 Anna Bagnato
Anna Bagnato Laura Rosanò
Laura Rosanò- Unit of Preclinical Models and New Therapeutic Agents, IRCCS-Regina Elena National Cancer Institute, Rome, Italy
Tumor cells acquire invasive and metastatic behavior by sensing changes in the localization and activation of signaling pathways, which in turn determine changes in actin cytoskeleton. The core-scaffold machinery associated to β-arrestin (β-arr) is a key mechanism of G-protein coupled receptors (GPCR) to achieve spatiotemporal specificity of different signaling complexes driving cancer progression. Within different cellular contexts, the scaffold proteins β-arr1 or β-arr2 may now be considered organizers of protein interaction networks involved in tumor development and metastatic dissemination. Studies have uncovered the importance of the β-arr engagement with a growing number of receptors, signaling molecules, cytoskeleton regulators, epigenetic modifiers, and transcription factors in GPCR-driven tumor promoting pathways. In many of these molecular complexes, β-arrs might provide a physical link to active dynamic cytoskeleton, permitting cancer cells to adapt and modify the tumor microenvironment to promote the metastatic spread. Given the complexity and the multidirectional β-arr-driven signaling in cancer cells, therapeutic targeting of specific GPCR/β-arr molecular mechanisms is an important avenue to explore when considering future new therapeutic options. The focus of this review is to integrate the most recent developments and exciting findings of how highly connected components of β-arr-guided molecular connections to other pathways allow precise control over multiple signaling pathways in tumor progression, revealing ways of therapeutically targeting the convergent signals in patients.
Introduction
G protein-coupled receptors (GPCRs) constitute the largest family among the membrane proteins, playing an important role not only in mediating physiological function but also controlling the recruitment and activation of intracellular molecules associated with human diseases, including cancer (Lagerstrom and Schioth, 2008; Hauser et al., 2017; Insel et al., 2018). Agonist-activated GPCRs couple to heterotrimeric G proteins, thus facilitating exchange of GDP by GTP in the Gα subunits followed by dissociation from the βγ dimers and transiently interacting with specific effectors to trigger canonical signal transduction cascades. The phosphorylation of GPCRs by G protein-coupled receptor kinases (GRKs), a subfamily of AGC (protein kinase A/G/C-like) kinases originally identified as inhibitors of GPCR signaling, promotes the recruitment to the phosphorylated receptor of the cytosolic proteins β-arrestins (β-arrs), leading to GPCR desensitization (Gurevich et al., 2012).
In the classical view, GPCR signaling is mediated by G proteins, while β-arr recruitment is associated with signaling desensitization of GPCR. In this old view, the competition between G proteins and β-arrs, β-arr1 or β-arr2, for an activated GPCR determines the signal termination, hampering further G protein signaling (Luttrell and Lefkowitz, 2002; Sente et al., 2018). Also, β-arrs promote clathrin-dependent-endocytosis of activated GPCR (Luttrell and Lefkowitz, 2002; Sente et al., 2018). One of the most important breakthroughs in GPCR field during the past decades was that the signal transduction of GPCRs is also strictly linked to β-arr. Beyond their known roles in GPCR desensitization/internalization, β-arrs have been implicated in the control of multiple outcomes, acting as multifunctional scaffold proteins and signaling transducers, crucial for intracellular signal propagation and amplification, and governing different cellular effects (Miller and Lefkowitz, 2001; DeFea, 2008; Shukla et al., 2011; Peterson and Luttrell, 2017). Almost two decades after the first evidence, GPCR field now encompasses an extensive knowledge that β-arrs integrate signals arising from GPCR with intrinsic cellular pathways in human disease, initiating waves of intracellular signaling in a G protein-independent manner and allowing the discovery of new therapies targeting selectively β-arr-mediated circuits, known as biased arrestin-biased agonism (Smith et al., 2018). Moreover, since β-arr-biased signaling requires phosphorylation of GPCRs by GRKs to promote high-affinity binding of β-arr to GPCRs and GRK subtypes might have preferential phosphorylation and trigger unique conformational changes in GPCRs, studies of β-arr-biased signaling might consider also the involvement of GRKs in cancer-related signaling pathways (Heitzler et al., 2012). In this regard, different isoforms of GRKs are able to modulate the response to many GPCRs involved in tumoral signaling via its direct interaction with other components of transduction cascades, as well outlined in a recent review (Nogués et al., 2018). Therefore, GRKs would also be considered critical to control the fate of β-arr-dependent signaling of GPCRs and as potential therapeutic targets in cancer.
Recent pharmacological studies on the paradigm of biased agonists, where a particular biased ligand can generate a GPCR conformation able to lead to a distinct functional outcome, usually either G-protein or β-arr-dependent signaling but not both, suggest that current GPCR-based therapeutics could be improved by increasing anticancer efficacy (Smith et al., 2018). Moreover, computational and atomic level dynamic simulation approaches provided new details linking phosphorylation of GPCR, β-arr interactions, and β-arr-dependent signaling, supporting the “barcode hypothesis,” in which distinct patterns of GPCR phosphorylation trigger specific conformational states of β-arr with specific functional outcomes (Srivastava et al., 2015). In addition, remarkable advances in the GPCR structural biology field deeply demonstrated that specific ligands, by stabilizing particular sets of conformations and permitting the interaction with specific effectors, might achieve specific efficacies for selected signaling pathway (Rosenbaum et al., 2009). Recently, this conceptual framework has been refined, whereby the activated GPCR might lead the formation of a “supercomplex,” where GPCR and β-arr1 form a unique signaling module with G-protein (Marshall, 2016; Thomsen et al., 2016). These findings support the hypothesis of a new way to signal, by concomitant binding of G proteins and β-arr to activated receptors, further providing an additional paradigm in GPCR-driven signaling transduction.
β-Arrestins as Scaffold Proteins in GPCR Signaling
In cancer cells and in a cell context- and cancer type-dependent manner, the pools of β-arr-dependent multiprotein complexes can be found localized to different intracellular compartments, as bound to the cytoskeleton, as endocytic adapters acting on specific signalosomes in endosomes and interacting with signaling proteins involved in gene transcription, protein ubiquitination, and cytoskeletal remodeling, among others (Ma and Pei, 2007; Sobolesky and Moussa, 2013; McGovern and DeFea, 2014; Black et al., 2016; Jean-Charles et al., 2016; Rosanò and Bagnato, 2016; Chaturvedi et al., 2018; Eichel and von Zastrow, 2018; Song et al., 2018). β-arr-dependent multiprotein complexes, transducing the GPCR signals, regulate the functionality of different tyrosine kinase receptor family members and directly control cytosolic, cytoskeletal remodeling or nuclear signaling components of pathways relevant for tumor growth, invasiveness, and metastatic progression (Figure 1). Through these functions, both β-arrs foster a plethora of signaling pathways, including members of the mitogen-activated protein kinase (MAPK), AKT, PI3K, Wnt, Hedgehog, E3 ubiquitin ligases, PTEN, nuclear factor-kB, and regulators of small GTPase activity. To expand the intracellular communication, agonists of GPCRs can activate tyrosine kinase receptors (RTK), through a signal cross talk. This can occur via a mechanism by a GPCR-mediated activation of proteases operating the ectodomain shedding of a membrane bound pro-ligand, such as heparin-binding epidermal growth factor (Hb-EGF), or by the intercellular activity of GPCR-activated tyrosine kinase, completely independent of ligand binding (Rosanò and Bagnato, 2016; Crudden et al., 2018). Moreover, accumulating evidence recognizes that the transactivation of RTKs by GPCRs is not unidirectional, as the cross talk between RTKs and GPCRs is reciprocal, GPCRs can be activated by RTKs, and β-arr can be used by RTKs, as in the case of insulin-like growth factor type 1 receptor (Girnita et al., 2005, 2007; Zheng et al., 2012; Crudden et al., 2018) or platelet-derived growth factor receptors (Pyne and Pyne, 2017). In both mechanisms, it is well known that some GPCRs use β-arr to execute and transduce this cross talk between GPCRs and RTKs, governing multiple cellular processes in cancer invasion and metastasis. Proteomic studies in cancer cells demonstrated a very impressive diversity of signaling cascade molecules, which can be engaged by β-arrs for a positive or negative signaling regulation (Xiao et al., 2007; Parisis et al., 2013; Xiao and Sun, 2018), underscoring the importance of GPCR-driven β-arrs in shaping and fine-tuning signaling in cancer progression.
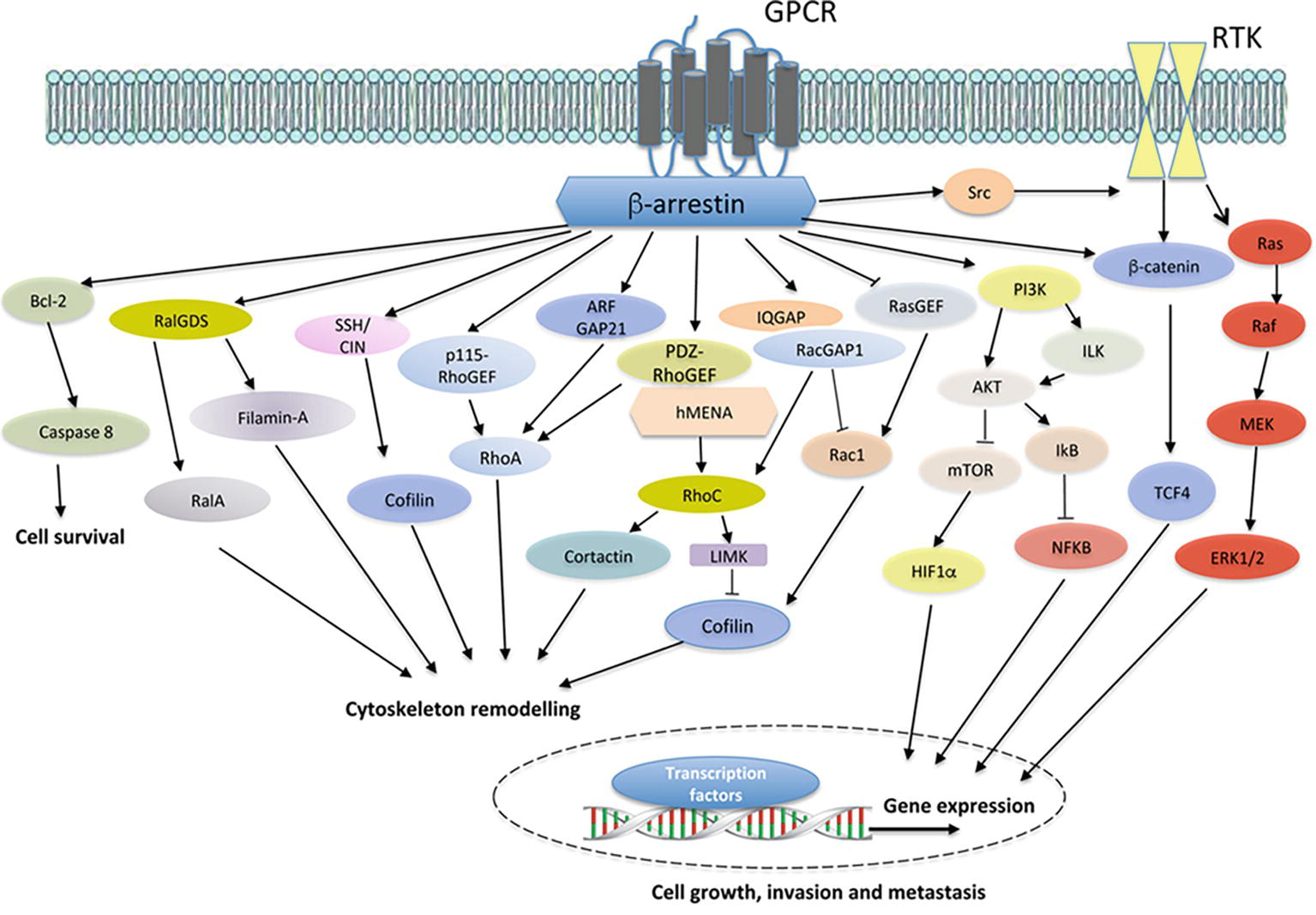
Figure 1. Model of GPCR/β-arr-dependent signal pathways controlling cell survival, cytoskeleton remodeling, and gene expression, leading to enhanced cell growth, invasion and metastasis. In different cancer cells, the binding of ligands to cognate GPCRs leads to the recruitment of β-arrestin (β-arr), which might activate diverse signal-transduction pathways, including Bcl2 and downstream caspase 8. The crosstalk with receptor tyrosine kinases (RTKs), through the recruitment and activation of Src, results in downstream pathway activation, such as members of the Ras/Raf/MEK/ERK family and β-catenin/TCF4. Moreover, GPCR stimulation activates PI3K, leading to AKT/integrin-linked kinase (ILK) signaling and mTOR inhibition. Beyond the cytosolic functions, β-arr might regulate hypoxia-inducible factor 1α (HIF1α) at the levels of transcription, leading to nuclear entry and binding to hypoxia-response elements and the gene transcription. Similarly, β-arr might activate nuclear factor-κB (NF-κB) signaling via inhibition of NF-κB inhibitor (IκB), resulting in the dissociation and subsequent nuclear localization of active NF-κB. At the same time, the interaction of β-arr with actin regulators, such as Filamin-A and LIMK, and ser/thr phosphatases, such as SSH and CIN, leads to enhanced cofilin activity in actin cytoskeleton effects. In addition, GPCR activation might promote the interaction between β-arr and either RHO-guanine nucleotide exchange factors (RHO-GEFs), such as p115RhoGEF or PDZ-RhoGEF, or Rho GTPase-activating proteins (RhoGAPs), such as ARF-GAP21, to activate RhoA GTPase and regulate actin remodeling. β-arr can also bind RalGDS to activate RalA GTPase in cytoskeletal reorganization. Moreover, the interaction of β-arr1 with PDZ-RhoGEF and members of ENA/VASP family, hMENA, might lead to RhoC GTPase activation, causing LIMK-dependent cofilin inhibition and cortactin activation, enhancing invasive behavior. At the same time, β-arr might bind IQGAP1 and RacGAP1, leading to the suppression of Rac1 activity and favoring activation of RhoC and invadopodia functions. The inhibition of β-arr-dependent RASGFR2 activates Rac1 promoting actin polymerization through cofilin activity. β-arr acts as hub regulating several cellular processes related to cancer progression via its interaction with different components of transduction cascades.
β-arrs are expressed in human tumors and mediate multiprotein signaling complexes, in which β-arr acts as membrane, cytosolic, or nuclear scaffold and signal transducer, culminating in multifaceted signaling processes, such as cell growth and proliferation, drug resistance, cell migration, invasion, and metastasis (Ma and Pei, 2007; Sobolesky and Moussa, 2013; Black et al., 2016; Jean-Charles et al., 2016; Rosanò and Bagnato, 2016; Chaturvedi et al., 2018; Song et al., 2018) (Table 1). In this review, we summarize new specific routes of β-arr-mediated signaling of GPCR in cancer, focusing on invasive behavior, sustained by complex machinery that includes physical interaction with adaptor proteins.

Table 1. β-arrestins in cancer.
β-arr1 as Scaffold for Cytoskeleton Remodeling in Tumor Cell Motility
Cell motility is a complex process in which cells change shape following activation of signaling pathways that control cytoskeleton dynamics and the turnover of cell-matrix and cell-cell junctions (Friedl and Alexander, 2011; Lambert et al., 2017). The primary driving force guiding cell motility is linked to actin assembly/disassembly within cells, in which different proteins might regulate the major steps in actin remodeling, as activation of proteins breaking the existing filaments into smaller fragments to create free barbed ends and protein nucleators facilitating association of actin monomers into filaments and membrane protrusions. Some of these processes depend on environmental cues, including ligand-dependent activation of GPCR, and their spatial/temporal regulation is mediated by proteins interacting directly or indirectly with the actin and microtubule cytoskeleton elements, in proximity with signaling proteins. During tumor cell migration, chemotaxis, and metastasis, β-arrs mediate GPCR-driven effects on actin cytoskeleton remodeling by orchestrating activation and localization of selected proteins at the leading edge and generating the necessary forces for movement (Zoudilova et al., 2007, 2010; Min and Defea, 2011; Ma et al., 2014). Although the first evidence showed that β-arr-MAPK complexes control actin cytoskeletal reorganization at the leading edge during cell migration (Sun et al., 2002; Ge et al., 2004; Décaillot et al., 2011), many other studies highlighted that β-arrs function as master signalosome scaffold for specific cytoskeleton-related signaling molecules, including c-Src, filamin, cofilin, and small monomeric GTPases, to connect GPCRs to the cytoskeleton and cell shape changes (Bhattacharya et al., 2002; Barnes et al., 2005; Hunton et al., 2005; Buchanan et al., 2006; Scott et al., 2006; Zoudilova et al., 2007, 2010; Li et al., 2009; Rosanò et al., 2009; Godin et al., 2010; Min and Defea, 2011; Ma et al., 2012; Semprucci et al., 2016; Shishkin et al., 2016; Tocci et al., 2016). Cofilin is considered as one of the primary actin filament severing proteins, operating a rapidly disassembling of existing filaments and promoting generation of actin filament extension and the formation of the leading edge during chemotaxis (Shishkin et al., 2016). The function of β-arr, by interacting with cofilin and phosphatases/proteases or by mediating the activity of small GTPases, is a prerequisite for controlling phosphorylation/inactivation-dependent cofilin activity (Zoudilova et al., 2007, 2010; Min and Defea, 2011). Growing evidence demonstrated that β-arr regulates the activity of RhoA, in the family of Rho GTPases, by interacting with guanine nucleotide exchange factors (GEFs), such as p115RhoGEF and PDZ-RhoGEF, or GTPase activating protein (GAPs), such as ARFGAP21, in regulating stress fiber assembly/disassembly, or with Ral-GDS to regulate RalA for controlling membrane ruffling and cell migration, as observed for activation of cognate receptors for angiotensin AT1A (ATII1A), beta-2 adrenergic (β2AR), formyl-Met-Leu-Phe, lysophosphatidic acid (LPA), or endothelin-1 (ET-1) (Bhattacharya et al., 2002; Barnes et al., 2005; Hunton et al., 2005; Li et al., 2009; Godin et al., 2010; Ma et al., 2012; Semprucci et al., 2016; Tocci et al., 2016). Other studies showed that GPCR-dependent cytoskeletal rearrangement and membrane protrusion formation might depend on the interaction of β-arr with the actin-binding protein filamin A (Scott et al., 2006).
The dissemination of cancer cells from primary tumors and their seeding in the metastatic niche often involves the local movement of tumor cells and their penetration into surrounding tissue, which require dramatic reorganization of the cell cytoskeleton and remodeling of extracellular matrix (ECM) (Friedl and Alexander, 2011; Eddy et al., 2017; Lambert et al., 2017; Mrkonjic et al., 2017; Paterson and Courtneidge, 2018). Therefore, invading cells adopt mesenchymal, elongated morphology with focalized proteolytic activities toward ECM, by forming invadopodia (Eddy et al., 2017; Mrkonjic et al., 2017; Paterson and Courtneidge, 2018). In this context, it has been demonstrated that β-arr-related molecular complexes might govern invasive and metastatic behavior by promoting invadopodia formation. The activation of GPCRs, such as ET-1R, initiates downstream signaling cascade and formation of protein complexes mediating actin rearrangement leading to invadopodia formation and ECM degradation, where β-arr1 regulates the spatial distribution of actin cytoskeleton and actin regulators (Bagnato and Rosanò, 2016; Semprucci et al., 2016). This cascade, starting with the interaction of β-arr1 with PDZ-RhoGEF, controlling the spatial distribution of RhoC GTPase and cofilin pathway, represents a critical route by which the tumor cells form active invadopodia, and acquires maximally proinvasive capabilities (Semprucci et al., 2016). Following studies demonstrated that β-arr1-associated molecular complexes during invasive protrusions involve members of the ENA/VASP family, known to regulate the actin-based motility of various cell types, and in particular, hMENA and the invasive isoform hMENAΔv6 (Krause et al., 2003; Gertler and Condeelis, 2011; Di Modugno et al., 2012, 2018). Inputs derived from ET-1R promote the formation of a signaling platform containing β-arr1-hMENA/hMENAΔv6/PDZ-RhoGEF converging on RhoC pathway, favoring pericellular matrix degradation, and conferring also a fitness advantage to tumor cells to breach the endothelial barrier and start the transendothelial migration process (Di Modugno et al., 2018). More recently, a deep understanding of how protein complexes are assembled into a functional unit to enhance specific signaling pathways revealed a new mechanistic link between β-arr1 and the integrin-related protein IQ-domain GTPase-activating protein 1 (IQGAP1), downstream of ET-1R signaling, in shaping cytoskeleton remodeling and invadopodia-dependent ECM degradation (Chellini et al., 2018). Specifically, IQGAP1/β-arr1 acts as small GTPase scaffolding platform, as RacGAP1, to promote Rac1 inhibition and concomitant RhoA,C activation, suggesting that ET-1R-guided β-arr1 interactions determine the convergence and activation/inhibition of specific signals for invadopodia, such as Rho GTPases, where IQGAP1 helps to define the discrete locations and/or time (Chellini et al., 2018). In line with these findings, the activation of the GPCR kisspeptin receptor promotes invadopodia formation in human breast cancer cells via β-arr2/ERK (Goertzen et al., 2016). Concordantly, new findings demonstrated a functional link between the tumor suppressor PTEN, scaffolding function of β-arr1, ARHGAP21 and cytoskeletal rearrangements, in driving evolution of 3D morphology phenotypes mimicking colorectal cancer in early step of metastatization (Jagan et al., 2013; Javadi et al., 2017). These new findings disclose so far an unexpected role of β-arr capable to rewire the GPCR signaling networks and activate specific machinery for changing shape, generating invasive protrusions, and remodeling ECM in invasive and metastatic cancer cells, and update the signaling paradigm that targeting GPCR/β-arr1 pathways can represent a possible route of therapeutic intervention (Rosanò et al., 2013; Bagnato and Rosanò, 2016; Goertzen et al., 2016; Semprucci et al., 2016; Chellini et al., 2018; Di Modugno et al., 2018).
Nuclear function of β-arr1 in Cancer
Among the non-canonical functions of β-arr, many studies demonstrated that nuclear β-arr1 might generate coordinated transcriptional responses to environmental changes, uncovering additional functions of β-arr1 in tumor progression (Kang et al., 2005; Shi et al., 2007; Hoeppner et al., 2012; Yang et al., 2015). To dissect a genomic landscape of β-arr1 in cancer and find direct transcriptional targets, an integrated whole-genome ChIP-Seq analysis and gene expression profiling have been performed in prostate cancer cells exposed to pseudohypoxia, a condition that mimics hypoxia that is frequently encountered within solid tumors. The results of this study revealed a partial overlap between β-arr1 and p300 acetyltransferase-binding sites in the same gene-proximal regions and the presence of non-overlapping sites, suggesting a double-hedged sword of β-arr1 in modulating gene, dependently or independently of p300 (Zecchini et al., 2014). A functional analysis of β-arr1 transcriptome also revealed an enrichment of genes involved in cellular metabolism and the cell cycle, with an overlap with the hypoxia-induced factor-1α (HIF-1α) transcriptome, including known HIF1α target genes involved in angiogenesis and aerobic glycolysis (Shenoy et al., 2012; Zecchini et al., 2014). Concordantly, in ovarian cancer, the activation of ET-1R, by mimicking hypoxia, promotes the interaction between β-arr1/p300 and HIF-1α, enhancing the transcription of genes, such as ET-1 and VEGF, required for tumor cell invasion and proangiogenic effects, operating a self-amplifying HIF-1α-mediated transcription of genes that sustain metastatic process (Cianfrocca et al., 2016). The findings further supporting the nuclear role of β-arr1/p300 in maintaining a more aggressive phenotype demonstrated the interplay with Wnt/β-catenin signaling (Chen et al., 2001; Bryja et al., 2007; Bonnans et al., 2012; Rosanò et al., 2013, 2014; Duan et al., 2016). Downstream of ET-1R activation, β-arr1/p300/β-catenin pathway, also represents a novel bypass mechanism through which this receptor is linked to chemoresistance, cancer stem cells like phenotype, and metastatic behavior (Rosanò et al., 2014). In both androgen-dependent and castration-resistant prostate cancer cells, β-arr1 enhances the binding of androgen receptor (AR) to androgen response elements, favoring cell proliferation, growth, and invasion, as well as in vivo tumor formation, local invasion, and distant metastasis (Purayil et al., 2015). Moreover, nuclear β-arr1 suppresses RasGRF2 gene expression through promoter hypermethylation, with consequent controlling of Rac1/cofilin pathways (Ma et al., 2014). An important interplay between nuclear β-arr1 and E2F transcription factor has been demonstrated in non-small cell lung cancers, contributing to the growth and progression of this tumor (Dasgupta et al., 2011; Perumal et al., 2014; Pillai et al., 2015). In myeloid leukemia where β-arr mediates the initiation and maintenance of tumor cells (Fereshteh et al., 2012; Kotula et al., 2014), the interaction of β-arr1 with the DNA-binding Enhancer of Zeste Homologue 2 (EZH2) protein mediates BCR/ABL histone acetylation during tumor progression (Qin et al., 2014). In addition, it has been proved that the self-renewal ability of the leukemia initiating cell-enriched subpopulation is linked to the ability of β-arr1 to promote the activity of the DNA methyltransferase 1 on PTEN promoter region, thus reducing the expression of PTEN (Shu et al., 2015). At the same time, nuclear β-arr1 represses the senescence of leukemic cells by interaction with hTERT, thus enhancing telomerase activity and telomere length (Liu et al., 2017), providing novel insights into the β-arr1-mediated regulation of leukemic cells. In Sonic Hedgehog medulloblastoma, where aberrant Sonic Hedgehog/Gli (Hh/Gli) signaling pathway is a critical regulator of tumor initiation and progression, β-arr1 promotes p300-mediated acetylation of Gli1 inhibiting its function, acting as negative regulators of self-renewal (Miele et al., 2017). All these findings, and in particular the specific contributions of β-arr1 for acetylation/methylation mechanisms or interactions with transcriptional factors or regulators, establish a new paradigm in multimodality of β-arr1 in controlling gene expression in cancer. However, their integration will have to be complemented with other studies in specific tumors and cell types, occurring during tumor development and metastasis. Future research will need to address whether similar mechanisms might occur for other GPCRs and open new ways to understand new nuclear interactions of β-arr1 in cancer and to obtain the effective knowledge of how β-arr1 is complicit in the epigenetic control of cancer progression.
Role of β-arr2 in Cancer Progression
Although β-arr1 and β-arr2 show high degree of sequence and structural similarity and functional overlap (Srivastava et al., 2015), emerging evidences establish an involvement of β-arr2 in cancer growth and progression, with contradictory results. Previous studies demonstrated that β-arr2 depletion promoted tumor growth and angiogenesis in a murine model of lung cancer (Raghuwanshi et al., 2008) and that low expression of β-arr2 is significantly associated with aggressive pathologic features and is predictive of poor patient prognosis, as observed in lung and hepatocellular carcinoma (Sun et al., 2016; Cong et al., 2017). In prostate cancer, β-arr2 inhibits cell viability and proliferation by downregulation of FOXO1 and represses AR signaling, and AR expression/activity negatively correlates with β-arr2 expression (Lakshmikanthan et al., 2009; Duan et al., 2015). By contrast, other results are consistent with the idea that β-arr2 action provides a supportive role in the development of human tumors, and β-arr2 is overexpressed in different human tumors, including breast and renal cell carcinoma, correlating with advanced stage and decreased patient survival, and mediates different tumor-promoting effects, such as cell migration and invasion (Sun et al., 2002; Ge et al., 2004; Alemayehu et al., 2013; Masannat et al., 2018). In both myeloid leukemia and ovarian cancer cells, the cross-signaling between β-arr2 and Wnt controls cell proliferation and metastasis through the interaction with c-Src followed by EGFR transactivation (Luttrell et al., 1999; Rosanò et al., 2009). The interaction of β-arr2 with c-Src is also implicated in regulating cell cycle progression and metastatic tumor growth in mice and further expanding the role of β-arr2 (Zhang et al., 2011). Very recently, a new role of β-arr2 has been linked to Hh signaling and medulloblastoma tumorigenesis, controlling SuFu-Gli3 complex, as a major control node in Hh signaling. In particular, it has been demonstrated that in the absence of Hh signaling the interaction of β-arr2 with the E3 ligase Itch and Suppressor of Fused (SuFu), a tumour suppressor gene, promotes the processing of Gli3 transcription factor into a cleaved repressor form, GLI3R, unveiling a new role of β-arr2 in controlling the immunosuppressive function of SuFu and maintaining the signaling off in the absence of ligand (Infante et al., 2018). These results further point out that function specialization of β-arr isoforms might exist in cancer, implying that different roles of β-arr2 function may be cell context- and cancer type-dependent, and that many other studies are needed to fully understand the mechanisms underlying the role of β-arr2 in cancer.
Future Exploration of Targeting Convergent GPCR/β-arr-Dependent Mechanisms in Cancer
The role of β-arr-mediated network in executing the functional consequences of GPCR signaling in tumor cells is providing new insight into the mechanisms that underlie cell migration and the acquisition of invasive traits. Besides the complexity of GRK-mediated signaling that could allow compensatory networks to strengthen cancer progression suggesting the feasibility of therapeutic strategies using GRK inhibitors (Nogués et al., 2017, 2018), the central theme that has emerged from this review is the critical role of GPCR/β-arr-driven signaling in rewiring the complex signaling network sustaining cancer progression. This indicates that targeting GPCR/β-arr pathways might represent a new route of therapeutic intervention by developing precision medicines with tailored efficacy profiles for cancer-specific context. The existence of biased ligands that can transduce intracellular signaling from a GPCR by favoring either the G-protein or the β-arr-mediated signaling pathways strongly leads to the development of signal-biased drugs (Smith et al., 2018; Xiao and Sun, 2018). In this context, findings obtained by specifically targeting ET-1R/β-arr1-driven pathways by using small molecule ET-1R antagonist demonstrated a potential therapeutic approach for controlling metastatic progression (Rosanò et al., 2014; Cianfrocca et al., 2016, 2017; Semprucci et al., 2016; Chellini et al., 2018; Di Modugno et al., 2018). In addition to directly targeting GPCRs, emerging data suggest the possibility to selective disruption of β-arr interactions by inhibiting the linked functional responses, as an RNA aptamer targeting βarr2 and a synthetic intrabody fragment recognizing βarr, or by specifically interfering with GPCR/β-arr interaction, revealing new ways of therapeutically targeting β-arr-driven convergent signals in patients to hamper metastatic dissemination (Chaturvedi et al., 2018).
Conclusions
Recent technological advancements in structural and cell biology have provided crucial insights into the molecular mechanisms of GPCR signaling mediated by both β-arrs and G-proteins, shedding additional light on the dynamic assembly and disassembly of GPCR signaling complexes (Alvarez-Curto et al., 2016; O’Hayre et al., 2017; Eichel et al., 2018; Grundmann et al., 2018; Gurevich and Gurevich, 2018; Gutkind and Kostenis, 2018; Latorraca et al., 2018; Luttrell et al., 2018). In these new advancements, studies limited to ERK signaling by using genome editing to modulate G protein or β-arr expression and/or function suggest that β-arrs, rather than being active GPCR transducers, are critical initiator of G-protein-mediating signaling cascade, acting as “rheostat” (Gutkind and Kostenis, 2018), uncovering new role of β-arr as dictating factors of G-protein-dependent signaling activation.
Although aspects of the GPCR/β-arr signaling network had been established previously, the novelty of the recent studies highlighted in this review is the ability of β-arr to orchestrate a complex signaling network that specifically controls in a fine-tuning manner, the time, intensity, and space of GPCR-mediated signaling flow to regulate distinct steps of tumor invasion, extravasation, and metastatic spread. Looking forward, a better understanding of how the different types of GPCRs contribute to β-arr-driven signaling activation in tumor cells and by tumor microenvironment and their interaction with other surface receptors is needed. These studies should consider the role of mechanical forces imposed by the ECM and tissue microenvironment in GPCR/β-arr-mediated actin cytoskeleton remodeling and should attempt to delineate the specific contributions of different effectors (such as Rho GTPase family members) to promote cytoskeleton effects related to cell motility and invasiveness in the context of GPCR/β-arr signaling (Figure 1). However, remains to be learned both about how β-arr-1 mediates gene expression changes to execute the GPCR-induced pro-metastatic effects in tumor cells and about how β-arr-1/-2 and G-protein-mediated effects may differ in this regard. The possibility that cytoplasmic and nuclear β-arrs are regulated by several cues and contribute to unexplored relevant aspects of tumor progression should also be considered. The impact of this work is likely to be substantial, given the intense interest in targeting GPCR/β-arr signaling as a therapeutic approach to inhibiting metastatic progression in cancer patients and highlighting the need for translation of preclinical insights into clinical applications. Considering the role of GPCR/β-arr-driven signaling in cancer progression and many efforts in using GPCR antagonists to dampen specifically β-arr-dependent signaling, other studies are needed not only to rewire the complexities of β-arr signaling networks and the functional effects in cancer but also to strongly improve therapeutic targeting of GPCR in cancer. In this regard, additional studies using adequate patient-derived models to further analyze the blockade of potential β-arr-dependent signaling machinery are needed to instruct treatment options in the clinic.
Author Contributions
Both authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by Associazione Italiana Ricerca sul Cancro (AIRC) to AB (AIRC IG 18382) and LR (AIRC IG 21372).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully acknowledge all members of the laboratory for their constant support and enthusiasm, and Maria Vincenza Sarcone for secretarial assistance.
References
Alemayehu, M., Dragan, M., Pape, C., Siddiqui, I., Sacks, D. B., Di Guglielmo, G. M., et al. (2013). β-arrestin2 regulates lysophosphatidic acid-induced human breast tumor cell migration and invasion via Rap1 and IQGAP1. PLoS One 8:e56174. doi: 10.1371/journal.pone.0056174
Alvarez, C. J., Lodeiro, M., Theodoropoulou, M., Camiña, J. P., Casanueva, F. F., and Pazos, Y. (2009). Obestatin stimulates Akt signalling in gastric cancer cells through beta-arrestin-mediated epidermal growth factor receptor transactivation. Endocr. Relat. Cancer 16, 599–611. doi: 10.1677/ERC-08-0192
Alvarez-Curto, E., Inoue, A., Jenkins, L., Raihan, S. Z., Prihandoko, R., Tobin, A. B., et al. (2016). Targeted elimination of G proteins and arrestins defines their specific contributions to both intensity and duration of G protein-coupled receptor signalling. J. Biol. Chem. 291, 27147–27159. doi: 10.1074/jbc.M116.754887
Bagnato, A., and Rosanò, L. (2016). Endothelin-1 receptor drives invadopodia: exploiting how β-arrestin-1 guides the way. Small GTPases 3, 1–5. doi: 10.1080/21541248.2016.1235526
Barnes, W. G., Reiter, E., Violin, J. D., Ren, X. R., Milligan, G., and Lefkowitz, R. J. (2005). Beta-arrestin 1 and galphaq/11 coordinately activate RhoA and stress fiber formation following receptor stimulation. J. Biol. Chem. 280, 8041–8050. doi: 10.1074/jbc.M412924200
Bhattacharya, M., Anborgh, P. H., Babwah, A. V., Dale, L. B., Dobransky, T., Benovic, J. L., et al. (2002). Beta-arrestins regulate a Ral-GDS Ral effector pathway that mediates cytoskeletal reorganization. Nat. Cell Biol. 4, 547–555. doi: 10.1038/ncb821
Black, J. B., Premont, R. T., and Daaka, Y. (2016). Feedback regulation of G protein-coupled receptor signalling by GRKs and arrestins. Semin. Cell Dev. Biol. 50, 95–104. doi: 10.1016/j.semcdb.2015.12.015
Bonnans, C., Flacelière, M., Grillet, F., Dantec, C., Desvignes, J. P., Pannequin, J., et al. (2012). Essential requirement for β-arrestin2 in mouse intestinal tumors with elevated Wnt signalling. Proc. Natl. Acad. Sci. U. S. A. 109, 3047–3052. doi: 10.1073/pnas.1109457109
Bryja, V., Gradl, D., Schambony, A., Arenas, E., Schulte, G., Fereshteh, M., et al. (2007). Beta-arrestin is a necessary component of Wnt/beta-catenin signalling in vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A. 104, 6690–6695. doi: 10.1073/pnas.0611356104
Buchanan, F. G., Gorden, D. L., Matta, P., Shi, Q., Matrisian, L. M., and DuBois, R. N. (2006). Role of beta-arrestin 1 in the metastatic progression of colorectal cancer. Proc. Natl. Acad. Sci. U. S. A. 103, 1492–1497. doi: 10.1073/pnas.0510562103
Chaturvedi, M., Schilling, J., Beautrait, A., Bouvier, M., Benovic, J. L., and Shukla, A. K. (2018). Emerging paradigm of intracellular targeting of G protein-coupled receptors. Trends Biochem. Sci. 43, 533–546. doi: 10.1016/j.tibs.2018.04.003
Chellini, L., Caprara, V., Spadaro, F., Sestito, R., Bagnato, A., and Rosanò, L. (2018). Regulation of extracellular matrix degradation and metastatic spread by IQGAP1 through endothelin-1 receptor signalling in ovarian cancer. Matrix Biol. S0945-053X(18)30303-2. doi: 10.1016/j.matbio.2018.10.005
Chen, W., Hu, L. A., Semenov, M. V., Yanagawa, S., Kikuchi, A., Lefkowitz, R. J., et al. (2001). beta-arrestin1 modulates lymphoid enhancer factor transcriptional activity through interaction with phosphorylated dishevelled proteins. Proc. Natl. Acad. Sci. U. S. A. 98, 14889–14894. doi: 10.1073/pnas.211572798
Cianfrocca, R., Rosanò, L., Spinella, F., Di Castro, V., Natali, P. G., and Bagnato, A. (2010). Beta-arrestin-1 mediates the endothelin-1-induced activation of Akt and integrin-linked kinase. Can. J. Physiol. Pharmacol. 88, 796–801. doi: 10.1139/Y10-052
Cianfrocca, R., Tocci, P., Semprucci, E., Spinella, F., Di Castro, V., Bagnato, A., et al. (2014). β-arrestin 1 is required for endothelin-1-induced NF-κB activation in ovarian cancer cells. Life Sci. 118, 179–184. doi: 10.1016/j.lfs.2014.01.078
Cianfrocca, R., Tocci, P., Rosanò, L., Caprara, V., Sestito, R., Di Castro, V., et al. (2016). Nuclear β-arrestin1 is a critical cofactor of hypoxia-inducible factor-1α signalling in endothelin-1-induced ovarian tumor progression. Oncotarget 7, 17790–17804. doi: 10.18632/oncotarget.746
Cianfrocca, R., Rosanò, L., Tocci, P., Sestito, R., Caprara, V., Di Castro, V., et al. (2017). Blocking endothelin-1-receptor/β-catenin circuit sensitizes to chemotherapy in colorectal cancer. Cell Death Differ. 24, 1811–1820. doi: 10.1038/cdd.2017.121
Cong, L., Qiu, Z., Zhao, Y., Wang, W. B., Wang, C. X., Shen, H. C., et al. (2017). Loss of β-arrestin-2 and activation of CXCR2 correlate with lymph node metastasis in non-small cell lung cancer. J. Cancer 8, 2785–2792. doi: 10.7150/jca.19631
Crudden, C., Shibano, T., Song, D., Suleymanova, N., Girnita, A., and Girnita, L. (2018). Blurring boundaries: receptor tyrosine kinases as functional G protein-coupled receptors. Int. Rev. Cell Mol. Biol. 339, 1–40. doi: 10.1016/bs.ircmb.2018.02.006
Dasgupta, P., Rizwani, W., Pillai, S., Davis, R., Banerjee, S., Hug, K., et al. (2011). ARRB1-mediated regulation of E2F target genes in nicotine-induced growth of lung tumors. J. Natl. Cancer Inst. 103, 317–333. doi: 10.1093/jnci/djq541
Décaillot, F. M., Kazmi, M. A., Lin, Y., Ray-Saha, S., Sakmar, T. P., and Sachdev, P. (2011). CXCR7/CXCR4 heterodimer constitutively recruits beta-arrestin to enhance cell migration. J. Biol. Chem. 286, 32188–32197. doi: 10.1074/jbc.M111.277038
DeFea, K. (2008). β-arrestins and heterotrimeric G-proteins: collaborators and competitors in signal transduction. Br. J. Pharmacol. 153, 298–309. doi: 10.1038/sj.bjp.0707508
Di Modugno, F., Iapicca, P., Boudreau, A., Mottolese, M., Terrenato, I., Perracchio, L., et al. (2012). Splicing program of human MENA produces a previously undescribed isoform associated with invasive, mesenchymal-like breast tumors. Proc. Natl. Acad. Sci. U. S. A. 109, 19280–19285. doi: 10.1073/pnas.1214394109
Di Modugno, F., Caprara, V., Chellini, L., Tocci, P., Spadaro, F., Ferrandina, G., et al. (2018). hMENA is a key regulator in endothelin-1/β-arrestin1-induced invadopodial function and metastatic process. Proc. Natl. Acad. Sci. U. S. A. 115, 3132–3137. doi: 10.1073/pnas.1715998115
Duan, X., Kong, Z., Liu, Y., Zeng, Z., Li, S., Wu, W., et al. (2015). β-arrestin2 contributes to cell viability and proliferation via the down-regulation of FOXO1 in castration-resistant prostate cancer. J. Cell. Physiol. 230, 2371–2381. doi: 10.1002/jcp.24963
Duan, X., Zhang, T., Kong, Z., Mai, X., Lan, C., Chen, D., et al. (2016). β-arrestin1 promotes epithelial-mesenchymal transition via modulating GSK-3β/β-catenin pathway in prostate cancer cells. Biochem. Biophys. Res. Commun. 479, 204–210. doi: 10.1016/j.bbrc.2016.09.039
Eddy, R. J., Weidmann, M. D., Sharma, V. P., and Condeelis, J. S. (2017). Tumor cell invadopodia: invasive protrusions that orchestrate metastasis. Trends Cell Biol. 27, 595–607. doi: 10.1016/j.tcb.2017.03.003
Eichel, K., Jullié, D., Barsi-Rhyne, B., Latorraca, N. R., Masureel, M., Sibarita, J. B., et al. (2018). Catalytic activation of β-arrestin by GPCRs. Nature 557, 381–386. doi: 10.1038/s41586-018-0079-1
Eichel, K., and von Zastrow, M. (2018). Subcellular organization of GPCR signalling. Trends Pharmacol. Sci. 39, 200–208. doi: 10.1016/j.tips.2017.11.009
Fereshteh, M., Ito, T., Kovacs, J. J., Zhao, C., Kwon, H. Y., Tornini, V., et al. (2012). β-arrestin2 mediates the initiation and progression of myeloid leukemia. Proc. Natl. Acad. Sci. U. S. A. 109, 12532–12537. doi: 10.1073/pnas.1209815109
Ferrandino, F., Bernardini, G., Tsaouli, G., Grazioli, P., Campese, A. F., Noce, C., et al. (2018). Intrathymic Notch3 and CXCR4 combinatorial interplay facilitates T-cell leukemia propagation. Oncogene 37, 6285–6298. doi: 10.1038/s41388-018-0401-2
Friedl, P., and Alexander, S. (2011). Cancer invasion and the microenvironment: plasticity and reciprocity. Cell 147, 992–1009. doi: 10.1016/j.cell.2011.11.016
Ge, L., Shenoy, S. K., Lefkowitz, R. J., and DeFea, K. (2004). Constitutive protease-activated receptor-2-mediated migration of MDA MB-231 breast cancer cells requires both beta-arrestin-1 and -2. J. Biol. Chem. 279, 55419–55424. doi: 10.1074/jbc.M410312200
Gertler, F., and Condeelis, J. (2011). Metastasis: tumor cells becoming MENAcing. Trends Cell Biol. 21, 81–90. doi: 10.1016/j.tcb.2010.10.001
Girnita, L., Shenoy, S. K., Sehat, B., Vasilcanu, R., Girnita, A., Lefkowitz, R. J., et al. (2005). {beta}-Arrestin is crucial for ubiquitination and down-regulation of the insulin-like growth factor-1 receptor by acting as adaptor for the MDM2 E3 ligase. J. Biol. Chem. 280, 24412–24419.
Girnita, L., Shenoy, S. K., Sehat, B., Vasilcanu, R., Vasilcanu, D., Girnita, A., et al. (2007). Beta-arrestin and Mdm2 mediate IGF-1 receptor-stimulated ERK activation and cell cycle progression. J. Biol. Chem. 282, 11329–11338.
Godin, C. M., Ferreira, L. T., Dale, L. B., Gros, R., Cregan, S. P., and Ferguson, S. S. (2010). The small GTPase Ral couples the angiotensin II type 1 receptor to the activation of phospholipase C-delta 1. Mol. Pharmacol. 77, 388–395. doi: 10.1124/mol.109.061069
Goertzen, C. G., Dragan, M., Turley, E., Babwah, A. V., and Bhattacharya, M. (2016). KISS1R signalling promotes invadopodia formation in human breast cancer cell via β-arrestin2/ERK. Cell. Signal. 28, 165–176. doi: 10.1016/j.cellsig.2015.12.010
Grundmann, M., Merten, N., Malfacini, D., Inoue, A., Preis, P., Simon, K., et al. (2018). Lack of beta-arrestin signalling in the absence of active G proteins. Nat. Commun. 9:341. doi: 10.1038/s41467-017-02661-3
Gurevich, E. V., Tesmer, J. J., Mushegian, A., and Gurevich, V. V. (2012). G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol. Ther. 133, 40–69. doi: 10.1016/j.pharmthera.2011.08.001
Gurevich, V. V., and Gurevich, E. V. (2018). Arrestins and G proteins in cellular signalling: the coin has two sides. Sci. Signal. 11:eaav1646. doi: 10.1126/scisignal.aav1646
Gutkind, J. S., and Kostenis, E. (2018). Arrestins as rheostats of GPCR signalling. Nat. Rev. Mol. Cell Biol. 19, 615–616. doi: 10.1038/s41580-018-0041-y
Hauser, A. S., Attwood, M. M., Rask-Andersen, M., Schiöth, H. B., and Gloriam, D. E. (2017). Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov. 16, 829–842. doi: 10.1038/nrd.2017.178
Heinrich, E. L., Lee, W., Lu, J., Lowy, A. M., and Kim, J. (2012). Chemokine CXCL12 activates dual CXCR4 and CXCR7-mediated signalling pathways in pancreatic cancer cells. J. Transl. Med. 10:68. doi: 10.1186/1479-5876-10-68
Heitzler, D., Durand, G., Gallay, N., Rizk, A., Ahn, S., Kim, J., et al. (2012). Competing G protein-coupled receptor kinases balance G protein and β-arrestin signalling. Mol. Syst. Biol. 8:590. doi: 10.1038/msb.2012.22
Hoeppner, C. Z., Cheng, N., and Ye, R. D. (2012). Identification of and its functional implications. J. Biol. Chem. 287, 8932–8943. doi: 10.1074/jbc.M111.294058
Hunton, D. L., Barnes, W. G., Kim, J., Ren, X. R., Violin, J. D., Reiter, E., et al. (2005). Beta-arrestin 2-dependent angiotensin II type 1A receptor-mediated pathway of chemotaxis. Mol. Pharmacol. 67, 1229–1236. doi: 10.1124/mol.104.006270
Infante, P., Faedda, R., Bernardi, F., Bufalieri, F., Lospinoso Severini, L., Alfonsi, R., et al. (2018). Itch/β-arrestin2-dependent non-proteolytic ubiquitylation of SuFu controls Hedgehog signalling and medulloblastoma tumorigenesis. Nat. Commun. 9, 976. doi: 10.1038/s41467-018-03339-0
Insel, P. A., Sriram, K., Wiley, S. Z., Wilderman, A., Katakia, T., McCann, T., et al. (2018). GPCRomics: GPCR expression in cancer cells and tumors identifies new, potential biomarkers and therapeutic targets. Front. Pharmacol. 9:431. doi: 10.3389/fphar.2018.00431
Jagan, I. C., Deevi, R. K., Fatehullah, A., Topley, R., Eves, J., Stevenson, M., et al. (2013). PTEN phosphatase-independent maintenance of glandular morphology in a predictive colorectal cancer model system. Neoplasia 15, 1218–1230. doi: 10.1593/neo.121516
Javadi, A., Deevi, R. K., Evergren, E., Blondel-Tepaz, E., Baillie, G. S., Scott, M. G., et al. (2017). PTEN controls glandular morphogenesis through a juxtamembrane β-Arrestin1/ARHGAP21 scaffolding complex. elife 6:e24578. doi: 10.7554/eLife.24578
Jean-Charles, P. Y., Freedman, N. J., and Shenoy, S. K. (2016). Cellular roles of beta-arrestins as substrates and adaptors of ubiquitination and deubiquitination. Prog. Mol. Biol. Transl. Sci. 141, 339–369. doi: 10.1016/bs.pmbts.2016.04.003
Jin, G., Westphalen, C. B., Hayakawa, Y., Worthley, D. L., Asfaha, S., Yang, X., et al. (2013). Progastrin stimulates colonic cell proliferation via CCK2R- and β-arrestin-dependent suppression of BMP2. Gastroenterology 145, 820–830. doi: 10.1053/j.gastro.2013.07.034
Jing, X., Zhang, H., Hu, J., Su, P., Zhang, W., Jia, M., et al. (2015). β-arrestin is associated with multidrug resistance in breast cancer cells through regulating MDR1 gene expression. Int. J. Clin. Exp. Pathol. 8, 1354–1363.
Kang, J., Shi, Y., Xiang, B., Qu, B., Su, W., Zhu, M., et al. (2005). A nuclear function of beta-arrestin1 in GPCR signalling: regulation of histone acetylation and gene transcription. Cell 123, 833–847. doi: 10.1016/j.cell.2005.09.011
Kong, Z., Deng, T., Zhang, M., Zhao, Z., Liu, Y., Luo, L., et al. (2018). β-arrestin1-medieated inhibition of FOXO3a contributes to prostate cancer cell growth in vitro and in vivo. Cancer Sci. 109, 1834–1842. doi: 10.1016/j.cellsig.2015.01.001
Kotula, J. W., Sun, J., Li, M., Pratico, E. D., Fereshteh, M. P., Ahrens, D. P., et al. (2014). Targeted disruption of β-arrestin 2-mediated signalling pathways by aptamer chimeras leads to inhibition of leukemic cell growth. PLoS One 9:e93441. doi: 10.1371/journal.pone.0093441
Krause, M., Dent, E. W., Bear, J. E., Loureiro, J. J., and Gertler, F. B. (2003). Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu. Rev. Cell Dev. Biol. 19, 541–564. doi: 10.1146/annurev.cellbio.19.050103.103356
Lagerstrom, M. C., and Schioth, H. B. (2008). Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug Discov. 7, 339–357. doi: 10.1038/nrd2518
Lakshmikanthan, V., Zou, L., Kim, J. I., Michal, A., Nie, Z., Messias, N. C., et al. (2009). Identification of betaArrestin2 as a corepressor of androgen receptor signalling in prostate cancer. Proc. Natl. Acad. Sci. U. S. A. 106, 9379–9384. doi: 10.1073/pnas.0900258106
Lambert, A. W., Pattabiraman, D. R., and Weinberg, R. A. (2017). Emerging biological principles of metastasis. Cell 168, 670–691. doi: 10.1016/j.cell.2016.11.037
Latorraca, N. R., Wang, J. K., Bauer, B., Townshend, R. J. L., Hollingsworth, S. A., Olivieri, J. E., et al. (2018). Molecular mechanism of GPCR-mediated arrestin activation. Nature 557, 452–456. doi: 10.1038/s41586-018-0077-3
Li, T. T., Alemayehu, M., Aziziyeh, A. I., Pape, C., Pampillo, M., Postovit, L. M., et al. (2009). Beta-arrestin/Ral signalling regulates lysophosphatidic acid-mediated migration and invasion of human breast tumor cells. Mol. Cancer Res. 7, 1064–1077. doi: 10.1158/1541-7786.MCR-08-0578
Liu, S., Liu, H., Qin, R., Shu, Y., Liu, Z., Zhang, P., et al. (2017). The cellular senescence of leukemia-initiating cells from acute lymphoblastic leukemia is postponed by β-Arrestin1 binding with P300-Sp1 to regulate hTERT transcription. Cell Death Dis. 8:e2756. doi: 10.1038/cddis.2017
Luttrell, L. M., Ferguson, S. S., Daaka, Y., Miller, W. E., Maudsley, S., Della Rocca, G. J., et al. (1999). Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science 283, 655–661. doi: 10.1126/science.283.5402.655
Luttrell, L. M., and Lefkowitz, R. J. (2002). The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 115, 455–465.
Luttrell, L. M., Wang, J., Plouffe, B., Smith, J. S., Yamani, L., Kaur, S., et al. (2018). Manifold roles of β-arrestins in GPCR signalling elucidated with siRNA and CRISPR/Cas9. Sci. Signal. 11:eaat7650. doi: 10.1126/scisignal.aat7650
Ma, L., and Pei, G. (2007). Beta-arrestin signalling and regulation of transcription. J. Cell Sci. 120, 213–218. doi: 10.1242/jcs.03338
Ma, X., Espana-Serrano, L., Kim, W., Thayele Purayil, H., Nie, Z., and Daaka, Y. (2014). βArrestin1 regulates the guanine nucleotide exchange factor RasGRF2 expression and the small GTPase Rac-mediated formation of membrane protrusion and cell motility. J. Biol. Chem. 289, 13638–13650. doi: 10.1074/jbc.M113.511360
Ma, X., Zhao, Y., Daaka, Y., and Nie, Z. (2012). Acute activation of β2-adrenergic receptor regulates focal adhesions through βArrestin2- and p115RhoGEF protein-mediated activation of RhoA. J. Biol. Chem. 287, 18925–18936. doi: 10.1074/jbc.M112.352260
Marshall, F. H. (2016). Visualizing GPCR ‘megaplexes’ which enable sustained intracellular signalling. Trends Biochem. Sci. 41, 985–986. doi: 10.1016/j.tibs.2016.10.006
Masannat, J., Purayil, H. T., Zhang, Y., Russin, M., Mahmud, I., Kim, W., et al. (2018). βArrestin2 mediates renal cell carcinoma tumor growth. Sci. Rep. 8, 4879. doi: 10.1038/s41598-018-23212-w
McGovern, K. W., and DeFea, K. A. (2014). Molecular mechanisms underlying beta-arrestin-dependent chemotaxis and actin-cytoskeletal reorganization. Handb. Exp. Pharmacol. 219, 341–359. doi: 10.1007/978-3-642-41199-1_17
Miele, E., Po, A., Begalli, F., Antonucci, L., Mastronuzzi, A., Marras, C. E., et al. (2017). β-arrestin1-mediated acetylation of Gli1 regulates Hedgehog/Gli signalling and modulates self-renewal of SHH medulloblastoma cancer stem cells. BMC Cancer 17:488. doi: 10.1186/s12885-017-3477-0
Miller, W. E., and Lefkowitz, R. J. (2001). Expanding roles for beta-arrestins as scaffolds and adapters in GPCR signalling and trafficking. Curr. Opin. Cell Biol. 13, 139–145. doi: 10.1016/S0955-0674(00)00190-3
Min, J., and Defea, K. (2011). β-arrestin-dependent actin reorganization: bringing the right players together at the leading edge. Mol. Pharmacol. 80, 760–768. doi: 10.1124/mol.111.072470
Mrkonjic, S., Destaing, O., and Albiges-Rizo, C. (2017). Mechanotransduction pulls the strings of matrix degradation at invadosome. Matrix Biol. 57–58, 190–203. doi: 10.1016/j.matbio.2016.06.007
Nogués, L., Reglero, C., Rivas, V., Neves, M., Penela, P., and Mayor, F. Jr. (2017). G-protein-coupled receptor kinase 2 as a potential modulator of the hallmarks of cancer. Mol. Pharmacol. 91, 220–228. doi: 10.1124/mol.116.107185
Nogués, L., Palacios-García, J., Reglero, C., Rivas, V., Neves, M., Ribas, C., et al. (2018). G protein-coupled receptor kinases (GRKs) in tumorigenesis and cancer progression: GPCR regulators and signalling hubs. Semin. Cancer Biol. 48, 78–90. doi: 10.1016/j.semcancer.2017.04.013
O’Hayre, M., Eichel, K., Avino, S., Zhao, X., Steffen, D. J., Feng, X., et al. (2017). Genetic evidence that β-arrestins are dispensable for the initiation of β2-adrenergic receptor signalling to ERK. Sci. Signal. 10:eaal3395. doi: 10.1126/scisignal.aal3395
Parisis, N., Metodieva, G., and Metodiev, M. V. (2013). Pseudopodial and β-arrestin-interacting proteomes from migrating breast cancer cells upon PAR2 activation. J. Proteome 80, 91–106. doi: 10.1016/j.jprot.2012.12.024
Paterson, E. K., and Courtneidge, S. A. (2018). Invadosomes are coming: new insights into function and disease relevance. FEBS J. 285, 8–27. doi: 10.1111/febs.14123
Perumal, D., Pillai, S., Nguyen, J., Schaal, C., Coppola, D., and Chellappan, S. P. (2014). Nicotinic acetylcholine receptors induce c-Kit ligand/stem cell factor and promote stemness in an ARRB1/β-arrestin-1 dependent manner in NSCLC. Oncotarget 5, 10486–10502. doi: 10.18632/oncotarget.2395
Peterson, Y. K., and Luttrell, L. M. (2017). The diverse roles of arrestin scaffolds in G protein-coupled receptor signalling. Pharmacol. Rev. 69, 256–297. doi: 10.1124/pr.116.013367
Pillai, S., Trevino, J., Rawal, B., Singh, S., Kovacs, M., Li, X., et al. (2015). β-arrestin-1 mediates nicotine-induced metastasis throughE2F1 target genes that modulate epithelial-mesenchymal transition. Cancer Res. 75, 1009–1020. doi: 10.1158/0008-5472.CAN-14-0681
Pyne, N. J., and Pyne, S. (2017). Sphingosine 1-phosphate receptor 1 signalling in mammalian cells. Molecules 22, 344. doi: 10.3390/molecules22030344
Purayil, H. T., Zhang, Y., Dey, A., Gersey, Z., Espana-Serrano, L., and Daaka, Y. (2015). Arrestin 2 modulates androgen receptor activation. Oncogene 34, 3144–3151. doi: 10.1038/onc.2014.252
Qin, R., Li, K., Qi, X., Zhou, X., Wang, L., Zhang, P., et al. (2014). β-arrestin1 promotes the progression of chronic myeloid leukaemia byregulating BCR/ABL H4 acetylation. Br. J. Cancer 111, 568–576. doi: 10.1038/bjc.2014.335
Raghuwanshi, S. K., Nasser, M. W., Chen, X., Strieter, R. M., and Richardson, R. M. (2008). Depletion of beta-arrestin-2 promotes tumor growth and angiogenesis in a murine model of lung cancer. J. Immunol. 180, 5699–5706. doi: 10.4049/jimmunol.180.8.5699
Ren, W., Wang, T., He, X., Zhang, Q., Zhou, J., Liu, F., et al. (2018). β-arrestin2 promotes 5FU-induced apoptosis via the NF-κB pathway in colorectal cancer. Oncol. Rep. 39, 2711–2720. doi: 10.3892/or.2018.6340
Rosanò, L., and Bagnato, A. (2016). β-arrestin1 at the cross-road of endothelin-1 signalling in cancer. J. Exp. Clin. Cancer Res. 35:121. doi: 10.1186/s13046-016-0401-4
Rosanò, L., Cianfrocca, R., Masi, S., Spinella, F., Di Castro, V., Biroccio, A., et al. (2009). Beta-arrestin links endothelin A receptor to beta-catenin signalling to induce ovarian cancer cell invasion and metastasis. Proc. Natl. Acad. Sci. U. S. A. 106, 2806–2811. doi: 10.1073/pnas.0807158106
Rosanò, L., Cianfrocca, R., Tocci, P., Spinella, F., Di Castro, V., Spadaro, F., et al. (2012). β-arrestin-1 is a nuclear transcriptional regulator of endothelin-1-induced β-catenin signalling. Oncogene 32, 5066–5077. doi: 10.1038/onc.2012.527
Rosanò, L., Cianfrocca, R., Tocci, P., Spinella, F., Di Castro, V., Caprara, V., et al. (2014). Endothelin A receptor/β-arrestin signalling to the Wnt pathway renders ovarian cancer cells resistant to chemotherapy. Cancer Res. 74, 7453–7464. doi: 10.1158/0008-5472.CAN-13-3133
Rosanò, L., Spinella, F., and Bagnato, A. (2013). Endothelin 1 in cancer: biological implications and therapeutic opportunities. Nat. Rev. Cancer 13, 637–651. doi: 10.1038/nrc3546
Rosenbaum, D. M., Rasmussen, S. G., and Kobilka, B. K. (2009). The structure and function of G-protein-coupled receptors. Nature 459, 356–363. doi: 10.1038/nature08144
Scott, M. G., Pierotti, V., Storez, H., Lindberg, E., Thuret, A., Muntaner, O., et al. (2006). Cooperative regulation of extracellular signal-regulated kinase activation and cell shape change by filamin A and beta-arrestins. Mol. Cell. Biol. 26, 3432–3445. doi: 10.1128/MCB.26.9.3432-3445.2006
Semprucci, E., Tocci, P., Cianfrocca, R., Sestito, R., Caprara, V., Veglione, M., et al. (2016). Endothelin A receptor drives invadopodia function and cell motility through the β-arrestin/PDZ-RhoGEF pathway in ovarian carcinoma. Oncogene 35, 3432–3442. doi: 10.1038/onc.2015.403
Sente, A., Peer, R., Srivastava, A., Baidya, M., Lesk, A. M., Balaji, S. T., et al. (2018). Molecular mechanism of modulating arrestin conformation by GPCR phosphorylation. Nat. Struct. Mol. Biol. 25, 538–545. doi: 10.1038/s41594-018-0071-3
Shenoy, S. K., Han, S., Zhao, Y. L., Hara, M. R., Oliver, T., Cao, Y., et al. (2012). β-arrestin1 mediates metastatic growth of breast cancer cells by facilitating HIF-1-dependent VEGF expression. Oncogene 31, 282–292. doi: 10.1038/onc.2011.238
Shi, Y., Feng, Y., Kang, J., Liu, C., Li, Z., Li, D., et al. (2007). Critical regulation of CD4+ T cell survival and autoimmunity by beta-arrestin 1. Nat. Immunol. 8, 817–824. doi: 10.1038/ni1489
Shishkin, S., Eremina, L., Pashintseva, N., Kovalev, L., and Kovaleva, M. (2016). Cofilin-1 and other ADF/Cofilin superfamily members in human malignant xells. Int. J. Mol. Sci. 18:E10. doi: 10.3390/ijms18010010
Shu, Y., Zhou, X., Qi, X., Liu, S., Li, K., Tan, J., et al. (2015). β-Arrestin1 promotes the self-renewal of the leukemia-initiating cell enriched subpopulation in B-lineage acute lymphoblastic leukemia related to DNMT1 activity. Cancer Lett. 357, 170–178. doi: 10.1016/j.canlet.2014.11.025
Shukla, A. K., Xiao, K., and Lefkowitz, R. J. (2011). Emerging paradigms of β-arrestin-dependent seven transmembrane receptor signalling. Trends Biochem. Sci. 36, 457–469. doi: 10.1016/j.tibs.2011.06.003
Smith, J. S., Lefkowitz, R. J., and Rajagopal, S. (2018). Biased signalling: from simple switches to allosteric microprocessors. Nat. Rev. Drug Discov. 17, 243–260. doi: 10.1038/nrd.2017.229
Sobolesky, P. M., and Moussa, O. (2013). The role of β-arrestins in cancer. Prog. Mol. Biol. Transl. Sci. 118, 395–411. doi: 10.1016/B978-0-12-394440-5.00015-2
Song, Q., Ji, Q., and Li, Q. (2018). The role and mechanism of β-arrestins in cancer invasion and metastasis. Int. J. Mol. Med. 41, 631–639. doi: 10.3892/ijmm.2017.3288
Spinella, F., Caprara, V., Di Castro, V., Rosanò, L., Cianfrocca, R., Natali, P. G., et al. (2013). Endothelin-1 induces the transactivation of vascular endothelial growth factor receptor-3 and modulates cell migration and vasculogenic mimicry in melanoma cells. J. Mol. Med. 91, 395–405. doi: 10.1007/s00109-012-0956-2
Srivastava, A., Gupta, B., Gupta, C., and Shukla, A. K. (2015). Emerging functional divergence of β-arrestin isoforms in GPCR function. Trends Endocrinol. Metab. 26, 628–642. doi: 10.1016/j.tem.2015.09.001
Sun, Y., Cheng, Z., Ma, L., and Pei, G. (2002). Beta-arrestin2 is critically involved in CXCR4-mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation. J. Biol. Chem. 277, 49212–49219. doi: 10.1074/jbc.M207294200
Sun, W. Y., Hu, S. S., Wu, J. J., Huang, Q., Ma, Y., Wang, Q. T., et al. (2016). Down-regulation of β-arrestin2 promotes tumour invasion and indicates poor prognosis of hepatocellular carcinoma. Sci. Rep. 6:35609. doi: 10.1038/srep35609
Thomsen, A. R. B., Plouffe, B., Cahill, T. J., Shukla, A. K., Tarrasch, J. T., Dosey, A. M., et al. (2016). GPCR-G protein-β-arrestin super-complex mediates sustained G protein signalling. Cell 166, 907–919. doi: 10.1016/j.cell.2016.07.004
Tocci, P., Caprara, V., Cianfrocca, R., Sestito, R., Di Castro, V., Bagnato, A., et al. (2016). Endothelin-1/endothelin A receptor axis activates RhoA GTPase in epithelial ovarian cancer. Life Sci. 159, 49–54. doi: 10.1016/j.lfs.2016.01.008
Tsai, C. C., Chou, Y. T., and Fu, H. W. (2019). Protease-activated receptor 2 induces migration and promotes Slug-mediated epithelial-mesenchymal transition in lung adenocarcinoma cells. Biochim. Biophys. Acta, Mol. Cell Res. 1866, 486–503. doi: 10.1016/j.bbamcr.2018.10.011
Wang, W., Han, T., Tong, W., Zhao, J., and Qiu, X. (2018). Overexpression of GPR35 confers drug resistance in NSCLC cells by β-arrestin/Akt signalling. OncoTargets Ther. 11, 6249–6257. doi: 10.2147/OTT.S175606
Xiao, K., McClatchy, D. B., Shukla, A. K., Zhao, Y., Chen, M., Shenoy, S. K., et al. (2007). Functional specialization of β-arrestin interactions revealed by proteomic analysis. Proc. Natl. Acad. Sci. U. S. A. 104, 12011–12016. doi: 10.1073/pnas.0704849104
Xiao, K., and Sun, J. (2018). Elucidating structural and molecular mechanisms of β-arrestin-biased agonism at GPCRs via MS-based proteomics. Cell. Signal. 41, 56–64. doi: 10.1016/j.cellsig.2017.09.013
Yang, Y., Guo, Y., Tan, S., Ke, B., Tao, J., Liu, H., et al. (2015). β-Arrestin1 enhances hepatocellular carcinogenesis through inflammation-mediated Akt signalling. Nat. Commun. 6, 7369. doi: 10.1038/ncomms8369
Zajac, M., Law, J., Cvetkovic, D. D., Pampillo, M., McColl, L., Pape, C., et al. (2011). GPR54 (KISS1R) transactivates EGFR to promote breast cancer cell invasiveness. PLoS One 6:e21599. doi: 10.1371/journal.pone.0021599
Zecchini, V., Madhu, B., Russell, R., Pértega-Gomes, N., Warren, A., Gaude, E., et al. (2014). Nuclear ARRB1 induces pseudohypoxia and cellular metabolism reprogramming in prostate cancer. EMBO J. 33, 1365–1382. doi: 10.15252/embj.201386874
Zhang, P., He, X., Tan, J., Zhou, X., and Zou, L. (2011). β-arrestin2 mediates β-2 adrenergic receptor signalling inducing prostate cancer cell progression. Oncol. Rep. 26, 1471–1477. doi: 10.3892/or.2011.1417
Zhang, Y., Yang, C. Q., Gao, Y., Wang, C., Zhang, C. L., and Zhou, X. H. (2014). Knockdown of CXCR7 inhibits proliferation and invasion of osteosarcoma cells through inhibition of the PI3K/Akt and β-arrestin pathways. Oncol. Rep. 32, 965–972. doi: 10.3892/or.2014.3290
Zhao, M., Zhou, G., Zhang, Y., Chen, T., Sun, X., Stuart, C., et al. (2009). Beta-arrestin2 inhibits opioid-induced breast cancer cell death through Akt and caspase-8 pathways. Neoplasma 56, 108–113. doi: 10.4149/neo_2009_02_108
Zheng, H., Shen, H., Oprea, I., Worrall, C., Stefanescu, R., Girnita, A., et al. (2012). β-Arrestin-biased agonism as the central mechanism of action for insulin-like growth factor 1 receptor-targeting antibodies in Ewing’s sarcoma. Proc. Natl. Acad. Sci. U. S. A. 109, 20620–20625. doi: 10.1073/pnas.1216348110
Zoudilova, M., Kumar, P., Ge, L., Wang, P., Bokoch, G. M., and DeFea, K. A. (2007). Beta-arrestin-dependent regulation of the Cofilin pathway downstream of protease-activated receptor-2. J. Biol. Chem. 282, 20634–20646. doi: 10.1074/jbc.M701391200
Zoudilova, M., Min, J., Richards, H. L., Carter, D., Huang, T., and DeFea, K. A. (2010). Beta-Arrestins scaffold cofilin with chronophin to direct localized actin filament severing and membrane protrusions downstream of protease-activated receptor-2. J. Biol. Chem. 285, 14318–14329. doi: 10.1074/jbc.M109.055806
Keywords: cancer, β-arrestin, G protein-coupled receptors, cytoskeleton, motility
Citation: Bagnato A and Rosanò L (2019) New Routes in GPCR/β-Arrestin-Driven Signaling in Cancer Progression and Metastasis. Front. Pharmacol. 10:114. doi: 10.3389/fphar.2019.00114
Edited by:
Yuichi Hattori, University of Toyama, JapanReviewed by:
Martina Schmidt, University of Groningen, NetherlandsRichard T. Premont, Harrington Discovery Institute, United States
Marco Falasca, Curtin University, Australia
Copyright © 2019 Bagnato and Rosanò. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Bagnato, YW5uYXRlcmVzYS5iYWduYXRvQGlmby5nb3YuaXQ=
Laura Rosanò, bGF1cmEucm9zYW5vQGlmby5nb3YuaXQ=