 Jayne-Louise E. Pritchard
Jayne-Louise E. Pritchard Tracy A. O’Mara
Tracy A. O’Mara Dylan M. Glubb
Dylan M. Glubb- Department of Genetics and Computational Biology, Molecular Cancer Epidemiology, QIMR Berghofer Medical Research Institute, Brisbane, QLD, Australia
The development of new drugs has become challenging as the necessary investments in time and money have increased while drug approval rates have decreased. A potential solution to this problem is drug repositioning which aims to use existing drugs to treat conditions for which they were not originally intended. One approach that may enhance the likelihood of success is to reposition drugs against a target that has a genetic basis. The multitude of genome-wide association studies (GWASs) conducted in recent years represents a large potential pool of novel targets for drug repositioning. Although trait-associated variants identified from GWAS still need to be causally linked to a target gene, recently developed functional genomic techniques, databases, and workflows are helping to remove this bottleneck. The pre-clinical validation of repositioning against these targets also needs to be carefully performed to ensure that findings are not confounded by off-target effects or limitations of the techniques used. Nevertheless, the approaches described in this review have the potential to provide a faster, cheaper and more certain route to clinical approval.
Introduction
Over 6,000 human medical conditions have defined molecular phenotypes (Johns Hopkins University, 2017) but only ∼500 conditions have approved therapies (National Institutes of Health, 2015). Furthermore, many approved therapies have suboptimal efficacy or are accompanied by unacceptable toxicity. Despite scientific advancements, drug development remains challenging as development time and costs are increasing while drug success rates are low. Indeed, for every US dollar spent on research and development, the number of new drugs that are approved by the US Food and Drug Administration (FDA) has roughly halved every 9 years since 1950 (Scannell et al., 2012). The magnitude and duration of this phenomenon suggest that current approaches addressing the research and development productivity problem are having a weak effect. Not surprisingly, pharmaceutical companies often cannot afford to pursue development of promising drug candidates. It is apparent that alternative directions are required to address these critical issues. This review will focus on promising approaches to improve the success of therapeutic development by repositioning existing drugs against molecular targets identified from genetic studies.
Drug Repositioning
Drug repositioning, also known as drug repurposing, aims to use existing therapies or drugs that have stalled in development to treat conditions for which they were not originally intended. Given that in the US alone there are ∼3000 approved drugs (U.S. Department of Health and Human Services, 2017) and thousands more which have not reached clinical approval, drug repositioning supplies a vast armamentarium to expedite the development of new therapies. The development of a new drug takes on average 13–15 years and costs between US$2–3 billion (Nosengo, 2016) with only a ∼10% chance that a new therapy will be successfully approved by government regulatory agencies (Smietana et al., 2016). In contrast, drug repositioning represents increased efficiency and lower costs because candidates already have established safety profiles from Phase I clinical trials, with time to approval estimated at 6.5 years at an average cost of US$300 million (Nosengo, 2016). One of the most successful drug repositioning examples is thalidomide, a drug whose use was originally discontinued due to severe skeletal birth defects (Kim and Scialli, 2011). After repositioning, thalidomide and its derivatives are now indicated for the treatment of multiple myeloma and a skin condition related to leprosy with sales revenues of billions of dollars.
Identifying Drug Targets From Genetic Studies
An approach that may increase the likelihood of drug repositioning success is the use of genetic studies to identify “druggable” targets. Drugs that have been linked to disease traits through genetic studies are reported to be twice as likely to be clinically approved compared to drugs with no such links (Nelson et al., 2015). The advent of large-scale genetic studies, primarily involving genome-wide association studies (GWASs), has greatly advanced our knowledge of the genetic basis for many diseases (Visscher et al., 2017), allowing researchers to leverage this information to identify targets for therapy. Indeed, genetic studies have identified a large number of genes whose proteins are already targeted by drugs used in clinical practice. For example, the genes encoding drug targets of tamoxifen (ESR1) and aromatase inhibitors (CYP19A1) have been linked to genetic variation associated with risk for breast (Dunning et al., 2016) and endometrial cancer (Thompson et al., 2016), diseases that are treated using these drugs. Moreover, genetic studies are revealing novel drug targets such as PCSK9. It was initially reported that PCSK9 nonsense mutation carriers had lower plasma levels of LDL cholesterol and a significantly reduced risk of coronary heart disease (Horton et al., 2007). A common genetic variant (rs11206510) ∼10 kb upstream of PCSK9 was also subsequently found to associate with coronary heart disease (Schunkert et al., 2011). Based on these genetic findings, two human monoclonal antibodies have been developed to lower cholesterol by inhibiting PCSK9 (Markham, 2015; Paton, 2016) and one of these drugs was recently found to lower LDL cholesterol levels by ∼60% in a large clinical trial (Sabatine et al., 2017). Additionally, findings from genetic studies have led to drug repositioning as is demonstrated by secukinumab, an antibody therapy that targets IL-17A, which was originally tested for efficacy in the treatment of psoriasis, rheumatoid arthritis and uveitis (Hueber et al., 2010). However, IL-17A belongs to an immune axis with IL-23 (Gaffen et al., 2014) and the association of a variant (rs11209032) ∼15 kb downstream of the gene encoding the IL-23 receptor (IL23R) with ankylosing spondylitis (Burton et al., 2007) thus provided a rationale for repositioning secukinumab to treat this additional inflammatory disease.
Genome-Wide Association Studies (GWASS)
Although GWAS have transformed the study of common genetic variation over the last 10 years, there has been criticism of their limited clinical impact. However, sample sizes for many diseases have only recently reached sufficient size to detect significant numbers of genome-wide significant loci (Visscher et al., 2017). As of November 2017, the GWAS catalog contains ∼53,000 unique variant-trait associations for more than 800 human traits and diseases (MacArthur et al., 2017), likely representing a large number of genes that could provide targets for drug repositioning studies. While ∼10% of GWAS variants affect the coding sequence and, therefore, have a high probability of affecting the function of the gene or encoded protein in which they are located, the vast majority of GWAS variants are found in intergenic or intronic regions and their gene targets are less clear. These non-coding variants likely affect the trait of interest through regulation of gene expression, but determining their gene targets is a complex task because GWAS variants may only regulate the nearest gene one third of the time (Gusev et al., 2016; Zhu et al., 2016). Long-range chromatin looping interactions allow genetic variants to potentially regulate a large number of genes over megabase distances (Mifsud et al., 2015). Thus, assigning the gene nearest a GWAS variant as a target may lead to false assignment of causation. An example of this is studies that were conducted on FTO. Intronic variants in FTO had been associated with obesity and body mass index and FTO was thought to be the regulatory target of these variants (Dina et al., 2007; Frayling et al., 2007). However, it was later determined through functional genetic experiments and mouse knockout studies that IRX3, a gene distally located from the GWAS variants, was the likely causal gene (Ragvin et al., 2010; Smemo et al., 2014).
The complexity of determining the causal genes through which trait-association variants act has constituted a major roadblock in the clinical translation of GWAS findings. However, in recent years, workflows have been developed to establish these causal genes (Edwards et al., 2013) and much progress is being made toward systematically identifying these genes using new functional genomic techniques that assess chromatin interactions and gene expression associations.
Approaches to Identify the Target Genes of Trait-Associated Variants
Sophisticated computational approaches have been developed to identify disease-gene associations from GWAS data and include NetWAS which incorporates functional genomic data to identify tissue-specific gene networks (Greene et al., 2015). However, these bioinformatic tools require firstly assigning trait-associated variants to a gene. Experimental techniques such as chromatin Confirmation Capture (3C) and related high-throughput techniques (e.g., 5C, ChIA-PET, Hi-C) have been successful in identifying long-range chromatin interactions between genomic regions (Schmitt et al., 2016). These data are extremely useful in identifying interactions between GWAS loci and potential target genes (Michailidou et al., 2015; Mifsud et al., 2015; Cheng et al., 2016). However, large scale chromatin interaction assays which assess all possible interactions (e.g., Hi-C) are costly, with billions of sequencing reads required to ensure suitable resolution and confidence to accurately assess interactions. Fortunately, there are numerous databases housing publicly available data from chromatin confirmation experiments across multiple tissues and cell types (Table 1). These databases also provide for standardization of the complex analysis of such experiments. 3C also allows for the identification of allele-specific interactions between genes and genomic regions containing trait-associated alleles (Glubb et al., 2017), implicating the involvement of the trait-associated variant in the chromatin interaction. Importantly, bioinformatic tools have now been developed to systematically identify allele-specific interactions from large-scale 3C experiments (Servant et al., 2015; Li et al., 2017). Approaches have also been developed to integrate functional genomic data to predict interactions between genes and regulatory regions (Table 1; e.g., PreSTIGE and IM-PET). These approaches take advantage of the vast amount of public data provided by consortia such as ENCODE (Encode Project Consortium, 2012) and Roadmap Epigenomics Project (Chadwick, 2012), as well as data made publicly available by researchers, such as the Gene Expression Omnibus (Barrett et al., 2013). Moreover, the experiments required to generate data for these integrative approaches, commonly ChIP-seq and RNA-seq, can be performed at a fraction of the cost of a Hi-C experiment.
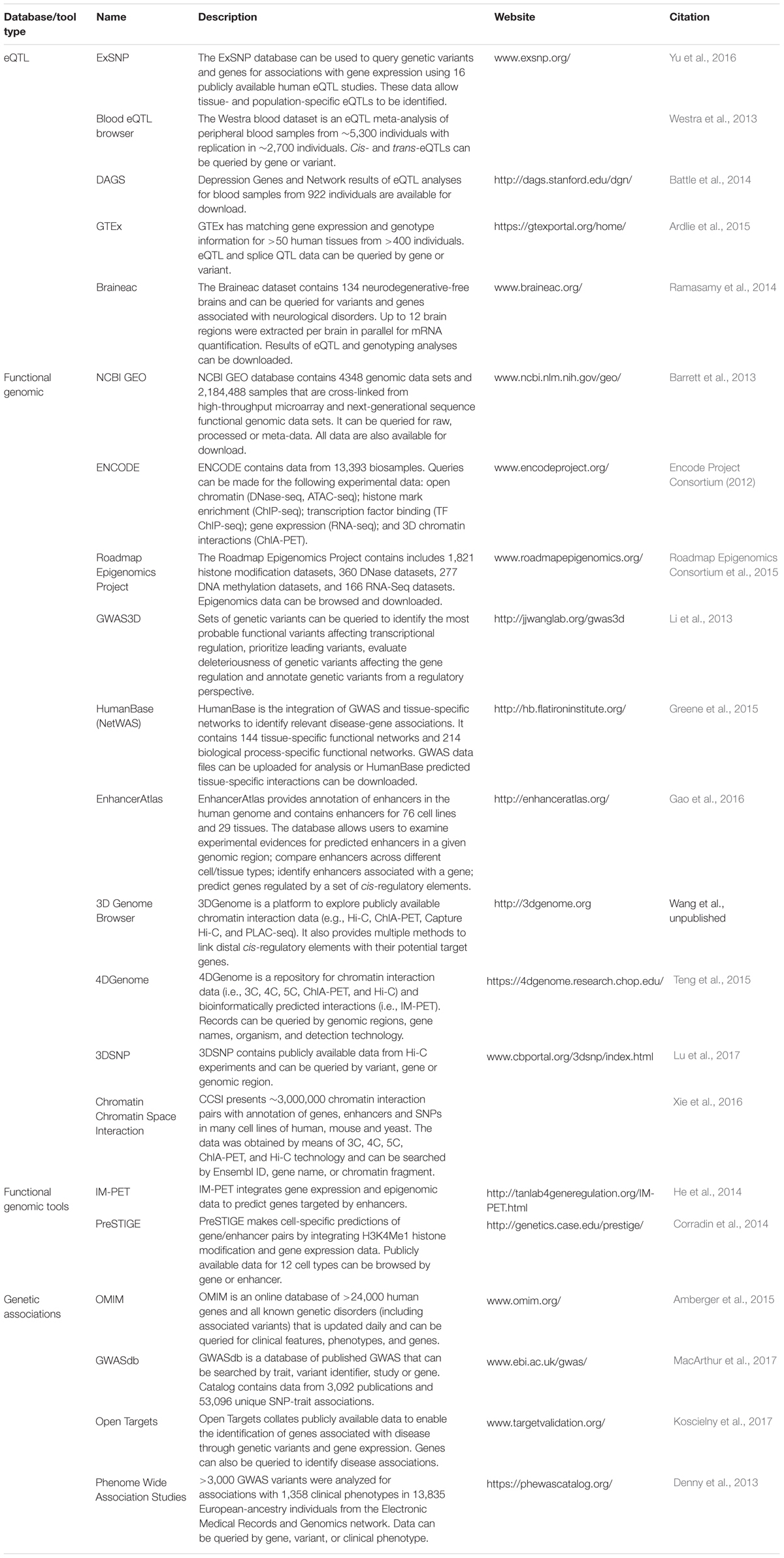
TABLE 1. Databases and tools for the identification of gene targets.
Identification of Causal Genes by Integrating Genotype and Gene Expression Data
The methods described above are useful in identifying target genes but it is still necessary to demonstrate the effect of trait-associated variants on target gene activity. The directionality of the effect is also crucial information that is used to inform the need for drugs with either antagonistic or agonistic actions on the target. In vitro experiments such as reporter gene assays can provide this information by identifying trait-associated variants that modulate the promoter activity of target genes through regulatory elements (Glubb et al., 2015, 2017). A powerful complementary approach is to link the genotype of trait-associated variants to gene expression using in vivo data, thus identifying target genes and the directionality of the effect of trait-variants. Expression quantitative trait loci (eQTL) analyses are useful in this regard as they can provide genome-wide lists of genetic variants that associate with gene expression in a particular tissue. There are now a number of eQTL databases (Table 1) that can be queried to determine if a trait-associated variant (or variants in linkage disequilibrium) associates with the expression of a specific gene. One of the most comprehensive of these in terms of the diversity of data is the Genotype-Tissue Expression (GTEx) project (Ardlie et al., 2015), which now has eQTL data available for 44 human tissues. Although some tissues currently have a relatively small number of samples (n < 100) and consequently suffer from low statistical power for eQTL analysis, data generation is ongoing (Ardlie et al., 2015). Furthermore, the GTEx project provides splicing QTL data, enabling the identification of genetic variants that associate with alternative gene transcripts. Other eQTL studies already include data from a large number of individuals providing statistical power to detect both cis- and trans-eQTLs with high confidence. For example, Westra et al. (2013) identified eQTLs using data from more than 5,000 blood samples (Table 1; Blood eQTL browser).
The linking of gene expression and genotype data can be applied at a multi-variant or gene-based level by combining genotype data to determine the cumulative effect of genetic variants on expression (Gamazon et al., 2015; Gusev et al., 2016). These data are used to predict gene expression levels in cohorts of genotyped individuals, allowing case-control transcriptome-wide association studies to examine whether the predicted gene expression associates with clinical phenotypes and the potentially causal genes identified could provide further targets for drug repositioning.
Tools for Identification of Drug Repositioning Candidates
Once evidence indicates that a gene is likely regulated by a trait-associated variant, the next step would be to assess whether an existing drug can be repositioned to target this gene or its encoded protein. Numerous databases can be accessed for this purpose, summarized in Table 2. These include databases that link drugs to genes through their known pharmacological targets (e.g., DrugBank and ChEMBL). However, many drugs have an array of off-target effects and these unintended pharmacological interactions are often not well known. Therefore, to identify additional pharmacological targets, other databases extract data from the literature that demonstrate the effects of drugs on gene/protein expression and function [e.g., Comparative Toxicogenomics Database (CTD)] or binding interactions between drugs and proteins [e.g., The Binding Database (BindingDB)].
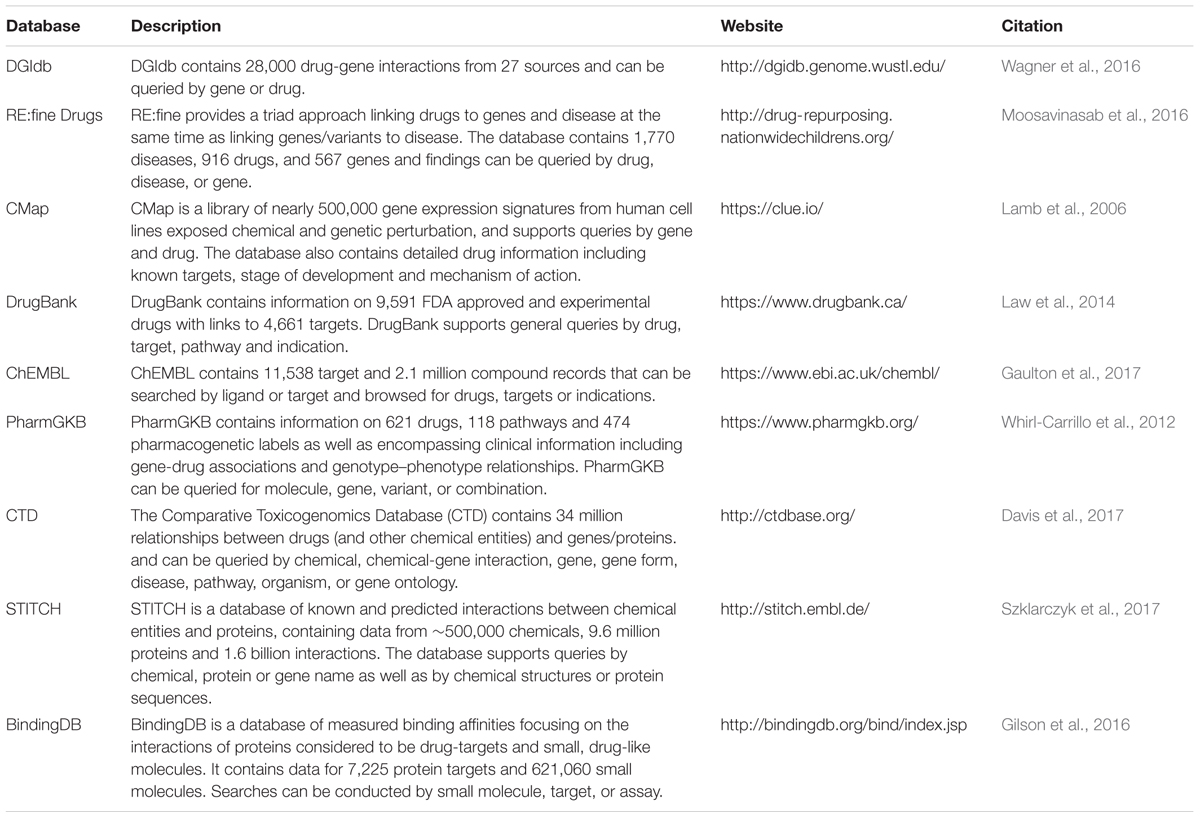
TABLE 2. Databases for the identification of drug candidates for repositioning.
A novel approach for identifying drug repositioning opportunities is provided through the Connectivity Map (CMap) database (Table 2). CMap is a resource that uses gene expression changes in response to drug treatment and gene perturbation (i.e., knockdown/overexpression) to find relationships between genes and drugs. CMap contains over one million gene expression signatures from the treatment of a variety of cell types with drug and gene perturbations. Differential expression signatures that arise from treatment can be compared to signatures in the database to perform both positive and negative correlations. These data could be applied to the identification of drug candidates for repositioning. For example, if a gene is known to be down-regulated by a trait-associated variant, a search can be performed to identify drugs that may have a beneficial expression signature, i.e., drugs with a similar signature to the opposite gene perturbation (in this case gene overexpression).
Pre-Clinical Validation of Drug Repositioning
Before clinical drug repositioning trials can be performed, pre-clinical studies are crucial to validate targeting of the gene or protein of interest and demonstrate a desired effect in cellular or animal models. Definitive proof that the target is necessary for the desired effect is not a trivial exercise and requires manipulation of the target in the model system, which is often accompanied with caveats (Kaelin, 2017). With the vast array of tools now available, it is now relatively straightforward to genetically manipulate targets by transcript overexpression/knockdown (e.g., cDNA clones and siRNA), gene knockout/knock-in and even by the introduction of gain or loss of function mutations (e.g., CRISPR/Cas9). These gene perturbation techniques can also be applied in a high throughput fashion to cellular or animal models using pooled cDNA, siRNA, or gRNA libraries to characterize gene function (Joung et al., 2017; Tsherniak et al., 2017) with image-based profiling providing the capability to measure multiple phenotypes at the same time (Caicedo et al., 2016). However, gene perturbation approaches themselves often have off-target effects that might confound findings and thus experiments need to be well controlled to ensure the correct interpretations are made (Kaelin, 2017). To unambiguously validate targets, rescue experiments are required. In these experiments, the desired phenotype is reversed (or rescued) by a drug resistant version of the target or reintroduction of the target through some means which is resistant to the original gene perturbation or ablation (Kaelin, 2017). Difficulties in reproducing experimental results between laboratories (Prinz et al., 2011) also highlights the need for multiple experimental lines of evidence and findings that are robust to different conditions and models.
The Identification of Further Diseases for Repositioning
Another method for identification of drug repositioning opportunities leverages the fact that genetic variation can have pleiotropic effects and associate with multiple clinical phenotypes. Therefore, a drug successfully repositioned using genetic data may be able to be repositioned for the treatment of further diseases if the underlying genetic variant(s) has a pleiotropic effect. A relatively new technique that can be applied to this is the phenome-wide association study (PheWAS), where a single variant is tested for association with a large number of phenotypes, enabling the identification of variants that confer susceptibility to multiple diseases (Denny et al., 2010). Databases that contain de-identified Electronic Medical Records (EMRs) are an efficient source of data for PheWAS (Manolio et al., 2009). EMR databases contain longitudinal health records that include prescription records, family histories, laboratory and image testing results, physician notes and, importantly, the International Classification of Disease codes (Hebbring, 2014). An additional approach would be to use genetic correlation analyses, such as LD-score regression, that use GWAS data to identify genetic similarities between diseases which could then provide an avenue for further repositioning.
Limitations of Drug Repositioning and Future Directions
Although drug repositioning appears to have many advantages over traditional drug development, there are some caveats. Firstly, there needs to be a drug that can be repositioned against the target of interest. This may not always be the case and, therefore, drug repositioning should be considered a complementary approach to the development of novel drugs. In terms of clinical trials, Phase I studies may still be necessary if an increased dosage of the repositioned drug is required, if a new drug delivery method is used, or if it the drug is intended to be used in a new population. Nevertheless, by repositioning a drug against a target on the basis of genetic evidence, the increased likelihood of approval may still offset the costs of Phase I trials. Intellectual property for drug repositioning needs to be considered as drug repositioning uses drugs that are already published cannot be patented because they have already been publicly disclosed. Lack of patentability reduces opportunity for profit dis-incentivizing pharmaceutical companies from pursuing that target. For example, generic drugs with well characterized safety profiles may appear amenable to drug repositioning, but a lack of intellectual property could prevent pharmaceutical companies from recouping costs spent on testing in clinical trials (Nosengo, 2016). However, repositioning drug candidates could be refined or modified to provide better targeting and thus generate new intellectual property. Current patent law regarding drug repositioning is complex and inconsistent and thus greater clarity and uniformity is required (Kremer and Jones, 2015). It is also important that exclusivity and patent strategies exist to provide incentives for pharmaceutical companies to invest in this area of research (Kremer and Jones, 2015). Furthermore, the drug repositioning process could be promoted by collaborative models involving academic researchers, pharmaceutical companies and other stake holders. For example, the MRC-Industry Asset Sharing Initiative1 and the NIH National Center for Advancing Translational Sciences (NIH-NCATS2) aim to deliver treatments and cures for disease to patients faster by improving the translational process.
Conclusion
Drug repositioning potentially provides a faster and cheaper approach to the development of new therapies and, if targets have a genetic basis, should carry less risk. Yet, a concerted effort still needs to be made to overcome the bottleneck of identifying targets from large-scale genetic studies and rigorous approaches need to be taken in the pre-clinical validation of drug repositioning to maximize likelihood of success in clinical studies.
Author Contributions
Conception or design of the work; the acquisition, analysis and interpretation of data for the work (J-LP, DG, and TO). Drafting the work or revising it critically for important intellectual content (J-LP, DG, and TO). Final approval of the version to be published (J-LP, DG, and TO). Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved (J-LP, DG, and TO).
Funding
This work was supported by QIMR Berghofer Medical Research Institute Near Miss Funding (DG). TO is supported by an NHMRC Early Career Fellowship (GNT1111246).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer SP and handling Editor declared their shared affiliation.
Acknowledgment
The authors thank Professor Mandy Spurdle for her guidance.
Footnotes
- ^ https://www.mrc.ac.uk/funding/browse/industry-asset-sharing-initiative/mrc-industry-asset-sharing-initiative/
- ^ https://ncats.nih.gov/
References
Amberger, J. S., Bocchini, C. A., Schiettecatte, F., Scott, A. F., and Hamosh, A. (2015). OMIM.org: online Mendelian Inheritance in Man (OMIM(R)), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 43, D789–D798. doi: 10.1093/nar/gku1205
Ardlie, K. G., Deluca, D. S., Segrè, A. V., Sullivan, T. J., Young, T. R., Gelfand, E. T., et al. (2015). The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660. doi: 10.1126/science.1262110
Barrett, T., Wilhite, S. E., Ledoux, P., Evangelista, C., Kim, I. F., Tomashevsky, M., et al. (2013). NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 41, D991–D995. doi: 10.1093/nar/gks1193
Battle, A., Mostafavi, S., Zhu, X., Potash, J. B., Weissman, M. M., McCormick, C., et al. (2014). Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Res. 24, 14–24. doi: 10.1101/gr.155192.113
Burton, P. R., Clayton, D. G., Cardon, L. R., Craddock, N., Deloukas, P., Duncanson, A., et al. (2007). Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat. Genet. 39, 1329–1337. doi: 10.1038/ng.2007.17
Caicedo, J. C., Singh, S., and Carpenter, A. E. (2016). Applications in image-based profiling of perturbations. Curr. Opin. Biotechnol. 39, 134–142. doi: 10.1016/j.copbio.2016.04.003
Chadwick, L. H. (2012). The NIH roadmap epigenomics program data resource. Epigenomics 4, 317–324. doi: 10.2217/epi.12.18
Cheng, T. H., Thompson, D. J., O’Mara, T. A., Painter, J. N., Glubb, D. M., Flach, S., et al. (2016). Five endometrial cancer risk loci identified through genome-wide association analysis. Nat. Genet. 48, 667–674. doi: 10.1038/ng.3562
Corradin, O., Saiakhova, A., Akhtar-Zaidi, B., Myeroff, L., Willis, J., Cowper-Sal-Lari, R., et al. (2014). Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits. Genome Res. 24, 1–13. doi: 10.1101/gr.164079.113
Davis, A. P., Grondin, C. J., Johnson, R. J., Sciaky, D., King, B. L., McMorran, R., et al. (2017). The comparative toxicogenomics database: update 2017. Nucleic Acids Res. 45, D972–D978. doi: 10.1093/nar/gkw838
Denny, J. C., Bastarache, L., Ritchie, M. D., Carroll, R. J., Zink, R., Mosley, J. D., et al. (2013). Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat. Biotechnol. 31, 1102–1111. doi: 10.1038/nbt.2749
Denny, J. C., Ritchie, M. D., Basford, M. A., Pulley, J. M., Bastarache, L., Brown-Gentry, K., et al. (2010). PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics 26, 1205–1210. doi: 10.1093/bioinformatics/btq126
Dina, C., Meyre, D., Gallina, S., Durand, E., Korner, A., Jacobson, P., et al. (2007). Variation in FTO contributes to childhood obesity and severe adult obesity. Nat. Genet. 39, 724–726. doi: 10.1038/ng2048
Dunning, A. M., Michailidou, K., Kuchenbaecker, K. B., Thompson, D., French, J. D., Beesley, J., et al. (2016). Breast cancer risk variants at 6q25 display different phenotype associations and regulate ESR1, RMND1 and CCDC170. Nat. Genet. 48, 374–386. doi: 10.1038/ng.3521
Edwards, S. L., Beesley, J., French, J. D., and Dunning, A. M. (2013). Beyond GWASs: illuminating the dark road from association to function. Am. J. Hum. Genet. 93, 779–797. doi: 10.1016/j.ajhg.2013.10.012
Encode Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. doi: 10.1038/nature11247
Frayling, T. M., Timpson, N. J., Weedon, M. N., Zeggini, E., Freathy, R. M., Lindgren, C. M., et al. (2007). A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316, 889–894. doi: 10.1126/science.1141634
Gaffen, S. L., Jain, R., Garg, A. V., and Cua, D. J. (2014). The IL-23–IL-17 immune axis: from mechanisms to therapeutic testing. Nat. Rev. Immunol. 14, 585–600. doi: 10.1038/nri3707
Gamazon, E. R., Wheeler, H. E., Shah, K. P., Mozaffari, S. V., Aquino-Michaels, K., Carroll, R. J., et al. (2015). A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 47, 1091–1098. doi: 10.1038/ng.3367
Gao, T., He, B., Liu, S., Zhu, H., Tan, K., and Qian, J. (2016). EnhancerAtlas: a resource for enhancer annotation and analysis in 105 human cell/tissue types. Bioinformatics 32, 3543–3551. doi: 10.1093/bioinformatics/btw495
Gaulton, A., Hersey, A., Nowotka, M., Bento, A. P., Chambers, J., Mendez, D., et al. (2017). The ChEMBL database in 2017. Nucleic Acids Res. 45, D945–D954. doi: 10.1093/nar/gkw1074
Gilson, M. K., Liu, T., Baitaluk, M., Nicola, G., Hwang, L., and Chong, J. (2016). BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 44, D1045–D1053. doi: 10.1093/nar/gkv1072
Glubb, D. M., Johnatty, S. E., Quinn, M. C. J., O’Mara, T. A., Tyrer, J. P., Gao, B., et al. (2017). Analyses of germline variants associated with ovarian cancer survival identify functional candidates at the 1q22 and 19p12 outcome loci. Oncotarget 8, 64670–64684. doi: 10.18632/oncotarget.18501
Glubb, D. M., Maranian, M. J., Michailidou, K., Pooley, K. A., Meyer, K. B., Kar, S., et al. (2015). Fine-scale mapping of the 5q11.2 breast cancer locus reveals at least three independent risk variants regulating MAP3K1. Am. J. Hum. Genet. 96, 5–20. doi: 10.1016/j.ajhg.2014.11.009
Greene, C. S., Krishnan, A., Wong, A. K., Ricciotti, E., Zelaya, R. A., Himmelstein, D. S., et al. (2015). Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet. 47, 569–576. doi: 10.1038/ng.3259
Gusev, A., Ko, A., Shi, H., Bhatia, G., Chung, W., Penninx, B. W., et al. (2016). Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 48, 245–252. doi: 10.1038/ng.3506
He, B., Chen, C., Teng, L., and Tan, K. (2014). Global view of enhancer–promoter interactome in human cells. Proc. Natl. Acad. Sci. U.S.A. 111, E2191–E2199. doi: 10.1073/pnas.1320308111
Hebbring, S. J. (2014). The challenges, advantages and future of phenome-wide association studies. Immunology 141, 157–165. doi: 10.1111/imm.12195
Horton, J. D., Cohen, J. C., and Hobbs, H. H. (2007). Molecular biology of PCSK9: its role in LDL metabolism. Trends Biochem. Sci. 32, 71–77. doi: 10.1016/j.tibs.2006.12.008
Hueber, W., Patel, D. D., Dryja, T., Wright, A. M., Koroleva, I., Bruin, G., et al. (2010). Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci. Transl. Med. 2:52ra72. doi: 10.1126/scitranslmed.3001107
Johns Hopkins University (2017). OMIM Gene Map Statistics [Online]. Available: http://www.omim.org/statistics/geneMap [accessed June 20, 2017].
Joung, J., Konermann, S., Gootenberg, J. S., Abudayyeh, O. O., Platt, R. J., Brigham, M. D., et al. (2017). Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 12, 828–863. doi: 10.1038/nprot.2017.016
Kaelin, W. G. Jr. (2017). Common pitfalls in preclinical cancer target validation. Nat. Rev. Cancer 17, 425–440. doi: 10.1038/nrc.2017.32
Kim, J. H., and Scialli, A. R. (2011). Thalidomide: the tragedy of birth defects and the effective treatment of disease. Toxicol. Sci. 122, 1–6. doi: 10.1093/toxsci/kfr088
Koscielny, G., An, P., Carvalho-Silva, D., Cham, J. A., Fumis, L., Gasparyan, R., et al. (2017). Open targets: a platform for therapeutic target identification and validation. Nucleic Acids Res. 45, D985–D994. doi: 10.1093/nar/gkw1055
Kremer, S., and Jones, R. (2015). Old Drugs, New Tricks: Patent Considerations in Drug Repurposing [Online]. Available at: http://www.lexology.com/library/detail.aspx?g=0e11981f-5268-4b0f-b6a6-1cb27e97e07c
Lamb, J., Crawford, E. D., Peck, D., Modell, J. W., Blat, I. C., Wrobel, M. J., et al. (2006). The connectivity map: using gene-expression signatures to connect small molecules, Genes, and disease. Science 313, 1929–1935. doi: 10.1126/science.1132939
Law, V., Knox, C., Djoumbou, Y., Jewison, T., Guo, A. C., Liu, Y., et al. (2014). DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 42, D1091–D1097. doi: 10.1093/nar/gkt1068
Li, G., Chen, Y., Snyder, M. P., and Zhang, M. Q. (2017). ChIA-PET2: a versatile and flexible pipeline for ChIA-PET data analysis. Nucleic Acids Res. 45:e4. doi: 10.1093/nar/gkw809
Li, M. J., Wang, L. Y., Xia, Z., Sham, P. C., and Wang, J. (2013). GWAS3D: detecting human regulatory variants by integrative analysis of genome-wide associations, chromosome interactions and histone modifications. Nucleic Acids Res. 41, W150–W158. doi: 10.1093/nar/gkt456
Lu, Y., Quan, C., Chen, H., Bo, X., and Zhang, C. (2017). 3DSNP: a database for linking human noncoding SNPs to their three-dimensional interacting genes. Nucleic Acids Res. 45, D643–D649. doi: 10.1093/nar/gkw1022
MacArthur, J., Bowler, E., Cerezo, M., Gil, L., Hall, P., Hastings, E., et al. (2017). The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 45, D896–D901. doi: 10.1093/nar/gkw1133
Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J., et al. (2009). Finding the missing heritability of complex diseases. Nature 461, 747–753. doi: 10.1038/nature08494
Markham, A. (2015). Evolocumab: first global approval. Drugs 75, 1567–1573. doi: 10.1007/s40265-015-0460-4
Michailidou, K., Beesley, J., Lindstrom, S., Canisius, S., Dennis, J., Lush, M. J., et al. (2015). Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat. Genet. 47, 373–380. doi: 10.1038/ng.3242
Mifsud, B., Tavares-Cadete, F., Young, A. N., Sugar, R., Schoenfelder, S., Ferreira, L., et al. (2015). Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet. 47, 598–606. doi: 10.1038/ng.3286
Moosavinasab, S., Patterson, J., Strouse, R., Rastegar-Mojarad, M., Regan, K., Payne, P. R., et al. (2016). ‘RE:fine drugs’: an interactive dashboard to access drug repurposing opportunities. Database (Oxford) 2016:baw083. doi: 10.1093/database/baw083
National Institutes of Health (2015). Transforming Translational Research. Rockville, MD: National Institutes of Health.
Nelson, M. R., Tipney, H., Painter, J. L., Shen, J., Nicoletti, P., Shen, Y., et al. (2015). The support of human genetic evidence for approved drug indications. Nat. Genet. 47, 856–860. doi: 10.1038/ng.3314
Paton, D. M. (2016). PCSK9 inhibitors: monoclonal antibodies for the treatment of hypercholesterolemia. Drugs Today (Barc) 52, 183–192. doi: 10.1358/dot.2016.52.3.2440527
Prinz, F., Schlange, T., and Asadullah, K. (2011). Believe it or not: how much can we rely on published data on potential drug targets? Nat. Rev. Drug Discov. 10, 712. doi: 10.1038/nrd3439-c1
Ragvin, A., Moro, E., Fredman, D., Navratilova, P., Drivenes, O., Engstrom, P. G., et al. (2010). Long-range gene regulation links genomic type 2 diabetes and obesity risk regions to HHEX, SOX4, and IRX3. Proc. Natl. Acad. Sci. U.S.A. 107, 775–780. doi: 10.1073/pnas.0911591107
Ramasamy, A., Trabzuni, D., Guelfi, S., Varghese, V., Smith, C., Walker, R., et al. (2014). Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat. Neurosci. 17, 1418–1428. doi: 10.1038/nn.3801
Roadmap Epigenomics Consortium, Kundaje, A., Meuleman, W., Ernst, J., Bilenky, M., Yen, A., et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. doi: 10.1038/nature14248
Sabatine, M. S., Giugliano, R. P., Keech, A. C., Honarpour, N., Wiviott, S. D., Murphy, S. A., et al. (2017). Evolocumab and clinical outcomes in patients with cardiovascular disease. N. Engl. J. Med. 376, 1713–1722. doi: 10.1056/NEJMoa1615664
Scannell, J. W., Blanckley, A., Boldon, H., and Warrington, B. (2012). Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug Discov. 11, 191–200. doi: 10.1038/nrd3681
Schmitt, A. D., Hu, M., and Ren, B. (2016). Genome-wide mapping and analysis of chromosome architecture. Nat. Rev. Mol. Cell Biol. 17, 743–755. doi: 10.1038/nrm.2016.104
Schunkert, H., Konig, I. R., Kathiresan, S., Reilly, M. P., Assimes, T. L., Holm, H., et al. (2011). Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat. Genet. 43, 333–338. doi: 10.1038/ng.784
Servant, N., Varoquaux, N., Lajoie, B. R., Viara, E., Chen, C.-J., Vert, J.-P., et al. (2015). HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16:259. doi: 10.1186/s13059-015-0831-x
Smemo, S., Tena, J. J., Kim, K. H., Gamazon, E. R., Sakabe, N. J., Gomez-Marin, C., et al. (2014). Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 507, 371–375. doi: 10.1038/nature13138
Smietana, K., Siatkowski, M., and Moller, M. (2016). Trends in clinical success rates. Nat. Rev. Drug Discov. 15, 379–380. doi: 10.1038/nrd.2016.85
Szklarczyk, D., Morris, J. H., Cook, H., Kuhn, M., Wyder, S., Simonovic, M., et al. (2017). The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 45, D362–D368. doi: 10.1093/nar/gkw937
Teng, L., He, B., Wang, J., and Tan, K. (2015). 4DGenome: a comprehensive database of chromatin interactions. Bioinformatics 31, 2560–2564. doi: 10.1093/bioinformatics/btv158
Thompson, D. J., O’Mara, T. A., Glubb, D. M., Painter, J. N., Cheng, T., Folkerd, E., et al. (2016). CYP19A1 fine-mapping and Mendelian randomization: estradiol is causal for endometrial cancer. Endocr. Relat. Cancer 23, 77–91. doi: 10.1530/ERC-15-0386
Tsherniak, A., Vazquez, F., Montgomery, P. G., Weir, B. A., Kryukov, G., Cowley, G. S., et al. (2017). Defining a cancer dependency map. Cell 170, 564–576.e16. doi: 10.1016/j.cell.2017.06.010
U.S. Department of Health and Human Services (2017). Drugs@FDA: FDA Approved Drug Products [Online]. Washington, DC: U.S. Department of Health and Human Services.
Visscher, P. M., Wray, N. R., Zhang, Q., Sklar, P., McCarthy, M. I., Brown, M. A., et al. (2017). 10 years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 101, 5–22. doi: 10.1016/j.ajhg.2017.06.005
Wagner, A. H., Coffman, A. C., Ainscough, B. J., Spies, N. C., Skidmore, Z. L., Campbell, K. M., et al. (2016). DGIdb 2.0: mining clinically relevant drug-gene interactions. Nucleic Acids Res. 44, D1036–D1044. doi: 10.1093/nar/gkv1165
Westra, H.-J., Peters, M. J., Esko, T., Yaghootkar, H., Schurmann, C., Kettunen, J., et al. (2013). Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243. doi: 10.1038/ng.2756
Whirl-Carrillo, M., McDonagh, E. M., Hebert, J. M., Gong, L., Sangkuhl, K., Thorn, C. F., et al. (2012). Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 92, 414–417. doi: 10.1038/clpt.2012.96
Xie, X., Ma, W., Songyang, Z., Luo, Z., Huang, J., Dai, Z., et al. (2016). CCSI: a database providing chromatin-chromatin spatial interaction information. Database (Oxford) 2016:bav124. doi: 10.1093/database/bav124
Yu, C. H., Pal, L. R., and Moult, J. (2016). Consensus genome-wide expression quantitative trait loci and their relationship with human complex trait disease. OMICS 20, 400–414. doi: 10.1089/omi.2016.0063
Keywords: drug repositioning, genetics, therapeutics, GWAS, eQTL, functional genomics
Citation: Pritchard J-LE, O’Mara TA and Glubb DM (2017) Enhancing the Promise of Drug Repositioning through Genetics. Front. Pharmacol. 8:896. doi: 10.3389/fphar.2017.00896
Received: 09 September 2017; Accepted: 22 November 2017;
Published: 06 December 2017.
Edited by:
Munir Pirmohamed, University of Liverpool, United KingdomReviewed by:
Sudeep Pushpakom, University of Liverpool, United KingdomKrishna Rani Kalari, Mayo Clinic, United States
Minoli A. Perera, Northwestern University, United States
Copyright © 2017 Pritchard, O’Mara and Glubb. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dylan M. Glubb, ZHlsYW4uZ2x1YmJAcWltcmJlcmdob2Zlci5lZHUuYXU=