as they negotiate this barrier, which influence their adhesive and migratory properties, effector functions (Luo et al., 2015) and survival (Filer et al., 2006; McGettrick et al., 2006) at the inflamed site. At the blood-tissue interface, selectivity between neutrophils and T-cells arises from the production and use of specific capture receptors (E-, P-selectins, low affinity α4β1-integrin), adhesion molecules (β2-, β1-integrins binding ICAM-1 or VCAM-1, respectively), chemokine and junctional molecule combinations (Reglero-Real et al., 2016). Tissue-specific “address-codes” (Parsonage et al., 2005) are created by the interactions of tissue-resident stromal cells with neighboring EC (McGettrick et al., 2012) and provide an extra level of complexity to the leukocyte adhesion cascade, controlling the number and type of leukocytes recruited during a given inflammatory event.
Chronic Inflammation: Dysregulation of Trafficking
Growing evidence indicates that leukocyte entry into, migration through and exit from peripherally inflamed tissues is changed to some degree in patients with IMIDs, and that these processes can differ between individual’s with the same clinical diagnosis, and over the life-time of disease [i.e., amongst different phases of disease, following therapeutic intervention; (Buckley and McGettrick, 2018)]. Susceptibility genes associated with IMIDs have been shown to directly influence leukocyte recruitment and migration. For instance, expression of the rheumatoid arthritis (RA) susceptibility variant of PTPTN22 (R620W) has been reported to increase the adhesive and migratory properties of murine T-cells (Burn et al., 2016) and human neutrophils (Bayley et al., 2015) in non-diseased models and subjects. Similarly alterations in the cellular metabolism of leukocytes [namely T-cells in RA (Shen et al., 2017) or monocytes in atherosclerosis] can render these cells hypermotile or overtly pro-inflammatory (Chimen et al., 2017). Additionally vascular EC from chronically inflamed tissues acquire pathogenic traits, including elevated expression of adhesion molecules (Jones et al., 1995; Salmi et al., 1997; Cañete et al., 2007). For instance, cultured rheumatoid synovial EC required minimum TNFα stimulation to recruit leukocytes (Abbot et al., 1999). Similarly, secretory smooth muscle cells associated with atherosclerotic plaques are able to prime neighboring blood vascular EC to recruit leukocytes in response to very low TNFα concentrations (Rainger and Nash, 2001). Pathogenic rheumatoid synovial fibroblasts overtly activate EC, leading to the inappropriate influx of leukocytes (Lally et al., 2005; Smith et al., 2008; McGettrick et al., 2009). These interactions evolve with the progression of RA (Filer et al., 2017). This will ultimately change the phenotype of EC (McGettrick et al., 2015) and therefore the types of leukocytes they recruited as the disease persists. Thus it is clear that IMIDs adversely affect key cellular components that control leukocyte migration. Whilst we currently are unable to modify the genetic background of a patient with IMIDs, targeting environmental alterations in key cellular components to trigger resolution pathways is a much needed strategy.
Triggering Resolution of Immune-Mediated Inflammatory Diseases
In humans, it is difficult to evaluate the impact of current IMID therapies on the process of leukocyte trafficking, with many studies only commenting on the changes in cell numbers in one tissue. A reduction in leukocyte numbers at an inflamed site due to drug treatment could arise from (i) reduced entry, (ii) enhanced clearance, (iii) promotion of exit, or (iv) retention in lymph nodes or other peripheral tissues that result in reduced numbers of leukocytes in the circulation (e.g., S1P inhibitors – see below). Indeed, anti-cytokine therapies systemically target key molecules utilized during leukocyte trafficking, including EC activation (TNFα, IL-1β) or EC-stroma crosstalk (e.g., IL-6). Raising the question – what properties should a pro-resolving agent have? If we specifically focus on the context of leukocyte trafficking, potential modes of action would include, limiting cellular infiltration; inducing apoptosis; modulating chemokine and cytokine gradients to promote egress and clearance; reprogramming of leukocytes (e.g., macrophage) phenotypes to induce suppressor cells and induction of tissue repair mechanisms (Sugimoto et al., 2016a).
Cytokines
The cytokine pathways promoting resolution are largely undefined thus far. Certain cytokines are considered to be anti-inflammatory, such as IL-10 and TGF-β, but does this mean they also induce resolution? IL-10 signaling caused the destabilization of TNFα and IL-1α mRNA, thereby reducing protein production in macrophages (Schaljo et al., 2009). Similarly, TGF-β can inhibit the translation of TNFα mRNA into protein in LPS-stimulated murine macrophages in vitro (Bogdan et al., 1992). Reduced TNFα levels at inflamed sites, as seen in patients treated with TNFα inhibitors, causes EC and stromal cells to revert to a resting-like phenotype, downregulating the expression of adhesion molecules and chemokines necessary to support leukocyte migration (Tak et al., 1996). Mice deficient in either IL-10 (Keubler et al., 2015) or TGF-β1 (Kulkarni and Karlsson, 1993; Dang et al., 1995; Letterio et al., 1996) have increased suspectibility to developing IMIDs. However, neither cytokine appeared to induce resolution when administered therapeutically in rodent models of IBD (Herfarth et al., 1998; Barbara et al., 2000; Kitani et al., 2000); and only TGF-β was shown to reduce leukocyte infiltration and disease severity (Kitani et al., 2000). Raising the question as to whether these cytokines can influence the migration of leukocytes to support resolution. IL-10 therapy has been reported to reduce the incidence of psoriasis relapse in a cohort of patients in remission (Friedrich et al., 2002), and induce clinical remission in ∼25% of patients with steroid-resistant Crohn’s disease when compared to placebo control (van Deventer et al., 1997). Whilst both cytokines modulate inflammation, their clinical potential as pro-resolving therapies has yet to be fully determined.
Chemokines
Chemokines are active regulators of leukocyte migration into and out of tissues, as well as playing a key role in the positioning of leukocytes within the inflamed site. The successes and failures of targeting the chemokine pathway to block their pro-inflammatory functions and interfere with leukocyte migration has been reviewed elsewhere (Asquith et al., 2015). Recently, an alternative means of targetting chemokines to induce resolution was described: pChemokines are short-chain peptides with high affinity for chemokine glycosaminoglycan binding domain, which enables them to act as competitive inhibitors for chemokine receptors (McNaughton et al., 2018). pCXCL8 treatment was able to reduce neutrophil migration across CXCL8-treated endothelium in vitro and limited the numbers of leukocytes infiltrating arthritic murine joints (McNaughton et al., 2018). pChemokines are potentially a promising new therapeutic option for IMIDs, limiting the inflammatory infiltrate. It remains to be seen whether pChemokines also display other pro-resolving mechanisms, such as inducing tissue repair or reprogramming of macrophages from classical to alternative activation.
Endogenous removal of chemokines, either by drainage through the lymphatics or by chemokine-scavenging atypical chemokines (ACKRs), is necessary to facilitate the removal of the inflammatory infiltrate during resolution (Bonecchi and Graham, 2016). The potential role of ACKRs in resolution has been reviewed elsewhere (Bonecchi and Graham, 2016). As an example, ACRK2 (also known as D6) deficient mice have increased chemokine expression in the kidney (Bideak et al., 2018) and skin (Jamieson et al., 2005), accompanied by accumulation of T-cells in these tissues and exacerbation of nephrotoxic nephritis and psoriasis. To date, it is unclear whether the functional properties of ACKR2 can be defined as pro-resolving rather than anti-inflammatory, and whether ACKR2 has utility as a therapeutic target. Nevertheless, it is possible that agents that manipulate the expression and/or sequestering properties of ACKRs may be able trigger the resolution process in patients with IMIDs. Further work in this area is urgently required.
Bioactive Pro-resolving Mediators – Resolvins, Lipoxins, Protectins, Maresin and Annexin A1
A variety of bioactive lipid mediators and proteins with pro-resolving properties have been identified, including lipoxins, resolvins, protectins and maresins (Serhan and Petasis, 2011), and subsequently shown to become dysregulated in patients with IMIDs contributing to pathology (Serhan, 2014; Brouwers et al., 2015). Circulating cytokine and chemokine levels can be directly modulated by such agents – for instance maresin can reduce IL-6, IL-1β (Marcon et al., 2013), and CCL2 levels (Martínez-Fernández et al., 2017), whilst annexin A1 (also known as lipocortin) is able to increase IL-10 production (Martínez-Fernández et al., 2017). Moreover, annexin A1, resolvins D1 and D2, and lipoxin A4 can all inhibit the expression of selectin molecules [e.g., P-selectin (Scalia et al., 1997), E-selectin (Chatterjee et al., 2014), or trigger L-selectin shedding (Strausbaugh and Rosen, 2001)] and also reduce β-integrin affinity states and their ability to cluster (Spite et al., 2009; Krishnamoorthy et al., 2010; Drechsler et al., 2015) on both leukocytes and on the endothelium. Reduced expression, activation and clustering of adhesion molecules, along with increased shedding will have considerable impact on the leukocyte recruitment cascade. Indeed, substantial evidence exists that pro-resolving lipid mediators, such as annexin A1, maresin-1, lipoxin A4, resolvin E1 and protectin D1 can inhibit neutrophil or monocyte infiltration into a variety of inflamed tissues, including mesentery (Lim et al., 1998), gut (Schwab et al., 2007), lung (Guido et al., 2013; Gong et al., 2014), brain (Gavins et al., 2012), atherosclerotic lesions (Drechsler et al., 2015), to promote resolution. Protectin D1, and to a lesser extent resolvin E1, are also able to enhance neutrophil and macrophage egress from inflamed cavities to neighboring lymphoid tissues (lymph node/spleen), further facilitating resolution through the removal of the microbial challenge via the lymphatics (Schwab et al., 2007). For further details, this topic is reviewed in depth elsewhere (Ortega-Gómez et al., 2013; Headland and Norling, 2015; Sugimoto et al., 2016b). Such data would indicate that these agents offer the potential to induce resolution in patients with IMIDs; however, the clinical efficacy of these agents has yet to be proven.
Sphingosine-1-Phosphate
Numerous pharmaceutical companies are currently interested in modifying the bioactivity of sphingosine-1-phosphate (S1P) (Dyckman, 2017), yet it remains unclear whether S1P functions as a pro-resolving or a pro-inflammatory lipid mediator. The most abundant store of S1P is found in the blood, where the majority is bound to plasma proteins reducing its bioavailability (Christoffersen et al., 2011). The two main consequences of this are: (i) a S1P concentration gradient between the blood and tissue (Pappu et al., 2007) and (ii) reduced S1P receptor (SIPR) expression on circulating leukocytes (Lo et al., 2005). However, re-expression of surface S1PR1 and S1PR4 is stimulated by chemokine-induced integrin activation of T-cells bound to inflamed blood vascular EC, sensitizing these cells to S1P (Chimen et al., 2015). Under these circumstances, locally released S1P was able to inhibit T-cell transendothelial migration, by reducing the affinity state of β2-integrins from high to low (Chimen et al., 2015). In this study, B-cells recruited to the inflamed EC and binding adiponectin secrete a novel 14 aa immunomodulatory peptide, called PEPITEM (PEPtide Inhibitor of Transendothelial Migration) (Chimen et al., 2015). PEPITEM binds to cadherin-15 on the endothelium triggering S1P production and release through the S1P transporter, SPSN2 (Chimen et al., 2015).
The inability to produce PEPITEM, and thus stimulate local S1P production, contributes to the inappropriate accumulation of T-cells in inflamed tissues in type-1-diabetes and RA (Chimen et al., 2015). Thus in this context S1P acts in an anti-inflammatory manner and could be an early initiator of the pro-resolving machinery. Mast cell derived S1P can also indirectly regulate leukocyte rolling triggering rapid mobilization of P-selectin to the EC surface in a S1PR3 dependent manner in response to tissue damage (Nussbaum et al., 2015). In a counter-regulatory manner, leukocyte rolling was enhanced in S1PR1 deficient mice, indicating that S1PR1 is inhibitory for leukocyte rolling (Nussbaum et al., 2015). Thus the actions of S1P may be subtly modified dependent on the different S1PR triggered.
In addition to regulating leukocyte entry into inflamed peripheral tissues, S1P has also been reported to influence the transit through and exit from these sites across lymphatic endothelium (Ledgerwood et al., 2007). For instance, S1P reportedly enables activated CD4+ T-cells (OT-II cells) to persist and move about within inflamed ear pinnae when the cells are injected directly into the tissue, whereby inhibiting S1P signaling with fingolimod reduced the speed at which T-cells cells traveled within the ear (Jaigirdar et al., 2017). Moreover, fingolimod significantly reduced the number of activated T-cells retained in the ear, suggesting that in the absence of S1P signals the activated T-cells were now able to migrated out of the tissue across the lymphatics (Jaigirdar et al., 2017). Similarly, S1P signaling through S1PR1 blocked T-cell migration across lymphatic endothelial cells of the footpad (Ledgerwood et al., 2007), strongly indicating that S1P signaling regulates T-cell exit of peripheral tissues. Overall it appears that S1P has dual roles in regulating leukocyte recruitment and migration, acting to both promote and inhibit it depending on context. The relationship between these properties and the resolution processes remains to be fully elucidated.
Dysregulation of S1P production, leading to higher S1P levels in chronically inflamed tissues is a shared feature of many IMIDs. For example, elevated levels of the enzyme (SPHK-1) necessary for S1P synthesis have been reported in rheumatoid synovium (Jaigirdar et al., 2017) and ulcerative colitis (Karuppuchamy et al., 2017), whilst high concentrations of S1P occur in broncholavage fluid (Ammit et al., 2001) and cerebrospinal fluid (Kułakowska et al., 2010) from asthma and multiple sclerosis (MS) patients. This tends to support the notion that the bioactivity of S1P is pro-inflammatory rather than pro-resolving. Indeed, inhibiting S1P signaling with FTY720 has protective effects when administered therapeutically in rodent models of MS (Sheridan and Dev, 2014), inflammatory arthritis (Matsuura et al., 2000; Wang et al., 2007; Tsunemi et al., 2010; Fujii et al., 2012; Han et al., 2015), and systemic lupus erythematosus (SLE) (Wenderfer et al., 2008). Moreover, fingolimod is an FDA-approved treatment for MS with clinical efficacy in reducing relapses in patients with relapsing remitting MS (Kappos et al., 2006; Singer, 2013), but not those with primary progressive MS (Lublin et al., 2016). Nevertheless, existing S1P modulators are known to induce lymphopenia (Wang et al., 2007; Tsunemi et al., 2010) by blocking lymphocyte exit from lymph nodes (Mandala et al., 2002; Chiba, 2005; Han et al., 2015) – thus indirectly reducing the circulating numbers of cells available to enter peripherally inflamed sites. This can result in a general immunosuppression in patients; increasing susceptibility to opportunistic infections, whilst reducing vaccine efficiency (Massberg and von Andrian, 2006). Moreover, it is highly probable that S1P modulators also interfere with the S1P-dependent migration of T-cell out of peripherally inflamed tissues across lymphatic endothelium (Ledgerwood et al., 2007), and thus maybe responsible for retention of cells at the inflamed site further exacerbating disease.
11-Beta HSD Enzymes in Regulating GC Function
Glucocorticoids (GCs) are steroid hormones responsible for regulating cellular metabolism, immune function, adhesion molecule expression, and leukocyte migration (Tomlinson and Stewart, 2001). Dexamethasone (a synthetic GC) reduced the expression of E-selectin on inflamed aortic EC, disrupting neutrophil migration (Brostjan et al., 1997). By contrast, dexamethasone enhanced CXCL12-induced chemotaxis of resting human T-cells in vitro (Ghosh et al., 2009). Blocking GC function with prophylactic administration of glucocorticoid receptor (GR) antagonists exacerbated neutrophil infiltration into the synovial of carrageenan-induced monoarthritis in rats (Leech et al., 1998). GCs also influence cell viability, promoting neutrophil survival (Cox, 1995; Ruiz et al., 2002), whilst stimulating eosinophil apoptosis (Druilhe et al., 2003). Importantly, GCs can indirectly promote the resolution of inflammation through the induction of annexin A1 on human neutrophils and monocytes (Goulding et al., 1990). Annexin A1 can disrupt neutrophil migration, causing adherent neutrophils to detach from inflamed mesenteric endothelium and re-enter the circulation (Lim et al., 1998) restoring tissue homeostasis. Synthetic GCs clearly elicit cell-type specific effects, eliciting more immunomodulatory rather than immunosuppressive effects and may even exacerbate inflammation. Yet they are commonly used to treat IMIDs [e.g., RA, MS, psoriasis (Coutinho and Chapman, 2011)], where prolonged use is associated with metabolic and endocrine dysregulation (Schäcke et al., 2002).
The predominately active GC in humans is cortisol, which upon binding to the cytosolic GR, modifies gene expression to promote an anti-inflammatory response (Schüle et al., 1990; De Bosscher et al., 1997). The local bioavailability of GC is regulated by metabolic enzymes, including the two isoforms of 11β-hydroxysteroid dehydrogenase [11β-HSD-1 and 11β-HSD-2; (Seckl and Walker, 2001; Tomlinson and Stewart, 2001)]. Residing in the lumen of the ER, 11β-HSD-1 primarily reduces cortisone (inactive GC) to cortisol (active GC) increasing local active GC concentrations, whilst 11β-HSD-2 catalyzes the reverse reaction – inactivating cortisol and reducing active GC levels (Albiston et al., 1994; Seckl and Walker, 2001; Tomlinson and Stewart, 2001). 11β-HSD-1 expression and activity are ubiquitious, albeit at varying amounts: high expression is found in GC-target tissues [e.g., liver and fat; (Seckl and Walker, 2001)] and much lower levels are seen in leukocytes (Thieringer et al., 2001; Chapman et al., 2009; Coutinho et al., 2016). In contrast, 11β-HSD-2 expression and activity are largely restricted to mineralocorticoid-target tissues, e.g., the kidneys, pancreas and large intestine (Albiston et al., 1994), and not found in leukocytes. Importantly, the expression and activity of 11β-HSD-1 is dynamically regulated during inflammation, where cytokines such as IL-1β (Sun and Myatt, 2003), IL-4 (Thieringer et al., 2001), and IL-13 (Thieringer et al., 2001) induce 11β-HSD-1 activity stimulating local increases in active GC which exert anti-inflammatory and pro-resolving effects. Interestingly, GC metabolism is skewed in patients with IMIDs, such as SLE (Ichikawa et al., 1997) and RA (Hardy et al., 2006), toward cortisol production and therefore should trigger GC-induced anti-inflammatory/pro-resolving pathways to dampen the inflammatory response. However, despite elevated plasma cortisol levels in patients with IMIDs, the anti-inflammatory/pro-resolving GC pathways are not obviously triggered. This discrepancy has been attributed to an insufficient levels of active GCs, as this deficiency can be overcome by administration of high-dose GC mimics to IMID patients (Straub and Cutolo, 2016). Thus the relationship between plasma cortisol and active GC is not strictly linear in chronic inflammation, opening avenues for further research into the dysregulation of GC metabolism.
11β-HSD-1 have also been reported to modulate leukocyte trafficking by influencing expression of chemokines and adhesion molecules (Wamil et al., 2011; Kipari et al., 2013; Mylonas et al., 2017). However, in vivo studies blocking 11β-HSD-1 function with chemical agents or in 11β-HSD-1-deficient (Hsd11b1-/-) mice have reported conflicting findings. In a model of acute thioglycollate-induced peritonitis in mice, augmented leukocyte recruitment was observed following prophylactic inhibition of local 11β-HSD-1 (Coutinho et al., 2016) and in Hsd11b1-/- mice (Coutinho et al., 2012). Similar findings were reported in carrageenan-induced pleurisy (Coutinho et al., 2012) and coronary artery ligation induced myocardial infarction (McSweeney et al., 2010) in Hsd11b1-/- mice, supporting the concept that 11β-HSD-1 functions to limit inflammation. In contrast, lower amounts of MCP-1 were released by adipocytes from Hsd11b1-/- mice on a high fat diet, resulting in fewer CD8+ T-cell and macrophage infiltrating mesenteric adipose tissue (Wamil et al., 2011). Similarly low VCAM-1 expression by aortic endothelial cells was attributed to the significant reduction in T-cell and macrophage within atherosclerotic plaques of Hsd11b1-/- mice on high fat diet (Kipari et al., 2013). These studies indicate that 11β-HSD-1 activity promotes leukocyte recruitment and hence inflammation. The field currently believes that the functional outcomes of 11β-HSD-1 activity, whether these be pro or anti-inflammatory, is governed by a mixture of cell-specific, tissue-specific and inflammatory context-specific factors. Therefore, it is impossible to say with any certainty that 11β-HSD-1 has pro-resolving properties and is a viable drug target without further studies in this area.
That said, phase 2 clinical trials examining the efficacy of 11β-HSD-1 selective inhibitors, such as INCB13739 in obesity-related inflammatory diseases are ongoing (Anagnostis et al., 2013), but as yet no candidate drug is in the pipeline for IMIDs. Nevertheless caution is required: 11β-HSD-1 down-regulators [e.g., glycyrrhizic acid and rosiglitazone (Mai et al., 2007; Wake et al., 2007)] are associated with increased risk of cardiovascular-associated morbidity (Nissen and Wolski, 2007), hypertension encephalopathy (Russo et al., 2000) and hypokalemic paralysis (Pant et al., 2010). Given the tissue-restricted expression patterns of 11β-HSD, there is growing excitement about the potential to specifically modulate local GC concentrations using tissue-specific targeted therapies. However, we do not fully understand the role of these enzymes in specific IMIDs. Critically, the effects of endogenous GC and synthetic mimics are context dependent based on cell-type and local environmental conditions creating a complex interplay between GC, 11β-HSD enzymes and local environment, which is not yet fully understood. Clarifying the role of 11β-HSD enzymes in different IMIDs will allow the anti-inflammatory and pro-resolution properties that they exert to be exploited to promote the resolution of inflammation.
Conclusion and Current Perceptions
The regulated movement of leukocytes into, through and out of peripheral tissues is vital in order to mediate tissue homeostasis in response to an inflammatory insult. We are expanding our understanding into how these processes are altered in the pathogenesis of IMIDs, and crucially the timing of such changes and their impact on the resolution of inflammation. With every step forward, key agents with the capacity to induce resolution and that may be amenable to therapeutic intervention become clearer. This represents an exciting new prospect that these novel drugs would actively target endogenous regulatory processes to reduce leukocyte entry into tissues and promote their clearance and egress to restore tissue homeostasis.
Author Contributions
HM wrote the first draft of the manuscript. SH, JL, FK, and MC wrote sections of the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
Funding
SH, JL, and FK were supported by MRC-Arthritis Research UK, Royal Society (RGF∖R1∖180008) and The Academy of Medical Sciences (SBF003∖1156) funded Ph.D. studentships, respectively. MC was supported by a Royal Society Dorothy Hodgkin Research fellowship (DH160044) and HM was supported by an Arthritis Research UK Career Development Fellowship (19899). Arthritis Research UK Rheumatoid Arthritis Pathogenesis Centre of Excellence (RACE) was part-funded by Arthritis Research UK (20298), this center is a collaboration between the Universities of Glasgow, Newcastle, and Birmingham. MRC-Arthritis Research UK Centre for Musculoskeletal Ageing Research (MR/P021220/1) is a collaboration between the Universities of Birmingham, Nottingham, and Oxford.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abbot, S. E., Whish, W. J., Jennison, C., Blake, D. R., and Stevens, C. R. (1999). Tumour necrosis factor alpha stimulated rheumatoid synovial microvascular endothelial cells exhibit increased shear rate dependent leucocyte adhesion in vitro. Ann. Rheum. Dis. 58, 573–581. doi: 10.1136/ard.58.9.573
Albiston, A. L., Obeyesekere, V. R., Smith, R. E., and Krozowski, Z. S. (1994). Cloning and tissue distribution of the human 11 beta-hydroxysteroid dehydrogenase type 2 enzyme. Mol. Cell. Endocrinol. 105, R11–R17.
Ammit, A. J., Hastie, A. T., Edsall, L. C., Hoffman, R. K., Amrani, Y., Krymskaya, V. P., et al. (2001). Sphingosine 1-phosphate modulates human airway smooth muscle cell functions that promote inflammation and airway remodeling in asthma. FASEB J. 15, 1212–1214. doi: 10.1096/fj.00-0742fje
Anagnostis, P., Katsiki, N., Adamidou, F., Athyros, V. G., Karagiannis, A., Kita, M., et al. (2013). 11beta-Hydroxysteroid dehydrogenase type 1 inhibitors: novel agents for the treatment of metabolic syndrome and obesity-related disorders? Metabolism 62, 21–33. doi: 10.1016/j.metabol.2012.05.002
Asquith, D. L., Bryce, S. A., and Nibbs, R. J. B. (2015). Targeting cell migration in rheumatoid arthritis. Curr. Opin. Rheumatol. 27, 204–211. doi: 10.1097/BOR.0000000000000150
Barbara, G., Xing, Z., Hogaboam, C., Gauldie, J., and Collins, S. (2000). Interleukin 10 gene transfer prevents experimental colitis in rats. Gut 46, 344–349. doi: 10.1136/gut.46.3.344
Bayley, R., Kite, K. A., McGettrick, H. M., Smith, J. P., Kitas, G. D., Buckley, C. D., et al. (2015). The autoimmune-associated genetic variant PTPN22 R620W enhances neutrophil activation and function in patients with rheumatoid arthritis and healthy individuals. Ann. Rheum. Dis. 74, 1588–1595. doi: 10.1136/annrheumdis-2013-204796
Bideak, A., Blaut, A., Hoppe, J. M., Müller, M. B., Federico, G., Eltrich, N., et al. (2018). The atypical chemokine receptor 2 limits renal inflammation and fibrosis in murine progressive immune complex glomerulonephritis. Kidney Int. 93, 826–841. doi: 10.1016/j.kint.2017.11.013
Bogdan, C., Paik, J., Vodovotz, Y., and Nathan, C. (1992). Contrasting mechanisms for suppression of macrophage cytokine release by transforming growth factor-beta and interleukin-10. J. Biol. Chem. 267, 23301–23308.
Bonecchi, R., and Graham, G. J. (2016). Atypical chemokine receptors and their roles in the resolution of the inflammatory response. Front. Immunol. 7:224. doi: 10.3389/fimmu.2016.00224
Brostjan, C., Anrather, J., Csizmadia, V., Natarajan, G., and Winkler, H. (1997). Glucocorticoids inhibit E-selectin expression by targeting NF-kappaB and not ATF/c-Jun. J. Immunol. 158, 3836–3844.
Brouwers, H., von Hegedus, J., Toes, R., Kloppenburg, M., and Ioan-Facsinay, A. (2015). Lipid mediators of inflammation in rheumatoid arthritis and osteoarthritis. Best Pract. Res. Clin. Rheumatol. 29, 741–755. doi: 10.1016/j.berh.2016.02.003
Buckley, C. D., and McGettrick, H. M. (2018). Leukocyte trafficking between stromal compartments: lessons from rheumatoid arthritis. Nat. Rev. Rheumatol. 14, 476–487. doi: 10.1038/s41584-018-0042-4
Burn, G. L., Cornish, G. H., Potrzebowska, K., Samuelsson, M., Griffi, J., Minoughan, S., et al. (2016). Superresolution imaging of the cytoplasmic phosphatase PTPN22 links integrin-mediated T cell adhesion with autoimmunity. Sci. Signal 9:ra99.
Cañete, J. D., Santiago, B., Cantaert, T., Sanmartí, R., Palacin, A., Celis, R., et al. (2007). Ectopic lymphoid neogenesis in psoriatic arthritis. Ann. Rheum. Dis. 66, 720–726. doi: 10.1136/ard.2006.062042
Chapman, K. E., Coutinho, A. E., Gray, M., Gilmour, J. S., Savill, J. S., and Seckl, J. R. (2009). The role and regulation of 11β-hydroxysteroid dehydrogenase type 1 in the inflammatory response. Mol. Cell. Endocrinol. 301, 123–131. doi: 10.1016/j.mce.2008.09.031
Chatterjee, A., Sharma, A., Chen, M., Toy, R., Mottola, G., and Conte, M. S. (2014). The pro-resolving lipid mediator maresin 1 (MaR1) attenuates inflammatory signaling pathways in vascular smooth muscle and endothelial cells. PLoS One 9:e0113480. doi: 10.1371/journal.pone.0113480
Chiba, K. (2005). FTY720, a new class of immunomodulator, inhibits lymphocyte egress from secondary lymphoid tissues and thymus by agonistic activity at sphingosine 1-phosphate receptors. Pharmacol. Ther. 108, 308–319. doi: 10.1016/j.pharmthera.2005.05.002
Chimen, M., McGettrick, H. M., Apta, B., Kuravi, S. J., Yates, C. M., Kennedy, A., et al. (2015). Homeostatic regulation of T cell trafficking by a B cell-derived peptide is impaired in autoimmune and chronic inflammatory disease. Nat. Med. 21, 467–475. doi: 10.1038/nm.3842
Chimen, M., Yates, C. M., McGettrick, H. M., Ward, L. S. C., Harrison, M. J., Apta, B., et al. (2017). Monocyte subsets co-regulate inflammatory responses by integrated signalling through TNF and IL-6 at the endothelial cell interface. J. Immunol. 198, 2834–2843. doi: 10.4049/jimmunol.1601281
Christoffersen, C., Obinata, H., Kumaraswamy, S. B., Galvani, S., Ahnström, J., Sevvana, M., et al. (2011). Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc. Natl. Acad. Sci. U.S.A. 108, 9613–9618. doi: 10.1073/pnas.1103187108
Coutinho, A. E., and Chapman, K. E. (2011). The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol. Cell. Endocrinol. 335, 2–13. doi: 10.1016/j.mce.2010.04.005
Coutinho, A. E., Gray, M., Brownstein, D. G., Salter, D. M., Sawatzky, D. A., Clay, S., et al. (2012). 11β-hydroxysteroid dehydrogenase type 1, but not type 2, deficiency worsens acute inflammation and experimental arthritis in mice. Endocrinology 153, 234–240. doi: 10.1210/en.2011-1398
Coutinho, A. E., Kipari, T. M. J., Zhang, Z., Esteves, C. L., Lucas, C. D., Gilmour, J. S., et al. (2016). 11β-hydroxysteroid dehydrogenase type 1 is expressed in neutrophils and restrains an inflammatory response in male mice. Endocrinology 157, 2928–2936. doi: 10.1210/en.2016-1118
Cox, G. (1995). Glucocorticoid treatment inhibits apoptosis in human neutrophils. Separation of survival and activation outcomes. J. Immunol. 154, 4719–4725.
Dang, H., Geiser, A. G., Letterio, J. J., Nakabayashi, T., Kong, L., Fernandes, G., et al. (1995). SLE-like autoantibodies and Sjögren’s syndrome-like lymphoproliferation in TGF-beta knockout mice. J. Immunol. 155, 3205–3212.
De Bosscher, K., Schmitz, M. L., Vanden Berghe, W., Plaisance, S., Fiers, W., and Haegeman, G. (1997). Glucocorticoid-mediated repression of nuclear factor-κBdependent transcription involves direct interference with transactivation. Proc. Natl. Acad. Sci. U.S.A. 94, 13504–13509. doi: 10.1073/pnas.94.25.13504
Drechsler, M., Jong, R. D., Rossaint, J., Viola, J. R., Leoni, G., Wang, J. M., et al. (2015). Annexin A1 counteracts chemokine-induced arterial myeloid cell recruitment. Circ. Res. 116, 827–835. doi: 10.1161/CIRCRESAHA.116.305825
Druilhe, A., Létuvé, S., and Pretolani, M. (2003). Glucocorticoid-induced apoptosis in human eosinophils: mechanisms of action. Apoptosis 8, 481–495. doi: 10.1023/A:1025590308147
Dyckman, A. J. (2017). Modulators of sphingosine-1-phosphate pathway biology: recent advances of sphingosine-1-phosphate receptor 1 (S1P1) agonists and future perspectives. J. Med. Chem. 60, 5267–5289. doi: 10.1021/acs.jmedchem.6b01575
Filer, A., Parsonage, G., Smith, E., Osborne, C., Thomas, A. M., Curnow, S. J., et al. (2006). Differential survival of leukocyte subsets mediated by synovial, bone marrow, and skin fibroblasts: site-specific versus activation-dependent survival of T cells and neutrophils. Arthritis Rheum. 54, 2096–2108. doi: 10.1002/art.21930
Filer, A., Ward, L. S. C., Kemble, S., Davies, C. S., Munir, H., Rogers, R., et al. (2017). Identification of a transitional fibroblast function in very early rheumatoid arthritis. Ann. Rheum. Dis. 76, 2105–2112. doi: 10.1136/annrheumdis-2017-211286
Friedrich, M., Döcke, W.-D., Klein, A., Philipp, S., Volk, H.-D., Sterry, W., et al. (2002). Immunomodulation by interleukin-10 therapy decreases the incidence of relapse and prolongs the relapse-free interval in psoriasis. J. Invest. Dermatol. 118, 672–677. doi: 10.1046/j.1523-1747.2002.01731.x
Fujii, Y., Hirayama, T., Ohtake, H., Ono, N., Inoue, T., Sakurai, T., et al. (2012). Amelioration of collagen-induced arthritis by a novel s1p1 antagonist with immunomodulatory activities. J. Immunol. 188, 206–215. doi: 10.4049/jimmunol.1101537
Fullerton, J. N., and Gilroy, D. W. (2016). Resolution of inflammation: a new therapeutic frontier. Nat. Rev. Drug Discov. 15, 551–567. doi: 10.1038/nrd.2016.39
Gavins, F. N. E., Hughes, E. L., Buss, N. A. P. S., Holloway, P. M., Getting, S. J., and Buckingham, J. C. (2012). Leukocyte recruitment in the brain in sepsis: involvement of the annexin 1-FPR2/ALX anti-inflammatory system. FASEB J. 26, 4977–4989. doi: 10.1096/fj.12-205971
Ghosh, M. C., Baatar, D., Collins, G., Carter, A., Indig, F., Biragyn, A., et al. (2009). Dexamethasone augments CXCR4-mediated signaling in resting human T cells via the activation of the Src kinase Lck. Blood 113, 575–584. doi: 10.1182/blood-2008-04-151803
Gong, J., Wu, Z. Y., Qi, H., Chen, L., and Li, H. B. (2014). Maresin 1 mitigates LPS-induced acute lung injury in mice. Br. J. Pharmacol. 171, 3539–3550. doi: 10.1111/bph.12714
Goulding, N. J., Godolphin, J. L., Sharland, P. R., Maddison, P. J., Sampson, M., Peers, S. H., et al. (1990). Anti-inflammatory lipocortin 1 production by peripheral blood leucocytes in response to hydrocortisone. Lancet 335, 1416–1418. doi: 10.1016/0140-6736(90)91445-G
Guido, B. C., Zanatelli, M., Tavares-de-Lima, W., Oliani, S. M., and Damazo, A. S. (2013). Annexin-A1 peptide down-regulates the leukocyte recruitment and up-regulates interleukin-10 release into lung after intestinal ischemia-reperfusion in mice. J. Inflamm. 10:10. doi: 10.1186/1476-9255-10-10
Han, Y., Li, X., Zhou, Q., Jie, H., Lao, X., Han, J., et al. (2015). FTY720 abrogates collagen-induced arthritis by hindering dendritic cell migration to local lymph nodes. J. Immunol. 195, 4126–4135. doi: 10.4049/jimmunol.1401842
Hardy, R. S., Filer, A., Cooper, M. S., Parsonage, G., Raza, K., and Hardie, D. L. (2006). Differential expression, function and response to inflammatory stimuli of 11beta-hydroxysteroid dehydrogenase type 1 in human fibroblasts: a mechanism for tissue-specific regulation of inflammation. Arthritis Res. Ther. 8:R108. doi: 10.1186/ar1993
Headland, S. E., and Norling, L. V. (2015). The resolution of inflammation: principles and challenges. Semin. Immunol. 27, 149–160. doi: 10.1016/j.smim.2015.03.014
Herfarth, H. H., Böcker, U., Janardhanam, R., and Sartor, R. B. (1998). Subtherapeutic corticosteroids potentiate the ability of interleukin 10 to prevent chronic inflammation in rats. Gastroenterology 115, 856–865. doi: 10.1016/S0016-5085(98)70257-4
Ichikawa, Y., Yoshida, K., Kawagoe, M., Saito, E., Abe, Y., Arikawa, K., et al. (1997). Altered equilibrium between cortisol and cortisone in plasma in thyroid dysfunction and inflammatory diseases. Metabolism 26, 989–997. doi: 10.1016/0026-0495(77)90016-6
Jaigirdar, S. A., Benson, R. A., Elmesmari, A., Kurowska-Stolarska, M. S., McInnes, I. B., Garside, P., et al. (2017). Sphingosine-1-phosphate promotes the persistence of activated CD4 T cells in inflamed sites. Front. Immunol. 8:1627. doi: 10.3389/fimmu.2017.01627
Jamieson, T., Cook, D. N., Nibbs, R. J. B., Rot, A., Nixon, C., McLean, P., et al. (2005). The chemokine receptor D6 limits the inflammatory response in vivo. Nat. Immunol. 6:403. doi: 10.1038/ni1182
Jones, S. C., Banks, R. E., Haidar, A., Gearing, A. J., Hemingway, I. K., Ibbotson, S. H., et al. (1995). Adhesion molecules in inflammatory bowel disease. Gut 36, 724–730. doi: 10.1136/gut.36.5.724
Kappos, L., Antel, J., Comi, G., Montalban, X., O’Connor, P., Polman, C. H., et al. (2006). Oral Fingolimod (FTY720) for relapsing multiple sclerosis. N. Engl. J. Med. 355, 1124–1140. doi: 10.1056/NEJMoa052643
Karuppuchamy, T., Behrens, E.-H., González-Cabrera, P., Sarkisyan, G., Gima, L., Boyer, J. D., et al. (2017). Sphingosine-1-phosphate receptor-1 (S1P(1)) is expressed by lymphocytes, dendritic cells, and endothelium and modulated during inflammatory bowel disease. Mucosal Immunol. 10, 162–171. doi: 10.1038/mi.2016.35
Keubler, L. M., Buettner, M., Häger, C., and Bleich, A. (2015). A multihit model: colitis lessons from the interleukin-10–deficient mouse. Inflamm. Bowel Dis. 21, 1967–1975. doi: 10.1097/MIB.0000000000000468
Kipari, T., Hadoke, P. W. F., Iqbal, J., Man, T.-Y., Miller, E., Coutinho, A. E., et al. (2013). 11β-hydroxysteroid dehydrogenase type 1 deficiency in bone marrow-derived cells reduces atherosclerosis. FASEB J. 27, 1519–1531. doi: 10.1096/fj.12-219105
Kitani, A., Fuss, I. J., Nakamura, K., Schwartz, O. M., Usui, T., and Strober, W. (2000). Treatment of Experimental (Trinitrobenzene Sulfonic Acid) Colitis by Intranasal Administration of Transforming Growth Factor (Tgf)-β1 Plasmid: TGF-β1–Mediated Suppression of T Helper Cell Type 1 Response Occurs by Interleukin (Il)-10 Induction and IL-12 Receptor β2 Chain Downregulation. J. Exp. Med. 192, 41–52. doi: 10.1084/jem.192.1.41
Krishnamoorthy, S., Recchiuti, A., Chiang, N., Yacoubian, S., Lee, C.-H., Yang, R., et al. (2010). Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc. Natl. Acad. Sci. U.S.A. 107, 1660–1665. doi: 10.1073/pnas.0907342107
Kułakowska, A., Żendzian-Piotrowska, M., Baranowski, M., Konończuk, T., Drozdowski, W., Górski, J., et al. (2010). Intrathecal increase of sphingosine 1-phosphate at early stage multiple sclerosis. Neurosci. Lett. 477, 149–152. doi: 10.1016/j.neulet.2010.04.052
Kulkarni, A. B., and Karlsson, S. (1993). Transforming growth factor-beta 1 knockout mice. A mutation in one cytokine gene causes a dramatic inflammatory disease. Am. J. Pathol. 143, 3–9.
Lally, F., Smith, E., Filer, A., Stone, M. A., Shaw, J. S., Buckley, C., et al. (2005). A novel mechanism of neutrophil recruitment in a coculture model of the rheumatoid synovium. Arthritis Rheum. 52, 3460–3469. doi: 10.1002/art.21394
Ledgerwood, L. G., Lal, G., Zhang, N., Garin, A., Esses, S. J., Ginhoux, F., et al. (2007). The sphingosine 1-phosphate receptor 1 causes tissue retention by inhibiting the entry of peripheral tissue T lymphocytes into afferent lymphatics. Nat. Immunol. 9, 42–53. doi: 10.1038/ni1534
Leech, M., Hutchinson, P., Holdsworth, S. R., and Morand, E. F. (1998). Endogenous glucocorticoids modulate neutrophil migration and synovial P-selectin but not neutrophil phagocytic or oxidative function in experimental arthritis. Clin. Exp. Immunol 112, 383–388. doi: 10.1046/j.1365-2249.1998.00601.x
Letterio, J. J., Geiser, A. G., Kulkarni, A. B., Dang, H., Kong, L., Nakabayashi, T., et al. (1996). Autoimmunity associated with TGF-beta1-deficiency in mice is dependent on MHC class II antigen expression. J. Clin. Invest. 98, 2109–2119. doi: 10.1172/JCI119017
Lim, L. H. K., Solito, E., Russo-Marie, F., Flower, R. J., and Perretti, M. (1998). Promoting detachment of neutrophils adherent to murine postcapillary venules to control inflammation: effect of lipocortin 1. Proc. Natl. Acad. Sci. U.S.A. 95, 14535–14539. doi: 10.1073/pnas.95.24.14535
Lo, C. G., Xu, Y., Proia, R. L., and Cyster, J. G. (2005). Cyclical modulation of sphingosine-1-phosphate receptor 1 surface expression during lymphocyte recirculation and relationship to lymphoid organ transit. J. Exp. Med. 201, 291–301. doi: 10.1084/jem.20041509
Lublin, F., Miller, D. H., Freedman, M. S., Cree, B. A. C., Wolinsky, J. S., Weiner, H., et al. (2016). Oral fingolimod in primary progressive multiple sclerosis (INFORMS): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet 387, 1075–1084. doi: 10.1016/S0140-6736(15)01314-8
Luo, D., McGettrick, H. M., Stone, P. C., Rainger, G. E., and Nash, G. B. (2015). The roles of integrins in function of human neutrophils after their migration through endothelium into interstitial matrix. PLoS One 10:e0118593. doi: 10.1371/journal.pone.0118593
Mai, K., Andres, J., Bobbert, T., Maser-Gluth, C., Möhlig, M., Bähr, V., et al. (2007). Rosiglitazone decreases 11β-hydroxysteroid dehydrogenase type 1 in subcutaneous adipose tissue. Clin. Endocrinol. 67, 419–425. doi: 10.1111/j.1365-2265.2007.02903.x
Mandala, S., Hajdu, R., Bergstrom, J., Quackenbush, E., Xie, J., Milligan, J., et al. (2002). Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 296, 346–349. doi: 10.1126/science.1070238
Marcon, R., Bento, A. F., Dutra, R. C., Bicca, M. A., Leite, D. F. P., and Calixto, J. B. (2013). Maresin 1, a proresolving lipid mediator derived from omega-3 polyunsaturated fatty acids, exerts protective actions in murine models of colitis. J. Immunol. 191, 4288–4298. doi: 10.4049/jimmunol.1202743
Martínez-Fernández, L., González-Muniesa, P., Laiglesia, L. M., Sáinz, N., Prieto-Hontoria, P. L., Escoté, X., et al. (2017). Maresin 1 improves insulin sensitivity and attenuates adipose tissue inflammation in ob/ob and diet-induced obese mice. FASEB J. 31, 2135–2145. doi: 10.1096/fj.201600859R
Massberg, S., and von Andrian, U. H. (2006). Fingolimod and sphingosine-1-phosphate — modifiers of lymphocyte migration. N. Engl. J. Med. 355, 1088–1091. doi: 10.1056/NEJMp068159
Matsuura, M., Imayoshi, T., and Okumoto, T. (2000). Effect of FTY720, a novel immunosuppressant, on adjuvant- and collagen-induced arthritis in rats. Int. J. Immunopharmacol. 22, 323–331. doi: 10.1016/S0192-0561(99)00088-0
McGettrick, H. M., Butler, L. M., Buckley, C. D., Rainger, G., and Nash, G. B. (eds) (2012). Tissue stroma as a regulator of leukocyte recruitment in inflammation. J. Leukoc. Biol. 91, 385–400. doi: 10.1189/jlb.0911458
McGettrick, H. M., Lord, J. M., Wang, K. Q., Rainger, G. E., Buckley, C. D., and Nash, G. B. (2006). Chemokine- and adhesion-dependent survival of neutrophils after transmigration through cytokine-stimulated endothelium. J. Leukoc. Biol. 79, 779–788. doi: 10.1189/jlb.0605350
McGettrick, H. M., Naame, N., He, S., Raza, K., Nash, G. B., Buckley, C. D., et al. (2015). Functional pathways in endothelial cells are differentially regulated by fibroblasts from patients with RA and resolving disease. Ann. Rheum. Dis. 74, A57–A57. doi: 10.1136/annrheumdis-2015-207259.132
McGettrick, H. M., Smith, E., Filer, A., Kissane, S., Salmon, M., Buckley, C. D., et al. (2009). Fibroblasts from different sites may promote or inhibit recruitment of flowing lymphocytes by endothelial cells. Eur. J. Immunol. 39, 113–125. doi: 10.1002/eji.200838232
McNaughton, E. F., Eustace, A. D., King, S., Sessions, R. B., Kay, A., Farris, M., et al. (2018). Novel anti-inflammatory peptides based on chemokine–glycosaminoglycan interactions reduce leukocyte migration and disease severity in a model of rheumatoid arthritis. J. Immunol. 200, 3201–3217. doi: 10.4049/jimmunol.1701187
McSweeney, S. J., Hadoke, P. W. F., Kozak, A. M., Small, G. R., Khaled, H., Walker, B. R., et al. (2010). Improved heart function follows enhanced inflammatory cell recruitment and angiogenesis in 11βHSD1-deficient mice post-MI. Cardiovasc. Res. 88, 159–167. doi: 10.1093/cvr/cvq149
Mellado, M., Cascio, G., Lucas, P., Pablos, J. L., Martínez-Muñoz, L., and Rodríguez-Frade, J. M. (2015). T cell migration in rheumatoid arthritis. Front. Immunol. 6:384. doi: 10.3389/fimmu.2015.00384
Mylonas, K. J., Turner, N. A., Bageghni, S. A., Kenyon, C. J., White, C. I., McGregor, K., et al. (2017). 11β-HSD1 suppresses cardiac fibroblast CXCL2, CXCL5 and neutrophil recruitment to the heart post MI. J. Endocrinol. 233, 315–327. doi: 10.1530/JOE-16-0501
Nissen, S. E., and Wolski, K. (2007). Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med. 356, 2457–2471. doi: 10.1056/NEJMoa072761
Nourshargh, S., and Alon, R. (2014). Leukocyte migration into inflamed tissues. Immunity 41, 694–707. doi: 10.1016/j.immuni.2014.10.008
Nussbaum, C., Bannenberg, S., Keul, P., Gräler, M. H., Gonçalves-de-Albuquerque, C. F., Korhonen, H., et al. (2015). Sphingosine-1-phosphate receptor 3 promotes leukocyte rolling by mobilizing endothelial P-selectin. Nat. Commun. 6:6416. doi: 10.1038/ncomms7416
Ortega-Gómez, A., Perretti, M., and Soehnlein, O. (2013). Resolution of inflammation: an integrated view. EMBO Mol. Med. 5, 661–674. doi: 10.1002/emmm.201202382
Pant, P., Nadimpalli, L., Singh, M., and Cheng, J. C. (2010). Quiz page june 2010: a case of severe hypokalemic paralysis and hypertension. Am. J. Kidney Dis. 55, A35–A37. doi: 10.1053/j.ajkd.2010.02.003
Pappu, R., Schwab, S. R., Cornelissen, I., Pereira, J. P., Regard, J. B., Xu, Y., et al. (2007). Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science 316, 295–298. doi: 10.1126/science.1139221
Parsonage, G., Filer, A. D., Haworth, O., Nash, G. B., Rainger, G. E., Salmon, M., et al. (2005). A stromal address code defined by fibroblasts. Trends Immunol. 26, 150–156. doi: 10.1016/j.it.2004.11.014
Rainger, G. E., and Nash, G. B. (2001). Cellular pathology of atherosclerosis: smooth muscle cells prime cocultured endothelial cells for enhanced leukocyte adhesion. Circ. Res. 88, 615–622. doi: 10.1161/01.RES.88.6.615
Reglero-Real, N., Colom, B., Bodkin, J. V., and Nourshargh, S. (2016). Endothelial cell junctional adhesion molecules: role and regulation of expression in inflammation. Arterioscler. Thromb. Vasc. Biol. 36, 2048–2057. doi: 10.1161/ATVBAHA.116.307610
Ruiz, L. M., Bedoya, G., Salazar, J., García de, O. D., and Patiño, P. J. (2002). Dexamethasone inhibits apoptosis of human neutrophils induced by reactive oxygen species. Inflammation 26, 215–222. doi: 10.1023/A:1019714618068
Russo, S., Mastropasqua, M., Mosetti, M. A., Persegani, C., and Paggi, A. (2000). Low doses of liquorice can induce hypertension encephalopathy. Am. J. Nephrol. 20, 145–148. doi: 10.1159/000013572
Salmi, M., Rajala, P., and Jalkanen, S. (1997). Homing of mucosal leukocytes to joints. Distinct endothelial ligands in synovium mediate leukocyte-subtype specific adhesion. J. Clin. Invest. 99, 2165–2172. doi: 10.1172/JCI119389
Scalia, R., Gefen, J., Petasis, N. A., Serhan, C. N., and Lefer, A. M. (1997). Lipoxin A4 stable analogs inhibit leukocyte rolling and adherence in the rat mesenteric microvasculature: role of P-selectin. Proc. Natl. Acad. Sci. U.S.A. 94, 9967–9972. doi: 10.1073/pnas.94.18.9967
Schäcke, H., Döcke, W.-D., and Asadullah, K. (2002). Mechanisms involved in the side effects of glucocorticoids. Pharmacol. Ther. 96, 23–43. doi: 10.1016/S0163-7258(02)00297-8
Schaljo, B., Kratochvill, F., Gratz, N., Sadzak, I., Sauer, I., Hammer, M., et al. (2009). Tristetraprolin is required for full anti-inflammatory response of murine macrophages to IL-10. J. Immunol. 183, 1197–1206. doi: 10.4049/jimmunol.0803883
Schüle, R., Rangarajan, P., Kliewer, S., Ransone, L. J., Bolado, J., Yang, N., et al. (1990). Functional antagonism between oncoprotein c-Jun and the glucocorticoid receptor. Cell 62, 1217–1226. doi: 10.1016/0092-8674(90)90397-W
Schwab, J. M., Chiang, N., Arita, M., and Serhan, C. N. (2007). Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 447, 869–874. doi: 10.1038/nature05877
Seckl, J. R., and Walker, B. R. (2001). Minireview: 11β-hydroxysteroid dehydrogenase type 1— A tissue-specific amplifier of glucocorticoid action. Endocrinology 142, 1371–1376. doi: 10.1210/endo.142.4.8114
Serhan, C. N. (2014). Novel pro-resolving lipid mediators in inflammation are leads for resolution physiology. Nature 510, 92–101. doi: 10.1038/nature13479
Serhan, C. N., and Petasis, N. A. (2011). Resolvins and protectins in inflammation-resolution. Chem. Rev. 111, 5922–5943. doi: 10.1021/cr100396c
Shen, Y., Wen, Z., Li, Y., Matteson, E. L., Hong, J., Goronzy, J. J., et al. (2017). Metabolic control of the scaffold protein TKS5 in tissue-invasive, proinflammatory T cells. Nat. Immunol. 18, 1025–1034. doi: 10.1038/ni.3808
Sheridan, G. K., and Dev, K. K. (2014). Targeting S1P receptors in experimental autoimmune encephalomyelitis in mice improves early deficits in locomotor activity and increases ultrasonic vocalisations. Sci. Rep. 4:5051. doi: 10.1038/srep05051
Singer, B. A. (2013). Fingolimod for the treatment of relapsing multiple sclerosis. Expert Rev. Neurother. 13, 589–602. doi: 10.1586/ern.13.52
Smith, E., McGettrick, H. M., Stone, M. A., Shaw, J. S., Middleton, J., Nash, G. B., et al. (2008). Duffy antigen receptor for chemokines and CXCL5 are essential for the recruitment of neutrophils in a multicellular model of rheumatoid arthritis synovium. Arthritis Rheum. 58, 1968–1973. doi: 10.1002/art.23545
Spite, M., Norling, L. V., Summers, L., Yang, R., Cooper, D., Petasis, N. A., et al. (2009). Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature 461, 1287–1291. doi: 10.1038/nature08541
Straub, R. H., and Cutolo, M. (2016). Glucocorticoids and chronic inflammation. Rheumatology 55, ii6–ii14. doi: 10.1093/rheumatology/kew348
Strausbaugh, H. J., and Rosen, S. D. (2001). A potential role for annexin 1 as a physiologic mediator of glucocorticoid-induced L-selectin shedding from myeloid cells. J. Immunol. 166, 6294–6300. doi: 10.4049/jimmunol.166.10.6294
Sugimoto, M. A., Sousa, L. P., Pinho, V., Perretti, M., and Teixeira, M. M. (2016a). Resolution of inflammation: What Controls Its Onset? Front. Immunol. 7:160. doi: 10.3389/fimmu.2016.00160
Sugimoto, M. A., Vago, J. P., Teixeira, M. M., and Sousa, L. P. (2016b). Annexin A1 and the resolution of inflammation: modulation of neutrophil recruitment, apoptosis, and clearance. J. immunol. Res. 2016, 8239258–8239258. doi: 10.1155/2016/8239258
Sun, K., and Myatt, L. (2003). Enhancement of glucocorticoid-induced 11β-hydroxysteroid dehydrogenase type 1 expression by proinflammatory cytokines in cultured human amnion fibroblasts. Endocrinology 144, 5568–5577. doi: 10.1210/en.2003-0780
Tak, P. P., Taylor, P. C., Breedveld, F. C., Smeets, T. J. M., Daha, M. R., Kluin, P. M., et al. (1996). Decrease in cellularity and expression of adhesion molecules by anti–tumor necrosis factor α monoclonal antibody treatment in patients with rheumatoid arthritis. Arthritis Rheum. 39, 1077–1081. doi: 10.1002/art.1780390702
Thieringer, R., Le Grand, C. B., Carbin, L., Cai, T.-Q., Wong, B., Wright, S. D., et al. (2001). 11β-Hydroxysteroid dehydrogenase type 1 is induced in human monocytes upon differentiation to macrophages. J. Immunol. 167, 30–35. doi: 10.4049/jimmunol.167.1.30
Tomlinson, J. W., and Stewart, P. M. (2001). Cortisol metabolism and the role of 11β-hydroxysteroid dehydrogenase. Best Pract. Res. Clin. Endocrinol. Metab. 15, 61–78. doi: 10.1053/beem.2000.0119
Tsunemi, S., Iwasaki, T., Kitano, S., Imado, T., Miyazawa, K., and Sano, H. (2010). Effects of the novel immunosuppressant FTY720 in a murine rheumatoid arthritis model. Clin. Immunol. 136, 197–204. doi: 10.1016/j.clim.2010.03.428
van Deventer, S. J., Elson, C. O., and Fedorak, R. N. (1997). Multiple doses of intravenous interleukin 10 in steroid-refractory Crohn’s disease. Crohn’s Disease Study Group. Gastroenterology 113, 383–389. doi: 10.1053/gast.1997.v113.pm9247454
Wake, D. J., Stimson, R. H., Tan, G. D., Homer, N. Z. M., Andrew, R., Karpe, F., et al. (2007). Effects of Peroxisome Proliferator-Activated Receptor-α and -γ Agonists on 11β-Hydroxysteroid Dehydrogenase Type 1 in Subcutaneous Adipose Tissue in Men. J. Clin. Endocrinol. Metab. 92, 1848–1856. doi: 10.1210/jc.2006-2713
Wamil, M., Battle, J. H., Turban, S., Kipari, T., Seguret, D., de Sousa Peixoto, R., et al. (2011). Novel fat depot–specific mechanisms underlie resistance to visceral obesity and inflammation in 11β-hydroxysteroid dehydrogenase type 1–deficient mice. Diabetes Metab. Res. Rev 60, 1158–1167. doi: 10.2337/db10-0830
Wang, F., Tan, W., Guo, D., and He, S. (2007). Reduction of CD4 positive T cells and improvement of pathological changes of collagen-induced arthritis by FTY720. Eur. J. Pharmacol. 573, 230–240. doi: 10.1016/j.ejphar.2007.07.029
Keywords: leukocytes, migration, inflammation, resolution, PEPITEM, sphingosine-1-phosphate, glucocorticoids, 11-beta hydroxysteroid dehydrogenase
Citation: Hopkin SJ, Lewis JW, Krautter F, Chimen M and McGettrick HM (2019) Triggering the Resolution of Immune Mediated Inflammatory Diseases: Can Targeting Leukocyte Migration Be the Answer? Front. Pharmacol. 10:184. doi: 10.3389/fphar.2019.00184
Received: 06 October 2018; Accepted: 14 February 2019;
Published: 01 March 2019.
Edited by:
Trinidad Montero-Melendez, Queen Mary University of London, United KingdomReviewed by:
Michael Hickey, Monash University, AustraliaDianne Cooper, Queen Mary University of London, United Kingdom
Copyright © 2019 Hopkin, Lewis, Krautter, Chimen and McGettrick. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Helen M. McGettrick, aC5tLm1jZ2V0dHJpY2tAYmhhbS5hYy51aw==
†These authors have contributed equally to this work
‡These authors jointly coordinated this work