 Siti Syazani Suhaimi1†
Siti Syazani Suhaimi1† Nurul-Syakima Ab Mutalib1*†
Nurul-Syakima Ab Mutalib1*† Sheau S. Khor2
Sheau S. Khor2 Reena Rahayu Md Zain3
Reena Rahayu Md Zain3 Saiful Effendi Syafruddin1
Saiful Effendi Syafruddin1 Nadiah Abu1Ahmad Zailani Hatta Mohd Dali3,4
Nadiah Abu1Ahmad Zailani Hatta Mohd Dali3,4 Rahman Jamal1*
Rahman Jamal1*- 1UKM Medical Molecular Biology Institute, Universiti Kebangsaan Malaysia, UKM Medical Center, Kuala Lumpur, Malaysia
- 2Thermo Fisher Scientific, Shah Alam, Malaysia
- 3Department of Pathology, Faculty of Medicine, Universiti Kebangsaan Malaysia, Kuala Lumpur, Malaysia
- 4Department of Obstetrics and Gynaecology, Faculty of Medicine, Universiti Kebangsaan Malaysia, Kuala Lumpur, Malaysia
Endometrioid endometrial cancer (EEC) is the commonest form of endometrial cancer and can be divided into estrogen receptor (ER) positive and negative subtypes. The mutational profiles of EEC have been shown to aid in tailoring treatment; however, little is known about the differences between the gene mutation profiles between these two subtypes. This study aims to investigate the gene mutation profile in ER positive and negative EEC, and to further elucidate the role of WHSC1 mutations in this cancer. EEC and normal endometrial tissues were obtained from 29 patients and subjected to next-generation sequencing (NGS) using Ion Ampliseq Comprehensive Cancer PanelTM targeting 409 cancer related. A total of 741 non-synonymous alterations were identified from 272 genes in ER positive subtype while 448 non-synonymous variants were identified from 221 genes in ER negative subtype. PTEN is the most frequently altered gene in ER positive subtype (64%, 7/11) while ARID1A is the most frequently altered gene in ER negative subtype (50%, 4/8). We also identified alterations in ERRB3 (36%, 4/11), GNAS (36%, 4/11), and WHSC1 (27%, 3/11) in the ER positive subtype. WHSC1 R1126H and L1268P were shown to significantly increase cell viability, proliferation, migration, and survival. In addition, reduction in ER expression sensitized EEC-1 cell with WHSC1 L1268P mutant to Fulvestrant treatment. We revealed the mutational spectra of ER positive and ER negative EEC that could lead to better understanding of the biological mechanisms of endometrial cancer and may ultimately result in improvement of treatment options and patient prognosis.
Introduction
Endometrial cancer is the sixth most common cancer diagnosed among women with approximately 320,000 new cases and 76,000 deaths worldwide each year (Ferlay et al., 2013). Early detection is common as the disease is symptomatic even at an early stage and therefore is often diagnosed at Stage I (Morice et al., 2016). Despite the fact that most cases are diagnosed early, the incidence and mortality rates for endometrial cancer have been rising in both developed as well as developing countries and are expected to rise further with the increasing aging population and high prevalence of obesity (Ferlay et al., 2015; Morice et al., 2016). Furthermore, although endometrial cancer is generally thought to be a cancer of the postmenopausal women, 14% of cases are diagnosed during premenopausal period with 5% of the patients younger than 40-year old (Duska et al., 2001).
In the past three decades, endometrial cancer has been broadly divided into two subtypes based on histological characteristics, expression of estrogen receptor (ER) and grade (Bokhman, 1983). Majority of endometrial cancer which are designated as Type I endometrioid endometrial cancer (EEC) account for more than 75% of all cases (Carlson et al., 2012), follow the estrogen-related pathway, and arise from hyperplastic endometrium background (Bansal et al., 2009). Frequently diagnosed in premenopausal and young postmenopausal women, Type I EEC is often low grade and well-differentiated thus carrying a favorable outcome (Garg and Soslow, 2014). The less common Type II non-endometrioid endometrial cancer (NEEC), which accounts for 10–20% of all cases, follows the estrogen unrelated pathway and arise in background of atrophic endometrium (Doll et al., 2008). This type has a poorer prognosis and usually presents at the advanced stage, especially in older postmenopausal women (Amant et al., 2005). NEEC is also associated with a high mortality, reduced survival rates, and tendency to recur. This dualistic categorization has been incorporated into clinical decision-making algorithms to define high-risk patients, yet its prognostic value remains limited because one-fifth of EEC (i.e., Type I) will eventually relapse, whereas half of NEEC (i.e., Type II) do not (Bokhman, 1983). Furthermore, 15–20% of EEC are high-grade lesions, and it is unclear where they fit into the dualistic model (Zannoni et al., 2013).
This dualistic model has also been supported by molecular studies where Type I EEC has been symbolized by frequent alterations in PTEN, PIK3CA, KRAS, CTNNB1, and ARID1A as well as defects in DNA mismatch repair (MMR) resulting in the microsatellite instability (MSI) phenotype (O’Hara and Bell, 2012). In contrast, mutations in TP53 and PP2R1A as well as a high expression of oncogene Her2/ERBB2 are the major genetic changes among Type II NEEC (Lax et al., 2000; O’Hara and Bell, 2012). However, this classification is controversial due to existence of a minority of endometrial cancer cases with overlapping clinical features, morphological and molecular characterization which represents a major obstacle to effective cancer treatment (Talhouk and McAlpine, 2016). For example, Type I EEC is not completely ER positive and loss of ER expression is correlated with aggressive behavior, high grade histology, and poor survival rate (Maniketh et al., 2014; Backes et al., 2016). In contrast to breast cancer, where the ER status (in addition to progesterone) is a pillar for its molecular and clinicopathological classification (Nadji et al., 2005), a comprehensive view of the mutation spectrum between ER positive and ER negative in the same molecular subtype (i.e., endometrioid) has not been fully elucidated. Therefore, the aim of this study is to characterize somatic gene alterations in ER positive and ER negative EEC using targeted deep sequencing of 409 cancer-related genes.
Materials and Methods
Clinical Specimen, DNA Extraction, and Quality Assessment
Fresh frozen tissues of EECs (n = 19) were obtained after hysterectomy from the patients admitted to the Universiti Kebangsaan Malaysia Medical Centre (UKMMC). The cancers were classified according to the World Health Organization (WHO) classification of tumors of the female reproductive system (Pecorelli, 2009). In addition, fresh frozen normal endometrium (n = 10) were obtained from patients surgically treated for non-malignant endometrial diseases. This study was approved by the Universiti Kebangsaan Malaysia Research Ethics Committee (UKM 1.5.3.5/244/AP-2012-011) and carried out in accordance with the approved guidelines. All patients provided written informed consent for their tissue samples to be used for research. Immunohistochemical staining for ER (antibody clone 1D5, DAKO, Carpinteria, CA, United States) and Hematoxylin and Eosin (H&E) staining was performed on the frozen sections and then reviewed by a pathologist. Immunohistochemical staining for ER was scored as positive if 1% or more of tumor nuclei were immunoreactive and negative if less than 1% of tumor cell nuclei were immunoreactive. Only cancer specimens containing more than 80% cancer cells and normal tissues with less than 20% necrosis were subjected to DNA isolation using the QIAamp® DNA Mini Kit (Qiagen, Valencia, CA, United States) following the manufacturer’s protocol.
Next-Generation Sequencing
The Ion AmpliseqTM Comprehensive Cancer Panel V2 (Life Technologies, Guilford, CT, United States), which covers 409 oncogenes and tumor suppressor genes, was used for library preparation (Supplementary Table S1) according to manufacturer’s instruction. The libraries were then normalized to 12–25 pM for template preparation on the Ion One Touch (Life Technologies, Guilford, CT, United States). The clonal amplification of the DNA libraries on the Ion Sphere Particles (ISPs) was carried out using emulsion PCR and the subsequent isolation of templated ISPs was performed using Ion OneTouch ES (Life Technologies, Guilford, CT, United States). Subsequently, next-generation sequencing (NGS) was performed on the Ion Torrent Personal Genome Machine (PGMTM) using 318TM chip and Ion Torrent PGM Sequencing 200 kit V2 (Life Technologies, Guilford, CT, United States).
Bioinformatics Analyses
Read Mapping and Variant Calling
Data from the sequencing runs were automatically transferred to the Torrent Server hosting the Torrent Suite Software v4.0.3. The readings were mapped to the reference genome (hg19) using the Torrent Mapping Alignment program (TMAP). Variant calling was generated using Torrent Variant Caller v 4.0 with low stringency settings (Life Technologies, Guilford, CT, United States).
Variant Annotation and Prioritization
The functional effects of the variants were further annotated using ANNOVAR (Wang et al., 2010) with respect to gene regions and filter-based annotations. Prediction on protein impact of variants was performed using SIFT, PolyPhen2 HDIV, PolyPhen2 HVAR, LRT, MutationTaster, MutationAssessor, FATHMM, GERP++, PhyloP, and SiPhy databases that are available in ANNOVAR. We classified SNVs as pathogenic if they were observed to be deleterious by three or more than three SNV protein prediction algorithms.
To evaluate which mutations could be actionable and to prioritize for the true somatic mutations, several additional filtering steps were performed. Details on filtrations steps are illustrated in Supplementary Figure S1. The alignment of each candidate variant was manually inspected to check for sequencing artifacts and alignment errors using the Integrated Genomic Viewer (IGV) (Thorvaldsdóttir et al., 2013). Oncoprint diagram and lollipop plot were created using Oncoprinter and Mutation Mapper tools, respectively (Cerami et al., 2012; Gao et al., 2013). The detected mutations were compared to those in available cancer databases from the COSMIC v82 (Forbes et al., 2017), MyCancerGenome (Van Allen et al., 2013), and International Cancer Genome Consortium (ICGC) database (Zhang et al., 2011). Cancer Genome Interpreter was used to classify driver mutation among the somatic alterations (Tamborero et al., 2018).
To identify relevant genes for further characterization, we prioritized the genes involved in endometrial cancer carcinogenesis using following strategies: (1) genes associated with EEC carcinogenesis and (2) genes relevant to ER signaling pathways [based on Kyoto Encyclopedia of Genes and Genomic (KEGG) estrogen signaling pathway (hsa04915)] (Kanehisa et al., 2016).
Sanger Sequencing for Validation
Identified variants were selected randomly for validation by Sanger sequencing. Primers for Sanger sequencing validation were designed using the IDT-DNA Primer Quest (Coralville, IA, United States). The PCR primers were described in Supplementary Table S2. PCR amplifications were performed using Applied Biosystem AmpliTaq® Gold 360 master Mix (Applied Biosystems, Foster City, CA, United States) following manufacturer’s instructions. Sanger sequencing was performed using BigDye Terminator v.3.1 Cycle Sequencing kits (Applied Biosystems, Foster City, CA, United States) on an ABI 3500 Genetic Analyzer platform (Life Technologies, Guilford, CT, United States).
Site-Directed Mutagenesis and Constructs
The wild-type plasmid construct of WHSC1 (RC212404) was obtained from OriGene Technologies (Rockville, MD, United States). Site-directed mutagenesis was performed using the Quick-ChangeTM Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, United States), according to the manufacturer’s instructions. The mutagenic primers were designed using QuickChange Primer Design Program and the sequences were provided in Supplementary Table S3.
Cell Lines and Transient Transfection
Endometrioid endometrial cancer-1 (ATCC® CRL-2923TM) and 293T (ATCC® CRL-3216TM) cells were maintained in RPMI 1640 and DMEM: F12 (both from Gibco, United States) supplemented with heat inactivated 10% fetal bovine serum (FBS; Gibco, United States). Twenty-four hours prior to transfection, cells were seeded in a 6-well plate with a density of 4 × 105 (EEC-1) and 6 × 105 (293T). Four micrograms of each construct was transfected using 8 μl Lipofectamine 2000 (Invitrogen, Carlsbad, CA, United States) according to the manufacturer’s instructions. Control cells were cells transfected with the empty vector pCMV6.
Gene Expression and Protein Analysis
Total RNA was isolated from the cell lines using the RNeasy Kit (Qiagen, Valencia, CA, United States) according to the manufacturer’s directions and cDNA was synthesized using High Capacity RNA to cDNA Kit (Applied Biosystem, Foster City, CA, United States). Quantitative real-time PCR was performed using TaqMan Fast Advanced Master Mix on the ABI 7500 Fast Real-Time PCR system (Applied Biosystem, Foster City, CA, United States). Relative expression was calculated using the ΔΔCt method (Livak and Schmittgen, 2001) with GAPDH as the housekeeping gene.
Protein lysate was resolved on a mini-protean TGX precast gel of 4–20% polyacrylamide (BioRad Laboratories, Hercules, CA, United States) and analyzed against the following primary antibodies; anti-DDK (Clone OTI4C5) (OriGene Technologies, Rockville, MD, United States), NSD2 (G12), ERα (HC-20), and anti-β-actin (sc-47778) (Santa Cruz Biotechnology Inc., Santa Cruz, CA, United States).
Cell-Based Assays
Cell viability was assessed using PrestoBlue® cell viability reagent (Invitrogen, Carlsbad, CA, United States), wound healing assay was performed using IBIDI Culture-Inserts (IBIDI GmbH, Martinsried, Germany), migration ability of the cells was assessed using QCMTM 24-well cell migration assay (Millipore, Billerica, MA, United States), and colony formation assay was performed by seeding 500 transfected cells in six-well plates followed by incubation at 37°C for 14 days and crystal violet staining. All assays were performed in triplicate.
BrdU Proliferation Assay
Endometrioid endometrial cancer-1 cell lines were seeded at 2 × 104 cells per well in 96-well plate and transfected with mutants or wild type of WHSC1 expression constructs before BrdU incorporation using BrdU Cell Proliferation Assay (Millipore, Temecula, CA, United States).
Luciferase Assay
To substantiate the importance of WHSC1 as a potential coactivator, dual luciferase assay was performed. We used Cignal ERE Reporter Assay kit from SABiosciences (Valencia, CA, United States). 293T cells were plated at a density of 3 × 105 cells per well (96-well plates) in phenol red-free DMEM: F12 with 10% v/v charcoal–dextran-treated FBS. The cells were then transfected with 100 ng of reporter plasmid, wild-type WHSC1 plasmid, and Renilla. Each sample was done in triplicate. The WHSC1 mutants were transfected in the same manner. After transfection for 24 h, the cells were stimulated with 10 nM E2 or vehicle (DMEM). The Dual-Glo Luciferase Assay System (Promega) was used according to the manufacturer’s instructions using VARIOLUX luminometer. Values were normalized to Renilla luciferase activity.
Results
Patient Demographic Data and Tumor Characteristic
Clinical specimens used in this study were collected from patients who were treated at the UKMMC. The information on the patient’s age, degree of differentiation, FIGO staging of tumor, metastasis, and ER status is summarized in Table 1. The majority of EEC in both groups were stage 1B 64% (7/11) in ER positive, 50% (4/8) in ER negative. While there are three cases of metastasis in ER negative = 37.5%, (3/8) has shown patients with advance disease.

TABLE 1. Patient demographics and tumor characteristics.
Mutations Analysis of Endometrial Cancer With Ion AmpliseqTM Cancer Panel
All patients from both groups have at least three alterations among the 409 genes screened. A total of 741 variants were identified in 272 genes in ER positive subtype [718 single-nucleotide variations (SNVs), 14 insertions/deletions (INDELs), and 9 block substitutions] (Figure 1A). While in ER negative subtype, a total of 448 variants altered in 221 different genes, including 421 SNVs, 21 INDELs, and 6 block substitutions (Figure 1B). We noted that mutations distribution of EECs in both subtypes were genomically heterogeneous. There were five highly mutated patients (median 178 per tumor, range from 97 to 330) and another 14 patients were non-highly mutated (median 11 mutations per tumor; range 3–42). We further investigated and found that hypermutated phenotype group could have a perturbed DNA repair system due to frequently altered DNA repair genes especially in MMR genes such as (i.e., MSH2, MSH6, and PMS1) (Supplementary Table S4).
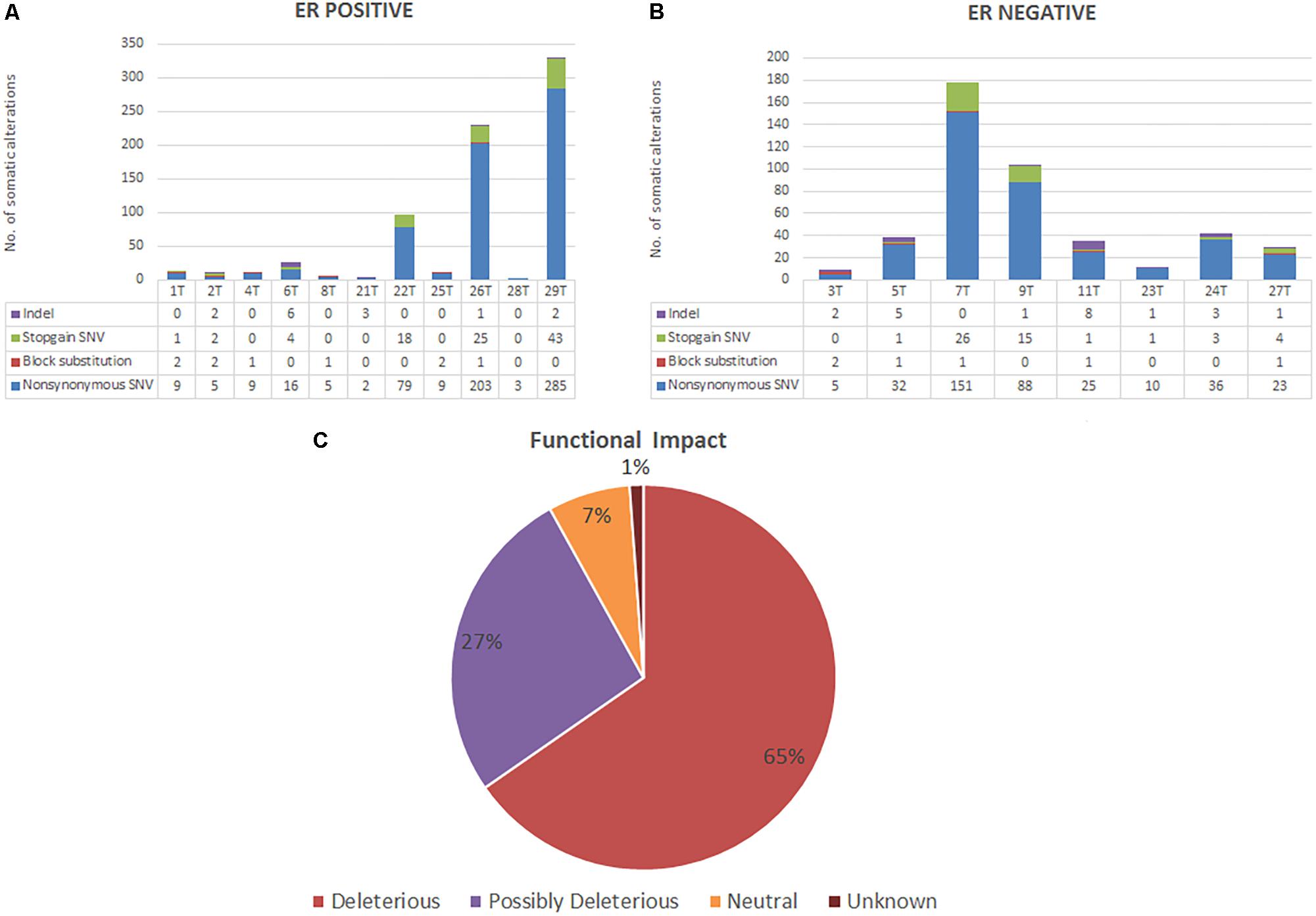
FIGURE 1. (A,B) Bar charts showing the number of somatic alterations identified in each patients according to respective groups. Indel in these bar charts including frameshift and non-frameshift substitution. (C) Pie chart showing the frequency of functional impact of gene mutations based on protein prediction algorithms in ANNOVAR.
Based on protein prediction algorithms among identified 1139 SNVs in both subtypes, 739 variants (65%) were predicted to have deleterious effects, 301 variants (27%) have possibly deleterious effects, while the other 78 variants (7%) has neutral or low protein impact (Figure 1C). We identified 134 out of 371 (36%) genes predicted as tumor driver which included a total of 295 alterations. From this 295 alteration, 178 (60%) were identified in ER positive whereby 9 out of 11 ER positive patients have at least two candidate driver alteration. Meanwhile in ER negative subtype, 117 (40%) alterations were identified in ER negative patients where all ER negative patients have at least three candidate driver alterations (Supplementary Table S5).
PTEN is the most frequently altered gene in ER positive subtype (64%, 7/11) while ARID1A is the most frequently altered gene in ER negative group (50%, 4/8) as shown in Figure 2A (Supplementary Tables S6, S7). Majority of PTEN alterations were missense and in-frame mutations. On the other hand, ARID1A mutations were truncating alterations including nonsense mutations and INDELs. Mutations in the ER signaling pathway is known to be important in endometrial cancer; therefore, we further examined the prevalence of mutations in the genes involved in ER signaling pathway. Overall, there are 38 genes out of 409 genes panel list of CCP known to be involved in ER signaling pathway (Supplementary Table S1). As shown in Figure 2B (Supplementary Tables S8, S9), ERBB3, GNAS, PIK3R1, and WHSC1 are the most frequently altered genes in ER positive subtype (36%, 4/11); (36%, 4/11); (27%, 3/11); and (27%, 3/11) while PIK3CA is the most frequently altered gene in ER negative group (50%, 4/8). Table 2 showed classification based on protein impact prediction and candidate driver mutations for each alteration in PTEN, ARID1A, PIK3CA, ERBB3, GNAS, PIK3R1, and WHSC1.
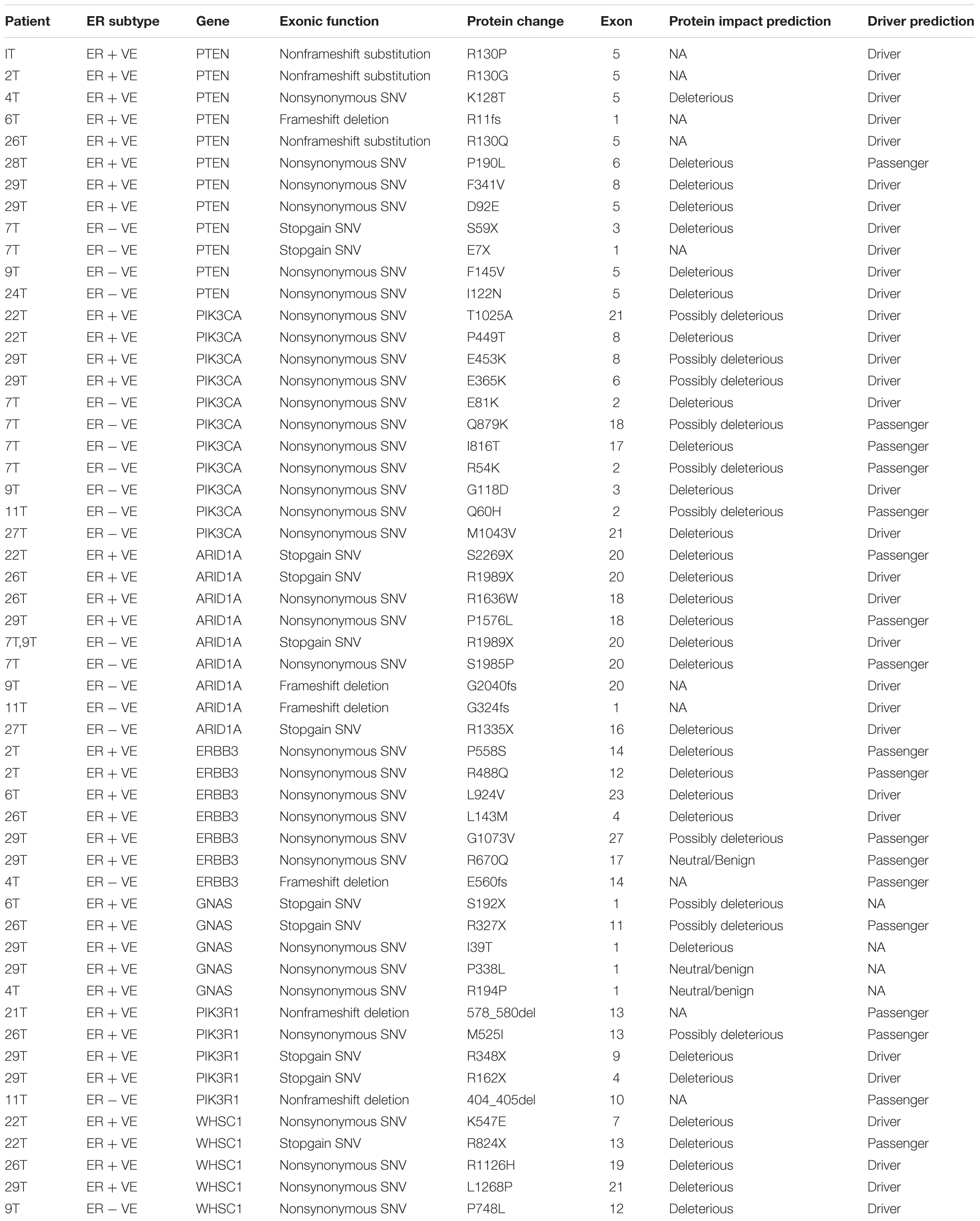
TABLE 2. Classification of somatic alterations in PTEN, PIK3CA, ARID1A, ERBB3, GNAS, PIK3R1, and WHSC1 genes.
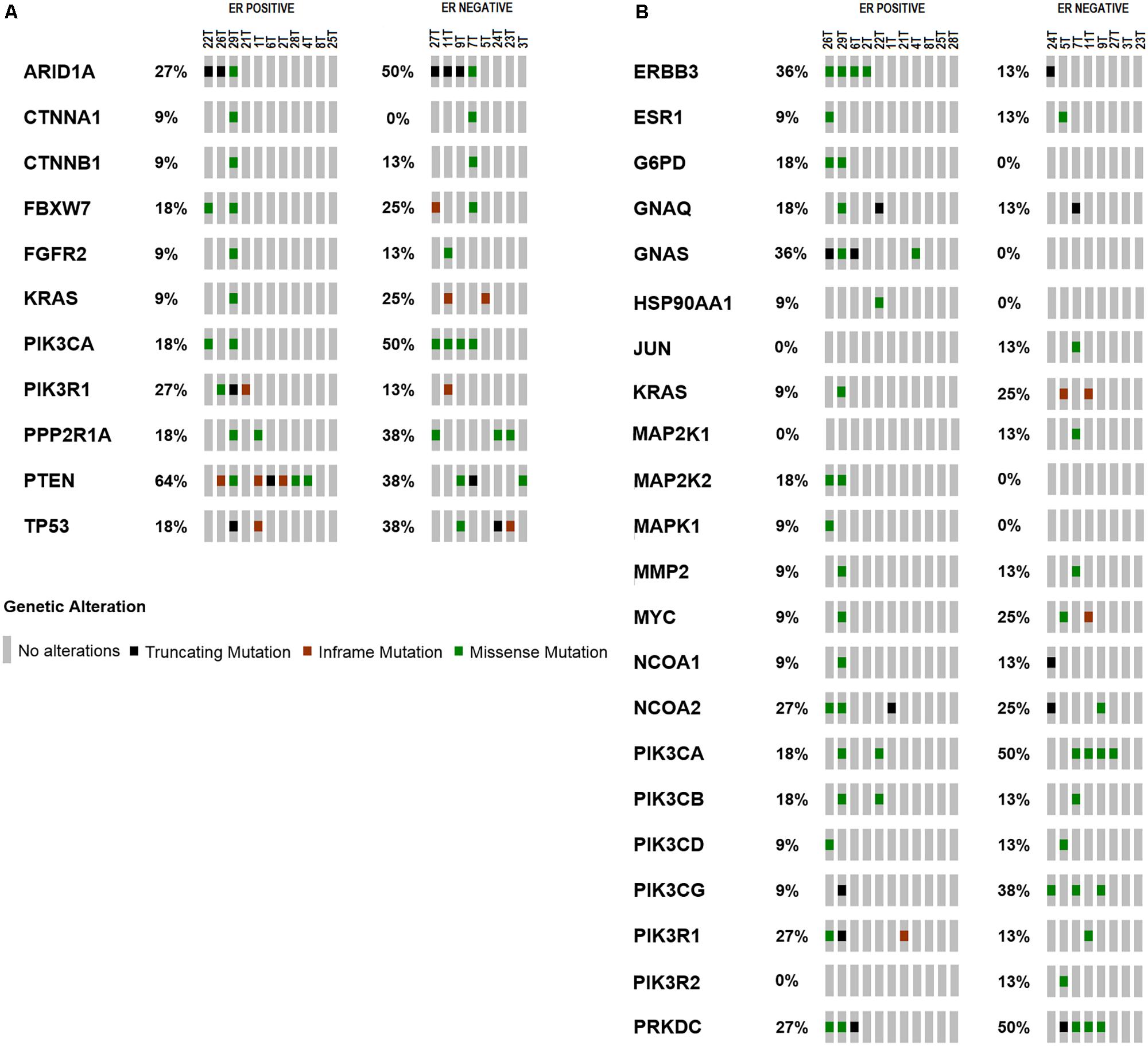
FIGURE 2. Frequency of mutations in (A) genes related in carcinogenesis of EEC. PTEN is the most frequently altered gene in ER positive subtype (64%, n = 11); ARID1A is the most frequently altered gene in ER negative group (63%, n = 8). (B) Frequency of mutations in genes relevant to ER signaling pathway compiled from literatures. ERBB3 and GNAS are the most frequently altered genes in ER positive subtype (36%, n = 11); PIK3CA is the most frequently altered gene in ER negative group (50%, n = 8).
Mutations Distribution in the Functional Domains of PTEN, ARID1A, ERBB3, and WHSC1
Mutations distribution in the functional domains of PTEN, ARID1A, and WHSC1 was illustrated in Supplementary Figure S2. Mutations in PTEN were localized to the catalytic phosphatase domain, with majority of mutants belongs to ER positive subtype group including the recurrent hotspot R130 residue, D92E, K128T (Supplementary Figure S2a). In addition, a frameshift deletion (R11fs) and two missense mutations (P190L and F341V) in C2 domain were also found in ER positive subtype. While in ER negative subtype group, two nonsense mutations (E7X and S59X) and two missense alterations (I122N and F145V) located on phosphatase domain have been identified. Most of these mutations have been reported via COSMIC v82, with the exception to R11fs, which was identified in our study.
Out of the 19 EECs sequenced, seven patients had nine ARID1A mutations (four mutations were identified in ER positive and five in ER negative EEC patients). Interestingly, recurrent hotspot mutation (R1989X) was observed in both ER positive and ER negative subtype group as shown in Supplementary Figure S2b. More ARID1A mutations were identified among ER negative subtype group, with mainly truncating type of mutations (nonsense and frameshift deletion) (G324fs, R1335X, R1989X, and G2040fs) and a missense mutation (S1985P). However, two nonsense mutations (R1989X and S2269X) and two missense mutations (P1576L and R1636W) which were located on non-functional domain of ARID1A have been identified in ER positive subtype. This study is the first to report the somatic G2040fs variant in EEC.
Higher mutation frequency of ERBB3 was observed in ER positive subtype (36%, 4/11) compared to ER negative subtype (18%, 1/8). As shown in Supplementary Figure S2c, the missense mutations in ER positive sample were mapped at functional domain of ERBB3 gene including two in extracellular domain (L143M and P558S) and one in tyrosine kinase domain (L924V), while the others mapped on non-functional domain (R488Q), (R670Q), and (G1073V). Nevertheless, the single frameshift deletion mutation (E560fs) reported in ER negative subtype was located at the extracellular domain IV of ERBB3. To date, most of the mutations we discovered in ERBB3 gene are novel, except for L143M, R488Q, and R670Q that have been recorded in Cosmic v82 and ICGC database, respectively.
WHSC1 was found to be altered in three ER positive patients and one ER negative patient. Majority of these somatic variations were distributed along the functional domain of WHSC1 as shown in Supplementary Figure S2d. All of the variants identified in this study are novel, with the exception of a mutation on the catalytic SET domain (R1126H) that has been identified in several cancers (Supplementary Table S10). One missense mutation K547E was identified in an area without a defined domain, while two missense mutations (P748L and L1268P) were located within the plant homeodomain (PHD) domains II and V, respectively. A nonsense mutation (R824X) was identified which was predicted to cause premature truncation of WHSC1 protein with a deletion of C terminal functional domains including PHD IV, proline–tryptophan–tryptophan–proline-2 (PWWP2), SET, and PHD V-CH5CH domain. The frequency and location of alterations in WHSC1 suggest that these cancer-associated mutations may have functional consequences.
Expression Analysis of Wild-Type and Mutant Constructs
The level of WHSC1 expression was firstly screened in several cell lines (Figure 3A) in order to gauge its expression. Upon transfection, WHSC1 mRNA levels were significantly reduced in the transfected cells with the mutant constructs K547E, R824X, and R1126H compared to the wild type indicating that these mutations affected the stability of the mRNA levels. However, the wild type and mutant L1268P were equally expressed in the transfected EEC-1 cell line (Figure 3B). This result was confirmed by the Western blot in EEC-1 and 293T cell lines (Figure 3C). Of note however, there is a second migrating band seen in L1268P in Western blot assay, which might explain the differences of mRNA of transcription level of L1268P and other mutant constructs.
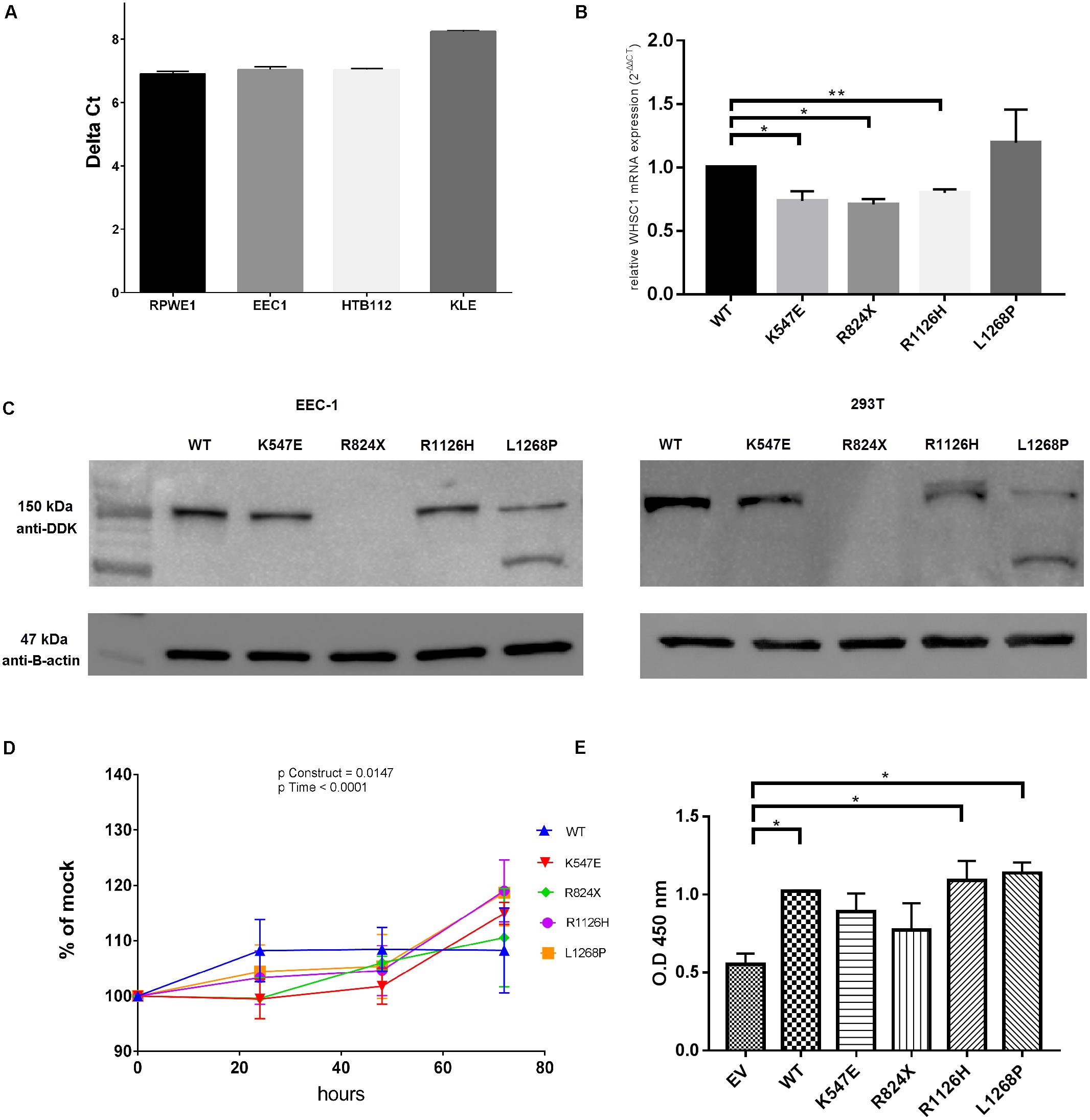
FIGURE 3. WHSC1 gene and protein analysis. (A) Quantification of mRNA from nuclear extract of normal prostate cell lines RPWE1 and endometrial cancer cell lines (EEC-1, HTB-112, KLE). (B) Quantification of mRNA expression of mutant WHSC1 in EEC-1 cell line. Relative WHSC1 mutants were calculated in comparison to WT WHSC1 48 h post-transfection by real-time PCR and normalized to GAPDH mRNA expression. Negative control (no template) was included in each experiment (mean ± SD, n = 2, ∗p < 0.05). The mRNA expression of WT was assigned as 1. (C) Western blot analysis shows protein expression of wild type and WHSC1 mutants in EEC-1 and 293T cell lines after 48 h post-transfection. Total protein harvested from EEC-1 cells and 293T cells solubilized in cell lysis buffer were analyzed using precast SDS 4–12% gels. The WT and mutants of WHSC1 (152.1 kDa) were detected using Anti-DDK Clone OTI4C5 (mouse origin). While β-actin (47 kDa) was detected using anti-β-actin (mouse origin) for positive control. Mutants WHSC1 promote cell growth. (D) Mutants WHSC1 promote cell viability. At 72 h, the mutants K547E, R1126H, and L1268P resulted in 15, 19, and 18% increase significantly in motility, respectively, over EV in EEC-1. Statistical significance in all cases was measured by Student’s t-test (∗p < 0.05), n = 3. Error bars represent average ± SD. Overall p-values were calculated by two-way ANOVA for time. (E) Mutants WHSC1 promote cell proliferation. BrdU colorimetric incorporation assay shows that wild type of WHSC1, R1126H, and L1268P significantly increased cell proliferation in EEC-1. Statistical significance in all cases was measured by Student’s t-test (∗p < 0.05), n = 2. Error bars represent average ± SD.
Assessment of Oncogenic Properties of WHSC1 Mutants
At 72 h, the mutants K547E, R1126H, and L1268P showed a 15 (p = 0.0002), 19 (p = 0.0043), and 18% (p = 0.0056) significant increase in cell proliferation, respectively, over the empty vector control in EEC-1 cell line (Figure 3D). The effect of the WHSC1 mutants on cell proliferation was further assessed by quantifying the activity of proliferating cells within 24 h after BrdU labeling. BrdU incorporation assay revealed significant increase in DNA synthesis in WHSC1 wild type (p = 0.0190), R1126H (p = 0.0341), and L1268P (p = 0.0136) mutants, relative to empty vector. In contrast, no significant effect on DNA synthesis was caused by the K547E and R824X mutants (Figure 3E).
To further address the role of constitutive activation of WHSC1 mutants on cell migration, we employed both the wound-healing and transwell migration assays. As shown in Figure 4A, the open area was rapidly covered by the mutant R1126H and L1268P in comparison with control (p = 0.0049 and p = 0.0040, respectively). The quantified open area in vector control cells reduced from 100 to only 76%, while R1126H mutant cells reduced from 100 to 95%, whereas L1268P mutant caused reduction from 100 to 94%. The wild type WHSC1 also showed significant wound closure from 100 to 85% (p = 0.0233). To further confirm migration ability of the mutants, we employed the transwell migration assay. Similarly, EEC-1 transfected with WHSC1 mutants R1126H and L1268P displayed a significant increase in number of migrated cells (Figure 4B) (p = 0.0102 and p = 0.0086, respectively). In this transwell migration assay, the wild-type WHSC1 also showed potential migration ability compared with vector alone control cells (p = 0.0233). In addition, only mutants R1126H and L1268P showed a significant increase in colony number compared to empty vector (Figure 4C) (p = 0.00416 and p = 0.0232, respectively).
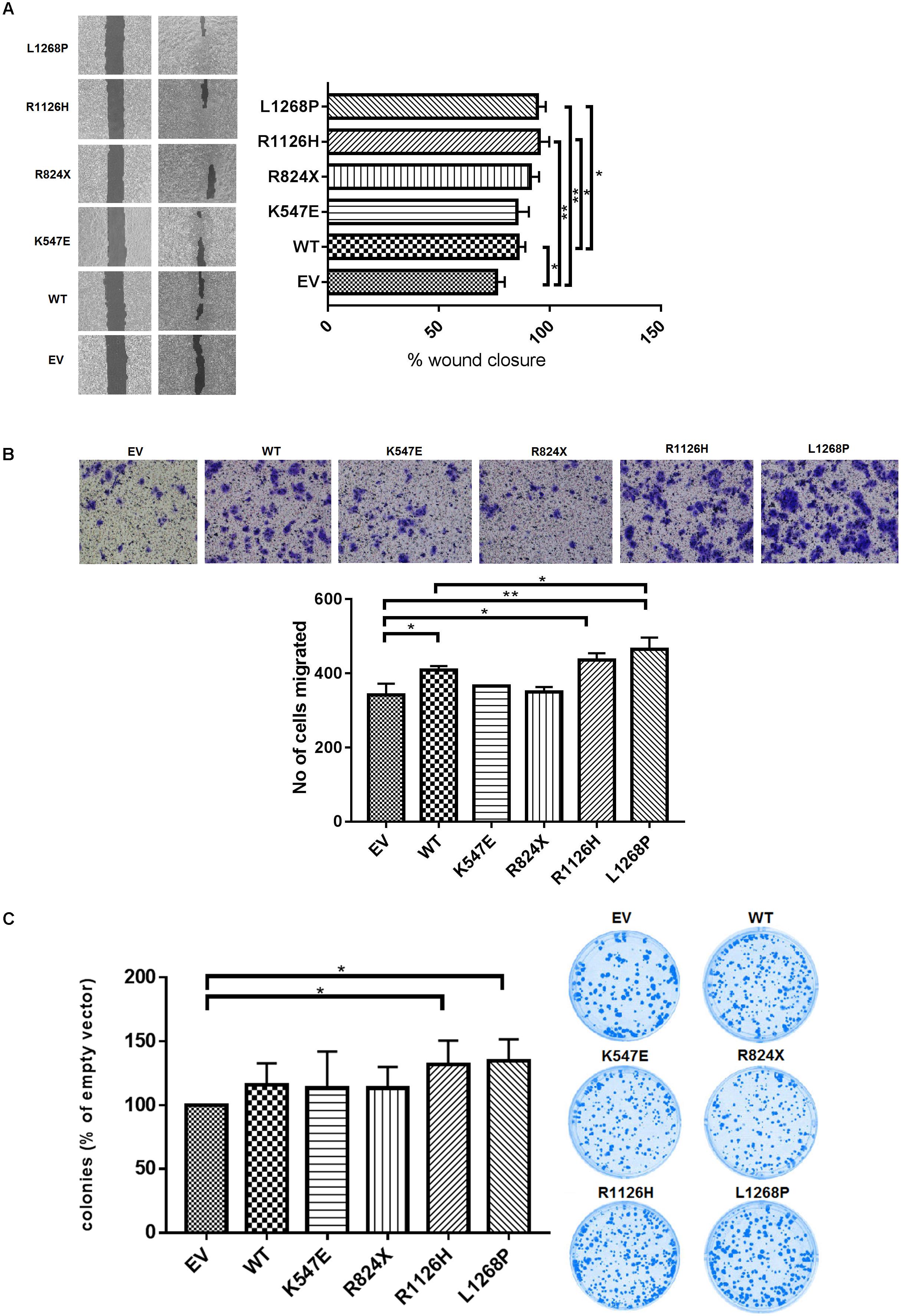
FIGURE 4. Effect of WHSC1 mutants on cell migration and clonogenic potential. (A) Wound healing assay displayed significant increase in average rate of wound closure at 72 h shown in R1126H and L1268P compared with EV and WT. (B) Transwell migration assay showed the numbers of migrated mutants R1126H and L1268P were significantly higher than EV. (C) Clonogenic survival was assessed by colony formation assay at day 14. The numbers of colonies were counted after crystal violet staining, and are expressed as percentage of cells expressing empty vector. There is significance of colony formation in R1126H and L1268P when compared with empty vector. No obvious effects were observed on the colony formation ability of EEC-1 cells after transfection with WT and mutants K547E and R824X. Statistical significance was measured by Student’s t-test (∗p < 0.05), n = 3. Error bars represent average ± SD.
WHSC1 Regulates ERα Gene Expression
Both R1126H and L1268P led to a significant increase of the coactivation potential in WHSC1 (Figure 5) albeit lower than the wild type WHSC1 when compared with empty vector. However, the truncated R824X mutant showed reduction of the coactivation potential of WHSC1 suggesting that the catalytic SET activity is required for its ability to stimulate estrogen-dependent gene transcription. When compared all mutants with the wild type, only L1268P signaling did not differ significantly.
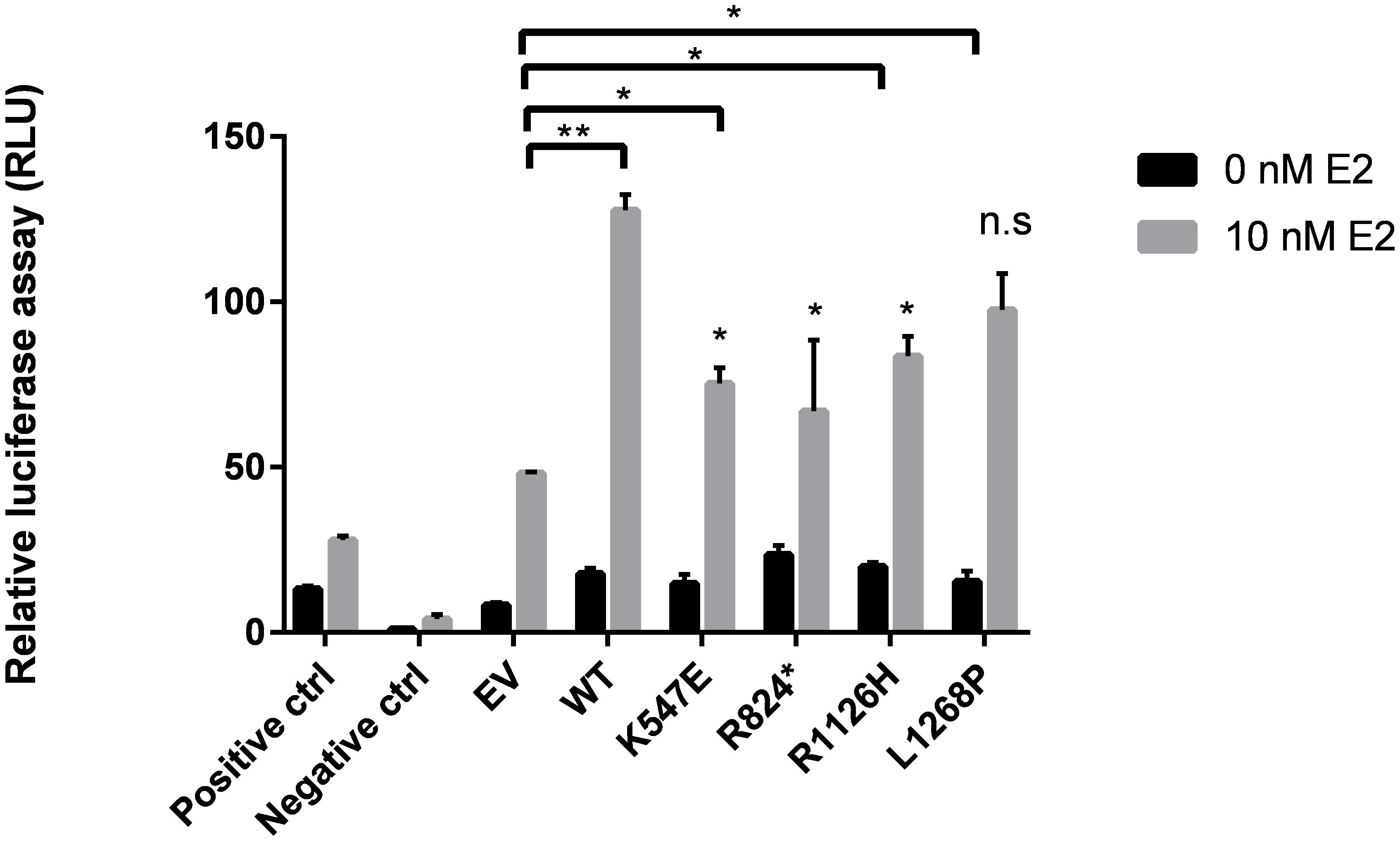
FIGURE 5. Effects of WHSC1 mutations on transcriptional activation of Estrogen Receptor Gene (ERα). WT WHSC1 showed increase in ERE-mediated luciferase activity relative to EV. K547E, R824X, and R1126H were associated with reduced ability to activate ER activity, compared to WT. Whereas L1268P activated ER activity comparable to WT (n.s.). Data are presented as mean values ± SD, n = 2.
Activating WHSC1 L1268P Is Sensitive to Fulvestrant
Fulvestrant acts as an antagonist to inhibit ER activity; therefore, we examine the effect of WHSC1 R1126H and L1268P on the dose-dependent inhibition of proliferation in EEC-1 cells. Fulvestrant was able to inhibit the activity of wild type and WHSC1 mutants. As shown in Figures 6A,B, L1268H had significantly greater antiproliferative effect only when compared with the empty vector with a 60% growth inhibition (p = 0.0281) compared to the wild type (54%) which was not significant (p = 0.362). The estrogen responsiveness was assayed as shown in Figure 6C, where we compared the ability of Fulvestrant to reduce ER protein levels in wild-type and mutants WHSC1, in the presence and absence of estrogen. We observed that Fulvestrant was able to downregulate the ER proteins of both wild type and mutants, with more reduction seen in the L1268P mutant’s ER proteins level. Upon treatment with estradiol and Fulvestrant, a clear reduction of steady-state L1268P WHSC1 mutant’s exogenous protein can be seen in the Western blot assay.
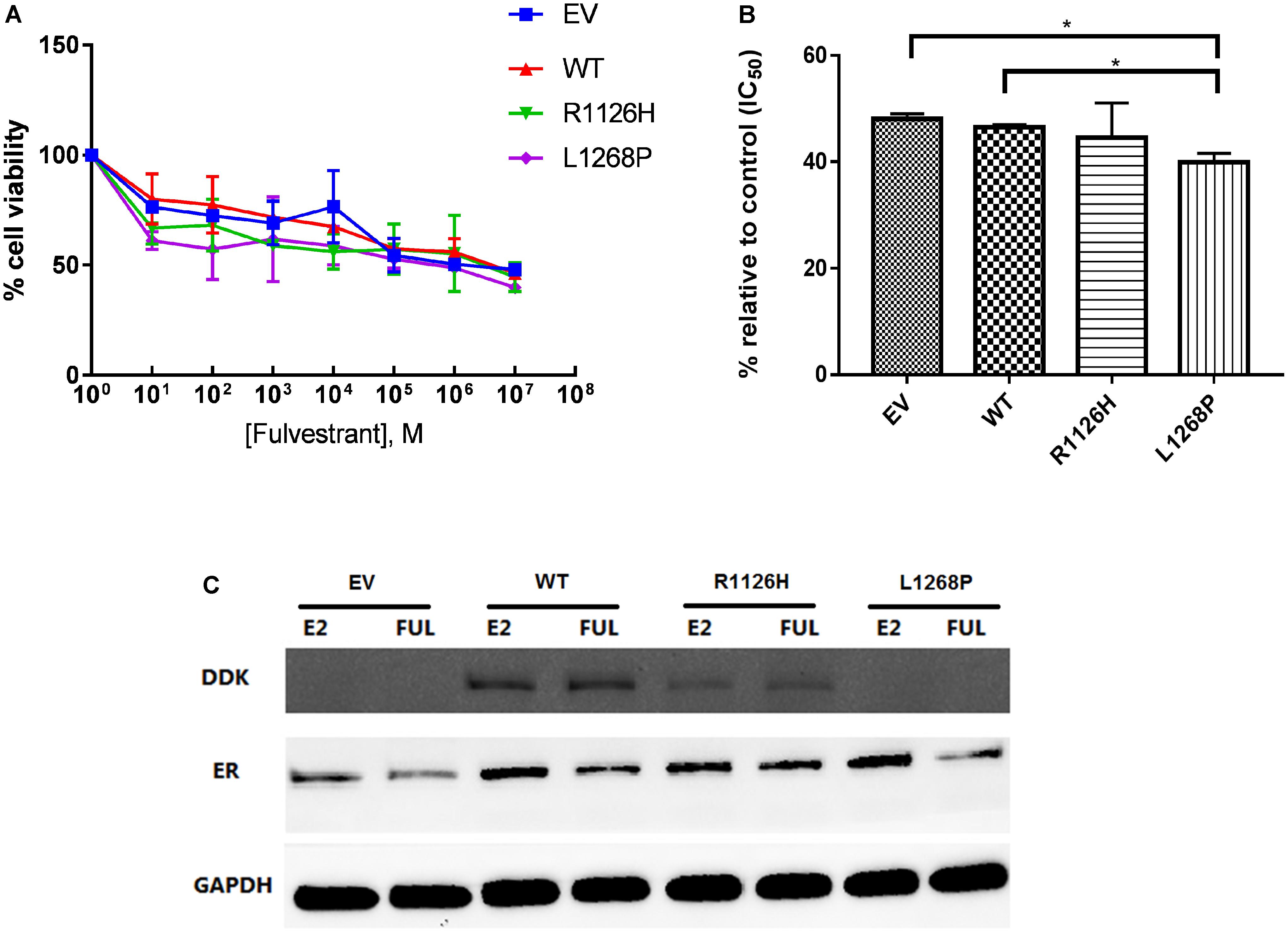
FIGURE 6. Effect of Fulvestrant on cell proliferation in mutants WHSC1 expressing EEC1 cells. (A) Treatment was performed with 10 nM E2 and various dosage of Fulvestrant for 24 h. (B) Quantification of proliferation after exposure to 10 nM E2 and 10-3 M Fulvestrant. The values represent the percentage of growing cells compared to the normalized control (100%). Results represent mean values ± SD, n = 2 performed in triplicate. WHSC1 mutants counteract the effect of ER inhibitor. (C) EEC-1 cells were transiently transfected with WT, R1126H, and L1268P mutant constructs and treated with 10 nM E2 or 100 nM of Fulvestrant for 24 h. Protein lysates were prepared and analyzed by Western Blot with indicated antibodies.
Discussion
Over the past few years emergence of novel technologies based on NGS have led to a new paradigm in understanding the mutational landscape of endometrial cancers. However, most studies were limited to either characterization of endometrial cancer without stratification based on grade and histological subtypes, or were focused on aggressive serous carcinoma subtype of endometrial cancer only (Kuhn et al., 2012; Le Gallo et al., 2012; Liang et al., 2012; Chang et al., 2017). Our study is not only the first report on the mutational landscape in ER positive and ER negative EEC, but also on the functional analysis of a potential gene candidate, WHSC1, involved in ER positive endometrioid tumorigenesis. Of the 11 ER positive and 8 ER negative EEC patients sequenced in our study, the mutation spectrum notably diverges among both subtypes.
Among known genes that have been reported to be altered in endometrial cancer in earlier studies, PTEN mutations occur frequently in ER positive subtype compared to ER negative subtype. In the TCGA study on EEC, PTEN mutations have also been associated with ER expression in endometrial cancer (The Cancer Genome Atlas Research Network et al., 2013). However, our study showed that PIK3CA mutations were comparable between both ER subtypes with more events occurring in the ER negative subtype compared to ER positive subtype. This is inconsistent with results from TCGA on breast cancer, which suggested the association between PIK3CA with ER positive expression (The Cancer Genome Atlas Network, 2012). From the 10 patients with PTEN mutation in our study, only 3 patients had mutation in PIK3CA, which is consistent with other investigators that claimed PIK3CA mutations are mutually exclusive with PTEN mutations, suggesting that carcinogenic signaling through phosphatidylinositol 3-kinase (PI3K) pathway is mediated either through inactivation of PTEN or activation of PIK3CA (Prat et al., 2007). ARID1A mutations were identified in both endometrial cancer subtypes but more commonly seen in the ER negative subtype. Almost all ARID1A mutants in our analysis coexist with PTEN and PIK3CA mutations, and it is postulated that these mutations together induce aberrant activation of PI3K and protein kinase B (AKT) phosphorylation that result in inhibition of cell survival and apoptosis (Liang et al., 2012).
PTEN is a 200-kb gene located on chromosome 10q23.3 that consists of nine exons coding for 403 amino acids yielding a 47 kDa protein (Li, 1997). This tumor suppressor gene plays an important role in the tumorigenesis of endometrial cancer, particularly in EEC (Djordjevic et al., 2012; O’Hara and Bell, 2012; The Cancer Genome Atlas Research Network et al., 2013). PTEN functions primarily as a lipid phosphatase via a dual specificity protein phosphatase dualistic domain (Maehama and Dixon, 1998). Remarkably, most reported mutations (R130, I122N, K128T) are clustered within the phosphatase signature motif between codon 122–132 that has been considered as the mutation hotspot site (Prasad et al., 2005; Bansal et al., 2009). In addition, mutations on the phosphatase binding loop D92E on this domain play critical roles as well (Giudice and Squarize, 2013). These mutants may abolish the phosphatase activity of the substrate phosphoinositide lipid, a key signaling component of PI3K pathway that regulate cell growth, proliferation, migration, and apoptosis (Fayard et al., 2010). In comparison, mutants located on C2 domain (P190L and F341V) could impair the phosphoinositide lipid binding function, ultimately its growth-suppressing activity despite retaining the catalytic phosphatase activity (Naguib et al., 2015). Other truncating mutations including nonsense and frameshift deletion (E7X, S59X, and R11fs) may result in functional inactivation of PTEN protein function, which leads to tumorigenesis (Xu et al., 2014). A number of PTEN missense mutations have been screened for its phosphatase activity which is important for its tumor suppression function, and majority of the mutants were shown to eliminate or reduce phosphatase activity (Han et al., 2000).
ARID1A mutations have been increasingly reported upon the emergence of NGS technology especially among various malignancies (Jones et al., 2012) including endometrial cancer (Liang et al., 2012). ARID1A is located on chromosome 1p36.11 and it contains 20 coding exons. The gene encodes a 24-kDa chromatin remodeling protein consisting of 2285 amino acids and a member of SWI/SNF family which plays a significant role in regulating the transcription of genes via ATP hydrolysis (Wilson and Roberts, 2011). Therefore, it is critical in regulating diverse cellular processes including DNA repair, differentiation, and development as well as tumor suppression (Lu and Allis, 2017). The ARIDIA mutations identified in our study are consistent with others regardless of the type of cancers, where most were truncating mutations that would result in loss of function, supporting its tumor suppressor role (Guan et al., 2012; Chang et al., 2015). Both nonsense and frameshift mutants (R1335X, R1989X, S2269X, G324fs, and G2040fs) would result in loss of function ARID1A protein by nonsense-mediated mRNA decay and degradation of misfolded truncated proteins. Thus, its tumor suppression activity is abolished due to disruption in CDKN1A and SMAD3 pathway which is related to cell cycle regulation and aberrant activation of PI3K/AKT pathway (Bosse et al., 2013). Missense mutations occur less frequently in ARID1A; hence, the functional manifestations are poorly understood (Bateman et al., 2016). Our study showed that the mutations commonly occurred in the ER negative subtype group, suggesting that inactivation of ARID1A might play important role in ER-mediated transcription in endometrial cancer. Moreover, the SWI/SNW complex has been reported to be vital for the transcriptional activation of ER (Belandia et al., 2002). Mutation in ARID1A is not that frequent in breast cancer (only ∼5%) (Jones et al., 2012) and there has been no reported association with ER status. However, low ARID1A expression has been found to associate with ER negativity of breast cancer (Zhang et al., 2012).
By focusing on genes involved in the ER signaling pathway, we identified relatively frequent mutations in ERBB3, GNAS, PI3KR1, and WHSC1 in ER positive subtype. Previous data by TCGA reported that 5.2% (16/373) of EEC have ERBB3 mutations (Cerami et al., 2012). This gene is located on 12q13.2 chromosome, consists of 28 exons, and encodes a protein composed of 1342 amino acids (Sithanandam and Anderson, 2008). ERBB3 encodes a member of EGFR family receptor tyrosine kinase, which is a well-known gene to be amplified and overexpressed in various cancers (Olayioye et al., 2000). With emergence of NGS, more somatic mutations in ERBB3 have gained attention. ERBB3 has been uncovered as a driver cancer gene in endometrial cancer through whole exome sequencing and loss of function screening by Liang et al. (2012). Using whole exome sequencing, ERBB3 mutations have been reported in colon, gastric, and gallbladder cancers (Jaiswal et al., 2013; Li et al., 2014). ERBB3 comprises of extracellular ligand-binding domains (I–IV), a tyrosine kinase domain, and a regulatory domain (Olayioye et al., 2000). In concordance with previous studies, the majority of mutations detected in our patients are located in the extracellular domain (L143M, P558S, E560fs) of the gene which are predicted to induce conformational changes, dysregulate ligand binding, or heterodimerization of ERBB3 with kinase-active ERBB2 (Jaiswal et al., 2013). Distinct from other EGFR members, ERBB3 has impaired tyrosine kinase activity; hence, it could be hypothesized that mutations mapped on this domain (L924V) could activate and stimulate protein tyrosine kinase (Jeong et al., 2006). These oncogenic mutations might promote tumorigenesis through aberrant activation of PI3K/AKT pathway, eliciting various biological responses including cell cycle progression, stimulation of cell migration, and invasion (Sithanandam and Anderson, 2008).
Perhaps the most promising gene from our study, based on mutation frequency and function, is WHSC1. It was more frequently mutated in the ER positive subtype. The WHSC1 gene encodes a histone methyltransferase, is classified as chromatin modification group, and plays an important role in regulating gene transcription (Li et al., 2009). A previous study showed that WHSC1 interacts with and regulate gene transcription activity of androgen receptors in prostate cancer (Kang et al., 2009) and ER in breast cancer (Feng et al., 2014), which provide a new basis for tumorigenesis in endocrine-related cancers. Hence, there is high possibility that WHSC1 is involved in development of endometrial cancer. WHSC1 is shown to be essential for ERα-dependent transcription induction in tamoxifen-resistant breast cancers, one of the potent coactivators in the ER pathway (Feng et al., 2014). They postulated that WHSC1 is recruited to the ER promoter by the bromodomain protein (BRD4) and direct H3K36 methylation that is responsible for promoting transcription initiation and elongation. Moreover, genetic alterations in the transcriptional coregulators of ER genes are likely to be the key players in the development of estrogen-dependent tumors through deregulation in the estrogen signaling pathway (Girault et al., 2006), further supporting our hypothesis for WHSC1 as the likely target gene in endometrial cancer.
WHSC1 (MMSET/NSD2) spans 120 kb, consists of 24 exons that encode a 1360 amino acid protein containing catalytic SET domain, and several chromatin-associated domains comprising a high mobility group (HMG) box which often representing a DNA-binding domain, two PWWP domains, five PHD zinc fingers defined as binding modules for methylated lysines, and C5HCH domain (Cys–His-Rich). Previous studies have almost exclusively focused on translocation and overexpression of WHSC1 while mutations in this gene have not yet been extensively characterized (Jaffe et al., 2013), even though the occurrence in a variety of cancers is increasingly reported (Huether et al., 2014; García-Carpizo et al., 2016). Based on our analysis, WHSC1 is frequently mutated in the ER positive subtype where four mutations (K574E, R1126H, R824X, and L1268P) were identified in three patients. We went on to study the role of these mutations in cell transformation by performing transient transfection in EEC-1 cell lines (endometrial cancer cell line expressing ER), followed by luciferase-based assay. Of the four mutations, we observed that only two WHSC1 mutations (R1126H and L1268P) resulted in increased cell proliferation, increased ability of the cells to migrate, and survive in clonogenic growth which represents gain of function phenotype. Both mutants are likely to exhibit increased level of methyltransferase activity of H3K36, which is generally thought to drive tumorigenesis, through activation of key genes (Rao et al., 2005). We postulate that the mutant R1126H which is located in the histone binding groove on SET domain, which is in proximity to the established hotspot mutants E1099K and D1125N, lead to a hyperactive WHSC1, causing an increase in methyltransferases activity and hence progression of malignancies (Jaffe et al., 2013). Since the mutant R1126H exhibited independent ER pathway activation, it may be involved in endometrial carcinogenesis through other pathways. Endometrial cancer may also be triggered through the transcriptional regulation of CCND1, the target gene of the β-catenin/Tcf-4 complex, through H3K36 methylation and activation of the WNT signaling pathway (Toyokawa et al., 2011). This was supported by the discovery of recurrent R1126H mutants in colorectal cancer (Supplementary Table S10), where WNT signaling is the hallmark among the many genetic events. The other pathway that this mutation might be involved in is the RAS pathway by altering the oncogenic RAS transcriptional responses resulting in increased methyltransferase activity (García-Carpizo et al., 2016). However, there is still the need to elucidate which one is the key event since epigenetic alterations usually mediate the transcription of a plethora of genes (Huang et al., 2013).
Hormonal therapy and chemotherapy are frequently used for advanced endometrial cancer, and there is no approved molecular targeted therapy so far (Temkin and Fleming, 2009). However, in one clinical trial, the use of Fulvestrant has resulted in positive response in some patients with advanced or recurrent endometrial cancer (Emons et al., 2013). Fulvestrant is a selective ER downregulator (SERD) which competitively inhibits the binding of estradiol to the ER and it degrades ER via the ubiquitin-proteasome (Berry et al., 2008). Our study also provides evidence that Fulvestrant has the ability to induce ER degradation and that it inhibits the proliferation of the transforming mutant L1268P cells, which is associated with increased sensitivity to this treatment. Hence, we propose that Fulvestrant the potential to inhibit the activity of ER signaling pathway in this particular mutant.
Conclusion
Using NGS, we have characterized the mutational landscape of different ER subtypes of EEC. We further demonstrated the role of the WHSC1 L1268P mutation in endometrial cancer carcinogenesis by activating the ER signaling pathway and its pivotal role in promoting cell proliferation, migration, and survival. The ER signaling pathway activation by WHSC1 LP1268H provides new evidence which is important for the future applications of targeted therapies for endometrial cancer carrying the WHSC1 L1268P mutations. A major limitation of our study is the small sample size, therefore further studies are warranted to explore the diagnostic and therapeutic potential of our discovery.
Ethics Statement
This study was carried out in accordance with the recommendations of Guidelines for ethical review of clinical research or research involving human subjects. The protocol was approved by the Universiti Kebangsaan Malaysia Research Ethics Committee (UKM 1.5.3.5/244/AP-2012-011). All subjects gave written informed consent in accordance with the Declaration of Helsinki.
Author Contributions
SSS performed the experiments and data analysis. N-SAM was involved in data interpretation, drafting the manuscript, and overseeing the experiments. SKS was heavily involved in data analysis. SES was involved in optimization of library preparation and sequencing. NA gave insight for the functional analyses. AMD is an endometrial surgeon involved in specimen collection and RMZ is a pathologist. RJ was involved in critical review of the manuscript. All authors read and approved the final manuscript.
Funding
This research was funded by a grant under the Projek Arus Perdana (UKM-AP-2012-011).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2018.00750/full#supplementary-material
FIGURE S1 | Variant analysis and prioritization workflow. Summary of our variant evaluation process for identifying candidate mutations.
FIGURE S2 | Mutations distribution across exon and functional domain of (a) PTEN, (b) ARID1A, (c) ERBB3, and (d) WHSC1. Any position with a mutation obtains a circle; the length of the line depends on the number of mutations detected at that codon. The gray bar represents the entire protein with the different amino acid positions (aa). The colored boxes are specific functional domains. On top of the lollipops variants are annotated as the amino-acid change at that specific site. Blue letter indicates mutation occur in ER positive subtype while green letter indicate mutation occur in ER negative subtype of our analysis.
TABLE S1 | Ion AmpliseqTM comprehensive cancer panel.
TABLE S2 | Primer’s sequence used for Sanger validation.
TABLE S3 | WHSC1 mutant primer for site-directed mutagenesis.
TABLE S4 | Somatic alterations among DNA repair gene in hypermutated patient sample.
TABLE S5 | Somatic alteration identified as driver mutations by Cancer Genome Interpreter.
TABLE S6 | Somatic alterations of ER positive subtypes in endometrioid endometrial cancer-related genes.
TABLE S7 | Somatic alteration of ER negative subtypes in endometrioid endometrial cancer-related genes.
TABLE S8 | Somatic alterations of ER positive subtype in genes relevant to estrogen signaling pathway.
TABLE S9 | Somatic alterations of ER negative subtype in genes relevant to estrogen signaling pathway.
TABLE S10 | Recurrent mutation site of WHSC1 in different cancers.
References
Amant, F., Moerman, P., Neven, P., Timmerman, D., Van Limbergen, E., and Vergote, I. (2005). Endometrial cancer. Lancet 366, 491–505. doi: 10.1016/S0140-6736(05)67063-8
Backes, F. J., Walker, C. J., Goodfellow, P. J., Hade, E. M., Agarwal, G., Mutch, D., et al. (2016). Estrogen receptor-alpha as a predictive biomarker in endometrioid endometrial cancer. Gynecol. Oncol. 141, 312–317. doi: 10.1016/j.ygyno.2016.03.006
Bansal, N., Yendluri, V., and Wenham, R. M. (2009). The molecular biology of endometrial cancers and the implications for pathogenesis, classification, and targeted therapies. Cancer Control 16, 8–13. doi: 10.1177/107327480901600102
Bateman, N. W., Shoji, Y., Conrads, K. A., Stroop, K. D., Hamilton, C. A., Darcy, K. M., et al. (2016). Identification and functional characterization of a novel bipartite nuclear localization sequence in ARID1A. Biochem. Biophys. Res. Commun. 469, 114–119. doi: 10.1016/j.bbrc.2015.11.080
Belandia, B., Orford, R. L., Hurst, H. C., and Parker, M. G. (2002). Targeting of SWI/SNF chromatin remodelling complexes to estrogen-responsive genes. EMBO J. 21, 4094–4103. doi: 10.1093/emboj/cdf412
Berry, N. B., Fan, M., and Nephew, K. P. (2008). Estrogen receptor-α hinge-region lysines 302 and 303 regulate receptor degradation by the proteasome. Mol. Endocrinol. 22, 1535–1551. doi: 10.1210/me.2007-0449
Bokhman, J. V. (1983). Two pathogenetic types of endometrial carcinoma. Gynecol. Oncol. 15, 10–17. doi: 10.1016/0090-8258(83)90111-7
Bosse, T., Haar, N. T., Seeber, L. M., Diest, P. J., Hes, F. J., Vasen, H. F. A., et al. (2013). Loss of ARID1A expression and its relationship with PI3K-Akt pathway alterations, TP53 and microsatellite instability in endometrial cancer. Mod. Pathol. 26, 1525–1535. doi: 10.1038/modpathol.2013.96
Carlson, M. J., Thiel, K. W., Yang, S., and Leslie, K. K. (2012). Catch it before it kills: progesterone, obesity, and the prevention of endometrial cancer. Discov. Med. 14, 215–222. doi: 10.1016/j.biotechadv.2011.08.021.Secreted
Cerami, E., Gao, J., Dogrusoz, U., Gross, B. E., Sumer, S. O., Aksoy, B. A., et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. doi: 10.1158/2159-8290.CD-12-0095
Chang, M. T., Asthana, S., Gao, S. P., Lee, B. H., Chapman, J. S., Kandoth, C., et al. (2015). Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 34, 155–163. doi: 10.1038/nbt.3391
Chang, Y. S., Huang, H., Da, Yeh, K. T., and Chang, J. G. (2017). Identification of novel mutations in endometrial cancer patients by whole-exome sequencing. Int. J. Oncol. 50, 1778–1784. doi: 10.3892/ijo.2017.3919
Djordjevic, B., Hennessy, B. T., Li, J., Barkoh, B. A., Luthra, R., Mills, G. B., et al. (2012). Clinical assessment of PTEN loss in endometrial carcinoma: immunohistochemistry outperforms gene sequencing. Mod. Pathol. 25, 699–708. doi: 10.1038/modpathol.2011.208
Doll, M., Abal, M., Monge, M., Gonzalez, S., Demajo, E., Colás, M., et al. (2008). Novel molecular profiles of endometrial cancer-new light through old windows. J. Steroid Biochem. Mol. Biol. 108, 221–229. doi: 10.1016/j.jsbmb.2007.09.020
Duska, L. R., Garrett, A., Rueda, B. R., Haas, J., Chang, Y., and Fuller, A. F. (2001). Endometrial cancer in women 40 years old or younger. Gynecol. Oncol. 83, 388–393. doi: 10.1006/gyno.2001.6434
Emons, G., Günthert, A., Thiel, F. C., Camara, O., Strauss, H. G., Breitbach, G. P., et al. (2013). Phase II study of fulvestrant 250 mg/month in patients with recurrent or metastatic endometrial cancer: a study of the Arbeitsgemeinschaft Gynäkologische Onkologie. Gynecol. Oncol. 129, 495–499. doi: 10.1016/j.ygyno.2013.02.039
Fayard, E., Xue, G., Parcellier, A., Bozulic, L., and Hemmings, B. A. (2010). Protein kinase B (PKB/Akt), a key mediator of the PI3K signaling pathway. Curr. Top. Microbiol. Immunol. 346, 31–56. doi: 10.1007/82-2010-58
Feng, Q., Zhang, Z., Shea, M. J., Creighton, C. J., Coarfa, C., Hilsenbeck, S. G., et al. (2014). An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res. 24, 809–819. doi: 10.1038/cr.2014.71
Ferlay, J., Soerjomataram, I., Dikshit, R., Eser, S., Mathers, C., Rebelo, M., et al. (2015). Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 136, E359–E386. doi: 10.1002/ijc.29210
Ferlay, J., Soerjomataram, I., Ervik, M., Dikshit, R., Eser, S., Mathers, C., et al. (2013). GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase. No. 11 [Internet]. Available at: http://globocan.iarc.fr
Forbes, S. A., Beare, D., Boutselakis, H., Bamford, S., Bindal, N., Tate, J., et al. (2017). COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 45, D777–D783. doi: 10.1093/nar/gkw1121
Gao, J., Aksoy, B. A., Dogrusoz, U., Dresdner, G., Gross, B., Sumer, S. O., et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6:pl1. doi: 10.1126/scisignal.2004088
García-Carpizo, V., Sarmentero, J., Han, B., Graña, O., Ruiz-Llorente, S., Pisano, D. G., et al. (2016). NSD2 contributes to oncogenic RAS-driven transcription in lung cancer cells through long-range epigenetic activation. Sci. Rep. 6:32952. doi: 10.1038/srep32952
Garg, K., and Soslow, R. A. (2014). Endometrial carcinoma in women aged 40 years and younger. Arch. Pathol. Lab. Med. 138, 335–342. doi: 10.5858/arpa.2012-0654-RA
Girault, I., Bièche, I., and Lidereau, R. (2006). Role of estrogen receptor α transcriptional coregulators in tamoxifen resistance in breast cancer. Maturitas 54, 342–351. doi: 10.1016/j.maturitas.2006.06.003
Giudice, F. S., and Squarize, C. H. (2013). The determinants of head and neck cancer: unmasking the PI3K pathway mutations. J. Carcinog. Mutagen. Suppl 5, 432–435. doi: 10.4172/2157-2518.S5-003
Guan, B., Gao, M., Wu, C.-H., Wang, T.-L., and Shih, I.-M. (2012). Functional analysis of In-frame Indel ARID1A mutations reveals new regulatory mechanisms of its tumor suppressor functions. Neoplasia 14, 986–993. doi: 10.1593/neo.121218
Han, S. Y., Kato, H., Kato, S., Suzuki, T., Shibata, H., Ishii, S., et al. (2000). Functional evaluation of PTEN missense mutations using in vitro phosphoinositide phosphatase assay. Cancer Res. 60, 3147–3151.
Huang, Z., Wu, H., Chuai, S., Xu, F., Yan, F., Englund, N., et al. (2013). NSD2 Is recruited through Its PHD domain to oncogenic gene loci to drive multiple myeloma. Cancer Res. 73, 6277–6288. doi: 10.1158/0008-5472.CAN-13-1000
Huether, R., Dong, L., Chen, X., Wu, G., Parker, M., Wei, L., et al. (2014). The landscape of somatic mutations in epigenetic regulators across 1,000 pediatric cancer genomes. Nat. Commun. 5:3630.
Jaffe, J. D., Wang, Y., Chan, H. M., Zhang, J., Huether, R., Kryukov, G. V., et al. (2013). Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat. Genet. 45, 1386–1393. doi: 10.1038/ng.2777
Jaiswal, B. S., Kljavin, N. M., Stawiski, E. W., Chan, E., Parikh, C., Durinck, S., et al. (2013). Oncogenic ERBB3 mutations in human cancers. Cancer Cell 23, 603–617. doi: 10.1016/j.ccr.2013.04.012
Jeong, E. G., Soung, Y. H., Woo, L. J., Hak, L. S., Woo, N. S., Lee, J. Y., et al. (2006). ERBB3 kinase domain mutations are rare in lung, breast and colon carcinomas. Int. J. Cancer 119, 2986–2987. doi: 10.1002/ijc.22257
Jones, S., Li, M., Williams Parsons, D., Zhang, X., Wesseling, J., Kristel, P., et al. (2012). Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types. Hum. Mutat. 33, 100–103. doi: 10.1002/humu.21633
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2016). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462. doi: 10.1093/nar/gkv1070
Kang, H. B., Choi, Y., Lee, J. M., Choi, K. C., Kim, H. C., Yoo, J. Y., et al. (2009). The histone methyltransferase, NSD2, enhances androgen receptor-mediated transcription. FEBS Lett. 583, 1880–1886. doi: 10.1016/j.febslet.2009.05.038
Kuhn, E., Wu, R. C., Guan, B., Wu, G., Zhang, J., Wang, Y., et al. (2012). Identification of molecular pathway aberrations in uterine serous carcinoma by genome-wide analyses. J. Natl. Cancer Inst. 104, 1503–1513. doi: 10.1093/jnci/djs345
Lax, S. F., Kendall, B., Tashiro, H., Slebos, R. J., and Hedrick, L. (2000). The frequency of p53, K- mutations, and microsatellite instability differs in uterine ras evidence endometrioid and serous carcinoma of distinct molecular genetic pathways. Cancer 88, 814–824. doi: 10.1002/(SICI)1097-0142(20000215)88:4<814::AID-CNCR12>3.0.CO;2-U
Le Gallo, M., O’Hara, A. J., Rudd, M. L., Urick, M. E., Hansen, N. F., O’Neil, N. J., et al. (2012). Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat. Genet. 44, 1310–1315. doi: 10.1038/ng.2455
Li, J. (1997). PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275, 1943–1947. doi: 10.1126/science.275.5308.1943
Li, M., Zhang, Z., Li, X., Ye, J., Wu, X., Tan, Z., et al. (2014). Whole-exome and targeted gene sequencing of gallbladder carcinoma identifies recurrent mutations in the ErbB pathway. Nat. Genet. 46, 872–876. doi: 10.1038/ng.3030
Li, Y., Trojer, P., Xu, C. F., Cheung, P., Kuo, A., Drury, W. J., et al. (2009). The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J. Biol. Chem. 284, 34283–34295. doi: 10.1074/jbc.M109.034462
Liang, H., Cheung, L. W. T., Li, J., Ju, Z., Yu, S., Stemke-Hale, K., et al. (2012). Whole-exome sequencing combined with functional genomics reveals novel candidate driver cancer genes in endometrial cancer. Genome Res. 22, 2120–2129. doi: 10.1101/gr.137596.112
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lu, C., and Allis, C. D. (2017). SWI/SNF complex in cancer. Nat. Genet. 49, 178–179. doi: 10.1038/ng.3779
Maehama, T., and Dixon, J. E. (1998). The tumor suppressor, PTEN/ MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 273, 13375–13379. doi: 10.1074/jbc.273.22.13375
Maniketh, I., Ravikumar, G., Crasta, J. A., Prabhu, R., and Vallikad, E. (2014). Estrogen and progesterone receptor expression in endometrioid endometrial carcinomas: a clinicopathological study. Middle East J. Cancer 5, 67–73.
Morice, P., Leary, A., Creutzberg, C., Abu-Rustum, N., and Darai, E. (2016). Endometrial cancer. Gynecol. Obstet. Fertil. 44, 239–243. doi: 10.1016/S0140-6736(15)00130-0
Nadji, M., Gomez-Fernandez, C., Ganjei-Azar, P., and Morales, A. R. (2005). Immunohistochemistry of estrogen and progesterone receptors reconsidered: experience with 5,993 breast cancers. Am. J. Clin. Pathol. 123, 21–27. doi: 10.1309/4WV7-9N2G-HJ3X-1841
Naguib, A., Bencze, G., Cho, H., Zheng, W., Tocilj, A., Elkayam, E., et al. (2015). PTEN functions by recruitment to cytoplasmic vesicles. Mol. Cell 58, 255–268. doi: 10.1016/j.molcel.2015.03.011
O’Hara, A. J., and Bell, D. W. (2012). The genomics and genetics of endometrial cancer. Adv. Genomics Genet. 2012, 33–47. doi: 10.2147/AGG.S28953
Olayioye, M. A., Neve, R. M., Lane, H. A., and Hynes, N. E. (2000). The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 19, 3159–3167. doi: 10.1093/emboj/19.13.3159
Pecorelli, S. (2009). FIGO committee on gynecology oncology revised FIGO staging for carcinoma of the vulva, cervix, and endometrium. Int. J. Gynecol. Obstet. 105, 103–104. doi: 10.1016/j.ijgo.2009.02.012
Prasad, M., Wang, H., Douglas, W., Barakat, R. R., and Ellenson, L. H. (2005). Molecular genetic characterization of tamoxifen-associated endometrial cancer. Gynecol. Oncol. 96, 25–31. doi: 10.1016/j.ygyno.2004.08.046
Prat, J., Gallardo, A., Cuatrecasas, M., and Catasús, L. (2007). Endometrial carcinoma: pathology and genetics. Pathology 39, 72–87. doi: 10.1080/00313020601136153
Rao, B., Shibata, Y., Strahl, B. D., and Lieb, J. D. (2005). Dimethylation of histone H3 at Lysine 36 demarcates regulatory and nonregulatory chromatin genome-wide. Mol. Cell Biol. 25, 9447–9459. doi: 10.1128/MCB.25.21.9447-9459.2005
Sithanandam, G., and Anderson, L. M. (2008). The ERBB3 receptor in cancer and cancer gene therapy. Cancer Gene Ther. 15, 413–448. doi: 10.1038/cgt.2008.15
Talhouk, A., and McAlpine, J. N. (2016). New classification of endometrial cancers: the development and potential applications of genomic-based classification in research and clinical care. Gynecol. Oncol. Res. Pract. 3:14. doi: 10.1186/s40661-016-0035-4
Tamborero, D., Rubio-Perez, C., Deu-Pons, J., Schroeder, M. P., Vivancos, A., Rovira, A., et al. (2018). Cancer genome interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 10:25. doi: 10.1186/s13073-018-0531-8
Temkin, S. M., and Fleming, G. (2009). Current treatment of metastatic endometrial cancer. Cancer Control 16, 38–45. doi: 10.1177/107327480901600106
The Cancer Genome Atlas Network (2012). Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. doi: 10.1038/nature11412
The Cancer Genome Atlas Research Network, Kandoth, C., Schultz, N., Cherniack, A. D., Akbani, R., Liu, Y., et al. (2013). Integrated genomic characterization of endometrial carcinoma. Nature 497, 67–73. doi: 10.1038/nature12113
Thorvaldsdóttir, H., Robinson, J. T., and Mesirov, J. P. (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192. doi: 10.1093/bib/bbs017
Toyokawa, G., Cho, H. S., Masuda, K., Yamane, Y., Yoshimatsu, M., Hayami, S., et al. (2011). Histone lysine methyltransferase Wolf-hirschhorn syndrome candidate 1 is involved in human carcinogenesis through regulation of the Wnt pathway. Neoplasia 13, 887–898. doi: 10.1593/neo.11048
Van Allen, E. M., Wagle, N., and Levy, M. A. (2013). Clinical analysis and interpretation of cancer genome data. J. Clin. Oncol. 31, 1825–1833. doi: 10.1200/JCO.2013.48.7215
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38:e164. doi: 10.1093/nar/gkq603
Wilson, B. G., and Roberts, C. W. M. (2011). SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 11, 481–492. doi: 10.1038/nrc3068
Xu, J., Li, Z., Wang, J., Chen, H., and Fang, J. Y. (2014). Combined PTEN mutation and protein expression associate with overall and disease-free survival of glioblastoma patients. Transl. Oncol. 7, 196–205. doi: 10.1016/j.tranon.2014.02.004
Zannoni, G. F., Monterossi, G., De Stefano, I., Gargini, A., Salerno, M. G., Farulla, I., et al. (2013). The expression ratios of estrogen receptor alpha (ERalpha) to estrogen receptor beta1 (ERbeta1) and ERalpha to ERbeta2 identify poor clinical outcome in endometrioid endometrial cancer. Hum. Pathol. 44, 1047–1054. doi: 10.1016/j.humpath.2012.09.007
Zhang, J., Baran, J., Cros, A., Guberman, J. M., Haider, S., Hsu, J., et al. (2011). International cancer genome consortium data portal-a one-stop shop for cancer genomics data. Database 2011:bar026. doi: 10.1093/database/bar026
Keywords: endometrial cancer, endometrioid subtype, next-generation sequencing, estrogen receptor, WHSC1, Fulvestrant
Citation: Suhaimi SS, Ab Mutalib N-S, Khor SS, Md Zain RR, Syafruddin SE, Abu N, Mohd Dali AZH and Jamal R (2018) Targeted Next-Generation Sequencing Identifies Actionable Targets in Estrogen Receptor Positive and Estrogen Receptor Negative Endometriod Endometrial Cancer. Front. Pharmacol. 9:750. doi: 10.3389/fphar.2018.00750
Received: 26 April 2018; Accepted: 20 June 2018;
Published: 13 July 2018.
Edited by:
Dong-Hua Yang, St. John’s University, United StatesReviewed by:
Kaijian Hou, American Diabetes Association, United StatesZuodong Qin, Hunan University of Science and Engineering, China
Copyright © 2018 Suhaimi, Ab Mutalib, Khor, Md Zain, Syafruddin, Abu, Mohd Dali and Jamal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nurul-Syakima Ab Mutalib, c3lha2ltYUBwcHVrbS51a20uZWR1Lm15; bnVydWxzeWFraW1hQGdtYWlsLmNvbQ== Rahman Jamal, cmFobWFuakBwcHVrbS51a20uZWR1Lm15
†Shared first authorship.