 Bin Lu
Bin Lu Ji Cao
Ji Cao Bo Yang
Bo Yang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 12 January 2018
Sec. Cancer Molecular Targets and Therapeutics
Volume 8 - 2017 | https://doi.org/10.3389/fphar.2017.00992
This article is part of the Research Topic Genetics Meets Drug Discovery View all 5 articles
Ferroptosis is a process driven by accumulated iron-dependent lipid ROS that leads to cell death, which is a distinct regulated cell death comparing to other cell death. The lethal metabolic imbalance resulted from GSH depletion or inactivation of glutathione peroxidase 4 is the executor of ferroptosis within the cancer cell. Small molecules-induced ferroptosis has a strong inhibition of tumor growth and enhances the sensitivity of chemotherapeutic drugs, especially in the condition of drug resistance. These evidences have highlighted the importance of ferroptosis in cancer therapeutics, but the roles of ferroptosis in tumorigenesis and development remain unclear. This article provides an overview of the mechanisms of ferroptosis, highlights the role of ferroptosis in cancer and discusses strategies for therapeutic modulation.
Ferroptosis, identified in 2012 by Dr. Brent R. Stockwell, is an iron-dependent form of non-apoptotic regulated cell death (Dixon et al., 2012). Ferroptosis is morphologically, biochemically, and genetically distinct from other well-known forms of cell death, including apoptosis, various forms of necrosis, and autophagy (Yagoda et al., 2007; Yang and Stockwell, 2008; Dixon et al., 2012; Fatokun et al., 2014). Apoptosis is a classical regulated cell death executed by dedicated pathways involving key pro-death effector proteins such as BCL2-associated X protein (BAX). However, ferroptosis is initialized through GSH depletion or inactivation of GPX4 activity (Dixon et al., 2012, 2014; Yang et al., 2014). During the ferroptosis, canonical hallmarks of apoptosis were not observed in several cancer cells, such as poly (ADP ribose) polymerase 1 (PARP1) cleavage, cytochrome c releasing from mitochondria, or pro-caspase-3 cleavage (Yagoda et al., 2007; Yang and Stockwell, 2008). Besides, there are special and distinct changes in mitochondrial morphology during the ferroptosis (Xie et al., 2016), mainly the loss of structural integrity, such as smaller than normal mitochondria with condensed mitochondrial membrane densities, reduced or absent mitochondria crista (Yagoda et al., 2007; Dixon et al., 2012) and outer mitochondrial membrane rupture (Friedmann Angeli et al., 2014). Thus, there are no cross-talk in the initial mechanisms or downstream evens between apoptosis and ferroptosis. So far, several groups have demonstrated that, in the early stage of ferroptosis, autophagy contributes to ferroptosis through providing available labile iron via NCOA4-mediated ferritinophagy (Gao et al., 2016; Hou et al., 2016). However, once ferroptosis is induced, especially in the later upstream events, autophagy maybe no longer a key regulator.
The overwhelming iron-dependent accumulation of lipid peroxidation products is considered the main killer in the ferroptotic process. Thus, small-molecule lipophilic antioxidants (e.g., ferrostatin-1 and α-tocopherol) and iron chelators (e.g., deferoxamine) can prevent ferroptotic cell death, whereas inhibitors of the other abovementioned forms of cell death cannot accomplish this feat. It is well-established that system Xc- (Dixon et al., 2014) and GPX4 (Yang et al., 2014) are key regulators of ferroptosis. Two classical compounds, erastin (Dolma et al., 2003) and (1S, 3R)-RSL3, can induce ferroptosis through the inhibition of system Xc- and GPX4, respectively. Cell death is crucial for normal development, homeostasis, and prevention of hyperproliferative diseases, such as cancer. Cancer cells can undergo several forms of regulated cell death during tumor development (e.g., apoptosis), and activation of regulated cell death is a primary and promising strategy for cancer therapy. Despite success in clinical cancer treatments, drug resistance to existing chemotherapeutic agents due to genetic alterations remains a problem. Intriguingly, erastin, a ferroptosis inducer, shows the ability to enhance the effectiveness of chemotherapy drugs (e.g., temozolomide, cisplatin, cytarabine/ara-C, and doxorubicin/Adriamycin) in certain cancer cells (Chen et al., 2015; Yu et al., 2015; Roh et al., 2016). Notably, RCCs, B cell-derived lymphomas and a subset of triple-negative breast cancer cell lines acquire a strong dependence on GPX4 and system Xc- (Conrad et al., 2016). These results indicate that inducing ferroptosis maybe a new therapeutic anticancer strategy in tumors, especially tumors containing the two-abovementioned metabolic signatures. p53 is a crucial tumor suppressor and is mutant in most cancer cells. Recently, the tumor-suppressive activity of p53 was partially attributed to its ability to induce ferroptosis via inhibiting system Xc- (Jiang et al., 2015); thus, ferroptosis may play a crucial role in tumorigenesis. In addition, some clinical drugs approved by the FDA [e.g., sorafenib (Dixon et al., 2014), sulfasalazine and ART] can also induce ferroptosis in several cancer cells, supporting the feasibility of using ferroptosis in preclinical and clinical settings. Taken together, ferroptosis shows an intimate association with tumors and cancer therapy, although it’s exact function in cancer remains unelucidated. In this review, we discuss the role of ferroptosis in tumorigenesis and cancer therapy, providing an improved understanding of ferroptotic mechanism-based therapeutic approaches.
Ferroptosis is initiated by the loss of GPX4 activity, which is mediated by two distinct mechanisms (Figure 1). The first mechanism is the inhibition of system Xc- (e.g., mediated by erastin), which indirectly inhibits GPX4 (Dixon et al., 2012, 2014). System Xc- is a cystine/glutamate antiporter that imports extracellular cystine in exchange for intracellular glutamate (Bridges et al., 2012). Cysteine, a reduced form of cystine, is a precursor for the synthesis of GSH. By using GSH as an essential cofactor, GPX4 exerts its phospholipid peroxidase activity to catalyze the reduction of lipid peroxides. Thus, the inhibition of system Xc- by small molecules causes GSH depletion and subsequently inactivates GPX4, ultimately leading to the accumulation of lethal lipid peroxides and the initiation of ferroptosis (Dixon et al., 2012, 2014; Yang et al., 2014). Notably, inhibition of γ-GCS (e.g., mediated by buthionine sulfoximine), which is the rate-limiting enzyme for the synthesis of GSH, is also sufficient to initiate ferroptosis (Yang et al., 2014). Another molecular mechanism of ferroptosis is the direct inhibition of GPX4 through the loss of its activity or promoting its degradation. The classical ferroptotic inducer (1S, 3R)-RSL3, can inhibit GPX4 enzymatic activity by covalently targeting its active site selenocysteine in an irreversible manner (Yang et al., 2014, 2016). FIN56, a new and specific ferrotosis inducer, is involved in decreasing GPX4 abundance. However, the detailed mechanism remains unclear, maybe related to the enzymatic activity of acetyl-CoA carboxylase (Shimada et al., 2016). Furthermore, genetic inhibition of GPX4 by siRNA was also sufficient to induce the accumulation of lipid ROS and ferroptotic cell death (Yang et al., 2014). In general, GPX4 is a central regulator of ferroptosis-triggering mechanisms. Given that the iron-dependent accumulation of lipid ROS kills cells undergoing ferroptosis, both iron metabolism and lipid peroxidation are two critical processes involved in the mechanism of ferroptosis. Therefore, the following contexts were mainly carried out in these two aspects (Figure 1).
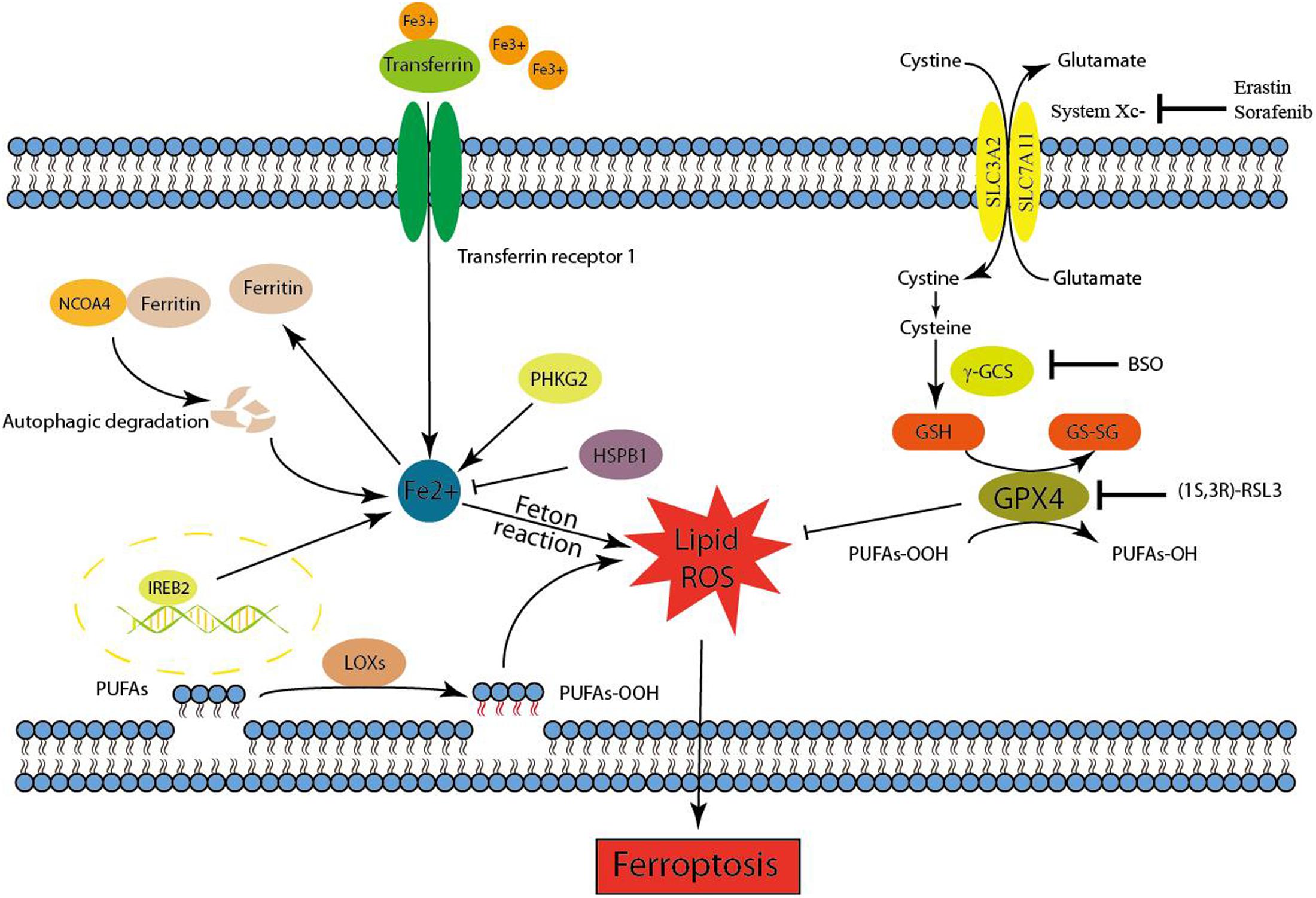
FIGURE 1. Mechanism of ferroptotic cell death. Ferroptosis is initiated by the inhibition of system Xc- or GPX4 activity, which ultimately leads to cell death. Lipid ROS maybe responsible for the ferroptotic process. On the one hand, the peroxidation of PUFAs is considered to be an important contributor. On the other hand, excess irons are the basis for ferroptosis execution.
Redox-active iron pools (i.e., Fe2+) are capable of directly catalyzing lipid peroxides to form damaging free radical via Fenton chemistry (Figure 1). Once lipid peroxides are not removed in time within the cell, constantly accumulative lipid alkoxy (RO ⋅) radicals will lead to ferroptosis ultimately. But this is just one of the reasons to explain why ferroptosis is dependent on iron rather than other metal ions.
Iron response element binding protein 2 (IREB2) has been identified as an essential gene for erastin-induced ferroptosis in HT-1080 and Calu-1 cells (Dixon et al., 2012). IREB2, also named IRP2, plays a crucial role in controlling intracellular and systemic iron homeostasis by post-transcriptionally controlling iron metabolism genes via the IRE–IRP system. These iron metabolism genes comprise ferritin [FTH1, FTL, TF, TfR1, Fpn, and DMT1 (Bogdan et al., 2016)]. Ferritin is the major intracellular iron storage protein, and the NCOA4-mediated autophagic degradation of ferritin (namely ferritinophagy) contributes to erastin-induced ferroptosis (Hou et al., 2016). Genetic inhibition of ATG5, ATG7, or NCOA4 decreased ferroptotic cell death, which was accompanied by decreased intracellular ferrous iron levels and lipid peroxidation end products (e.g., MDA). Moreover, the inhibition of lysosomal activity also decreased ferroptosis. Transferrin (TF), an iron-binding serum protein, can be transported into the cell via TfR1-mediated endocytosis (Bogdan et al., 2016). TF was identified as an executioner of serum-dependent cell death upon cystine starvation. Further experiments demonstrated that TF is an essential component for the induction of ferroptotic cell death. In addition, only TF transported into the cell with iron loaded by TfR1 can exert its corresponding ability (i.e., inducing ferroptosis) in certain contexts (Gao et al., 2015). In general, a certain amount of available labile iron is the basis for ferroptosis execution.
Heat shock protein family B (small) member 1 (HSPB1), a small heat shock protein, is a negative regulator of erastin-induced ferroptosis in vitro and in vivo (Sun et al., 2015). HSPB1 can downregulate TfR1-mediated iron uptake by stabilizing actin cytoskeleton, and HSPB1 overexpression decelerates transferrin endocytosis and recycling (Chen et al., 2006).
Heme oxygenase 1 (HO-1) plays a dual role in the regulation of ferroptosis. Increased expression of HO-1 is an important event in erastin-induced ferroptosis in HT-1080 fibrosarcoma cells, and HO-1 likely provides iron supplements for promoting ferroptosis (Kwon et al., 2015). Intriguingly, another study suggested that HO-1 negatively regulated erastin- or sorafenib-induced ferroptosis in HCC cells (Sun et al., 2016). HO-1 knockdown by shRNA enhanced growth inhibition in response to erastin and sorafenib in HCC cells. The role of HO-1 in ferroptosis maybe dependent on different pathological contexts, although the precise mechanism of this phenomenon needs to be studied further.
Phosphorylase kinase catalytic subunit gamma 2 (PHKG2) positively regulates ferroptosis through the modulation of available iron, and PHKG2 silencing may function as iron chelation (Yang et al., 2016). The detailed mechanism of PHKG2 in iron metabolism regulation is unknown and needs further investigation.
The overwhelming accumulation of lipid ROS is the ferroptosis executioner, which can be prevented by lipophilic antioxidants and iron chelators. NOXs provide a source of accumulated ROS in erastin-induced ferroptosis (Dixon et al., 2012). Indeed, the pharmacological inhibition of NOXs and the NADPH-generating PPP strongly rescued erastin-induced ferroptosis in Calu-1 cells. However, pharmacological inhibition of NOXs or PPP partially rescued erastin-induced ferroptosis in HT-1080 cells, especially loss of inhibition in high concentration of erastin. The contradictory results may due to different cell lines, reflecting that NOX/PPP pathway is likely to be a downstream consequence rather than an initiation factor. In addition to NOXs, cell membrane lipid peroxidation products are another source of ROS generation, which really drives ferroptosis execution.
In membrane lipid environments, PUFAs, but not monounsaturated FA, cholesterol, and cardiolipin, are specifically peroxidized in ferroptosis (Yang et al., 2016). Indeed, PUFAs [e.g., arachidonic acid (AA, 20:4n6)] and PUFA derivatives [e.g., linoleate (18:2n6)] were significantly decreased following erastin treatment in HT-1080 cells (Skouta et al., 2014). Numerous regulators and pathways involving the synthesis of FA, such as glutaminolysis (Gao et al., 2015), citrate synthase (Dixon et al., 2012), and acetyl-CoA carboxylases, are necessary for the execution of ferroptosis (Shimada et al., 2016). ACSL4 preferentially acylates AA, while LPCAT3 preferentially catalyzes the insertion of acylated AA into membrane phospholipids, ultimately leading to the conversion of lysoPC to PC (Dixon et al., 2015). These two genes are essential for the execution of ferroptosis induced by the inhibition of GPX4. Moreover, ACSF2 is also required for erastin-induced ferroptosis (Dixon et al., 2012). Taken together, these genes ensure adequate membrane lipid PUFA production to promote ferroptosis for subsequent lipid peroxidation and ROS generation.
Lipoxygenases can drive ferroptosis, mediated by system Xc- inhibition, by catalyzing the dioxygenation of membrane lipid PUFAs to produce fatty acid hydroperoxides (Yang et al., 2016). For example, Zileuton (Rossi et al., 2010), a 5-lipoxygenase inhibitor, confers neuroprotection against glutamate oxidative damage by inhibiting ferroptosis (Liu et al., 2015). Under normal conditions, fatty acid hydroperoxides are converted to fatty acid alcohols under GPX4 mediation. However, this process is blocked during ferroptosis because of GPX4 inactivation. Accumulated fatty acid hydroperoxides are further catalyzed into lipid peroxyl radicals by iron-mediated Fenton reactions, which are lethal to cells. The oxidative destruction of membrane lipid PUFAs increases with ferroptosis, as lysoPC accumulates and specific PUFAs (Skouta et al., 2014). A study showed that resistance to system Xc- inhibition dramatically increased the expression of AKR1C (Dixon et al., 2014), which can detoxify cytotoxic oxidative PUFA breakdown products (e.g., MDA and 4-HNE). However, the detailed mechanism underlying the direct inhibition of GPX4-induced ferroptosis is still enigmatic.
Cancer cells can undergo several regulated forms of cell death during tumor development, including apoptosis (Kerr et al., 1994), autophagy (Kondo et al., 2005; Levine, 2006), and necrosis (Amaravadi and Thompson, 2007). Unsurprisingly, ferroptosis also plays a role in the development of cancer and maybe a beneficial strategy for anticancer treatment.
Different lines of evidence suggest that ferroptosis plays a crucial role in the suppression of tumorigenesis. Specifically, GPX4 knockdown using siRNAs decreases the level of GPX4 protein, a central mediator of ferroptosis, inducing renal cell carcinoma cell death with accompanying lipid ROS generation (Yang et al., 2014). This process can be rescued by the iron chelator DFO and the antioxidant vitamin E. Moreover, inhibition of a member of cystine/glutamate antiporter, SLC7A11, can induce ferroptosis (Dixon et al., 2012). SLC7A11 is highly expressed in human tumors (Huang et al., 2005; Liu X. X. et al., 2011), and its overexpression rescues cancer cells from ferroptosis. Both the pharmacological and genetic inhibition of SLC7A11 induce ferroptotic cell death and enhance cisplatin cytotoxicity in cisplatin-resistant head and neck cancer (HNC) cells in vitro and in vivo (Roh et al., 2016). In addition, a recent study showed that p53 inhibits tumor growth partially by repressing the expression of SLC7A11 and subsequently inducing ferroptosis in addition to traditional p53-mediated functions, such as cell cycle arrest, senescence, and apoptosis (Jiang et al., 2015). Notably, p533KR, an acetylation-defective mutant that fails to induce cell cycle arrest, senescence and apoptosis, fully retains its ability to regulate SLC7A11 expression and induce ferroptosis upon ROS-induced stress (Jiang et al., 2015). p533KR mice did not succumb to the early-onset spontaneous thymic lymphoma formation that is commonly observed in p53-null mice (Li et al., 2012). Consistently, unlike p53-/-XRCC4-/- mice, p533KR/3KRXRCC4-/- mice did not succumb to pro-B cell lymphomas (Li et al., 2016). This suggests that p533KR retains its tumor-suppressive activity and that uncanonical functions of p53 play crucial roles in the suppression of tumorigenesis. Taken together, these findings indicate that ferroptosis serves as a critical barrier to cancer development.
Accumulating evidence has shown that the activation of ferroptosis contributes to anticancer treatment for several forms of human cancer (Table 1). Erastin, a RAS-selective lethal compound (Dolma et al., 2003) that triggers ferroptosis by directly inhibiting cystine/glutamate antiporter system Xc- activity, displays synthetic lethality in engineered cells. However, erastin does not show selective lethality in RAS-mutated cancer cell lines over RAS wild-type counterparts in a large, diverse panel of cell lines. Analysis of erastin sensitivity data from 117 cell lines and a 60-cancer cell line panel (NCI 60) harvested from eight diverse tissues (leukemia, lung, colon, CNS, melanoma, ovarian, kidney, and breast) revealed that diffuse, large B cell lymphomas and RCCs are particularly sensitive to ferroptosis (Yang et al., 2014). Moreover, low-cytotoxic doses of erastin remarkably enhance the anticancer activities of 2 first-line chemotherapy drugs, cytarabine and doxorubicin, in HL60 cells (Yu et al., 2015). Given that AML has a high recurrence rate, inducing ferroptosis may overcome drug resistance in AML cells.

TABLE 1. Ferroptosis relevance among various types of cancer.
Sorafenib, a multikinase inhibitor, is clinically approved for the treatment of renal cell carcinoma and other indications. Recently, sorafenib was identified as a ferroptosis inducer by inhibiting system Xc- function and triggering subsequent ferroptotic cell death (Dixon et al., 2014). However, acquired resistance to sorafenib has been found in HCC patients, which results in poor prognosis. Previous studies show that NRF2 (Sun et al., 2016) and MT-1G (Houessinon et al., 2016), two negative regulators of ferroptosis that function by blocking GSH depletion-mediated lipid peroxidation in HCC cells, play a central role in protecting HCC cells against ferroptosis and result in sorafenib resistance. Importantly, the genetic and pharmacological inhibition of NRF2 or MT-1G in HCC cells enhances the anticancer activity of sorafenib in vitro and in tumor xenograft models.
In addition, numerous molecules, such as RSL3, RSL5, ART (Eling et al., 2015), DHA (Lin et al., 2016), and a series of small-molecule inducers (FINs) (Weiwer et al., 2012; Yang et al., 2012), have been shown to introduce ferroptotic cell death in cancer cells. Co-treatment with the ferroptosis inhibitor ferrostatin-1 can block ART-induced lipid peroxidation and cell death and increase long-term cell survival and proliferation.
As discussed above, ferroptosis participates in cancer development and treatment response, contributing to tumor suppression activity. Although many experiments in vitro have shown that ferroptosis can be detected via several measuring methods, such as cell viability, iron level and ROS level, it would be harder to prove the presence of ferroptosis in vivo. So far, PTGS2, a gene encoding cyclooxygenase-2 (COX-2), is the unique widely used marker for erastin or RSL-3-induced ferroptosis in tumor (Yang et al., 2014).
Activation of regulated cell death is an anticancer treatment strategy. Despite success in clinical cancer treatment, drug resistance to existing chemotherapeutic agents due to genetic alterations remains a problem (Pommier et al., 2004; Liu L. et al., 2011; Sui et al., 2013). Considering the clinical limitations and the discovery of ferroptosis (Dixon et al., 2012), activation of a regulated non-apoptotic form of cell death, ferroptosis, may provide new drug targets.
As described above, compelling evidence suggests that the activation of ferroptosis is an effective, novel pathway for cancer intervention. In principle, ferroptosis activation could be achieved by (i) the identification of small molecules as ferroptosis inducers using systematic screening and structural basis or by (ii) the inhibition of key molecules related to ferroptosis, such as the cystine/glutamate antiporter SLC7A11. We will comment here on the avenues in which progress can be made in cancer treatment by ferroptosis activation.
Since ferroptosis was identified as a novel form of iron-dependent non-apoptotic cell death, it has been recognized as a new target of drug discovery in recent years. Using systematic screening, a number of small molecules, such as erastin (Dixon et al., 2012), sorafenib (Sun et al., 2016), ART (Eling et al., 2015), DHA (Lin et al., 2016), RSL3, and FINs, were found to induce cell death via ferroptosis. As described above in Section “Ferroptosis and Tumor Suppression,” the combination of erastin and sorafenib maybe a promising therapeutic strategy for cancer, especially in sorafenib-resistant contexts.
In addition, ART specifically induced ROS- and lysosomal iron-dependent cell death in PDAC cell lines (Eling et al., 2015). Co-treatment with the ferroptosis inhibitor ferrostatin-1 blocked ART-induced lipid peroxidation and cell death and increased long-term cell survival and proliferation. DHA specifically causes HNC cell death via contribution from both ferroptosis and apoptosis (Lin et al., 2016). Moreover, 10 different artemisinin derivatives were found to kill tumor cells not only by the induction of apoptosis, autophagy or necroptosis, as shown in the past, but also by ferroptosis (Ooko et al., 2015). In a larger screening to find ferroptosis-inducing compounds, a series of small-molecule inducers, named FINs, were discovered. FIN56 treatment resulted in loss of the GPX4 protein through post-translational degradation and blocked the mevalonate-derived production of lipophilic antioxidants, such as coenzyme Q10 (Shimada et al., 2016). However, the full therapeutic potential of this perspective needs to be explored in more detail in the future.
Given that (1) GPX4 is responsible for ferroptotic cell death induced by lipid peroxidation signaling and (2) knockdown of GPX4 expression using siRNA reagents is sufficient to induce ferroptosis (Yang et al., 2014), GPX4 inhibitors maybe potential candidates for cancer therapy. Considering the success of inhibiting GPX4 by RSL3, finding the structural basis underlying the RSL3 inhibition of GPX4 to design GPX4 inhibitors is a hot research topic. A recent study identified a ligand-binding site on GPX4, and covalently targeting the active site selenocysteine can inhibit GPX4 to induce ferroptosis (Yang et al., 2016). Ideally, a cocrystal structure of RSL3 bound to GPX4 would initiate a search for drug-like GPX4 inhibitors. Systematic screening for ferroptosis inducers and understanding the structural basis for interfering with the active site selenocysteine of GPX4 may confront tumor chemotherapy resistance. Meanwhile, we should consider that GPX4 is an essential enzyme for life and knockout of GPX4 in mice caused embryonic lethality between E7.5 and E8.5 (Yant et al., 2003). But it still could be true that cancer cells will be more sensitive to the inhibition of GPX4 comparing to the normal cells. Thus, the inhibition GPX4 still could be the potential therapeutic approach but the side effects caused by the loss of GPX4 should also be considered in future study.
So far researches on the safety or efficiency of ferroptotic drugs in vivo are limited, mainly in piperazine erastin (PE), (1S, 3R)-RSL3, and sorafenib. PE is an analog of erastin with better water solubility and metabolic stability, which is suitable for in vivo experiments (Yang et al., 2014). It has been demonstrated that ferroptosis induced by PE or (1S, 3R)-RSL3 prevents tumor growth in xenograft mouse tumor models. Besides, when loss of the retinoblastoma protein, sorafenib has a powerful effect on tumor regression in murine xenografts of HCC (Louandre et al., 2015). However, the above studies have not yet been involved in the evaluation of drug safety. A resolution of this matter will be required in future.
In addition to GPX4, inhibition of an increasing number of key molecules has been related to ferroptosis, which may induce cell death and eradicate chemotherapy/radiotherapy-resistant cancer cells.
Since the status of NRF2 is a key factor determining the therapeutic response to ferroptosis-targeted therapies in HCC cells (Sun et al., 2016), inhibiting the expression of NRF2 during ferroptosis-targeted therapies to enhance curative tumor effects is necessary. In addition, iron-enriched tumor environments as well as the use of sorafenib to treat RCCs in the clinic suggest that RCCs, especially chromophobe RCCs, could be targeted for ferroptosis induction (Kroll et al., 1984; Sato et al., 2005). A recent study demonstrated that autophagy contributes to ferroptosis by the degradation of ferritin in fibroblasts and cancer cells. Remarkably, NCOA4 was a cargo receptor for the selective autophagic turnover of ferritin (ferritinophagy) in ferroptosis (Hou et al., 2016). Consistently, overexpression of NCOA4 increased ferritin degradation and promoted ferroptosis.
Moreover, several studies have shown that pharmacological and genetic inhibition of the cystine/glutamate antiporter SLC7A11 significantly sensitized resistant HNC cells to cisplatin in vitro and in vivo, and SLC7A11-deficient mice developed normally and were healthy (Sato et al., 2005), suggesting that compounds targeting system Xc- with high specificity may have minimal side effects in preclinical and clinical settings. However, system Xc- is probably not an effective target for ferroptosis in some cancer, especially cancer cells can bypass their dependence on system Xc-. For example, when loss of cysteinyl-tRNA synthetase resulted in the accumulation of cystathionine, the transsulfuration pathway was activated and ultimately led to resistant to erastin-induced ferroptosis (Hayano et al., 2016). In addition, inhibition of the transsulfuration pathway resensitized cells to erastin. On another hand, system Xc- is also a promising target for some cancers, which depend on cysteine/cystine import system (system Xc-) due to defective transsulfuration pathway. For example, diffuse large B cell lymphoma were particularly sensitive to erastin-induced ferroptosis (Yang et al., 2014). Thus, whether system Xc- can become a therapeutic target of ferroptosis should be considered the genetic composition of cancer cells.
Collectively, inhibition of key ferroptotic molecules is a promising therapeutic strategy for the treatment of tumor, especially drug-resistant tumors.
The data described herein clearly show that ferroptosis is an iron-dependent form of non-apoptotic cell death and an ideal target for biochemical studies. ROS-induced ferroptosis contributes to tumor growth suppression and chemotherapy sensitivity. Because the activation of ferroptosis is widely recognized as a new target of drug discovery, an increasing number of small molecules have been identified to induce ferroptosis directly or indirectly by targeting iron metabolism and lipid peroxidation, and other studies have focused on the inhibition of key molecules related to ferroptosis. Meanwhile, whether compounds targeting ferroptotic cell death regulators have high specificity and minimal side effects in preclinical and clinical settings and what kinds of cancer prefer to be targeted by ferroptosis induction remain to be elucidated.
In summary, an improved understanding of the ferroptosis mechanism and the role of ferroptosis in cancer will create new opportunities for diagnosis and therapeutic intervention.
BY and JC conceived, designed the conception of review article, and made the amendments of the paper. BL conducted the paper. XC, MY, and QH collected the related research articles.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer RP and handling editor declared their shared affiliation.
This work was supported by grants from the National Natural Science Foundation of China (81625024 and 81402951), International Science and Technology Cooperation Program of China (2014DFE30050), and Department of Education of Zhejiang Province (Y201430401).
AA, arachidonic acid; ACSF2, acyl-CoA synthetase family member 2; ACSL4, acyl-CoA synthetase long-chain family member 4; AKR1C, aldo–keto reductase family 1 member C; AML, acute myeloid leukemia; ART, artesunate; ATG5, autophagy related 5; ATG7, autophagy related 7; DFO, desferrioxamine; DHA, dihydroartemisinin; DMT1, divalent metal transporter 1; FA, fatty acids; FDA, Food and Drug Administration; FINs, ferroptosis-inducing agents; Fpn, ferroportin; FTH1, ferritin heavy chain 1; FTL, ferritin light chain; γ-GCS, γ-glutamylcysteine synthetase; GPX4, glutathione peroxidase 4; HCC, hepatocellular carcinoma; 4-HNE, 4-hydroxynonenal; HO-1, heme oxygenase 1; HSPB1, heat shock protein family B (small) member 1; IREB2, iron response element binding protein 2; LOXs, lipoxygenases; LPCAT3, lysophosphatidylcholine acyltransferase 3; lysoPC, lysophosphatidylcholine; MDA, malondialdehyde; MT, metallothionein; NCOA4, nuclear receptor coactivator 4; NOXs, NAPDH oxidases; NRF2, nuclear factor, erythroid 2 like 2; PC, phosphatidylcholine; PDAC, prostate adenocarcinoma; PHKG2, phosphorylase kinase catalytic subunit gamma 2; PPP, pentose phosphate pathway; PUFAs, polyunsaturated fatty acids; RCCs, renal cell carcinomas; SLC7A11, solute carrier family 7 member 11; TF, Transferrin; TfR1, transferrin receptor 1.
Amaravadi, R. K., and Thompson, C. B. (2007). The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin. Cancer Res. 13, 7271–7279. doi: 10.1158/1078-0432.CCR-07-1595
Bogdan, A. R., Miyazawa, M., Hashimoto, K., and Tsuji, Y. (2016). Regulators of iron homeostasis: new players in metabolism, cell death, and disease. Trends Biochem. Sci. 41, 274–286. doi: 10.1016/j.tibs.2015.11.012
Bridges, R. J., Natale, N. R., and Patel, S. A. (2012). System xc(-) cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br. J. Pharmacol. 165, 20–34. doi: 10.1111/j.1476-5381.2011.01480.x
Chen, H., Zheng, C., Zhang, Y., Chang, Y. Z., Qian, Z. M., and Shen, X. (2006). Heat shock protein 27 downregulates the transferrin receptor 1-mediated iron uptake. Int. J. Biochem. Cell Biol. 38, 1402–1416. doi: 10.1016/j.biocel.2006.02.006
Chen, L., Li, X., Liu, L., Yu, B., Xue, Y., and Liu, Y. (2015). Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-gamma-lyase function. Oncol. Rep. 33, 1465–1474. doi: 10.3892/or.2015.3712
Conrad, M., Angeli, J. P., Vandenabeele, P., and Stockwell, B. R. (2016). Regulated necrosis: disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 15, 348–366. doi: 10.1038/nrd.2015.6
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. doi: 10.1016/j.cell.2012.03.042
Dixon, S. J., Patel, D. N., Welsch, M., Skouta, R., Lee, E. D., Hayano, M., et al. (2014). Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 3:e02523. doi: 10.7554/eLife.02523
Dixon, S. J., Winter, G. E., Musavi, L. S., Lee, E. D., Snijder, B., Rebsamen, M., et al. (2015). Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem. Biol. 10, 1604–1609. doi: 10.1021/acschembio.5b00245
Dolma, S., Lessnick, S. L., Hahn, W. C., and Stockwell, B. R. (2003). Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 3, 285–296. doi: 10.1016/S1535-6108(03)00050-3
Eling, N., Reuter, L., Hazin, J., Hamacher-Brady, A., and Brady, N. R. (2015). Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2, 517–532. doi: 10.18632/oncoscience.160
Fatokun, A. A., Dawson, V. L., and Dawson, T. M. (2014). Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 171, 2000–2016.
Friedmann Angeli, J. P., Schneider, M., Proneth, B., Tyurina, Y. Y., Tyurin, V. A., Hammond, V. J., et al. (2014). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180–1191. doi: 10.1038/ncb3064
Galmiche, A., Chauffert, B., and Barbare, J. C. (2014). New biological perspectives for the improvement of the efficacy of sorafenib in hepatocellular carcinoma. Cancer Lett. 346, 159–162. doi: 10.1016/j.canlet.2013.12.028
Gao, M., Monian, P., Pan, Q., Zhang, W., Xiang, J., and Jiang, X. (2016). Ferroptosis is an autophagic cell death process. Cell Res. 26, 1021–1032. doi: 10.1038/cr.2016.95
Gao, M., Monian, P., Quadri, N., Ramasamy, R., and Jiang, X. (2015). Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell 59, 298–308. doi: 10.1016/j.molcel.2015.06.011
Greenshields, A. L., Shepherd, T. G., and Hoskin, D. W. (2016). Contribution of reactive oxygen species to ovarian cancer cell growth arrest and killing by the anti-malarial drug artesunate. Mol. Carcinog. 56, 75–93. doi: 10.1002/mc.22474
Hasegawa, M., Takahashi, H., Rajabi, H., Alam, M., Suzuki, Y., Yin, L., et al. (2016). Functional interactions of the cystine/glutamate antiporter, CD44v and MUC1-C oncoprotein in triple-negative breast cancer cells. Oncotarget 7, 11756–11769. doi: 10.18632/oncotarget.7598
Hayano, M., Yang, W. S., Corn, C. K., Pagano, N. C., and Stockwell, B. R. (2016). Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 23, 270–278. doi: 10.1038/cdd.2015.93
Hou, W., Xie, Y., Song, X., Sun, X., Lotze, M. T., Zeh, H. J., et al. (2016). Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428. doi: 10.1080/15548627.2016.1187366
Houessinon, A., Francois, C., Sauzay, C., Louandre, C., Mongelard, G., Godin, C., et al. (2016). Metallothionein-1 as a biomarker of altered redox metabolism in hepatocellular carcinoma cells exposed to sorafenib. Mol. Cancer 15:38. doi: 10.1186/s12943-016-0526-2
Huang, Y., Dai, Z., Barbacioru, C., and Sadee, W. (2005). Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res. 65, 7446–7454. doi: 10.1158/0008-5472.CAN-04-4267
Jiang, L., Kon, N., Li, T., Wang, S. J., Su, T., Hibshoosh, H., et al. (2015). Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62. doi: 10.1038/nature14344
Kerr, J. F., Winterford, C. M., and Harmon, B. V. (1994). Apoptosis. Its significance in cancer and cancer therapy. Cancer 73, 2013–2026. doi: 10.1002/1097-0142(19940415)73:8<2013::AID-CNCR2820730802>3.0.CO;2-J
Kondo, Y., Kanzawa, T., Sawaya, R., and Kondo, S. (2005). The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 5, 726–734. doi: 10.1038/nrc1692
Kroll, M. H., Jiji, V., and Jiji, R. (1984). Microcytic hypochromic anemia associated with renal cell carcinoma. South Med. J. 77, 635–637. doi: 10.1097/00007611-198405000-00024
Kwon, M. Y., Park, E., Lee, S. J., and Chung, S. W. (2015). Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 6, 24393–24403. doi: 10.18632/oncotarget.5162
Levine, B. (2006). Unraveling the role of autophagy in cancer. Autophagy 2, 65–66. doi: 10.4161/auto.2.2.2457
Li, T., Kon, N., Jiang, L., Tan, M., Ludwig, T., Zhao, Y., et al. (2012). Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 149, 1269–1283. doi: 10.1016/j.cell.2012.04.026
Li, T., Liu, X., Jiang, L., Manfredi, J., Zha, S., and Gu, W. (2016). Loss of p53-mediated cell-cycle arrest, senescence and apoptosis promotes genomic instability and premature aging. Oncotarget 7, 11838–11849. doi: 10.18632/oncotarget.7864
Lin, R., Zhang, Z., Chen, L., Zhou, Y., Zou, P., Feng, C., et al. (2016). Dihydroartemisinin (DHA) induces ferroptosis and causes cell cycle arrest in head and neck carcinoma cells. Cancer Lett. 381, 165–175. doi: 10.1016/j.canlet.2016.07.033
Liu, L., Yang, M., Kang, R., Wang, Z., Zhao, Y., Yu, Y., et al. (2011). HMGB1-induced autophagy promotes chemotherapy resistance in leukemia cells. Leukemia 25, 23–31. doi: 10.1038/leu.2010.225
Liu, X. X., Li, X. J., Zhang, B., Liang, Y. J., Zhou, C. X., Cao, D. X., et al. (2011). MicroRNA-26b is underexpressed in human breast cancer and induces cell apoptosis by targeting SLC7A11. FEBS Lett. 585, 1363–1367. doi: 10.1016/j.febslet.2011.04.018
Liu, Y., Wang, W., Li, Y., Xiao, Y., Cheng, J., and Jia, J. (2015). The 5-lipoxygenase inhibitor zileuton confers neuroprotection against glutamate oxidative damage by inhibiting ferroptosis. Biol. Pharm. Bull. 38, 1234–1239. doi: 10.1248/bpb.b15-00048
Louandre, C., Marcq, I., Bouhlal, H., Lachaier, E., Godin, C., Saidak, Z., et al. (2015). The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 356, 971–977. doi: 10.1016/j.canlet.2014.11.014
Ma, S., Henson, E. S., Chen, Y., and Gibson, S. B. (2016). Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 7:e2307. doi: 10.1038/cddis.2016.208
Ooko, E., Saeed, M. E., Kadioglu, O., Sarvi, S., Colak, M., Elmasaoudi, K., et al. (2015). Artemisinin derivatives induce iron-dependent cell death (ferroptosis) in tumor cells. Phytomedicine 22, 1045–1054. doi: 10.1016/j.phymed.2015.08.002
Pommier, Y., Sordet, O., Antony, S., Hayward, R. L., and Kohn, K. W. (2004). Apoptosis defects and chemotherapy resistance: molecular interaction maps and networks. Oncogene 23, 2934–2949. doi: 10.1038/sj.onc.1207515
Roh, J. L., Kim, E. H., Jang, H. J., Park, J. Y., and Shin, D. (2016). Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett. 381, 96–103. doi: 10.1016/j.canlet.2016.07.035
Rossi, A., Pergola, C., Koeberle, A., Hoffmann, M., Dehm, F., Bramanti, P., et al. (2010). The 5-lipoxygenase inhibitor, zileuton, suppresses prostaglandin biosynthesis by inhibition of arachidonic acid release in macrophages. Br. J. Pharmacol. 161, 555–570. doi: 10.1111/j.1476-5381.2010.00930.x
Sato, H., Shiiya, A., Kimata, M., Maebara, K., Tamba, M., Sakakura, Y., et al. (2005). Redox imbalance in cystine/glutamate transporter-deficient mice. J. Biol. Chem. 280, 37423–37429. doi: 10.1074/jbc.M506439200
Sehm, T., Rauh, M., Wiendieck, K., Buchfelder, M., Eyupoglu, I. Y., and Savaskan, N. E. (2016). Temozolomide toxicity operates in a xCT/SLC7a11 dependent manner and is fostered by ferroptosis. Oncotarget 7, 74630–74647. doi: 10.18632/oncotarget.11858
Shimada, K., Skouta, R., Kaplan, A., Yang, W. S., Hayano, M., Dixon, S. J., et al. (2016). Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 12, 497–503. doi: 10.1038/nchembio.2079
Skouta, R., Dixon, S. J., Wang, J., Dunn, D. E., Orman, M., Shimada, K., et al. (2014). Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 136, 4551–4556. doi: 10.1021/ja411006a
Sui, X., Chen, R., Wang, Z., Huang, Z., Kong, N., Zhang, M., et al. (2013). Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 4:e838. doi: 10.1038/cddis.2013.350
Sun, X., Ou, Z., Chen, R., Niu, X., Chen, D., Kang, R., et al. (2016). Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63, 173–184. doi: 10.1002/hep.28251
Sun, X., Ou, Z., Xie, M., Kang, R., Fan, Y., Niu, X., et al. (2015). HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 34, 5617–5625. doi: 10.1038/onc.2015.32
Weiwer, M., Bittker, J. A., Lewis, T. A., Shimada, K., Yang, W. S., Macpherson, L., et al. (2012). Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg. Med. Chem. Lett. 22, 1822–1826. doi: 10.1016/j.bmcl.2011.09.047
Xie, Y., Hou, W., Song, X., Yu, Y., Huang, J., Sun, X., et al. (2016). Ferroptosis: process and function. Cell Death Differ. 23, 369–379. doi: 10.1038/cdd.2015.158
Yagoda, N., Von Rechenberg, M., Zaganjor, E., Bauer, A. J., Yang, W. S., Fridman, D. J., et al. (2007). RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447, 864–868. doi: 10.1038/nature05859
Yamaguchi, H., Hsu, J. L., Chen, C. T., Wang, Y. N., Hsu, M. C., Chang, S. S., et al. (2013). Caspase-independent cell death is involved in the negative effect of EGF receptor inhibitors on cisplatin in non-small cell lung cancer cells. Clin. Cancer Res. 19, 845–854. doi: 10.1158/1078-0432.CCR-12-2621
Yang, W. S., Kim, K. J., Gaschler, M. M., Patel, M., Shchepinov, M. S., and Stockwell, B. R. (2016). Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. U.S.A. 113, E4966–E4975. doi: 10.1073/pnas.1603244113
Yang, W. S., Shimada, K., Delva, D., Patel, M., Ode, E., Skouta, R., et al. (2012). Identification of simple compounds with microtubule-binding activity that inhibit cancer cell growth with high potency. ACS Med. Chem. Lett. 3, 35–38. doi: 10.1021/ml200195s
Yang, W. S., Sriramaratnam, R., Welsch, M. E., Shimada, K., Skouta, R., Viswanathan, V. S., et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. doi: 10.1016/j.cell.2013.12.010
Yang, W. S., and Stockwell, B. R. (2008). Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 15, 234–245. doi: 10.1016/j.chembiol.2008.02.010
Yant, L. J., Ran, Q., Rao, L., Van Remmen, H., Shibatani, T., Belter, J. G., et al. (2003). The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic. Biol. Med. 34, 496–502. doi: 10.1016/S0891-5849(02)01360-6
Keywords: ferroptosis, lipid ROS, iron metabolism, cancer therapeutics, tumorigenesis
Citation: Lu B, Chen XB, Ying MD, He QJ, Cao J and Yang B (2018) The Role of Ferroptosis in Cancer Development and Treatment Response. Front. Pharmacol. 8:992. doi: 10.3389/fphar.2017.00992
Received: 12 October 2017; Accepted: 22 December 2017;
Published: 12 January 2018.
Edited by:
Thales Papagiannakopoulos, New York University, United StatesReviewed by:
Chun Hei Antonio Cheung, National Cheng Kung University, TaiwanCopyright © 2018 Lu, Chen, Ying, He, Cao and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ji Cao, Y2Fvamk4OEB6anUuZWR1LmNu Bo Yang, eWFuZzkyNEB6anUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.