 Byron Carpenter
Byron Carpenter Guillaume Lebon
Guillaume Lebon
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 14 December 2017
Sec. Experimental Pharmacology and Drug Discovery
Volume 8 - 2017 | https://doi.org/10.3389/fphar.2017.00898
This article is part of the Research TopicPurinergic Pharmacology, Volume IView all 62 articles
Adenosine receptors (ARs) comprise the P1 class of purinergic receptors and belong to the largest family of integral membrane proteins in the human genome, the G protein-coupled receptors (GPCRs). ARs are classified into four subtypes, A1, A2A, A2B, and A3, which are all activated by extracellular adenosine, and play central roles in a broad range of physiological processes, including sleep regulation, angiogenesis and modulation of the immune system. ARs are potential therapeutic targets in a variety of pathophysiological conditions, including sleep disorders, cancer, and dementia, which has made them important targets for structural biology. Over a decade of research and innovation has culminated with the publication of more than 30 crystal structures of the human adenosine A2A receptor (A2AR), making it one of the best structurally characterized GPCRs at the atomic level. In this review we analyze the structural data reported for A2AR that described for the first time the binding of mode of antagonists, including newly developed drug candidates, synthetic and endogenous agonists, sodium ions and an engineered G protein. These structures have revealed the key conformational changes induced upon agonist and G protein binding that are central to signal transduction by A2AR, and have highlighted both similarities and differences in the activation mechanism of this receptor compared to other class A GPCRs. Finally, comparison of A2AR with the recently solved structures of A1R has provided the first structural insight into the molecular determinants of ligand binding specificity in different AR subtypes.
Key Concepts
1. A2AR crystallization: selection of different conformational states
Structure determination of A2AR required the application of novel protein engineering techniques to lock the receptor in defined conformational states and facilitate the growth of crystals that diffract to high resolution.
2. Structural determinants of A2AR ligand binding and selectivity
The atomic resolution structural features of A2AR that dictate which ligands it can bind and whether the ligands act as agonists, to promote signaling, or antagonists, to block signaling.
3. Ligand-induced activation of A2AR
The molecular changes that are induced in A2AR by agonist binding, which facilitate G protein coupling and ultimately signal transduction.
4. Structural diversity of the adenosine receptor family
The difference in the primary and ternary structure between the four AR subtypes that is ultimately responsible for their ligand-binding specificity and pharmacological profiles.
Purinergic signaling is predominantly mediated by extracellular purine nucleosides and nucleotides, including adenosine and adenosine triphosphate (ATP), but also by purine bases such as caffeine and xanthine. Purinergic receptors are integral membrane protein that are divided into three classes, P1 (better known as adenosine receptors), P2Y, and P2X (Burnstock, 2006). Both P1 and P2Y receptors belong to the G protein-coupled receptor (GPCR) family, whereas P2X receptors are ATP-gated ion channels. Adenosine receptors (ARs) are divided into four subtypes, A1, A2A, A2B, and A3 (Fredholm et al., 2001), which are broadly expressed in the central nervous system as well as peripheral tissues of the cardiovascular, respiratory, renal, and immune systems (Fredholm et al., 2001, 2011). Extracellular adenosine is the endogenous agonist for all ARs, however differences in the adenosine binding affinity, tissue distribution, expression level and G protein coupling preference between the subtypes gives each a distinct signaling profile (Cieslak et al., 2008; Fredholm, 2014). A1R and A3R predominantly activate heterotrimeric G proteins belonging to the Gαi/o family, which inhibit cAMP production by adenylate cyclase, in contrast A2AR and A2BR predominantly activate Gαs family members, which stimulate cAMP production (Jacobson and Gao, 2006). G protein βγ subunits also contribute to signaling through the mitogen-activated protein kinase (MAPK) and phospholipase C (PLC) pathways (Jacobson and Gao, 2006). ARs mediate the general cytoprotective functions associated with extracellular adenosine, with some of the key physiological processes regulated by individual subtypes being: sleep, vasoconstriction and inhibition of neurotransmitter release by A1R; sleep, angiogenesis, and immunosuppression by A2AR; vascular integrity and myocardial preconditioning by A2BR; mast cell regulation and myocardial preconditioning by A3R (Fredholm et al., 2011).
ARs have been proposed as potential targets in a wide variety of pathophysiological conditions, including arrhythmia, ischemia, sleep disorders, pain, dementia, Parkinson's, renal failure, asthma, type 2 diabetes, glaucoma, inflammation, and cancer (Jacobson and Gao, 2006; Cieslak et al., 2008; Sawynok, 2016). However, one of the challenges of therapeutic intervention has been targeting individual AR subtypes with sufficient specificity to limit off-target side effects (Chen et al., 2013). Medicinal chemistry approaches have been used to develop an array of compounds that exhibit improved subtype specificity (Müller and Jacobson, 2011), but very few have been approved for clinical use, due in part to the persistence of undesirable side effects (Chen et al., 2013; Glukhova et al., 2017). Further improvements in subtype specificity, coupled with the development of allosteric modulators (Gentry et al., 2015) that bind outside the orthosteric site, and biased ligands (Kenakin and Christopoulos, 2013) that can target a distinct signaling pathway associated with an individual AR subtype, may help to eliminate side effects entirely. Structure-based drug design, which involves in silico screening of vast compound libraries against experimentally determined receptor structures, offers huge potential for the development of a new generation of highly selective orthosteric, allosteric, and biased ligands, however, the difficulty of crystallizing GPCRs has, until recently, hindered this approach (Jazayeri et al., 2015).
Structure determination of GPCRs is notoriously challenging due to their conformationally dynamic nature and poor thermostability when extracted from the plasma membrane. During the past decade crystallization strategies have been developed that have revolutionized the structural biology of GPCRs, these include protein engineering approaches, such as fusion proteins (Cherezov et al., 2007; Chun et al., 2012), antibodies (Rasmussen et al., 2007, 2011a) and conformational thermostabilization (Magnani et al., 2008; Serrano-Vega et al., 2008; Shibata et al., 2009), as well as technical developments, such as the lipidic cubic phase (LCP) (Landau and Rosenbusch, 1996; Caffrey, 2015). Human A2AR has been at the forefront of this revolution and is now one of the best structurally characterized GPCRs, with more than 30 structures deposited in the protein data bank (PDB; Table 1). It is the only receptor for which structures of three distinct activation states have been reported, namely the inactive conformation bound to an antagonist or inverse agonist (Jaakola et al., 2008), an intermediate-active conformation bound to an agonist (Lebon et al., 2011b; Xu et al., 2011), and the active conformation bound to both an agonist and engineered G protein (Carpenter et al., 2016). Crystallization of the other AR subtypes has proved more difficult and it is only during the past year that structures of A1R have been published (Cheng et al., 2017; Glukhova et al., 2017). Significantly, these have provided the first atomic resolution insight in to the molecular determinants of ligand binding specificity in different AR subtypes.
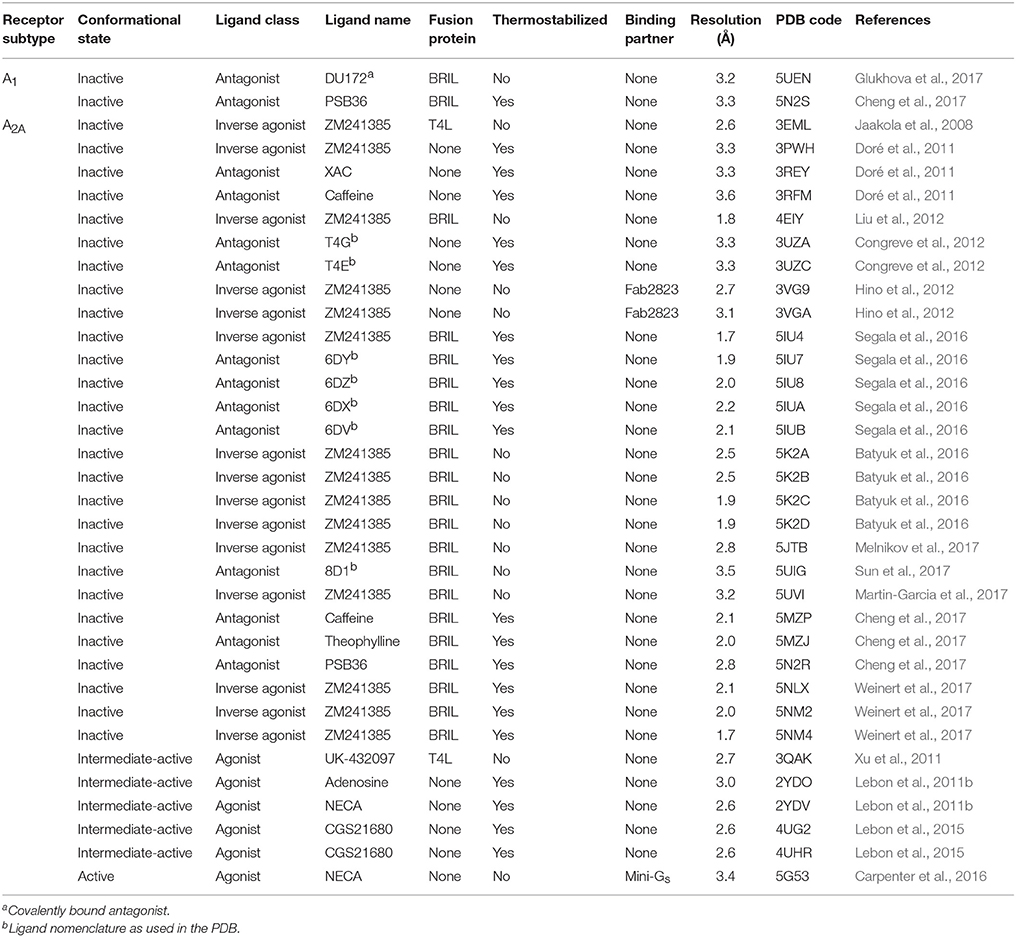
Table 1. Adenosine receptor X-ray crystal structures.
In this review we consolidate and analyze all of the structural information published during the past decade, which provides a near complete picture of A2AR activation. We compare the binding mode of antagonists, including the widely consumed stimulant caffeine (Doré et al., 2011; Cheng et al., 2017), and agonists, including the endogenous ligand adenosine (Lebon et al., 2011b). We highlight the agonist-induced conformational changes that activate A2AR (Lebon et al., 2011b; Xu et al., 2011), and the cooperative conformational changes induced by G protein coupling (Carpenter et al., 2016). Finally, we compare A2AR with the recently solved structures of A1R (Cheng et al., 2017; Glukhova et al., 2017) and discuss the current evidence for the molecular basis of ligand binding specificity in different AR subtypes.
GPCRs are challenging targets for structural studies for three main reasons. First, flexibility and conformationally dynamics play a central role in receptor activation by maintaining a dynamic equilibrium between different conformational states (Kobilka and Deupi, 2007). Ligand binding is often insufficient to trap the receptor in a distinct conformation (Manglik et al., 2015; Ye et al., 2016), which can perturb the growth of crystals that diffract to high resolution (Cherezov et al., 2007; Warne et al., 2008; Tate and Schertler, 2009). Second, GPCRs are highly unstable upon extraction from the membrane by detergent solubilization, which makes purification of the receptors both technically challenging and inefficient (Serrano-Vega et al., 2008). Third, class A receptors are compact proteins that typically have only minimal hydrophilic surface area capable of forming crystal contacts. Structure determination of virtually all GPCRs, including A2AR, has therefore required the development of novel protein engineering strategies (discussed below), crystallization techniques, including LCP (Landau and Rosenbusch, 1996; Xu et al., 2011; Caffrey, 2015), and data collection methods, including the use of micrometer-sized X-ray beams (Moukhametzianov et al., 2008) or serial crystallography (Weinert et al., 2017), in order to obtain well-diffracting crystals and collect high resolution diffraction data.
The first structure of A2AR was solved bound to the inverse agonist ZM241385 at 2.6 Å resolution (Jaakola et al., 2008). This structure was facilitated by a combined approach of using a high affinity ligand, which locks the receptor in its inactive state, and by replacing the third intracellular loop (ICL3) with a T4 lysozyme (T4L) fusion protein (Rosenbaum et al., 2007), which increases the hydrophilic surface area available for crystal contact formation (Figure 1A). This fusion protein strategy was subsequently modified to utilize apocytochrome b562RIL (BRIL) instead of T4L (Liu et al., 2012), which resulted in the solution of seven additional structures of A2AR bound to ZM241385 ranging in resolution from 3.2 to 1.8 Å (Liu et al., 2012; Batyuk et al., 2016; Martin-Garcia et al., 2017; Melnikov et al., 2017), and one structure bound to the antagonist 8D1 (Sun et al., 2017) (Table 1). Conformational thermostabilization, which utilizes alanine scanning mutagenesis to identify point mutations that stabilize the receptor in a particular conformational state and increase its thermostability in detergent (Magnani et al., 2008, 2016), was also applied to solve the structure of A2AR bound to ZM241385 (Doré et al., 2011). The construct A2AR-StaR2 contained eight thermostabilizing mutations (A54L2.52, T88A3.36, R107A3.55, K1224.43, L202A5.63, L235A6.37, V239A6.41, and S277A7.42; superscripts refer to Ballesteros–Weinstein numbering) (Ballesteros and Weinstein, 1995) that increased the stability of the receptor in the detergent dodecylmaltoside (DDM) by ~18°C. A2AR-StaR2 has since been crystallized bound to four different antagonists XAC, caffeine, T4G and T4E (Doré et al., 2011; Congreve et al., 2012). Conformational thermostabilization has also been used in combination with a BRIL fusion protein to facilitate the crystallization of A2AR bound to the antagonists 6DY, 6DZ, 6DX, 6DV, ZM241385, caffeine, theophylline, and PSB36 (Segala et al., 2016; Cheng et al., 2017). Furthermore, ZM241385-bound A2AR has been co-crystallized in complex with an antibody Fab fragment (Fab2823), which acts as an intracellular inverse agonist locking the receptor in its inactive conformation, and also helps to increase the hydrophilic protein surface available for crystal contact formation (Figure 1B) (Hino et al., 2012).
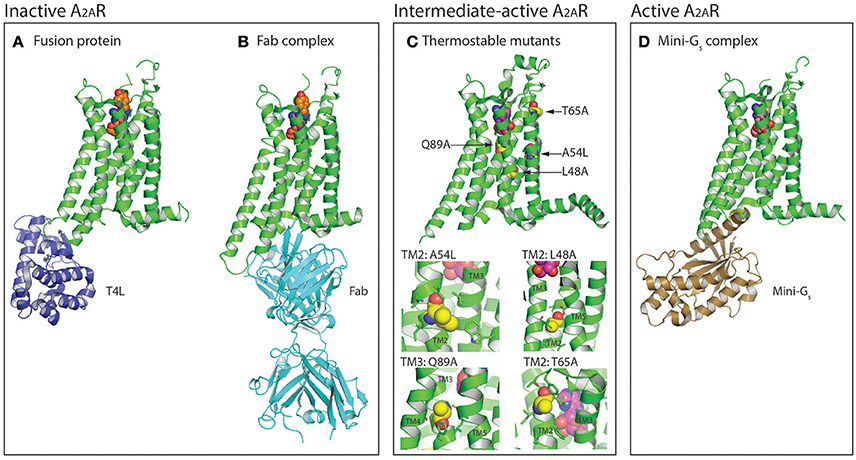
Figure 1. Protein engineering strategies used for the selection and crystallization of distinct A2AR conformations. (A) Crystal structure of A2AR (colored green) in the inactive conformation bound to the inverse agonist ZM241385 (shown as spheres, carbon atoms colored orange) with T4 lysozyme (T4L; colored blue) fused between the intracellular ends of H5 and H6 (PDB: 3EML) (Jaakola et al., 2008). The T4L fusion protein has also been substituted with apocytochrome b562RIL (BRIL; not shown) (Liu et al., 2012). (B) Structure of A2AR in the inactive conformation bound to ZM241385 (shown as spheres, carbon atoms colored orange) and an antibody Fab fragment (colored cyan) that acts as an intracellular inverse agonist (PDB: 3VG9 and 3VGA) (Hino et al., 2012). (C) Crystal structure of thermostabilized A2AR-GL26 in the intermediate-active conformation bound to the agonist adenosine (shown as spheres, carbon atoms colored magenta), the four thermostabilizing mutations (L48A2.46, A54L2.52, T65A2.63, and Q89A3.37) are shown as spheres and their carbon atoms colored yellow (PDB: 2YDO) (Lebon et al., 2011b). In the expanded view the residues with which the thermostabilizing mutations interact (distance <4 Å) are shown as sticks. Conformational thermostabilization has facilitated the crystallization of A2AR in both the agonist- and antagonist- bound conformations. (D) Structure of A2AR in the active conformation bound to NECA (shown as spheres, carbon atoms colored light pink) and an engineered G protein mini-Gs (colored brown; PDB: 5G53) (Carpenter et al., 2016). Figures were prepared using PyMOL (PyMOL™ Molecular Graphics System, Version 1.8.6.0).
The first agonist bound structure of A2AR was solved in complex with the synthetic agonist UK-432097, using the T4L fusion strategy (Xu et al., 2011). This ligand is large, approximately three times the molecular weight of adenosine, and imparts a significant increase in thermostability to the receptor. Crystallization of A2AR bound to smaller less stabilizing agonists required the application of the conformational thermostabilization methodology (Figure 1C). In this case the receptor was thermostabilized in the presence of the agonist NECA, and four mutations (L48A2.46, A54L2.52, T65A2.63, and Q89A3.37) were combined in the final construct (A2AR-GL26) (Lebon et al., 2011a). The stability of A2AR-GL26 was ~16°C higher than the wild type receptor in DDM, which facilitated structure determination of the receptor in complex with the small agonists adenosine and NECA (Lebon et al., 2011b), as well as the larger CGS21680 (Lebon et al., 2015). All four agonist-bound structures adopted a conformation that was different from the inactive state, but that did not fully resemble the active state of the β2 adrenergic receptor (β2AR) in complex with heterotrimeric Gs (Rasmussen et al., 2011b), this conformation was therefore defined as the intermediate-active state (Lebon et al., 2011b). Interestingly, the high affinity agonist UK-432097 was sufficiently stabilizing to allow the crystallization of non-thermostabilized A2AR in the intermediate-active state. This is likely due to the large size of the ligand, which results in it forming more extensive molecular contacts with the receptor, in particular UK-432097 forms four additional direct hydrogen bonds with A2AR compared to adenosine. In the case of small low affinity agonists, such as adenosine, conformational thermostabilization was absolutely necessary to facilitate structured determination.
The agonist-bound A2AR structures exhibited some characteristics of the active receptor (Lebon et al., 2012), however stabilizing the fully active conformation requires simultaneous binding of the agonist and G protein or a functional mimetic (Rasmussen et al., 2011a,b). Co-crystallization of GPCRs in complex with heterotrimeric G proteins is the ideal case for characterizing receptors in their active state, however this approach is challenging, due in part to the large size and conformationally dynamic nature of the G protein (Westfield et al., 2011). The development of nanobodies that act as surrogates of heterotrimeric G proteins has proved to be a powerful approach to crystallize receptors in their active conformation (Steyaert and Kobilka, 2011), however the disadvantage of this method is that nanobodies do not recapitulate the native GPCR–G protein interface. Structure determination of A2AR in its active conformation was achieved using a recently developed minimal G protein, which is composed of a single engineered domain from the Gαs subunit (Figure 1D) (Carpenter and Tate, 2016, 2017b,c). This mini G protein (mini-Gs) sufficiently stabilized NECA-bound A2AR in its fully active conformation to facilitate crystallization and structure determination at 3.4 Å resolution (Carpenter et al., 2016). This approach has now been applied to most heterotrimeric G proteins (Nehmé et al., 2017) and should play an important role solving high-resolution structures of other GPCRs in their active state (Strege et al., 2017).
The orthosteric binding site of A2AR can be defined by the residues involved in binding the endogenous agonist adenosine and the naturally occurring antagonist caffeine (see Figure 2 for the ligand structures and atom numbering, and Table 2 for A2AR-ligand contacts). Adenosine and caffeine share a common xanthine moiety that in both cases establishes van der Waals interactions with M1775.38, M2707.35 and I2747.39, and a π-stacking interaction with the aromatic ring of F168, which is part of the helical portion of extracellular loop 2 (ECL2; Figures 3, 4) (Doré et al., 2011; Lebon et al., 2011b; Cheng et al., 2017). N2536.55 forms hydrogen bonds with either the amine groups at positions C6 and N7 of the adenine moiety of adenosine (Lebon et al., 2011b) or O11/O13 from the xanthine heterocycle of caffeine, for which two distinct binding orientations have been observed (Cheng et al., 2017). The ribose moiety of adenosine forms van der Waals interactions with V843.32, L853.33, T883.36, W2466.48, and L2496.51; these residues form similar contacts with many antagonist. The main difference between the adenosine- and caffeine-binding modes is the formation of hydrogen bonds from the hydroxyl groups at positions C2 and C3 of the ribose moiety of adenosine to S2777.42 and H2787.43. The recent high-resolution structure of A2AR bound to caffeine (2.10 Å) did reveal a water-mediated contact between H2787.43 and O11 of caffeine (Cheng et al., 2017). However, the presence of this water molecule highlights the difference in the distance between these atoms compared to the adenosine-bound state, where a direct interaction is observed. This distance is reduced concomitantly with the conformational changes in H3 and H7 associated with agonist binding to the receptor (Figure 5) (Lebon et al., 2012). All agonists that have been co-crystallized with A2AR engage H3 through T883.36 and H7 through S2777.42/H2787.43 (Table 2). Some antagonists do interact with either T883.36 or S2777.42/H2787.43, but never at the same time, which suggests that the simultaneous engagement of these residues in H3 and H7 may be a key determinant of agonist activity.
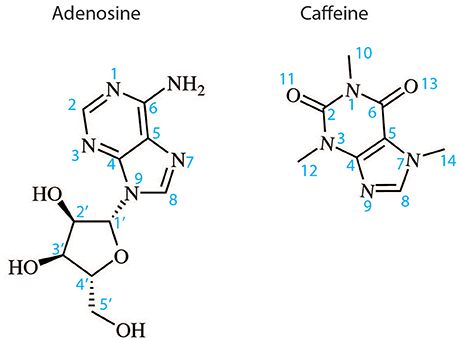
Figure 2. Two dimensional structures of the endogenous A2AR agonist adenosine and the naturally occurring antagonist caffeine. The atom numbering indicated is used in the text to describe the binding of these ligands and their derivatives to A2AR.
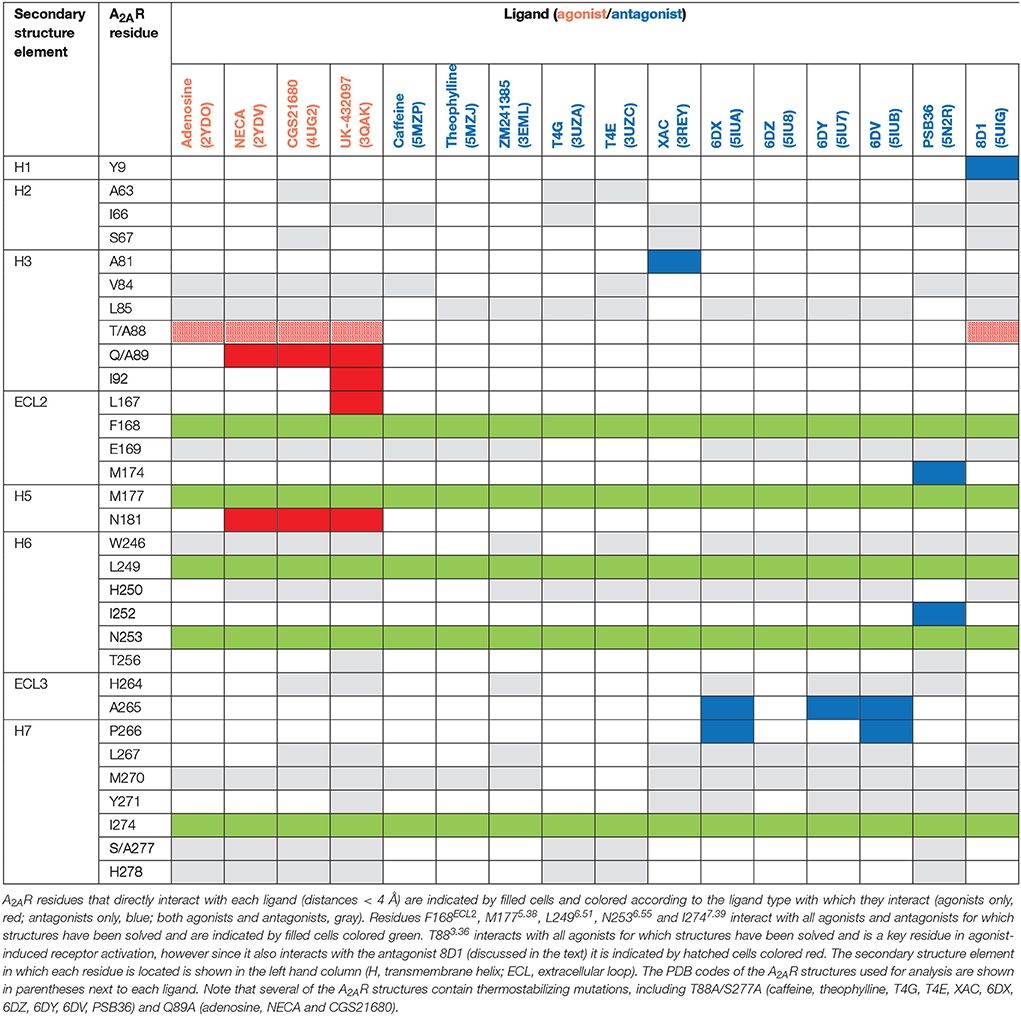
Table 2. A2AR-ligand interactions.
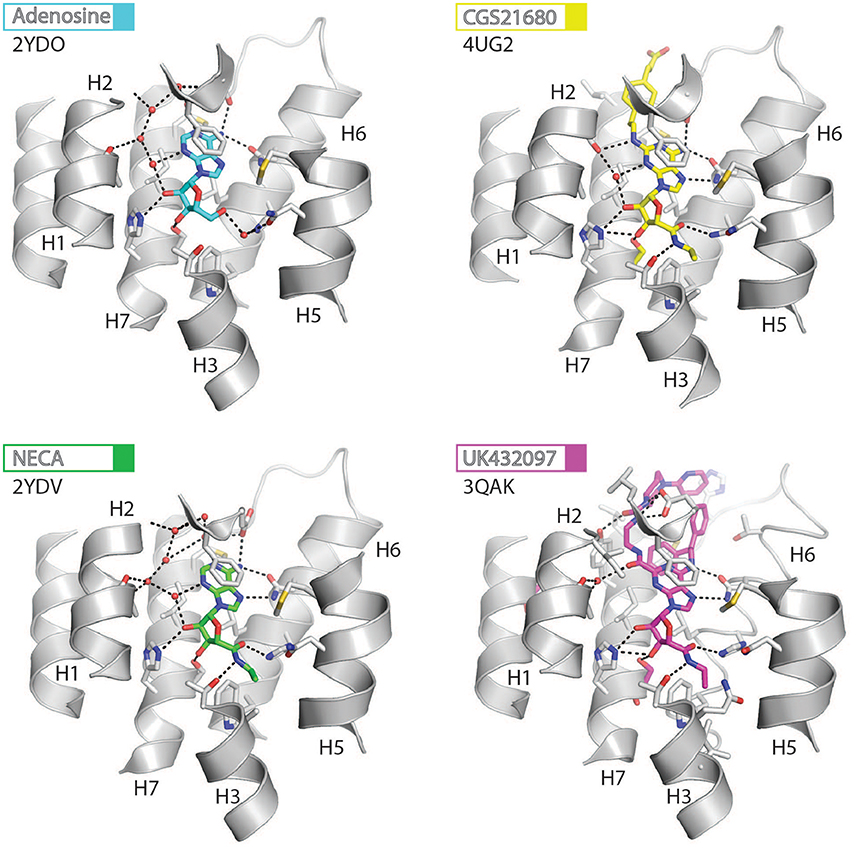
Figure 3. High-resolution view of the agonist-binding site of human A2AR. Four different agonists have been co-crystallized with A2AR in the intermediate-active conformation. The receptor is shown as cartoons and colored gray, residues and side chains that interact with the ligand are shown as sticks and colored by element (carbon, gray; nitrogen, blue; oxygen, red; sulfur, yellow). The ligands are shown as sticks and their carbon atoms are colored to match their labels, PDB codes are shown in the figure. Polar contacts are represented as dashed lines and water molecules are shown as red spheres.
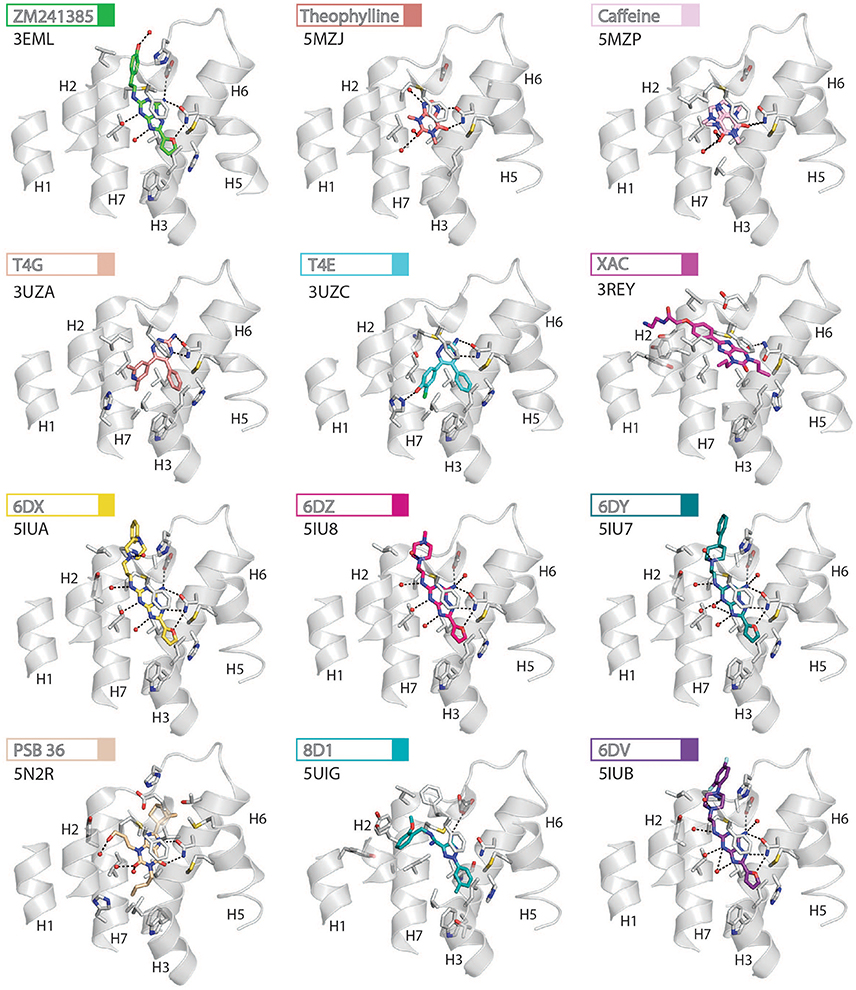
Figure 4. High-resolution view of the antagonist- or inverse agonist- binding site of human A2AR. Twelve different antagonists or inverse agonists have been co-crystallized with A2AR, in the inactive conformation. The receptor is shown as cartoons and colored gray, residues and side chains that interact with the ligand are shown as sticks and colored by element (carbon, gray; nitrogen, blue; oxygen, red; sulfur, yellow). The ligands are shown as sticks and their carbon atoms are colored to match their labels, PDB codes are shown in the figure. Polar contacts are represented as dashed lines and water molecules are shown as red spheres. Note that for caffeine the two distinct binding orientations that were observed in the structure are overlaid.
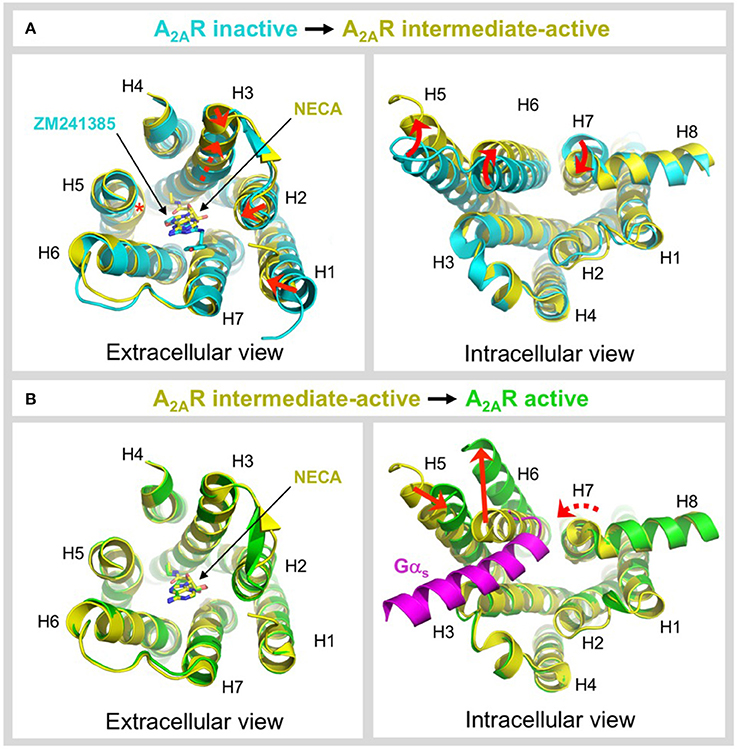
Figure 5. Ligand-induced activation of A2AR. (A) Conformational changes associated with agonist-induced transition from the inactive state (colored cyan) to the intermediate-active state (colored yellow; PDB: 2YDV) (Lebon et al., 2011b). Two different inactive state structures were used for the alignments, the extracellular view uses 4EIY (Liu et al., 2012) because it is the highest resolution non-thermostabilized structure available, the intracellular view uses 3PWH (Doré et al., 2011) because it was crystallized without a fusion protein in ICL3. The extracellular view shows the 1–2 Å inward shifts in the ends of H1, H2, and H3 (indicated by a red arrows), the 2 Å translocation of H3 along its axis (indicated by a dashed red arrow), and the inward bulge in H5 (indicated by a red asterisk). The inverse agonist ZM241385 (colored cyan) and agonist NECA (colored yellow) are shown as sticks. The intracellular view shows the combined rotational and lateral movements in H5, H6, and H7 (indicated by curved red arrows). H5 and H6 are displaced outwards by 7 and 5 Å, respectively, while H7 moves inwards in by 4 Å. (B) Conformational changes associated with G protein-induced transition from the intermediate-active state (colored yellow; PDB: 2YDV) (Lebon et al., 2011b) to the active state (colored green; PDB: 5G53) (Carpenter et al., 2016). No significant changes are observed on the extracellular side of the receptor, and the position of NECA (shown as sticks) is essentially identical in both states (Carpenter et al., 2016). On the intracellular side of the receptor an outward movement of H6 by 14 Å (indicated by a red arrow) is required to accommodate binding of mini-Gs (colored magenta). H5 moves inwards by 5 Å (indicated by a red arrow) and forms direct contacts the α5 helix of mini-Gs. H7 undergoes a rotation, without significant lateral movement (indicated by a curved and dashed red arrow), which reorients the H7-H8 boundary to interact with the C-terminus of mini-Gs (Carpenter et al., 2016). Note that for clarity, ECL2 has been omitted from the extracellular view of all alignments.
Structures of A2AR have been solved in complex with three high-affinity synthetic agonists NECA, CGS21680 and UK-432097 (Lebon et al., 2011b, 2015; Xu et al., 2011). These agonists share a core adenosine moiety, the binding mode of which is very similar to adenosine itself, with the ribose group establishing hydrogen bonds with S2777.42 and H2787.43. For all three ligands the N-ethylcarboxyamido tail at position C5′ of the ribose ring extends deep into the binding pocket. The nitrogen and oxygen atoms form polar contacts with T883.36 and H2506.52, respectively, and the substituent is further stabilized by van der Waals interactions with N1815.42, W2466.48 and to a lesser extent Q/A3.37. In contrast, the OH group at this position of adenosine interacts with N1815.42 and H2506.52 through water-mediated interactions and M1775.38 through a van der Waals interaction. The major difference between the binding mode of these agonists can be seen in the extracellular loops of the receptor. NECA and adenosine are both stabilized by a hydrogen bond with E169ECL2 (Lebon et al., 2011b); this residue also forms a salt bridge with H264ECL3, which closes the top of the binding pocket and is known to affect ligand binding kinetics (Guo et al., 2016; Segala et al., 2016). CGS21680 takes advantage of similar hydrogen bond between the amine group at position C6 of adenine moiety and E169ECL2 (Lebon et al., 2015). The larger (2-carboxyethyl)phenylethylamino substituent at the C2 position protrudes outside the binding pocket and is stabilized by van der Waals interactions with E169ECL2, H264ECL3, and L2677.32 as well as a hydrogen bond with S672.65. As a consequence, the extracellular end of H2 is displaced inward compared to other agonist-bound structures, reducing the volume of the binding pocket. UK-432097 has two substituents on the adenine moiety, that make it an even larger molecule than CGS21680. The consequence is that the bulky 2-(3-[1-(pyridine-2-yl)piperidin-4-yl]ureido)ethylcarboxamido substitution at position C2 displaces ECL3 away from the binding pocket, which induces rotamer changes in E169ECL2 and H264ECL3 and breaks the salt bridge between these side chains (Xu et al., 2011). The urea group forms two hydrogen bonds with E169ECL2 as well as a hydrogen bond and van der Waals interactions with Y2717.36 on its opposite side. The pyridinyl-piperidine group, which extends furthest from the binding pocket, is stabilized by van der Waals interactions with L2677.32 and H264ECL3.
Like agonists, antagonists can exploit subsidiary binding sites by expanding their contact surface outside the orthosteric binding pocket. Starting with ZM241385, the trizolotriazine ring occupies the orthosteric binding site, and is surrounded by F168ECL2, L2496.51, M2707.35, I2747.39. Two residues, E169ECL2 and N2536.55, form hydrogen bonds with the amine group of the ZM241385 heterocycle and an additional hydrogen bond is established between N2536.55 and the oxygen of the furan ring. The formation of van der Waals interactions with M1775.38, W2466.48, L2496.51, and H2506.52 stabilize H5 and H6 against the furan ring. ZM241385 explores the chemical space outside the orthosteric site by taking advantage of the cavity on the extracellular surface of the receptor. Two distinct orientations have been observed for the phenol ring of ZM241385, in the first conformation the salt bridge between E169ECL2 and H264ECL3 is intact and the phenol ring forms van der Waals interactions with H264ECL3, L2677.32 and M2707.35 (Jaakola et al., 2008). Interestingly, the phenylethylamine group of ZM241385 adopts a binding mode similar to the (2-carboxyethyl)phenylethylamino substituent of CGS21680 (Lebon et al., 2015). The second conformation was observed in a thermostabilized receptor structure where the salt bridge between E169ECL2 and H264ECL3 is broken (Doré et al., 2011). In this case the phenol moiety is pointing toward H1 and H2 engaging S672.65, Y2717.36, and I2747.39 through van der Waals interactions and A632.61 through a hydrogen bond. Both Y91.35 and Y2717.36 sidechains adopt different rotamers in order to accommodate the phenol substituent in this pose, demonstrating that this subsidiary pocket is conformationally malleable. The xanthine derivative XAC occupies the orthosteric pocket with the xanthine ring adopting a similar position to caffeine and the trizolotriazine heterocycle of ZM241385 (Doré et al., 2011). However, the xanthine core is branched at position N1 and N3; the N3 propyl substituent forms van der Waals interactions with I662.64 and A813.29 that are not observed for ZM241385. On the extracellular surface of the receptor the phenoxy-acetamide tail of XAC adopts a pose that is similar to the phenol group of ZM241385 in the thermostabilized receptor structure and is engaged by S672.65, L2677.32, M2707.35, Y2717.36, and I2747.39. However, none of these interactions are unique to XAC since they are all observed for ZM241385 in either of its two poses. The recent structure of 8D1 (also known as compound-1) bound to A2AR provides another example of a ligand that exploits this subsidiary binding site (Sun et al., 2017). The methoxy phenyl substituent of 8D1 occupies the extracellular pocket formed by Y91.35, A632.61, I662.64, S672.65, L2677.32, M2707.35, Y2717.36, and I2747.39 in a similar way to XAC and ZM241385. In this case a unique interaction is observed between 8D1 and Y91.35 that is not observed for any other ligand (Table 2). Thus, several different ligands and chemical substituents have been shown to take advantage of the subsidiary binding pocket located between H1, H2, and H7 on the extracellular surface of the receptor.
Cavities identified from high-resolution crystal structures provide valuable information for structure-based drug discovery. This can be best illustrated by the study of Congreve et al. who have reported the discovery of 1,2,4-triazine derivatives as A2AR antagonists by exploiting structural data (Congreve et al., 2012). The authors optimized a series of compounds and hypothesized that 1,2,4-triazine derivatives may occupy the same area of the binding pocket as the ribose moiety of agonists. Solution of the corresponding ligand–receptor structures showed that the triazine ring of T4E or T4G sits in a pocket similar to other ligands, and forms van der Waals interactions with F168ECL2 and two hydrogen bonds with N2536.55. The phenol substituent forms van der Waals interactions with L853.33, M1775.38, W2466.48 L2496.51, and H2506.52. As predicted the dimethyl-pyridine group of T4G occupies the ribose pocket and the phenolic hydroxyl group of T4E establishes a hydrogen bond with H2787.43, but importantly the ligands retain their antagonistic properties.
Crystallographic studies have also highlighted the role that ECL2 and ECL3 play in ligand binding, specifically the effect of the salt bridge between E169ECL2 and H264ECL3 (Lebon et al., 2015; Guo et al., 2016; Segala et al., 2016). These two residue as well as the salt bridge that they form, stabilize several ligands in the binding pocket, as described above for ZM241385. The phenol ring of the ZM241385 has recently been replaced by a set of larger substitutions and their structures have been solved in complex with A2AR at high resolution (Segala et al., 2016). The substitutions were reported to affect the residence time of the ligands, with the ligand 6DV (also known as 12x) displaying the slowest off-rate. This observation is also in agreement with the agonist-bound structures where the salt bridge closes the binding site for adenosine, NECA and CGS21680 (Lebon et al., 2011b, 2015). Mutation of either E169ECL2 or H264ECL3 has been shown to impair the potency of NECA whereas only mutation of H264ECL3 affected CGS21680 (Lebon et al., 2015). The lower potency of agonists on these mutant receptors might be a consequence of a faster off-rate in absence of the salt bridge, however it appears that the stabilizing effect of the salt bridge can be compensated for by extended molecular contact formed by large molecules, such as CGS21680.
GPCRs exist in dynamic equilibrium between several discrete conformational states that are separated by energy barriers (Manglik et al., 2015; Ye et al., 2016). The inactive and active states are well-conserved between GPCRs (Rosenbaum et al., 2009; Carpenter and Tate, 2017a), however a number of intermediate conformations have been identified that appear to be more divergent (Lebon et al., 2011b; Xu et al., 2011; White et al., 2012; Manglik et al., 2015; Ye et al., 2016). Agonist binding to the receptor is one of the key events required to overcome the energy barrier of activation and increase occupancy of the conformational state(s) that are capable of binding heterotrimeric G proteins (Manglik et al., 2015; Ye et al., 2016; Prosser et al., 2017). A2AR is one of the only receptors for which an intermediate-active agonist-bound state has been crystallized (Lebon et al., 2011b; Xu et al., 2011). This structure has provided unique insight into the molecular changes that occur during two key activation events, namely agonist-induced transition from the inactive to intermediate-active state and G protein-induced transition from the intermediate-active to active state. The structures used for comparison are the inactive state bound to the inverse agonist ZM241385 (Doré et al., 2011; Liu et al., 2012), the intermediate-active state bound to the agonist NECA (Lebon et al., 2011b) and the active state bound to NECA and mini-Gs (Carpenter et al., 2016), see Figure 5. Agonist binding to A2AR triggers a series of conformational changes, most notably within the ligand-binding pocket and on the intracellular side of the receptor (Figure 5A). In the ligand binding pocket the most significant changes are a 2 Å translocation of H3 along its axis that is necessary to prevent steric clashes of V843.32 and L853.33 with NECA, the formation of contacts between the ribose moiety of the agonist and residues S2777.42 and H2787.43 in H7 that are completely absent in the inverse agonist-bound state, and an inward bulge in H5 that disrupts the local helix geometry and shifts C1855.46 toward the core of the receptor by 4 Å (Lebon et al., 2011b). Notably, the bulge in H5 has a knock-on effect on the position of H2506.52, which is shifted toward the ligand by 2 Å, a movement that would be sterically forbidden if the inverse agonist were bound (Lebon et al., 2011b, 2012; Xu et al., 2011). This bulge is also observed in other GPCR structures suggesting it is one of the key event in activation (Venkatakrishnan et al., 2013). Agonist binding causes only subtle conformational changes on the extracellular side of the receptor, including 1-2 Å inward shifts in the ends of H1, H2, and H3. More significant changes are observed on the intracellular side, which are thought to prime the receptor for G protein coupling (Venkatakrishnan et al., 2016), specifically combined rotations and lateral movements in the cytoplasmic ends of H5, H6, and H7, outwards by 7 and 5 Å for H5 and H6, respectively, and inwards by 4 Å for H7 (Figure 5A) (Lebon et al., 2011b; Xu et al., 2011). The ionic lock between R1023.50 and E2286.30, which is often engaged in inactive GPCR structures, is broken by the reorientation of H6.
Sodium ions (Na+) act as negative allosteric modulators of many class A GPCRs, typically stabilizing the ligand-free and antagonist-bound states, thereby imposing an energy barrier on receptor activation (Katritch et al., 2014). The high-resolution model of A2AR in its inactive state was the first GPCR structure to reveal the mode of Na+ binding (Liu et al., 2012). In A2AR the Na+-binding pocket is composed of residues D522.50, S913.39, T883.36, W2466.48, N2807.45, and S2817.46; both D522.50 and S913.39 form direct polar interactions with Na+, whereas interactions with the other residues are mediated through a network of ordered water molecules (Liu et al., 2012; Gutiérrez-de-Terán et al., 2013). The conformational changes induced by agonist binding, particularly the rotation and inward shift of H7 and the outward shift of H6, cause the Na+-binding pocket to collapse, potentiating the displacement of Na+, thereby allowing the agonist to overcome the negative allosteric effect of Na+ and activate the receptor (Gutiérrez-de-Terán et al., 2013). Interestingly, the intermediate-active conformation is not observed in crystal structures of agonist-bound β1AR (Warne et al., 2011) or β2AR (Rosenbaum et al., 2011). Recent 19F nuclear magnetic resonance (19F NMR) studies have confirmed the existence of an additional active-like state in A2AR compared to β2AR (Manglik et al., 2015; Ye et al., 2016; Prosser et al., 2017), which may correlate with the intermediate-active conformation observed in the crystal structures, thus highlighting differences in the energy landscape of activation between these receptors.
In contrast to the widely distributed effects of agonist binding, G protein-induced conformational changes are confined to the intracellular side of the receptor (Figure 5B). G protein coupling has been reported to increase the agonist-binding affinity of A2AR between 10- and 40-fold (Murphree et al., 2002; Carpenter et al., 2016), yet it is striking that the conformation of the residues in the ligand-binding pocket and the position of the agonist are essentially identical in both the intermediate-active and active states (Carpenter et al., 2016). The most likely explanation is that in the intermediate-active structure the ligand-binding pocket has already adopted the high-affinity conformation (Carpenter et al., 2016). However, it cannot be discounted that the relatively small increase in the agonist-binding affinity of A2AR does not result from direct changes to the ligand-binding pocket, for example, a reduction in the conformational dynamics of the receptor caused by G protein binding could decrease the off-rate of the agonist, thus increasing its affinity (Carpenter and Tate, 2017a). On the intracellular side of the receptor the pivotal event is a 14 Å outward movement of the cytoplasmic end of H6 to accommodate binding of the α5 helix from mini-Gs. The outward movement of H6 triggers both a 5 Å inward movement of H5, positioning it to interact with the α5 helix of the G protein, and a rotation within H7 that reorients the H7-H8 boundary to form extensive contacts with the C-terminus of mini-Gs (Carpenter et al., 2016). These helix movements also result in reorientation of R1023.50, Y1975.58, and Y2887.53 side chains within the core of the receptor, which likely stabilizes the active state, and which is one of the signatures of a GPCR in its active conformation (Carpenter and Tate, 2017a). It is clear that the intermediate-active state of A2AR is incompatible with G protein binding in its final orientation, i.e., that observed in the A2AR–mini-Gs structure (Carpenter et al., 2016), due to a large sterically forbidden clash between the α5 helix of mini-Gs and H6 of the receptor (Figure 5B). However, it is possible that the intermediate-active state of A2AR may be responsible for recognition of the G protein through an initial docking interaction, before cooperative conformational changes trigger nucleotide dissociation from the G protein and drive the receptor into its active conformation (Rasmussen et al., 2011b; Flock et al., 2015; Carpenter et al., 2016).
The amino acid sequence of the four AR subtypes is relatively poorly conserved, A2AR shares only 49, 56, and 39% identity with A1R, A2BR, and A3R, respectively (aligned over residues 1-312 of A2AR). This means that, despite there being a wealth of structural data available for A2AR, it has proved challenging to homology model other AR subtypes with sufficient accuracy for structure-based drug design applications (Glukhova et al., 2017). It is only during the past year that structures of an AR other than A2AR have been published, namely two structures of A1R bound to the xanthine antagonists DU172 (Glukhova et al., 2017) and PSB36 (Cheng et al., 2017). The two A1R structures are closely related and align with an RMSD of 0.6 Å (over 235 Cα atoms), they also align well with the ZM241385-bound structure of A2AR (Liu et al., 2012), with RMSDs of 0.8 Å (over 238 Cα atoms) and 1.0 Å (over 234 Cα atoms) for the DU172- and PSB36- bound structures, respectively. The intracellular side of both A1R structures strongly resemble the ZM241385-bound A2AR structure (3PWH), which was crystallized without a fusion protein in ICL3 (Doré et al., 2011), and the ionic lock between residues R1053.50 and E2296.30 is engaged in both cases (Cheng et al., 2017; Glukhova et al., 2017). The organization of the sodium-binding site is also well-conserved between A1R and A2AR, which supports mutagenesis data that indicated the negative allosteric effect of sodium on A1R was mediated through this site (Barbhaiya et al., 1996), although neither A1R structure was of sufficient resolution to conclusively model the sodium ion.
The most striking differences between A1R and A2AR are the conformational variations in extracellular ends of H1, H2, H3, and H7 and the orientation of ECL2. In the DU172-bound structure H3 is displaced inwards by 4 Å, and H1, H2, and H7 are displaced outwards by 5, 4, and 4 Å, respectively (Figure 6A). The outward movements in H1, H2, and H7 are required to accommodate the benzene sulfonate group of DU172, which is covalently linked to Y2717.36, and result in both the expansion of the orthosteric site and the formation of a secondary allosteric pocket (Glukhova et al., 2017). Direct comparison of A1R and A2AR bound to PSB36, which does not contain the benzene sulfonate substituent, also reveals a partial expansion of the orthosteric pocket (Cheng et al., 2017), indicating that this region of A1R is indeed more conformationally malleable than that of A2AR. Intriguingly, it has been suggested that the conformational rearrangements in H1, H2, and H3 in A1R may be a direct result of the different disulphide bond structure of ECL2 (Glukhova et al., 2017). In both A1R structures ECL2 adopts a similar conformation, which is different from that observed in any of the published A2AR structures (Figure 6B). The helical segment in ECL2 of A1R is extended by five residues and is positioned almost perpendicular to the transmembrane helices, compared to the near parallel arrangement in A2AR (Cheng et al., 2017; Glukhova et al., 2017). There is only a single disulphide bond in ECL2 of A1R compared to three within the same region of A2AR, which results in it adopting an extended conformation (Figure 6B). This appears to reduce conformational constraints on the extracellular ends of H2 and H3 allowing them to be displaced outwards, which in turn influences the positioning of H1. The displacement of H7 may also be linked to structural divergence in the extracellular loops, in this case a single amino acid truncation in ECL3 of A1R has been proposed to induce the outward tilt observed in the structures (Cheng et al., 2017; Glukhova et al., 2017).
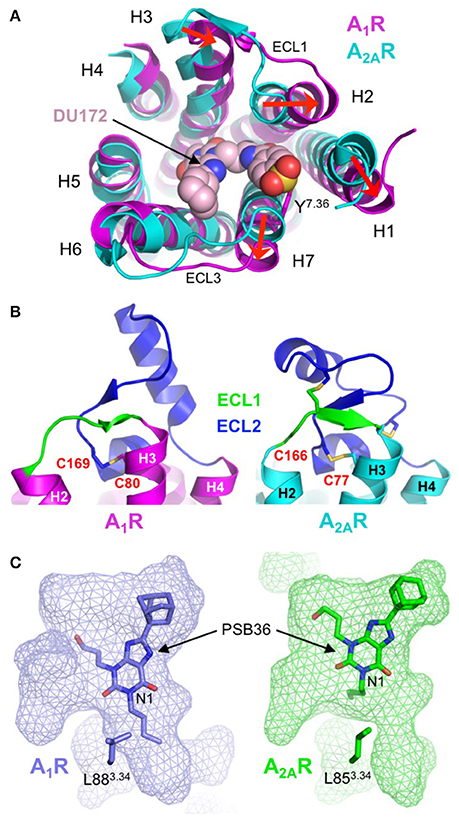
Figure 6. Structural diversity of the adenosine receptor family. (A) Extracellular view of the conformational differences between DU172-bound A1R (colored magenta; PDB: 5UEN) (Glukhova et al., 2017) and ZM241385-bound A2AR (colored cyan; PDB: 4EIY) (Liu et al., 2012). The extracellular ends of H1, H2 and H7 are displaced outwards by 5, 4, and 4 Å, respectively and H3 is displaced inwards by 4 Å (indicated by red arrows). The antagonist DU172 (shown as spheres) is covalently attached to Y2717.36 (shown as sticks) through a benzene sulfonate linkage. Note that for clarity, ECL2 has been omitted from the alignment. (B) Differential conformations of ECL1 (colored green) and ECL2 (colored blue) in A1R (colored magenta; PDB: 5UEN) (Glukhova et al., 2017) and A2AR (colored cyan; PDB: 4EIY) (Liu et al., 2012), disulphide bonds are shown as sticks. In A1R ECL2 adopts an extended conformation that is stabilized by a single disulphide bond (C803.25-C169ECL2). In contrast, ECL2 from A2AR adopts a compact conformation that is stabilized by three disulphide bond, one of which (C773.25-C166ECL2) is conserved in A1R. (C) Surface representation of the ligand-binding pocket (rendered as semi-transparent mesh) in A1R (colored blue; PDB: 5N2R) (Cheng et al., 2017) and A2AR (colored green; PDB: 5N2S) (Cheng et al., 2017), which highlights differences in both the topology of the orthosteric site and the binding orientation of PSB36 (shown as sticks). The butyl substituent at position N1 of the xanthine core of PSB36 fits into a channel between H3, H5, and H6 in A1R, this channel is constricted in A2AR due to the different orientation of L853.34 (shown as sticks), which results in the ligand binding in a different orientation.
What do these structures tell us about the molecular determinants of ligand-binding specificity in different AR subtypes? First, sequence differences in the binding pocket do not appear to be the main determinant of ligand-binding specificity in ARs. The orthosteric binding pocket of A1R and A2AR in the PSB36-bound structures differ by only four residues V622.57, N702.65, E170ECL2, and T2707.35 (corresponding to A592.57, S672.65, L170ECL2, and M2707.35 in A2AR), and of these, only T/M2707.35 form direct contacts with the ligand (Cheng et al., 2017). Mutagenesis studies have shown that residue 2707.35 is important in ligand-binding specificity, introducing the T270M mutation into A1R resulted in decreased binding affinity of A1R-specific ligands and increased binding affinity of A2AR-specific ligands, the reverse effect was observed when the M270T mutation was introduced into A2AR (Cheng et al., 2017; Glukhova et al., 2017). However, the positioning of this residue on the extracellular end of H7, at the perimeter of the binding pocket, suggests its main role is to act as a “gatekeeper” that regulates ligand access to the orthosteric site (Glukhova et al., 2017). Second, the topology of the binding pocket appears to play a central role in ligand-binding specificity. As described above the disulphide bond structure of ECL1 and ECL2 in A1R increases mobility in H1, H2 and H3, which causes an expansion of the binding pocket that is required to accommodate the benzene sulfonate group of the A1R-selective ligand DU172 (Figure 6A) (Glukhova et al., 2017). Binding pocket topology is also the predominant factor in the differential binding modes of PSB36 between A1R and A2AR (Figure 6C). PSB36 binds 2 Å deeper in the orthosteric pocket of A1R due to the presence of a channel between H3, H5 and H6 that can accommodate the butyl substituent at position N1 of the xanthine core (Figure 6C) (Cheng et al., 2017). This channel is created by a 2 Å displacement of L883.34 in A1R that appears to be the result of an upstream proline residue (P863.31), which is located outside the binding pocket, distorting the helix geometry in this region of H3. A proline at this position is unique to A1R and helps to explain why substitutions at position N1 of the xanthine core contribute to A1R selectivity (Cheng et al., 2017). Thus, the amino acid sequence both inside and outside the ligand-binding site, the extracellular loop structure and the topology of the binding pocket play interconnected roles in governing the ligand binding affinity and kinetics that are ultimately responsible for the functional selectivity of AR subtypes.
A decade of research and innovation has culminated in the crystallization of more than 30 structures of human A2AR in complex with one inverse agonist, 11 antagonists and four agonists, as well as an engineered G protein. These structures represent the inactive, intermediate-active and active conformational states, and A2AR remains the only receptor for which this complete series of structures has been reported. Most of the structure were obtained using high affinity ligands, such as ZM241385, XAC, NECA, UK-432097, CGS21680. However, the application of conformational thermostabilization has also facilitated structure determination of the receptor in complex with lower affinity ligands, including the endogenous agonist adenosine and the natural plant-derived antagonists caffeine and theophylline. Structural characterization of the ligand-binding pocket of A2AR has provide novel insight into the binding modes of different classes of ligands; the ribose moiety has been identified as a key component of agonists that helps to stabilize the intermediate-active state before the receptor can adopt the fully active conformation upon G protein coupling. The chemical diversity of compounds co-crystallized with A2AR has also revealed how some ligands can exploit subsidiary binding sites on the extracellular surface of the receptor, as exemplified by the antagonists XAC, ZM241385, 8D1, T4G, and T4E and agonists CGS21680 and UK-432097.
High-resolution structures have not only provided a clear picture of the ligand-binding pocket, but have also highlighted the impact of receptor flexibility, notably in ECL2 and ECL3, on the mode and kinetics of ligand binding. Furthermore, the recently solved structures of A1R revealed that binding pocket topology and extracellular loop structure are two of the most important factors affecting the ligand binding specificity of different AR subtypes. These observations highlight the challenges of homology modeling GPCRs, since differential extracellular loop structures, global helix movements and changes in binding pocket topology are more difficult to model than amino acid substitutions within the orthosteric site. Thus, continued efforts to experimentally determine structures of all four AR subtypes in the three distinct activation states are essential to maximize the potential of structure based drug design for this family of receptors. Interestingly, despite the fact that all ARs are known to signal through G protein-independent pathways, no biased ligands have thus far been reported for A2AR (Verzijl and Ijzerman, 2011). Functional selectivity has now been observed in A1R, A2BR, and A3R (Gao et al., 2014; Baltos et al., 2016a,b), which suggests that biased ligands could also be developed for A2AR. However, it is also possible that subtle differences in the energy landscape and mechanism of A2AR activation may mean that biased signaling is less pronounced for this receptor. Therefore, at present A2AR is not an ideal model for studying functional selectivity, however structural insight from other GPCRs that have been co-crystallized with biased agonists, such as β1AR (Warne et al., 2012), may yet help to facilitate the design biased ligands for A2AR.
Finally, how will the wealth of high-quality structural data reported for A2AR shape the future of drug development for this receptor? Structural based design has already been used to develop novel A2AR antagonists, including a 1,2,4-triazine derivative that is a preclinical candidate for the treatment of Parkinson's disease (Congreve et al., 2012). Further application of this approach will likely result in the identification of additional novel compounds, both agonists and antagonists, and will also facilitate the derivatization and optimization of existing ligands. The identification of the subsidiary binding pocket on the extracellular surface of A2AR is an important step toward the design of allosteric modulators. Exploitation of this pocket has thus far been achieved using orthosteric ligands with large substituents that extend outside the orthosteric site, however in the future it may also be possible to target this region using purely allosteric ligands. Such compounds have the potential to modulate the properties of orthosteric ligands, and could thus be used to fine tune adenosine signaling through individual AR subtypes.
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
BC was supported by funding from a BBSRC/EPSRC grant awarded to WISB (BB/M017982/1). GL was supported by the ATIP-AVENIR program, the CNRS, INSERM and Montpellier University. We thank Chris Tate for comments on the manuscript.
Ballesteros, J. A., and Weinstein, H. (1995). Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 25, 366–428. doi: 10.1016/S1043-9471(05)80049-7
Baltos, J. A., Gregory, K. J., White, P. J., Sexton, P. M., Christopoulos, A., and May, L. T. (2016a). Quantification of adenosine A(1) receptor biased agonism: implications for drug discovery. Biochem. Pharmacol. 99, 101–112. doi: 10.1016/j.bcp.2015
Baltos, J. A., Paoletta, S., Nguyen, A. T., Gregory, K. J., Tosh, D. K., Christopoulos, A., et al. (2016b). Structure-activity analysis of biased agonism at the human Adenosine A3 receptor. Mol. Pharmacol. 90, 12–22. doi: 10.1124/mol.116.103283
Barbhaiya, H., McClain, R., Ijzerman, A., and Rivkees, S. A. (1996). Site-directed mutagenesis of the human A1 adenosine receptor: influences of acidic and hydroxy residues in the first four transmembrane domains on ligand binding. Mol. Pharmacol. 50, 1635–1642.
Batyuk, A., Galli, L., Ishchenko, A., Han, G. W., Gati, C., Popov, P. A., et al. (2016). Native phasing of x-ray free-electron laser data for a G protein-coupled receptor. Sci. Adv. 2:e1600292. doi: 10.1126/sciadv.1600292
Burnstock, G. (2006). Purinergic signalling. Br. J. Pharmacol. 147(Suppl. 1), S172–S181. doi: 10.1038/sj.bjp.0706429
Caffrey, M. (2015). A comprehensive review of the lipid cubic phase or in meso method for crystallizing membrane and soluble proteins and complexes. Acta Crystallogr. F Struct. Biol. Commun. 71(Pt. 1), 3–18. doi: 10.1107/S2053230X14026843
Carpenter, B., and Tate, C. G. (2016). Engineering a minimal G protein to facilitate crystallisation of G protein-coupled receptors in their active conformation. Protein Eng. Des. Sel. 29, 583–594. doi: 10.1093/protein/gzw049
Carpenter, B., and Tate, C. G. (2017a). Active state structures of G protein-coupled receptors highlight the similarities and differences in the G protein and arrestin coupling interfaces. Curr. Opin. Struct. Biol. 45, 124–132. doi: 10.1016/j.sbi.2017.04.010
Carpenter, B., and Tate, C. G. (2017b). Expression and purification of mini G proteins from Escherichia coli. Bio Protoc. 7:e2235. doi: 10.21769/BioProtoc.2235
Carpenter, B., and Tate, C. G. (2017c). Expression, purification and crystallisation of the Adenosine A2A receptor bound to an engineered mini G protein. Bio Protoc. 7:e2234. doi: 10.21769/BioProtoc.2234
Carpenter, B., Nehmé, R., Warne, T., Leslie, A. G., and Tate, C. G. (2016). Structure of the adenosine A2A receptor bound to an engineered G protein. Nature 536, 104–107. doi: 10.1038/nature18966
Chen, J. F., Eltzschig, H. K., and Fredholm, B. B. (2013). Adenosine receptors as drug targets–what are the challenges? Nat. Rev. Drug Discov. 12, 265–286. doi: 10.1038/nrd3955
Cheng, R. K. Y., Segala, E., Robertson, N., Deflorian, F., Doré, A. S., Errey, J. C., et al. (2017). Structures of human A1 and A2A adenosine receptors with xanthines reveal determinants of selectivity. Structure 25:e1274. doi: 10.1016/j.str.2017.06.012
Cherezov, V., Rosenbaum, D. M., Hanson, M. A., Rasmussen, S. G., Thian, F. S., Kobilka, T. S., et al. (2007). High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318, 1258–1265. doi: 10.1126/science.1150577
Chun, E., Thompson, A. A., Liu, W., Roth, C. B., Griffith, M. T., Katritch, V., et al. (2012). Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure 20, 967–976. doi: 10.1016/j.str.2012.04.010
Cieslak, M., Komoszynski, M., and Wojtczak, A. (2008). Adenosine A(2A) receptors in Parkinson's disease treatment. Purinergic Signal. 4, 305–312. doi: 10.1007/s11302-008-9100-8
Congreve, M., Andrews, S. P., Doré, A. S., Hollenstein, K., Hurrell, E., Langmead, C. J., et al. (2012). Discovery of 1,2,4-triazine derivatives as adenosine A2A antagonists using structure based drug design. J. Med. Chem. 55, 1898–1903. doi: 10.1021/jm201376w
Doré, A. S., Robertson, N., Errey, J. C., Ng, I., Hollenstein, K., Tehan, B., et al. (2011). Structure of the adenosine A2A receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure 19, 1283–1293. doi: 10.1016/j.str.2011.06.014
Flock, T., Ravarani, C. N. J., Sun, D., Venkatakrishnan, A. J., Kayikci, M., Tate, C. G., et al. (2015). Universal allosteric mechanism for Galpha activation by GPCRs. Nature 524, 173–179. doi: 10.1038/nature14663
Fredholm, B. B. (2014). Adenosine–a physiological or pathophysiological agent? J. Mol. Med. 92, 201–206. doi: 10.1007/s00109-013-1101-6
Fredholm, B. B., IJzerman, A. P., Klotz, K. N., and Linden, J. (2001). International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 53, 527–552.
Fredholm, B. B., IJzerman, A. P., Jacobson, K. A., Linden, J., and Müller, C. E. (2011). International union of basic and clinical pharmacology. LXXXI. nomenclature and classification of adenosine receptors–an update. Pharmacol. Rev. 63, 1–34. doi: 10.1124/pr.110.003285
Gao, Z. G., Balasubramanian, R., Kiselev, E., Wei, Q., and Jacobson, K. A. (2014). Probing biased/partial agonism at the G protein-coupled A2B adenosine receptor. Biochem. Pharmacol. 90, 297–306. doi: 10.1016/j.bcp.2014.05.008
Gentry, P. R., Sexton, P. M., and Christopoulos, A. (2015). Novel allosteric modulators of G Protein-coupled receptors. J. Biol. Chem. 290, 19478–19488. doi: 10.1074/jbc.R115.662759
Glukhova, A., Thal, D. M., Nguyen, A. T., Vecchio, E. A., Jörg, M., Scammells, P. J., et al. (2017). Structure of the Adenosine A1 Receptor reveals the basis for subtype selectivity. Cell 168, 867–877.e813. doi: 10.1016/j.cell.2017.01.042
Guo, D., Pan, A. C., Dror, R. O., Mocking, T., Liu, R., Heitman, L. H., et al. (2016). Molecular basis of ligand dissociation from the Adenosine A2A receptor. Mol. Pharmacol. 89, 485–491. doi: 10.1124/mol.115.102657
Gutiérrez-de-Terán, H., Massink, A., Rodríguez, D., Liu, W., Han, G. W., Joseph, J. S., et al. (2013). The role of a sodium ion binding site in the allosteric modulation of the A2A adenosine G protein-coupled receptor. Structure 21, 2175–2185. doi: 10.1016/j.str.2013.09.020
Hino, T., Arakawa, T., Iwanari, H., Yurugi-Kobayashi, T., Ikeda-Suno, C., Nakada-Nakura, Y., et al. (2012). G-protein-coupled receptor inactivation by an allosteric inverse-agonist antibody. Nature 482, 237–240. doi: 10.1038/nature10750
Jaakola, V. P., Griffith, M. T., Hanson, M. A., Cherezov, V., Chien, E. Y., Lane, J. R., et al. (2008). The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 322, 1211–1217. doi: 10.1126/science.1164772
Jacobson, K. A., and Gao, Z. G. (2006). Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov. 5, 247–264. doi: 10.1038/nrd1983
Jazayeri, A., Dias, J. M., and Marshall, F. H. (2015). From G protein-coupled receptor structure resolution to rational drug design. J. Biol. Chem. 290, 19489–19495. doi: 10.1074/jbc.R115.668251
Katritch, V., Fenalti, G., Abola, E. E., Roth, B. L., Cherezov, V., and Stevens, R. C. (2014). Allosteric sodium in class A GPCR signaling. Trends Biochem. Sci. 39, 233–244. doi: 10.1016/j.tibs.2014.03.002
Kenakin, T., and Christopoulos, A. (2013). Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat. Rev. Drug Discov. 12, 205–216. doi: 10.1038/nrd3954
Kobilka, B. K., and Deupi, X. (2007). Conformational complexity of G-protein-coupled receptors. Trends Pharmacol. Sci. 28, 397–406. doi: 10.1016/j.tips.2007.06.003
Landau, E. M., and Rosenbusch, J. P. (1996). Lipidic cubic phases: a novel concept for the crystallization of membrane proteins. Proc. Natl. Acad. Sci. U.S.A. 93, 14532–14535. doi: 10.1073/pnas.93.25.14532
Lebon, G., Bennett, K., Jazayeri, A., and Tate, C. G. (2011a). Thermostabilisation of an agonist-bound conformation of the human adenosine A2A receptor. J. Mol. Biol. 409, 298–310. doi: 10.1016/j.jmb.2011.03.075
Lebon, G., Edwards, P. C., Leslie, A. G., and Tate, C. G. (2015). Molecular determinants of CGS21680 binding to the human Adenosine A2A receptor. Mol. Pharmacol. 87, 907–915. doi: 10.1124/mol.114.097360
Lebon, G., Warne, T., and Tate, C. G. (2012). Agonist-bound structures of G protein-coupled receptors. Curr. Opin. Struct. Biol. 22, 482–490. doi: 10.1016/j.sbi.2012.03.007
Lebon, G., Warne, T., Edwards, P. C., Bennett, K., Langmead, C. J., Leslie, A. G., et al. (2011b). Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 474, 521–525. doi: 10.1038/nature10136
Liu, W., Chun, E., Thompson, A. A., Chubukov, P., Xu, F., Katritch, V., et al. (2012). Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337, 232–236. doi: 10.1126/science.1219218
Magnani, F., Serrano-Vega, M. J., Shibata, Y., Abdul-Hussein, S., Lebon, G., Miller-Gallacher, J., et al. (2016). A mutagenesis and screening strategy to generate optimally thermostabilized membrane proteins for structural studies. Nat. Protoc. 11, 1554–1571. doi: 10.1038/nprot.2016.088
Magnani, F., Shibata, Y., Serrano-Vega, M. J., and Tate, C. G. (2008). Co-evolving stability and conformational homogeneity of the human adenosine A2A receptor. Proc. Natl. Acad. Sci. U.S.A. 105, 10744–10749. doi: 10.1073/pnas.0804396105
Manglik, A., Kim, T. H., Masureel, M., Altenbach, C., Yang, Z., Hilger, D., et al. (2015). Structural insights into the dynamic process of beta2-Adrenergic receptor signaling. Cell 161, 1101–1111. doi: 10.1016/j.cell.2015.04.043
Martin-Garcia, J. M., Conrad, C. E., Nelson, G., Stander, N., Zatsepin, N. A., Zook, J., et al. (2017). Serial millisecond crystallography of membrane and soluble protein microcrystals using synchrotron radiation. IUCrJ 4, 439–454. doi: 10.1107/S205225251700570X
Melnikov, I., Polovinkin, V., Kovalev, K., Gushchin, I., Shevtsov, M., Shevchenko, V., et al. (2017). Fast iodide-SAD phasing for high-throughput membrane protein structure determination. Sci. Adv. 3:e1602952. doi: 10.1126/sciadv.1602952
Moukhametzianov, R., Burghammer, M., Edwards, P. C., Petitdemange, S., Popov, D., Fransen, M., et al. (2008). Protein crystallography with a micrometre-sized synchrotron-radiation beam. Acta Crystallogr. D Biol. Crystallogr. 64(Pt. 2), 158–166. doi: 10.1107/S090744490705812X
Müller, C. E., and Jacobson, K. A. (2011). Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim. Biophys. Acta 1808, 1290–1308. doi: 10.1016/j.bbamem.2010.12.017
Murphree, L. J., Marshall, M. A., Rieger, J. M., MacDonald, T. L., and Linden, J. (2002). Human A2A adenosine receptors: high-affinity agonist binding to receptor-G protein complexes containing Gbeta(4). Mol. Pharmacol. 61, 455–462. doi: 10.1124/mol.61.2.455
Nehmé, R., Carpenter, B., Singhal, A., Strege, A., Edwards, P. C., White, C. F., et al. (2017). Mini-G proteins: novel tools for studying GPCRs in their active conformation. PLoS ONE 12:e0175642. doi: 10.1371/journal.pone.0175642
Prosser, R. S., Ye, L., Pandey, A., and Orazietti, A. (2017). Activation processes in ligand-activated G protein-coupled receptors: a case study of the adenosine A2A receptor. Bioessays 39:1700072. doi: 10.1002/bies.201700072
Rasmussen, S. G., Choi, H. J., Fung, J. J., Pardon, E., Casarosa, P., Chae, P. S., et al. (2011a). Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 469, 175–180. doi: 10.1038/nature09648
Rasmussen, S. G., Choi, H. J., Rosenbaum, D. M., Kobilka, T. S., Thian, F. S., Edwards, P. C., et al. (2007). Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature 450, 383–387. doi: 10.1038/nature06325
Rasmussen, S. G., DeVree, B. T., Zou, Y., Kruse, A. C., Chung, K. Y., Kobilka, T. S., et al. (2011b). Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549–555. doi: 10.1038/nature10361
Rosenbaum, D. M., Cherezov, V., Hanson, M. A., Rasmussen, S. G., Thian, F. S., Kobilka, T. S., et al. (2007). GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science 318, 1266–1273. doi: 10.1126/science.1150609
Rosenbaum, D. M., Rasmussen, S. G., and Kobilka, B. K. (2009). The structure and function of G-protein-coupled receptors. Nature 459, 356–363. doi: 10.1038/nature08144
Rosenbaum, D. M., Zhang, C., Lyons, J. A., Holl, R., Aragao, D., Arlow, D. H., et al. (2011). Structure and function of an irreversible agonist-beta(2) adrenoceptor complex. Nature 469, 236–240. doi: 10.1038/nature09665
Sawynok, J. (2016). Adenosine receptor targets for pain. Neuroscience 338, 1–18. doi: 10.1016/j.neuroscience.2015.10.031
Segala, E., Guo, D., Cheng, R. K., Bortolato, A., Deflorian, F., Doré, A. S., et al. (2016). Controlling the dissociation of ligands from the Adenosine A2A receptor through modulation of salt bridge strength. J. Med. Chem. 59, 6470–6479. doi: 10.1021/acs.jmedchem.6b00653
Serrano-Vega, M. J., Magnani, F., Shibata, Y., and Tate, C. G. (2008). Conformational thermostabilization of the beta1-adrenergic receptor in a detergent-resistant form. Proc. Natl. Acad. Sci. U.S.A. 105, 877–882. doi: 10.1073/pnas.0711253105
Shibata, Y., White, J. F., Serrano-Vega, M. J., Magnani, F., Aloia, A. L., Grisshammer, R., et al. (2009). Thermostabilization of the neurotensin receptor NTS1. J. Mol. Biol. 390, 262–277. doi: 10.1016/j.jmb.2009.04.068
Steyaert, J., and Kobilka, B. K. (2011). Nanobody stabilization of G protein-coupled receptor conformational states. Curr. Opin. Struct. Biol. 21, 567–572. doi: 10.1016/j.sbi.2011.06.011
Strege, A., Carpenter, B., Edwards, P. C., and Tate, C. G. (2017). Strategy for the thermostabilization of an agonist-bound GPCR coupled to a G protein. Methods Enzymol. 594, 243–264. doi: 10.1016/bs.mie.2017.05.014
Sun, B., Bachhawat, P., Chu, M. L., Wood, M., Ceska, T., Sands, Z. A., et al. (2017). Crystal structure of the adenosine A2A receptor bound to an antagonist reveals a potential allosteric pocket. Proc. Natl. Acad. Sci. U.S.A. 114, 2066–2071. doi: 10.1073/pnas.1621423114
Tate, C. G., and Schertler, G. F. (2009). Engineering G protein-coupled receptors to facilitate their structure determination. Curr. Opin. Struct. Biol. 19, 386–395. doi: 10.1016/j.sbi.2009.07.004
Venkatakrishnan, A. J., Deupi, X., Lebon, G., Heydenreich, F. M., Flock, T., Miljus, T., et al. (2016). Diverse activation pathways in class A GPCRs converge near the G-protein-coupling region. Nature 536, 484–487. doi: 10.1038/nature19107
Venkatakrishnan, A. J., Deupi, X., Lebon, G., Tate, C. G., Schertler, G. F., and Babu, M. M. (2013). Molecular signatures of G-protein-coupled receptors. Nature 494, 185–194. doi: 10.1038/nature11896
Verzijl, D., and Ijzerman, A. P. (2011). Functional selectivity of adenosine receptor ligands. Purinergic Signal. 7, 171–192. doi: 10.1007/s11302-011-9232-0
Warne, T., Edwards, P. C., Leslie, A. G., and Tate, C. G. (2012). Crystal structures of a stabilized beta1-adrenoceptor bound to the biased agonists bucindolol and carvedilol. Structure 20, 841–849. doi: 10.1016/j.str.2012.03.014
Warne, T., Moukhametzianov, R., Baker, J. G., Nehmé, R., Edwards, P. C., Leslie, A. G., et al. (2011). The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature 469, 241–244. doi: 10.1038/nature09746
Warne, T., Serrano-Vega, M. J., Baker, J. G., Moukhametzianov, R., Edwards, P. C., Henderson, R., et al. (2008). Structure of a β1-adrenergic G-protein-coupled receptor. Nature 454, 486–491. doi: 10.1038/nature07101
Weinert, T., Olieric, N., Cheng, R., Brünle, S., James, D., Ozerov, D., et al. (2017). Serial millisecond crystallography for routine room-temperature structure determination at synchrotrons. Nat. Commun. 8:542. doi: 10.1038/s41467-017-00630-4
Westfield, G. H., Rasmussen, S. G., Su, M., Dutta, S., DeVree, B. T., Chung, K. Y., et al. (2011). Structural flexibility of the G alpha s alpha-helical domain in the β2-adrenoceptor Gs complex. Proc. Natl. Acad. Sci. U.S.A. 108, 16086–16091. doi: 10.1073/pnas.1113645108
White, J. F., Noinaj, N., Shibata, Y., Love, J., Kloss, B., Xu, F., et al. (2012). Structure of the agonist-bound neurotensin receptor. Nature 490, 508–513. doi: 10.1038/nature11558
Xu, F., Wu, H., Katritch, V., Han, G. W., Jacobson, K. A., Gao, Z. G., et al. (2011). Structure of an agonist-bound human A2A adenosine receptor. Science 332, 322–327. doi: 10.1126/science.1202793
Keywords: GPCR, adenosine, structural biology, G protein, drugs, x-ray diffraction
Citation: Carpenter B and Lebon G (2017) Human Adenosine A2A Receptor: Molecular Mechanism of Ligand Binding and Activation. Front. Pharmacol. 8:898. doi: 10.3389/fphar.2017.00898
Received: 24 October 2017; Accepted: 24 November 2017;
Published: 14 December 2017.
Edited by:
Kenneth A. Jacobson, National Institutes of Health (NIH), United StatesReviewed by:
Fei Xu, ShanghaiTech University, ChinaCopyright © 2017 Carpenter and Lebon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Byron Carpenter, Yi5jYXJwZW50ZXJAd2Fyd2ljay5hYy51aw==
Guillaume Lebon, Z2xlYm9uQGlnZi5jbnJzLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.