 Sun-Mi Yun
Sun-Mi Yun Seok-Ho Kim
Seok-Ho Kim Eun-Hee Kim
Eun-Hee Kim
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pharmacol. , 27 February 2019
Sec. Gastrointestinal and Hepatic Pharmacology
Volume 10 - 2019 | https://doi.org/10.3389/fphar.2019.00162
Inflammatory bowel disease is known as the most chronic inflammatory disorder in colon, which subsequently progresses to intestinal obstruction and fistula formation. Many studies to date for the treatment of IBD have been focused on inflammation. However, most of the anti-inflammatory agents do not have anti-fibrotic effects and could not relieve intestinal stricture in IBD patients. Because preventing or reversing intestinal fibrosis in IBD is a major therapeutic target, we analyzed the papers focusing on TGF-β signaling in intestinal fibrosis. TGF-β is a good candidate to treat the intestinal fibrosis in IBD which involves TGF-β signaling pathway, EMT, EndMT, ECM, and other regulators. Understanding the mechanism involved in TGF-β signaling will contribute to the treatment and diagnosis of intestinal fibrosis occurring in IBD as well as the understanding of the molecular mechanisms underlying the pathogenesis.
Intestinal fibrosis is an unsolved issue in IBD, such as UC, and CD. An acute inflammation can be recovered from quickly, but chronic inflammation due to severe and repetitive tissue injury will progress to irreversible fibrosis with a poor regenerative capacity. It gives rise to accumulation of excessive connective tissues, eventually resulting in further complications including epilepsy, kidney disease, idiopathic pulmonary fibrosis, and heart failure. Until recently, researchers have been focusing on studying the mechanisms of inflammation to alleviate and to inhibit intestinal fibrosis. However, anti-inflammatory agents had various problems and limitations to relieve or to treat fibrosis in IBD. Therefore, in order to treat fibrotic diseases, it should no longer be focused only on inflammation and should explore the new approaches to anti-fibrotic mechanisms.
Transforming growth factor-β has a critical role in cellular responses, such as development, proliferation and differentiation. TGF-β is the master regulator to drive fibrosis in all organs including the intestine (Clouthier et al., 1997; Sime et al., 1997; Pucilowska et al., 2000; Sato et al., 2003; Rieder and Fiocchi, 2008, 2009; Wynn, 2008; Fiocchi and Lund, 2011; Mifflin et al., 2011; Speca et al., 2012). Canonical TGF-β signaling via Smads has a central role in the progression of fibrosis (Roberts et al., 2003). Nevertheless, the mechanism of TGF-β signaling in intestinal fibrosis is still not fully understood.
In this review, we analyzed clinical and therapeutic limitations for treatment of intestinal fibrosis and focused on anti-fibrotic mechanisms rather than on anti-inflammation, particularly investigating the molecular mechanism of TGF-β signaling in fibrosis occurring in IBD. Comprehension of the downstream mediators which regulate the TGF-β signaling pathway will contribute to the development of diagnostic and therapeutic agents for intestinal fibrosis in IBD.
Approximately 50% of CD patients suffer from fibrotic strictures and 75% of them will eventually undergo surgery (Siassi et al., 2007; Rieder and Fiocchi, 2009; Latella and Papi, 2012; Rieder et al., 2016). Interestingly, intestinal obstruction appeared in patients with extensive bowel resection as well as in CD patients, suggesting it cannot be completely cured by surgery and should consider new therapeutic strategies in addition to surgical treatment.
Various agents, such as salicylates, steroids, immunosuppressive drugs, and biological therapies have been used for the treatment of IBD. Such treatments may reduce the inflammatory symptoms, but do not relieve the fibrotic obstruction (Dignass et al., 2010; Van Assche et al., 2010; Latella et al., 2011; Caprilli et al., 2012). Moreover, some anti-inflammatory drugs may exacerbate fibrosis. 5-Aminosalicylic acid is known as an anti-inflammatory drug, but its anti-fibrotic effect has not yet been studied. Corticosteroids, effective agents in controlling inflammation, showed anti-fibrotic effects in various fibrotic diseases such as systemic sclerosis (Badea et al., 2009), idiopathic pulmonary fibrosis (Rogliani et al., 2008) and retroperitoneal fibrosis (Vaglio et al., 2007), with the exception of intestinal fibrosis (Graham et al., 1995). However, steroids can cause serious side effects when used for a long time. Immunosuppressive agents including azathioprine, 6-mercaptopurine, cyclosporine, and methotrexate have shown to have an anti-inflammatory effect in several chronic inflammatory diseases such as IBD. Some of the drugs have anti-fibrotic effect in pulmonary fibrosis (Dheda et al., 2004) and retroperitoneal fibrosis (Warnatz et al., 2005), however, have no significant effect on the treatment of intestinal fibrosis in IBD (Cosnes et al., 2011; Latella and Papi, 2012). Many of these anti-inflammatory treatments such as anti-TNF agents may have the effect of reducing inflammation, but they may not prevent or reverse fibrosis in IBD patients (Sorrentino et al., 2006). It suggests the possibility of new mechanisms regulating fibrosis different from the inflammation control mechanism. (Rieder and Fiocchi, 2009; Latella et al., 2013).
Despite the continuous efforts to find effective mechanisms and treatments, neither IBD progression nor surgical rates have significantly reduced (Cosnes et al., 2005; Spinelli et al., 2010; Luna et al., 2011). Especially, the mechanism of fibrosis in IBD is still not studied much compared to fibrosis occurring in other organs including the liver, lung, and kidney (Latella et al., 2014). Therefore, understanding the mechanism of TGF-β signaling may be key to alleviating and treating intestinal fibrosis.
Transforming growth factor-β signaling pathway is known to play a multifunctional role in the regulation of embryonic development, immunity, carcinogenesis, inflammation, and fibrosis. In normal conditions, TGF-β signaling maintains tissue homeostasis via the regulation of cell proliferation. On the other hand, TGF-β switches its function to accelerate the progress and the development of diseases such as cancer and fibrosis in abnormal conditions.
Transforming growth factor-β binds to betaglycan, also known as the type III TGF-β receptor, subsequently forming a heteromeric complex with TGF-β type II receptor. Binding of the ligands to type II receptors recruits and activates the type I receptor via the interaction between type II and type I receptors. The activated TGF-β receptors phosphorylate Smad2 and Smad3 heterodimer which interact with Smad4. The Smad2/3-Smad4 complexes translocate from the cytosol to the nucleus and bind to the additional DNA-binding cofactors to regulate specific TGF-β target genes such as plasminogen activator inhibitor-1, fibronectin, and collagen type I (Katuri et al., 2006; Schultze-Mosgau et al., 2006).
The expression of TGF-β and its receptors was increased in intestinal cells of patients with IBD, in particular in CD patients (Babyatsky et al., 1996). Also, overexpression of TGF-β in the colons of mice caused colonic fibrosis (Vallance et al., 2005). TGF-β2 is one of the most potent inducer of scarring and the TGF-β2 neutralizing antibody (Lerdelimumab) inhibited scarring after glaucoma surgery in rabbits (Mead et al., 2003). The overexpression of TGF-β2 might contribute to fibrosis by increasing ECM deposition. On the other hand, loss of Smad3 reduced tissue fibrosis in several organs, such as skin (Lakos et al., 2004), kidney (Inazaki et al., 2004), lung (Zhao et al., 2002), liver (Lee et al., 2016), and intestine (Zanninelli et al., 2006; Latella et al., 2009). Therefore, therapeutic agents targeting TGF-β for various fibrotic diseases have been investigated (Varga and Pasche, 2008; Hawinkels and Ten Dijke, 2011). Inhibitors of the TGF-β receptor, which are antibodies, that inhibit the binding of the ligand to the receptor, and antisense oligonucleotides that reduce the expression of TGF-β have been developed (Table 1). TGF-β signaling is negatively regulated by inhibitory Smad6 and Smad7 (Massague et al., 2005). Nevertheless, Smad7 is overexpressed in chronic inflammation and precancerous conditions in the colonic mucosa. Inhibition of Smad7 using antisense oligonucleotides decreased inflammation in an animal model of TNBS-induced colitis (Boirivant et al., 2006) and showed clinical remission in patients with CD (Monteleone et al., 2012). An oral antisense oligonucleotide of Smad7 called mongersen showed clinical effects on CD treatment. However, Monteleone and colleagues observed intestinal obstruction in the patients treated with 40 mg of mongersen (Monteleone et al., 2015; Vermeire, 2015). It might be caused by intestinal fibrosis, because TGF-β signaling promotes fibrosis by the synthesis and accumulation of collagen. Accordingly, Smad7 may not be a good remedy for treating intestinal fibrosis and further investigation of the Smad7 mechanism on the progression of intestinal fibrosis in IBD patients is needed.
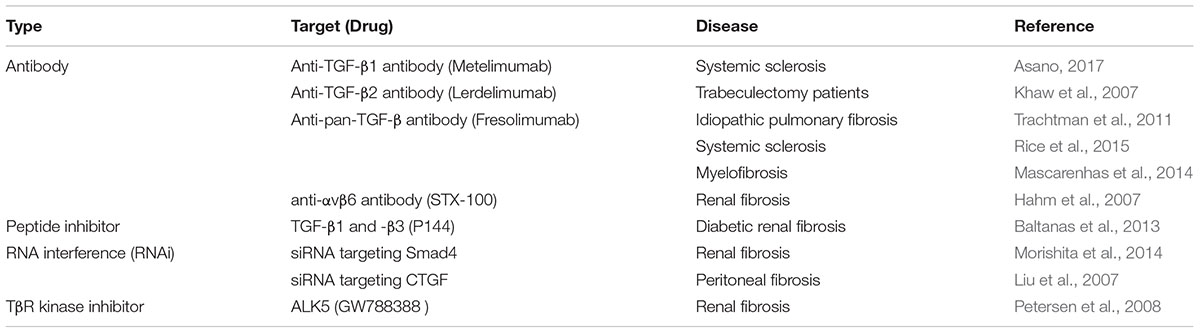
Table 1. Therapeutic agents targeting TGF-β in various fibrotic diseases.
Epithelial-to-mesenchymal transition and EndMT lead to fibrosis. EMT is the main mechanism of development, cancer progression, and fibrosis (Kalluri and Neilson, 2003). During the process of EMT, epithelial cells changed to spindle-shape morphology, lost epithelial molecules such as E-cadherin, and gained mesenchymal molecules such as vimentin and alpha-smooth muscle actin (α-SMA). Epithelial markers and mesenchymal markers were regulated in the progression of intestinal fibrosis both in vivo and in vitro (Rousseaux and Desreumaux, 2006; Flier et al., 2010). These changes are mediated primarily by canonical TGF-β pathways including Smad3 but also by non-canonical TGF-β pathways including mitogen-activated protein kinase signaling and Wnt/β-catenin signaling (Bakin et al., 2002; Li et al., 2004; Wang D. et al., 2011). EMT has been described in many fibrotic diseases such as renal, pulmonary, and liver fibrosis (Kalluri and Neilson, 2003; Rastaldi, 2006; Willis and Borok, 2007; Zeisberg and Kalluri, 2008). Therefore, EMT-regulating genes can be the strategic target for intestinal fibrosis. Recently, peroxisome proliferator-activated receptor gamma (PPAR-γ) modulator, GED-0507-34 Levo, reduced EMT progression by reducing EMT-related genes in chronic colitis-associated fibrosis animal models (Di Gregorio et al., 2017).
Transforming growth factor-β is a critical inducer in EndMT as in EMT (van Meeteren and ten Dijke, 2012). EndMT also caused exaggerated myofibroblast accumulation and extracellular matrix production in several organs (Piera-Velazquez et al., 2011). TGF-β can induce collagen accumulation in connective tissues as well as morphological changes that produce differentiated cells and activated fibroblasts (Zeisberg et al., 2003; Lamouille et al., 2014). Endothelial-specific depletion of Tgfr2 inhibited EndMT in regulating fibrotic responses to renal injury in mice (Xavier et al., 2015). The direct correlation between EndMT and IBD-related fibrosis has not yet been reported, whereas TGF-β and EndMT related genes including collagen I alpha 2 are reported to be abundant in the intestine of IBD (Burke et al., 2011; Sadler et al., 2013; Scharl et al., 2015). In this regard, EndMT can also contribute to intestinal fibrosis through differentiation of fibroblasts in IBD.
Excessive production and deposition of ECM was induced in the inflammatory response and the intestinal fibrosis by activating myofibroblasts which are cells located between fibroblasts and smooth muscle cells (Rieder and Fiocchi, 2009; Speca et al., 2012). The myofibroblasts are implicated in wound healing and fibrosis. These cells induce the production of type I and type III collagens and the expression of α-SMA, and reduce the expression of ECM-degradative enzymes (Desmouliere and Gabbiani, 1995; Krieg et al., 2007). Many growth factors (PDGF, epidermal growth factor, insulin-like growth factors, and CTGF) and cytokines (IL-1 and IL-13) including TGF-β stimulate ECM synthesis through local fibroblasts leading to fibrosis (Barrientos et al., 2008). Particularly, the expression of CTGF regulated by TGF-β contributed to the progression of fibrosis (Grotendorst, 1997). Smooth muscle cells were differentiated into myofibroblasts in the condition of chronic inflammation or fibrosis (Rieder and Fiocchi, 2008, 2009). These cells actively accelerate fibrosis in IBD by inducing the production of collagen and matrix metalloproteinases (MMPs) due to stimulation of inflammatory mediators such as TGF-β. MMPs play a role in cell migration and invasion by ECM degradation in the immune response and fibrotic response as well as in physiologic function of normal cells. Therefore, regulatory factors to control ECM were focused as a therapeutic target in intestinal fibrogenesis (Luna et al., 2011). Holvoet and colleagues (2017) showed that inhibiting Rho kinases activity by administration of AMA0825 prevented and resolved intestinal fibrosis in experimental murine models and CD patient samples through inhibition of myofibroblast accumulation, expression of pro-fibrotic factors, and accumulation of ECM. In addition, Rho kinases inhibition reversed the established fibrosis in a chronic animal model and obstructed ex vivo pro-fibrotic protein secretion from stenotic CD biopsies (Holvoet et al., 2017). Although AMA0825 treatment did not have anti-inflammatory effects, combining AMA0825 with anti-TNF antibody in the adoptive T-cell transfer model for intestinal fibrosis could not only prevent the accumulation of fibrotic tissues but could also ameliorate inflammation. Therefore, AMA0825 may be highly valued as an additional therapeutic agent for existing anti-inflammatory drugs for CD.
The coagulation response appears at the early stage of the wound healing mechanism which corresponds to acute inflammation. Activated platelets release growth factors including PDGF and TGF-β1, which stimulate ECM synthesis by local fibroblasts (Barrientos et al., 2008). Some publications have reported that PDGF is implicated in pulmonary, renal, and hepatic fibrosis. However, a role of PDGF in intestinal fibrosis is still unclear (Valatas et al., 2017). The thrombospondin1 (TSP1), the first discovered in the α-granules released after platelet activation, plays an important role in tissue repair (Murphy-Ullrich and Mosher, 1985). Interestingly, TSP1 is well known to activate the latent form of TGF-β1 (Lawler, 2000; Sweetwyne and Murphy-Ullrich, 2012). Treatment with intraperitoneal injections of a peptide which blocks TSP1 binding and TSP1-dependent TGF-β1 activation reduced cardiac fibrosis (Belmadani et al., 2007). Moreover, several studies have shown that treatment with TSP1 antagonist peptides can prevent bleomycin-induced lung fibrosis in mice and reduce the activation of TGF-β (Yehualaeshet et al., 2000; Chen et al., 2009; Ezzie et al., 2011). Treatment with DSS induced a more severe colitis in TSP1-/- mice, however, ABT-510, a peptide derived from TSP1 consistently diminished angiogenesis and bleeding by DSS challenge (Punekar et al., 2008). Although there is no report that TSP1 is directly involved in intestinal fibrosis, these reports suggest that TSP1 may be an attractive therapeutic target for intestinal fibrosis.
Macrophage induces TGF-β1 production in the early wound-healing response. Therefore, differentiated macrophages increased the production of PPAR-α and PPAR-γ. Recent study reported that macrophages regulate inflammation and fibrogenesis in IBD. Particularly, PPAR-γ is not only associated with adipogenesis but also with inflammation control (Rousseaux and Desreumaux, 2006). The activation of PPAR-γ with the interaction of its ligands inhibits TGF-β/Smad3 pathway by antagonizing Smad3 and by reducing CTGF expression (Zhang et al., 2009). PPAR-γ agonists attached to these receptors can prevent fibrosis induced by activated macrophages or by TGF-β1 through inhibiting fibroblast migration and proliferation (Ricote et al., 1998; Odegaard et al., 2007; Kulkarni et al., 2011).
Interleukin-17 has a critical role in promoting inflammation by increasing the production of chemokines to recruit and to activate granulocytes (Maloy, 2008). It reduced myofibroblast migration and increased the expression of MMPs and collagen production. IL-17A-mediated fibrosis required TGF-β in an animal model of pulmonary fibrosis (Wilson et al., 2010). It induced the recruitment of neutrophil, resulting in the progression of tissue damage and fibrosis in the airways (Laan et al., 1999). IL-17A expression was increased in fibrotic CD tissues compared with non-fibrotic CD tissues (Biancheri et al., 2013). In addition, its expression is associated with many types of fibrosis, such as pulmonary fibrosis (Wilson et al., 2010), myocardial fibrosis (Feng et al., 2009), hepatic fibrosis (Wang L. et al., 2011), and intestinal fibrosis (Bamba et al., 2003; Biancheri et al., 2013).
Interleukin-13 plays a role as a profibrotic mediator in the development of several fibrosis such as chronic asthma (Kumar et al., 2002), lung fibrosis (Kolodsick et al., 2004; Murray et al., 2008), systemic sclerosis (Fuschiotti, 2011), skin fibrosis (Oh et al., 2011), and ulcerative colitis (Heller et al., 2002). In the progression of fibrosis, it induces the differentiation of fibroblasts to myofibroblasts and deposition of collagens (Wynn, 2007, 2008). Also, IL-13 induced the production and secretion of TGF-β via IL-13 signaling in monocytes and macrophages (Fichtner-Feigl et al., 2006, 2008). Blockade of the IL-13 signaling leads to the inhibition of TGF-β production, resulting in the suppression of collagen deposition and the reduction of fibrotic progression. IL-13 binding to the IL-13 receptor accelerated intestinal fibrosis development in TNBS-induced chronic colitis by inducing TGF-β. Mechanistically, it has been hypothesized that IL-13 induces fibrosis by activating TGF-β (Lee et al., 2001). However, according to the research, IL-13 can act as an inducer of fibrosis independently from TGF-β by directly regulating the proliferative properties of fibroblasts, epithelial cells, and smooth muscle cells (Lee et al., 2001; Kuperman et al., 2002).
One cytokine involved in TGF-β signaling is IL-1β, a potent proinflammatory mediator, which induces EMT and myofibroblast activation through a TGF-β1-mediated mechanism (Fan et al., 2001), resulting in the development of fibrosis in IBD. Another cytokine involved in TGF-β signaling is IFN-γ, a cytokine that plays a critical role in innate and adaptive immunity, activates macrophages and induces Class II major histocompatibility complex molecule expression. It inhibits the TGF-β-induced phosphorylation of Smad3 and the activation of TGF-β target genes by inducing the expression of Smad7 (Ulloa et al., 1999). Moreover, IFN-γ regulates fibrogenesis in IBD through the reduction of fibroblast proliferation and collagen synthesis in activated myofibroblasts (Gurujeyalakshmi and Giri, 1995; Leeb et al., 2003).
The TGF-β signaling is known to be involved in the onset and the progression of many diseases such as cancer, immune diseases and fibrosis. Numerous publications have displayed that molecules related to TGF-β signaling were implicated in fibrosis (Figure 1). Many attempts have been made to develop inhibitors of TGF-β signaling pathway as a treatment. However, blockage of TGF-β1 can have high risk and problems because it is involved in other cellular functions such as differentiation, proliferation, transformation, and immune system. Additionally, the deficiency of TGF-β, Smad2, and Smad4 is lethal in vivo (Kulkarni et al., 1993; Nomura and Li, 1998). Despite the risk, TGF-β is still an important target in the progression of intestinal fibrosis because it correlates with the complex and diverse signaling pathways regulating the mechanism of the progression of intestinal fibrosis in IBD (Table 2). Therefore, TGF-β signaling is a potential strategy to treat and alleviate fibrosis in several fibrotic diseases including IBD (Speca et al., 2012). The challenge moving forward is to elucidate the complex mechanism of TGF-β signaling with relation to the other various signaling pathways and to find effective therapeutic agents targeting TGF-β signaling in the regulation of intestinal fibrosis in IBD.
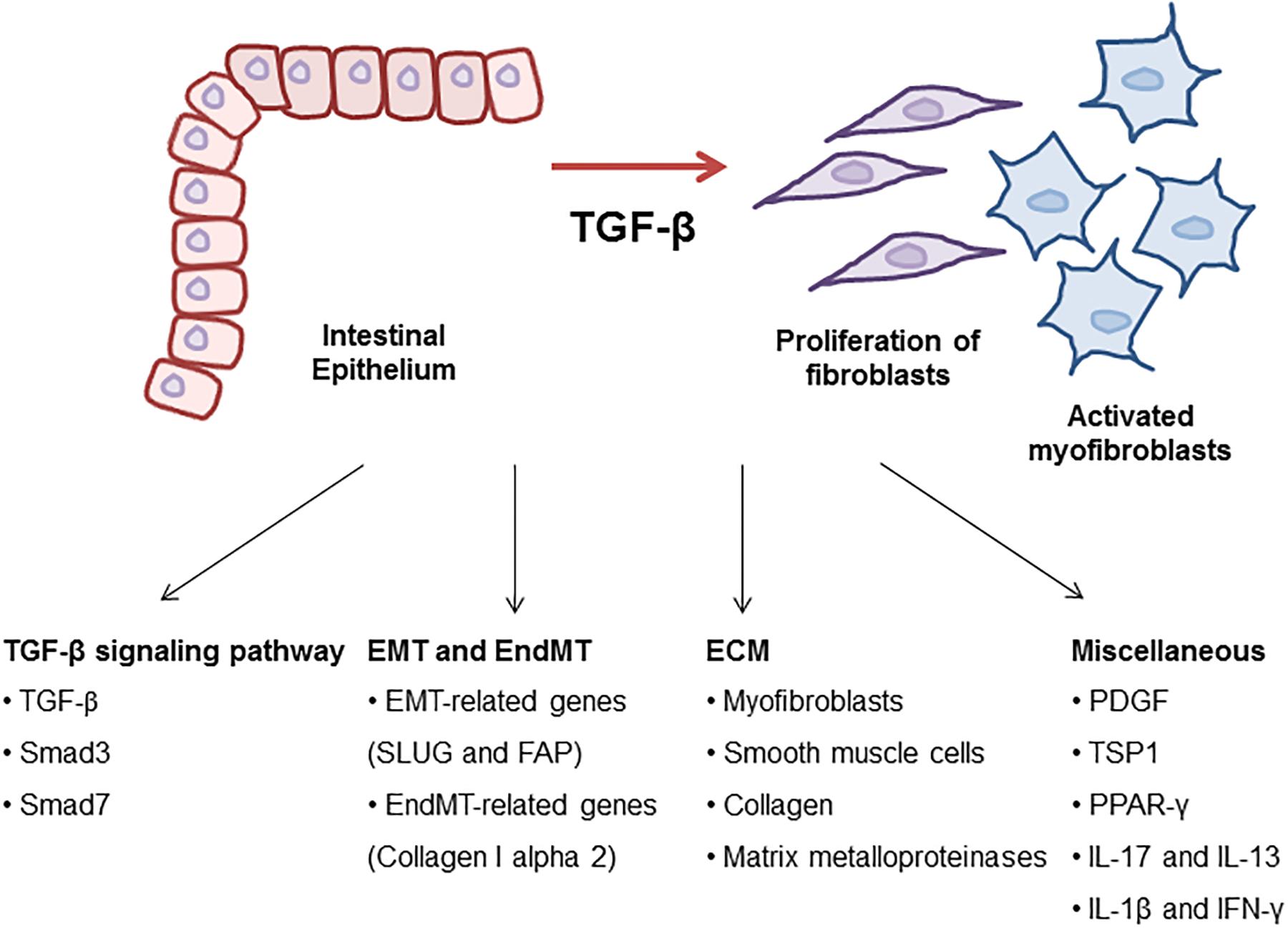
Figure 1. Molecular mechanism of TGF-β signaling in intestinal fibrosis. TGF-β is a key regulator to drive intestinal fibrosis occurring in IBD. In the progression of fibrosis, TGF-β signaling-related genes including TGF-β, Smad3, and Smad7 are upregulated in patients with IBD. TGF-β induces EMT and EndMT-regulating genes resulting in the progression of intestinal fibrosis. Also, ECM synthesis, including the production of collagen and MMPs, is a characteristic of intestinal strictures and luminal stenosis and is a major therapeutic challenge for IBD. Many growth factors such as TGF-β induced ECM deposition by local fibroblasts and myofibroblasts differentiated by fibroblasts. Other cytokines (IL-17, IL-13, IL-1β, and IFN-γ) and TSP1 play roles as profibrotic mediators through the activation of fibroblasts in the development of intestinal fibrosis.
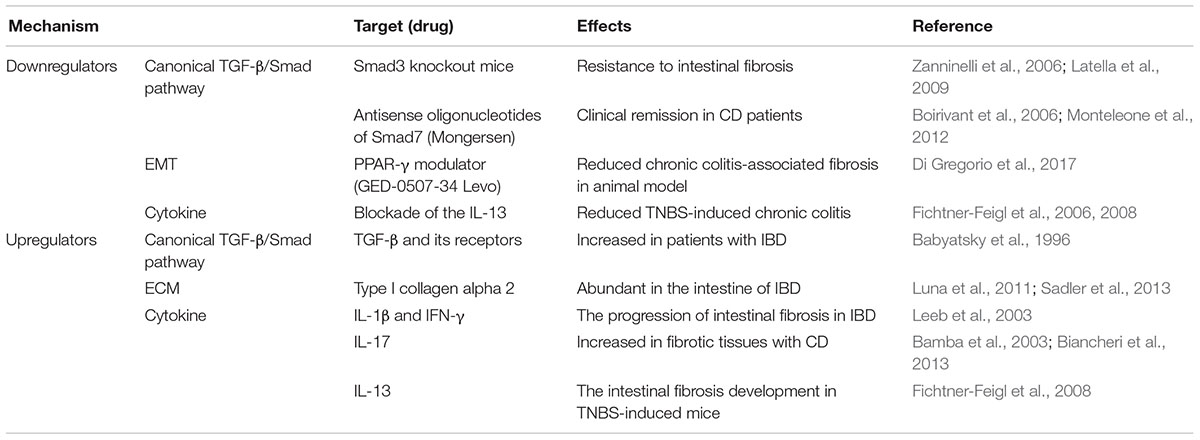
Table 2. A summary of the regulators of TGF-β signaling pathway in intestinal fibrosis.
S-MY prepared the manuscript. S-HK reviewed the drafts and provided important information. E-HK conceived the idea, reviewed the drafts, and provided important information for the completion of this manuscript. All authors contributed to the writing and final approval of the manuscript.
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea Government (Ministry of Education) (No. 2015R1D1A1A059949).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
α-SMA, alpha-smooth muscle actin; CD, Crohn’s disease; CTGF, connective tissue growth factor; DSS, dextran sulfate sodium; ECM, extracellular matrix; EMT, epithelial-to-mesenchymal transition; EndMT, endothelial-to-mesenchymal transition; IBD, inflammatory bowel disease; IFN-γ, interferon-gamma; IL, interleukin; MMPs, matrix metalloproteinases; PDGF, platelet-derived growth factor; PPAR, peroxisome proliferator-activated receptor; TGF-β, transforming growth factor-beta; TNBS, trinitrobenzene sulfonic acid; TNF, tumor necrosis factor; TSP1, thrombospondin1; UC, ulcerative colitis.
Asano, Y. (2017). Recent advances in the treatment of skin involvement in systemic sclerosis. Inflamm. Regen. 37:12. doi: 10.1186/s41232-017-0047-4
Babyatsky, M. W., Rossiter, G., and Podolsky, D. K. (1996). Expression of transforming growth factors alpha and beta in colonic mucosa in inflammatory bowel disease. Gastroenterology 110, 975–984. doi: 10.1053/gast.1996.v110.pm8613031
Badea, I., Taylor, M., Rosenberg, A., and Foldvari, M. (2009). Pathogenesis and therapeutic approaches for improved topical treatment in localized scleroderma and systemic sclerosis. Rheumatology 48, 213–221. doi: 10.1093/rheumatology/ken405
Bakin, A. V., Rinehart, C., Tomlinson, A. K., and Arteaga, C. L. (2002). p38 mitogen-activated protein kinase is required for TGFbeta-mediated fibroblastic transdifferentiation and cell migration. J. Cell Sci. 115, 3193–3206.
Baltanas, A., Miguel-Carrasco, J. L., San Jose, G., Cebrian, C., Moreno, M. U., Dotor, J., et al. (2013). A synthetic peptide from transforming growth factor-beta(1) type III receptor inhibits NADPH oxidase and prevents oxidative stress in the kidney of spontaneously hypertensive rats. Antioxid. Redox Signal. 19, 1607–1618. doi: 10.1089/ars.2012.4653
Bamba, S., Andoh, A., Yasui, H., Araki, Y., Bamba, T., and Fujiyama, Y. (2003). Matrix metalloproteinase-3 secretion from human colonic subepithelial myofibroblasts: role of interleukin-17. J. Gastroenterol. 38, 548–554. doi: 10.1007/s00535-002-1101-8
Barrientos, S., Stojadinovic, O., Golinko, M. S., Brem, H., and Tomic-Canic, M. (2008). Growth factors and cytokines in wound healing. Wound Repair Regen. 16, 585–601. doi: 10.1111/j.1524-475X.2008.00410.x
Belmadani, S., Bernal, J., Wei, C. C., Pallero, M. A., Dell’italia, L., Murphy-Ullrich, J. E., et al. (2007). A thrombospondin-1 antagonist of transforming growth factor-beta activation blocks cardiomyopathy in rats with diabetes and elevated angiotensin II. Am. J. Pathol. 171, 777–789. doi: 10.2353/ajpath.2007.070056
Biancheri, P., Pender, S. L., Ammoscato, F., Giuffrida, P., Sampietro, G., Ardizzone, S., et al. (2013). The role of interleukin 17 in Crohn’s disease-associated intestinal fibrosis. Fibrogenesis Tissue Repair 6:13. doi: 10.1186/1755-1536-6-13
Boirivant, M., Pallone, F., Di Giacinto, C., Fina, D., Monteleone, I., Marinaro, M., et al. (2006). Inhibition of Smad7 with a specific antisense oligonucleotide facilitates TGF-beta1-mediated suppression of colitis. Gastroenterology 131, 1786–1798. doi: 10.1053/j.gastro.2006.09.016
Burke, J. P., Cunningham, M. F., Sweeney, C., Docherty, N. G., and O’connell, P. R. (2011). N-cadherin is overexpressed in Crohn’s stricture fibroblasts and promotes intestinal fibroblast migration. Inflamm. Bowel Dis. 17, 1665–1673. doi: 10.1002/ibd.21543
Caprilli, R., Latella, G., and Frieri, G. (2012). Treatment of inflammatory bowel diseases: to heal the wound or to heal the sick? J. Crohns Colitis 6, 621–625. doi: 10.1016/j.crohns.2012.02.009
Chen, Y., Wang, X., Weng, D., Tian, L., Lv, L., Tao, S., et al. (2009). A TSP-1 synthetic peptide inhibits bleomycin-induced lung fibrosis in mice. Exp. Toxicol. Pathol. 61, 59–65. doi: 10.1016/j.etp.2008.04.010
Clouthier, D. E., Comerford, S. A., and Hammer, R. E. (1997). Hepatic fibrosis, glomerulosclerosis, and a lipodystrophy-like syndrome in PEPCK-TGF-beta1 transgenic mice. J. Clin. Invest. 100, 2697–2713. doi: 10.1172/JCI119815
Cosnes, J., Gower-Rousseau, C., Seksik, P., and Cortot, A. (2011). Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology 140, 1785–1794. doi: 10.1053/j.gastro.2011.01.055
Cosnes, J., Nion-Larmurier, I., Beaugerie, L., Afchain, P., Tiret, E., and Gendre, J. P. (2005). Impact of the increasing use of immunosuppressants in Crohn’s disease on the need for intestinal surgery. Gut 54, 237–241. doi: 10.1136/gut.2004.045294
Desmouliere, A., and Gabbiani, G. (1995). Myofibroblast differentiation during fibrosis. Exp. Nephrol. 3, 134–139.
Dheda, K., Lalloo, U. G., Cassim, B., and Mody, G. M. (2004). Experience with azathioprine in systemic sclerosis associated with interstitial lung disease. Clin. Rheumatol. 23, 306–309. doi: 10.1007/s10067-004-0906-7
Di Gregorio, J., Sferra, R., Speca, S., Vetuschi, A., Dubuquoy, C., Desreumaux, P., et al. (2017). Role of glycogen synthase kinase-3beta and PPAR-gamma on epithelial-to-mesenchymal transition in DSS-induced colorectal fibrosis. PLoS One 12:e0171093. doi: 10.1371/journal.pone.0171093
Dignass, A., Van Assche, G., Lindsay, J. O., Lemann, M., Soderholm, J., Colombel, J. F., et al. (2010). The second European evidence-based consensus on the diagnosis and management of Crohn’s disease: current management. J. Crohns Colitis 4, 28–62. doi: 10.1016/j.crohns.2009.12.002
Ezzie, M. E., Piper, M. G., Montague, C., Newland, C. A., Opalek, J. M., Baran, C., et al. (2011). Thrombospondin-1-deficient mice are not protected from bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 44, 556–561. doi: 10.1165/rcmb.2009-0019OC
Fan, J. M., Huang, X. R., Ng, Y. Y., Nikolic-Paterson, D. J., Mu, W., Atkins, R. C., et al. (2001). Interleukin-1 induces tubular epithelial-myofibroblast transdifferentiation through a transforming growth factor-beta1-dependent mechanism in vitro. Am. J. Kidney Dis. 37, 820–831. doi: 10.1016/S0272-6386(01)80132-3
Feng, W., Li, W., Liu, W., Wang, F., Li, Y., and Yan, W. (2009). IL-17 induces myocardial fibrosis and enhances RANKL/OPG and MMP/TIMP signaling in isoproterenol-induced heart failure. Exp. Mol. Pathol. 87, 212–218. doi: 10.1016/j.yexmp.2009.06.001
Fichtner-Feigl, S., Strober, W., Geissler, E. K., and Schlitt, H. J. (2008). Cytokines mediating the induction of chronic colitis and colitis-associated fibrosis. Mucosal Immunol. 1(Suppl. 1), S24–S27. doi: 10.1038/mi.2008.41
Fichtner-Feigl, S., Strober, W., Kawakami, K., Puri, R. K., and Kitani, A. (2006). IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat. Med. 12, 99–106. doi: 10.1038/nm1332
Fiocchi, C., and Lund, P. K. (2011). Themes in fibrosis and gastrointestinal inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G677–G683. doi: 10.1152/ajpgi.00104.2011
Flier, S. N., Tanjore, H., Kokkotou, E. G., Sugimoto, H., Zeisberg, M., and Kalluri, R. (2010). Identification of epithelial to mesenchymal transition as a novel source of fibroblasts in intestinal fibrosis. J. Biol. Chem. 285, 20202–20212. doi: 10.1074/jbc.M110.102012
Fuschiotti, P. (2011). Role of IL-13 in systemic sclerosis. Cytokine 56, 544–549. doi: 10.1016/j.cyto.2011.08.030
Graham, M. F., Willey, A., Adams, J., and Diegelmann, R. F. (1995). Corticosteroids increase procollagen gene expression, synthesis, and secretion by human intestinal smooth muscle cells. Gastroenterology 109, 1454–1461. doi: 10.1016/0016-5085(95)90630-4
Grotendorst, G. R. (1997). Connective tissue growth factor: a mediator of TGF-beta action on fibroblasts. Cytokine Growth Factor Rev. 8, 171–179. doi: 10.1016/S1359-6101(97)00010-5
Gurujeyalakshmi, G., and Giri, S. N. (1995). Molecular mechanisms of antifibrotic effect of interferon gamma in bleomycin-mouse model of lung fibrosis: downregulation of TGF-beta and procollagen I and III gene expression. Exp. Lung Res. 21, 791–808. doi: 10.3109/01902149509050842
Hahm, K., Lukashev, M. E., Luo, Y., Yang, W. J., Dolinski, B. M., Weinreb, P. H., et al. (2007). Alphav beta6 integrin regulates renal fibrosis and inflammation in Alport mouse. Am. J. Pathol. 170, 110–125. doi: 10.2353/ajpath.2007.060158
Hawinkels, L. J., and Ten Dijke, P. (2011). Exploring anti-TGF-beta therapies in cancer and fibrosis. Growth Factors 29, 140–152. doi: 10.3109/08977194.2011.595411
Heller, F., Fuss, I. J., Nieuwenhuis, E. E., Blumberg, R. S., and Strober, W. (2002). Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL-13-producing NK-T cells. Immunity 17, 629–638. doi: 10.1016/S1074-7613(02)00453-3
Holvoet, T., Devriese, S., Castermans, K., Boland, S., Leysen, D., Vandewynckel, Y. P., et al. (2017). Treatment of intestinal fibrosis in experimental inflammatory bowel disease by the pleiotropic actions of a local rho kinase inhibitor. Gastroenterology 153, 1054–1067. doi: 10.1053/j.gastro.2017.06.013
Inazaki, K., Kanamaru, Y., Kojima, Y., Sueyoshi, N., Okumura, K., Kaneko, K., et al. (2004). Smad3 deficiency attenuates renal fibrosis, inflammation,and apoptosis after unilateral ureteral obstruction. Kidney Int. 66, 597–604. doi: 10.1111/j.1523-1755.2004.00779.x
Kalluri, R., and Neilson, E. G. (2003). Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Invest. 112, 1776–1784. doi: 10.1172/JCI20530
Katuri, V., Tang, Y., Li, C., Jogunoori, W., Deng, C. X., Rashid, A., et al. (2006). Critical interactions between TGF-beta signaling/ELF, and E-cadherin/beta-catenin mediated tumor suppression. Oncogene 25, 1871–1886. doi: 10.1038/sj.onc.1209211
Khaw, P., Grehn, F., Hollo, G., Overton, B., Wilson, R., Vogel, R., et al. (2007). A phase III study of subconjunctival human anti-transforming growth factor beta(2) monoclonal antibody (CAT-152) to prevent scarring after first-time trabeculectomy. Ophthalmology 114, 1822–1830. doi: 10.1016/j.ophtha.2007.03.050
Kolodsick, J. E., Toews, G. B., Jakubzick, C., Hogaboam, C., Moore, T. A., Mckenzie, A., et al. (2004). Protection from fluorescein isothiocyanate-induced fibrosis in IL-13-deficient, but not IL-4-deficient, mice results from impaired collagen synthesis by fibroblasts. J. Immunol. 172, 4068–4076. doi: 10.4049/jimmunol.172.7.4068
Krieg, T., Abraham, D., and Lafyatis, R. (2007). Fibrosis in connective tissue disease: the role of the myofibroblast and fibroblast-epithelial cell interactions. Arthritis Res. Ther. 9(Suppl. 2), S4. doi: 10.1186/ar2188
Kulkarni, A. A., Thatcher, T. H., Olsen, K. C., Maggirwar, S. B., Phipps, R. P., and Sime, P. J. (2011). PPAR-gamma ligands repress TGFbeta-induced myofibroblast differentiation by targeting the PI3K/Akt pathway: implications for therapy of fibrosis. PLoS One 6:e15909. doi: 10.1371/journal.pone.0015909
Kulkarni, A. B., Huh, C. G., Becker, D., Geiser, A., Lyght, M., Flanders, K. C., et al. (1993). Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. U.S.A. 90, 770–774. doi: 10.1073/pnas.90.2.770
Kumar, R. K., Herbert, C., Yang, M., Koskinen, A. M., Mckenzie, A. N., and Foster, P. S. (2002). Role of interleukin-13 in eosinophil accumulation and airway remodelling in a mouse model of chronic asthma. Clin. Exp. Allergy 32, 1104–1111. doi: 10.1046/j.1365-2222.2002.01420.x
Kuperman, D. A., Huang, X., Koth, L. L., Chang, G. H., Dolganov, G. M., Zhu, Z., et al. (2002). Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat. Med. 8, 885–889. doi: 10.1038/nm734
Laan, M., Cui, Z. H., Hoshino, H., Lotvall, J., Sjostrand, M., Gruenert, D. C., et al. (1999). Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J. Immunol. 162, 2347–2352.
Lakos, G., Takagawa, S., Chen, S. J., Ferreira, A. M., Han, G., Masuda, K., et al. (2004). Targeted disruption of TGF-beta/Smad3 signaling modulates skin fibrosis in a mouse model of scleroderma. Am. J. Pathol. 165, 203–217. doi: 10.1016/S0002-9440(10)63289-0
Lamouille, S., Xu, J., and Derynck, R. (2014). Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15, 178–196. doi: 10.1038/nrm3758
Latella, G., Caprilli, R., and Travis, S. (2011). In favour of early surgery in Crohn’s disease: a hypothesis to be tested. J. Crohns Colitis 5, 1–4. doi: 10.1016/j.crohns.2010.11.003
Latella, G., and Papi, C. (2012). Crucial steps in the natural history of inflammatory bowel disease. World J. Gastroenterol. 18, 3790–3799. doi: 10.3748/wjg.v18.i29.3790
Latella, G., Rogler, G., Bamias, G., Breynaert, C., Florholmen, J., Pellino, G., et al. (2014). Results of the 4th scientific workshop of the ECCO (I): pathophysiology of intestinal fibrosis in IBD. J. Crohns Colitis 8, 1147–1165. doi: 10.1016/j.crohns.2014.03.008
Latella, G., Sferra, R., Speca, S., Vetuschi, A., and Gaudio, E. (2013). Can we prevent, reduce or reverse intestinal fibrosis in IBD? Eur. Rev. Med. Pharmacol. Sci. 17, 1283–1304.
Latella, G., Vetuschi, A., Sferra, R., Zanninelli, G., D’angelo, A., Catitti, V., et al. (2009). Smad3 loss confers resistance to the development of trinitrobenzene sulfonic acid-induced colorectal fibrosis. Eur. J. Clin. Invest. 39, 145–156. doi: 10.1111/j.1365-2362.2008.02076.x
Lawler, J. (2000). The functions of thrombospondin-1 and-2. Curr. Opin. Cell Biol. 12, 634–640. doi: 10.1016/S0955-0674(00)00143-5
Lee, J. H., Kaminski, N., Dolganov, G., Grunig, G., Koth, L., Solomon, C., et al. (2001). Interleukin-13 induces dramatically different transcriptional programs in three human airway cell types. Am. J. Respir. Cell Mol. Biol. 25, 474–485. doi: 10.1165/ajrcmb.25.4.4522
Lee, J. I., Wright, J. H., Johnson, M. M., Bauer, R. L., Sorg, K., Yuen, S., et al. (2016). Role of Smad3 in platelet-derived growth factor-C-induced liver fibrosis. Am. J. Physiol. Cell Physiol. 310, C436–C445. doi: 10.1152/ajpcell.00423.2014
Leeb, S. N., Vogl, D., Gunckel, M., Kiessling, S., Falk, W., Goke, M., et al. (2003). Reduced migration of fibroblasts in inflammatory bowel disease: role of inflammatory mediators and focal adhesion kinase. Gastroenterology 125, 1341–1354. doi: 10.1016/j.gastro.2003.07.004
Li, J. H., Wang, W., Huang, X. R., Oldfield, M., Schmidt, A. M., Cooper, M. E., et al. (2004). Advanced glycation end products induce tubular epithelial-myofibroblast transition through the RAGE-ERK1/2 MAP kinase signaling pathway. Am. J. Pathol. 164, 1389–1397. doi: 10.1016/S0002-9440(10)63225-7
Liu, F. Y., Xiao, L., Peng, Y. M., Duan, S. B., Liu, H., Liu, Y. H., et al. (2007). Inhibition effect of small interfering RNA of connective tissue growth factor on the expression of vascular endothelial growth factor and connective tissue growth factor in cultured human peritoneal mesothelial cells. Chin. Med. J. (Engl.) 120, 231–236. doi: 10.1097/00029330-200702010-00012
Luna, J., Masamunt, M. C., Lawrance, I. C., and Sans, M. (2011). Mesenchymal cell proliferation and programmed cell death: key players in fibrogenesis and new targets for therapeutic intervention. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G703–G708. doi: 10.1152/ajpgi.00504.2010
Maloy, K. J. (2008). The interleukin-23/Interleukin-17 axis in intestinal inflammation. J. Intern. Med. 263, 584–590. doi: 10.1111/j.1365-2796.2008.01950.x
Mascarenhas, J., Li, T., Sandy, L., Newsom, C., Petersen, B., Godbold, J., et al. (2014). Anti-transforming growth factor-beta therapy in patients with myelofibrosis. Leuk. Lymphoma 55, 450–452. doi: 10.3109/10428194.2013.805329
Massague, J., Seoane, J., and Wotton, D. (2005). Smad transcription factors. Genes Dev. 19, 2783–2810. doi: 10.1101/gad.1350705
Mead, A. L., Wong, T. T., Cordeiro, M. F., Anderson, I. K., and Khaw, P. T. (2003). Evaluation of anti-TGF-beta2 antibody as a new postoperative anti-scarring agent in glaucoma surgery. Invest. Ophthalmol. Vis. Sci. 44, 3394–3401. doi: 10.1167/iovs.02-0978
Mifflin, R. C., Pinchuk, I. V., Saada, J. I., and Powell, D. W. (2011). Intestinal myofibroblasts: targets for stem cell therapy. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G684–G696. doi: 10.1152/ajpgi.00474.2010
Monteleone, G., Fantini, M. C., Onali, S., Zorzi, F., Sancesario, G., Bernardini, S., et al. (2012). Phase I clinical trial of Smad7 knockdown using antisense oligonucleotide in patients with active Crohn’s disease. Mol. Ther. 20, 870–876. doi: 10.1038/mt.2011.290
Monteleone, G., Neurath, M. F., Ardizzone, S., Di Sabatino, A., Fantini, M. C., Castiglione, F., et al. (2015). Mongersen, an oral SMAD7 antisense oligonucleotide, and Crohn’s disease. N. Engl. J. Med. 372, 1104–1113. doi: 10.1056/NEJMoa1407250
Morishita, Y., Yoshizawa, H., Watanabe, M., Ishibashi, K., Muto, S., Kusano, E., et al. (2014). siRNAs targeted to Smad4 prevent renal fibrosis in vivo. Sci. Rep. 4:6424. doi: 10.1038/srep06424
Murphy-Ullrich, J. E., and Mosher, D. F. (1985). Localization of thrombospondin in clots formed in situ. Blood 66, 1098–1104.
Murray, L. A., Argentieri, R. L., Farrell, F. X., Bracht, M., Sheng, H., Whitaker, B., et al. (2008). Hyper-responsiveness of IPF/UIP fibroblasts: interplay between TGFbeta1, IL-13 and CCL2. Int. J. Biochem. Cell Biol. 40, 2174–2182. doi: 10.1016/j.biocel.2008.02.016
Nomura, M., and Li, E. (1998). Smad2 role in mesoderm formation, left-right patterning and craniofacial development. Nature 393, 786–790. doi: 10.1038/31693
Odegaard, J. I., Ricardo-Gonzalez, R. R., Goforth, M. H., Morel, C. R., Subramanian, V., Mukundan, L., et al. (2007). Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 447, 1116–1120. doi: 10.1038/nature05894
Oh, M. H., Oh, S. Y., Yu, J., Myers, A. C., Leonard, W. J., Liu, Y. J., et al. (2011). IL-13 induces skin fibrosis in atopic dermatitis by thymic stromal lymphopoietin. J. Immunol. 186, 7232–7242. doi: 10.4049/jimmunol.1100504
Petersen, M., Thorikay, M., Deckers, M., Van Dinther, M., Grygielko, E. T., Gellibert, F., et al. (2008). Oral administration of GW788388, an inhibitor of TGF-beta type I and II receptor kinases, decreases renal fibrosis. Kidney Int. 73, 705–715. doi: 10.1038/sj.ki.5002717
Piera-Velazquez, S., Li, Z., and Jimenez, S. A. (2011). Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 179, 1074–1080. doi: 10.1016/j.ajpath.2011.06.001
Pucilowska, J. B., Williams, K. L., and Lund, P. K. (2000). Fibrogenesis. IV. Fibrosis and inflammatory bowel disease: cellular mediators and animal models. Am. J. Physiol. Gastrointest Liver Physiol. 279, G653–G659. doi: 10.1152/ajpgi.2000.279.4.G653
Punekar, S., Zak, S., Kalter, V. G., Dobransky, L., Punekar, I., Lawler, J. W., et al. (2008). Thrombospondin 1 and its mimetic peptide ABT-510 decrease angiogenesis and inflammation in a murine model of inflammatory bowel disease. Pathobiology 75, 9–21. doi: 10.1159/000113790
Rastaldi, M. P. (2006). Epithelial-mesenchymal transition and its implications for the development of renal tubulointerstitial fibrosis. J. Nephrol. 19, 407–412.
Rice, L. M., Padilla, C. M., Mclaughlin, S. R., Mathes, A., Ziemek, J., Goummih, S., et al. (2015). Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J. Clin. Invest. 125, 2795–2807. doi: 10.1172/JCI77958
Ricote, M., Li, A. C., Willson, T. M., Kelly, C. J., and Glass, C. K. (1998). The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 391, 79–82. doi: 10.1038/34178
Rieder, F., and Fiocchi, C. (2008). Intestinal fibrosis in inflammatory bowel disease – Current knowledge and future perspectives. J. Crohns Colitis 2, 279–290. doi: 10.1016/j.crohns.2008.05.009
Rieder, F., and Fiocchi, C. (2009). Intestinal fibrosis in IBD–a dynamic, multifactorial process. Nat. Rev. Gastroenterol. Hepatol. 6, 228–235. doi: 10.1038/nrgastro.2009.31
Rieder, F., Latella, G., Magro, F., Yuksel, E. S., Higgins, P. D., Di Sabatino, A., et al. (2016). European crohn’s and colitis organisation topical review on prediction, diagnosis and management of fibrostenosing crohn’s disease. J. Crohns Colitis 10, 873–885. doi: 10.1093/ecco-jcc/jjw055
Roberts, A. B., Russo, A., Felici, A., and Flanders, K. C. (2003). Smad3: a key player in pathogenetic mechanisms dependent on TGF-beta. Ann. N. Y. Acad. Sci. 995, 1–10. doi: 10.1111/j.1749-6632.2003.tb03205.x
Rogliani, P., Mura, M., Assunta Porretta, M., and Saltini, C. (2008). New perspectives in the treatment of idiopathic pulmonary fibrosis. Ther. Adv. Respir Dis. 2, 75–93. doi: 10.1177/1753465808089363
Rousseaux, C., and Desreumaux, P. (2006). [The peroxisome-proliferator-activated gamma receptor and chronic inflammatory bowel disease (PPARgamma and IBD)]. J. Soc. Biol. 200, 121–131. doi: 10.1051/jbio:2006015
Sadler, T., Scarpa, M., Rieder, F., West, G., and Stylianou, E. (2013). Cytokine-induced chromatin modifications of the type I collagen alpha 2 gene during intestinal endothelial-to-mesenchymal transition. Inflamm. Bowel Dis. 19, 1354–1364. doi: 10.1097/MIB.0b013e318281f37a
Sato, M., Muragaki, Y., Saika, S., Roberts, A. B., and Ooshima, A. (2003). Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Invest. 112, 1486–1494. doi: 10.1172/JCI19270
Scharl, M., Huber, N., Lang, S., Furst, A., Jehle, E., and Rogler, G. (2015). Hallmarks of epithelial to mesenchymal transition are detectable in Crohn’s disease associated intestinal fibrosis. Clin. Transl. Med. 4:1. doi: 10.1186/s40169-015-0046-5
Schultze-Mosgau, S., Kopp, J., Thorwarth, M., Rodel, F., Melnychenko, I., Grabenbauer, G. G., et al. (2006). Plasminogen activator inhibitor-I-related regulation of procollagen I (alpha1 and alpha2) by antitransforming growth factor-beta1 treatment during radiation-impaired wound healing. Int. J. Radiat. Oncol. Biol. Phys. 64, 280–288. doi: 10.1016/j.ijrobp.2005.09.006
Siassi, M., Weiger, A., Hohenberger, W., and Kessler, H. (2007). Changes in surgical therapy for Crohn’s disease over 33 years: a prospective longitudinal study. Int. J. Colorectal. Dis. 22, 319–324. doi: 10.1007/s00384-006-0150-5
Sime, P. J., Xing, Z., Graham, F. L., Csaky, K. G., and Gauldie, J. (1997). Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J. Clin. Invest. 100, 768–776. doi: 10.1172/JCI119590
Sorrentino, D., Avellini, C., Beltrami, C. A., Pasqual, E., and Zearo, E. (2006). Selective effect of infliximab on the inflammatory component of a colonic stricture in Crohn’s disease. Int. J. Colorectal. Dis. 21, 276–281. doi: 10.1007/s00384-005-0739-0
Speca, S., Giusti, I., Rieder, F., and Latella, G. (2012). Cellular and molecular mechanisms of intestinal fibrosis. World J. Gastroenterol. 18, 3635–3661. doi: 10.3748/wjg.v18.i28.3635
Spinelli, A., Correale, C., Szabo, H., and Montorsi, M. (2010). Intestinal fibrosis in Crohn’s disease: medical treatment or surgery? Curr. Drug Targets 11, 242–248. doi: 10.2174/138945010790309984
Sweetwyne, M. T., and Murphy-Ullrich, J. E. (2012). Thrombospondin1 in tissue repair and fibrosis: TGF-beta-dependent and independent mechanisms. Matrix Biol. 31, 178–186. doi: 10.1016/j.matbio.2012.01.006
Trachtman, H., Fervenza, F. C., Gipson, D. S., Heering, P., Jayne, D. R., Peters, H., et al. (2011). A phase 1, single-dose study of fresolimumab, an anti-TGF-beta antibody, in treatment-resistant primary focal segmental glomerulosclerosis. Kidney Int. 79, 1236–1243. doi: 10.1038/ki.2011.33
Ulloa, L., Doody, J., and Massague, J. (1999). Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway. Nature 397, 710–713. doi: 10.1038/17826
Vaglio, A., Palmisano, A., Corradi, D., Salvarani, C., and Buzio, C. (2007). Retroperitoneal fibrosis: evolving concepts. Rheum. Dis. Clin. North Am. 33, 803–817, vi–vii. doi: 10.1016/j.rdc.2007.07.013
Valatas, V., Filidou, E., Drygiannakis, I., and Kolios, G. (2017). Stromal and immune cells in gut fibrosis: the myofibroblast and the scarface. Ann. Gastroenterol. 30, 393–404. doi: 10.20524/aog.2017.0146
Vallance, B. A., Gunawan, M. I., Hewlett, B., Bercik, P., Van Kampen, C., Galeazzi, F., et al. (2005). TGF-beta1 gene transfer to the mouse colon leads to intestinal fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 289, G116–G128. doi: 10.1152/ajpgi.00051.2005
Van Assche, G., Dignass, A., Reinisch, W., Van Der Woude, C. J., Sturm, A., De Vos, M., et al. (2010). The second European evidence-based Consensus on the diagnosis and management of Crohn’s disease: special situations. J. Crohns Colitis 4, 63–101. doi: 10.1016/j.crohns.2009.09.009
van Meeteren, L. A., and ten Dijke, P. (2012). Regulation of endothelial cell plasticity by TGF-beta. Cell Tissue Res. 347, 177–186. doi: 10.1007/s00441-011-1222-6
Varga, J., and Pasche, B. (2008). Antitransforming growth factor-beta therapy in fibrosis: recent progress and implications for systemic sclerosis. Curr. Opin. Rheumatol. 20, 720–728. doi: 10.1097/BOR.0b013e32830e48e8
Vermeire, S. (2015). Oral SMAD7 antisense drug for Crohn’s disease. N. Engl. J. Med. 372, 1166–1167. doi: 10.1056/NEJMe1415053
Wang, D., Dai, C., Li, Y., and Liu, Y. (2011). Canonical Wnt/beta-catenin signaling mediates transforming growth factor-beta1-driven podocyte injury and proteinuria. Kidney Int. 80, 1159–1169. doi: 10.1038/ki.2011.255
Wang, L., Chen, S., and Xu, K. (2011). IL-17 expression is correlated with hepatitis Brelated liver diseases and fibrosis. Int. J. Mol. Med. 27, 385–392. doi: 10.3892/ijmm.2011.594
Warnatz, K., Keskin, A. G., Uhl, M., Scholz, C., Katzenwadel, A., Vaith, P., et al. (2005). Immunosuppressive treatment of chronic periaortitis: a retrospective study of 20 patients with chronic periaortitis and a review of the literature. Ann. Rheum. Dis. 64, 828–833. doi: 10.1136/ard.2004.029793
Willis, B. C., and Borok, Z. (2007). TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease. Am. J. Physiol. Lung Cell Mol. Physiol. 293, L525–L534. doi: 10.1152/ajplung.00163.2007
Wilson, M. S., Madala, S. K., Ramalingam, T. R., Gochuico, B. R., Rosas, I. O., Cheever, A. W., et al. (2010). Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J. Exp. Med. 207, 535–552. doi: 10.1084/jem.20092121
Wynn, T. A. (2007). Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Invest. 117, 524–529. doi: 10.1172/JCI31487
Wynn, T. A. (2008). Cellular and molecular mechanisms of fibrosis. J. Pathol. 214, 199–210. doi: 10.1002/path.2277
Xavier, S., Vasko, R., Matsumoto, K., Zullo, J. A., Chen, R., Maizel, J., et al. (2015). Curtailing endothelial TGF-beta signaling is sufficient to reduce endothelial-mesenchymal transition and fibrosis in CKD. J. Am. Soc. Nephrol. 26, 817–829. doi: 10.1681/ASN.2013101137
Yehualaeshet, T., O’connor, R., Begleiter, A., Murphy-Ullrich, J. E., Silverstein, R., and Khalil, N. (2000). A CD36 synthetic peptide inhibits bleomycin-induced pulmonary inflammation and connective tissue synthesis in the rat. Am. J. Respir. Cell Mol. Biol. 23, 204–212. doi: 10.1165/ajrcmb.23.2.4089
Zanninelli, G., Vetuschi, A., Sferra, R., D’angelo, A., Fratticci, A., Continenza, M. A., et al. (2006). Smad3 knock-out mice as a useful model to study intestinal fibrogenesis. World J. Gastroenterol. 12, 1211–1218. doi: 10.3748/wjg.v12.i8.1211
Zeisberg, M., Hanai, J., Sugimoto, H., Mammoto, T., Charytan, D., Strutz, F., et al. (2003). BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 9, 964–968. doi: 10.1038/nm888
Zeisberg, M., and Kalluri, R. (2008). Fibroblasts emerge via epithelial-mesenchymal transition in chronic kidney fibrosis. Front. Biosci. 13:6991–6998. doi: 10.2741/3204
Zhang, G. Y., Cheng, T., Zheng, M. H., Yi, C. G., Pan, H., Li, Z. J., et al. (2009). Activation of peroxisome proliferator-activated receptor-gamma inhibits transforming growth factor-beta1 induction of connective tissue growth factor and extracellular matrix in hypertrophic scar fibroblasts in vitro. Arch. Dermatol. Res. 301, 515–522. doi: 10.1007/s00403-009-0959-1
Keywords: transforming growth factor-β, inflammatory bowel disease, intestinal fibrosis, epithelial-to-mesenchymal transition, endothelial-to-mesenchymal transition, extracellular matrix
Citation: Yun S-M, Kim S-H and Kim E-H (2019) The Molecular Mechanism of Transforming Growth Factor-β Signaling for Intestinal Fibrosis: A Mini-Review. Front. Pharmacol. 10:162. doi: 10.3389/fphar.2019.00162
Received: 09 November 2018; Accepted: 11 February 2019;
Published: 27 February 2019.
Edited by:
Luca Antonioli, University of Pisa, ItalyReviewed by:
Raquel Abalo, Universidad Rey Juan Carlos, SpainCopyright © 2019 Yun, Kim and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seok-Ho Kim, a3NoMzQxMEBjaGEuYWMua3I= Eun-Hee Kim, ZWhraW1AY2hhLmFjLmty
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.