 Mariachiara Zuccarini1,2*†
Mariachiara Zuccarini1,2*† Patricia Giuliani1,2†
Patricia Giuliani1,2† Monica Frinchi3
Monica Frinchi3 Giuseppa Mudò3Rosa Maria Serio4
Giuseppa Mudò3Rosa Maria Serio4 Natale Belluardo3
Natale Belluardo3 Silvana Buccella1,2
Silvana Buccella1,2 Marzia Carluccio1,2
Marzia Carluccio1,2 Daniele F. Condorelli5
Daniele F. Condorelli5 Francesco Caciagli1,2*
Francesco Caciagli1,2* Renata Ciccarelli1,2
Renata Ciccarelli1,2 Patrizia Di Iorio1,2
Patrizia Di Iorio1,2- 1Department of Medical, Oral and Biotechnological Sciences, Università degli Studi “G. d’Annunzio” Chieti-Pescara, Chieti, Italy
- 2Aging Research Center, Ce.S.I., “G. d’Annunzio” University Foundation, Chieti, Italy
- 3Department of Experimental Biomedicine and Clinical Neurosciences, University of Palermo, Palermo, Italy
- 4Department of Biological, Chemical and Pharmaceutical Sciences and Technologies (STEBICEF), University of Palermo, Palermo, Italy
- 5Department of Bio-Medical Sciences, University of Catania, Catania, Italy
Mounting evidence suggests that the guanine-based purines stand out as key player in cell metabolism and in several models of neurodegenerative disorders, such as Parkinson’s and Alzheimer’s diseases. Guanosine (GUO) and guanine (GUA) are extracellular signaling molecules derived from the breakdown of the correspondent nucleotide, GTP, and their intracellular and extracellular levels are regulated by the fine-tuned activity of two major enzymes, purine nucleoside phosphorylase (PNP) and guanine deaminase (GDA). Noteworthy, GUO and GUA, seem to play opposite roles in the modulation of cognitive functions, such as learning and memory. Indeed GUO, despite exerting neuroprotective, anti-apoptotic and neurotrophic effects, causes a decay of cognitive activities, whereas GUA administration in rats results in working memory improvement (prevented by L-NAME pre-treatment). This study was designed to investigate, in a model of SH-SY5Y neuroblastoma cell line, the signal transduction pathway activated by extracellular GUA. Altogether, our results showed that: (i) in addition to an enhanced phosphorylation of ASK1, p38 and JNK, likely linked to a non-massive and transient ROS production, the PKB/NO/sGC/cGMP/PKG/ERK cascade seems to be the main signaling pathway elicited by extracellular GUA; (ii) the activation of this pathway occurs in a pertussis-toxin sensitive manner, thus suggesting the involvement of a putative G protein coupled receptor; (iii) the GUA-induced NO production, strongly reduced by cell pre-treatment with L-NAME, is negatively modulated by the EPAC-cAMP-CaMKII pathway, which causes the over-expression of GDA that, in turn, reduces the levels of GUA. These molecular mechanisms activated by GUA may be useful to support our previous observation showing that GUA improves learning and memory functions through the stimulation of NO signaling pathway, and underscore the therapeutic potential of oral administration of guanine for treating memory-related disorders.
Introduction
Guanine-based purines are known to play crucial role in the modulation of neurotransmission and neuropathologies (Ciccarelli et al., 2001; Boison, 2011; Bettio et al., 2016; Di Liberto et al., 2016). In particular, the purine nucleoside Guanosine (GUO), which is mostly released from astrocytes under pathological conditions (i.e., hypoxic or hypoglycemic stress), is thought to exert both neurotrophic and neuroprotective effects (Di Iorio et al., 2001, 2004; Giuliani et al., 2012, 2015; Lanznaster et al., 2016); indeed, it oversees neuronal development and synaptic activity, and protects neuronal and glial cells against oxidative stress and excitotoxicity (Neary, 1996; Schmidt et al., 2007; Tarozzi et al., 2010; Quincozes-Santos et al., 2014; Bellaver et al., 2015; Ribeiro, 2016; Thomaz et al., 2016). Furthermore, in rats, GUO administration during pre-training displays amnesic effect on inhibitory avoidance task (Roesler et al., 2000; Vinadé et al., 2004; Saute et al., 2006). At present, much less is known about the effects that Guanine (GUA) exerts in the central nervous system. Intracellular GUA derives from guanosine triphosphate (GTP) breakdown and represents the starting point of reactions deputed to maintain intracellular levels of GTP (purine salvage pathway). When intracellular levels of GUA are excessive, it may be transported outside the cells by specific transmembrane nucleobases transporters, although most of the extracellular GUA derives from the breakdown of the released GTP and it is generated by GUO in a reaction catalyzed by the purine nucleoside phosphorylase (PNP) (Rathbone et al., 2008; Giuliani et al., 2016, 2017; Peña-Altamira et al., 2017). On the contrary, GUA degradation to xanthine (Xan) is mediated by Guanine deaminase (GDA) or cypin (Miyamoto et al., 1982), which has been regarded as one of the “intrinsic factors” that regulate dendrite morphology together with the small GTPases RhoA, Rac1, the β-catenin (Yu and Malenka, 2004), PSD-95 (Charych et al., 2006) and the calcium/calmodulin-dependent protein kinase II (CAMKII) (Fink et al., 2003). CaMKII is a synaptic signaling molecule that plays a crucial role during long-term memory formation (Lucchesi et al., 2011) and its endogenous inhibitors CaMK2N1 and CaMK2N2 are highly expressed during memory consolidation (Lepicard et al., 2006). Cyclic AMP-CREB axis is implicated in learning and memory processes and has been shown to activate CaMKII.
In a previous work (Giuliani et al., 2012), we reported the effects of GUO and GUA on learning and memory in a model of passive avoidance task in rats. In that study, the oral administration of GUO exerted amnesic activity on inhibitory avoidance task and was unable to prevent the amnesic effect caused by N-omega-nitro-l-arginine methyl ester (L-NAME), a non-specific NOS inhibitor known to reduce the capability of treated animals to acquire or retain information in several learning tasks. Conversely, the administration of GUA counteracted the L-NAME-mediated amnesic effects, by increasing the step-through latency either when it was given in the learning phase or during the memory consolidation phase.
In addition to GUO and GUA, another guanine-based purine has been correlated to changes in memory processes, namely cyclic guanosine monophosphate (cGMP), which exerts memory-enhancing effect through the modulation of NMDA receptors and the glutamate-nitric oxide (NO) pathway (Cabrera-Pastor et al., 2016) or via NOS-soluble guanylyl cyclase (sGC)-cGMP- protein kinase G (PKG) pathway (Friebe and Koesling, 2003; Boess et al., 2004; Masood et al., 2009; Bollen et al., 2014; Lueptow et al., 2015). Noteworthy, there is a large body of evidence confirming the existence of a cross-talk between NO and ERK signaling pathways during memory formation and learning processes (Moosavi et al., 2014). Indeed, it has been shown that ERK represents a crucial downstream mediator of NO in the brain (Chien et al., 2008) and that the blockage of NO-cGMP-PKG prevents the activation of ERK mediated by high-frequency stimulation-(HFS) (Ping and Schafe, 2010).
Based on the above mentioned mnesic effects elicited, in vivo, by GUA, and several findings showing that NO-cGMP-PKG-ERK signaling pathway is positively correlated with enhancement of memory formation (Adams and Sweatt, 2002; Davis and Laroche, 2006; Giovannini, 2006; Philips et al., 2007; Chien et al., 2008; Adaikkan and Rosenblum, 2012), in this study we aimed to:
(a) verify, by using human neuroblastoma cell line SH-SY5Y, if cGMP and NO-PKG-ERK signaling pathway resulted to be activated upon cell exposure to GUA;
(b) assess whether the activation of this signaling pathway may involve the extracellular GUA interaction with a new putative receptor.
Materials and Methods
Materials and Chemicals
The human neuroblastoma cell line SH-SY5Y was purchased from European Collection of Authenticated Cell Culture (ECACC, Salisbury, United Kingdom); Guanine, Guanosine, Nutrient Mixture F-12 Ham, Minimum Essential Medium Eagle (MEM), Non-Essential Amino Acids (NEAA), L-Glutamine, Trypsin-EDTA, Pertussis toxin from Bordetella pertussis (PTX), 3-Isobutyl-1-methylxanthine (IBMX), 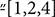 Oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), 8-(4-Chloro-phenylthio)-2′-O-methyladenosine 3′,5′-cyclic monophosphate monosodium hydrate (8-pCPT-2′-O-Me-cAMP), N-[2-[N-(4-Chlorocinnamyl)-N-methylaminomethyl]phenyl]-N-(2-hydroxyethyl)-4-methoxybenzenesulfonamide phosphate salt, N-[2-[[[3-(4′-Chlorophenyl)-2-propenyl]methylamino]methyl]phenyl]-N-(2-hydroxyethyl)-4′-methoxybenzenesulfonamide phosphate salt (KN-93), Propentofylline, S-(4-Nitrobenzyl)-6-thioinosine (NBTI), 2′,7′-Dichlorofluorescin diacetate (H2DCF-DA), dimethylsulfoxide (DMSO), Ionomycin, trypsin/EDTA, EDTA, EGTA, HEPES, Phosphate Buffer Solution (PBS), dithiothreitol (DTT), NADPH, calmodulin, CaCl2, tetrahydrobiopterin and the cationic exchange resin Dowex AG50WX-8, N-(1-naphthylethylenediamine) dihydrochloride, were purchased from Sigma (Milan, Italy); NG-Nitro-L-arginine methyl ester hydrochloride (L-NAME), MSC 20329644, GF109203X, 10-DEBC hydrochloride, Dipyridamole and LY 294002 hydrochloride were purchased from Tocris (Milan, Italy); Penicillin-streptomycin and Heat-inactivated fetal bovine serum (FBS) were purchased from Gibco® (Thermo Fischer Scientific, Monza, Italy); Phospho-ASK1, Phospho-p38 MAPK, Phospho-SAPK/JNK, Phospho-PKC (pan), Phospho-Akt, Phospho-p44/42 MAPK (Erk1/2), β-Actin, secondary anti-rabbit IgG HRP-linked antibody were purchased from Cell Signaling Technology (Cell Signaling, Leiden, Netherlands); PNP and Guanase Deaminase antibodies were purchased from Novus Biologicals (Space Import-Export, Milan, Italy).
Oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), 8-(4-Chloro-phenylthio)-2′-O-methyladenosine 3′,5′-cyclic monophosphate monosodium hydrate (8-pCPT-2′-O-Me-cAMP), N-[2-[N-(4-Chlorocinnamyl)-N-methylaminomethyl]phenyl]-N-(2-hydroxyethyl)-4-methoxybenzenesulfonamide phosphate salt, N-[2-[[[3-(4′-Chlorophenyl)-2-propenyl]methylamino]methyl]phenyl]-N-(2-hydroxyethyl)-4′-methoxybenzenesulfonamide phosphate salt (KN-93), Propentofylline, S-(4-Nitrobenzyl)-6-thioinosine (NBTI), 2′,7′-Dichlorofluorescin diacetate (H2DCF-DA), dimethylsulfoxide (DMSO), Ionomycin, trypsin/EDTA, EDTA, EGTA, HEPES, Phosphate Buffer Solution (PBS), dithiothreitol (DTT), NADPH, calmodulin, CaCl2, tetrahydrobiopterin and the cationic exchange resin Dowex AG50WX-8, N-(1-naphthylethylenediamine) dihydrochloride, were purchased from Sigma (Milan, Italy); NG-Nitro-L-arginine methyl ester hydrochloride (L-NAME), MSC 20329644, GF109203X, 10-DEBC hydrochloride, Dipyridamole and LY 294002 hydrochloride were purchased from Tocris (Milan, Italy); Penicillin-streptomycin and Heat-inactivated fetal bovine serum (FBS) were purchased from Gibco® (Thermo Fischer Scientific, Monza, Italy); Phospho-ASK1, Phospho-p38 MAPK, Phospho-SAPK/JNK, Phospho-PKC (pan), Phospho-Akt, Phospho-p44/42 MAPK (Erk1/2), β-Actin, secondary anti-rabbit IgG HRP-linked antibody were purchased from Cell Signaling Technology (Cell Signaling, Leiden, Netherlands); PNP and Guanase Deaminase antibodies were purchased from Novus Biologicals (Space Import-Export, Milan, Italy).
Cell Culture
The human neuroblastoma cells, SH-SY5Y, were cultured in 75 cm2 flasks in a 1:1 mixture of F-12 nutrient mixture (Ham 12) and Eagle’s MEM (EBSS) supplemented with 2 mM Glutamine, 1% Non-Essential Amino Acids (NEAA), 15% Foetal Bovine Serum (FBS) and 100 units/mL penicillin and 100 μg/mL streptomycin and maintained at 37°C in 5% CO2, humified air.
For the evaluation of PNP and GDA release, cell medium was removed and replaced by serum free-medium and maintained in humified atmosphere, 5% CO2, 37°C. At the end of the experiment, aliquots (2 mL) of the culture medium were collected, placed in suitable devices (Amikon Ultra 2 mL, cutoff 10 K, Merck Millipore, Germany) and centrifuged following the manufacturer’s instruction, in order to concentrate culture media containing the enzymes.
For the evaluation of purine release, cell medium was removed and replaced with Krebs-HEPES buffer (15 m M HEPES, pH 7.4, 120 mM NaCl, 4 mM KCl, 1.2 mM MgSO4, 1 mM CaCl2), and 10 mM D-glucose oxygenated (95% O2/5% CO2). After 30 min, the cells were incubated for further 30 min with the same buffer containing 2.5 μM GUO combined with 0.675 μM of [3H]GUO (specific activity 5.3 Ci/mmol; Movarek Biochemicals). At the end of this incubation period, cells were washed twice with unlabeled Krebs-HEPES buffer and maintained in this medium in standard condition (37°C, 5% CO2). When used, purine uptake inhibitors were added to Krebs-HEPES just after the incubation with labeled GUO. At the end of the experiment, an aliquot of the culture medium was collected and immediately heat-inactivated for 5 min at 70°C to avoid any further enzymatic degradation of the released purine. Samples were, then, centrifuged, filtered with 0.2 μm filters (Millipore, Vimodrone, Italy) and stored at 80°C before HPLC analysis.
For Immunoblot assays, SH-SY5Y cells were subcultured in 100 × 20-mm Petri Dishes (BD Falcon) at a seeding density of 2 × 105 per dish (for each sample two dishes were pulled together) and grown until 80% confluence. Before all experiments, cells were starved for 24 h in medium containing 0.1% FBS.
HPLC Method for the Evaluation of Purine Levels in the Extracellular Milieu
According to the method previously described (Giuliani et al., 2012, 2017), purines were measured by an Agilent 1100 series HPLC system (Agilent Technologies, Santa Clara, CA, United States), by using, for the separation of the compounds, a reverse phase analytical column (LiChrospher 100 RP-18 5 μm in LiChroCART 125-4, Merck) and a 15-min linear gradient [from 100% of buffer A (60 mM KH2PO4 and 5 mM tetrabutylammonium phosphate, pH 6.0) to 100% solvent B (30% methanol plus 70% buffer A)] at a flow rate of 1.5 mL/min. The detection of unlabeled compounds was achieved using a Diode Array Detector (Agilent Technologies, Santa Clara, CA, United States) with wavelength set at 254 nm for all the substances except Uric Acid (UAc), which was 290 nm. Released purines were identified and quantified by comparison with pure external standards. Since many purine compounds are present in the extracellular milieu at concentrations below the UV detection limit, the HPLC system was equipped with an online radiochemical detector (FLO-ONE 500 TR, Packard Instruments) for the concurrent measurement of radiolabeled purine present in the outflow from the Diode Array Detector, in order to improve the sensibility of the analysis. Thus, the HPLC effluent was mixed with the liquid scintillation cocktail (Ultima-FloM, Perkin-Elmer) at a flow ratio of 2:1 and passed through a 500 μL detector flow cell. Radio-chromatograms were integrated and each radioactive peak was quantified.
Measurement of Enzyme Activity in the Extracellular Milieu
PNP activity was measured in an assay buffer containing 50 mM HEPES, pH 7.0, 50 mM inorganic phosphate (Pi), used as co-substrate, and 200 μM GUO used as substrate, whereas the mixture used to evaluate GDA activity consisted of 100 mM TrisHCl pH 8 plus 200 μM GUA used as substrate. These enzymatic reactions were started by adding aliquot of the concentrated extracellular culture medium. The mixtures were then incubated by shaking at 37°C for 15 min, to evaluate PNP activity, and for 60 min, to determine GDA activity. The reactions were stopped by heating the mixture at 70°C for 5 min and the precipitated proteins were removed by centrifugation. The enzyme activity was determined by quantifying the rate of conversion of GUO to GUA, for PNP, or the conversion of GUA to XAN for GDA, using the HPLCmethod previously described (Giuliani et al., 2016). In this case, the Agilent HPLC was equipped with a thermostated column compartment, a diode array detector, and a fluorescence detector (Agilent Technologies). Briefly, separation was achieved using a Phenomenex Kinetex pentafluorophenyl analytical column (5 μm pore size, 100 Å particle size, 250 × 4.6 mm; Phenomenex INC) at 35°C. Separation was carried out with a 15-min non-linear gradient elution (flow rate 1 mL/min) using a mobile phase composed of 0.1% (v/v) formic acid in water (solution A) and methanol (solution B). The fluorescent GUO and GUA were monitored at an excitation wavelength of 260 nm and an emission wavelength of 375 nm, whereas for the non-fluorescent compounds, i.e., XAN and UAc, the UV detector was set at 254 and 290 nm respectively. Allsubstances were identified and quantified by comparison with pure external standards. Enzyme activity was expressed as Unit (U) present in the total medium, being 1 U of enzyme the amount of enzyme that converts 1 μmole of substrateinto product per min.
Cell Viability Assay
Cell death was monitored by using the CytoTox-96 assay (Promega Italia, Milan, Italy) that allows to evaluate the lactate dehydrogenase (LDH) activity. The assay is based on a 30-min coupled enzymatic assay, catalyzed by released LDH, which results in conversion of a tetrazolium salt, 2-p-(iodophenyl)-3-(p-nitrophenyl)-5-phenyltetrazolium chloride (INT), into a red formazan product. SH-SY5Y cells were seeded in 96-well plates at 5 × 103 cells/well of confluence and incubated for 2 days. For all samples, the cell culture medium was replaced with Krebs-HEPES buffer (15 mM HEPES, pH 7.4, 120 mM NaCl, 4 mM KCl, 1.2 mM MgSO4, 1 mM CaCl2) with or without 50 μM GUA (0–12 h). At the end of exposure, the Lysis solution was added for 45 min to control wells for the determination of maximum LDH release. Afterward, 50 μL of collected media were transferred to a fresh 96-well (enzymatic assay) plate, together with 50 μL of Substrate buffer containing 0.7 mM p-iodonitrotetrazolium Violet, 50 mM L-lactic acid, 0.3 mM phenazine methoxysulfate, 0.4 mM NAD and 0.2 M Tris-HCl pH 8.0. Finally, the plate was protected from light and incubated for 30 min at RT. The absorbance was recorded at 490 nm of wavelength in a microplate reader after adding the Stop Solution. LDH activity was expressed as the proportion of LDH released into the culture medium compared to the total amount of LDH present in cells lysates and calculated as follows: (medium absorbance value – white absorbance value)/(medium absorbance + lysate absorbance) × 100.
Immunoblot
SH-SY5Y cells were seeded overnight onto 100 mm Petri Dishes (BD Falcon) at 2.0 × 105 cells/dish in 6 mL of 1:1 mixture of F-12 nutrient mixture (Ham 12) and Eagle’s MEM (EBSS) supplemented with 2 mM Glutamine, 1% Non Essential Amino Acids (NEAA), 15% Foetal Bovine Serum (FBS) and 100 units/mL penicillin and 100 μg/mL streptomycin. After 24-h starvation, cells were submitted to different treatments in MEM supplemented with 0.5% FBS and 1% Penicillin/Streptomycin. After treatment, cells were washed twice with ice cold 1× PBS (Sigma–Aldrich), lysed with RIPA Buffer (Sigma–Aldrich) containing 150 mM NaCl, 10 mM EDTA, 1% NP40, 0.5% deoxycholic acid, 0.1% SDS, and 50 mM Tris, pH 7.5, supplemented with 1% Protease Inhibitor Cocktail (Sigma–Aldrich), scraped off, pulled, and clarified by centrifugation at 12.500 × g for 20 min, 4°C. Before performing Immunoblot, a sample buffer (5× Laemmli buffer with 10% mercaptoethanol) was added to melted lysates 1:4. Protein concentrations were obtained using the Bio-Rad Protein Assay (Bio-Rad Laboratories, Hercules, CA, United States) based on the Bradford method. An equal amount of 50–70 μg of protein was resolved by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). The resolved proteins were transferred onto a nitrocellulose membrane and then incubated with blocking buffer 1× TBS containing 0.1% Tween-20 (TBST) and 3% BSA or 5% non-fat dry milk for 2 h, RT, and subsequently probed with specific primary antibody at 4°C, overnight. After washing with TBST, the membrane was further probed with corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies at RT for 1 h. Membranes were finally washed, before subjecting them to ECL Plus Immunoblot Detection Reagent (Amersham, GE Healthcare). The immunoreactive bands were visualized under a chemiluminescence detection system (UVItec, Cambridge, United Kingdom). Band intensity data were obtained using Quantity One software (Bio-Rad Laboratories). Blotting membranes were stripped and re-probed with anti-actin antibody as equal loading control. Estimates of phosphorylated proteins are presented as densitometric ratios, normalized to the corresponding total protein content. Apart from PNP antibody (1:500), all primary antibodies [Phospho-ASK1 (Ser83), Phospho-p38 MAPK (Thr180/Tyr182), Phospho-SAPK/JNK (Thr183/Tyr185), Phospho-PKC (Ser660), Phospho-Akt (Thr450), Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204), Guanase Deaminase, β-Actin] were diluted 1:1000 in 3% BSA/1× TBS/0.1% Tween 20 or 2.5% non-fat dry milk/1× TBS/0.1% Tween 20. The secondary antibody was used at 1:2500 dilution in 3% BSA/1× TBS/0.1% Tween 20 or 2.5% non-fat dry milk/1× TBS/0.1% Tween 20.
Measurement of Cellular Reactive Oxygen Species (ROS)
The amount of intracellular reactive oxygen species (ROS) was measured by using the probe H2DCF-DA (Ha et al., 1997), which diffuses into the cells and is oxidized to the green fluorescent compound 2′,7′-dichlorofluorescein (DCF) upon reaction with intracellular hydrogen peroxide or low-molecular-weight hydroperoxides. Cells were seeded at 1 × 106 cells/well in 6-well culture plates and incubated overnight. After exposure to different concentration of GUA for 30 min, cells were incubated with 5 μM H2DCF-DA for 30 min, in the dark, at 37°C. At the end of incubation, the cells were washed with PBS and fluorescence was measured at an excitation wavelength of 480 nm and an emission wavelength of 540 nm in a fluorescence microplate reader (Thermo Fischer Scientific, Monza, Italy). ROS production was determined by analyzing DCF fluorescence normalized for total protein content. The fluorescence intensity was proportional to the amount of ROS produced by cells.
Determination of Nitric Oxide Synthase (NOS) Activity
Nitric oxide synthase activity was measured from the conversion of L-[3 H]-arginine to L-[3 H]-citrulline based on the method of Bredt et al. (1991) with modifications. SH-SY5Y cells were grown overnight in 6-well plates. After 24-h starvation, cells were exposed for 30 min to 50 μM GUA, 5 μM L-NAME or 2 μM Ionomycin, the latter used as positive control. When used in combination, L-NAME was administered 15 min before cell exposure to GUA or Ionomycin. Thereafter, cells were washed three times with ice-cold 1X PBS, scraped in 1X PBS containing 1 mM EDTA, and centrifuged for 10 min at 1200 g. The pellets were resuspended in a reaction buffer containing 50 mM Hepes, 1 mM EDTA, 1mM DTT (pH 7.2) and sonicated on ice with two 10 s bursts. The reaction was started by addition to samples of reaction mixture [1 mM NADPH, 1 nmol/l calmodulin, 1.25 mM CaCl2, 3 μM tetrahydrobiopterin, 2.5 μCi/μl of L-[3H]arginine (Perkin Elmer, Boston, MA, United States, specific activity 42.6 Ci/mmol), unlabelled arginine]. After an incubation of 15 min at 37°C, the assay was stopped by adding 20 mM Hepes-Na containing 2 mM EDTA and 2 mM EGTA (pH 5.5), and the reaction mixture was applied to 2-ml columns of Dowex AG50WX-8 (Na+ form), which were eluted with 4 ml of water. The radioactivity corresponding to the [3H]-citrulline was measured by liquid scintillation analyzer (Tris-Carb 2100 TR, Perkin Elmer) and normalized for extract protein content determined with Bradford method. NOS activity was expressed as pmoles citrulline/min/mg cell protein.
Statistical Analysis
Data are represented as means ± standard error of mean (SEM). Comparisons among experimental groups were performed by Student t-test or by two-way ANOVA followed by Sidak’s multiple comparisons test using GraphPad Prism 6.01 (San Diego, CA, United States), as indicated. Statistical difference was accepted when P < 0.05. All experiments were performed at least three times.
Results
The Levels of Guanine-Based Purines in SH-SY5Y Culture Media Are Controlled by Specific Nucleobase and Nucleoside Transporters and by the Presence of Purine-Converting Enzymes
A hallmark of several neurodegenerative diseases is the activation of neuronal and glial cells and the following induction of oxidative stress and neuronal toxicity. In the attempt to investigate neuronal response to GUA exposure, we used SH-SY5Y, a human derived neuroblastoma cell, which has been often used as a model to study molecular mechanisms associated to ROS production, apoptosis and amyloid-β-induced neuronal toxicity in Alzheimer’s disease (Tarozzi et al., 2010; Ali-Rahmani et al., 2014; Puangmalai et al., 2017; Modi et al., 2017).
We firstly measured both the intracellular and extracellular levels of purines in cultured SH-SY5Y cells, in resting conditions. In the SH-SH5Y lysates, the levels of Guanine-based nucleotides prevailed over the correspondent nucleobases and the inhibition of the uptake, performed by treating cell culture with a cocktail of inhibitors of nucleoside and nucleobase transmembrane transporters (100 μM propentofylline, 10 μM NBTI, 10 μM dypiridamole) did not caused any significant effect (Figure 1A). An opposite trend was observed in the culture medium, wherein the uptake inhibitors significantly increased extracellular levels of GUA, Xan and UAc [two-way ANOVA analysis and Sidak’s multiple comparisons test: P < 0,005] (Figure 1B). This suggested that the levels of extracellular guanine-based purines depended both on the balance between nucleotide release and nucleoside uptake and on the presence, in the culture medium, of extracellular purine-converting enzymes. Indeed, by using Immunoblot analysis and HPLC for the measurement of the enzyme activities (Giuliani et al., 2017), we confirmed the presence in the culture medium of PNP and GDA (Figure 1C). Both the enzymes tended to accumulate in the culture medium over the time (from 2 up to 12 h) following a similar trend even though at different levels (PNP amount was about 7–10 fold higher than GDA). This event was not associated with significant cell damage or death, as demonstrated by the constant and minor presence of LDH, measured during the same period in the culture media (Figure 1C).
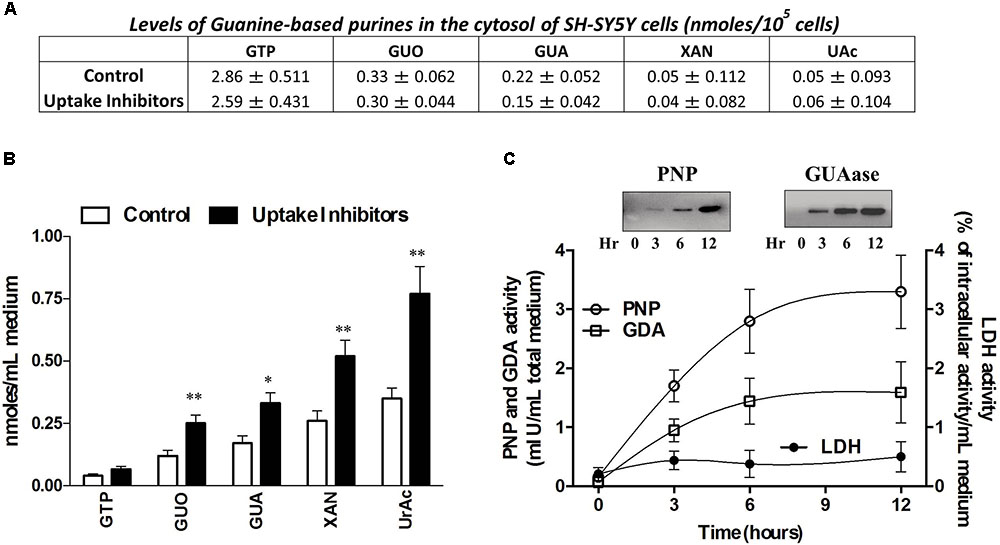
FIGURE 1. SH-SY5Y neuroblastoma cells release guanine-based purines, PNP and GDA in the culture medium. (A) HPLC analysis of intracellular levels of Guanine-based purines in SH-SY5Y cells. (B) HPLC analysis of extracellular levels of guanine-based purines at rest (control) and in the presence of the inhibitors of cell uptake. SH-SY5Y were incubated with 2.5 μM guanosine (GUO) combined with 0.675 μM of [3H]GUO, the latter used as tracer. Values are expressed as nmoles/mL of culture medium, and represent the mean ± SEM of five independent experiments. ∗p < 0.05; ∗∗p < 0.01: statistical significance versus untreated cells (control) (Student’s t-test). (C) Evaluation of the presence and activity of purine nucleoside phosphorylase (PNP) and guanine deaminase (GDA) in the culture medium of SH-SY5Y cells. (C1) Representative Immunoblots of PNP and GDA expression in SH-SY5Y culture medium. After 24 h incubation, SH-SY5Y cell culture medium was collected after 3, 6, and 12 h, concentrated and analyzed for PNP and GDA expression. (C2) HPLC analysis of PNP and GDA activity evaluated up to 12 h. PNP activity was assayed by using 200 μM guanosine (GUO) as substrate plus 50 mM Pi as co-substrate for 15 min at 37°C, whereas GDA activity was measured by using 100 mM TrisHCl pH 8 plus 200 μM GUA as substrate for 60 min at 37°C. Values are expressed as milli-International Units (mIU) of enzyme per total culture medium and represent the mean ± SEM of three independent experiments, run in duplicate. (C3) Evaluation of cell viability of SH-SY5Y cells by LDH assay. Values are expressed as the percentage of the intracellular LDH activity that was determined after cell lysis, and represent the mean ± SEM of five different experiments.
Guanine Increases ASK1, p38, JNK, PKC and PKB Phosphorylation and This Effect Is Prevented, Except for PKB, by Cell Treatment with a Cocktail of Inhibitors of the Uptake
Cell exposure to exogenous GUA, for 30 min, elicited a non-massive and dose-dependent production of ROS. This occurred without any significant collateral LDH production (Figure 2). A similar effect has been found in different glial cells (astrocytes or microglial cells, data not shown), wherein ROS production seemed to be even greater than that elicited in SH-SY5Y cells. The ROS production has been often associated with an increased phosphorylation of ASK1, p38 and JNK (Shen and Liu, 2006; Jiang et al., 2017) and it has been reported that the inhibition of c-Jun N-terminal Kinase (JNK) in SH-SY5Y cells, prevented 6-Hydroxydopamine-induced ROS production and toxicity (Feng et al., 2013). Based on this evidence, we investigated which signaling transduction pathways were involved in GUA-induced effect. SH-SY5Y cells were exposed to 50 μM GUA, the half maximal effective concentration, for 10 min, and the expression of p-ASK1, p-p38, p-JNK, PKC and PKB was analyzed by Immunoblot. The short-term exposure of cells to GUA significantly increased the phosphorylation of all the mentioned kinases. The two-way ANOVA analysis of their expression showed an effect of cell exposure to GUA [F(1,12) = 27.53, P = 0.0002 for p-ASK1; F(1,12) = 16.89, P = 0.0014 for p-38; F(1,12) = 41.35, P = 0.0001 for p-JNK; F(1,12) = 16.09, P = 0.0017 for PKC and F(1,12) = 27.07, P = 0.0002 for PKB] (Figures 3A,B). Cell pre-treatment with the previously mentioned cocktail of inhibitors of both nucleoside and nucleobase transmembrane transporters, 30 min before cell exposure to GUA, did not modify the basal phosphorylation level of all kinases but strongly reduced GUA-induced phosphorylation of ASK1, p38, JNK and PKC, [two-way ANOVA: F(1,12) = 4.77, P = 0.045 for p-ASK1; F(1,12) = 13.07, P = 0.0035 for p-JNK; F(1,12) = 7.20, P = 0.019 for PKC with interaction values between factors of F(1,12) = 13.86, P = 0.0029; F(1,12) = 15.51, P = 0.002 and F(1,12) = 22.49, P = 0.0005, respectively], revealing that the effect of GUA on these pathways was mainly intracellular (Figures 3A,B). Conversely, GUA-induced phosphorylation of PKB was not modified, [two-way ANOVA: F(1,12) = 0.6131, P = 0.4488 and no effect of the interaction between both factors F(1,12) = 0.01, P = 0.9212], thus suggesting that the activation of this pathway mainly depends on the extracellular activity of GUA (Figure 3C).
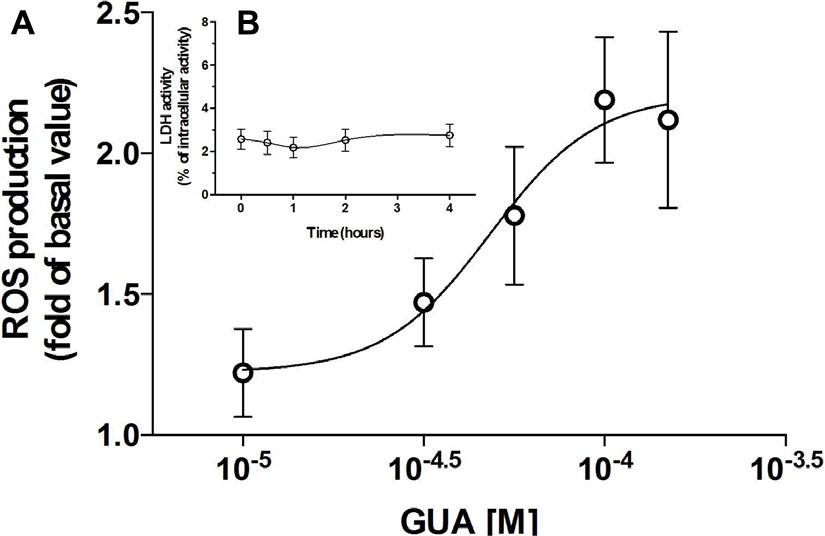
FIGURE 2. Guanine (GUA) induces ROS production in SH-SY5Y neuroblastoma cells in a dose-dependent manner. (A) SH-SY5Y cells were incubated for 30 min with different concentrations of GUA (0–100 μM). The levels of ROS are presented as folds of control and represent the mean ± SEM of five different experiments. (B) Evaluation of cell viability of SH-SY5Y cells exposed to 50 μM GUA up to 4 h. Values are expressed as the percentage of the intracellular LDH activity that was determined after cell lysis and represent the mean ± SEM of five different experiments.
FIGURE 3. Guanine induces ASK1, p38, JNK, PKC and PKB phosphorylation in SH-SY5Y neuroblastoma cells. SH-SY5Y cells were cultured for 24 h and incubated for 10 min with 50 μM GUA, in the absence or presence of a cocktail of inhibitors of both nucleoside and nucleobase transmembrane transporters (100 μM Propentofylline, 10 μM NBTI, 10 μM Dypiridamole). Representative Immunoblot analysis of (A1) p-ASK1 (Thr845), (A2) p-p38, (A3) p-JNK, (B) PKC, (C) p-PKB, with the respective β-actin as loading control, and the correspondent quantitative data of densitometric analysis. Each column represents the mean ± SEM of four independent experiments, and it is expressed as relative protein expression normalized to β-actin. Student’s t-test: ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, vs. untreated cells (Basal); #P < 0.05, ##P < 0.01, vs. GUA-treated cells.
PI3K-PKB-ERK Is the Main Signaling Pathway Activated by Extracellular GUA in SH-SY5Y Cells
Due to the evidence that ERK stands out as a key player in the modulation of several memory processes (Giovannini, 2006) and that, in different experimental models, it represents the downstream effector of ROS-induced phosphorylation of ASK1, p38 and JNK (Lee and Cheong, 2017), we sought to determine the relevance of each of these pathways on GUA-induced effect, by selectively blocking ASK1 or PKC, two upstream kinases in ERK signaling. Immunoblot analysis of p-ERK1/2 expression revealed that GUA elicited a dose-dependent phosphorylation of ERK (10-100 μM), [two-way ANOVA analysis and Sidak’s multiple comparisons test: P < 0,005 for GUA 50 and 100 μM] and the time course [two-way ANOVA analysis: F(1,24) = 74.57, P < 0,0001 with interaction values between factors of F(2,24) = 5.053, P = 0.0147] showed that the maximum effect of the purine base was achieved in 10 min and decreased after 30 min (Figure 4). The inhibition of ASK1 or PKC by cell pre-treatment with 10 μM MSC2032964A or 1 μM GF109203X for 30 min, respectively, did not affect basal ERK phosphorylation, and only in part reduced that induced by GUA [two-way ANOVA: GUA effect F(1,16) = 4.543, P < 0.0489, ASK1 inhibitor effect F(1,16) = 37.66, P < 0.0001 with interaction values between factors of F(3,32) = 4.449, P = 0.0499; GUA effect F(1,16) = 3.220, P < 0.0917, PKC inhibitor effect F(1,16) = 39.35, P < 0.0001 with interaction values between factors of F(3,32) = 4.504, P = 0.0498]. Conversely, the inhibition of PKB by cell pre-treatment with 10 μM 10-DEBC dihydrochloride, as well as the inhibition of the upstream PI3K by 25 μM LY294002, did not modify the basal p-ERK expression and strongly reduced that elicited by GUA [two-way ANOVA: GUA effect F(1,16) = 10.84, P < 0.0046, 10-DEBC effect F(1,16) = 22.53, P < 0.0002 with interaction values between factors of F(3,32) = 13.82, P = 0.0019; GUA effect F(1,16) = 6.456, P < 0.0218, LY294002 effect F(1,16) = 20.12, P < 0.0004 with interaction values between factors of F(3,32) = 8.564, P = 0.0098] (Figures 5A,B). This suggests that, in SH-SY5Y cells, PI3-PKB-ERK signaling cascade is likely the main pathway activated by extracellular GUA.
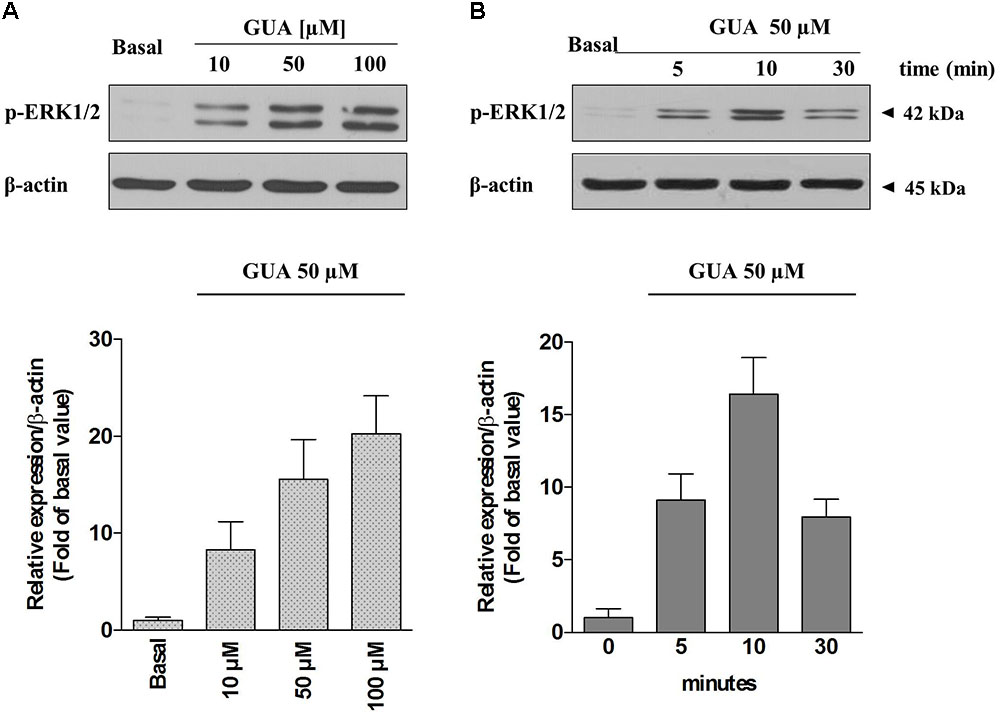
FIGURE 4. Guanine induces ERK1 phosphorylation in SH-SY5Y neuroblastoma cells in a dose-dependent manner. Representative Immunoblot analysis of ERK1/2 expression in SH-SY5Y cells, with β-actin as loading control, and the correspondent quantitative data of densitometric analysis. (A) Cells were cultured for 24 h and exposed for 10 min to different concentrations of GUA (0–100 μM). (B) The time course of activation of ERK1/2 induced by GUA. Cells were cultured for 24 h and exposed to 50 μM GUA for different time points (0–30 min). Each column represents the mean ± SEM of at least four independent experiments, and it is expressed as relative protein expression normalized to β-actin.
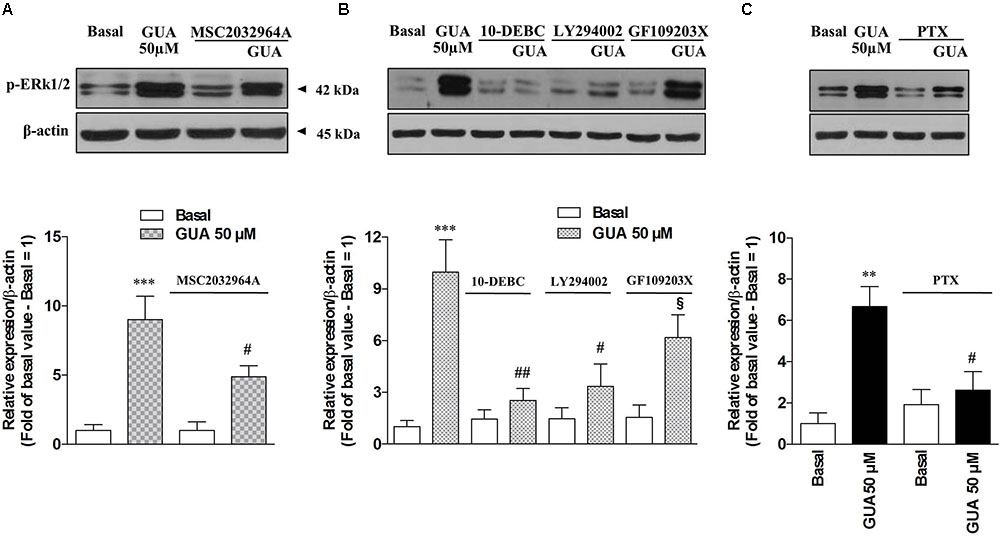
FIGURE 5. Guanine induces ERK1 phosphorylation in SH-SY5Y neuroblastoma cells via PI3K-PKB-ERK signaling pathway in a PTX-sensitive manner. Representative Immunoblot analysis of ERK1/2 expression in SH-SY5Y cells, with β-actin as loading control, and the correspondent quantitative data of densitometric analysis. Cells were cultured for 24 h and exposed for 10 min (A,B) or for 4 h (C) to 50 μM GUA, in the absence or presence of (A) ASK1 inhibitor, 10 μM MSC2032964A, (B) PKC inhibitor (1 μM GF109203X), PKB inhibitor (10 μM 10-DEBC dihydrochloride), PI3K inhibitor (25 μM LY294002), (C) 200 ng/mL Pertussis Toxin (PTX.) Each column represents the mean ± SEM of at least four independent experiments, and it is expressed as relative protein expression normalized to β-actin. Student’s t-test: ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, vs. untreated cells (Basal); #P < 0.05, ##P < 0.01, vs. GUA-treated cells, §P < 0.05, vs. GUA- and GF109203X-treated cells.
GUA Activates PKB-ERK Signaling Pathway in a Pertussis-Toxin Sensitive Manner
We, then, inquired whether GUA effect may be ascribed to an eventual interaction with a new putative receptor. For this purpose, SH-SY5Y cells were treated for 4 h with 200 ng/mL Pertussis Toxin (PTX), a specific inhibitor of Gi/Go-proteins, and the expression of p-ERK 1/2 was analyzed by Immunoblot. PTX did not affect basal ERK1/2 phosphorylation but significantly reduced that induced by GUA [two-way ANOVA: F(1,16) = 9.795, P = 0.0065 with interaction values between factors of F(1,16) = 16.09, P = 0.0010], thus supporting the involvement of a Gi protein-coupled receptor in the effect of GUA (Figure 5C).
Extracellular GUA Stimulates the PI3K-PKB Phosphorylation and NO Production and Activates the Downstream sGC-cGMP-PKG-ERK Pathway
Nitric oxide (NO) is known to induce ERK phosphorylation via sGC-cGMP-PKG pathway, (Prickaerts et al., 2001; Suvarna and O’Donnell, 2002; Boess et al., 2004; Rutten et al., 2007; Chien et al., 2008; Masood et al., 2009; Xu et al., 2013; Bollen et al., 2014; Matsumoto et al., 2016). Hence, we evaluated the involvement of NO system on GUA-induced ERK phosphorylation, by selectively blocking every step of this cascade and by analyzing p-ERK1/2 expression by Immunoblot. As expected, phosphodiesterase inhibitor, 10 μM IBMX, strongly enhanced GUA-induced ERK phosphorylation [two-way ANOVA: IBMX effect F(1,16) = 67.23, P < 0.0001]. Most importantly, NOS inhibitor, 1 mM L-NAME, completely prevented ERK1/2 phosphorylation elicited by GUA [two-way ANOVA: L-NAME effect F(1,16) = 16.82, P < 0.0008]. The cell co-treatment with IBMX and L-NAME partially restored GUA-induced ERK phosphorylation, thus confirming the presence of other pathways converging on cGMP activation. The sGC inhibitor, 10 μM ODQ, likewise L-NAME, completely abrogated GUA-mediated ERK phosphorylation [two-way ANOVA: ODQ effect F(1,16) = 25.33, P < 0.0001]. Of note, among the possible pathways involved in the PKB-ERK signaling, extracellular GUA stimulates the PI3K-PKB phosphorylation and NO production, thus evoking the downstream activation of sGC-cGMP-PKG pathway (Figure 6). Corroborating with these findings, PDE inhibitors have been shown to improve cognitive skills and memory formation in rodents (Reneerkens et al., 2009). The mechanism likely involves the induction of cAMP-protein kinase A (PKA)-cAMP responsive element-binding protein (CREB) and cGMP-PKG-CREB signaling pathways (Rutten et al., 2007), which are both associated with late-phase of Long-Term Potentiation (LTP).
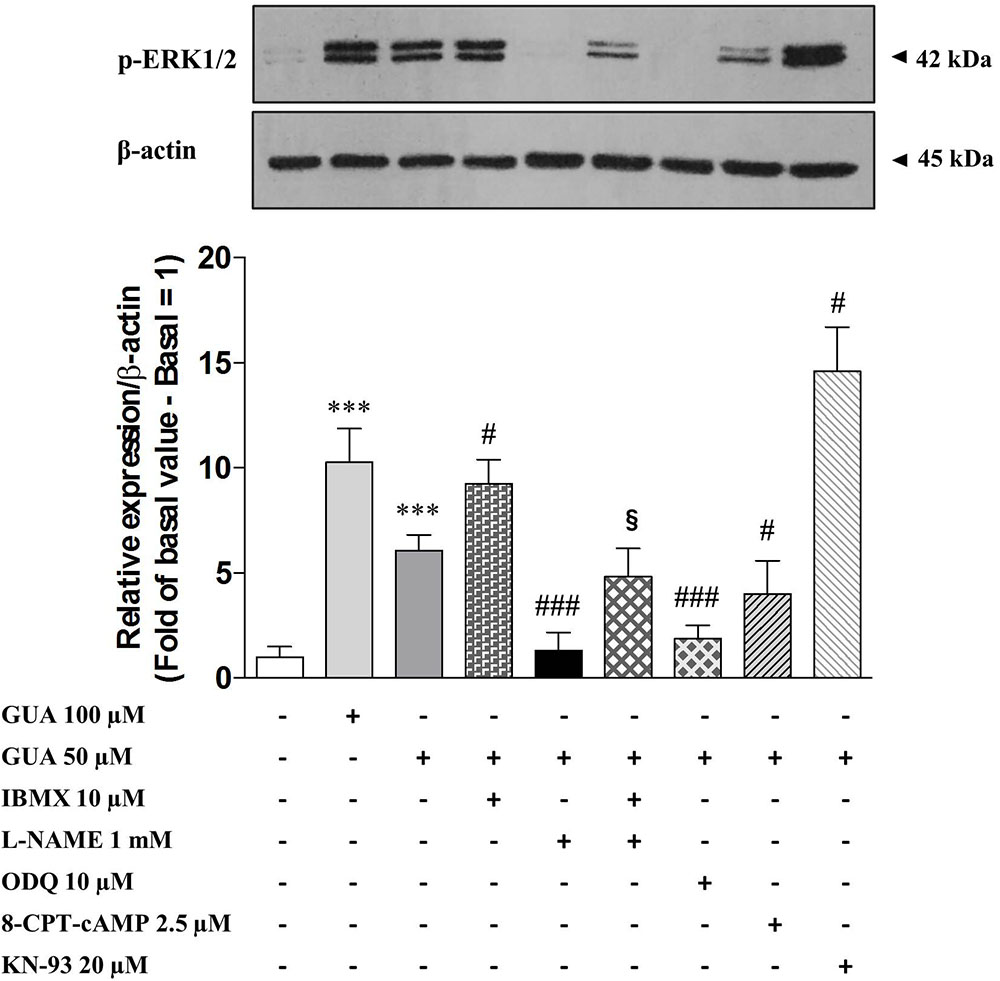
FIGURE 6. Guanine activates sGC-cGMP-PKG-ERK signaling pathway in SH-SY5Y neuroblastoma cells. Representative Immunoblot analysis of ERK1/2 expression in SH-SY5Y cells, with β-actin as loading control, and the correspondent quantitative data of densitometric analysis. Cells were cultured for 24 h and exposed for 30 min to 100 μM GUA, 50 μM GUA, a phosphodiesterase inhibitor (10 μM IBMX), a NOS inhibitor (1 mM L-NAME), a sGC inhibitor (10 μM ODQ), an EPAC specific cAMP analog (2,5 μM 8-CPT-cAMP), a CaMKII inhibitor (20 μM KN-93). Each column represents the mean ± SEM of at least three independent experiments, and it is expressed as relative protein expression normalized to β-actin. Student’s t-test: ∗∗∗P < 0.001, vs. untreated cells (Basal); #P < 0.05, ###P < 0.001, vs. GUA-treated cells, §P < 0.05, vs. GUA- and L-NAME-treated cells.
The Activation of the cAMP-Epac-CaMKII Pathway Influences NOS Activity
In an attempt to investigate whether the activation of collateral signaling pathway was able to affect the NO-sGC-cGMP-PKG-ERK cascade, we examined the possible role of cAMP-Epac-CaMKII pathway. CaMKII (Ca2+/calmodulin-dependent protein kinases II) is highly expressed in hippocamapal neurons and is involved in the glutamate-mediated LTP phase, wherein two major events occur: (i) Ca2+ enters the cell through NMDA channels and activates CaMKII that, in turn, recruits AMPA receptors to the plasma membrane; (ii) Ca2+ increases cAMP that activates ERK signaling (Giovannini, 2006; Miyamoto, 2006).
It has been recently proposed that extracellular cGMP regulates the glutamate-NO-cGMP pathway in a learning task, and this modulation resulted to be biphasic and relied on an inverse correlation between CaMKII and NOS activity (Moosavi et al., 2014; Cabrera-Pastor et al., 2016).
In our study, Immunoblot analysis of p-ERK showed that 2,5 μM 8-CPT-cAMP, an EPAC specific cAMP analog, inhibited GUA-induced ERK phosphorylation [two-way ANOVA: effect of 8-CPT-cAMP F(1,16) = 18.16, P < 0.0006], although this result has to be further elucidated by using an EPAC-specific inhibitor. The inhibitor of CaMKII, 20 μM KN-93, caused an opposite effect and significantly enhanced p-ERK expression [two-way ANOVA: effect of KN-93 F(1,16) = 30.80, P < 0.0001] (Figure 6). Therefore, we hypothesized that the functional interplay between NOS activity and CaMKII phosphorylation, observed in our experimental model, might have similar features to the above-mentioned glutamate-NO-cGMP pathway.
GUA Enhances NO Production in SH-SY5Y Cells and Pre-treatment with L-NAME Prevents This Effect
Finally, in order to compare the present data with those previously obtained in vivo (Giuliani et al., 2012), wherein L-NAME was able to prevent the GUA-mediated mnesic effect, we evaluated the effects of GUA on NO production. Cell exposure to 50 μM GUA for 30 min significantly increased NO production [two-way ANOVA: GUA effect F(1,16) = 6.451, P < 0.0218, L-NAME effect F(1,16) = 6.451, P < 0.0218 with interaction values between factors of F(1,16) = 4.681, P = 0.0460] (Figure 7). A similar effect has been obtained when SH-SY5Y cells were challenged with 2 μM ionomycin, a calcium ionophore that induces NO production mainly via mobilization of intracellular Ca2+ [two-way ANOVA: GUA effect F(1,16) = 5.523, P < 0.0319, ionomycin effect F(1,16) = 5.523, P < 0.0319 with interaction values between factors of F(1,16) = 4.190, P = 0.0575]. Cell pre-treatment (15 min before exposure to GUA or ionomycin) with 5 μM L-NAME, which per se did not affect the NO production of SH-SY5Y cells, strongly reduced the NO production induced by either GUA or ionomycin (Figure 7).
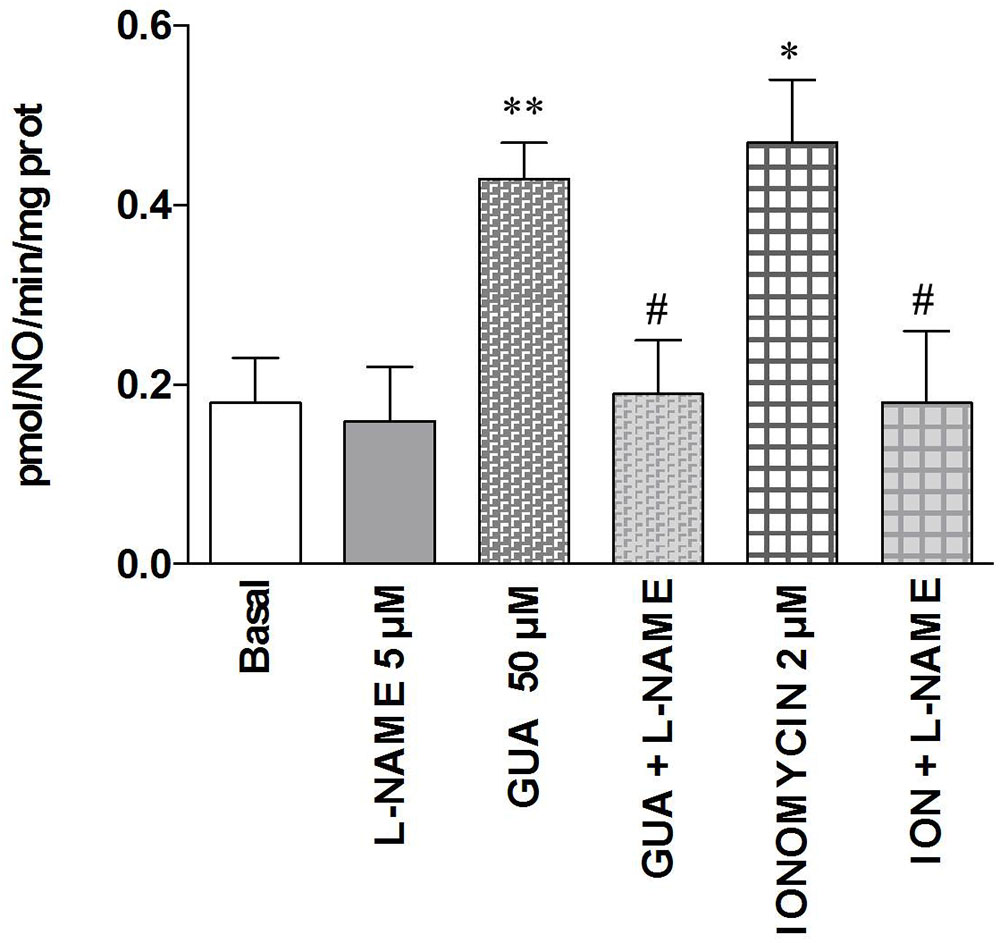
FIGURE 7. Guanine increases NO production in SH-SY5Y neuroblastoma cells. SH-SY5Y cells were treated for 30 min with 5 μM L-NAME or 50 μM GUA (alone or in combination), 2 μM ionomycin (ION), alone or in combination with 5 μM L-NAME. NOS activity was determined by the conversion of L-[3H]-arginine into L-[3H]-citrulline and expressed as pmol/NO/min/mg prot tot. Data were expressed as means ± SEM of four different experiments. Student’s t-test: ∗P < 0.05, ∗∗P < 0.01, vs. untreated cells (Basal); #P < 0.05, vs. GUA-treated cells.
Discussion
This work provides new insights on the transduction pathways involved in neuronal plasticity, in particular on the putative mechanism responsible of GUA-induced mnesic effects.
The major findings of the present study, performed in a model of neuronal-like cells (SH-SY5Y cells), are the following: (i) in resting condition, GUA is present in the intercellular milieu and derives from GTP breakdown, promoted by the activity of purine-converting enzymes (i.e., PNP and GDA) released in the culture medium; (ii) the inhibition of specific nucleoside and nucleobase transmembrane transporters enhances the extracellular levels of GUO, GUA and XAN; (iii) the addition of GUA to the culture medium caused a non-massive and dose-dependent production of ROS, and promotes the phosphorylation of ASK1, p38, JNK, PKC and PKB; (iv) extracellular GUA stimulates the PI3K-PKB phosphorylation and NO production, and activates the downstream sGC-cGMP-PKG-ERK pathway in a pertussis-toxin sensitive manner; (v) GUA-induced NO production is negatively modulated by the EPAC-cAMP-CAMKII pathway.
To better understand the results of this study, it has to be emphasized that, both in vitro and in vivo (Rathbone et al., 2008; Giuliani et al., 2012), the system of the extracellular guanine-based purines, together with a physiological and concerted activity of the enzymes regulating their metabolism (in particular PNP and GDA), is basically oriented to generate an higher amount of GUA rather than GUO or XAN; moreover, GUA stimulates the cGMP formation through NO production, which is known to modulate a broad range of effects in the CNS, such as neuronal development and synaptic plasticity (Prast and Philippu, 2001).
The evidence of the involvement of NO-cGMP pathway in some forms of learning and memory has been worked out by many authors (Erceg et al., 2005; Piedrafita et al., 2007; Ben Aissa et al., 2016; Cabrera-Pastor et al., 2016; Zhou et al., 2017). In several brain areas (i.e., hippocampus, cerebellum and striatum), increased levels of cGMP seemed to improve learning and memory consolidation, mainly during early stages of memory formation (immediately after training). This data is confirmed by full amnesia for inhibitory avoidance task when rats were treated with LY 83583, a soluble guanylate cyclase inhibitor, immediately after training (Bernabeu et al., 1997). However, not all signal transduction pathways, leading to cGMP formation, cause beneficial effects on cognitive functions. For example, cGMP has been found to mediate the stimulation of dendritic number and branching as well as the neurite elongation. This event likely occurs as a consequence of an interplay between cGMP and the activity of extrinsic/external factors such as neurotrophins (Tian et al., 2017) that, usually, stimulate cAMP formation. Cyclic nucleotides are differently involved in the memory consolidation process, since it has been reported that cGMP regulates the early consolidation phase, whereas cAMP is implicated in late processes of memory formation (Izquierdo et al., 2006; Rutten et al., 2007; Bollen et al., 2014). In this functional interplay, a significant role seems to be played by the guanine-based purines, in particular by GUO and GUA. Indeed, several studies reported that GUO causes memory impairment (Roesler et al., 2000; Vinadé et al., 2003, 2004) but, at the same time and sinergistically with NGF, it promotes neurite outgrowth in PC12 cells via activation of heme-oxygenase and cGMP formation (Gysbers and Rathbone, 1996; Bau et al., 2005; Tomaselli et al., 2005). Cell exposure to neurotrophic factors such as NGF, for more than 24 h, promotes the phosphorylation of PKC-Ras-MAPK (Kumar et al., 2005) and, in turn, increases the mRNA levels of GDA, responsible of GUA degradation (Rathbone et al., 1999). This enzyme favors dendrite branching and elongation by directly binding tubulin heterodimers, thus promoting the microtubule assembly via cytoskeletal rearrangement (Akum et al., 2004). Therefore, the synergistic activity of neurotrophins with agents promoting neuritogenesis, such as GUO, explains why the neurogenic activity of GUO, despite being linked to an increased cGMP formation, is associated with an amnesic effect. Conversely, GUA promotes the early stages of memory formation in rats (Giuliani et al., 2012) and its effect seems to be linked to the NO-cGMP signaling pathway. In line with it, we observed that L-NAME prevented memory consolidation caused by the administration of GUA in vivo, in a model of passive avoidance task (Giuliani et al., 2012). In the same study, the administration of GUA, 15 min before treatment with L-NAME, prevented the amnesic effect of the NOS inhibitor.
The involvement of the NO-cGMP-PKG-ERK signaling pathway in synaptic plasticity has been extensively reported (Chien et al., 2008; Ota et al., 2010; Bartus et al., 2013; Matsumoto et al., 2016). However, the role of NO in each step of learning appears controversial and it might be task-dependent. ERK cascade is positively implicated in the development of fear conditioning, conditioned taste aversion memory, spatial memory, step-down inhibitory avoidance and object recognition memory (Giovannini, 2006; Chen et al., 2017; Vithayathil et al., 2017). In our study, this pathway resulted to be activated upon cell exposure to GUA. Specifically, extracellular GUA stimulated the PI3K-PKB phosphorylation and NO production, and activated the downstream sGC-cGMP-PKG-ERK pathway. It is feasible that GUA-induced ROS production, with the subsequent phosphorylation of ASK1, p38 and JNK, may contribute to the cGMP formation trough other pathways than NO, as confirmed by our data, wherein, in cells pre-treated with L-NAME, the inhibition of phosphodiesterases caused a limited increase in GUA-induced ERK1/2 phosphorylation.
Noteworthy, if the role of cGMP in learning and memory is important, much more valuable is that its mnesic effect is mediated by NO production. Indeed, the increase of the intracellular levels of cGMP induced by GUO, through the activation of HEME-oxygenase and independent of the NO production, is important for the dendrite and neurite outgrowth but it is associated with amnesia (Vinadé et al., 2004; Bau et al., 2005).
We, then, took into account the existence of a more complex network, wherein the NO production is eventually under the control of other molecular mechanisms and, among them, we investigated the EPAC-CAMKII system. CaMKII has a prominent role in memory formation (LTM) (Tan, 2007; Takao et al., 2010; Giese and Mizuno, 2013; Nakamura et al., 2017). For this purpose, we took advantage of a recent study, where they showed that the activation of CAMKII, upon stimulation of NMDA receptors, inhibits the production of NO that functions as a retrograde signal able to modulate the glutamatergic system (Cabrera-Pastor et al., 2016). Noteworthy, in our model, the activity of GUA was similar to that of glutamate, since we observed that the stimulation of the EPAC-CAMKII pathway inhibited the NO-mediated phosphorylation of ERK1/2 induced by GUA. The blockage of this pathway by using a CaMKII inhibitor, KN-93, amplified GUA effect. Consistent with it, two endogenous CaMKII inhibitors (CaMK2N1 and CaMK2N2) have been shown to prevent memory loss after retrieval (Vigil et al., 2017). However, it should be pointed out that the influence exerted by CaMKII on the phosphorylation of ERK is not univocal. Indeed, it has been reported that: (i) in vascular smooth muscle cells the CaMKII inhibitor KN-93 caused an H2O2-mediated reduction of ERK1/2 and PKB phosphorylation (Robison et al., 2007; Bouallegue et al., 2009); (ii) the presynaptic injection of a CaMKII inhibitor blocked LTP and neurotransmitter release induced by either the NO donor or the PKG activator (Feil and Kleppisch, 2008).
Finally, we speculated that the extracellular effect of GUA may be mediated by a membrane receptor at present not well identified. In this study, cell treatment with pertussis toxin strongly reduced the GUA-evoked ERK1/2 phosphorylation, thus indicating that this putative new GUA receptor is likely coupled with Gi/o proteins.
Guanine-based purines, in particular GUO, seems to bind to metabotropic receptors and many of their effects are mediated through G-protein-dependent signaling pathways; it has been shown, for instance, that the pertussis toxin-mediated inhibition of Gi/Go-protein reverses some of the effects of GUO on cell viability and glutamate uptake in hippocampal slices (Traversa et al., 2003; Dal-Cim et al., 2012, 2013).
Several years ago, our group found a specific [3H]-guanosine binding site in rat brain membranes, compatible with an unknown G protein-coupled receptor (Traversa et al., 2002, 2003; Di Liberto et al., 2012; Grillo et al., 2012). In order to provide insight into the characteristics of the binding site, we evaluated the relative abilities of purine analogs to displace [3H]GUO. Binding data revealed that all the adenine-based purines as well as GTP, GDP and GMP were ineffective in displacing [3H]GUO. The 6-Thio-GUO or 6-keto-GUO derivatives resulted to be as effective as GUO in displacing [3H]GUO. On the contrary, the binding affinity was strongly reduced when the 6-amino or 2-amino derivatives were assayed.
These findings seem to be compatible with a membrane binding site, expressed in the rat brain, which, in addition to GUO, may interact with GUA, although with a lower affinity. At present, we are carrying out a study to individuate and functionally characterize this new putative receptor.
At the same time, it cannot be excluded an interaction of GUA with another receptor functionally different from G protein-coupled receptors. Indeed, it is well known that compounds that are structurally very similar to GUO and GUA represent the main agonists of several Toll-like receptors (i.e., TLR9, 7 and 8) (Lee et al., 2003; Yu et al., 2017).
In this context, it has been recently reported that the stimulation of some subtypes of Toll-Like receptors (TLR9, 7 and 8) in microglial cells leads to cognitive improvements and ameliorates the vascular amyloid pathology in triple transgenic mice expressing human Swedish K670N/M671L and vasculotropic Dutch/Iowa E693Q/D694N mutations and exhibiting early cerebral microvascular accumulation of Aβ (Scholtzova et al., 2017). Interestingly, some guanine-based purines and their modified derivatives have been recently recognized as endogenous ligands for TLRs, especially 7 and 9 subtypes (Shibata et al., 2016; Abdul-Cader et al., 2017). In this regard, in order to eliminate the effects potentially mediated by TLRs, in our study we used cultured SH-SY5Y cells, wherein the expression of TLR 7/8 and 9 is not reported in literature.
Conclusion
Targeting NO-cGMP-PKG-ERK signaling pathway may represent an interesting approach for the development of new drugs in the treatment of memory dysfunctions occurring in neurodegenerative and psychiatric diseases, among others Alzheimer’s disease and dementia. Initial promising findings in this direction have been reported regarding the use of PDE inhibitors (Kumar and Singh, 2017; Prickaerts et al., 2017) or sGC stimulator (Montfort et al., 2017) in the treatment of neuroinflammatory and neuropathological conditions.
It is plausible to expect that, beyond the above-mentioned guanine-derivatives, the administration of GUA itself (Giuliani et al., 2012), due to its long half-life in vivo, may elicit molecular changes that underlie synaptic alterations and memory formation through a putative receptor that could represent a new pharmacological target. This may serve the purpose of avoiding a major challenge, that is to discriminate the numerous downstream effectors ensuing the activation of NO-cGMP-ERK signaling pathway, thus bypassing the sophisticated network of different and multifunctional protein kinases.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This study was partially supported by funds to PDI from the Italian Ministry of Education, University and Research (MIUR).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abdul-Cader, M. S., Ahmed-Hassan, H., Amarasinghe, A., Nagy, E., Sharif, S., and Abdul-Careem, M. F. (2017). Toll-like receptor (TLR)21 signalling-mediated antiviral response against avian influenza virus infection correlates with macrophage recruitment and nitric oxide production. J. Gen. Virol. 98, 1209–1223. doi: 10.1099/jgv.0.000787
Adaikkan, C., and Rosenblum, K. (2012). The role of protein phosphorylation in the gustatory cortex and amygdala during taste learning. Exp. Neurobiol. 21, 37–51. doi: 10.5607/en.2012.21.2.37
Adams, J. P., and Sweatt, J. D. (2002). Molecular psychology: roles for the ERK MAP kinase cascade in memory. Annu. Rev. Pharmacol. Toxicol. 42, 135–163. doi: 10.1146/annurev.pharmtox.42.082701.145401
Akum, B. F., Chen, M., Gunderson, S. I., Riefler, G. M., Scerri-Hansen, M. M., and Firestein, B. L. (2004). Cypin regulates dendrite patterning in hippocampal neurons by promoting microtubule assembly. Nat. Neurosci. 7, 145–152. doi: 10.1038/nn1179
Ali-Rahmani, F., Grigson, P. S., Lee, S., Neely, E., Connor, J. R., and Schengrund, C. L. (2014). H63D mutation in hemochromatosis alters cholesterol metabolism and induces memory impairment. Neurobiol. Aging 35, 1511.e1–1511.e12. doi: 10.1016/j.neurobiolaging.2013.12.014
Bartus, K., Pigott, B., and Garthwaite, J. (2013). Cellular targets of nitric oxide in the hippocampus. PLoS One 8:e57292. doi: 10.1371/journal.pone.0057292
Bau, C., Middlemiss, P. J., Hindley, S., Jiang, S., Ciccarelli, R., Caciagli, F., et al. (2005). Guanosine stimulates neurite outgrowth in PC12 cells via activation of heme oxygenase and cyclic GMP. Purinergic Signal. 1, 161–172. doi: 10.1007/s11302-005-6214-0
Bellaver, B., Souza, D. G., Bobermin, L. D., Goncalves, C. A., Souza, D. O., and Quincozes-Santos, A. (2015). Guanosine inhibits LPS-induced pro- inflammatory response and oxidative stress in hippocampal astrocytes through the heme oxygenase-1 pathway. Purinergic Signal. 11, 571–580. doi: 10.1007/s11302-015-9475-2
Ben Aissa, M., Lee, S. H., Bennett, B. M., and Thatcher, G. R. (2016). Targeting NO/cGMP signaling in the CNS for neurodegeneration and Alzheimer’s disease. Curr. Med. Chem. 23, 2770–2788. doi: 10.2174/0929867323666160812145454
Bernabeu, R., Schroder, N., Quevedo, J., Cammarota, M., Izquierdo, I., and Medina, J. H. (1997). Further evidence for the involvement of a hippocampal cGMP/cGMP-dependent protein kinase cascade in memory consolidation. Neuroreport 8, 2221–2224. doi: 10.1097/00001756-199707070-00026
Bettio, L. E., Gil-Mohapel, J., and Rodrigues, A. L. (2016). Guanosine and its role in neuropathologies. Purinergic Signal. 12, 411–426. doi: 10.1007/s11302-016-9509-4
Boess, F. G., Hendrix, M., van der Staay, F. J., Erb, C., Schreiber, R., van Staveren, W., et al. (2004). Inhibition of phosphodiesterase 2 increases neuronal cGMP, synaptic plasticity and memory performance. Neuropharmacology 47, 1081–1092. doi: 10.1016/j.neuropharm.2004.07.040
Boison, D. (2011). Modulators of nucleoside metabolism in the therapy of brain diseases. Curr. Top. Med. Chem. 11, 1068–1086. doi: 10.2174/156802611795347609
Bollen, E., Puzzo, D., Rutten, K., Privitera, L., De Vry, J., Vanmierlo, T., et al. (2014). Improved long-term memory via enhancing cGMP-PKG signaling requires cAMP-PKA signaling. Neuropsychopharmacology 39, 2497–2505. doi: 10.1038/npp.2014.106
Bouallegue, A., Pandey, N. R., and Srivastava, A. K. (2009). CaMKII knockdown attenuates H2O2-induced phosphorylation of ERK1/2, PKB/Akt, and IGF-1R in vascular smooth muscle cells. Free Radic. Biol. Med. 47, 858–866. doi: 10.1016/j.freeradbiomed.2009.06.022
Bredt, D. S., Hwang, P. M., Glatt, C. E., Lowenstein, C., Reed, R. R., and Snyder, S. H. (1991). Cloned and expressed nitric oxide synthase structurally resembles cytochrome P-450 reductase. Nature 351, 714–718. doi: 10.1038/351714a0
Cabrera-Pastor, A., Malaguarnera, M., Taoro-Gonzalez, L., Llansola, M., and Felipo, V. (2016). Extracellular cGMP modulates learning biphasically by modulating glycine receptors, CaMKII and glutamate-nitric oxide-cGMP pathway. Sci. Rep. 6:33124. doi: 10.1038/srep33124
Charych, E. I., Akum, B. F., Goldberg, J. S., Jörnsten, R. J., Rongo, C., Zheng, J. Q., et al. (2006). Activity-independent regulation of dendrite patterning by postsynaptic density protein PSD-95. J. Neurosci. 26, 10164–10176. doi: 10.1523/JNEUROSCI.2379-06.2006
Chen, X., Wang, X., Tang, L., Wang, J., Shen, C., Liu, J., et al. (2017). Nhe5 deficiency enhances learning and memory via upregulating Bdnf/TrkB signaling in mice. Am. J. Med. Genet. B Neuropsychiatr. Genet. 174, 828–838. doi: 10.1002/ajmg.b.32600
Chien, W. L., Liang, K. C., and Fu, W. M. (2008). Enhancement of active shuttle avoidance response by the NO-cGMP-PKG activator YC-1. Eur. J. Pharmacol. 590, 233–240. doi: 10.1016/j.ejphar.2008.06.040
Ciccarelli, R., Ballerini, P., Sabatino, G., Rathbone, M. P., D’Onofrio, M., Caciagli, F., et al. (2001). Involvement of astrocytes in purine-mediated reparative processes in the brain. Int. J. Dev. Neurosci. 19, 395–414. doi: 10.1016/S0736-5748(00)00084-8
Dal-Cim, T., Ludka, F. K., Martins, W. C., Reginato, C., Parada, E., Egea, J., et al. (2013). Guanosine controls inflammatory pathways to afford neuroprotection of hippocampal slices under oxygen and glucose deprivation conditions. J. Neurochem. 126, 437–450. doi: 10.1111/jnc.12324
Dal-Cim, T., Molz, S., Egea, J., Parada, E., Romero, A., Budni, J., et al. (2012). Guanosine protects human neuroblastoma SH-SY5Y cells against mitochondrial oxidative stress by inducing heme oxigenase- 1 via PI3K/Akt/GSK-3beta pathway. Neurochem. Int. 61, 397–404. doi: 10.1016/j.neuint.2012.05.021
Davis, S., and Laroche, S. (2006). Mitogen-activated protein kinase/extracellular regulated kinase signalling and memory stabilization: a review. Genes Brain Behav. 5, 61–72. doi: 10.1111/j.1601-183X.2006.00230.x
Di Iorio, P., Ballerini, P., Traversa, U., Nicoletti, F., D’Alimonte, I., Kleywegt, S., et al. (2004). The antiapoptotic effect of guanosine is mediated by the activation of the PI 3-kinase/AKT/PKB pathway in cultured rat astrocytes. Glia 46, 356–368. doi: 10.1002/glia.20002
Di Iorio, P., Caciagli, F., Giuliani, P., Ballerini, P., Ciccarelli, R., Sperling, O., et al. (2001). Purine nucleosides protect injured neurons and stimulate neuronal regeneration by intracellular and membrane receptor-mediated mechanisms. Drug Dev. Res. 52, 303–315. doi: 10.1002/ddr.1128
Di Liberto, V., Garozzo, R., Grillo, M., Mudo, G., Caciagli, F., Condorelli, D. F., et al. (2012). Identification of GPR23/LPA4 as a candidate G protein-coupled receptor for guanosine. Acta Physiol. 206(Suppl. 692):O.16. doi: 10.1089/adt.2009.0261
Di Liberto, V., Mudò, G., Garozzo, R., Frinchi, M., Fernandez-Dueñas, V., Di Iorio, P., et al. (2016). The guanine-based purinergic system: the tale of an orphan neuromodulation. Front. Pharmacol. 7:158. doi: 10.3389/fphar.2016.00158
Erceg, S., Monfort, P., Hernández-Viadel, M., Rodrigo, R., Montoliu, C., and Felipo, V. (2005). Oral administration of sildenafil restores learning ability in rats with hyperammonemia and with portacaval shunts. Hepatology 41, 299–306. doi: 10.1002/hep.20565
Feil, R., and Kleppisch, T. (2008). NO/cGMP-dependent modulation of synaptic transmission. Handb. Exp. Pharmacol. 184, 529–560. doi: 10.1007/978-3-540-74805-2_16
Feng, Y., Chambers, J. W., Iqbal, S., Koenig, M., Park, H., Cherry, C., et al. (2013). A small molecule bidentate-binding dual inhibitor probe of the LRRK2 and JNK kinases. ACS Chem. Biol. 8, 1747–1754. doi: 10.1021/cb3006165
Fink, C. C., Bayer, K. U., Myers, J. W., Ferrell, J. E. Jr., Schulman, H., and Meyer, T. (2003). Selective regulation of neurite extension and synapse formation by the beta but not the alpha isoform of CaMKII. Neuron 39, 283–297. doi: 10.1016/S0896-6273(03)00428-8
Friebe, A., and Koesling, D. (2003). Regulation of nitric oxide-sensitive guanylyl cyclase. Circ. Res. 93, 96–105. doi: 10.1161/01.RES.0000082524.34487.31
Giese, K. P., and Mizuno, K. (2013). The roles of protein kinases in learning and memory. Learn. Mem. 20, 540–552. doi: 10.1101/lm.028449.112
Giovannini, M. G. (2006). The role of the extracellular signal-regulated kinase pathway in memory encoding. Rev. Neurosci. 17, 619–634. doi: 10.1515/REVNEURO.2006.17.6.619
Giuliani, P., Ballerini, P., Buccella, S., Ciccarelli, R., Rathbone, M. P., Romano, S., et al. (2015). Guanosine protects glial cells against 6-hydroxydopamine toxicity. Adv. Exp. Med. Biol. 837, 23–33. doi: 10.1007/5584_2014_73
Giuliani, P., Buccella, S., Ballerini, P., Ciccarelli, R., D’Alimonte, I., Cicchitti, S., et al. (2012). Guanine-based purines modulate the effect of L-NAME on learning and memory in rats. Panminerva Med. 54, 53–58.
Giuliani, P., Zuccarini, M., Buccella, S., Pena-Altamira, L. E., Polazzi, E., Virgili, M., et al. (2017). Evidence for purine nucleoside phosphorylase (PNP) release from rat C6 glioma cells. J. Neurochem. 141, 208–221. doi: 10.1111/jnc.14004
Giuliani, P., Zuccarini, M., Buccella, S., Rossini, M., D’Alimonte, I., Ciccarelli, R., et al. (2016). Development of a new HPLC method using fluorescence detection without derivatization for determining purine nucleoside phosphorylase activity in human plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 1009–1010, 114–121. doi: 10.1016/j.jchromb.2015.12.012
Grillo, M., di Liberto, V., Garozzo, R., Mudo, G., Caciagli, F., Condorelli, D. F., et al. (2012). Brain expression and 3H-guanosine binding analysis of novel G protein- coupled receptor for guanosine (GPR23/LPA4). Acta Physiol. 206(Suppl. 692):P4.19.
Gysbers, J. W., and Rathbone, M. P. (1996). Neurite outgrowth in PC12 cells is enhanced by guanosine through both cAMP-dependent and -independent mechanisms. Neurosci. Lett. 220, 175–178. doi: 10.1016/S0304-3940(96)13253-5
Ha, H. C., Woster, P. M., Yager, J. D., and Casero, R. A. (1997). The role of polyamine catabolism in polyamine analogue-induced programmed cell death. Proc. Natl. Acad. Sci. U.S.A. 94, 11557–11562. doi: 10.1073/pnas.94.21.11557
Izquierdo, I., Bevilaqua, L. R., Rossato, J. I., Bonini, J. S., Medina, J. H., and Cammarota, M. (2006). Different molecular cascades in different sites of the brain control memory consolidation. Trends Neurosci. 29, 496–505. doi: 10.1016/j.tins.2006.07.005
Jiang, W., Chen, Y., Li, B., and Gao, S. (2017). DBA-induced caspase-3-dependent apoptosis occurs through mitochondrial translocation of cyt-c in the rat hippocampus. Mol. Biosyst. 3, 1863–1873. doi: 10.1039/c7mb00246g
Kumar, A., and Singh, N. (2017). Inhibitor of phosphodiestearse-4 improves memory deficits, oxidative stress, neuroinflammation and neuropathological alterations in mouse models of dementia of Alzheimer’s type. Biomed. Pharmacother. 88, 698–707. doi: 10.1016/j.biopha.2017.01.059
Kumar, V., Zhang, M. X., Swank, M. W., Kunz, J., and Wu, G. Y. (2005). Regulation of dendritic morphogenesis by Ras–PI3K–Akt–mTOR and Ras–MAPK signaling pathways. J. Neurosci. 25, 11288–11299. doi: 10.1523/JNEUROSCI.2284-05.2005
Lanznaster, D., Dal-Cim, T., Piermartiri, T. C. B., and Tasca, C. I. (2016). Guanosine: a neuromodulator with therapeutic potential in brain disorders. Aging Dis. 7, 657–679. doi: 10.14336/AD.2016.0208
Lee, D. S., and Cheong, S. H. (2017). Taurine have neuroprotective activity against oxidative damage-induced HT22 cell death through heme oxygenase-1 pathway. Adv. Exp. Med. Biol. 975, 159–171. doi: 10.1007/978-94-024-1079-2_14
Lee, J., Chuang, T. H., Redecke, V., She, L., Pitha, P. M., Carson, D. A., et al. (2003). Molecular basis for the immunostimulatory activity of guanine nucleoside analogs: activation of Toll-like receptor 7. Proc. Natl. Acad. Sci. U.S.A. 100, 6646–6651. doi: 10.1073/pnas.0631696100
Lepicard, E. M., Mizuno, K., Antunes-Martins, A., von Hertzen, L. S., and Giese, K. P. (2006). An endogenous inhibitor of calcium/calmodulin-dependent kinase II is up-regulated during consolidation of fear memory. Eur. J. Neurosci. 23, 3063–3070. doi: 10.1111/j.1460-9568.2006.04830.x
Lucchesi, W., Mizuno, K., and Giese, K. P. (2011). Novel insights into CaMKII function and regulation during memory formation. Brain Res. Bull. 85, 2–8. doi: 10.1016/j.brainresbull.2010.10.009
Lueptow, L. M., Zhan, C. G., and O’Donnell, J. M. (2015). Cyclic GMP–mediated memory enhancement in the object recognition test by inhibitors of phosphodiesterase-2 in mice. Psychopharmacology 233, 447–456. doi: 10.1007/s00213-015-4129-1
Masood, A., Huang, Y., Hajjhussein, H., Xiao, L., Li, H., Wang, W., et al. (2009). Anxiolytic effects of phosphodiesterase-2 inhibitors associated with increased cGMP signaling. J. Pharmacol. Exp. Ther. 331, 690–699. doi: 10.1124/jpet.109.156729
Matsumoto, Y., Matsumoto, C. S., Takahashi, T., and Mizunami, M. (2016). Activation of NO-cGMP signaling rescues age-related memory impairment in crickets. Front. Behav. Neurosci. 10:166. doi: 10.3389/fnbeh.2016.00166
Miyamoto, E. (2006). Molecular mechanism of neuronal plasticity: induction and maintenance of long-term potentiation in the hippocampus. J. Pharmacol. Sci. 100, 433–442. doi: 10.1254/jphs.CPJ06007X
Miyamoto, S., Ogawa, H., Shiraki, H., and Nakagawa, H. (1982). Guanine deaminase from rat brain. Purification, characteristics, and contribution to ammoniagenesis in the brain. J. Biochem. 91, 167–176. doi: 10.1093/oxfordjournals.jbchem.a133673
Modi, K. K., Jana, A., Ghosh, S., Watson, R., and Pahan, K. (2017). A physically-modified saline suppresses neuronal apoptosis, attenuates tau phosphorylation and protects memory in an animal model of Alzheimer’s disease. PLoS One 12:e0180602. doi: 10.1371/journal.pone.0103606
Montfort, W. R., Wales, J. A., and Weichsel, A. (2017). Structure and activation of soluble guanylyl cyclase, the nitric oxide sensor. Antioxid. Redox Signal. 26, 107–121. doi: 10.1089/ars.2016.6693
Moosavi, M., Abbasi, L., Zarifkar, A., and Rastegar, K. (2014). The role of nitric oxide in spatial memory stages, hippocampal ERK and CaMKII phosphorylation. Pharmacol. Biochem. Behav. 122, 164–172. doi: 10.1016/j.pbb.2014.03.021
Nakamura, T. Y., Nakao, S., Nakajo, Y., Takahashi, J. C., Wakabayashi, S., and Yanamoto, H. (2017). Possible signaling pathways mediating neuronal calcium sensor-1-dependent spatial learning and memory in mice. PLoS One 12:e0170829. doi: 10.1371/journal.pone.0170829
Neary, J. T. (1996). Trophic actions of extracellular nucleotides and nucleosides on glial and neuronal cells. Trends Neurosci. 19, 13–18. doi: 10.1016/0166-2236(96)81861-3
Ota, K. T., Monsey, M. S., Wu, M. S., Young, G. J., and Schafe, G. E. (2010). Synaptic plasticity and NO-cGMP-PKG signaling coordinately regulate ERK-driven gene expression in the lateral amygdala and in the auditory thalamus following Pavlovian fear conditioning. Learn. Mem. 17, 221–235. doi: 10.1101/lm.1592510
Peña-Altamira, L. E., Polazzi, E., Giuliani, P., Beraudi, A., Massenzio, F., Mengoni, I., et al. (2017). Release of soluble and vesicular purine nucleoside phosphorylase from rat astrocytes and microglia induced by pro-inflammatory stimulation with extracellular ATP via P2X7 receptors. Neurochem. Int. doi: 10.1016/j.neuint.2017.10.010 [Epub ahead of print].
Philips, G. T., Tzvetkova, E. I., and Carew, T. J. (2007). Transient mitogen-activated protein kinase activation is confined to a narrow temporal window required for the induction of two-trial long-term memory in Aplysia. J. Neurosci. 27, 13701–13705. doi: 10.1523/JNEUROSCI.4262-07.2007
Piedrafita, B., Cauli, O., Montoliu, C., and Felipo, V. (2007). The function of the glutamate-nitric oxide-cGMP pathway in brain in vivo and learning ability decrease in parallel in mature compared with young rats. Learn. Mem. 14, 254–258. doi: 10.1101/lm.541307
Ping, J., and Schafe, G. E. (2010). The NO-cGMP-PKG signaling pathway coordinately regulates ERK and ERK-driven gene expression at pre- and postsynaptic sites following LTP-inducing stimulation of thalamo-amygdala synapses. Neural Plast. 2010:540940. doi: 10.1155/2010/540940
Prast, H., and Philippu, A. (2001). Nitric oxide as modulator of neuronal function. Prog. Neurobiol. 64, 51–68. doi: 10.1016/S0301-0082(00)00044-7
Prickaerts, J., de Vente, J., Honig, W., Steinbusch, H. W. M., and Blokland, A. (2001). cGMP, but not cAMP, in rat hippocampus is involved in early stages of object memory consolidation. Eur. J. Pharmacol. 436, 83–87. doi: 10.1016/S0014-2999(01)01614-4
Prickaerts, J., Heckman, P. R. A., and Blokland, A. (2017). Investigational phosphodiesterase inhibitors in phase I and phase II clinical trials for Alzheimer’s disease. Expert Opin. Investig. Drugs 26, 1033–1048. doi: 10.1080/13543784.2017.1364360
Puangmalai, N., Thangnipon, W., Soi-Ampornkul, R., Suwanna, N., Tuchinda, P., and Nobsathian, S. (2017). Neuroprotection of N-benzylcinnamide on scopolamine-induced cholinergic dysfunction in human SH-SY5Y neuroblastoma cells. Neural Regen. Res. 12, 1492–1498. doi: 10.4103/1673-5374.215262
Quincozes-Santos, A., Bobermin, L. D., Souza, D. G., Bellaver, B., Goncalves, C. A., and Souza, D. O. (2014). Guanosine protects C6 astroglial cells against azide-induced oxidative damage: a putative role of heme-oxygenase 1. J. Neurochem. 130, 61–74. doi: 10.1111/jnc.12694
Rathbone, M., Pilutti, L., Caciagli, F., and Jiang, S. (2008). Neurotrophic effects of extracellular guanosine. Nucleosides Nucleotides Nucleic Acids 27, 666–672. doi: 10.1080/15257770802143913
Rathbone, M. P., Middlemiss, P. J., Gysbers, J. W., Andrew, C., Herman, M. A., Reed, J. K., et al. (1999). Trophic effects of purines in neurons and glial cells. Prog. Neurobiol. 59, 663–690. doi: 10.1016/S0301-0082(99)00017-9
Reneerkens, O. A. H., Rutten, K., Steinbusch, H. W. M., Blokland, A., and Prickaerts, J. (2009). Selective phosphodiesterase inhibitors: a promising target for cognition enhancement. Psychopharmacology 202, 419–443. doi: 10.1007/s00213-008-1273-x
Ribeiro, F. (2016). Purine nucleosides in neuroregeneration and neuroprotection. Neuropharmacology 104, 226–242. doi: 10.1016/j.neuropharm.2015.11.006
Robison, A. J., Winder, D. G., Colbran, R. J., and Bartlett, R. K. (2007). Oxidation of calmodulin alters activation and regulation of CaMKII. Biochem. Biophys. Res. Commun. 356, 97–101. doi: 10.1016/j.bbrc.2007.02.087
Roesler, R., Vianna, M. R., Lara, D. R., Izquierdo, I., Schmidt, A. P., and Souza, D. O. (2000). Guanosine impairs inhibitory avoidance performance in rats. Neuroreport 11, 2537–2540. doi: 10.1097/00001756-200008030-00038
Rutten, K., Prickaerts, J., Hendrix, M., van der Staay, F. J., Şik, A., and Blokland, A. (2007). Time-dependent involvement of cAMP and cGMP in consolidation of object memory: studies using selective phosphodiesterase type 2, 4 and 5 inhibitors. Eur. J. Pharmacol. 558, 107–112. doi: 10.1016/j.ejphar.2006.11.041
Saute, J. A., da Silveira, L. E., Soares, F. A., Martini, L. H., Souza, D. O., and Ganzella, M. (2006). Amnesic effect of GMP depends on its conversion to guanosine. Neurobiol. Learn. Mem. 85, 206–212. doi: 10.1016/j.nlm.2005.10.006
Schmidt, A. P., Lara, D. R., and Souza, D. O. (2007). Proposal of a guanine-based purinergic system in the mammalian central nervous system. Pharmacol. Ther. 116, 401–416. doi: 10.1016/j.pharmthera.2007.07.004
Scholtzova, H., Do, E., Dhakal, S., Sun, Y., Liu, S., Mehta, P. D., et al. (2017). Innate immunity stimulation via toll-like receptor 9 ameliorates vascular amyloid pathology in Tg-SwDI mice with associated cognitive benefits. J. Neurosci. 37, 936–959. doi: 10.1523/JNEUROSCI.1967-16.2016
Shen, H. M., and Liu, Z. G. (2006). JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic. Biol. Med. 40, 928–939. doi: 10.1016/j.freeradbiomed.2005.10.056
Shibata, T., Ohto, U., Nomura, S., Kibata, K., Motoi, Y., Zhang, Y., et al. (2016). Guanosine and its modified derivatives are endogenous ligands for TLR7. Int. Immunol. 28, 211–222. doi: 10.1093/intimm/dxv062
Suvarna, N. U., and O’Donnell, J. M. (2002). Hydrolysis of N-methyl-D-aspartate receptor-stimulated cAMP and cGMP by PDE4 and PDE2 phosphodiesterases in primary neuronal cultures of rat cerebral cortex and hippocampus. J. Pharmacol. Exp. Ther. 302, 249–256. doi: 10.1124/jpet.302.1.249
Takao, K., Tanda, K., Nakamura, K., Kasahara, J., Nakao, K., Katsuki, M., et al. (2010). Comprehensive behavioral analysis of calcium/calmodulin-dependent protein kinase IV knockout mice. PLoS One 5:e9460. doi: 10.1371/journal.pone.0009460
Tan, S. E. (2007). Roles of hippocampal nitric oxide and calcium/calmodulin-dependent protein kinase II in inhibitory avoidance learning in rats. Behav. Pharmacol. 18, 29–38. doi: 10.1097/FBP.0b013e3280142636
Tarozzi, A., Merlicco, A., Morroni, F., Bolondi, C., Di Iorio, P., Ciccarelli, R., et al. (2010). Guanosine protects human neuroblastoma cells from oxidative stress and toxicity induced by amyloid-beta peptide oligomers. J. Biol. Regul. Homeost. Agents 24, 297–306.
Thomaz, D. T., Dal-Cim, T. A., Martins, V. C., Cunha, M. P., Lanznaster, D., de Bem, A. F., et al. (2016). Guanosine prevents nitroxidative stress and recovers mitochondrial membrane potential disruption in hippocampal slices subjected to oxygen/glucose deprivation. Purinergic Signal. 12, 707–718. doi: 10.1007/s11302-016-9534-3
Tian, X., Yan, H., Li, J., Wu, S., Wang, J., and Fan, L. (2017). Neurotrophin promotes neurite outgrowth by inhibiting Rif GTPase activation downstream of MAPKs and PI3K signaling. Int. J. Mol. Sci. 18:148. doi: 10.3390/ijms18010148
Tomaselli, B., Podhraski, V., Heftberger, V., Bock, G., and Baier-Bitterlich, G. (2005). Purine nucleoside-mediated protection of chemical hypoxia-induced neuronal injuries involves p42/44 MAPK activation. Neurochem. Int. 46, 513–521. doi: 10.1016/j.neuint.2005.02.003
Traversa, U., Bombi, G., Camaioni, E., Macchiarulo, A., Costantino, G., Palmieri, C., et al. (2003). Rat brain guanosine binding site. Biological studies and pseudo- receptor construction. Bioorg. Med. Chem. 11, 5417–5425. doi: 10.1016/j.bmc.2003.09.043
Traversa, U., Bombi, G., Di Iorio, P., Ciccarelli, R., Werstiuk, E. S., and Rathbone, M. P. (2002). Specific [3H]-guanosine binding sites in rat brain membranes. Br. J. Pharmacol. 135, 969–976. doi: 10.1038/sj.bjp.0704542
Vigil, F. A., Mizuno, K., Lucchesi, W., Valls-Comamala, V., and Giese, K. P. (2017). Prevention of long-term memory loss after retrieval by an endogenous CaMKII inhibitor. Sci. Rep. 7:4040. doi: 10.1038/s41598-017-04355-8
Vinadé, E. R., Izquierdo, I., Lara, D. R., Schmidt, A. P., and Souza, D. O. (2004). Oral administration of guanosine impairs inhibitory avoidance performance in rats and mice. Neurobiol. Learn. Mem. 81, 137–143. doi: 10.1016/j.nlm.2003.12.003
Vinadé, E. R., Schmidt, A. P., Frizzo, M. E., Izquierdo, I., Elisabetsky, E., and Souza, D. O. (2003). Chronically administered guanosine is anticonvulsant, amnesic and anxiolytic in mice. Brain Res. 977, 97–102. doi: 10.1016/S0006-8993(03)02769-0
Vithayathil, J., Pucilowska, J., Friel, D., and Landreth, G. E. (2017). Chronic impairment of ERK signaling in glutamatergic neurons of the forebrain does not affect spatial memory retention and LTP in the same manner as acute blockade of the ERK pathway. Hippocampus 27, 1239–1249. doi: 10.1002/hipo.22769
Xu, Y., Pan, J., Chen, L., Zhang, C., Sun, J., Li, J., et al. (2013). Phosphodiesterase-2 inhibitor reverses corticosterone-induced neurotoxicity and related behavioural changes via cGMP/PKG dependent pathway. Int. J. Neuropsychopharmacol. 16, 835–847. doi: 10.1017/S146114571200065X
Yu, C., An, M., Li, M., and Liu, H. (2017). Immunostimulatory properties of lipid modified CpG oligonucleotides. Mol. Pharm. 14, 2815–2823. doi: 10.1021/acs.molpharmaceut.7b00335
Yu, X., and Malenka, R. C. (2004). Multiple functions for the cadherin/catenin complex during neuronal development. Neuropharmacology 47, 779–786. doi: 10.1016/j.neuropharm.2004.07.031
Keywords: guanine, L-NAME, nitric oxide, cGMP, ERK, SH-SY5Y cell line
Citation: Zuccarini M, Giuliani P, Frinchi M, Mudò G, Serio RM, Belluardo N, Buccella S, Carluccio M, Condorelli DF, Caciagli F, Ciccarelli R, and Di Iorio P (2018) Uncovering the Signaling Pathway behind Extracellular Guanine-Induced Activation of NO System: New Perspectives in Memory-Related Disorders. Front. Pharmacol. 9:110. doi: 10.3389/fphar.2018.00110
Received: 15 November 2017; Accepted: 31 January 2018;
Published: 21 February 2018.
Edited by:
Kenneth A. Jacobson, National Institutes of Health (NIH), United StatesReviewed by:
Martina Schmidt, University of Groningen, NetherlandsFabio Tascedda, University of Modena and Reggio Emilia, Italy
Copyright © 2018 Zuccarini, Giuliani, Frinchi, Mudò, Serio, Belluardo, Buccella, Carluccio, Condorelli, Caciagli, Ciccarelli, and Di Iorio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesco Caciagli, ZnJhbmNlc2NvLmNhY2lhZ2xpQHVuaWNoLml0 Mariachiara Zuccarini, bWFyaWFjaGlhcmEuenVjY2FyaW5pQHVuaWNoLml0
†These authors are co-first authors.