 Todd A. Johnson
Todd A. Johnson H. A. Jinnah
H. A. Jinnah Naoyuki Kamatani
Naoyuki Kamatani- 1StaGen Co., Ltd., Tokyo, Japan
- 2Departments of Neurology and Human Genetics, Emory University School of Medicine, Atlanta, GA, United States
Germline mutations in cellular-energy associated genes have been shown to lead to various monogenic disorders. Notably, mitochondrial disorders often impact skeletal muscle, brain, liver, heart, and kidneys, which are the body’s top energy-consuming organs. However, energy-related dysfunctions have not been widely seen as causes of common diseases, although evidence points to such a link for certain disorders. During acute energy consumption, like extreme exercise, cells increase the favorability of the adenylate kinase reaction 2-ADP -> ATP+AMP by AMP deaminase degrading AMP to IMP, which further degrades to inosine and then to purines hypoxanthine -> xanthine -> urate. Thus, increased blood urate levels may act as a barometer of extreme energy consumption. AMP deaminase deficient subjects experience some negative effects like decreased muscle power output, but also positive effects such as decreased diabetes and improved prognosis for chronic heart failure patients. That may reflect decreased energy consumption from maintaining the pool of IMP for salvage to AMP and then ATP, since de novo IMP synthesis requires burning seven ATPs. Similarly, beneficial effects have been seen in heart, skeletal muscle, or brain after treatment with allopurinol or febuxostat to inhibit xanthine oxidoreductase, which catalyzes hypoxanthine -> xanthine and xanthine -> urate reactions. Some disorders of those organs may reflect dysfunction in energy-consumption/production, and the observed beneficial effects related to reinforcement of ATP re-synthesis due to increased hypoxanthine levels in the blood and tissues. Recent clinical studies indicated that treatment with xanthine oxidoreductase inhibitors plus inosine had the strongest impact for increasing the pool of salvageable purines and leading to increased ATP levels in humans, thereby suggesting that this combination is more beneficial than a xanthine oxidoreductase inhibitor alone to treat disorders with ATP deficiency.
Introduction
In society and industry, three categories are important: things, information, and energy. Similarly, those three should also be important in biology at the cellular level. Since many diseases are caused by cellular shortages of chemicals (things) and DNA (information), there should also be diseases caused by a shortage of cellular ATP (energy). Based on the June 2018 Integrated Mitochondrial Protein Index from MitoMiner (Smith and Robinson, 2016), over 1,600 proteins coded by chromosomal genes are known to be associated with mitochondria. Since humans have about 20,000 genes (Harrow et al., 2012), that means that about 8% of all genes are associated with mitochondria and with energy. It is of interest that at a societal scale, about 8% of world-wide economic activity is also energy-related (Institute for Energy Research [IER], 2010).
Mitochondria and the glycolytic pathway are responsible for cellular energy production, and germline mutations in the genes associated with them have been shown to lead to various monogenic disorders. Of note, mitochondrial disorders often result in dysfunctions or symptoms involving one or more of the top energy consuming organs (Wallace, 1999), which are skeletal muscle, brain, liver, heart, and kidneys (349.7, 316.8, 278, 136.4, and 129 kcal/day, respectively) (Wang et al., 2010). Especially, heart, skeletal muscle, and the brain are often observed as targets of mitochondrial disorders, but dysfunction has also been observed in the other high-energy utilizing organs such as the liver (Lee and Sokol, 2007) and kidneys (Rahman and Hall, 2013; Che et al., 2014; Emma et al., 2016; Emma and Salviati, 2017). Additionally, mutations in genes related to mitochondrial function sometimes lead to pathology in other small organs/tissues that utilize large amounts of energy relative to their size or weight such as inner-ear hair cells (Hinson et al., 2007; Tiede et al., 2009; Yan et al., 2017) or structures of the eye such as retina, optic nerves, and extraocular muscles (Barot et al., 2011; Schrier and Falk, 2011).
Although rare genetic diseases such as mitochondrial diseases are known to be associated with energy, for common diseases, energy shortage has not been studied as extensively as other potential causes. For example, titles/abstracts in PubMed that mentioned etiology/causes and Alzheimer’s disease (AD; n = 6318), Parkinson’s disease (PD; n = 4602), heart disease (heart or cardiac disease/failure, heart; n = 20643), or diabetes (n = 22698) only included energy related terms (cellular energy, energetics, bioenergetics, energy and mitochondria, mitochondrial function, mitochondrial dysfunction) for a small percentage of papers (AD = 3.7%, PD = 7.9%, heart = 0.6%, diabetes = 1.3%). Notably, for years, amyloid, tau, and alpha-synuclein hypotheses have dominated AD and PD research (Zhang et al., 1989; Stefanis, 2012; Ozansoy and Başak, 2013), but recently, researchers have suggested that mitochondrial or bioenergetic dysfunction may be related to etiology of AD or PD (Winklhofer and Haass, 2010; Wellstead and Cloutier, 2011; Desler et al., 2017; Onyango et al., 2017; Swerdlow et al., 2017).
Cellular Energy-Charge and ATP Turnover
Adenosine triphosphate (ATP) is known as the “energy currency” of the cell, and central to use of that currency is the system’s ability to generate and maintain levels of what is known as the “energy charge,” the ratio of the concentrations [ATP+0.5∗ADP]/[ATP+ADP+AMP] (Chapman and Atkinson, 1973). Although mitochondrial and glycolytic pathways are used to produce energy from molecules such as sugars, proteins, and fatty acids, instantaneous energy needs are satisfied first through the phosphocreatine (PCr) shuttle (Guimarães-Ferreira, 2014) and then through the combined efforts of AMP deaminase (AMPD), AMP-activated protein kinase (AMPK), and adenylate kinase (AK) (Panayiotou et al., 2014). AMPK acts as a form of energy charge sensor (Hardie et al., 2016), which regulates AMPD activity, while AMPD deaminates AMP to IMP to maintain higher values of the energy charge (Lanaspa et al., 2012; Plaideau et al., 2014; Lanaspa et al., 2015) and favor the forward AK reaction that produces ATP and AMP from two ADP molecules (Figure 1; Saks et al., 2014). IMP may then be degraded to inosine via 5′-nucleotidase and then to hypoxanthine (Hx) by purine nucleotide phosphorylase (PNP) and potentially further degraded to xanthine (X) and uric acid (UA) through xanthine oxidoreductase (XOR) (Figure 1; Maiuolo et al., 2016). Thus, such purine molecules form the scaffold of the key molecule for storing cellular energy.
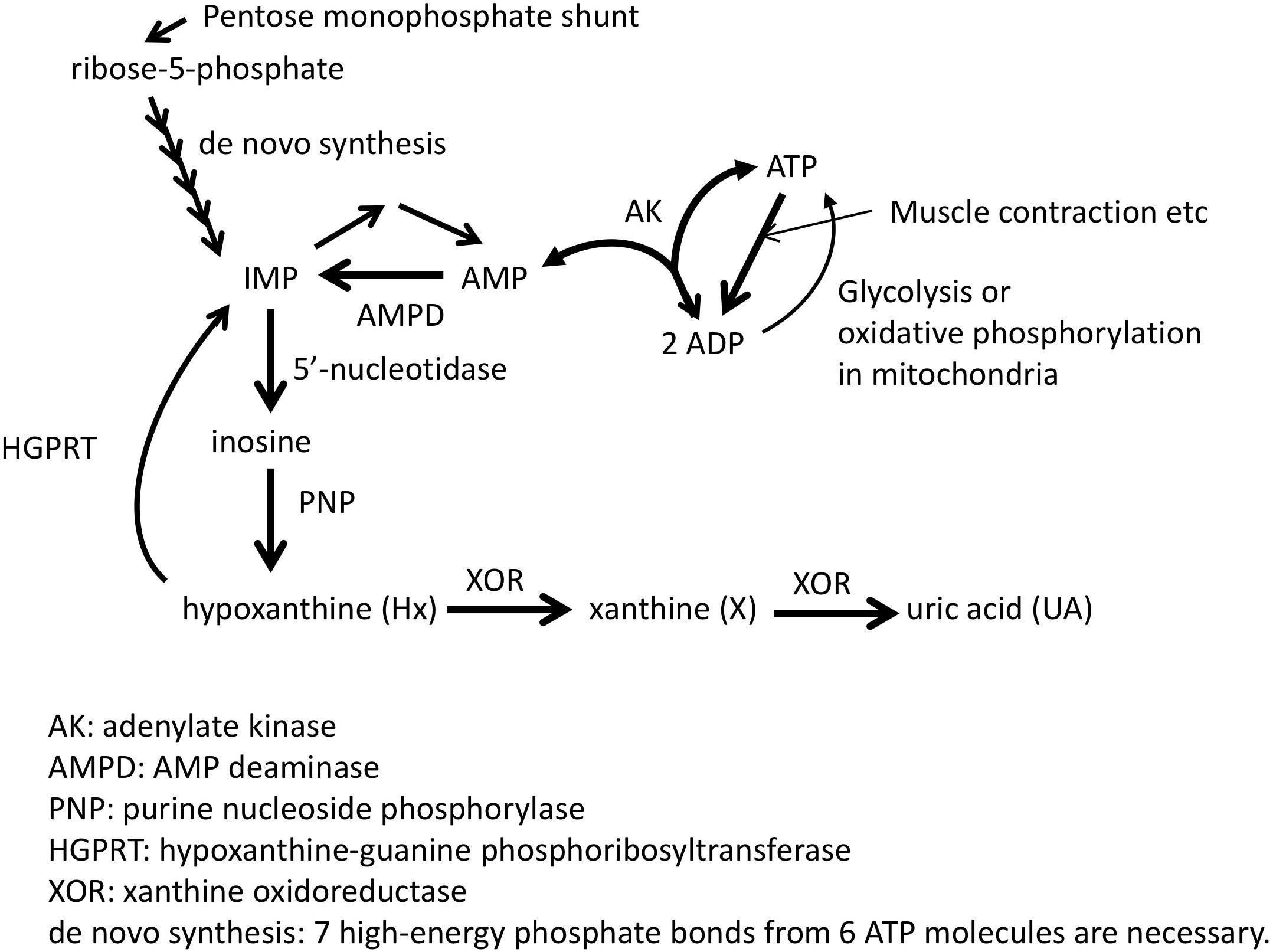
Figure 1. Pathway related to ATP synthesis and degradation. Adapted from Kamatani et al. (2017) with permission of the journal.
Considering Differences Between Humans and Animal Models
When comparing and interpreting results from studies based on animal-models versus those from human subjects, researchers should consider both differences in metabolic rates and biochemical pathways that exist between species. While safety is of paramount importance, not accounting for such differences may also potentially lead one not to consider developing a drug based on phenomena observed in animal models that do not apply to humans.
One notable difference relates to Kleiber’s Law, which states that an organism’s resting energy expenditure (REE) relates to its mass (M) as REE∝M3/4 (Kleiber, 1932; Brody, 1945). Thus, REE per-unit-mass (REE/M) for small animals is much greater than that of large animals; REE/M is about eight times greater in mice (196 kcal/kg per day) than in humans (24.8 kcal/kg per day) (Wang et al., 2012). Commensurate with a smaller animal’s need to generate energy and therefore resynthesize ATP at a much faster rate, other studies found that excretion of purine degradation products was about seven-times higher in dogs and 40-times higher in rats than in humans (Hitchings, 1966).
Another notable difference in purine metabolism relates to the different end-products of purine degradation. Guanosine and adenosine nucleotide degradation pathways converge on X, which is then degraded in most mammals to slightly soluble UA by XOR and readily soluble allantoin by urate oxidase (UOX) (Johnson et al., 2009). However, a string of UOX genetic mutations in the ape-lineage finally led to its pseudogenization in humans (Kratzer et al., 2014), meaning that the terminal end-product of purine degradation is different in humans (UA) and typical animal models (allantoin). At least partly due to their increased metabolic rate, UOX knockout in mice results in extreme hyperuricemia and is highly lethal (Wu et al., 1994), while it is generally benign in humans (who are all UOX knockouts). In real-world terms, this is an example of the importance of considering human/animal-model differences, because drugs used to treat hyperuricemia such as allopurinol and febuxostat resulted in renal calculi formation in mice and rats but were not expected to cause such problems in humans (Horiuchi et al., 1999).
Taken together, these observations and results suggest that both similarities and differences between biochemical and metabolic pathways need to be considered when using animal models to extrapolate about biologic and clinical phenomena in humans.
Xanthine Oxidoreductase (XOR): A Key Enzyme in Purine Salvage
Xanthine oxidoreductase is a homodimeric metallo-flavoprotein that has in its N-terminal region two iron-sulfur redox centers, at its C-terminal domain a catalytic center with the molybdenum cofactor molybdopterin (Moco), and the intervening region a flavin-adenine dinucleotide (FAD) binding domain (Battelli et al., 2014). Research almost 100 years ago measured XOR activity in cow milk, and showed in various rat tissues that it had highest activity in liver and spleen, intermediate activity in kidney and lungs, and little to no activity in muscle (Morgan et al., 1922; Massey and Harris, 1997).
Localization of XOR in Human Tissues, Cell-Types, and Blood
Several lines of evidence can be used to understand the presence/absence of XOR in various human tissues and cell types. First, we examined results from several human immunohistochemistry (IHC) studies that investigated XOR protein localization using anti-XOR antibodies (Abs) (Hellsten-Westing, 1993; Moriwaki et al., 1996; Linder et al., 1999, 2005; Martin et al., 2004). Second, we analyzed gene expression data for XDH from the GTEx Portal browser1 (Supplementary Table S1; n = 53; GTEx Consortium, 2017) and the FANTOM5 human Phase 1and 2 promoterome2 (Supplementary Table S2; n = 1829; Hon et al., 2017; Lizio et al., 2015, 2017), and for comparison with a commonly analyzed model species, for mouse Xdh data from the Comprehensive Mouse Transcriptomic BodyMap3 (Supplementary Table S3; n = 17; Li et al., 2017) and the FANTOM5 mouse promoterome4 (Supplementary Table S4; n = 1195; Abugessaisa et al., 2017; Noguchi et al., 2017). Examination of the human and mouse FANTOM5 datasets, which assayed large numbers of tissues and cell-types, suggested that expression within a species can vary by tissue sub-location and also across cell-types within a tissue. Finally, data from the Human Proteome Map provides additional evidence for XOR protein in various tissues (Kim et al., 2014). Human tissue expression levels from GTEx Portal RNA-seq data (Figure 2A) closely mirrored those observed in protein expression data from the Human Proteome Map (Figure 2B; Kim et al., 2014).
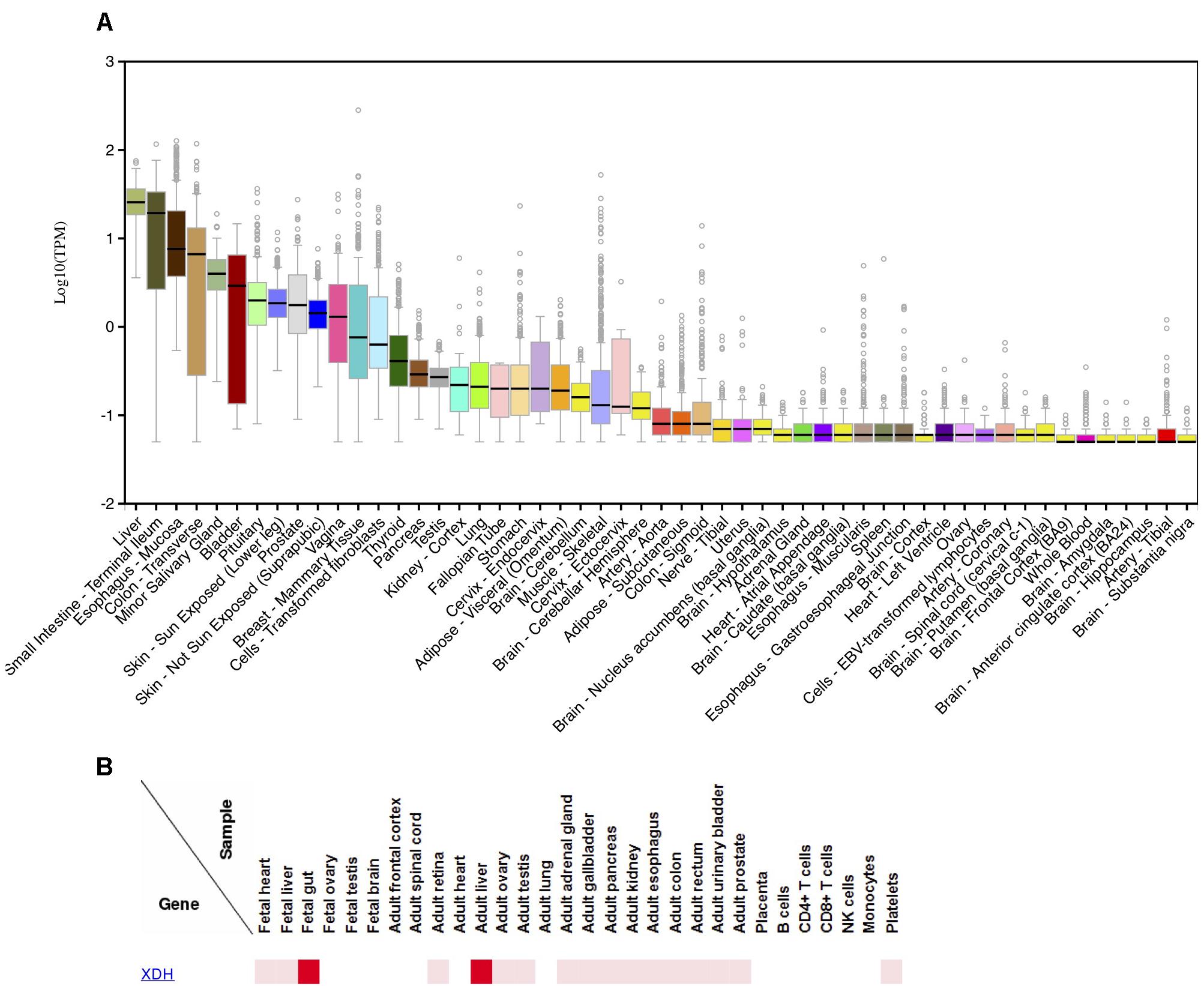
Figure 2. Expression of XDH in various human tissues.(A) Distribution of log10 (TPM) of XDH RNA-seq expression levels in 53 human tissues; produced 9/25/2018 on the GTEx Portal5. (B) Relative XDH protein expression levels in human tissues and cell-lines; produced 10/11/2018 on the Human Proteome Map web-site6.
Among multi-tissue IHC analyses, Linder et al. (1999) found the highest XOR staining in liver periportal hepatocytes and Kupffer cells, various proximal intestine enterocytes and goblet cells, and lactating mammary gland ductal epithelium. FANTOM5 data agreed with those findings, with liver, small-intestine/intestine, and mammary gland tissues or cell-types among high expressing samples in both human and mouse adult datasets. While small-intestine was top ranked in human FANTOM5, in the mouse, intestinal mucosa had six times the relative expression levels compared to whole adult intestine, suggesting that expression is greater in certain intestinal cell subsets. Moriwaki et al. (1996) found moderate to high levels of XOR in epithelium from various tissues including small and large intestine, trachea, bile duct, esophagus, tongue, breast mammary tissue, and lung bronchi. In human FANTOM5, 41% of human epithelial samples (30/73) expressed XDH, including a majority of bronchial (4/7) and all small-airway (3/3) epithelial samples.
Both Moriwaki et al. (1996) and Linder et al. (1999) found that brain tissues were negative for XOR expression. In agreement, FANTOM5 human data showed no XDH expression in human brain samples (brain, adult samples: 0/2; Astrocyte-cerebellum: 0/3; Astrocyte-cerebral cortex: 0/3; cerebellum, adult 0/3; cerebral meninges: 0/1; hippocampus: 0/4; substantia nigra: 0/4), and all thirteen of GTEx Portal brain tissues had median TPM values of 0 or if expressed, were less than 0.5% that of liver.
In skeletal muscle, Hellsten-Westing (1993) and Linder et al. (1999) did not detect XOR in myocytes, and in human gene expression data, human skeletal muscle samples displayed no or low XDH expression, with no expression in adult FANTOM5 samples (0/6) and GTEx samples having a very low median TPM of 0.08 (0.31% of liver). In contrast to human data, the FANTOM5 mouse biceps femoris muscle sample showed strong expression, with a relative-log expression (RLE) of 52.0 (19.6% of that in liver). In heart, Linder et al. (1999) found no staining for XOR across all cell-types, Hellsten-Westing (1993) found no staining of myocytes, while Moriwaki et al. (1996) reported weak staining of cardiac myocytes. Both GTEx heart tissues (Atrial appendage; Left ventricle) showed a very low median TPM of 0.01 (0.04% of liver), and FANTOM5 human heart samples and cardiac-related cell-types showed no XDH expression (heart, adult diseased: 0/2; heart, adult, pool: 0/1; heart, fetal, pool: 0/1; Cardiac myocytes: 0/3; Fibroblast-Cardiac: 0/6; mesenchymal precursor cell-cardiac: 0/4). In contrast, mouse BodyMap heart tissue and FANTOM5 cardiac myocytes displayed high Xdh expression (BodyMap: mean FPKM 18.47 or 18.8% of small intestine; FANTOM5: 45.6-181.4 RLE or 17–68% of liver).
In both skeletal muscle and heart, Hellsten-Westing (1993) identified capillary endothelium as weakly positive and smooth muscle cells of arterioles and venules as strongly positive for XOR, respectively. Linder et al. (1999) and Moriwaki et al. (1996) also made the general observation that XOR stained capillary endothelium, but Linder et al. (1999) did not observe staining of heart blood vessels, and the authors pointed to a lack of knowledge as to whether vascular endothelial cells expressed XDH mRNA, or rather, if XOR from the blood was bound to sulfated glycosaminoglycans on the endothelial cell membrane (Adachi et al., 1993; Radi et al., 1997). To answer that question, we examined mouse and human FANTOM5, and while the mouse data provided some support for localized XOR expression, with the single mouse endothelial sample (hepatic Sinusoidal Endothelial Cells) displaying very strong Xdh levels (89.6% of liver), no human vascular endothelial cell samples expressed XDH (Aortic:0/3; Artery 0/3; Microvascular 0/3; Vein 0/3; Umbilical vein: 0/3; Vein: 0/3). Similarly, Xdh was strongly expressed in the single mouse aortic smooth muscle cells sample, but human vascular smooth muscle samples overall lacked XDH expression (Aortic: 1/4; Brachiocephalic: 0/3; Brain vascular 0/3; Carotid: 0/3; Coronary artery: 0/3; Pulmonary artery: 0/3; Subclavian artery: 0/3; Umbilical artery: 0/4; Umbilical vein: 0/3).
Similar to our findings for endothelial and vascular smooth muscle cells, previous reports described XOR as a regulator of adipose tissue differentiation and found that mouse white adipose tissue (WAT) had ∼30% higher expression levels compared to liver (Cheung et al., 2007; Murakami et al., 2014). In contrast, GTEx human subcutaneous and visceral fat tissue had low median expression of 0.03 TPM and 0.14 TPM, respectively, (0.12 and 0.55%, of liver) and none of FANTOM5 adult adipose tissue samples or mesenchymal adipose related cells (precursors, stem cells, and during adipogenic induction) showed detectable XDH expression.
Circulating XOR as a Source of Endothelial Protein-Binding/Activity
Since liver is one of the top sites of XOR activity in humans, damage to the liver has been suspected to allow XOR to enter the circulation (Tan et al., 1993; Martin et al., 2004). In tests of hepatotoxic agents in animal models, it was shown that XOR is released into the blood (Giler et al., 1976, 1977; Zima et al., 1993; Zima et al., 2001). Additionally, since ischemia-reperfusion (I-R) can result in injury to organs, researchers investigated I-R induced in animal models as well as I-R in human subjects undergoing surgery. In rabbits, occlusion of the thoracic aorta resulted in four-fold increases in plasma levels of liver enzymes (AST, LDH) and XOR during post-ischemia reperfusion, suggesting that ischemia caused liver damage and leakage of various cellular enzymes, including active XOR, accompanied restored blood flow (Nielsen et al., 1994). In similar human surgeries, circulating XOR was increased (∼two-fold increase), likely from damaged liver and intestine due to the aortic cross-clamp procedure, and was suggested as a source of ROS activity that could cause damage in downstream organs in the circulation that normally lack XOR expression such as the heart or lungs (Tan et al., 1995).
However, within normal human plasma, XOR levels have historically been described as undetectable or very-low (Fried and Fried, 1974). Using a new more sensitive assay (Murase et al., 2016), a recent study found XOR activity in plasma from 282 Japanese subjects of 32 (19–58) pmol/h/ml (median and interquartile range) (Furuhashi et al., 2018). However, our calculations show that plasma XOR at those concentrations would only account for a negligible amount: about 0.09% of daily production [(32 × 10-12 moles/h/ml∗5000 ml∗24 h/day∗168.1103 g/mole)/0.7 g/day]. Thus, under certain pathologic conditions, XOR levels may substantially increase and lead to downstream effects (Battelli et al., 2014), but under normal physiologic conditions, the amount of circulating XOR is notably low and unlikely to elicit local pathology.
Xanthine Dehydrogenase Is the Predominant Form of XOR
Depending on the species, the XOR enzyme may degrade Hx to X through either xanthine dehydrogenase (XDH) or xanthine oxidase (XO) enzymatic activities (Stirpe and Della Corte, 1969):
XDH:
XO:
The mechanism of transition between XDH and XO activities has been described as dependent on either proteolytic nicking or disulfide bonds formed between cysteine residues in two parts of the enzyme structure (amino acids residues: Cys535-Cys992 and the C-terminal Cys1316-Cys1324) (Della Corte and Stirpe, 1968; Corte and Stirpe, 1972). Oxidation of Cys535-Cys992 to form a disulfide bond occurs at a fast pace, while that of the C-terminal residues occurs much more slowly (Nishino et al., 2015). A clearer picture of the conversion mechanism was identified by Nishino and collegues using X-ray crystallography of modified versions of the enzyme that lock one or the other activities in place (Nishino et al., 2008b) or that removed the C-terminal peptide of the enzyme (Nishino and Okamoto, 2015). Those experiments showed that the C-terminal peptide in the structure undergoes insertion into the FAD cavity, and that that conformation maintains XDH activity. From that, it was proposed that interactions with cell membranes, such as in capillary endothelium, by the C-terminal peptide may facilitate conversion to the XO form. Although the analyses of various O2 levels and cultured human bronchial epithelial cells showed that XOR activity increased with decreasing O2 levels, XO as a percent of total XOR activity (XO/XOR) was reported to remain unchanged, and XO/XOR was about 20% at each oxygen level (Linder et al., 2003).
Early studies that examined XDH to XO conversion did so using tissue homogenates (Corte and Stirpe, 1968; Stirpe and Della Corte, 1969; Engerson et al., 1987; McKelvey et al., 1988), and one of the more comprehensive reports found that in rat lung, kidney, liver, and heart, XO/XOR was between 10 and 40% if homogenates were processed and assayed quickly (Engerson et al., 1987). However, it was noted that prolonged time in culture, especially under conditions simulating ischemia, resulted in XO/XOR between 50 and 100%. In human normal liver homogenates, XO/XOR of about 20–25% was observed, while in patients with liver cirrhosis, it increased to between 35 and 45% (Stirpe et al., 2002).
Since the XO/XOR ratio increased with longer incubation times of the tissue homogenate, one study tried to obtain true in situ values by examining XDH and XO activities via histochemical analysis of rat liver sections and found that XO activity only represented about 4% of total activity, which suggested that XDH represented the physiologically predominant XOR form (Frederiks and Bosch, 1996). In addition, they found that under ischemic conditions, XO/XOR did not increase, but rather, that it slightly decreased (Frederiks et al., 1993; Frederiks and Bosch, 1996). To understand whether a “xanthine oxidase activating factor” was present in homogenized versus intact tissue, another report found that freeze-thaw cycling of tissue samples released a mitochondrial protease that caused irreversible XO conversion (Saksela et al., 1999). Taken together, such past reports suggest that XDH represents the physiologically normal activity of XOR, even under certain pathologic conditions.
Purine Salvage and the “Circulating Hypoxanthine Pool”
Purine molecules form the scaffold for the high-energy phosphates such as ATP and GTP, and they can be either produced de novo or salvaged from nucleobases or nucleosides that have either been previously synthesized and undergone partial degradation or obtained from the diet (Ipata et al., 2011). However, since de novo synthesis expends energy in the form of 7 high-energy phosphate bonds from 6 ATP molecules to regenerate IMP (Figure 1; Garrett and Grisham, 2016), energy-intensive tissues such as skeletal muscle, heart muscle cells, and brain neurons extensively use salvage pathways to maintain their purine levels (Manfredi and Holmes, 1985; Ipata, 2011). In that process, three purine nucleobases can be salvaged by two different enzymes, with adenine phosphoribosyltransferase (APRT) converting adenine (Ade) to AMP, or hypoxanthine-guanine phosphoribosyltransferase (HGPRT; gene HPRT1) converting Guanine (Gua) and Hx to GMP and IMP, respectively. After the salvage of Hx, IMP may then be converted by Adenylosuccinate synthetase (ADSS; genes ADSS and ADSSL1) to S-AMP (adenylosuccinate) which is then converted by Adenylosuccinate lyase (ADSL) to AMP.
That process of purine salvage and its importance was proposed over 40 years ago, and analyses at the time showed that a substantial fraction (∼95%) of the body’s production and intake of purines is salvaged and recycled from Hx back into nucleotides (Murray et al., 1970; Murray, 1971). Analyses of purine metabolism showed that plasma and urine purine levels varied with different levels of physical activity, and Harkness et al. (1983) observed that acute muscular exercise resulted in dramatically increased plasma Hx levels, and from that, they proposed that the release of Hx and its transport between tissues could be considered as a “circulating hypoxanthine pool” (Harkness et al., 1983). Later, analyses indicated that the total purine loss (i.e., excreted in 24 h urine samples) increased with the number of severe acute exercise sessions (Stathis et al., 1999), and Hellsten et al. (1998) showed that after intense exercise, Hx and UA in the blood increased during recovery, and that while salvage pathways recovered IMP still present in the muscle into ATP, Hx that had been released into the blood appeared not to be salvaged, but rather, Hx was degraded into UA by the liver, returned to the circulation, and then taken up by the muscle tissue. A later study by Stathis et al. (2005) investigated whether treatment with an XOR inhibitor (allopurinol) would attenuate total purine output and allow for greater Hx salvage by the muscle after intense exercise, but allopurinol administration was found to actually increase total purine loss. That study also illustrated that Ino and its metabolite Hx can be readily transported out of muscle cells once IMP is de-phosphorylated by 5′-nucleotidase, and that the increased concentrations of Ino and Hx in the blood may then be readily removed into the urine by the kidneys. The high-transfer potential of Ino and Hx out of exercised muscle likely reflects the action of equilibrative nucleoside transporters ENT1 (SLC29A1) and ENT2 (SLC29A2) that passively move nucleosides and nucleobases out of cellular compartments with high metabolite concentrations (Boswell-Casteel and Hays, 2017).
Much of our early knowledge on the existence of purine salvage originated from research on patients with Lesch-Nyhan’s Disease (LND), which results in “mental retardation, spastic cerebral palsy, choreoathetosis, uric acid urinary stones, and self-destructive biting of fingers and lips” (Hamosh, 2017). LND is caused by genetic mutations in the HPRT1 gene that result in a deficiency in HGPRT enzyme function (Seegmiller et al., 1967; Nguyen and Nyhan, 2016). Thus, LND patients are not able to salvage Gua and Hx to GMP and IMP, respectively, and they display increased levels of purine metabolites including Hx, UA, X, and Ino in the blood (Jiménez et al., 1989a). Analysis of the distribution of purine metabolites in the plasma and urine through use of radioactive tracers suggested that purine salvage was a normal mechanism for Hx reutilization in humans (Edwards et al., 1979), and other researchers showed that purines produced in the liver were taken up by erythrocytes and transported to other tissues (Pritchard et al., 1975). Such results, taken together with the severe neurological symptoms that accompany the disease played a key role in showing the importance of purine salvage pathways in normal functioning of the brain.
Although ENT1 and ENT2 have been proposed as the transporters for Hx salvage, their respective Km values for Hx are quite high (ENT1 Km = 6.0 ± 0.5 mM; ENT2 Km = 1.5 ± 0.2 mM) compared to typical plasma Hx concentrations (∼0.46 μM; see Section 2.8, below) (Yao et al., 2011), suggesting that some other more efficient mechanism existed for the transport of Hx across membranes. Thus, Bone et al. (2010, 2015) identified the existence of a nucleobase transporter in studies of Hx transport in cardiac microvascular endothelial cells (MVEC) (Bone and Hammond, 2007) and in MVEC from ENT1-/- mice). Although unable to identify the actual gene or protein involved, they termed the putative transporter as equilibrative nucleobase transporter 1 (ENBT1). That name was later used by Furukawa et al. (2015) who identified solute carrier family 43 member 3 (SLC43A3) as able to uptake nucleobases such as adenine, guanine, and hypoxanthine, but not nucleosides. Interestingly, they also showed that nucleobase import was greater when cells expressed both SLC43A3 and APRT (for adenine import) or HGPRT (for guanine import), suggesting that the nucleobase transporter and salvage enzyme work together in a cooperative fashion, with the final imported product being the nucleotides.
Interestingly, an earlier study by Wallgard et al. (2008) had looked for genes that were differentially expressed in microvascular endothelial cells, and they found SLC43A3 differentially enriched in brain microvessels versus rest-of-brain tissue. In a review of FANTOM5, we found that all of twenty-one human endothelial cell samples expressed SLC43A3. As Furukawa had found that the Km for Hx was 1.32 μM, which is close to reported normal Hx plasma concentrations, these results suggest that SLC43A3 functions in the microvasculature of various tissues to facilitate purine uptake and salvage. Interestingly, all FANTOM5 skeletal muscle samples also expressed the gene, suggesting a role for nucleobase import and salvage despite the contradictory results from research involving intense muscular exercise.
Serum Urate Is a Biomarker of ATP Consumption
If that purine salvage process fails to salvage Hx back into high-energy phosphate molecules, and instead, XOR degrades it to X, then X can only be further degraded by XOR to UA (Figure 1). The degradation process may take place within cells if they express XOR or following excretion of Hx from a cell when it traverses organs with XOR activity such as the liver or endothelium. Consequently, processes maintaining cellular energy charge may degrade the molecular scaffold for ATP into a form (UA) that cannot be salvaged back into energy storing molecules. As described above, after degradation to UA, new purine molecules would need to be salvaged from dietary intake or resynthesized de novo at an energy expense of 7 high-energy phosphate bonds from 6 ATP molecules to regenerate IMP (Figure 1; Garrett and Grisham, 2016).
Previous results suggest that serum UA is a biomarker of ATP consumption, since rapid ATP consumption can lead to purine degradation and an increased rate of UA production in humans. For example, intense muscular exercise (Hellsten et al., 1999; Stathis et al., 1994, 1999), fructose challenge (Budillon et al., 1992; Donderski et al., 2015), alcohol intake (Lieber, 1965; Schmidt et al., 2013), and high brain activity (Salvadore et al., 2010; Goodman et al., 2016) can lead to energy crisis and resultant hyperuricemia. In addition, vascular regions undergoing ischemia-reperfusion may produce increased purine degradation products; a sign that a local energy crisis has occurred. For example, one study investigated the difference in the concentrations of adenine nucleotide degradation products between the aortic root (Ao) and the coronary sinus (Cs), before, during, and after cross-clamping of the aorta during cardiopulmonary bypass (Lazzarino et al., 1994). The authors found that pre-clamping Cs blood was slightly increased for Ado, Ino, Hx,and X (< 1 μmol/L), while UA was increased > 10 μmol/L, showing that XOR was present within the coronary vasculature. The Cs-Ao difference for purine degradation products increased 2–3 fold during the ischemic period, and decreased afterward. In addition, malondialdehyde, a marker of oxidative stress, significantly increased while the aorta was clamped, but there was no apparent correlation with post-operative outcomes, suggesting that oxidative stress in such pathologic situations may not be as important as is often thought.
In contrast, UA production is low in infirm aged people (Beberashvili et al., 2015, 2016), those with low nutrition (Beberashvili et al., 2016), and patients with certain neurodegenerative disorders (Rinaldi et al., 2003; de Lau et al., 2005). For multiple sclerosis (MS), decreased plasma/serum UA levels were observed in multiple studies (Druloviæ et al., 2001; Sotgiu et al., 2002; Liu et al., 2012; Moccia et al., 2015), but some studies have also observed that UA levels were increased compared to control subjects (Amorini et al., 2009; Tavazzi et al., 2011). Tavazzi et al. (2011) identified relapsing-remitting MS patients as having lower UA levels compared to secondary progressive or primary progressive MS patients, and the authors have described serum purine metabolite concentrations as related to an imbalance in energy production (Lazzarino et al., 2017).
Inhibited Purine Degradation Maintains ATP Levels
Evidence suggests that inhibition of purine degradation leads to alterations of bioenergetic pathways and conserved ATP levels. Data that supports this conclusion is found in genetic deficiencies of the AMPD gene family, as well as from clinical and experimental analyses in various organ systems. AMPD enzymes catalyze the conversion of AMP to IMP (Figure 1), and expression of the three AMPD genes, AMPD1, AMPD2, and AMPD3, are differently expressed in various organs and in various types of cells (Morisaki et al., 1990; Mahnke-Zizelman and Sabina, 1992).
AMPD1 is very strongly expressed in skeletal muscle and diaphragm (Morisaki et al., 1990), and it has been examined extensively for the impact of a non-sense mutation that is common in European ancestry populations but rare in most other world-wide population samples (rs17602729; Gln45Ter; C34T; AFT-EUR = 0.123 in 1000 Genomes)7 Originally, deficiency in AMPD1 due to rs17602729 homozygosity (TT) was identified as a putative cause of myopathy (Fishbein et al., 1978). However, greater than 1% of European ancestry populations are AMPD1 deficient, and many of those subjects are asymptomatic, which suggests a heterogeneity of its effects or that other factors might be involved (Verzijl et al., 1998; Sabina and Holmes, 2001; Hanisch et al., 2008). Studies of elite athletes found significantly less C34T carriers compared to controls, with more skewed genotype frequencies in sprint/power athletes versus endurance and mixed-event athletes, and there appeared to be a deficit of TT individuals (Rubio et al., 2005; Muniesa et al., 2010; Tsianos et al., 2010; Ciȩszczyk et al., 2011, 2012; Ginevičienė et al., 2014; Grealy et al., 2015). However, endurance athletes who had reached the elite level who were heterozygous appeared to generally perform comparably to CC individuals (Rubio et al., 2005; Tsianos et al., 2010). Among various exercise studies of normal and AMPD1 deficient subjects (Norman et al., 2001; Tarnopolsky et al., 2001; De Ruiter et al., 2002; Fischer et al., 2007; Norman et al., 2008), the study with the largest sample-size (nCC = 89; nCT = 38; nTT = 12) (Fischer et al., 2007), which performed a short-term high-intensity Wingate cycling test, found that TT subjects exhibited decreased mean-power output and increased fatigue. However, compared to CC and CT individuals, they did not experience depletion of ATP at the end of the test (Norman et al., 2001). In heart failure patients, genetic AMPD1 deficient subjects exhibited decreased systolic blood pressure (de Groote et al., 2006; Safranow et al., 2009; Tousoulis et al., 2014; Feng et al., 2017), an increase in cardiac output (left ventricular ejection fraction) (Gastmann et al., 2004; Collins et al., 2006; Safranow et al., 2009; Feng et al., 2017), and improved heart failure prognosis (Loh et al., 1999; Gastmann et al., 2004). Interestingly, AMPD1 was also reported as a putative target of the diabetes drug metformin (Cheng et al., 2014), and AMPD1 deficient individuals have been reported to have decreased rates of diabetes (Safranow et al., 2009).
AMPD2 is most prominently expressed in non-skeletal muscle tissues, including brain, liver, and thymus (Morisaki et al., 1990). Genetic AMPD2 deficiency leads to increased ATP but a decrease of GTP and has been reported to lead to rare central nervous system (CNS) disorders (Akizu et al., 2013; Novarino et al., 2014).
AMPD3 is most strongly expressed in erythrocytes but is also found in other tissues, including skeletal muscle. Genetic AMPD3 deficiency has been observed, and it has been reported to lead to a 1.5 fold increase of ATP in erythrocytes without any health problem (Ogasawara et al., 1987; Yamada et al., 1994). Studies of AMPD3 deficient mice have also reported increased ATP levels (Cheng et al., 2012; Daniels et al., 2013; O’Brien et al., 2015).
In addition to genetic deficiencies of the AMPD isozymes, experimental studies have shown that perturbation of certain pathways can modulate cellular energy through AMPD. For instance, under conditions that mimic oxidative stress in studies of erythrocyte energy metabolism, Tavazzi et al. (2000, 2001) found that AMPD activity increased when incubated with H2O2 or NaNO2). Those studies suggest that changes to the local erythrocyte environment may result in deficient energy states due to purine loss. Another study by the same group investigated the effect of angiotensin II (AII) on matrix metalloproteinase activity (MMP) in the canine heart. AII administration increased MMP activity, as well as chamber diastolic stiffening and MMP activity, and decreased tissue bioenergetics, while treatment with an MMP inhibitor was shown to decrease the effects of AII and reduce purine loss, likely through inhibition of AMPD activity (Paolocci et al., 2006).
XOR Inhibitor Is Beneficial for Diseases of Heart, Skeletal Muscle and Brain
As noted above, heart, skeletal muscle, and brain are among the organs requiring the largest amounts of energy and that are typical targets of mitochondrial diseases, and interestingly, recent human and animal studies have shown that XOR inhibitors (XOIs) such as allopurinol and febuxostat are useful for the treatment of disorders of heart, skeletal muscle and brain.
XOR Inhibitor Alone Is Good for the Heart
In animals, at least 11 papers reported that heart failure and ischemic heart disease were improved by the administration of allopurinol or febuxostat (DeWall et al., 1971; Arnold et al., 1980; Hopson et al., 1995; Pérez et al., 1998; Ekelund et al., 1999; Khatib et al., 2001; Ukai et al., 2001; Stull et al., 2004; Hou et al., 2006; Xu et al., 2008; Zhao et al., 2008; Yao and Seko, 2017; El-Bassossy et al., 2018). In humans, at least 20 papers, including randomized controlled trials (RCTs) and meta-analyses, reported that heart failure and ischemic heart disease were improved by the administration of allopurinol or febuxostat (Saugstad, 1996; Cappola et al., 2001; Gavin and Struthers, 2005; Pacher et al., 2006; Noman et al., 2010; Rentoukas et al., 2010; Thanassoulis et al., 2010; Kelkar et al., 2011; Gotsman et al., 2012; Hirsch et al., 2012; Agarwal and Messerli, 2013; Agarwal et al., 2013; Larsen et al., 2016; MacIsaac et al., 2016; Singh and Yu, 2016, 2017; Ansari-Ramandi et al., 2017; Foody et al., 2017; Singh et al., 2017; Bredemeier et al., 2018). Because of febuxostat’s more recent approval, most reports analyzed only allopurinol.
XOR Inhibitor Alone Is Good for Skeletal Muscle
Administration of allopurinol was reported to improve the effect of rehabilitation in the elderly (Beveridge et al., 2013), prevent muscle damage due to intense exercise such as playing soccer (Sanchis-Gomar et al., 2015) or crewing on an America’s Cup yacht (Barrios et al., 2011), and reduce muscle atrophy due to disuse (Ferrando et al., 2018). The Ferrando et al. (2018) study also confirmed an inhibitory effect of allopurinol on disuse muscle atrophy in experiments using mice. In addition to those studies, allopurinol has also been shown to reduce muscle fiber damage in a mouse model of chemically induced rhabdomyolysis (de Bragança et al., 2017) and increase maximal isometric force produced by muscles in aged mice (Ryan et al., 2011).
XOR Inhibitor Alone Is Good for the Brain
One hypothesis regarding the etiology of neurodegenerative disorders has been that UA has a protective effect. That was based on studies that showed that subjects with higher serum UA levels had lower hazard ratios (HR) or inverse correlation for PD (Alonso et al., 2007; Schwarzschild et al., 2008; Ascherio et al., 2009; Johansen et al., 2009; McFarland et al., 2013), AD (Maesaka et al., 1993; McFarland et al., 2013; Al-khateeb et al., 2015; Lu et al., 2016), amyotrophic lateral sclerosis (ALS) (Abraham and Drory, 2014), and multiple-sclerosis (MS) (Liu et al., 2012). Based on those observations, several clinical trials have been designed to test the effect of Ino supplementation in neurodegenerative disorders, with the expectation being that Ino degradation in the body would increase blood and cerebrospinal fluid (CSF) levels of UA to presumably more protective levels. Three trials examined its use in MS patients, and while one suggested some effect (Markowitz et al., 2009), the other two that had larger sample-sizes showed no benefit of the Ino treatment (Gonsette et al., 2010; Muñoz García et al., 2015). More recently, the SURE-PD study was initiated to investigate Ino treatment in PD patients (The Parkinson Study Group SURE-PD Investigators et al., 2014). SURE-PD has released a proof-of-principal report that shows that Ino increases UA levels and that it is generally tolerated, but the study showed contradictory efficacy results depending on the PD measure examined, and additionally, the study was not powered to determine efficacy of the treatment.
In contradiction to those studies, recent analyses of large clinical databases suggest that the hypothesis that UA exerts a protective effect against neurodegenerative disorders is mis-directed. First, a 2015 Taiwanese study by Hong et al. (2015) reported that gout patients have lower risk of dementia than controls. At first glance, that would agree with the concept that UA exerts a protective effect. However, their results also indicated that gout patients treated with UA lowering drugs had lower risks of both vascular and non-vascular dementias. Second, a more recent report by Singh and Cleveland (2018a) analyzed United States Medicare data to examine the incidence of dementia with respect to use of XOIs and reported a lower occurrence of dementia in individuals receiving either high-dose allopurinol or 40 mg/day febuxostat. That is, compared with allopurinol less than 200 mg/day, higher allopurinol doses (200 to 299 and at least 300 mg/day) and febuxostat 40 mg/day dose were each associated with lower HRs for dementia: 0.80 (95% CI 0.64, 0.98), 0.59 (95% CI 0.50, 0.71), and 0.64 (95% CI 0.47, 0.86), respectively. In a companion study, Singh and Cleveland (2018b) also reported that use of UA lowering drugs was not associated with an increase in the rate of dementia in older adults. which suggests that UA itself does not have a protective effect against dementia. In addition, experimental animal studies have also shown the beneficial effects of XOIs on neurodegenerative disorders. Kato et al. (2016) demonstrated that XOIs delayed the pathological process of ALS in model mice (Nishino et al., 2008a), and the same authors have also reported that XOIs delayed the pathological process of AD in model mice and inhibited development of senile plaques and neurofibrillary tangles (Nishino et al., 2016).
Mechanism of the Beneficial Effects of XOR Inhibitors on Heart, Skeletal Muscle, and Brain
Within the various reports on XOIs′ beneficial effects, several hypotheses have been raised. Namely, the mechanism of action is likely related to: (1) UA reduction, (2) reactive oxygen species (ROS) suppression, or (3) ATP enhancement.
Evidence suggests that XOIs′ UA lowering effect is unlikely to be the mechanism of those beneficial effects. In animals such as mice and rats, serum UA levels are very low even without XOI treatment (Watanabe et al., 2014), and therefore, the benefits of XOR inhibition that were observed in animal models are unlikely to be due to its further lowering. Also, of importance to brain-related studies, CSF UA concentrations are very low compared to those in blood, with human CSF levels about 5% of those seen in plasma samples ([UACSF]∼12.8 μM; [UAPlasma]∼250 μM) (Harkness, 1988; Jiménez et al., 1989b). Those low levels are related to two things. First, as introduced above, XOR expression in brain tissue is very low, with human and mice brains having 0.8% and 0.44% of levels seen in the highest XOR expressing tissues (Supplementary Tables S1–S4). Second, the blood-brain barrier (BBB) and blood-cerebral spinal fluid barrier have been reported to be only weakly permeable to UA (Jiménez et al., 1989a; Redzic et al., 2001). Considering that evidence, UA reduction in serum is unlikely to affect the brain.
Although many reports hypothesized that the suppression of ROS is the mechanism of the beneficial effects of XOIs, that possibility also appears quite unlikely, since while heart, skeletal muscle and brain are among the organs that consume the largest amounts of energy, they are also among those with the lowest amounts of XOR in humans (Figure 2A; Linder et al., 1999), with XOR activity mostly limited to the vascular endothelium of those tissues (Kelley, 2015). Also, recent reports have shown that inhibition of ROS by scavengers is often harmful because minimal levels of ROS are useful for signal transduction and cancer inhibition (Diebold and Chandel, 2016; Scialò et al., 2016; Quijano et al., 2016). The high ATP consumption that occurs in those organs should also coincide with high ROS production, since ROS is produced along with the consumption of oxygen. However, ROS are produced mostly in mitochondria, and based on a rough calculation of the daily ratio of moles UA excreted to moles O2 consumed (UAg.excr = 0.75 g, UAmole.excr∼2.97 × 10-3 moles; 22 moles O2 per day) (Ng et al., 1984; Engelberg, 1996), oxygen consumption by mitochondria is at least 5,000 fold higher than that consumed by XOR. Thus, ROS related damage is more likely to stem from mitochondria than from XOR.
Therefore, most of the ROS produced would stem from mitochondria and thereby can never be suppressed by XOIs. Of course, this may suggest that mitochondria may be damaged by ROS and that that damage leads to ATP depletion. In fact, the association of ROS with various diseases including neurodegenerative disorders may be through the damage of mitochondria by excessive ROS. However, that does not support inhibition of ROS production as the reason for the beneficial effects of XOR inhibition that has been observed in previous studies.
Based on those observations, it is unlikely that ROS inhibition is the reason for XOIs′ beneficial effects. Rather, ATP enhancement through enhanced purine salvage is the more likely mechanism. Support for ATP enhancement as the mechanism can be found in experimental studies using in vitro and animal models as well as human clinical studies.
One such study in the early 2000s of allopurinol’s impact on mitochondrial function examined rat livers under ischemic/hypoxic conditions and found that decreases in ATP levels after ischemia/reperfusion were attenuated with allopurinol treatment (Jeon et al., 2001). Another study around the same time-period examined prepared rat hearts under hypoxic conditions, and the authors reported that both ATP and total adenine nucleotide pools were enhanced after allopurinol administration (Khatib et al., 2001). Later, a report on canines examined the impact of XOIs on cardiac bioenergetics under conditions simulating myocardial ischemia, with subjects examined at rest and then with/without XOI under either basal cardiac work (BCW) or high cardiac work (HCW) after catecholamine administration (Lee et al., 2011). Without XOI, the ratio of PCr/ATP and ADP levels were unchanged at BCW, but the PCr/ATP ratio decreased and levels of ADP increased at HCW. In contrast, with XOI administration, the ratio of PCr/ATP increased and levels of ADP decreased at BCW, while at HCW the increase in ADP levels was attenuated compared to no XOI. Similarly, a study of non-ischemic cardiomyopathy patients showed using 31P magnetic resonance spectroscopy (31P-MRS) that allopurinol increased the flux through creatine kinase (CK) and increased the rate of ATP synthesis as well as decreased ADP levels (Hirsch et al., 2012). In a neurological study of hypoxia/ischemia in piglets, authors reported that allopurinol pre-treatment attenuated a decrease in the PCr/Pi ratio, which is another measure of cellular energy status (Peeters-Scholte et al., 2003). Further, a study of potential causes of MS that used a mouse model of experimental autoimmune encephalomyelitis (EAE) reported that XO played a role in mechanisms leading to demyelination, and that EAE progression was reduced with XOI treatment (Honorat et al., 2013). A follow-up study examined NeuroA2 cells treated with rotenone, which causes dysfunction of mitochondrial electron transport and reduces ATP levels, and found that XOI administration to rotenone treated in vitro culture increased cellular ATP levels (Honorat et al., 2017).
Hypoxanthine Is Important in Neurodegenerative Disorders
It has been reported that serum UA is reduced in patients with neurodegenerative disorders such as PD (Havelund et al., 2017; Sakuta et al., 2017; Wen et al., 2017), ALS (Abraham and Drory, 2014; Paganoni et al., 2018; Zhang et al., 2018), and AD (Euser et al., 2009; Lu et al., 2016), and as mentioned above, many researchers consider that these observations represent a direct protective role of UA suppressing ROS in the brain (Kori et al., 2016). However, since XOR is not expressed in the brain, and UA levels in the brain are naturally very low, it is more likely that low UA in neurodegenerative disorder patients reflects physiologic and biochemical changes that result in decreased energy production and low ATP consumption.
Such a hypothesis is supported by FDG-PET studies of the brain, which use the glucose analog FDG (fluoro-deoxy glucose) to assay metabolic activity through regional glucose uptake. That glucose uptake represents some glycolytic but predominantly mitochondrial activity in the brain for production of ATP (Mosconi, 2013). FDG-PET studies have identified increased regions of hypometabolism, and thus reduced ATP consumption, in the brains of patients with certain neurodegenerative diseases such as AD (Johnson et al., 2012; Lin and Rothman, 2014). Recently, a European task-force recommended use of FDG-PET in answering a number of clinical questions including the differential diagnosis of AD and PD from similar pathologies (Nobili et al., 2018).
If ATP consumption in the brain is reduced in such diseases, the UA precursor Hx should also be low. In line with that, a recent meta-analysis found that a number of metabolomic studies of PD patients identified decreases of compounds either related to energy production or purine-related pathways (Havelund et al., 2017). Among the latter, one report found that Hx levels in PD patients’ plasma were decreased by 41% while UA was decreased by 14% (Johansen et al., 2009). In addition to that data, a proteomics study targeting hundreds of proteins for incidental AD in the longitudinal Framingham Offspring Cohort identified elevations of anthranic acid and glutamic acid and reductions of Hx and taurine (Chouraki et al., 2017), and a MALDI-MSI analysis targeted at hundreds of proteins in Alzheimer’s model mice detected elevations of Hx and taurine (Esteve et al., 2017). Although those results are suggestive of a relationship between purine metabolism and the neurological diseases, we should also note that another potential cause of reduced Hx levels could be decreased physical activity that might accompany PD and AD.
It is of interest that both Hx and taurine were found among hundreds of compounds whose concentrations were different between disease and control subjects. However, in the mouse model, both Hx and taurine were increased, while they were decreased in human AD patients. This may reflect a difference as to the pathogenic mechanisms for AD between mice and humans, with a common facet of both being that unfolding and degradation of mis-folded/abnormal proteins such as amyloid or tau by proteasomes carries a high energy cost (Benaroudj et al., 2003; Peth et al., 2013). In the case of the mouse model, excessive production of misfolded proteins requires the expenditure of large amounts of ATP in order to degrade them, which is evidenced by increased purine degradation products such as Hx. In contrast, in humans the decreased levels of degradation products from energy production and purine pathways reflect energy production dysfunctions that result in a reduced capacity to degrade abnormal proteins. Thus, the elevation of Hx in the model mice and its reduction in the human patients can be explained by the different mechanisms by which amyloid beta and tau accumulation occur between model mice and human disease.
The mechanism by which we propose that XOIs enhance ATP is illustrated in Figure 3, which suggests that the blood concentration of Hx plays an important role. Since XOR exists mostly in liver in humans, the inhibition of XOR leads to the elevation of Hx in the liver, and eventually in the blood. As noted above, for each molecule of purine that is lost, seven high-energy phosphate bonds are needed for the de novo synthesis of IMP. Since elevated Hx saves purines for salvage, XOI should lead to the enhancement of ATP. Support for that was seen in animal models, such that when a XOI alone was administered to mice or rats, marked elevation of Hx occurred (Schmidt et al., 2009; Szasz et al., 2013; Kato et al., 2016). Similarly, in humans, allopurinol treatment of cancer patients before chemotherapy treatment was shown to increase Hx levels by about 1.5-fold (Wung and Howell, 1984).
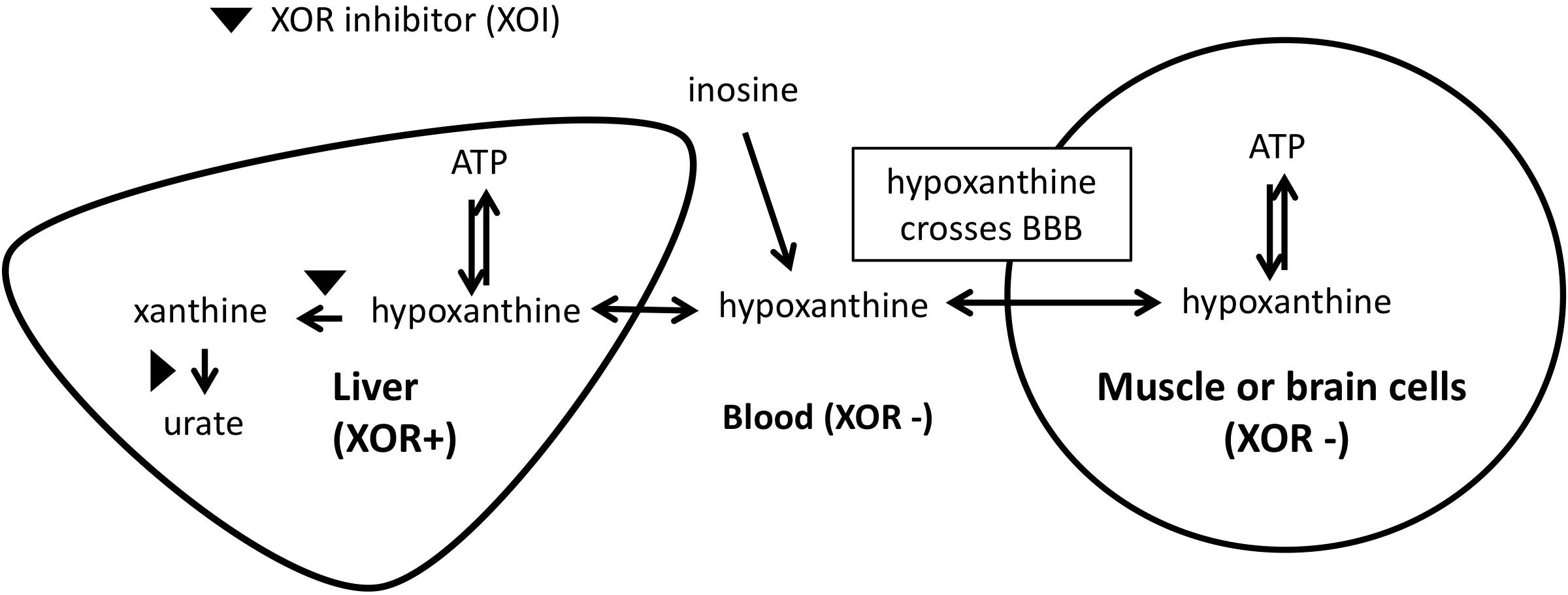
Figure 3. Mechanism of ATP enhancement by XOI or XOI+ inosine.
With respect to brain levels of Hx, under normal physiologic conditions the mean concentration of Hx in CSF was between 1.8 and 2.0 μM from two reports (Harkness and Lund, 1983; Amorini et al., 2009). In blood, reported mean plasma/serum levels for Hx have varied widely as 0.46 μM (Wung and Howell, 1980), 1.5μM (Harkness and Lund, 1983), 4 μM (Amorini et al., 2009), and 32.4 μM (Psychogios et al., 2011), with the most representative value coming from the Wung and Howell (1980) report, which showed clearly that measured Hx concentrations in plasma/serum increases with time between blood draw and isolation of the plasma/serum. Based on those values, CSF in non-diseased individuals appears to be about three to 4-fold enriched for Hx compared to plasma. That enrichment within CSF (Harkness and Lund, 1983) presumably relates to the lack of XOR in brain. Since Hx is known to cross the BBB (Jiménez et al., 1989a), excess Hx produced in the CNS would be expected to move to the blood. However, with XOI treatment, blood levels of Hx should become elevated, and the BBB’s permeability to Hx should allow it to move into the CNS. Alternatively, the less favorable transport kinetics should reduce its movement from the CNS to the main circulation. An analogous situation was observed in a previous study of LND patients who are deficient for HGPRT, the enzyme that salvages Hx to IMP, and thus lack a key node of the purine salvage pathway (Figure 1; Jiménez et al., 1989a). Hx in HGPRT(-) patients was elevated 3.9-times and 6.4-times compared to their reported normal plasma and CSF, respectively, and Hx in HGPRT(-) patients’ CSF was 2.3-times more than in plasma ([HxNormal-CSF]∼2.7 μM, [HxNormal-plasma]∼1.9 μM; [HxHGPRT(-)-CSF]∼17.2 μM; [HxHGPRT(-)-plasma]∼7.3 μM). Interestingly, treatment with an XOI (Allopurinol) increased Hx in the plasma and CSF to roughly equal levels ([HxHGPRT(-)-Allop-CSF]∼35.0 μM; [HxHGPRT(-)-Allop-plasma]∼38.62 μM), suggesting that as Hx could not be degraded by the liver due to XOR inhibition, its levels built-up in the blood and finally equilibrated with those in CSF.
Provision of a Hypoxanthine Source in Addition to an XOR Inhibitor May Further Enhance ATP
The reports described above have shown that XOIs can have a beneficial effect on various diseases, but it may be possible to enhance the effect by simultaneous administration of a Hx source. As noted above, when a XOI alone was administered to mice or rats, a marked elevation of Hx occurred, but mice and rats produce over 25 times more UA per unit body weight as compared to humans (Horiuchi et al., 1999; Hosoyamada et al., 2016). Those higher rates of UA production occur because of the higher rate of energy generation and ATP consumption in small versus large mammals. Due to the difference in purine flux within the body, the substantial elevation of Hx in humans that would be needed to increase purine salvage may not be achieved by only inhibiting XOR. Similarly, provision of additional purines alone would not be beneficial, since oral purines will be substantially broken down to UA in the digestive tract or during absorption. Therefore, treatment using a combination of an XOI with additional purines would be expected to increase purine salvage, decrease energy expenditures by reducing de novo purine synthesis, and achieve better enhancement of cellular ATP.
Combination Treatment With Administration of an XOR Inhibitor and Inosine
To examine that last question, clinical studies were performed using healthy human subjects. Treatment with high-dose XOI alone produced only a slight increase in levels of Hx and ATP, while treatment with Ino alone did not increase Hx or ATP (Kamatani et al., 2017). In contrast, only the combination of an XOI and Ino was observed to substantially increase blood levels of Hx and ATP in the human subjects. In an additional study, it was observed that Ino alone could enhance ATP in human erythrocytes incubated in saline, likely due to the lack of XOR in erythrocytes (Kamatani et al., 2018).
Based on those results, Kamatani et al. (2019) performed a small clinical study of the combined use of an XOI and Ino was performed in two mitochondrial disease patients. After treatment, brain natriuretic peptide decreased by 31% for one patient with mitochondrial myopathy, and for the other patient, who had mitochondrial diabetes, insulinogenic index was raised 3.1-fold. In addition to that small study, the combination therapy is currently under evaluation in a mid-sized study of 30 PD patients.
Therapy combining an XOI and Ino is similar to the treatment of deficiency for carbamoyl-phosphate synthetase 2 (CAD). Through treatment with oral uridine, CNS symptoms were dramatically improved (Koch et al., 2017).
Conclusion
Over many years of research, a number of different groups have examined the effects of XOI treatment on disorders of various organ systems, especially of the heart, skeletal muscles, and brain. With the combination drug, it is possible that the disease status of patients with many such disorders will be improved in the future.
Author Contributions
TJ, HJ, and NK contributed to the conception of the article. TJ analyzed the external data. TJ and NK wrote the first draft of the manuscript. All authors contributed to manuscript revision, and read and approved the submitted version.
Conflict of Interest Statement
This work was performed under the auspices of StaGen Co., Ltd. TJ and NK are employees of StaGen Co., Ltd. NK holds stock in and is Chairman of StaGen Co., Ltd., and is also a Director of the Tsukuba International Clinical Pharmacology Clinic. StaGen Co., Ltd., has received a Japanese patent and has pending patent applications in other jurisdictions for use of XOI+inosine as an ATP enhancement therapy.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge the Genotype-Tissue Expression (GTEx) and FANTOM5 projects for data that was summarized in this report. GTEx was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from the GTEx V7 Portal on 9/25/2018 and from the FANTOM5 Zenbu browser on 10/01/2018.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2019.00098/full#supplementary-material
Footnotes
- ^Downloaded on 9/25/2018 from: https://gtexportal.org/home/gene/XDH
- ^Downloaded on 1/7/2019 from the track “FANTOM5 CAGE phase 1and2 human tracks pooled filtered with 3 or more tags per library and rle normalized” for the human XDH promoter region from: http://fantom.gsc.riken.jp/zenbu/gLyphs/#config=ZCsN4JmAA9wiyJYsBEdfeC;loc=hg19::chr2:31637532..31637635+
- ^Li et al. (2017) Supplementary Table S6.
- ^Downloaded on 1/7/2019 from the track “FANTOM5 CAGE Phase 1 and 2 mouse tracks pooled filtered with 3 or more tags per library and rule normalized” for the mouse Xdh promoter region from: http://fantom.gsc.riken.jp/zenbu/gLyphs/#config=DmO_zYuHPxvu83HbOzew1C;loc=mm9::chr17:74299467..74299576+
- ^https://gtexportal.org/home/gene/XDH
- ^http://www.humanproteomemap.org
- ^https://www.ensembl.org/Homo_sapiens/Variation/Population?db=core;r=1:114692936-114693936;v=rs17602729;vdb=variation;vf=502602520
References
Abraham, A., and Drory, V. E. (2014). Influence of serum uric acid levels on prognosis and survival in amyotrophic lateral sclerosis: a meta-analysis. J. Neurol. 261, 1133–1138. doi: 10.1007/s00415-014-7331-x
Abugessaisa, I., Noguchi, S., Hasegawa, A., Harshbarger, J., Kondo, A., Lizio, M., et al. (2017). FANTOM5 CAGE profiles of human and mouse reprocessed for GRCh38 and GRCm38 genome assemblies. Sci. Data 4:170107. doi: 10.1038/sdata.2017.107
Adachi, T., Fukushima, T., Usami, Y., and Hirano, K. (1993). Binding of human xanthine oxidase to sulphated glycosaminoglycans on the endothelial-cell surface. Biochem. J. 289, 523–527. doi: 10.1042/bj2890523
Agarwal, V., Hans, N., and Messerli, F. H. (2013). Effect of allopurinol on blood pressure: a systematic review and meta-analysis. J. Clin. Hypertens. 15, 435–442. doi: 10.1111/j.1751-7176.2012.00701.x
Agarwal, V., and Messerli, F. (2013). Effect of allopurinol on blood pressure (author response to Allopurinol on hypertension: insufficient evidence to recommend). J. Clin. Hypertens. 15:701. doi: 10.1111/jch.12152
Akizu, N., Cantagrel, V., Schroth, J., Cai, N., Vaux, K., McCloskey, D., et al. (2013). AMPD2 regulates GTP synthesis and is mutated in a potentially treatable neurodegenerative brainstem disorder. Cell 154, 505–517. doi: 10.1016/j.cell.2013.07.005
Al-khateeb, E., Althaher, A., Al-khateeb, M., Al-Musawi, H., Azzouqah, O., Al-Shweiki, S., et al. (2015). Relation between uric acid and Alzheimer’s disease in elderly Jordanians. J. Alzheimers Dis. 44, 859–865. doi: 10.3233/JAD-142037
Alonso, A., Rodríguez, L. A., Logroscino, G., and Hernán, M. A. (2007). Gout and risk of Parkinson disease: a prospective study. Neurology 69, 1696–1700. doi: 10.1212/01.wnl.0000279518.10072.df
Amorini, A. M., Petzold, A., Tavazzi, B., Eikelenboom, J., Keir, G., Belli, A., et al. (2009). Increase of uric acid and purine compounds in biological fluids of multiple sclerosis patients. Clin. Biochem. 42, 1001–1006. doi: 10.1016/j.clinbiochem.2009.03.020
Ansari-Ramandi, M. M., Maleki, M., Alizadehasl, A., Amin, A., Taghavi, S., Alemzadeh-Ansari, M. J., et al. (2017). Safety and effect of high dose allopurinol in patients with severe left ventricular systolic dysfunction. J. Cardiovasc. Thorac. Res. 9, 102–107. doi: 10.15171/jcvtr.2017.17
Arnold, W. L., DeWall, R. A., Kezdi, P., and Zwart, H. H. (1980). The effect of allopurinol on the degree of early myocardial ischemia. Am. Heart J. 99, 614–624. doi: 10.1016/0002-8703(80)90736-X
Ascherio, A., LeWitt, P. A., Xu, K., Eberly, S., Watts, A., Matson, W. R., et al. (2009). Urate as a predictor of the rate of clinical decline in Parkinson disease. Arch. Neurol. 66, 1460–1468. doi: 10.1001/archneurol.2009.247
Barot, M., Gokulgandhi, M. R., and Mitra, A. K. (2011). Mitochondrial dysfunction in retinal diseases. Curr. Eye Res. 36, 1069–1077. doi: 10.3109/02713683.2011.607536
Barrios, C., Hadala, M., Almansa, I., Bosch-Morell, F., Palanca, J. M., and Romero, F. J. (2011). Metabolic muscle damage and oxidative stress markers in an America’s Cup yachting crew. Eur. J. Appl. Physiol. 111, 1341–1350. doi: 10.1007/s00421-010-1762-6
Battelli, M. G., Bolognesi, A., and Polito, L. (2014). Pathophysiology of circulating xanthine oxidoreductase: new emerging roles for a multi-tasking enzyme. Biochim. Biophys. Acta 1842, 1502–1517. doi: 10.1016/j.bbadis.2014.05.022
Beberashvili, I., Erlich, A., Azar, A., Sinuani, I., Feldman, L., Gorelik, O., et al. (2016). Longitudinal study of serum uric acid, nutritional status, and mortality in maintenance hemodialysis patients. Clin. J. Am. Soc. Nephrol. 11, 1015–1023. doi: 10.2215/CJN.10400915
Beberashvili, I., Sinuani, I., Azar, A., Shapiro, G., Feldman, L., Stav, K., et al. (2015). Serum uric acid as a clinically useful nutritional marker and predictor of outcome in maintenance hemodialysis patients. Nutrition 31, 138–147. doi: 10.1016/j.nut.2014.06.012
Benaroudj, N., Zwickl, P., Seemüller, E., Baumeister, W., and Goldberg, A. L. (2003). ATP hydrolysis by the proteasome regulatory complex PAN serves multiple functions in protein degradation. Mol. Cell. 11, 69–78. doi: 10.1016/S1097-2765(02)00775-X
Beveridge, L. A., Ramage, L., McMurdo, M. E., George, J., and Witham, M. D. (2013). Allopurinol use is associated with greater functional gains in older rehabilitation patients. Age Ageing 42, 400–404. doi: 10.1093/ageing/aft046
Bone, D. B., Antic, M., Quinonez, D., and Hammond, J. R. (2015). Hypoxanthine uptake by skeletal muscle microvascular endothelial cells from equilibrative nucleoside transporter 1 (ENT1)-null mice: effect of oxidative stress. Microvasc. Res. 98, 16–22. doi: 10.1016/j.mvr.2014.11.005
Bone, D. B., Choi, D. S., Coe, I. R., and Hammond, J. R. (2010). Nucleoside/nucleobase transport and metabolism by microvascular endothelial cells isolated from ENT1-/- mice. Am. J. Physiol. Heart Circ. Physiol. 299, H847–H856. doi: 10.1152/ajpheart.00018.2010
Bone, D. B., and Hammond, J. R. (2007). Nucleoside and nucleobase transporters of primary human cardiac microvascular endothelial cells: characterization of a novel nucleobase transporter. Am. J. Physiol. Heart Circ. Physiol. 293, H3325–H3332. doi: 10.1152/ajpheart.01006.2007
Boswell-Casteel, R. C., and Hays, F. A. (2017). Equilibrative nucleoside transporters-A review. Nucleosides Nucleotides Nucleic Acids 36, 7–30. doi: 10.1080/15257770.2016.1210805
Bredemeier, M., Lopes, L. M., Eisenreich, M. A., Hickmann, S., Bongiorno, G. K., d’Avila, R., et al. (2018). Xanthine oxidase inhibitors for prevention of cardiovascular events: a systematic review and meta-analysis of randomized controlled trials. BMC Cardiovasc. Disord. 18:24. doi: 10.1186/s12872-018-0757-9
Budillon, G., Citarella, C., Loguercio, C., Nardone, G., Sicolo, P., and Del Vecchio Blanco, C. (1992). Hyperuricemia induced by fructose load in liver cirrhosis. Ital. J. Gastroenterol. 24, 373–377.
Cappola, T. P., Kass, D. A., Nelson, G. S., Berger, R. D., Rosas, G. O., Kobeissi, Z. A., et al. (2001). Allopurinol improves myocardial efficiency in patients with idiopathic dilated cardiomyopathy. Circulation 104, 2407–2411. doi: 10.1161/hc4501.098928
Chapman, A. G., and Atkinson, D. E. (1973). Stabilization of adenylate energy charge by the adenylate deaminase reaction. J. Biol. Chem. 248, 8309–8312.
Che, R., Yuan, Y., Huang, S., and Zhang, A. (2014). Mitochondrial dysfunction in the pathophysiology of renal diseases. Am. J. Physiol. Ren. Physiol. 306, F367–F378. doi: 10.1152/ajprenal.00571.2013
Cheng, J., Morisaki, H., Toyama, K., Ikawa, M., Okabe, M., and Morisaki, T. (2012). AMPD3-deficient mice exhibit increased erythrocyte ATP levels but anemia not improved due to PK deficiency. Genes Cells 17, 913–922. doi: 10.1111/gtc.12006
Cheng, J., Morisaki, H., Toyama, K., Sugimoto, N., Shintani, T., Tandelilin, A., et al. (2014). AMPD1: a novel therapeutic target for reversing insulin resistance. BMC Endocr. Disord. 14:96. doi: 10.1186/1472-6823-14-96
Cheung, K. J., Tzameli, I., Pissios, P., Rovira, I., Gavrilova, O., Ohtsubo, T., et al. (2007). Xanthine oxidoreductase is a regulator of adipogenesis and PPARgamma activity. Cell Metab 5, 115–128. doi: 10.1016/j.cmet.2007.01.005
Chouraki, V., Preis, S. R., Yang, Q., Beiser, A., Li, S., Larson, M. G., et al. (2017). Association of amine biomarkers with incident dementia and Alzheimer’s disease in the framingham study. Alzheimers Dement. 13, 1327–1336. doi: 10.1016/j.jalz.2017.04.009
Ciȩszczyk, P., Ostanek, M., Leoñska-Duniec, A., Sawczuk, M., Maciejewska, A., Eider, J., et al. (2012). Distribution of the AMPD1 C34T polymorphism in Polish power-oriented athletes. J. Sports Sci. 30, 31–35. doi: 10.1080/02640414.2011.623710
Ciȩszczyk, P., Eider, J., Ostanek, M., Leoñska-Duniec, A., Ficek, K., Kotarska, K., et al. (2011). Is the C34T polymorphism of the AMPD1 gene associated with athlete performance in rowing. Int. J. Sports Med. 32, 987–991. doi: 10.1055/s-0031-1283186
Collins, R. P., Palmer, B. R., Pilbrow, A. P., Frampton, C. M., Troughton, R. W., Yandle, T. G., et al. (2006). Evaluation of AMPD1 C34T genotype as a predictor of mortality in heart failure and post-myocardial infarction patients. Am. Heart J. 152, 312–320. doi: 10.1016/j.ahj.2005.12.015
Corte, E. D., and Stirpe, F. (1968). Regulation of xanthine oxidase in rat liver: modifications of the enzyme activity of rat liver supernatant on storage at 20 degrees. Biochem. J. 108, 349–351. doi: 10.1042/bj1080349
Corte, E. D., and Stirpe, F. (1972). The regulation of rat liver xanthine oxidase. Involvement of thiol groups in the conversion of the enzyme activity from dehydrogenase (type D) into oxidase (type O) and purification of the enzyme. Biochem. J. 126, 739–745. doi: 10.1042/bj1260739
Daniels, I. S., O Brien, W. G., Nath, V., Zhao, Z., and Lee, C. C. (2013). AMP deaminase 3 deficiency enhanced 5’-AMP induction of hypometabolism. PLoS One 8:e75418. doi: 10.1371/journal.pone.0075418
de Bragança, A. C., Moreau, R. L. M., de Brito, T., Shimizu, M. H. M., Canale, D., de Jesus, D. A., et al. (2017). Ecstasy induces reactive oxygen species, kidney water absorption and rhabdomyolysis in normal rats. Effect of N-acetylcysteine and Allopurinol in oxidative stress and muscle fiber damage. PLoS One 12:e0179199. doi: 10.1371/journal.pone.0179199
de Groote, P., Lamblin, N., Helbecque, N., Mouquet, F., Hermant, X., Amouyel, P., et al. (2006). The impact of the AMPD1 gene polymorphism on exercise capacity, other prognostic parameters, and survival in patients with stable congestive heart failure: a study in 686 consecutive patients. Am. Heart J. 152, 736–741. doi: 10.1016/j.ahj.2006.04.003
de Lau, L. M., Koudstaal, P. J., Hofman, A., and Breteler, M. M. (2005). Serum uric acid levels and the risk of Parkinson disease. Ann. Neurol. 58, 797–800. doi: 10.1002/ana.20663
De Ruiter, C. J., May, A. M., van Engelen, B. G., Wevers, R. A., Steenbergen-Spanjers, G. C., and de Haan, A. (2002). Muscle function during repetitive moderate-intensity muscle contractions in myoadenylate deaminase-deficient Dutch subjects. Clin Sci 102, 531–539. doi: 10.1042/cs1020531
Della Corte, E., and Stirpe, F. (1968). The regulation of rat-liver xanthine oxidase: Activation by proteolytic enzymes. FEBS Lett. 2, 83–84. doi: 10.1016/0014-5793(68)80107-3
Desler, C., Lillenes, M. S., Tønjum, T., and Rasmussen, L. J. (2017). The role of mitochondrial dysfunction in the progression of Alzheimer’s disease. Curr. Med. Chem. doi: 10.2174/0929867324666170616110111 [Epub ahead of print].
DeWall, R. A., Vasko, K. A., Stanley, E. L., and Kezdi, P. (1971). Responses of the ischemic myocardium to allopurinol. Am. Heart J. 82, 362–370. doi: 10.1016/0002-8703(71)90302-4
Diebold, L., and Chandel, N. S. (2016). Mitochondrial ROS regulation of proliferating cells. Free Radic. Biol. Med. 100, 86–93. doi: 10.1016/j.freeradbiomed.2016.04.198
Donderski, R., Miśkowiec-Wiśniewska, I., Kretowicz, M., Grajewska, M., Manitius, J., Kamiñska, A., et al. (2015). The fructose tolerance test in patients with chronic kidney disease and metabolic syndrome in comparison to healthy controls. BMC Nephrol. 16:68. doi: 10.1186/s12882-015-0048-y
Druloviæ, J., Dujmoviæ, I., Stojsavljeviæ, N., Mesaros, S., Andjelkoviæ, S., Miljkoviæ, D., et al. (2001). Uric acid levels in sera from patients with multiple sclerosis. J. Neurol. 248, 121–126.
Edwards, N. L., Recker, D., and Fox, I. H. (1979). Overproduction of uric acid in hypoxanthine-guanine phosphoribosyltransferase deficiency. Contribution by impaired purine salvage. J. Clin. Invest. 63, 922–930. doi: 10.1172/JCI109392
Ekelund, U. E., Harrison, R. W., Shokek, O., Thakkar, R. N., Tunin, R. S., Senzaki, H., et al. (1999). Intravenous allopurinol decreases myocardial oxygen consumption and increases mechanical efficiency in dogs with pacing-induced heart failure. Circ. Res. 85, 437–445.
El-Bassossy, H. M., Mahmoud, M. F., and Eid, B. G. (2018). The vasodilatory effect of allopurinol mediates its antihypertensive effect: effects on calcium movement and cardiac hemodynamics. Biomed. Pharmacother. 100, 381–387. doi: 10.1016/j.biopha.2018.02.033
Emma, F., Montini, G., Parikh, S. M., and Salviati, L. (2016). Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat. Rev. Nephrol. 12, 267–280. doi: 10.1038/nrneph.2015.214
Emma, F., and Salviati, L. (2017). Mitochondrial cytopathies and the kidney. Nephrol. Ther. 13(Suppl. 1), S23–S28. doi: 10.1016/j.nephro.2017.01.014
Engelberg, J. (1996). How many pounds of oxygen do we “eat” each day. Am. J. Physiol. 271, S43–S44. doi: 10.1152/advances.1996.271.6.S43
Engerson, T. D., McKelvey, T. G., Rhyne, D. B., Boggio, E. B., Snyder, S. J., and Jones, H. P. (1987). Conversion of xanthine dehydrogenase to oxidase in ischemic rat tissues. J. Clin. Invest. 79, 1564–1570. doi: 10.1172/JCI112990
Esteve, C., Jones, E. A., Kell, D. B., Boutin, H., and McDonnell, L. A. (2017). Mass spectrometry imaging shows major derangements in neurogranin and in purine metabolism in the triple-knockout 3 × Tg Alzheimer mouse model. Biochim. Biophys. Acta 1865, 747–754. doi: 10.1016/j.bbapap.2017.04.002
Euser, S. M., Hofman, A., Westendorp, R. G., and Breteler, M. M. (2009). Serum uric acid and cognitive function and dementia. Brain 132, 377–382. doi: 10.1093/brain/awn316
Feng, A. F., Liu, Z. H., Zhou, S. L., Zhao, S. Y., Zhu, Y. X., and Wang, H. X. (2017). Effects of AMPD1 gene C34T polymorphism on cardiac index, blood pressure and prognosis in patients with cardiovascular diseases: a meta-analysis. BMC Cardiovasc. Disord. 17:174. doi: 10.1186/s12872-017-0608-0
Ferrando, B., Gomez-Cabrera, M. C., Salvador-Pascual, A., Puchades, C., Derbré, F., Gratas-Delamarche, A., et al. (2018). Allopurinol partially prevents disuse muscle atrophy in mice and humans. Sci. Rep. 8:3549. doi: 10.1038/s41598-018-21552-1
Fischer, H., Esbjörnsson, M., Sabina, R. L., Strömberg, A., Peyrard-Janvid, M., and Norman, B. (2007). AMP deaminase deficiency is associated with lower sprint cycling performance in healthy subjects. J. Appl. Physiol. 103, 315–322. doi: 10.1152/japplphysiol.00185.2007
Fishbein, W. N., Armbrustmacher, V. W., and Griffin, J. L. (1978). Myoadenylate deaminase deficiency: a new disease of muscle. Science 200, 545–548. doi: 10.1126/science.644316
Foody, J., Turpin, R. S., Tidwell, B. A., Lawrence, D., and Schulman, K. L. (2017). Major cardiovascular events in patients with gout and associated cardiovascular disease or heart failure and chronic kidney disease initiating a xanthine oxidase inhibitor. Am. Health Drug Benefits 10, 393–401.
Frederiks, W. M., and Bosch, K. S. (1996). The proportion of xanthine oxidase activity of total xanthine oxidoreductase activity in situ remains constant in rat liver under various (patho)physiological conditions. Hepatology 24, 1179–1184. doi: 10.1002/hep.510240533
Frederiks, W. M., Marx, F., and Kooij, A. (1993). The effect of ischaemia on xanthine oxidase activity in rat intestine and liver. Int. J. Exp. Pathol. 74, 21–26.
Fried, R., and Fried, L. W. (1974). “Xanthine oxidase (xanthine dehydrogenase),” in Methods of Enzymatic Analysis, ed. H.-U. Bergmeyer (New York, NY: Academic Press, Inc.), 2.
Furuhashi, M., Matsumoto, M., Murase, T., Nakamura, T., Higashiura, Y., Koyama, M., et al. (2018). Independent links between plasma xanthine oxidoreductase activity and levels of adipokines. J. Diabetes Investig. doi: 10.1111/jdi.12982 [Epub ahead of print].
Furukawa, J., Inoue, K., Maeda, J., Yasujima, T., Ohta, K., Kanai, Y., et al. (2015). Functional identification of SLC43A3 as an equilibrative nucleobase transporter involved in purine salvage in mammals. Sci. Rep. 5:15057. doi: 10.1038/srep15057
Garrett, R. H., and Grisham, C. M. (2016). The Synthesis and Degradation of Nucleotides Biochemistry. Boston: Cengage Learning, 927–956.
Gastmann, A., Sigusch, H. H., Henke, A., Reinhardt, D., Surber, R., Gastmann, O., et al. (2004). Role of adenosine monophosphate deaminase-1 gene polymorphism in patients with congestive heart failure (influence on tumor necrosis factor-alpha level and outcome). Am. J. Cardiol. 93, 1260–1264. doi: 10.1016/j.amjcard.2004.02.011
Gavin, A. D., and Struthers, A. D. (2005). Allopurinol reduces B-type natriuretic peptide concentrations and haemoglobin but does not alter exercise capacity in chronic heart failure. Heart 91, 749–753. doi: 10.1136/hrt.2004.040477
Giler, S., Ventura, E., Levy, E., Urca, I., Sperling, O., and de Vries, A. (1976). Elevation of serum xanthine oxidase following halothane anesthesia in the rat. Experientia 32, 620–621. doi: 10.1007/BF0199019
Giler, S. H., Sperling, O., Ventura, E., Levy, E., Urca, I., and De Vries, A. (1977). Effect of methoxyflurane anesthesia on serum uric acid in man. Biomedicine 27, 13–15.
Ginevičienė, V., Jakaitienë, A., Pranculis, A., Milašius, K., Tubelis, L., and Utkus, A. (2014). AMPD1 rs17602729 is associated with physical performance of sprint and power in elite Lithuanian athletes. BMC Genet. 15:58. doi: 10.1186/1471-2156-15-58
Gonsette, R. E., Sindic, C., D’hooghe, M. B., De Deyn, P. P., Medaer, R., Michotte, A., et al. (2010). Boosting endogenous neuroprotection in multiple sclerosis: the association of inosine and interferon beta in relapsing- remitting multiple sclerosis (ASIIMS) trial. Mult. Scler. 16, 455–462. doi: 10.1177/1352458509360547
Goodman, A. M., Wheelock, M. D., Harnett, N. G., Mrug, S., Granger, D. A., and Knight, D. C. (2016). The hippocampal response to psychosocial stress varies with salivary uric acid level. Neuroscience 339, 396–401. doi: 10.1016/j.neuroscience.2016.10.002
Gotsman, I., Keren, A., Lotan, C., and Zwas, D. R. (2012). Changes in uric acid levels and allopurinol use in chronic heart failure: association with improved survival. J. Card. Fail. 18, 694–701. doi: 10.1016/j.cardfail.2012.06.528
Grealy, R., Herruer, J., Smith, C. L., Hiller, D., Haseler, L. J., and Griffiths, L. R. (2015). Evaluation of a 7-gene genetic profile for athletic endurance phenotype in ironman championship triathletes. PLoS One 10:e0145171. doi: 10.1371/journal.pone.0145171
GTEx Consortium. (2017). Genetic effects on gene expression across human tissues. Nature 550, 204–213. doi: 10.1038/nature24277
Guimarães-Ferreira, L. (2014). Role of the phosphocreatine system on energetic homeostasis in skeletal and cardiac muscles. Einstein 12, 126–131. doi: 10.1590/S1679-45082014RB2741
Hamosh, A. (2017). Data from Lesch-Nyhan Syndrome; LNS. Available at: https://www.omim.org/entry/300322
Hanisch, F., Joshi, P., and Zierz, S. (2008). AMP deaminase deficiency in skeletal muscle is unlikely to be of clinical relevance. J. Neurol. 255, 318–322. doi: 10.1007/s00415-008-0530-6
Hardie, D. G., Schaffer, B. E., and Brunet, A. (2016). AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell. Biol. 26, 190–201. doi: 10.1016/j.tcb.2015.10.013
Harkness, R. A. (1988). Hypoxanthine, xanthine and uridine in body fluids, indicators of ATP depletion. J. Chromatogr. 429, 255–278. doi: 10.1016/S0378-4347(00)83873-6
Harkness, R. A., and Lund, R. J. (1983). Cerebrospinal fluid concentrations of hypoxanthine, xanthine, uridine and inosine: high concentrations of the ATP metabolite, hypoxanthine, after hypoxia. J. Clin. Pathol. 36, 1–8. doi: 10.1136/jcp.36.1.1
Harkness, R. A., Simmonds, R. J., and Coade, S. B. (1983). Purine transport and metabolism in man: the effect of exercise on concentrations of purine bases, nucleosides and nucleotides in plasma, urine, leucocytes and erythrocytes. Clin. Sci. 64, 333–340. doi: 10.1042/cs0640333
Harrow, J., Frankish, A., Gonzalez, J. M., Tapanari, E., Diekhans, M., Kokocinski, F., et al. (2012). GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774. doi: 10.1101/gr.135350.111
Havelund, J. F., Heegaard, N. H. H., Færgeman, N. J. K., and Gramsbergen, J. B. (2017). Biomarker research in parkinson’s disease using metabolite profiling. Metabolites 7:42. doi: 10.3390/metabo7030042
Hellsten, Y., Richter, E. A., Kiens, B., and Bangsbo, J. (1999). AMP deamination and purine exchange in human skeletal muscle during and after intense exercise. J. Physiol. 520(Pt 3), 909–920. doi: 10.1111/j.1469-7793.1999.00909.x
Hellsten, Y., Sjödin, B., Richter, E. A., and Bangsbo, J. (1998). Urate uptake and lowered ATP levels in human muscle after high-intensity intermittent exercise. Am. J. Physiol. 274, E600–E606. doi: 10.1152/ajpendo.1998.274.4.E600
Hellsten-Westing, Y. (1993). Immunohistochemical localization of xanthine oxidase in human cardiac and skeletal muscle. Histochemistry 100, 215–222. doi: 10.1007/BF0026909
Hinson, J. T., Fantin, V. R., Schönberger, J., Breivik, N., Siem, G., McDonough, B., et al. (2007). Missense mutations in the BCS1L gene as a cause of the Björnstad syndrome. N. Engl. J. Med. 356, 809–819. doi: 10.1056/NEJMoa055262
Hirsch, G. A., Bottomley, P. A., Gerstenblith, G., and Weiss, R. G. (2012). Allopurinol acutely increases adenosine triphospate energy delivery in failing human hearts. J. Am. Coll. Cardiol. 59, 802–808. doi: 10.1016/j.jacc.2011.10.895
Hitchings, G. H. (1966). Effects of allopurinol in relation to purine biosynthesis. Ann. Rheum. Dis. 25, 601–607. doi: 10.1136/ard.25.Suppl_6.601
Hon, C. C., Ramilowski, J. A., Harshbarger, J., Bertin, N., Rackham, O. J., Gough, J., et al. (2017). An atlas of human long non-coding RNAs with accurate 5’ ends. Nature 543, 199–204. doi: 10.1038/nature21374
Hong, J. Y., Lan, T. Y., Tang, G. J., Tang, C. H., Chen, T. J., and Lin, H. Y. (2015). Gout and the risk of dementia: a nationwide population-based cohort study. Arthritis Res. Ther. 17:139. doi: 10.1186/s13075-015-0642-1
Honorat, J. A., Kinoshita, M., Okuno, T., Takata, K., Koda, T., Tada, S., et al. (2013). Xanthine oxidase mediates axonal and myelin loss in a murine model of multiple sclerosis. PLoS One 8:e71329. doi: 10.1371/journal.pone.0071329
Honorat, J. A., Nakatsuji, Y., Shimizu, M., Kinoshita, M., Sumi-Akamaru, H., Sasaki, T., et al. (2017). Febuxostat ameliorates secondary progressive experimental autoimmune encephalomyelitis by restoring mitochondrial energy production in a GOT2-dependent manner. PLoS One 12:e0187215. doi: 10.1371/journal.pone.0187215
Hopson, S. B., Lust, R. M., Sun, Y. S., Zeri, R. S., Morrison, R. F., Otaki, M., et al. (1995). Allopurinol improves myocardial reperfusion injury in a xanthine oxidase-free model. J. Natl. Med. Assoc. 87, 480–484.
Horiuchi, H., Ota, M., Kobayashi, M., Kaneko, H., Kasahara, Y., Nishimura, S., et al. (1999). A comparative study on the hypouricemic activity and potency in renal xanthine calculus formation of two xanthine oxidase/xanthine dehydrogenase inhibitors: TEI-6720 and allopurinol in rats. Res. Commun. Mol. Pathol. Pharmacol. 104, 307–319.
Hosoyamada, M., Tsurumi, Y., Hirano, H., Tomioka, N. H., Sekine, Y., Morisaki, T., et al. (2016). Urat1-Uox double knockout mice are experimental animal models of renal hypouricemia and exercise-induced acute kidney injury. Nucleosides Nucleotides Nucleic Acids 35, 543–549. doi: 10.1080/15257770.2016.1143559
Hou, M., Hu, Q., Chen, Y., Zhao, L., Zhang, J., and Bache, R. J. (2006). Acute effects of febuxostat, a nonpurine selective inhibitor of xanthine oxidase, in pacing induced heart failure. J. Cardiovasc. Pharmacol. 48, 255–263. doi: 10.1097/01.fjc.0000249961.61451.da
Institute for Energy Research [IER] (2010). A Primer on Energy and the Economy: Energy’s Large Share of the Economy Requires Caution in Determining Policies That Affect It. Available at: https://www.instituteforenergyresearch.org/uncategorized/a-primer-on-energy-and-the-economy-energys-large-share-of-the-economy-requires-caution-in-determining-policies-that-affect-it/ [accessed August 30, 2018].
Ipata, P. L. (2011). Origin, utilization, and recycling of nucleosides in the central nervous system. Adv. Physiol. Educ. 35, 342–346. doi: 10.1152/advan.00068.2011
Ipata, P. L., Camici, M., Micheli, V., and Tozz, M. G. (2011). Metabolic network of nucleosides in the brain. Curr. Top. Med. Chem. 11, 909–922. doi: 10.2174/156802611795347555
Jeon, B. R., Yeom, D. H., and Lee, S. M. (2001). Protective effect of allopurinol on hepatic energy metabolism in ischemic and reperfused rat liver. Shock 15, 112–117.
Jiménez, M. L., Puig, J. G., Mateos, F. A., Ramos, T. H., Castroviejo, I. P., and Vázquez, J. O. (1989a). Hypoxanthine and xanthine transport through the blood-brain barrier in hypoxanthine phosphoribosyltransferase (HPRT) deficiency. Adv. Exp. Med. Biol. 253A, 173–179. doi: 10.1007/978-1-4684-5673-8_28
Jiménez, M. L., Puig, J. G., Mateos, F. A., Ramos, T. H., Melián, J. S., Nieto, V. G., et al. (1989b). Increased purine nucleotide degradation in the central nervous system (CNS) in PRPP synthetase superactivity. Adv. Exp. Med. Biol. 253A, 9–13. doi: 10.1007/978-1-4684-5673-8_2
Johansen, K. K., Wang, L., Aasly, J. O., White, L. R., Matson, W. R., Henchcliffe, C., et al. (2009). Metabolomic profiling in LRRK2-related Parkinson’s disease. PLoS One 4:e7551. doi: 10.1371/journal.pone.0007551
Johnson, K. A., Fox, N. C., Sperling, R. A., and Klunk, W. E. (2012). Brain imaging in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006213. doi: 10.1101/cshperspect.a006213
Johnson, R. J., Sautin, Y. Y., Oliver, W. J., Roncal, C., Mu, W., Gabriela Sanchez-Lozada, L., et al. (2009). Lessons from comparative physiology: could uric acid represent a physiologic alarm signal gone awry in western society. J. Comp. Physiol. B 179, 67–76. doi: 10.1007/s00360-008-0291-7
Kamatani, N., Hashimoto, M., Sakurai, K., Gokita, K., Yoshihara, J., Sekine, M., et al. (2017). Clinical studies on changes in purine compounds in blood and urine by the simultaneous administration of febuxostat and inosine, or by single administration of each. Gout Nucleic Acid Metab. 41, 171–181. doi: 10.6032/gnam.41.171
Kamatani, N., Kushiyama, A., Toyo-oka, L., and Toyo-oka, T. (2019). Treatment of two mitochondrial disease patients with a combination of febuxostat and inosine that enhances cellular ATP. J. Hum. Genet. doi: 10.1038/s10038-018-0558-0 [Epub ahead of print].
Kamatani, N., Taniguchi, A., Yamaoka, N., Kanno, H., Furihata, K., Fukuuchi, T., et al. (2018). In vitro enhancement of ATP in human erythrocytes from a healthy subject and two patients with thalassemia and hemoglobinopathy. Gout Nucleic Acid Metab. 42:59. doi: 10.6032/gnam.42.59
Kato, S., Kato, M., Kusano, T., and Nishino, T. (2016). New strategy that delays progression of amyotrophic lateral sclerosis in G1H-G93A transgenic mice: oral administration of xanthine oxidoreductase inhibitors that are not substrates for the purine salvage pathway. J. Neuropathol. Exp. Neurol. 75, 1124–1144. doi: 10.1093/jnen/nlw088
Kelkar, A., Kuo, A., and Frishman, W. H. (2011). Allopurinol as a cardiovascular drug. Cardiol. Rev. 19, 265–271. doi: 10.1097/CRD.0b013e318229a908
Kelley, E. E. (2015). A new paradigm for XOR-catalyzed reactive species generation in the endothelium. Pharmacol. Rep. 67, 669–674. doi: 10.1016/j.pharep.2015.05.004
Khatib, S. Y., Farah, H., and El-Migdadi, F. (2001). Allopurinol enhances adenine nucleotide levels and improves myocardial function in isolated hypoxic rat heart. Biochemistry 66, 328–333.
Kim, M. S., Pinto, S. M., Getnet, D., Nirujogi, R. S., Manda, S. S., Chaerkady, R., et al. (2014). A draft map of the human proteome. Nature 509, 575–581. doi: 10.1038/nature13302
Koch, J., Mayr, J. A., Alhaddad, B., Rauscher, C., Bierau, J., Kovacs-Nagy, R., et al. (2017). CAD mutations and uridine-responsive epileptic encephalopathy. Brain 140, 279–286. doi: 10.1093/brain/aww300
Kori, M., Aydın, B., Unal, S., Arga, K. Y., and Kazan, D. (2016). Metabolic biomarkers and neurodegeneration: a pathway enrichment analysis of alzheimer’s disease, parkinson’s disease, and amyotrophic lateral sclerosis. OMICS 20, 645–661. doi: 10.1089/omi.2016.0106
Kratzer, J. T., Lanaspa, M. A., Murphy, M. N., Cicerchi, C., Graves, C. L., Tipton, P. A., et al. (2014). Evolutionary history and metabolic insights of ancient mammalian uricases. Proc. Natl. Acad. Sci. U.S.A. 111, 3763–3768. doi: 10.1073/pnas.1320393111
Lanaspa, M. A., Cicerchi, C., Garcia, G., Li, N., Roncal-Jimenez, C. A., Rivard, C. J., et al. (2012). Counteracting roles of AMP deaminase and AMP kinase in the development of fatty liver. PLoS One 7:e48801. doi: 10.1371/journal.pone.0048801
Lanaspa, M. A., Epperson, L. E., Li, N., Cicerchi, C., Garcia, G. E., Roncal-Jimenez, C. A., et al. (2015). Opposing activity changes in AMP deaminase and AMP-activated protein kinase in the hibernating ground squirrel. PLoS One 10:e0123509. doi: 10.1371/journal.pone.0123509
Larsen, K. S., Pottegård, A., Lindegaard, H. M., and Hallas, J. (2016). Effect of allopurinol on cardiovascular outcomes in hyperuricemic patients: a cohort study. Am. J. Med. 129, 299.e2–306.e2. doi: 10.1016/j.amjmed.2015.11.003
Lazzarino, G., Amorini, A. M., Petzold, A., Gasperini, C., Ruggieri, S., Quartuccio, M. E., et al. (2017). Serum compounds of energy metabolism impairment are related to disability, disease course and neuroimaging in multiple sclerosis. Mol. Neurobiol. 54, 7520–7533. doi: 10.1007/s12035-016-0257-9
Lazzarino, G., Raatikainen, P., Nuutinen, M., Nissinen, J., Tavazzi, B., Di Pierro, D., et al. (1994). Myocardial release of malondialdehyde and purine compounds during coronary bypass surgery. Circulation 90, 291–297. doi: 10.1161/01.CIR.90.1.291
Lee, J., Hu, Q., Mansoor, A., Kamdar, F., and Zhang, J. (2011). Effect of acute xanthine oxidase inhibition on myocardial energetics during basal and very high cardiac workstates. J. Cardiovasc. Transl. Res. 4, 504–513. doi: 10.1007/s12265-011-9276-0
Lee, W. S., and Sokol, R. J. (2007). Liver disease in mitochondrial disorders. Semin. Liver Dis. 27, 259–273. doi: 10.1055/s-2007-985071
Li, B., Qing, T., Zhu, J., Wen, Z., Yu, Y., Fukumura, R., et al. (2017). A comprehensive mouse transcriptomic bodymap across 17 tissues by RNA-seq. Sci. Rep. 7:4200. doi: 10.1038/s41598-017-04520-z
Lieber, C. S. (1965). Hyperuricemia induced by alcohol. Arthritis Rheum. 8, 786–798. doi: 10.1002/art.1780080442
Lin, A. L., and Rothman, D. L. (2014). What have novel imaging techniques revealed about metabolism in the aging brain. Future Neurol. 9, 341–354. doi: 10.2217/fnl.14.13
Linder, N. (2005). Expression and Regulation of Human Xanthine Oxidoreductase. Available at: http://urn.fi/URN:ISBN:952-10-2277-9
Linder, N., Lundin, J., Isola, J., Lundin, M., Raivio, K. O., and Joensuu, H. (2005). Down-regulated xanthine oxidoreductase is a feature of aggressive breast cancer. Clin. Cancer Res. 11, 4372–4381. doi: 10.1158/1078-0432.CCR-04-2280
Linder, N., Martelin, E., Lapatto, R., and Raivio, K. O. (2003). Posttranslational inactivation of human xanthine oxidoreductase by oxygen under standard cell culture conditions. Am. J. Physiol. Cell. Physiol. 285, C48–C55. doi: 10.1152/ajpcell.00561.2002
Linder, N., Rapola, J., and Raivio, K. O. (1999). Cellular expression of xanthine oxidoreductase protein in normal human tissues. Lab. Invest. 79, 967–974.
Liu, B., Shen, Y., Xiao, K., Tang, Y., Cen, L., and Wei, J. (2012). Serum uric acid levels in patients with multiple sclerosis: a meta-analysis. Neurol. Res. 34, 163–171. doi: 10.1179/1743132811Y.0000000074
Lizio, M., Harshbarger, J., Abugessaisa, I., Noguchi, S., Kondo, A., Severin, J., et al. (2017). Update of the FANTOM web resource: high resolution transcriptome of diverse cell types in mammals. Nucleic Acids Res. 45, D737–D743. doi: 10.1093/nar/gkw995
Lizio, M., Harshbarger, J., Shimoji, H., Severin, J., Kasukawa, T., Sahin, S., et al. (2015). Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 16:22. doi: 10.1186/s13059-014-0560-6
Loh, E., Rebbeck, T. R., Mahoney, P. D., DeNofrio, D., Swain, J. L., and Holmes, E. W. (1999). Common variant in AMPD1 gene predicts improved clinical outcome in patients with heart failure. Circulation 99, 1422–1425. doi: 10.1161/01.CIR.99.11.1422
Lu, N., Dubreuil, M., Zhang, Y., Neogi, T., Rai, S. K., Ascherio, A., et al. (2016). Gout and the risk of Alzheimer’s disease: a population-based, BMI-matched cohort study. Ann. Rheum. Dis. 75, 547–551. doi: 10.1136/annrheumdis-2014-206917
MacIsaac, R. L., Salatzki, J., Higgins, P., Walters, M. R., Padmanabhan, S., Dominiczak, A. F., et al. (2016). Allopurinol and Cardiovascular outcomes in adults with hypertension. Hypertension 67, 535–540. doi: 10.1161/HYPERTENSIONAHA.115.06344
Maesaka, J. K., Wolf-Klein, G., Piccione, J. M., and Ma, C. M. (1993). Hypouricemia, abnormal renal tubular urate transport, and plasma natriuretic factor(s) in patients with Alzheimer’s disease. J. Am. Geriatr. Soc. 41, 501–506. doi: 10.1111/j.1532-5415.1993.tb01885.x
Mahnke-Zizelman, D. K., and Sabina, R. L. (1992). Cloning of human AMP deaminase isoform E cDNAs. Evidence for a third AMPD gene exhibiting alternatively spliced 5’-exons. J. Biol. Chem. 267, 20866–20877.
Maiuolo, J., Oppedisano, F., Gratteri, S., Muscoli, C., and Mollace, V. (2016). Regulation of uric acid metabolism and excretion. Int. J. Cardiol. 213, 8–14. doi: 10.1016/j.ijcard.2015.08.109
Manfredi, J. P., and Holmes, E. W. (1985). Purine salvage pathways in myocardium. Annu. Rev. Physiol. 47, 691–705. doi: 10.1146/annurev.ph.47.030185.003355
Markowitz, C. E., Spitsin, S., Zimmerman, V., Jacobs, D., Udupa, J. K., Hooper, D. C., et al. (2009). The treatment of multiple sclerosis with inosine. J. Altern. Complement. Med. 15, 619–625. doi: 10.1089/acm.2008.0513
Martin, H. M., Moore, K. P., Bosmans, E., Davies, S., Burroughs, A. K., Dhillon, A. P., et al. (2004). Xanthine oxidoreductase is present in bile ducts of normal and cirrhotic liver. Free Radic. Biol. Med. 37, 1214–1223. doi: 10.1016/j.freeradbiomed.2004.06.045
Massey, V., and Harris, C. M. (1997). Milk xanthine oxidoreductase: the first one hundred years. Biochem. Soc. Trans. 25, 750–755. doi: 10.1042/bst0250750
McFarland, N. R., Burdett, T., Desjardins, C. A., Frosch, M. P., and Schwarzschild, M. A. (2013). Postmortem brain levels of urate and precursors in Parkinson’s disease and related disorders. Neurodegener. Dis. 12, 189–198. doi: 10.1159/000346370
McKelvey, T. G., Höllwarth, M. E., Granger, D. N., Engerson, T. D., Landler, U., and Jones, H. P. (1988). Mechanisms of conversion of xanthine dehydrogenase to xanthine oxidase in ischemic rat liver and kidney. Am. J. Physiol. 254, G753–G760. doi: 10.1152/ajpgi.1988.254.5.G753
Moccia, M., Lanzillo, R., Palladino, R., Russo, C., Carotenuto, A., Massarelli, M., et al. (2015). Uric acid: a potential biomarker of multiple sclerosis and of its disability. Clin. Chem. Lab. Med. 53, 753–759. doi: 10.1515/cclm-2014-0744
Morgan, E. J., Stewart, C. P., and Hopkins, F. G. (1922). On the anaerobic and aerobic oxidation of xanthin and hypoxanthin by tissues and by milk. Proc. R. Soc. Lond. B 94, 109–131. doi: 10.1098/rspb.1922.0047
Morisaki, T., Sabina, R. L., and Holmes, E. W. (1990). Adenylate deaminase. A multigene family in humans and rats. J. Biol. Chem. 265, 11482–11486.
Moriwaki, Y., Yamamoto, T., Yamaguchi, K., Suda, M., Yamakita, I., and Takahashi, S. (1996). Immunohistochemical localization of xanthine oxidase in human tissues. Acta Histochem. Cytochem. 29, 153–162. doi: 10.1267/ahc.29.153
Mosconi, L. (2013). Glucose metabolism in normal aging and Alzheimer’s disease: methodological and physiological considerations for PET studies. Clin. Transl. Imaging 1, 217–233. doi: 10.1007/s40336-013-0026-y
Muniesa, C. A., González-Freire, M., Santiago, C., Lao, J. I., Buxens, A., Rubio, J. C., et al. (2010). World-class performance in lightweight rowing: is it genetically influenced? A comparison with cyclists, runners and non-athletes. Br. J. Sports Med. 44, 898–901. doi: 10.1136/bjsm.2008.051680
Muñoz García, D., Midaglia, L., Martinez Vilela, J., Marín Sánchez, M., López González, F. J., Arias Gómez, M., et al. (2015). Associated Inosine to interferon: results of a clinical trial in multiple sclerosis. Acta Neurol. Scand. 131, 405–410. doi: 10.1111/ane.12333
Murakami, N., Ohtsubo, T., Kansui, Y., Goto, K., Noguchi, H., Haga, Y., et al. (2014). Mice heterozygous for the xanthine oxidoreductase gene facilitate lipid accumulation in adipocytes. Arterioscler. Thromb. Vasc. Biol. 34, 44–51. doi: 10.1161/ATVBAHA.113.302214
Murase, T., Nampei, M., Oka, M., Miyachi, A., and Nakamura, T. (2016). A highly sensitive assay of human plasma xanthine oxidoreductase activity using stable isotope-labeled xanthine and LC/TQMS. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 1039, 51–58. doi: 10.1016/j.jchromb.2016.10.033
Murray, A. W. (1971). The biological significance of purine salvage. Annu. Rev. Biochem. 40, 811–826. doi: 10.1146/annurev.bi.40.070171.004115
Murray, A. W., Elliott, D. C., and Atkinson, M. R. (1970). Nucleotide biosynthesis from preformed purines in mammalian cells: regulatory mechanisms and biological significance. Prog. Nucleic Acid Res. Mol. Biol. 10, 87–119. doi: 10.1016/S0079-6603(08)60562-0
Ng, R. H., Menon, M., and Ladenson, J. H. (1984). Collection and handling of 24-hour urine specimens for measurement of analytes related to renal calculi. Clin. Chem. 30, 467–471.
Nguyen, K. V., and Nyhan, W. L. (2016). Mutation in the human HPRT1 gene and the lesch-nyhan syndrome. Nucleosides Nucleotides Nucleic Acids 35, 426–433. doi: 10.1080/15257770.2015.1098660
Nielsen, V. G., Weinbroum, A., Tan, S., Samuelson, P. N., Gelman, S., and Parks, D. A. (1994). Xanthine oxidoreductase release after descending thoracic aorta occlusion and reperfusion in rabbits. J. Thorac. Cardiovasc. Surg. 107, 1222–1227. doi: 10.1016/S0022-5223(94)70041-9
Nishino, T., Kato, S., Kato, M., Suzuki, H., and Okamoto, K. (2016). Drug for Preventing and/or Treating Dementia. WIPO Pat. Appl. PCT/JP2016/055226. Geneva: World Intellectual Property Organization.
Nishino, T., and Okamoto, K. (2015). Mechanistic insights into xanthine oxidoreductase from development studies of candidate drugs to treat hyperuricemia and gout. J Biol Inorg Chem 20, 195–207. doi: 10.1007/s00775-014-1210-x
Nishino, T., Abe, Y., and Kato, S. (2008a). Therapeutic Agent for Amyotrophic Lateral Sclerosis. U.S. Patent No 8,318,792. Washington, DC: U.S. Patent and Trademark Office.
Nishino, T., Okamoto, K., Eger, B. T., Pai, E. F., and Nishino, T. (2008b). Mammalian xanthine oxidoreductase - mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J. 275, 3278–3289. doi: 10.1111/j.1742-4658.2008.06489.x
Nishino, T., Okamoto, K., Kawaguchi, Y., Matsumura, T., Eger, B. T., Pai, E. F., et al. (2015). The C-terminal peptide plays a role in the formation of an intermediate form during the transition between xanthine dehydrogenase and xanthine oxidase. FEBS J. 282, 3075–3090. doi: 10.1111/febs.13277
Nobili, F., Arbizu, J., Bouwman, F., Drzezga, A., Agosta, F., Nestor, P., et al. (2018). European Association of Nuclear Medicine and European Academy of Neurology recommendations for the use of brain 18 F-fluorodeoxyglucose positron emission tomography in neurodegenerative cognitive impairment and dementia: Delphi consensus. Eur. J. Neurol. 25, 1201–1217. doi: 10.1111/ene.13728
Noguchi, S., Arakawa, T., Fukuda, S., Furuno, M., Hasegawa, A., Hori, F., et al. (2017). FANTOM5 CAGE profiles of human and mouse samples. Sci. Data 4:170112. doi: 10.1038/sdata.2017.112
Noman, A., Ang, D. S., Ogston, S., Lang, C. C., and Struthers, A. D. (2010). Effect of high-dose allopurinol on exercise in patients with chronic stable angina: a randomised, placebo controlled crossover trial. Lancet 375, 2161–2167. doi: 10.1016/S0140-6736(10)60391-1
Norman, B., Nygren, A. T., Nowak, J., and Sabina, R. L. (2008). The effect of AMPD1 genotype on blood flow response to sprint exercise. Eur. J. Appl. Physiol. 103, 173–180. doi: 10.1007/s00421-008-0683-0
Norman, B., Sabina, R. L., and Jansson, E. (2001). Regulation of skeletal muscle ATP catabolism by AMPD1 genotype during sprint exercise in asymptomatic subjects. J. Appl. Physiol. 91, 258–264. doi: 10.1152/jappl.2001.91.1.258
Novarino, G., Fenstermaker, A. G., Zaki, M. S., Hofree, M., Silhavy, J. L., Heiberg, A. D., et al. (2014). Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 343, 506–511. doi: 10.1126/science.1247363
O’Brien, W. G., Berka, V., Tsai, A. L., Zhao, Z., and Lee, C. C. (2015). CD73 and AMPD3 deficiency enhance metabolic performance via erythrocyte ATP that decreases hemoglobin oxygen affinity. Sci. Rep. 5:13147. doi: 10.1038/srep13147
Ogasawara, N., Goto, H., Yamada, Y., Nishigaki, I., Itoh, T., Hasegawa, I., et al. (1987). Deficiency of AMP deaminase in erythrocytes. Hum. Genet 75, 15–18. doi: 10.1007/BF0027383
Onyango, I. G., Khan, S. M., and Bennett, J. P. (2017). Mitochondria in the pathophysiology of Alzheimer’s and Parkinson’s diseases. Front. Biosci. 22, 854–872.
Ozansoy, M., and Başak, A. N. (2013). The central theme of Parkinson’s disease: α-synuclein. Mol. Neurobiol. 47, 460–465. doi: 10.1007/s12035-012-8369-3
Pacher, P., Nivorozhkin, A., and Szabó, C. (2006). Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol. Pharmacol. Rev. 58, 87–114. doi: 10.1124/pr.58.1.6
Paganoni, S., Nicholson, K., Chan, J., Shui, A., Schoenfeld, D., Sherman, A., et al. (2018). Urate levels predict survival in amyotrophic lateral sclerosis: analysis of the expanded Pooled resource open-access ALS clinical trials database. Muscle Nerve 57, 430–434. doi: 10.1002/mus.25950
Panayiotou, C., Solaroli, N., and Karlsson, A. (2014). The many isoforms of human adenylate kinases. Int. J. Biochem. Cell. Biol. 49, 75–83. doi: 10.1016/j.biocel.2014.01.014
Paolocci, N., Tavazzi, B., Biondi, R., Gluzband, Y. A., Amorini, A. M., Tocchetti, C. G., et al. (2006). Metalloproteinase inhibitor counters high-energy phosphate depletion and AMP deaminase activity enhancing ventricular diastolic compliance in subacute heart failure. J. Pharmacol. Exp. Ther. 317, 506–513. doi: 10.1124/jpet.105.099168
Peeters-Scholte, C., Braun, K., Koster, J., Kops, N., Blomgren, K., Buonocore, G., et al. (2003). Effects of allopurinol and deferoxamine on reperfusion injury of the brain in newborn piglets after neonatal hypoxia-ischemia. Pediatr. Res. 54, 516–522. doi: 10.1203/01.PDR.0000081297.53793.C6
Pérez, N. G., Gao, W. D., and Marbán, E. (1998). Novel myofilament Ca2+-sensitizing property of xanthine oxidase inhibitors. Circ. Res. 83, 423–430. doi: 10.1161/01.RES.83.4.423
Peth, A., Nathan, J. A., and Goldberg, A. L. (2013). The ATP costs and time required to degrade ubiquitinated proteins by the 26 S proteasome. J. Biol. Chem. 288, 29215–29222. doi: 10.1074/jbc.M113.482570
Plaideau, C., Lai, Y. C., Kviklyte, S., Zanou, N., Löfgren, L., Andersén, H., et al. (2014). Effects of pharmacological AMP deaminase inhibition and Ampd1 deletion on nucleotide levels and AMPK activation in contracting skeletal muscle. Chem. Biol. 21, 1497–1510. doi: 10.1016/j.chembiol.2014.09.013
Pritchard, J. B., O’Connor, N., Oliver, J. M., and Berlin, R. D. (1975). Uptake and supply of purine compounds by the liver. Am. J. Physiol. 229, 967–972. doi: 10.1152/ajplegacy.1975.229.4.967
Psychogios, N., Hau, D. D., Peng, J., Guo, A. C., Mandal, R., Bouatra, S., et al. (2011). The human serum metabolome. PLoS One 6:e16957. doi: 10.1371/journal.pone.0016957
Quijano, C., Trujillo, M., Castro, L., and Trostchansky, A. (2016). Interplay between oxidant species and energy metabolism. Redox Biol. 8, 28–42. doi: 10.1016/j.redox.2015.11.010
Radi, R., Rubbo, H., Bush, K., and Freeman, B. A. (1997). Xanthine oxidase binding to glycosaminoglycans: kinetics and superoxide dismutase interactions of immobilized xanthine oxidase-heparin complexes. Arch. Biochem. Biophys. 339, 125–135. doi: 10.1006/abbi.1996.9844
Rahman, S., and Hall, A. M. (2013). Mitochondrial disease–an important cause of end-stage renal failure. Pediatr. Nephrol. 28, 357–361. doi: 10.1007/s00467-012-2362-y
Redzic, Z. B., Segal, M. B., Gasic, J. M., Markovic, I. D., Vojvodic, V. P., Isakovic, A., et al. (2001). The characteristics of nucleobase transport and metabolism by the perfused sheep choroid plexus. Brain Res. 888, 66–74. doi: 10.1016/S0006-8993(00)03006-7
Rentoukas, E., Tsarouhas, K., Tsitsimpikou, C., Lazaros, G., Deftereos, S., and Vavetsi, S. (2010). The prognostic impact of allopurinol in patients with acute myocardial infarction undergoing primary percutaneous coronary intervention. Int. J. Cardiol. 145, 257–258. doi: 10.1016/j.ijcard.2009.08.037
Rinaldi, P., Polidori, M. C., Metastasio, A., Mariani, E., Mattioli, P., Cherubini, A., et al. (2003). Plasma antioxidants are similarly depleted in mild cognitive impairment and in Alzheimer’s disease. Neurobiol. Aging 24, 915–919. doi: 10.1016/S0197-4580(03)00031-9
Rubio, J. C., Martín, M. A., Rabadán, M., Gómez-Gallego, F., San Juan, A. F., Alonso, J. M., et al. (2005). Frequency of the C34T mutation of the AMPD1 gene in world-class endurance athletes: does this mutation impair performance. J. Appl. Physiol. 98, 2108–2112. doi: 10.1152/japplphysiol.01371.2004
Ryan, M. J., Jackson, J. R., Hao, Y., Leonard, S. S., and Alway, S. E. (2011). Inhibition of xanthine oxidase reduces oxidative stress and improves skeletal muscle function in response to electrically stimulated isometric contractions in aged mice. Free Radic. Biol. Med. 51, 38–52. doi: 10.1016/j.freeradbiomed.2011.04.002
Sabina, R. L., and Holmes, E. W. (2001). “Myoadenylate deaminase deficiency,” in The Metabolic & Molecular Bases of Inherited Disease, ed. C. R. Scriver (New York, NY: McGraw-Hill).
Safranow, K., Czyzycka, E., Binczak-Kuleta, A., Rzeuski, R., Skowronek, J., Wojtarowicz, A., et al. (2009). Association of C34T AMPD1 gene polymorphism with features of metabolic syndrome in patients with coronary artery disease or heart failure. Scand. J. Clin. Lab. Invest. 69, 102–112. doi: 10.1080/00365510802430964
Saks, V., Schlattner, U., Tokarska-Schlattner, M., Wallimann, T., Bagur, R., Zorman, S., et al. (2014). “Systems level regulation of cardiac energy fluxes via metabolic cycles: role of creatine, phosphotransfer pathways, and AMPK signaling,” in Systems Biology of Metabolic and Signaling Networks Energy, Mass and Information Transfer, eds M. A. Aon, V. Saks, and U. Schlattner (Berlin: Springer-Verlag), 261–322.
Saksela, M., Lapatto, R., and Raivio, K. O. (1999). Irreversible conversion of xanthine dehydrogenase into xanthine oxidase by a mitochondrial protease. FEBS Lett. 443, 117–120. doi: 10.1016/S0014-5793(98)01686-X
Sakuta, H., Suzuki, K., Miyamoto, T., Miyamoto, M., Numao, A., Fujita, H., et al. (2017). Serum uric acid levels in Parkinson’s disease and related disorders. Brain Behav. 7:e00598. doi: 10.1002/brb3.598
Salvadore, G., Viale, C. I., Luckenbaugh, D. A., Zanatto, V. C., Portela, L. V., Souza, D. O., et al. (2010). Increased uric acid levels in drug-naïve subjects with bipolar disorder during a first manic episode. Prog. Neuropsychopharmacol. Biol. Psychiatry 34, 819–821. doi: 10.1016/j.pnpbp.2010.02.027
Sanchis-Gomar, F., Pareja-Galeano, H., Gomez-Cabrera, M. C., Candel, J., Lippi, G., Salvagno, G. L., et al. (2015). Allopurinol prevents cardiac and skeletal muscle damage in professional soccer players. Scand. J. Med. Sci. Sports 25, e110–e115. doi: 10.1111/sms.12213
Saugstad, O. D. (1996). Role of xanthine oxidase and its inhibitor in hypoxia: reoxygenation injury. Pediatrics 98, 103–107.
Schmidt, A. P., Böhmer, A. E., Antunes, C., Schallenberger, C., Porciúncula, L. O., Elisabetsky, E., et al. (2009). Anti-nociceptive properties of the xanthine oxidase inhibitor allopurinol in mice: role of A1 adenosine receptors. Br. J. Pharmacol. 156, 163–172. doi: 10.1111/j.1476-5381.2008.00025.x
Schmidt, J. A., Crowe, F. L., Appleby, P. N., Key, T. J., and Travis, R. C. (2013). Serum uric acid concentrations in meat eaters, fish eaters, vegetarians and vegans: a cross-sectional analysis in the EPIC-Oxford cohort. PLoS One 8:e56339. doi: 10.1371/journal.pone.0056339
Schrier, S. A., and Falk, M. J. (2011). Mitochondrial disorders and the eye. Curr. Opin. Ophthalmol. 22, 325–331. doi: 10.1097/ICU.0b013e328349419d
Schwarzschild, M. A., Schwid, S. R., Marek, K., Watts, A., Lang, A. E., Oakes, D., et al. (2008). Serum urate as a predictor of clinical and radiographic progression in Parkinson disease. Arch. Neurol. 65, 716–723. doi: 10.1001/archneur.2008.65.6.nct70003
Scialò, F., Sriram, A., Fernández-Ayala, D., Gubina, N., Lõhmus, M., Nelson, G., et al. (2016). Mitochondrial ROS produced via reverse electron transport extend animal lifespan. Cell Metab. 23, 725–734. doi: 10.1016/j.cmet.2016.03.009
Seegmiller, J. E., Rosenbloom, F. M., and Kelley, W. N. (1967). Enzyme defect associated with a sex-linked human neurological disorder and excessive purine synthesis. Science 155, 1682–1684. doi: 10.1126/science.155.3770.1682
Singh, J. A., and Cleveland, J. D. (2018a). Comparative effectiveness of allopurinol versus febuxostat for preventing incident dementia in older adults: a propensity-matched analysis. Arthritis Res. Ther. 20:167. doi: 10.1186/s13075-018-1663-3
Singh, J. A., and Cleveland, J. D. (2018b). Use of urate-lowering therapies is not associated with an increase in the risk of incident dementia in older adults. Ann. Rheum. Dis. 77, 1243–1245. doi: 10.1136/annrheumdis-2017-212094
Singh, J. A., Ramachandaran, R., Yu, S., and Curtis, J. R. (2017). Allopurinol use and the risk of acute cardiovascular events in patients with gout and diabetes. BMC Cardiovasc. Disord. 17:76. doi: 10.1186/s12872-017-0513-6
Singh, J. A., and Yu, S. (2016). Allopurinol reduces the risk of myocardial infarction (MI) in the elderly: a study of medicare claims. Arthritis Res. Ther. 18:209. doi: 10.1186/s13075-016-1111-1
Singh, J. A., and Yu, S. (2017). Allopurinol and the risk of atrial fibrillation in the elderly: a study using Medicare data. Ann. Rheum. Dis. 76, 72–78. doi: 10.1136/annrheumdis-2015-209008
Smith, A. C., and Robinson, A. J. (2016). Mitominer v3.1, an update on the mitochondrial proteomics database. Nucleic Acids Res. 44, D1258–D1261. doi: 10.1093/nar/gkv1001
Sotgiu, S., Pugliatti, M., Sanna, A., Sotgiu, A., Fois, M. L., Arru, G., et al. (2002). Serum uric acid and multiple sclerosis. Neurol. Sci. 23, 183–188. doi: 10.1007/s100720200059
Stathis, C. G., Carey, M. F., and Snow, R. J. (2005). The influence of allopurinol on urinary purine loss after repeated sprint exercise in man. Metabolism 54, 1269–1275. doi: 10.1016/j.metabol.2005.02.004
Stathis, C. G., Febbraio, M. A., Carey, M. F., and Snow, R. J. (1994). Influence of sprint training on human skeletal muscle purine nucleotide metabolism. J. Appl. Physiol. 76, 1802–1809. doi: 10.1152/jappl.1994.76.4.1802
Stathis, C. G., Zhao, S., Carey, M. F., and Snow, R. J. (1999). Purine loss after repeated sprint bouts in humans. J. Appl. Physiol. 87, 2037–2042. doi: 10.1152/jappl.1999.87.6.2037
Stefanis, L. (2012). α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2:a009399. doi: 10.1101/cshperspect.a009399
Stirpe, F., and Della Corte, E. (1969). The regulation of rat liver xanthine oxidase. Conversion in vitro of the enzyme activity from dehydrogenase (type D) to oxidase (type O). J. Biol. Chem. 244, 3855–3863.
Stirpe, F., Ravaioli, M., Battelli, M. G., Musiani, S., and Grazi, G. L. (2002). Xanthine oxidoreductase activity in human liver disease. Am. J. Gastroenterol. 97, 2079–2085. doi: 10.1111/j.1572-0241.2002.05925.x
Stull, L. B., Leppo, M. K., Szweda, L., Gao, W. D., and Marbán, E. (2004). Chronic treatment with allopurinol boosts survival and cardiac contractility in murine postischemic cardiomyopathy. Circ. Res. 95, 1005–1011. doi: 10.1161/01.RES.0000148635.73331.c5
Swerdlow, R. H., Koppel, S., Weidling, I., Hayley, C., Ji, Y., and Wilkins, H. M. (2017). Mitochondria, cybrids, aging, and Alzheimer’s Disease. Prog. Mol. Biol. Transl. Sci. 146, 259–302. doi: 10.1016/bs.pmbts.2016.12.017
Szasz, T., Davis, R. P., Garver, H. S., Burnett, R. J., Fink, G. D., and Watts, S. W. (2013). Long-term inhibition of xanthine oxidase by febuxostat does not decrease blood pressure in deoxycorticosterone acetate (DOCA)-salt hypertensive rats. PLoS One 8:e56046. doi: 10.1371/journal.pone.0056046
Tan, S., Gelman, S., Wheat, J. K., and Parks, D. A. (1995). Circulating xanthine oxidase in human ischemia reperfusion. South Med. J. 88, 479–482.
Tan, S., Yokoyama, Y., Dickens, E., Cash, T. G., Freeman, B. A., and Parks, D. A. (1993). Xanthine oxidase activity in the circulation of rats following hemorrhagic shock. Free Radic. Biol. Med. 15, 407–414. doi: 10.1016/0891-5849(93)90040-2
Tarnopolsky, M. A., Parise, G., Gibala, M. J., Graham, T. E., and Rush, J. W. (2001). Myoadenylate deaminase deficiency does not affect muscle anaplerosis during exhaustive exercise in humans. J. Physiol. 533, 881–889. doi: 10.1111/j.1469-7793.2001.t01-1-00881.x
Tavazzi, B., Amorini, A. M., Fazzina, G., Di Pierro, D., Tuttobene, M., Giardina, B., et al. (2001). Oxidative stress induces impairment of human erythrocyte energy metabolism through the oxygen radical-mediated direct activation of AMP-deaminase. J. Biol. Chem. 276, 48083–48092. doi: 10.1074/jbc.M101715200
Tavazzi, B., Batocchi, A. P., Amorini, A. M., Nociti, V., D’Urso, S., Longo, S., et al. (2011). Serum metabolic profile in multiple sclerosis patients. Mult. Scler. Int. 2011:167156. doi: 10.1155/2011/167156
Tavazzi, B., Di Pierro, D., Amorini, A. M., Fazzina, G., Tuttobene, M., Giardina, B., et al. (2000). Energy metabolism and lipid peroxidation of human erythrocytes as a function of increased oxidative stress. Eur. J. Biochem. 267, 684–689. doi: 10.1046/j.1432-1327.2000.01042.x
Thanassoulis, G., Brophy, J. M., Richard, H., and Pilote, L. (2010). Gout, allopurinol use, and heart failure outcomes. Arch. Intern. Med. 170, 1358–1364. doi: 10.1001/archinternmed.2010.198
The Parkinson, Study Group Sure-Pd Investigators, Schwarzschild, M. A., Ascherio, A., Beal, M. F., Cudkowicz, M. E., et al. (2014). Inosine to increase serum and cerebrospinal fluid urate in Parkinson disease: a randomized clinical trial. JAMA Neurol. 71, 141–150. doi: 10.1001/jamaneurol.2013.5528
Tiede, L., Steyger, P. S., Nichols, M. G., and Hallworth, R. (2009). Metabolic imaging of the organ of corti–a window on cochlea bioenergetics. Brain Res. 1277, 37–41. doi: 10.1016/j.brainres.2009.02.052
Tousoulis, D., Kioufis, S., Siasos, G., Oikonomou, E., Zaromitidou, M., Maniatis, K., et al. (2014). The impact of AMPD1 gene polymorphism on vascular function and inflammation in patients with coronary artery disease. Int. J. Cardiol. 172, e516–e518. doi: 10.1016/j.ijcard.2014.01.078
Tsianos, G. I., Evangelou, E., Boot, A., Zillikens, M. C., van Meurs, J. B., Uitterlinden, A. G., et al. (2010). Associations of polymorphisms of eight muscle- or metabolism-related genes with performance in Mount Olympus marathon runners. J. Appl. Physiol. 108, 567–574. doi: 10.1152/japplphysiol.00780.2009
Ukai, T., Cheng, C. P., Tachibana, H., Igawa, A., Zhang, Z. S., Cheng, H. J., et al. (2001). Allopurinol enhances the contractile response to dobutamine and exercise in dogs with pacing-induced heart failure. Circulation 103, 750–755. doi: 10.1161/01.CIR.103.5.750
Verzijl, H. T., van Engelen, B. G., Luyten, J. A., Steenbergen, G. C., van den Heuvel, L. P., ter Laak, H. J., et al. (1998). Genetic characteristics of myoadenylate deaminase deficiency. Ann. Neurol. 44, 140–143. doi: 10.1002/ana.410440124
Wallace, D. C. (1999). Mitochondrial diseases in man and mouse. Science 283, 1482–1488. doi: 10.1126/science.283.5407.1482
Wallgard, E., Larsson, E., He, L., Hellström, M., Armulik, A., Nisancioglu, M. H., et al. (2008). Identification of a core set of 58 gene transcripts with broad and specific expression in the microvasculature. Arterioscler. Thromb. Vasc. Biol. 28, 1469–1476. doi: 10.1161/ATVBAHA.108.165738
Wang, Z., Ying, Z., Bosy-Westphal, A., Zhang, J., Schautz, B., Later, W., et al. (2010). Specific metabolic rates of major organs and tissues across adulthood: evaluation by mechanistic model of resting energy expenditure. Am. J. Clin. Nutr. 92, 1369–1377. doi: 10.3945/ajcn.2010.29885
Wang, Z., Zhang, J., Ying, Z., and Heymsfield, S. B. (2012). Organ-tissue level model of resting energy expenditure across mammals: new insights into Kleiber’s Law. ISRN Zool. 2012:673050. doi: 10.5402/2012/673050
Watanabe, T., Tomioka, N. H., Watanabe, S., Tsuchiya, M., and Hosoyamada, M. (2014). False in vitro and in vivo elevations of uric acid levels in mouse blood. Nucleosides Nucleotides Nucleic Acids 33, 192–198. doi: 10.1080/15257770.2013.865742
Wellstead, P., and Cloutier, M. (2011). An energy systems approach to Parkinson’s disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 3, 1–6. doi: 10.1002/wsbm.107
Wen, M., Zhou, B., Chen, Y. H., Ma, Z. L., Gou, Y., Zhang, C. L., et al. (2017). Serum uric acid levels in patients with Parkinson’s disease: a meta-analysis. PLoS One 12:e0173731. doi: 10.1371/journal.pone.0173731
Winklhofer, K. F., and Haass, C. (2010). Mitochondrial dysfunction in Parkinson’s disease. Biochim. Biophys. Acta 1802, 29–44. doi: 10.1016/j.bbadis.2009.08.013
Wu, X., Wakamiya, M., Vaishnav, S., Geske, R., Montgomery, C., Jones, P., et al. (1994). Hyperuricemia and urate nephropathy in urate oxidase-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 91, 742–746. doi: 10.1073/pnas.91.2.742
Wung, W. E., and Howell, S. B. (1980). Simultaneous liquid chromatography of 5-fluorouracil, uridine, hypoxanthine, xanthine, uric acid, allopurinol, and oxipurinol in plasma. Clin. Chem. 26, 1704–1708.
Wung, W. E., and Howell, S. B. (1984). Hypoxanthine concentrations in normal subjects and patients with solid tumors and leukemia. Cancer Res. 44, 3144–3148.
Xu, X., Hu, X., Lu, Z., Zhang, P., Zhao, L., Wessale, J. L., et al. (2008). Xanthine oxidase inhibition with febuxostat attenuates systolic overload-induced left ventricular hypertrophy and dysfunction in mice. J. Card. Fail. 14, 746–753. doi: 10.1016/j.cardfail.2008.06.006
Yamada, Y., Goto, H., Murase, T., and Ogasawara, N. (1994). Molecular basis for human erythrocyte AMP deaminase deficiency: screening for the major point mutation and identification of other mutations. Hum. Mol. Genet. 3, 2243–2245. doi: 10.1093/hmg/3.12.2243
Yan, D., Xiang, G., Chai, X., Qing, J., Shang, H., Zou, B., et al. (2017). Screening of deafness-causing DNA variants that are common in patients of European ancestry using a microarray-based approach. PLoS One 12:e0169219. doi: 10.1371/journal.pone.0169219
Yao, S. Y., Ng, A. M., Cass, C. E., Baldwin, S. A., and Young, J. D. (2011). Nucleobase transport by human equilibrative nucleoside transporter 1 (hENT1). J. Biol. Chem. 286, 32552–32562. doi: 10.1074/jbc.M111.236117
Yao, T., and Seko, Y. (2017). Effects of febuxostat and inosine on rat myocardial ischemia/reperfusion injury. Cardiovasc. Disord. Med. 2. doi: 10.15761/cdm.1000146
Zhang, F., Zhang, Q., Ke, Y., Hao, J., Lu, L., Lu, N., et al. (2018). Serum uric acid levels in patients with amyotrophic lateral sclerosis: a meta-analysis. Sci. Rep. 8:1100. doi: 10.1038/s41598-018-19609-2
Zhang, H., Sternberger, N. H., Rubinstein, L. J., Herman, M. M., Binder, L. I., and Sternberger, L. A. (1989). Abnormal processing of multiple proteins in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 86, 8045–8049. doi: 10.1073/pnas.86.20.8045
Zhao, L., Roche, B. M., Wessale, J. L., Kijtawornrat, A., Lolly, J. L., Shemanski, D., et al. (2008). Chronic xanthine oxidase inhibition following myocardial infarction in rabbits: effects of early versus delayed treatment. Life Sci. 82, 495–502. doi: 10.1016/j.lfs.2007.12.010
Zima, T., Fialová, L., Mestek, O., Janebová, M., Crkovská, J., Malbohan, I., et al. (2001). Oxidative stress, metabolism of ethanol and alcohol-related diseases. J. Biomed. Sci. 8, 59–70. doi: 10.1159/000054014
Keywords: adenosine triphosphate, cellular energetics, hypoxanthine (PubChem COD: 790), inosine (PubChem CID: 6021), purines, xanthine oxidoreductase inhibitors, cardiovascular diseases, CNS diseases
Citation: Johnson TA, Jinnah HA and Kamatani N (2019) Shortage of Cellular ATP as a Cause of Diseases and Strategies to Enhance ATP. Front. Pharmacol. 10:98. doi: 10.3389/fphar.2019.00098
Received: 08 November 2018; Accepted: 24 January 2019;
Published: 19 February 2019.
Edited by:
Luis Rivas, Spanish National Research Council (CSIC), SpainReviewed by:
Christos George Stathis, Victoria University, AustraliaGiuseppe Lazzarino, Università degli Studi di Catania, Italy
Copyright © 2019 Johnson, Jinnah and Kamatani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Todd A. Johnson, dG9kZC5qb2huc29uQHN0YWdlbi5jby5qcA==