 Allan K. N. Alencar
Allan K. N. Alencar Guilherme C. Montes
Guilherme C. Montes Eliezer J. Barreiro
Eliezer J. Barreiro Gisele Zapata-Sudo
Gisele Zapata-Sudo
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 04 December 2017
Sec. Experimental Pharmacology and Drug Discovery
Volume 8 - 2017 | https://doi.org/10.3389/fphar.2017.00858
This article is part of the Research TopicPurinergic Pharmacology, Volume IView all 62 articles
Pulmonary arterial hypertension (PAH) is a clinical condition characterized by pulmonary arterial remodeling and vasoconstriction, which promote chronic vessel obstruction and elevation of pulmonary vascular resistance. Long-term right ventricular (RV) overload leads to RV dysfunction and failure, which are the main determinants of life expectancy in PAH subjects. Therapeutic options for PAH remain limited, despite the introduction of prostacyclin analogs, endothelin receptor antagonists, phosphodiesterase type 5 inhibitors, and soluble guanylyl cyclase stimulators within the last 15 years. Through addressing the pulmonary endothelial and smooth muscle cell dysfunctions associated with PAH, these interventions delay disease progression but do not offer a cure. Emerging approaches to improve treatment efficacy have focused on beneficial actions to both the pulmonary vasculature and myocardium, and several new targets have been investigated and validated in experimental PAH models. Herein, we review the effects of adenosine and adenosine receptors (A1, A2A, A2B, and A3) on the cardiovascular system, focusing on the A2A receptor as a pharmacological target. This receptor induces pulmonary vascular and heart protection in experimental models, specifically models of PAH. Targeting the A2A receptor could potentially serve as a novel and efficient approach for treating PAH and concomitant RV failure. A2A receptor activation induces pulmonary endothelial nitric oxide synthesis, smooth muscle cell hyperpolarization, and vasodilation, with important antiproliferative activities through the inhibition of collagen deposition and vessel wall remodeling in the pulmonary arterioles. The pleiotropic potential of A2A receptor activation is highlighted by its additional expression in the heart tissue, where it participates in the regulation of intracellular calcium handling and maintenance of heart chamber structure and function. In this way, the activation of A2A receptor could prevent the production of a hypertrophic and dysfunctional phenotype in animal models of cardiovascular diseases.
Pulmonary hypertension (PH) refers to a complex group of cardiopulmonary diseases that may lead to right-sided heart failure and reduced life expectancy (Galie et al., 2016). Pulmonary arterial hypertension (PAH) represents group 1 within the PH classification system (Simonneau et al., 2013). PAH is characterized by precapillary PH (i.e., mean pulmonary arterial pressure ≥ 25 mmHg and normal pulmonary capillary wedge pressure ≤ 15 mmHg), as a consequence of exacerbated remodeling and hypertrophy of the walls of the pulmonary arteries (PAs) (Galie et al., 2016). The most severe form of PH, PAH has a prevalence of 15 cases per million adult population, incidence of 2.4 cases per million adult population per year (McGoon et al., 2013), and mean survival rate of 2.8 years in the absence of specific treatments (McGoon et al., 2013; Simonneau et al., 2013).
Current knowledge on PAH pathophysiology highlights microenvironmental changes in the pulmonary vessels as the initial cause of the disease. Crosstalk among endothelial cells (ECs), pulmonary arterial smooth muscle cells (PASMCs), myofibroblasts, pericytes, and circulating immunologic cells triggers PAH pathogenesis (Guignabert et al., 2015). Elevated pulmonary vascular resistance (PVR) causes insult to the right ventricular (RV) myocardium, with subsequent activation of neurohormonal, immunological, and mechanical-stretch signaling (Voelkel et al., 2006). The poor prognosis of PAH is mainly a consequence of long-term pressure overload in the RV chamber, which initially responds with adaptive myocardial hypertrophy. This response is followed by progressive contractile dysfunction, global heart failure, and premature death (Voelkel et al., 2006; Sztrymf et al., 2010).
Treatment of patients with PAH is palliative. Available agents include lung vasodilators, such as phosphodiesterase type 5 inhibitors, endothelin receptor antagonists, prostacyclin analogs, and guanylyl cyclase stimulators (Galie et al., 2016). Lung transplantation is an important strategy in eligible subjects with advanced PAH who are refractory to drug intervention (Galie et al., 2016). Current treatments of PAH simply control the intense PA vasoconstriction, but recent advances in understanding disease pathophysiology have driven efforts to develop new pharmacological strategies targeting the irreversible remodeling of the pulmonary vascular bed. Favorable actions in the hypertrophied and dysfunctional RV are required for better outcomes, and there is a clear need for pleiotropic targets with beneficial roles throughout the cardiopulmonary system (pulmonary vessel and cardiac cells).
Adenosine has a well-known pulmonary vasodilator effect with a rapid onset of action (Reeves et al., 1991; Nootens et al., 1995). Adenosine use is recommended as an alternative strategy in pulmonary vasoreactivity testing for identification of patients suitable for high-dose calcium channel blocker treatment (Galie et al., 2016). Patients suffering from PAH exhibit low adenosine levels in the pulmonary circulation, suggesting that this nucleoside plays a role in PAH pathophysiology. The reduction of the adenosine release on pulmonary vascular endothelium is consequent to the endothelial dysfunction that is present in PAH. Low levels in PAH may also be explained by increased activity of the adenosine deaminase in the pulmonary circulation (Saadjian et al., 1999), which was found in rats with hemolysis-associated pulmonary hypertension (Tofovic et al., 2009). However, clinical implications of the adenosine deaminase or its inhibitors in PAH remain unclear. Adenosine promotes various beneficial effects in the heart (Shryock and Belardinelli, 1997) and has been defined as a retaliatory metabolic product (Newby et al., 1985).
Long-term use of adenosine in PAH and RV failure has been limited by its extremely short half-life (Nootens et al., 1995) and adverse effects by non-selective activation of its four receptor subtypes. Nevertheless, adenosine receptors (ARs) promote multiple salutary actions in cardiac and vascular cells, including ECs, fibroblasts, and myocytes (McIntosh and Lasley, 2012; Headrick et al., 2013). Immune cells express ARs to respond to the modulatory effects of adenosine in an inflammatory environment (Hasko et al., 2008). Researchers from several fields are considering the possibility of targeting ARs as a therapeutic approach in various clinical conditions, including cerebral and cardiac diseases, sleep disorders, immune and inflammatory disorders, and cancer (Jacobson and Gao, 2006; Hasko et al., 2008; McIntosh and Lasley, 2012; Chen et al., 2013; Headrick et al., 2013).
This review compiles findings on the specific roles of each AR subtype in the cardiovascular system. The work focuses on AR activities in the small circulation, specifically in PAH pathogenesis, including the aberrant cellular proliferation and influx of inflammatory cells in and around various components of the vascular wall, as well as the distinct roles of ARs in RV cells. The main goal of this review is to answer an intriguing question: Does a specific AR exist that could be considered as a future target with pleiotropic potential in the whole cardiopulmonary system of patients with PAH?
Before addressing the potential benefits of targeting specific ARs for the treatment of PAH, we must understand the intrinsic pathophysiological mechanisms of this disease. Dynamic interactions among cells in the pulmonary vascular bed are the main determinant of cellular behavior (e.g., proliferation, apoptosis, differentiation, migration, and survival) in PA walls. Abnormal cell communication can lead to development of PAH (Eddahibi et al., 2006; Xu and Mao, 2011; Guignabert and Dorfmuller, 2013; Guignabert et al., 2015). Crosstalk between PA cells is regulated through direct cell–cell contact and release of bioactive factors, such as EC-produced paracrine factors that influence the proliferation of PASMCs and fibroblasts (Eddahibi et al., 2006; Sakao et al., 2009; Amabile et al., 2013).
Below a cross-sectional diameter of 500 μm (lobular to intra-acinar level), the medial layer of the PAs is typically composed solely of smooth muscle cells (SMCs), due to loss of elastic fibers. Beyond this level, the PAs leave their adjacent airways and become barely muscularized precapillary arterioles (Guignabert and Dorfmuller, 2013). Vascular remodeling in the lungs of PAH patients initially begins in this type of arteriole and presents as PASMC hyperproliferation, underlining the importance of this cell type in PAH pathogenesis (Schermuly et al., 2011; Guignabert and Dorfmuller, 2013). Early lesions correspond to hypertrophic and hyperplastic PASMCs within the tunica media (Guignabert and Dorfmuller, 2013). The abnormal proliferation of PASMCs during PAH development occurs in response to the signaling of growth factors, including potent mitogens and chemoattractants for vascular cells, which bind to and activate surface tyrosine kinase receptors (TKRs) (Schermuly et al., 2011).
Pulmonary ECs play a crucial role during the formation and maturation of blood vessels, producing and releasing growth factors that recruit and stabilize all vascular cells (Folkman and D’Amore, 1996; Hanahan, 1997). Eddahibi et al. (2006) showed that EC-induced PASMC growth was greater in tissues from PAH patients than from controls. Dysregulation of this process and excessive release of growth factors by ECs are intrinsic abnormalities linked to PAH pathogenesis. Indeed, abnormal communication between ECs and other vascular cells in PAH may occur due to loss-of-function of the endothelium.
Among different factors, bone morphogenic protein (BMP) signaling regulates pulmonary EC survival and differentiation (Teichert-Kuliszewska et al., 2006). Patients with PAH show abnormal BMP signaling, linked to mutations of bone morphogenetic protein receptor-2 (BMPR2) in ECs (Teichert-Kuliszewska et al., 2006). These mutations could lead to deleterious consequences in ECs and PASMCs. Loss of BMPR2 signaling in ECs may increase apoptosis in response to environmental stress and injury, particularly at the level of the distal PAs (Teichert-Kuliszewska et al., 2006). This apoptosis could be an initiating mechanism for PAH by leading to vessel obliteration due to degeneration of EC structures (Zhao et al., 2005). Excessive loss of ECs promotes the development of apoptosis-resistant and hyperproliferative ECs, which are characteristic features of later stages of PAH (Voelkel et al., 2002).
The initial trigger for pulmonary EC injury is unknown, although some events, such as shear stress, local inflammation, genetic predisposition, toxins, and reactive oxygen species (ROS)-induced cell damage, might be important inducers of endothelial dysfunction (Amabile et al., 2013). Exuberant proliferation of ECs leads to formation of plexiform lesions. These dynamic networks of vascular channels formed by the monoclonal proliferation of ECs (St Croix and Steinhorn, 2016) are a morphologic hallmark of severe PAH. A single plexiform lesion can occlude the entire length of an affected vessel (Cool et al., 1999). However, debate surrounds the functional importance of these lesions in the context of PAH, as it remains unclear if they have a role in disease progression or are simply a morphologic indicator of irreversible, end-stage disease (Tuder et al., 2007; Abe et al., 2010; Jonigk et al., 2011; St Croix and Steinhorn, 2016).
In PASMCs, signaling by BMPR2 is necessary for the control of cell proliferation and differentiation (Upton et al., 2008; Garcia de Vinuesa et al., 2016). Together with EC-induced cell proliferation and medial hypertrophy, BMPR2 mutations in PASMCs participate in the process of initiating or maintaining pulmonary vessel hypertrophy in PAH patients (Zhang et al., 2003).
Fibroblasts play important roles in PAH pathogenesis by responding to injury and chemoattraction via endothelium-derived growth factors. Rapid migration of fibroblasts to the injured vessel leads to formation of the neointimal layer and, most importantly, fibroblast transdifferentiation into other cell types, including myofibroblasts – an abnormal type of PASMC that contributes to muscularization of the distal vessels (Sartore et al., 2001; Sisbarro et al., 2005; Sakao et al., 2009).
Taken together, these findings indicate that any effect on BMPR2 signaling may trigger abnormal communication between ECs, PASMCs, and fibroblasts via growth factors. Subsequently, these growth factors trigger hyperproliferation and differentiation of cells, formation of plexiform lesions, obliteration and fibrosis of the vessels, and increases of PVR and pulmonary vascular pressure.
In an elegant review, Schermuly et al. (2011) summarized the growth factors that are most clearly implicated in PAH pathogenesis. The review showed increased signaling of the following molecules in injured pulmonary vascular cells: vascular endothelial growth factor (VEGF), transforming growth factor alpha and beta (TGF-α and TGF-β), platelet-derived growth factor (PDGF), and hepatocyte growth factor (HGF) (Schermuly et al., 2011). All of these growth factors bind to TKRs, resulting in multi-phosphorylation of tyrosine residues for the assembly of downstream signaling molecules that are recruited to the receptor and activated in response to agonist stimulation (Lemmon and Schlessinger, 2010). Subsets of intracellular signaling components influenced by growth factor receptor activation are intertwined in a complex network. Examples of intracellular molecules activated by TKRs are phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt), mitogen-activated protein kinases (Ras/Raf/MAPK/ERK), protein kinase C (PKC), Janus kinase (JAK), signal transducer and activator of transcription (STAT), and cyclins (a family of proteins that control cell cycle progression by activating cyclin-dependent kinases) (Koledova and Khalil, 2006; Lemmon and Schlessinger, 2010).
Roles of TKRs and their antagonists in PAH have been reviewed, and cardiac safety issues of these molecules have been raised, especially in patients with heart disease that is intrinsically related to pulmonary vascular disease (Godinas et al., 2013). The large blockage spectrum and lack of selectivity of TKR antagonists would provide unexpected toxicities, including injury to the pulmonary vasculature. Furthermore, the benefit-to-risk ratio of available TKR antagonists is low in the context of PAH (Godinas et al., 2013). Thus, new approaches for PAH treatment should be aimed at ensuring a low toxicity profile, in addition to the high anti-proliferative efficacy.
Accumulating evidence shows that RhoA, a member of the Ras homolog gene family, and its downstream effectors, the Rho kinases (ROCKs), mediate the pathogenesis of PAH through their pro-proliferative contributions (Nagaoka et al., 2006; Guilluy et al., 2009; Connolly and Aaronson, 2011). ROCK downregulates expression of anti-proliferative molecules, leading to acceleration of cell cycle progression and vascular cell hyperproliferation (Laufs et al., 1999; Sawada et al., 2000). Distal muscularization of pulmonary vessels has been linked to amplification of the RhoA/ROCK pathway in PASMCs (Emerson et al., 1999; Fagan et al., 1999; Aznar and Lacal, 2001; Keil et al., 2002).
Another important factor involved in endothelium-smooth muscle interactions, serotonin makes a major contribution to PASMC hyperplasia in PAH (Eddahibi et al., 2006). Serotonin-mediated PASMC proliferation was greater in mice with genetically mutated BMPR2 (Long et al., 2006). The BMP and serotonin mechanisms are convergent in PASMCs; both stimulate RhoA/ROCK signaling downstream for a pro-proliferative phenotype (Liu et al., 2009). These previous reports suggest that vascular cell proliferation induced by downstream growth factors could be attenuated, in part, through inhibition of RhoA/ROCK signaling.
The RhoA/ROCK pathway is of interest to pharmaceutical companies due to the pleiotropic effects of its inhibitors in numerous diseases (Feng et al., 2016). Fasudil is a ROCK inhibitor that has shown salutary effects in the cardiopulmonary system of PAH patients and in different experimental models of the disease. These effects derive from its anti-proliferative profile in the pulmonary vessels, which it achieves by reducing PVR and RV overload (Mouchaers et al., 2010; Raja, 2012; Fukumoto et al., 2013; Jiang et al., 2014; Xiao et al., 2015; Gupta et al., 2017). Nevertheless, the cardioprotective potential of this pharmacological modality is not completely clear, as the effect of fasudil on reducing PAH-related changes in the RV may be an indirect response of the lower afterload in the right heart chamber. In addition to the beneficial effects of ROCK inhibitors in the left heart (Demiryurek et al., 2005; Takeshima et al., 2012), their direct effects should be investigated in the RV cells, because the structures, hemodynamics, and functions of both ventricles are distinct.
Secondarily to the pro-proliferative phenotype in pulmonary vascular cells, damaged crosstalk between ECs and PASMCs can exacerbate PA vasoconstriction as a consequence of an imbalance in the production of endothelium-derived vasodilator [nitric oxide (NO) and prostacyclin] and constrictor factors (endothelin-1, serotonin, and angiotensin II). Crosstalk dysfunction is a pivotal element in the development and progression of the disease (Guignabert et al., 2015).
Furthermore, PAH is among many pathophysiologic conditions that are linked to inflammation (Schermuly et al., 2011). Inflammation has a pivotal role during development of PAH in humans and in animal models of the disease. Some patients with PAH present increased levels of tumor necrosis factor alpha (TNF-α), interleukins 12 and 6 (IL-12 and IL-6), and interferon-γ, associated with other inflammatory signals, such as plasma cell dyscrasia polyneuropathy. Induction of PAH in rodents by chronic hypoxia or monocrotaline leads to an increase in the number of inflammatory cells in the lungs (Minamino et al., 2001; Soon et al., 2010; Bi et al., 2013). Vascular and inflammatory cells are important local sources of cytokines and chemokines, which can trigger pulmonary vascular remodeling in PAH (Sutendra et al., 2011; Rabinovitch et al., 2014). Histopathological studies have demonstrated the presence of complement system components, autoantibodies, and inflammatory cells (neutrophils) in the vessel lumen, which can bind to the endothelium and may infiltrate the medial muscular layer. Inflammatory infiltrate in the neointimal layer is composed of T- and B-lymphocytes, with macrophages, mast cells, and dendritic cells present in the adventitial layer. Lymphoid follicles, characterized by T cells, B cells, and plasmacytoid dendritic cells (APCs), are found in the periadventitial space (Stacher et al., 2012; Rabinovitch et al., 2014). Thus, reversion of the pulmonary vessel injury in PAH requires an anti-inflammatory profile.
Adenosine is a purine nucleoside that is widely distributed in all tissues. Structurally, adenosine is formed by the linking of adenine to a ribose sugar molecule. Synthesis of adenosine is increased after myocardial infarction (MI) (Daly, 1982; Hasko and Cronstein, 2004; Asakura et al., 2007; Hisatome, 2007). Adenosine is synthesized inside and outside the cells for immediate use and is not stored in vesicles for future release. For these reasons, this molecule is an autacoid, similar to several biological transmitters (Newby and Holmquist, 1981; Deussen et al., 1989; Deussen and Schrader, 1991; Decking et al., 1997; Zimmermann, 2000).
Extracellular production of adenosine depends on a cascade reaction initiated by ATP and ADP as substrates (Dunwiddie et al., 1997). ATP and ADP are converted to AMP by catalysis of ectonucleoside triphosphate diphosphohydrolases (NTPDases 1, 2, 3, and 8) on the cell surface, followed by AMP hydrolysis to adenosine by ecto-5′-nucleotidase (NTE5/CD73) (Ballard, 1970; Headrick and Willis, 1990; Decking et al., 1997; Bak and Ingwall, 1998; Gustafson and Kroll, 1998). Intracellular adenosine might be provided through other signaling processes. One of these events is the catabolism of ATP from stressed cells under hypoxia (Ham and Evans, 2012). This condition might be responsible for the perturbed recycling of adenosine and ATP, and the increased activity of specific phosphatases leads to excessive levels of adenosine (Spychala, 2000). Another source of intracellular adenosine is the cytosolic 5′-nucleotidase-mediated metabolism (dephosphorylation) of AMP, which is formed from the degradation of cyclic AMP (cAMP) by phosphodiesterases. Intracellular adenosine is also importantly generated by the conversion of S-adenosylhomocysteine to adenosine and homocysteine, catalyzed by S-adenosylhomocysteine hydrolase (Kloor and Osswald, 2004; Hermes et al., 2007; Antonioli et al., 2015).
Adenosine is converted, extra- or intracellularly, to inosine by adenosine deaminase (Ford et al., 2000; Singh and Sharma, 2000; Antonioli et al., 2012). Phosphorylation of adenosine to AMP by adenosine kinase reduces the intracellular concentration of adenosine (De Jong, 1977; Drabikowska et al., 1985; Pak et al., 1994; Boison, 2013). The adenosine concentration depends on two families of transmembrane proteins, concentrative and equilibrative nucleoside transporters, which facilitate the movement of nucleosides, nucleobases, and their analogs across the cell membrane. There are four isoforms of human equilibrative nucleoside transporters, but only ENT1 and ENT2 are expressed in tissues throughout the body (King et al., 2006; Playa et al., 2014). The adenosine transport process usually follows gradients from high to low concentration, with no energy spending (Chen et al., 2013). The extracellular adenosine concentration varies substantially between tissue types (Ramakers et al., 2008). The local adenosine concentration may be increased from the nano- to the micromolar range, signaling tissue injury, by extreme pathophysiological cell activation (seizure) or insults due to ischemia, trauma, inflammation, or cancer (Fredholm et al., 2001; Linden, 2001; Flamand et al., 2006; Hasko et al., 2008; Antonioli et al., 2012, 2013; Hasko and Cronstein, 2013).
Drury and Szent-Gyorgyi (1929) described adenosine as a potent vasodilator-like molecule. In the cardiovascular system, adenosine offers protective effects against episodes of angina pectoris, preconditioning, and ischemia/reperfusion (I/R) injury (Olafsson et al., 1987; Lasley and Mentzer, 1992; Woolfson et al., 1996; Miura et al., 2000). Adenosine mediates its physiological effects on tissue regeneration and repair by binding to and activating a family of G-protein coupled receptors, denoted P1 purinoceptors and adenosine receptors A1, A2A, A2B, and A3 (Ralevic and Burnstock, 1998; Merighi et al., 2015), all expressed in the cardiovascular system (Headrick et al., 2013).
The transduction pathways involved in adenosine signaling through its receptors in the cardiovascular system are complex. A1 receptor may couple to different G-proteins in order to regulate adenylyl cyclase (AC), phospholipase C (PLC), Ca2+ and K+ channel functions, and possesses a high affinity for adenosine (Fredholm et al., 2000). When it is coupled to Gi protein it inhibits AC and modulates K+ and Ca2+ channels activities. But it may also couple with Gs to activate AC, or with Gq/11 to stimulate PLC enzyme and, subsequently, the inositol triphosphate (IP3) and diacylglycerol (DAG) production (Headrick et al., 2013). It was also described that A1 receptor promotes K+ efflux via βγ subunit of G-protein inwardly rectifying channels (GIRK/KIR3) (Kurachi et al., 1986; Kirsch et al., 1988; Belardinelli et al., 1995; Mubagwa and Flameng, 2001).
The A2A receptor is very sensitive to adenosine (Fredholm et al., 2000). This receptor couples with Gs to activate AC and increase cAMP levels in the cardiovascular system and it interacts with A1 receptor, dopamine D2, metabotropic glutamate 5, NMDA and cannabinoid CB1 receptor in other organs, but the specific functions and occurrence of such interactions remains unknown (Headrick et al., 2013).
Adenosine has the lowest affinity for the A2B receptor subtype (Fredholm et al., 2000). A2B signaling is mediated via Gs protein to stimulate AC activity, but it also may couple to Gq/11 protein to activate PLC (Headrick et al., 2013). It was reported that A2B receptor might also modulate the arachidonic acid cascade (Donoso et al., 2005). Additionally, A2B signaling can increase endothelial NO bioavailability with subsequent KATP channel opening in rodent vascular cells (Morrison et al., 2002; Hinschen et al., 2003). A2B may also couple to p38-MAPK in coronary vessels (Teng et al., 2005).
The A3 receptor is the newest subtype that was identified (Fredholm et al., 2000). Similarly to A1 receptor, it can couple to Gi protein to inhibit AC activity, and also to Gq/11 to modulate PLC and Ca2+ intracellular handling (Headrick et al., 2013). Involvement of A2A and A2B receptor-mediated vasodilation has been reported in several vessels, including the muscular arteries [mesenteric (Hiley et al., 1995), renal (Rump et al., 1999), and coronary arteries (Flood and Headrick, 2001)] and elastic arteries, including the aorta of several species [guinea pigs (Stoggall and Shaw, 1990), rats (Prentice and Hourani, 1996), and hamsters (Prentice and Hourani, 2000)]. Adenosine relaxes precontracted isolated PA rings, an effect that probably occur via A2A and A2B receptor activation (El-Kashef et al., 1999). Some investigators have suggested that vascular relaxation in response to A2A activation may be independent of ECs (Pearl, 1994), whereas others have shown substantial EC involvement (Martin and Potts, 1994). Resolving this controversy, researchers demonstrated that A2A is located in both the vascular endothelium and vascular SMCs (Leal et al., 2008), and that its activation is involved in vasodilation (Lewis et al., 1994; Prentice and Hourani, 1996).
Activation of the Gs protein-coupled endothelial A2A receptor triggers NO release by activating the AC-protein kinase A (PKA) pathway (Ikeda et al., 1997; Ray and Marshall, 2006). Activation of A2A in vascular SMCs increases formation of cAMP and activation of PKA, which leads to phosphorylation and opening of potassium channels. This effect, in turn, causes hyperpolarization and vasodilatation (Ko et al., 2008). In contrast, the A1 and A3 receptors negatively modulate A2A-B receptor-induced vasodilation (Talukder et al., 2002; Mustafa et al., 2009; Ponnoth et al., 2009). The A1 receptor plays a negative role in regulating blood pressure, causes contraction of vascular smooth muscle, and decreases coronary blood flow (Ponnoth et al., 2009). These findings indicate that adenosine can act as a vasoconstrictor or a vasodilator, depending on its interaction with specific receptor subtypes, plasma levels, and tissue localization. Differences in the adenosine regulation of the pulmonary vascular tonus might be explained, in part, due to the higher affinity of this nucleoside for the A1 receptor (promotes pulmonary vasoconstriction) than for A2B receptor (promotes pulmonary vasodilation). Saadjian et al. (1999) have described a correlation of the progression of PH and the lower level of adenosine observed in patients. Low adenosine levels in lungs from PH patients are likely to stimulate A1 more than A2B receptors, thus contributing to the vasoconstriction in those subjects.
The pulmonary circulation is one of the few regions in the body where activation of ARs has a dual function (i.e., promoting both contraction and relaxation) that depends on the basal tone of the blood vessel (Cheng et al., 1996). In pulmonary vessels, A1 receptors are stimulated under circumstances of low vasculature tone, thereby promoting vasoconstriction; when vessel tone is high, A2 receptor subtypes are activated and promote vasodilation (Tabrizchi and Bedi, 2001). We can extrapolate these findings to the context of PAH. In terms of the induction of pulmonary vessel relaxation, ARs in the large PAs (vs. small arteries) were reported to be the A2B subtype (vs. A2A subtype) (Tabrizchi and Bedi, 2001). This information is relevant in the pathological environment of PAH, as the disease begins in the distal microvasculature. Thus, we may assume that activation of A2 receptor subtypes in the pulmonary vessels could be targeted to reduce the extremely high vascular tone in PAH patients.
Pharmacological manipulation of adenosine signaling is of great interest for numerous cardiovascular conditions. Several potential agonists or antagonists of ARs are being studied in experimental models of left ventricle (LV) ischemia and dysfunction and systemic hypertension. Whether the development of selective and potent ligands for ARs could be an interesting treatment alternative in the context of PAH and RV failure remains speculative.
Repair of the pulmonary vessel cells in patients with PAH could be promoted by inhibition of the migration and/or proliferation of ECs, PASMCs, and/or fibroblasts. In this section, we describe recent findings for each AR subtype regarding their roles as targets to control PA wall remodeling and subsequent hypertrophy.
Several studies demonstrated the presence of ARs in lung in different species. For example, A1, A2B, and A3 receptors were detected in rat airway SMCs (Michoud et al., 2002). In humans, levels of A2B transcripts were the highest in bronchial SMCs; A1 and A2A transcripts were detected as well, but A3 transcripts were below the detection limit (Zhong et al., 2004). Immunohistochemical analyses of human lung parenchyma demonstrated expression of A2A and A3 receptors in bronchiolar and alveolar epithelial cells, SMCs in bronchiolar and vessel walls, and ECs in the PAs (Varani et al., 2006). In contrast, A2B receptor was expressed only in mast cells and macrophages, and A1 receptor was expressed only in a few alveolar macrophages (Varani et al., 2006). Differences in AR expression between rodents and humans might be due to the cell- and tissue-specific effects of hypoxia on their expression (Fozard and Hannon, 2000).
Few studies have addressed specific roles of the A1 receptor in the growth regulation of pulmonary vascular cells. This receptor subtype is down regulated in the pulmonary vasa vasorum ECs of calves with experimentally induced neonatal PH and in these cells the A1 receptor activation leads to actin cytoskeletal remodeling and a barrier formation in vasa vasorum. A1 activation in ECs could be targeted with the goal of reducing neovascularization and function of the vasa vasorum, indirectly contributing to the integrity of the pulmonary vasculature by preventing the triggering of inflammation (Umapathy et al., 2013). Nevertheless, A1 receptor could be only a vascular bed-specific target for PAH in advanced stages, by blocking vasa vasorum expansion in large pulmonary vessels. Although the knowledge of the influence of A1 receptor in animal model of PAH, this receptor is poorly expressed in human pulmonary vascular cells (Varani et al., 2006). Thus, it could be considered that A1 receptor may not be relevant to the progression of HAP, but it is important further evaluation to characterize specific functions of this adenosine receptor subtype in the small lung vasculature from PAH patients, since in some pathological conditions the adenosine receptors pattern might be changed.
A2A is the most well-described AR subtype in the pulmonary circulation and in the context of PAH. Using an A2A receptor knockout (KO) mouse model, Xu et al. (2011) provided the first evidence of the critical contribution of A2A to PAH development. At a postnatal age of 14–16 weeks, A2A KO mice exhibited hemodynamic, histological, and ultrastructural characteristics suggestive of PAH. These changes included increases in RV systolic pressure, RV mass, and wall area and thickness, cellular proliferation in pulmonary resistance vessels, activation and hypertrophy of the PASMCs and ECs, and collagen deposition in the PA wall adventitia (Xu et al., 2011). The spontaneous PAH and altered PA remodeling were supported by the anatomical localization of A2A in the vasculature, further demonstrating the functional activation of A2A in ECs. These findings suggest that the effect of adenosine in PAH is likely mediated by the A2A receptor in pulmonary vessels (Xu et al., 2011).
Recently, the same research group showed that A2A KO mice exhibited key pathogenic characteristics of PAH, including muscularization of the pulmonary arterioles, PA remodeling, lumen narrowing, proliferation of pulmonary vascular ECs and SMCs, excessive hypertrophy of fibroblasts, and collagen deposition. A2A KO mice overexpressed RhoA and ROCK mRNA and protein. As mentioned above, activation of RhoA/ROCK signaling may cause pulmonary vascular remodeling and development of PAH. Thus, this experimental study provides sufficient evidence for validation of the A2A receptor as an anti-remodeling target in the pulmonary circulation (Shang et al., 2015). As such, this receptor may be a promising target for PAH therapy in the future (Antoniu, 2012). We agree with the authors on the need to confirm the specific downstream biochemical pathways that lead to inhibition of RhoA/ROCK signaling by A2A receptor activation.
Salidroside, an active ingredient isolated from Rhodiola rosea, has multiple pharmacological activities, including anti-inflammatory, antioxidation, antistress, anticancer, and immune-enhancing effects. Salidroside effectively inhibited chronic hypoxia-induced PAH and PA remodeling by increasing A2A expression and enhancing A2A-related mitochondria-dependent apoptosis. The authors suggested that it could be targeted for reducing vessel wall remodeling and hypertrophy in PAH (Huang et al., 2015).
New compounds of the class N-acylhydrazones were designed and synthetized to act as agonists of AR. The lead compound for these new derivatives was named LASSBio-294, which showed beneficial activity in rat model of MI (Costa et al., 2010; da Silva et al., 2014) via activation of the A2A receptor (da Silva et al., 2017). LASSBio-294 has shown low efficacy in the rat model of PAH (data not published) which leads to explore the influence of adding the methyl group in the N-acylhydrazone function as a chemical strategy to optimize its pharmacological properties. Thus, among the new series of methylated N-acylhydrazone derivatives, LASSBio-1359 (EC50 = 6.6 ± 1.7 × 10-6 M) (Alencar et al., 2013) and LASSBio-1386 (EC50 = 6.8 ± 0.6 × 10-6 M) (Alencar et al., 2014), showed a more potent vasodilator effect in rat PAs than LASSBio-294 (EC50 = 7.0 ± 0.7 × 10-2 M). Those data reinforced the fact that the N-methyl group markedly changes the conformation of N-acylhydrazone function in the pulmonary vascular target. It was also previously described the importance of the presence of a small lipophilic group in substitution of the amide group of the N-acylhydrazone moiety to the increased cardiovascular effects (Silva et al., 2005).
Both compounds, LASSBio-1359 and LASSBio-1386, promoted PA vasodilation by the activation of the A2A receptor, since their vascular effects were reduced by the selective antagonist of the A2A receptor, ZM 241385 (Alencar et al., 2013, 2014). The binding assays for both substances confirmed their higher selectivity for the A2A receptor subtype than for others (e.g., A1, A2B, and A3). LASSBio-1359 (10 μM), significantly inhibited the binding of agonist to the A2A receptor (CGS21680) by an average of 78.6%, while for other receptors it did not surpass 30% inhibition (Alencar et al., 2013). Furthermore, computational docking studies demonstrated how LASSBio-1359 and LASSBio-1386 might interact with the amino acid residues of the A2A receptor crystal structure compared to the agonist CGS21680, supporting the fact that they were ligands of A2A receptors (Alencar et al., 2013, 2014).
The involvement of the A2A receptor in the regulation of cardiopulmonary physiology was investigated after administration of LASSBio-1359 and LASSBio-1386 in rats with monocrotaline-induced PAH (Alencar et al., 2013, 2014). Animals with monocrotaline-induced PAH, exhibited intense pulmonary microvessel remodeling and hypertrophy and when treated with LASSBio-1386 showed reductions of the proliferative changes in the pulmonary arterioles and pulmonary vascular remodeling. It was also observed a downregulation of the A2A receptor in pulmonary tissue and RV tissue from rats with PAH (Alencar et al., 2014). Another A2A receptor ligand, LASSBio-1359, induced pulmonary vascular relaxation and promoted recovery of endothelial function in PA rings from rats with PAH. Monocrotaline-induced PAH produced fibromuscular hypertrophy and hyperplasia in the arteriole walls which in turn increased the RV systolic pressure and led to RV hypertrophy. However, daily oral treatment with the A2A agonist abolished the increased RV overload and reduced vessel wall hypertrophy. Importantly, LASSBio-1359 exhibited satisfactory efficacy through long-term oral administration regimens in the cardiopulmonary system of rats with PAH, with no side effects in the systemic circulation, such as hypotension or a compensatory increase in heart rate (Alencar et al., 2013).
Stimulation of the A2A receptor effectively reduced neointimal layer formation in a murine model of carotid artery ligation (McPherson et al., 2001). Collagen deposition and wall thickening were increased in the adventitial layer of PA walls from A2A KO mice (Xu et al., 2011). Therefore, we speculate that in addition to its anti-inflammatory effects (addressed later in this review), A2A exhibits anti-remodeling activity in the pulmonary vascular bed, through which it can reduce fibroblast migration and transdifferentiation into myofibroblasts during PAH pathogenesis. This possibility was supported by our study showing that the A2A receptor agonist LASSBio-1359 reduced collagen deposition in the pulmonary arterioles of PAH rats (Alencar et al., 2013). Figure 1 shows an overview of the beneficial effects of A2A receptor in the pathogenesis of PAH.
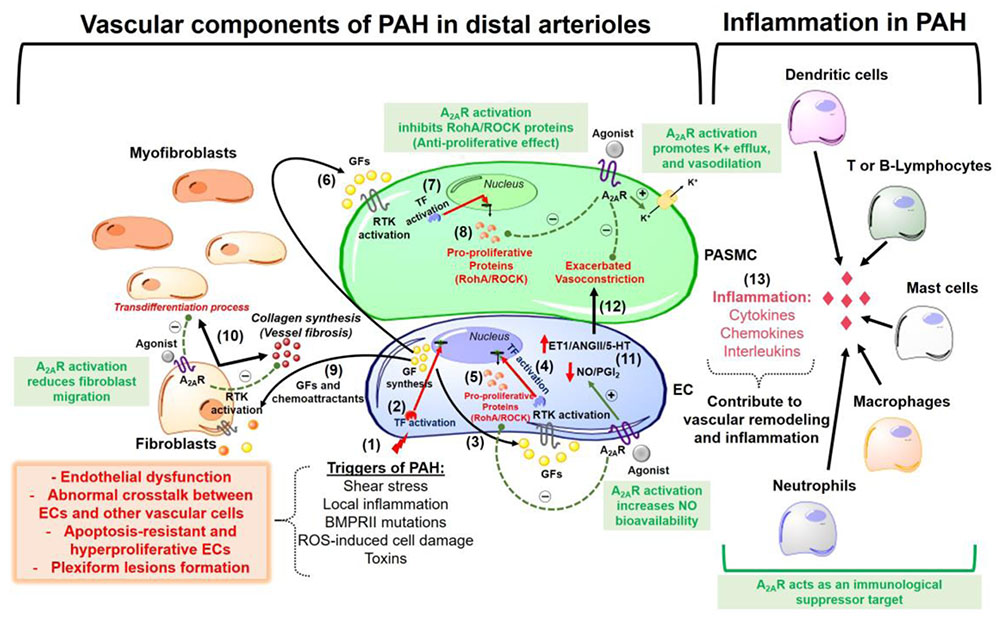
FIGURE 1. Overview of pathophysiological mechanisms responsible for pulmonary arterial hypertension (PAH) development in lung pre-capillary arterioles. The beneficial effects of the adenosine A2A receptor activation are highlighted in the green boxes. The events are sequentially numbered: (1) Triggers of PAH initially promote endothelial cells (EC) injury and dysfunction. The apoptosis-resistant ECs develop a hyperproliferative phenotype with a subsequent formation of obstructive plexiform lesions and this might be explained by (2) activation of transcription factors (TF) that promote a nuclear codification for an abnormal synthesis of growth factors (GF) and chemoattractants. (3) These GFs act activate receptors tyrosine kinase (RTK) in the ECs (autocrine signaling). (4) activation of RTK in ECs promotes gene transcription for the synthesis of (5) pro-proliferative proteins (e.g., RhoA/ROCK proteins). (6) ECs also release GFs that act in the pulmonary arterial smooth muscle cells (PASMC) triggering a paracrine signaling which is similar to what happens after activation of RTKs in ECs (steps 7 and 8) for the synthesis of pro-proliferative proteins. (9) Endothelial-derived chemoattractants and GFs also are responsible by fibroblasts migration and (10) collagen deposition (vessel wall fibrosis) with subsequent transdifferentiation into myofibroblasts. Other pathogenic characteristics of PAH are (11) increased endothelial production of vasoconstrictor molecules [endothelin-1 (ET-1), angiotensin II (AngII) and serotonin (5-HT)] and decreased release of vasodilators as nitric oxide (NO) and prostacyclin (PGI2), an event that is responsible by the (12) exacerbated vasoconstriction in the disease progression. (13) An inflammatory environment is formed by recruitment of several immunological cells, all of which releasing molecules as cytokines, chemokines, and interleukins that contribute to vascular remodeling and lung vessel injury through inflammation. A2AR, adenosine A2A receptor; ROS, reactive oxygen species; RhoA/ROCK, pro-proliferative Rho kinases.
Of the four ARs, A2B has emerged as the receptor that regulates many of the adenosine-driven remodeling responses seen in chronic lung diseases (Karmouty-Quintana et al., 2013b). Although there have been no studies of the A2B receptor in in vivo animal models or clinical PAH, there have been some studies of its roles in PH due to lung fibrosis or chronic obstructive pulmonary disease (COPD).
Pulmonary hypertension is a common and deadly complication of interstitial lung disease (Behr and Ryu, 2008). Genetic removal of the A2B receptor or treatment with its selective antagonist attenuated vascular remodeling in a mouse model of PH related to lung fibrosis. Karmouty-Quintana et al. (2012) proposed that A2B receptor activation can promote the release of endothelin-1 and IL-6 from ECs and PASMCs, respectively, potentiating vessel wall remodeling and evolution to a PH phenotype. These authors later demonstrated an upregulation of the adenosine axis in lungs from patients with PH secondary to idiopathic pulmonary fibrosis, leading to enhanced accumulation of adenosine and expression of A2B. The authors stated that, under acute conditions, hyperactivation of the A2B receptor by adenosine is protective and leads to lung tissue repair. In humans with lung fibrosis, however, sustained activation of the receptor is deleterious and contributes to development of pulmonary vascular remodeling and PH (Garcia-Morales et al., 2016).
Varani et al. (2006) reported that the A2B receptor is expressed only in mast and macrophage immunologic cells of human lung. Recently, researchers found that conditional deletion of the A2B receptor from myeloid cells was able to alter the phenotype of macrophages and dampen the development of fibrosis in a mouse model of lung injury-induced PH. The authors suggested a role for A2B-expressing myeloid cells as important regulators of fibrosis. These findings open the possibility of selectively blocking this AR subtype on macrophages as a novel therapeutic strategy for lung fibrosis and PH secondary to chronic lung diseases (Karmouty-Quintana et al., 2015).
Patients with COPD frequently develop PAH, characterized by extensive remodeling of the pulmonary vasculature due to increased proliferation of PASMCs and ECs, muscularization of previously non-muscular arteries, increased vascular tone, and formation of complex vascular lesions. Alveolar hypoxia, inflammation, and emphysema in patients with COPD contribute to remodeling, vasoconstriction, and reduction of the vascular bed (Chaouat et al., 2008). Patients with both COPD and PH present remodeled vessels, characterized by increased smooth muscle and collagen deposition. Patient lung sections had elevated A2B transcript levels, which were significantly correlated with increased PA pressures. Findings from this study suggest a role for A2B receptor antagonism as treatment for PH secondary to COPD (Karmouty-Quintana et al., 2013a).
On the other hand, in an in vitro model of cultured PASMCs under hypoxic conditions, Qian et al. (2013) showed that activation of the A2B receptor induced antiproliferative effects. In another report, A2B receptor stimulation counteracted PDGF-induced proliferation of human coronary SMCs through activating the exchange protein activated by cAMP (Mayer et al., 2011). Antiproliferative effects mediated by A2B receptors have been observed in studies of rat and human aortic and pre-glomerular SMCs and in human coronary SMCs (Dubey et al., 1996, 1998, 2010; Jackson et al., 2010, 2011). In aortic-cultured SMCs, the proliferation-inducing transcription factor B-Myb elevated endogenous A2B receptor mRNA and receptor activity levels, which, in turn, decreased cell proliferation (St Hilaire et al., 2008). Accordingly, we assume that it would be of great importance to evaluate the specific roles of the A2B receptor subtype and its agonists in both clinical and pre-clinical PAH experiments for further considerations about using this receptor as a target for the disease treatment. What would be an innovative strategy, antagonism or agonism of the A2B receptor in PAH patients? This question might give a clue for the basic and clinical science about discovering any beneficial strategy concerning A2B receptor for the treatment of PAH and its symptoms.
Regarding the specific roles of the A3 receptor in the proliferation of pulmonary vessel cells in the context of PAH, we did not find consistent studies to discuss in this review. Nevertheless, experiments performed in aortas from A3 receptor-deficient mice showed that this receptor has a role in increasing the proliferation of vascular SMCs (Jones et al., 2004). Another study demonstrated that A3 receptor activation in cultured human coronary SMCs was coupled to SMC proliferation via activation of phospholipase C and MAPK, with subsequent induction of the early growth response proteins EGR2 and EGR3 (Hinze et al., 2012).
There is divergent information regarding the A1 receptor in the inflammatory process. In several animal models of inflammation, A1 receptor mediated anti-inflammatory effect (Liao et al., 2003; Lee et al., 2004; Tsutsui et al., 2004; Kim et al., 2008), however, A1 receptor is also implicated with altered vascular response and systemic inflammation in an allergic mouse model of asthma (Ponnoth et al., 2010). Moreover, the block of A1 receptor attenuated endotoxin-induced lung injury in cats (Neely et al., 1997). Immune cells appear to exert activity mediated by activation of the A1 receptor because its activation may promote neutrophils adherence to endothelial cell and chemeotaxis (Barletta et al., 2012). Neutrophils are cells recruited to tissue in response to pathogen (host defense) or some inflammatory disease (Barletta et al., 2012). In early stage of inflammation the concentration of adenosine is low which may evoke neutrophil recruitment (Cronstein et al., 1990).
Evidences have suggested that anti-inflammatory effect via activation of A2A receptor is a result of a positive stimulation of adenylate cyclase (AC) system with increased PKA levels and subsequently decrease NF-κB signaling through lower release of pro-inflammatory cytokines, such as TNF-α and IL-1β. Moreover, to decrease the release of interleukins and chemokines, the A2A receptors activation inhibits events that occur during an immune response such as antigen presentation, adhesion and trafficking cellular, cell proliferation (Milne and Palmer, 2011; Headrick et al., 2013; Geldenhuys et al., 2017). A2A receptors are expressed in CD4+ T lymphocytes cells which are inhibited in the heart after infarction of myocardium being the primarily target to A2A agonist modulating a protective effect in heart (Yang et al., 2006). Furthermore, A2A agonists reduce CD3+ T lymphocytes and neutrophils which could inhibit the cellular adhesion consequently no transmigration across endothelial cell occurs (Yang et al., 2005; Hasko and Pacher, 2008; Barletta et al., 2012). Macrophage is a defense cell that can also migrate to inflammatory location and its regulation might be through adenosine receptor activation. There are two phenotypes of macrophage: phenotype 1 (M1) which is characterized by expression of several inflammatory cytokines (TNF-α, IL-1β, IL-6, and IL-12) and chemokines ensuring an inflammatory profile and phenotype 2 (M2) which controls inflammation and improves tissue healing. Stimulation of A2A receptors might promote the change in the macrophage phenotype 1 to type 2 (Hasko and Cronstein, 2013; Cronstein and Sitkovsky, 2017) promoting reduction of inflammation.
The A2B receptors are preferentially coupled to stimulatory G protein (Fredholm et al., 2001), but also couple to Gq protein leading to the regulation of intracellular calcium levels (Feoktistov and Biaggioni, 1997; Linden et al., 1999). Activation of A2B receptors might mediate mast cell function, increasing degranulation and stimulation of IL-8 secretion (Feoktistov and Biaggioni, 1995, 1996, 1997). In addition, the A2B receptors activation increases IL-6 production by pulmonary fibroblasts, leading to the formation of myofibroblasts that deposit extracellular matrix (Hasko et al., 2008). In contrast, A2B receptors might modulate beneficial anti-inflammatory effect through the inhibition of neutrophils activity (Barletta et al., 2012). Similarly to A2A receptors, A2B receptors when stimulated produce a change in the macrophage profile from phenotype 1 to phenotype 2.
The A3 receptors are widely expressed on immune cells and, upon binding their agonists, can activate phospholipase C and mediate inhibition of the PI3K/Akt and NF-kB signaling pathways to suppress production of TNF-α, IL-12, and IL-6 (Fishman et al., 2006, 2012; Varani et al., 2010b; Lee et al., 2011; Faas et al., 2017).
In an important review, Voelkel et al. (2006) described the most relevant structural and functional changes to occur in the RV chamber subsequent to long-term pressure overload due to elevated PVR. The initial response of the right heart is myocardial remodeling and hypertrophy to compensate for the elevated postload, accompanied by progressive contractile dysfunction. Finally, the chamber dilates to allow compensatory preload and to maintain stroke volume despite the reduced systolic function. Patients may develop clinical evidence of RV failure, including elevated filling pressure, diastolic dysfunction, and reduced cardiac output. The LV develops diastolic dysfunction due to the increased size and pressure overload of the RV, such that PAH results in global heart failure (Voelkel et al., 2006). Maladaptive RV remodeling and subsequent hypertrophy might be accelerated by neurohormonal signaling, oxidative stress of cardiac cells, metabolic changes, and myocardial inflammation, which affect the cardiomyocytes, cardiac fibroblasts, and coronary vascular cells (Ryan and Archer, 2014). Thus, we propose that a cardioprotective target should be beneficial by acting throughout all of the cardiac cells.
In the heart, adenosine might modulate the growth and death of cardiomyocytes, cardiac fibroblasts, ECs and SMCs, as well as affect the extracellular matrix (Headrick et al., 2013). One important study evaluated the impact of chronic heart failure on the adenosine system and the effects of its stimulation on disease development in humans.
Patients with moderate heart failure who were treated with dipyridamole, an adenosine uptake inhibitor, exhibited reduced AR expression, increased adenosine levels, and symptom improvement (Asakura et al., 2007). Hence, disturbances of the adenosine system might contribute to development of chronic heart failure.
Although preclinical and clinical studies have shown benefits of activating the A1 receptor in the context of LV dysfunction and failure (Ely and Berne, 1992; Headrick et al., 2003; Liao et al., 2003; Peart and Headrick, 2007; Chuo et al., 2016; Dinh et al., 2017), there is a lack of data regarding the effects of this receptor subtype in experimental models of PAH-induced RV impairment and global heart failure. We may discuss that the current unclear specific roles of this adenosine receptor subtype in the context of PAH should be the cause of researchers to consider it of low relevance.
Adenosine plays roles in the inflammatory process of LV heart failure. In cardiomyocytes from patients with LV heart failure, treatment with adenosine or the selective A2 receptor agonist DPMA decreased TNF-α expression levels by 40% or 87%, respectively (Wagner et al., 1998). Activation of the A2A receptor mediated the inflammatory process via activation of PKA, which, in turn, inhibited synthesis of proinflammatory molecules, such as TNF-α and IL-1β (Varani et al., 2010a; Impellizzeri et al., 2011).
The A2A receptor is expressed in mast cells (Marquardt, 1994), neutrophils (Fredholm et al., 1996), and CD4+ T cells (Koshiba et al., 1999). Xu et al. (1996) showed that adult rat ventricular myocytes express adenosine A2A receptor messenger RNA and through an immunoblotting technique, it was demonstrated that they express A2A receptor protein (Kilpatrick et al., 2002). Although numerous in vivo studies of the A2A receptor implicate its anti-inflammatory effects, some studies have suggested that its cardioprotective activities could be due, at least in part, to direct myocardial effects (Woodiwiss et al., 1999; Monahan et al., 2000; Chandrasekera et al., 2010; McIntosh and Lasley, 2012). Constitutive overexpression of the A2A receptor in young mice with LV dysfunction was associated with increases in cardiac contractility, heart rate, and LV mass (Chan et al., 2008).
Cardiac overexpression of the A2A receptor showed protective effects by attenuating fibrosis and improving cardiac function in a mouse model of heart failure (Hamad et al., 2012). Another work showed that this receptor is expressed in cardiac fibroblasts, where it has an antifibrotic role (Sassi et al., 2014).
As addressed earlier in this review, our laboratory has synthesized two new molecules, LASSBio-1359 and LASSBio-1386, from the lead compound LASSBio-294. Using in silico approaches, we identified a putative docking pose of this compound class at the A2A receptor. In vitro assays of all three compounds indicate A2A receptor agonist activity, although second messenger assays and full binding curves were not performed. The compounds exhibited vascular benefits and cardioprotective activities when administered chronically in rats with MI-induced LV heart failure (Costa et al., 2010; da Silva et al., 2014, 2017) or PAH-induced RV heart failure (Alencar et al., 2013, 2014). LASSBio-294 was chronically administered to normotensive and spontaneous hypertensive rats 4 weeks after MI. In addition to showing inotropic and lusitropic activities, this drug decreased cardiac remodeling, reduced cell infiltration, and improved Ca2+ influx into the sarcoplasmic reticulum in two different studies (Costa et al., 2010; da Silva et al., 2017). In a third work, chronic administration of LASSBio-294 to rats 4 weeks after MI reduced exercise intolerance by recovering Ca2+ homeostasis in the skeletal muscle. LASSBio-294 prevented cellular infiltration into the skeletal muscle, similarly to what was described for cardiac muscle. We have proven that the beneficial effects of LASSBio-294 occur through its activation of the A2A receptor (da Silva et al., 2017) in the cardiac and skeletal muscles (da Silva et al., 2014) and the subsequent increase of cAMP levels.
Given its salutary effects in animal models of left heart disease, we hypothesized that the A2A receptor should show cardioprotective potential in both LV and RV tissues. We chronically administered our new adenosine A2A agonists LASSBio-294 (data not published) LASSBio-1359 and LASSBio-1386 to rats with monocrotaline-induced RV failure (Alencar et al., 2013, 2014). Our new derivatives showed beneficial effects on the regulation of right heart physiology, as depicted by echocardiographic evaluations after long-term A2A receptor activation (2 weeks of oral treatment with both substances) in monocrotaline-induced RV failure. Treatment of RV failure rats with LASSBio-1386 significantly improved their exercise capacity compared to control animals. Finally, we demonstrated, for the first time, that rats with RV failure had reduced A2A receptor levels in RV tissue, which were correlated with reductions of SERCA2 content and Ca2+-ATPase activity. Activation of SERCA2 is one of the most important processes to regulate cardiomyocyte relaxation (Katz, 2006). SERCA induces Ca2+ uptake from the cytosol to the sarcoplasmic reticulum lumen, activating cell relaxation and promoting restock of Ca2+ for the next cardiac contraction. Elevated SERCA2 activity and density lead to an increased potential for the next cardiac cycle (Periasamy and Huke, 2001). These findings suggest the beneficial effects of A2A receptor-mediated signaling on global cardiac function. Using echocardiography, we confirmed the impairment of systolic function in PAH rats (Alencar et al., 2014). These data are consistent with previous reports showing an impaired pattern of this receptor in different experimental models of LV disease.
Additionally, we may discuss that the reduced expression of A2A receptor in the cardiopulmonary tissues in rats with PAH-induced RV failure may be a consequence of the chronic state of the disease. Possibly, in earlier stage of PAH, the A2A receptor expression could be unchanged or increased as a physiological compensatory mechanism. However, further studies are necessary to determine this receptor subtype levels during all stages of the disease in preclinical models. Despite the reduced expression of the A2A receptor in the cardiopulmonary system on late stage of PAH, lower levels of the receptor were still detected and LASSBio-1359 and LASSBio-1386 could bind and activate these receptors, ameliorating PAH.
No study has investigated whether the A2B receptor plays a role in RV function or in the pathophysiology of PAH-induced RV failure. However, this receptor is highly expressed in cardiac fibroblasts. Despite displaying antifibrotic potential in several in vitro studies, most in vivo animal models have described this receptor as being an inducer of cardiac remodeling and fibrosis. In their review, Vecchio et al. (2017) proposed that some proinflammatory mechanism may underlie this profibrotic activity of the A2B receptor. A2B receptor promoted a moderate increase in cardiac contractility in ex vivo mouse hearts (Chandrasekera et al., 2010). A later study showed that A2B receptor prevented mitochondrial oxidative stress by decreasing superoxide generation, but its effects were simultaneously potentiated by activation of the A2A receptor. Xu et al. (2017) concluded that the A2A and A2B receptors act together in the regulation of heart metabolism.
Given its high expression in mast cells, neutrophils, eosinophils, and other inflammatory cells, A3 receptor has been speculated as a potential target for the treatment of ischemic conditions, glaucoma, asthma, arthritis, cancer, and other inflammation-related disorders (Gessi et al., 2008). Low-level expression of A3 receptor in the heart provided effective protection against ischemic injury without detectable adverse effects, whereas overexpression led to development of dilated cardiomyopathy. Expression of this receptor was below the limits of detection of radioligand binding or northern blot (Black et al., 2002). Combined with the lack of works addressing the potential of A3 receptor in PAH and RV failure, we do not have enough arguments to assume that the synthesis of new ligands for this adenosine target could represent a suitable strategy to treat the disease in the future.
Over the past 20 years, scientists in medicinal chemistry have generated agonists and antagonists with high affinity and high selectivity for human variants of each of the four ARs. Moreover, agonist and antagonist ligands containing positron-emitting radioisotopes have been developed to monitor the in vivo occupancy of ARs in humans (Muller and Jacobson, 2011; Chen et al., 2013). As such, the lack of selective ligands of ARs is not a limiting factor for research and drug development, as has been the case for some other G-protein coupled receptors. Furthermore, researchers continue their efforts to develop novel adenosine ligands with refined structure-activity relationships, improved in vivo biodistribution, and tissue selectivity, which are crucial to druggability (Muller and Jacobson, 2011; Chen et al., 2013). A bigger problem in this process has been the broad distribution of the ARs. A possible approach for achieving tissue selectivity could be the use of partial agonists that would predominantly act where there are a high number of so-called “spare” receptors (Chen et al., 2013), as we also commented earlier in this review.
Advances in our understanding of the pathogenetic role of adenosine in PAH may soon be translated into effective treatment options. There is a complex interplay among the different distribution patterns and/or affinities of the four AR subtypes in specific cell types at different stages of disease. As such, combinations of selective antagonist/agonists for the different AR subtypes will likely be required to obtain reasonable clinical efficacy. Alternatively, controlling the factors involved in driving adenosine concentrations in tissue may be important.
Regarding the roles of A1, A2B, and A3 adenosine receptor subtypes, with this review we are assuming the need of closely studying each one in specific pre-clinical models of PAH for further discussions on their use as targets to treat this deleterious cardiopulmonary disease in the clinical field.
Nevertheless, data discussed in this review indicate a role for the A2A receptor in mediating beneficial effects, such as pulmonary vascular relaxation, reduction of pulmonary vessel and RV hypertrophy, amelioration of RV dysfunction, and exercise capacity, in rats with PAH. We have briefly described the importance of the adenosine system, specifically the A2A receptor, as a new target for the treatment of PAH. Several factors remain to be explored. For example, global and local A2A receptor function should be investigated in human PAH. The site of action of these drugs should be carefully determined through their administration to animals with cell-specific receptor deletions. Adenosine signaling and the effects of each drug over the specific disease course (e.g., acute and chronic stages of PAH and RV failure) should be carefully monitored in clinical studies. Finally, the possibility of combining direct A2A receptor actions with drugs targeting other pathways and/or targets should be examined. Such explorations could uncover new therapeutic strategies, which are greatly needed for patients with PAH.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by Conselho Nacional de Desenvolvimento Cientifico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Programa de Apoio a Núcleos de Experiência (PRONEX), Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Instituto Nacional de Ciência e Tecnologia INCT-INOFAR (Proc. 465249/2014-0).
Abe, K., Toba, M., Alzoubi, A., Ito, M., Fagan, K. A., Cool, C. D., et al. (2010). Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation 121, 2747–2754. doi: 10.1161/CIRCULATIONAHA.109.927681
Alencar, A. K., Pereira, S. L., da Silva, F. E., Mendes, L. V., Cunha Vdo, M., Lima, L. M., et al. (2014). N-acylhydrazone derivative ameliorates monocrotaline-induced pulmonary hypertension through the modulation of adenosine AA2R activity. Int. J. Cardiol. 173, 154–162. doi: 10.1016/j.ijcard.2014.02.022
Alencar, A. K., Pereira, S. L., Montagnoli, T. L., Maia, R. C., Kummerle, A. E., Landgraf, S. S., et al. (2013). Beneficial effects of a novel agonist of the adenosine A2A receptor on monocrotaline-induced pulmonary hypertension in rats. Br. J. Pharmacol. 169, 953–962. doi: 10.1111/bph.12193
Amabile, N., Guignabert, C., Montani, D., Yeghiazarians, Y., Boulanger, C. M., and Humbert, M. (2013). Cellular microparticles in the pathogenesis of pulmonary hypertension. Eur. Respir. J. 42, 272–279. doi: 10.1183/09031936.00087212
Antonioli, L., Blandizzi, C., Csoka, B., Pacher, P., and Hasko, G. (2015). Adenosine signalling in diabetes mellitus–pathophysiology and therapeutic considerations. Nat. Rev. Endocrinol. 11, 228–241. doi: 10.1038/nrendo.2015.10
Antonioli, L., Blandizzi, C., Pacher, P., and Hasko, G. (2013). Immunity, inflammation and cancer: a leading role for adenosine. Nat. Rev. Cancer 13, 842–857. doi: 10.1038/nrc3613
Antonioli, L., Colucci, R., La Motta, C., Tuccori, M., Awwad, O., Da Settimo, F., et al. (2012). Adenosine deaminase in the modulation of immune system and its potential as a novel target for treatment of inflammatory disorders. Curr. Drug Targets 13, 842–862. doi: 10.2174/138945012800564095
Antoniu, S. A. (2012). Targeting RhoA/ROCK pathway in pulmonary arterial hypertension. Expert Opin. Ther. Targets 16, 355–363. doi: 10.1517/14728222.2012.671811
Asakura, M., Asanuma, H., Kim, J., Liao, Y., Nakamaru, K., Fujita, M., et al. (2007). Impact of adenosine receptor signaling and metabolism on pathophysiology in patients with chronic heart failure. Hypertens Res. 30, 781–787. doi: 10.1291/hypres.30.781
Aznar, S., and Lacal, J. C. (2001). Rho signals to cell growth and apoptosis. Cancer Lett 165, 1–10. doi: 10.1016/S0304-3835(01)00412-8
Bak, M. I., and Ingwall, J. S. (1998). Regulation of cardiac AMP-specific 5′-nucleotidase during ischemia mediates ATP resynthesis on reflow. Am. J. Physiol. 274(4 Pt 1), C992–C1001.
Ballard, F. J. (1970). Adenine nucleotides and the adenylate kinase equilibrium in livers of foetal and newborn rats. Biochem. J. 117, 231–235. doi: 10.1042/bj1170231
Barletta, K. E., Ley, K., and Mehrad, B. (2012). Regulation of neutrophil function by adenosine. Arterioscler. Thromb. Vasc. Biol. 32, 856–864. doi: 10.1161/ATVBAHA.111.226845
Behr, J., and Ryu, J. H. (2008). Pulmonary hypertension in interstitial lung disease. Eur. Respir. J. 31, 1357–1367. doi: 10.1183/09031936.00171307
Belardinelli, L., Shryock, J. C., Song, Y., Wang, D., and Srinivas, M. (1995). Ionic basis of the electrophysiological actions of adenosine on cardiomyocytes. FASEB J. 9, 359–365.
Bi, L. Q., Zhu, R., Kong, H., Wu, S. L., Li, N., Zuo, X. R., et al. (2013). Ruscogenin attenuates monocrotaline-induced pulmonary hypertension in rats. Int. Immunopharmacol. 16, 7–16. doi: 10.1016/j.intimp.2013.03.010
Black, R. G. Jr., Guo, Y., Ge, Z. D., Murphree, S. S., Prabhu, S. D., Jones, W. K., et al. (2002). Gene dosage-dependent effects of cardiac-specific overexpression of the A3 adenosine receptor. Circ. Res. 91, 165–172. doi: 10.1161/01.RES.0000028007.91385.EE
Boison, D. (2013). Adenosine kinase: exploitation for therapeutic gain. Pharmacol. Rev. 65, 906–943. doi: 10.1124/pr.112.006361
Chan, T. O., Funakoshi, H., Song, J., Zhang, X. Q., Wang, J., Chung, P. H., et al. (2008). Cardiac-restricted overexpression of the A(2A)-adenosine receptor in FVB mice transiently increases contractile performance and rescues the heart failure phenotype in mice overexpressing the A(1)-adenosine receptor. Clin. Transl. Sci. 1, 126–133. doi: 10.1111/j.1752-8062.2008.00027.x
Chandrasekera, P. C., McIntosh, V. J., Cao, F. X., and Lasley, R. D. (2010). Differential effects of adenosine A2a and A2b receptors on cardiac contractility. Am. J. Physiol. Heart Circ. Physiol. 299, H2082–H2089. doi: 10.1152/ajpheart.00511.2010
Chaouat, A., Naeije, R., and Weitzenblum, E. (2008). Pulmonary hypertension in COPD. Eur. Respir. J. 32, 1371–1385. doi: 10.1183/09031936.00015608
Chen, J. F., Eltzschig, H. K., and Fredholm, B. B. (2013). Adenosine receptors as drug targets–what are the challenges? Nat. Rev. Drug Discov. 12, 265–286. doi: 10.1038/nrd3955
Cheng, D. Y., DeWitt, B. J., Suzuki, F., Neely, C. F., and Kadowitz, P. J. (1996). Adenosine A1 and A2 receptors mediate tone-dependent responses in feline pulmonary vascular bed. Am. J. Physiol. 270(1 Pt 2), H200–H207.
Chuo, C. H., Devine, S. M., Scammells, P. J., Krum, H., Christopoulos, A., May, L. T., et al. (2016). VCP746, a novel A1 adenosine receptor biased agonist, reduces hypertrophy in a rat neonatal cardiac myocyte model. Clin. Exp. Pharmacol. Physiol 43, 976–982. doi: 10.1111/1440-1681.12616
Connolly, M. J., and Aaronson, P. I. (2011). Key role of the RhoA/Rho kinase system in pulmonary hypertension. Pulm. Pharmacol. Ther. 24, 1–14. doi: 10.1016/j.pupt.2010.09.001
Cool, C. D., Stewart, J. S., Werahera, P., Miller, G. J., Williams, R. L., Voelkel, N. F., et al. (1999). Three-dimensional reconstruction of pulmonary arteries in plexiform pulmonary hypertension using cell-specific markers. Evidence for a dynamic and heterogeneous process of pulmonary endothelial cell growth. Am. J. Pathol. 155, 411–419. doi: 10.1016/S0002-9440(10)65137-1
Costa, D. G., da Silva, J. S., Kummerle, A. E., Sudo, R. T., Landgraf, S. S., Caruso-Neves, C., et al. (2010). LASSBio-294, A compound with inotropic and lusitropic activity, decreases cardiac remodeling and improves Ca(2)(+) influx into sarcoplasmic reticulum after myocardial infarction. Am. J. Hypertens 23, 1220–1227. doi: 10.1038/ajh.2010.157
Cronstein, B. N., Daguma, L., Nichols, D., Hutchison, A. J., and Williams, M. (1990). The adenosine/neutrophil paradox resolved: human neutrophils possess both A1 and A2 receptors that promote chemotaxis and inhibit O2 generation, respectively. J. Clin. Invest. 85, 1150–1157. doi: 10.1172/JCI114547
Cronstein, B. N., and Sitkovsky, M. (2017). Adenosine and adenosine receptors in the pathogenesis and treatment of rheumatic diseases. Nat. Rev. Rheumatol. 13, 41–51. doi: 10.1038/nrrheum.2016.178
da Silva, J. S., Gabriel-Costa, D., Sudo, R. T., Wang, H., Groban, L., Ferraz, E. B., et al. (2017). Adenosine A2A receptor agonist prevents cardiac remodeling and dysfunction in spontaneously hypertensive male rats after myocardial infarction. Drug Des. Devel. Ther. 11, 553–562. doi: 10.2147/DDDT.S113289
da Silva, J. S., Pereira, S. L., Maia Rdo, C., Landgraf, S. S., Caruso-Neves, C., Kummerle, A. E., et al. (2014). N-acylhydrazone improves exercise intolerance in rats submitted to myocardial infarction by the recovery of calcium homeostasis in skeletal muscle. Life Sci 94, 30–36. doi: 10.1016/j.lfs.2013.11.012
Daly, J. W. (1982). Adenosine receptors: targets for future drugs. J. Med. Chem. 25, 197–207. doi: 10.1021/jm00345a001
De Jong, J. W. (1977). Partial purification and properties of rat-heart adenosine kinase. Arch. Int. Physiol. Biochim. 85, 557–569. doi: 10.3109/13813457709069872
Decking, U. K., Schlieper, G., Kroll, K., and Schrader, J. (1997). Hypoxia-induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circ. Res. 81, 154–164. doi: 10.1161/01.RES.81.2.154
Demiryurek, S., Kara, A. F., Celik, A., Babul, A., Tarakcioglu, M., and Demiryurek, A. T. (2005). Effects of fasudil, a Rho-kinase inhibitor, on myocardial preconditioning in anesthetized rats. Eur. J. Pharmacol. 527, 129–140. doi: 10.1016/j.ejphar.2005.10.018
Deussen, A., Lloyd, H. G., and Schrader, J. (1989). Contribution of S-adenosylhomocysteine to cardiac adenosine formation. J. Mol. Cell Cardiol. 21, 773–782. doi: 10.1016/0022-2828(89)90716-5
Deussen, A., and Schrader, J. (1991). Cardiac adenosine production is linked to myocardial pO2. J. Mol. Cell Cardiol. 23, 495–504. doi: 10.1016/0022-2828(91)90173-J
Dinh, W., Albrecht-Kupper, B., Gheorghiade, M., Voors, A. A., van der Laan, M., and Sabbah, H. N. (2017). Partial adenosine A1 agonist in heart failure. Handb. Exp. Pharmacol. 243, 177–203. doi: 10.1007/164_2016_83
Donoso, M. V., Lopez, R., Miranda, R., Briones, R., and Huidobro-Toro, J. P. (2005). A2B adenosine receptor mediates human chorionic vasoconstriction and signals through arachidonic acid cascade. Am. J. Physiol. Heart Circ. Physiol. 288, H2439–H2449. doi: 10.1152/ajpheart.00548.2004
Drabikowska, A. K., Halec, L., and Shugar, D. (1985). Purification and properties of adenosine kinase from rat liver: separation from deoxyadenosine kinase activity. Z. Naturforsch. C 40, 34–41.
Drury, A. N., and Szent-Gyorgyi, A. (1929). The physiological activity of adenine compounds with especial reference to their action upon the mammalian heart. J. Physiol. 68, 213–237. doi: 10.1113/jphysiol.1929.sp002608
Dubey, R. K., Gillespie, D. G., Mi, Z., and Jackson, E. K. (1998). Adenosine inhibits growth of human aortic smooth muscle cells via A2B receptors. Hypertension 31(1 Pt 2), 516–521. doi: 10.1161/01.HYP.31.1.516
Dubey, R. K., Gillespie, D. G., Osaka, K., Suzuki, F., and Jackson, E. K. (1996). Adenosine inhibits growth of rat aortic smooth muscle cells. Possible role of A2b receptor. Hypertension 27(3 Pt 2), 786–793. doi: 10.1161/01.HYP.27.3.786
Dubey, R. K., Rosselli, M., Gillespie, D. G., Mi, Z., and Jackson, E. K. (2010). Extracellular 3′,5′-cAMP-adenosine pathway inhibits glomerular mesangial cell growth. J. Pharmacol. Exp. Ther. 333, 808–815. doi: 10.1124/jpet.110.166371
Dunwiddie, T. V., Diao, L., and Proctor, W. R. (1997). Adenine nucleotides undergo rapid, quantitative conversion to adenosine in the extracellular space in rat hippocampus. J. Neurosci. 17, 7673–7682.
Eddahibi, S., Guignabert, C., Barlier-Mur, A. M., Dewachter, L., Fadel, E., Dartevelle, P., et al. (2006). Cross talk between endothelial and smooth muscle cells in pulmonary hypertension: critical role for serotonin-induced smooth muscle hyperplasia. Circulation 113, 1857–1864. doi: 10.1161/CIRCULATIONAHA.105.591321
El-Kashef, H., Elmazar, M. M., Al-Shabanah, O. A., and Al-Bekairi, A. M. (1999). Effect of adenosine on pulmonary circulation of rabbits. Gen. Pharmacol. 32, 307–313. doi: 10.1016/S0306-3623(98)00184-0
Ely, S. W., and Berne, R. M. (1992). Protective effects of adenosine in myocardial ischemia. Circulation 85, 893–904. doi: 10.1161/01.CIR.85.3.893
Emerson, M., Momi, S., Paul, W., Alberti, P. F., Page, C., and Gresele, P. (1999). Endogenous nitric oxide acts as a natural antithrombotic agent in vivo by inhibiting platelet aggregation in the pulmonary vasculature. Thromb. Haemost. 81, 961–966.
Faas, M.M., Saez, T., and de Vos, P. (2017). Extracellular ATP and adenosine: the Yin and Yang in immune responses? Mol. Aspects Med. 55, 9–19. doi: 10.1016/j.mam.2017.01.002
Fagan, K. A., Fouty, B. W., Tyler, R. C., Morris, K. G. Jr., Hepler, L. K., Sato, K., et al. (1999). The pulmonary circulation of homozygous or heterozygous eNOS-null mice is hyperresponsive to mild hypoxia. J. Clin. Invest. 103, 291–299. doi: 10.1172/JCI3862
Feng, Y., LoGrasso, P. V., Defert, O., and Li, R. (2016). Rho kinase (ROCK) inhibitors and their therapeutic potential. J. Med. Chem. 59, 2269–2300. doi: 10.1021/acs.jmedchem.5b00683
Feoktistov, I., and Biaggioni, I. (1995). Adenosine A2b receptors evoke interleukin-8 secretion in human mast cells. An enprofylline-sensitive mechanism with implications for asthma. J. Clin. Invest. 96, 1979–1986. doi: 10.1172/JCI118245
Feoktistov, I., and Biaggioni, I. (1996). Role of adenosine in asthma. Drug Dev. Res. 39, 333–336. doi: 10.1002/(SICI)1098-2299(199611/12)39:3/4<333::AID-DDR14>3.0.CO;2-2
Fishman, P., Bar-Yehuda, S., Liang, B. T., and Jacobson, K. A. (2012). Pharmacological and therapeutic effects of A3 adenosine receptor agonists. Drug Discov. Today 17, 359–366. doi: 10.1016/j.drudis.2011.10.007
Fishman, P., Bar-Yehuda, S., Madi, L., Rath-Wolfson, L., Ochaion, A., Cohen, S., et al. (2006). The PI3K-NF-kappaB signal transduction pathway is involved in mediating the anti-inflammatory effect of IB-MECA in adjuvant-induced arthritis. Arthritis Res. Ther. 8:R33. doi: 10.1186/ar1887
Flamand, N., Lefebvre, J., Lapointe, G., Picard, S., Lemieux, L., Bourgoin, S. G., et al. (2006). Inhibition of platelet-activating factor biosynthesis by adenosine and histamine in human neutrophils: involvement of cPLA2alpha and reversal by lyso-PAF. J. Leukoc. Biol. 79, 1043–1051. doi: 10.1189/jlb.1005614
Flood, A., and Headrick, J. P. (2001). Functional characterization of coronary vascular adenosine receptors in the mouse. Br. J. Pharmacol. 133, 1063–1072. doi: 10.1038/sj.bjp.0704170
Folkman, J., and D’Amore, P. A. (1996). Blood vessel formation: what is its molecular basis? Cell 87, 1153–1155. doi: 10.1016/S0092-8674(00)81810-3
Ford, H. Jr., Dai, F., Mu, L., Siddiqui, M. A., Nicklaus, M. C., Anderson, L., et al. (2000). Adenosine deaminase prefers a distinct sugar ring conformation for binding and catalysis: kinetic and structural studies. Biochemistry 39, 2581–2592. doi: 10.1021/bi992112c
Fozard, J. R., and Hannon, J. P. (2000). Species differences in adenosine receptor-mediated bronchoconstrictor responses. Clin. Exp. Allergy 30, 1213–1220. doi: 10.1046/j.1365-2222.2000.00894.x
Fredholm, B. B., Arslan, G., Halldner, L., Kull, B., Schulte, G., and Wasserman, W. (2000). Structure and function of adenosine receptors and their genes. Naunyn Schmiedebergs Arch. Pharmacol. 362, 364–374. doi: 10.1007/s002100000313
Fredholm, B. B., IJzerman, AP., Jacobson, K. A., Klotz, K. N., and Linden, J. (2001). International union of pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 53, 527–552.
Fredholm, B. B., Zhang, Y., and van der Ploeg, I. (1996). Adenosine A2A receptors mediate the inhibitory effect of adenosine on formyl-Met-Leu-Phe-stimulated respiratory burst in neutrophil leucocytes. Naunyn Schmiedebergs Arch. Pharmacol. 354, 262–267. doi: 10.1007/BF00171056
Fukumoto, Y., Yamada, N., Matsubara, H., Mizoguchi, M., Uchino, K., Yao, A., et al. (2013). Double-blind, placebo-controlled clinical trial with a rho-kinase inhibitor in pulmonary arterial hypertension. Circ. J. 77, 2619–2625. doi: 10.1253/circj.CJ-13-0443
Galie, N., Humbert, M., Vachiery, J. L., Gibbs, S., Lang, I., Torbicki, A., et al. (2016). 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart J. 37, 67–119. doi: 10.1093/eurheartj/ehv317
Garcia de Vinuesa, A., Abdelilah-Seyfried, S., Knaus, P., Zwijsen, A., and Bailly, S. (2016). BMP signaling in vascular biology and dysfunction. Cytokine Growth Factor Rev. 27, 65–79. doi: 10.1016/j.cytogfr.2015.12.005
Garcia-Morales, L. J., Chen, N. Y., Weng, T., Luo, F., Davies, J., Philip, K., et al. (2016). Altered hypoxic-adenosine axis and metabolism in group III pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 54, 574–583. doi: 10.1165/rcmb.2015-0145OC
Geldenhuys, W. J., Hanif, A., Yun, J., and Nayeem, M. A. (2017). Exploring adenosine receptor ligands: potential role in the treatment of cardiovascular diseases. Molecules 22:E917. doi: 10.3390/molecules22060917.
Gessi, S., Merighi, S., Varani, K., Leung, E., Mac Lennan, S., and Borea, P. A. (2008). The A3 adenosine receptor: an enigmatic player in cell biology. Pharmacol. Ther. 117, 123–140. doi: 10.1016/j.pharmthera.2007.09.002
Godinas, L., Guignabert, C., Seferian, A., Perros, F., Bergot, E., Sibille, Y., et al. (2013). Tyrosine kinase inhibitors in pulmonary arterial hypertension: a double-edge sword? Semin. Respir. Crit. Care Med. 34, 714–724. doi: 10.1055/s-0033-1356494
Guignabert, C., and Dorfmuller, P. (2013). Pathology and pathobiology of pulmonary hypertension. Semin. Respir. Crit. Care Med. 34, 551–559. doi: 10.1055/s-0033-1356496
Guignabert, C., Tu, L., Girerd, B., Ricard, N., Huertas, A., Montani, D., et al. (2015). New molecular targets of pulmonary vascular remodeling in pulmonary arterial hypertension: importance of endothelial communication. Chest 147, 529–537. doi: 10.1378/chest.14-0862
Guilluy, C., Eddahibi, S., Agard, C., Guignabert, C., Izikki, M., Tu, L., et al. (2009). RhoA and Rho kinase activation in human pulmonary hypertension: role of 5-HT signaling. Am. J. Respir. Crit. Care Med. 179, 1151–1158. doi: 10.1164/rccm.200805-691OC
Gupta, N., Rashid, J., Nozik-Grayck, E., McMurtry, I. F., Stenmark, K. R., and Ahsan, F. (2017). Cocktail of superoxide dismutase and fasudil encapsulated in targeted liposomes slows PAH progression at a reduced dosing frequency. Mol. Pharm 14, 830–841. doi: 10.1021/acs.molpharmaceut.6b01061
Gustafson, L. A., and Kroll, K. (1998). Downregulation of 5′-nucleotidase in rabbit heart during coronary underperfusion. Am. J. Physiol. 274(2 Pt 2), H529–H538.
Ham, J., and Evans, B. A. (2012). An emerging role for adenosine and its receptors in bone homeostasis. Front. Endocrinol. 3:113. doi: 10.3389/fendo.2012.00113
Hamad, E. A., Zhu, W., Chan, T. O., Myers, V., Gao, E., Li, X., et al. (2012). Cardioprotection of controlled and cardiac-specific over-expression of A(2A)-adenosine receptor in the pressure overload. PLOS ONE 7:e39919. doi: 10.1371/journal.pone.0039919
Hanahan, D. (1997). Signaling vascular morphogenesis and maintenance. Science 277, 48–50. doi: 10.1126/science.277.5322.48
Hasko, G., and Cronstein, B. (2013). Regulation of inflammation by adenosine. Front. Immunol. 4:85. doi: 10.3389/fimmu.2013.00085
Hasko, G., and Cronstein, B. N. (2004). Adenosine: an endogenous regulator of innate immunity. Trends Immunol. 25, 33–39. doi: 10.1016/j.it.2003.11.003
Hasko, G., Linden, J., Cronstein, B., and Pacher, P. (2008). Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat. Rev. Drug Discov. 7, 759–770. doi: 10.1038/nrd2638
Hasko, G., and Pacher, P. (2008). A2A receptors in inflammation and injury: lessons learned from transgenic animals. J. Leukoc. Biol. 83, 447–455. doi: 10.1189/jlb.0607359
Headrick, J. P., Ashton, K. J., Rose’meyer, R. B., and Peart, J. N. (2013). Cardiovascular adenosine receptors: expression, actions and interactions. Pharmacol. Ther. 140, 92–111. doi: 10.1016/j.pharmthera.2013.06.002
Headrick, J. P., Hack, B., and Ashton, K. J. (2003). Acute adenosinergic cardioprotection in ischemic-reperfused hearts. Am. J. Physiol. Heart Circ. Physiol. 285, H1797–H1818. doi: 10.1152/ajpheart.00407.2003
Headrick, J. P., and Willis, R. J. (1990). Adenosine formation and energy metabolism: a 31P-NMR study in isolated rat heart. Am. J. Physiol. 258(3 Pt 2), H617–H624.
Hermes, M., Osswald, H., and Kloor, D. (2007). Role of S-adenosylhomocysteine hydrolase in adenosine-induced apoptosis in HepG2 cells. Exp. Cell Res. 313, 264–283. doi: 10.1016/j.yexcr.2006.10.003
Hiley, C. R., Bottrill, F. E., Warnock, J., and Richardson, P. J. (1995). Effects of pH on responses to adenosine, CGS 21680, carbachol and nitroprusside in the isolated perfused superior mesenteric arterial bed of the rat. Br. J. Pharmacol. 116, 2641–2646. doi: 10.1111/j.1476-5381.1995.tb17220.x
Hinschen, A. K., Rose’Meyer, R. B., and Headrick, J. P. (2003). Adenosine receptor subtypes mediating coronary vasodilation in rat hearts. J. Cardiovasc. Pharmacol. 41, 73–80. doi: 10.1097/00005344-200301000-00010
Hinze, A. V., Mayer, P., Harst, A., and von Kugelgen, I. (2012). Adenosine A(3) receptor-induced proliferation of primary human coronary smooth muscle cells involving the induction of early growth response genes. J. Mol. Cell Cardiol. 53, 639–645. doi: 10.1016/j.yjmcc.2012.08.003
Hisatome, I. (2007). Adenosine and cardioprotection in chronic heart failure: genes and protein expression. Hypertens. Res. 30, 757–758. doi: 10.1291/hypres.30.757
Huang, X., Zou, L., Yu, X., Chen, M., Guo, R., Cai, H., et al. (2015). Salidroside attenuates chronic hypoxia-induced pulmonary hypertension via adenosine A2a receptor related mitochondria-dependent apoptosis pathway. J. Mol. Cell Cardiol. 82, 153–166. doi: 10.1016/j.yjmcc.2015.03.005
Ikeda, U., Kurosaki, K., Ohya, K., and Shimada, K. (1997). Adenosine stimulates nitric oxide synthesis in vascular smooth muscle cells. Cardiovasc. Res. 35, 168–174. doi: 10.1016/S0008-6363(97)00068-0
Impellizzeri, D., Di Paola, R., Esposito, E., Mazzon, E., Paterniti, I., Melani, A., et al. (2011). CGS 21680, an agonist of the adenosine (A2A) receptor, decreases acute lung inflammation. Eur. J. Pharmacol. 668, 305–316. doi: 10.1016/j.ejphar.2011.06.049
Jackson, E. K., Ren, J., and Gillespie, D. G. (2011). 2′,3′-cAMP, 3′-AMP, and 2′-AMP inhibit human aortic and coronary vascular smooth muscle cell proliferation via A2B receptors. Am. J. Physiol. Heart Circ. Physiol. 301, H391–H401. doi: 10.1152/ajpheart.00336.2011
Jackson, E. K., Ren, J., Gillespie, D. G., and Dubey, R. K. (2010). Extracellular 2,3-cyclic adenosine monophosphate is a potent inhibitor of preglomerular vascular smooth muscle cell and mesangial cell growth [corrected]. Hypertension 56, 151–158. doi: 10.1161/HYPERTENSIONAHA.110.152454
Jacobson, K. A., and Gao, Z. G. (2006). Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov. 5, 247–264. doi: 10.1038/nrd1983
Jiang, X., Wang, Y. F., Zhao, Q. H., Jiang, R., Wu, Y., Peng, F. H., et al. (2014). Acute hemodynamic response of infused fasudil in patients with pulmonary arterial hypertension: a randomized, controlled, crossover study. Int. J. Cardiol. 177, 61–65. doi: 10.1016/j.ijcard.2014.09.101
Jones, M. R., Zhao, Z., Sullivan, C. P., Schreiber, B. M., Stone, P. J., Toselli, P. A., et al. (2004). A(3) adenosine receptor deficiency does not influence atherogenesis. J. Cell. Biochem. 92, 1034–1043. doi: 10.1002/jcb.20122
Jonigk, D., Golpon, H., Bockmeyer, C. L., Maegel, L., Hoeper, M. M., Gottlieb, J., et al. (2011). Plexiform lesions in pulmonary arterial hypertension composition, architecture, and microenvironment. Am. J. Pathol. 179, 167–179. doi: 10.1016/j.ajpath.2011.03.040
Karmouty-Quintana, H., Philip, K., Acero, L. F., Chen, N. Y., Weng, T., Molina, J. G., et al. (2015). Deletion of ADORA2B from myeloid cells dampens lung fibrosis and pulmonary hypertension. FASEB J. 29, 50–60. doi: 10.1096/fj.14260182
Karmouty-Quintana, H., Weng, T., Garcia-Morales, L. J., Chen, N. Y., Pedroza, M., Zhong, H., et al. (2013a). Adenosine A2B receptor and hyaluronan modulate pulmonary hypertension associated with chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 49, 1038–1047. doi: 10.1165/rcmb.2013-0089OC
Karmouty-Quintana, H., Xia, Y., and Blackburn, M. R. (2013b). Adenosine signaling during acute and chronic disease states. J. Mol. Med. 91, 173–181. doi: 10.1007/s00109-013-0997-1
Karmouty-Quintana, H., Zhong, H., Acero, L., Weng, T., Melicoff, E., West, J. D., et al. (2012). The A2B adenosine receptor modulates pulmonary hypertension associated with interstitial lung disease. FASEB J. 26, 2546–2557. doi: 10.1096/fj.11-200907
Keil, A., Blom, I. E., Goldschmeding, R., and Rupprecht, H. D. (2002). Nitric oxide down-regulates connective tissue growth factor in rat mesangial cells. Kidney Int. 62, 401–411. doi: 10.1046/j.1523-1755.2002.00462.x
Kilpatrick, E. L., Narayan, P., Mentzer, R. M. Jr., and Lasley, R. D. (2002). Cardiac myocyte adenosine A2a receptor activation fails to alter cAMP or contractility: role of receptor localization. Am. J. Physiol. Heart Circ. Physiol. 282,H1035–H1040. doi: 10.1152/ajpheart.00808.2001
Kim, J., Kim, M., Song, J. H., and Lee, H. T. (2008). Endogenous A1 adenosine receptors protect against hepatic ischemia reperfusion injury in mice. Liver Transpl. 14, 845–854. doi: 10.1002/lt.21432
King, A. E., Ackley, M. A., Cass, C. E., Young, J. D., and Baldwin, S. A. (2006). Nucleoside transporters: from scavengers to novel therapeutic targets. Trends Pharmacol. Sci. 27, 416–425. doi: 10.1016/j.tips.2006.06.004
Kirsch, G. E., Yatani, A., Codina, J., Birnbaumer, L., and Brown, A. M. (1988). Alpha-subunit of Gk activates atrial K+ channels of chick, rat, and guinea pig. Am. J. Physiol. 254(6 Pt 2), H1200–H1205.
Kloor, D., and Osswald, H. (2004). S-Adenosylhomocysteine hydrolase as a target for intracellular adenosine action. Trends Pharmacol. Sci. 25, 294–297. doi: 10.1016/j.tips.2004.04.004
Ko, E. A., Han, J., Jung, I. D., and Park, W. S. (2008). Physiological roles of K+ channels in vascular smooth muscle cells. J. Smooth Muscle Res. 44, 65–81. doi: 10.1540/jsmr.44.65
Koledova, V. V., and Khalil, R. A. (2006). Ca2+, calmodulin, and cyclins in vascular smooth muscle cell cycle. Circ. Res. 98, 1240–1243. doi: 10.1161/01.RES.0000225860.41648.63
Koshiba, M., Rosin, D. L., Hayashi, N., Linden, J., and Sitkovsky, M. V. (1999). Patterns of A2A extracellular adenosine receptor expression in different functional subsets of human peripheral T cells. Flow cytometry studies with anti-A2A receptor monoclonal antibodies. Mol. Pharmacol. 55, 614–624.
Kurachi, Y., Nakajima, T., and Sugimoto, T. (1986). On the mechanism of activation of muscarinic K+ channels by adenosine in isolated atrial cells: involvement of GTP-binding proteins. Pflugers. Arch. 407, 264–274. doi: 10.1007/BF00585301
Lasley, R. D., and Mentzer, R. M. Jr. (1992). Adenosine improves recovery of postischemic myocardial function via an adenosine A1 receptor mechanism. Am. J. Physiol. 263(5 Pt 2), H1460–H1465.
Laufs, U., Marra, D., Node, K., and Liao, J. K. (1999). 3-Hydroxy-3-methylglutaryl-CoA reductase inhibitors attenuate vascular smooth muscle proliferation by preventing rho GTPase-induced down-regulation of p27(Kip1). J. Biol. Chem. 274, 21926–21931. doi: 10.1074/jbc.274.31.21926
Leal, S., Sa, C., Goncalves, J., Fresco, P., and Diniz, C. (2008). Immunohistochemical characterization of adenosine receptors in rat aorta and tail arteries. Microsc. Res. Tech. 71, 703–709. doi: 10.1002/jemt.20609
Lee, H. S., Chung, H. J., Lee, H. W., Jeong, L. S., and Lee, S. K. (2011). Suppression of inflammation response by a novel A(3) adenosine receptor agonist thio-Cl-IB-MECA through inhibition of Akt and NF-kappaB signaling. Immunobiology 216, 997–1003. doi: 10.1016/j.imbio.2011.03.008
Lee, H. T., Gallos, G., Nasr, S. H., and Emala, C. W. (2004). A1 adenosine receptor activation inhibits inflammation, necrosis, and apoptosis after renal ischemia-reperfusion injury in mice. J. Am. Soc. Nephrol. 15, 102–111. doi: 10.1097/01.ASN.0000102474.68613.AE
Lemmon, M. A., and Schlessinger, J. (2010). Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134. doi: 10.1016/j.cell.2010.06.011
Lewis, C. D., Hourani, S. M., Long, C. J., and Collis, M. G. (1994). Characterization of adenosine receptors in the rat isolated aorta. Gen. Pharmacol. 25, 1381–1387. doi: 10.1016/0306-3623(94)90162-7
Liao, Y., Takashima, S., Asano, Y., Asakura, M., Ogai, A., Shintani, Y., et al. (2003). Activation of adenosine A1 receptor attenuates cardiac hypertrophy and prevents heart failure in murine left ventricular pressure-overload model. Circ. Res. 93, 759–766. doi: 10.1161/01.RES.0000094744.88220.62
Linden, J. (2001). Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annu. Rev. Pharmacol. Toxicol. 41, 775–787. doi: 10.1146/annurev.pharmtox.41.1.775
Linden, J., Thai, T., Figler, H., Jin, X., and Robeva, A. S. (1999). Characterization of human A(2B) adenosine receptors: radioligand binding, western blotting, and coupling to G(q) in human embryonic kidney 293 cells and HMC-1 mast cells. Mol. Pharmacol. 56, 705–713.
Liu, Y., Ren, W., Warburton, R., Toksoz, D., and Fanburg, B. L. (2009). Serotonin induces Rho/ROCK-dependent activation of Smads 1/5/8 in pulmonary artery smooth muscle cells. FASEB J. 23, 2299–2306. doi: 10.1096/fj.08-127910
Long, L., MacLean, M. R., Jeffery, T. K., Morecroft, I., Yang, X., Rudarakanchana, N., et al. (2006). Serotonin increases susceptibility to pulmonary hypertension in BMPR2-deficient mice. Circ. Res. 98, 818–827. doi: 10.1161/01.RES.0000215809.47923.fd
Marquardt, D. L. (1994). Adenosine and other mast cell preformed mediators. Allergy Proc. 15, 125–127. doi: 10.2500/108854194778702883
Martin, P. L., and Potts, A. A. (1994). The endothelium of the rat renal artery plays an obligatory role in A2 adenosine receptor-mediated relaxation induced by 5′-N-ethylcarboxamidoadenosine and N6-cyclopentyladenosine. J. Pharmacol. Exp. Ther. 270, 893–899.
Mayer, P., Hinze, A. V., Harst, A., and von Kugelgen, I. (2011). A(2)B receptors mediate the induction of early genes and inhibition of arterial smooth muscle cell proliferation via Epac. Cardiovasc. Res. 90, 148–156. doi: 10.1093/cvr/cvq371
McGoon, M. D., Benza, R. L., Escribano-Subias, P., Jiang, X., Miller, D. P., Peacock, A. J., et al. (2013). Pulmonary arterial hypertension: epidemiology and registries. J. Am. Coll. Cardiol. 62(25 Suppl), D51–D59. doi: 10.1016/j.jacc.2013.10.023
McIntosh, V. J., and Lasley, R. D. (2012). Adenosine receptor-mediated cardioprotection: are all 4 subtypes required or redundant? J. Cardiovasc. Pharmacol. Ther. 17, 21–33. doi: 10.1177/1074248410396877
McPherson, J. A., Barringhaus, K. G., Bishop, G. G., Sanders, J. M., Rieger, J. M., Hesselbacher, S. E., et al. (2001). Adenosine A(2A) receptor stimulation reduces inflammation and neointimal growth in a murine carotid ligation model. Arterioscler. Thromb. Vasc. Biol. 21, 791–796. doi: 10.1161/01.ATV.21.5.791
Merighi, S., Borea, P. A., and Gessi, S. (2015). Adenosine receptors and diabetes: focus on the A2B adenosine receptor subtype. Pharmacol. Res. 99, 229–236. doi: 10.1016/j.phrs.2015.06.015
Michoud, M. C., Napolitano, G., Maghni, K., Govindaraju, V., Cogo, A., and Martin, J. G. (2002). Effects of extracellular triphosphate nucleotides and nucleosides on airway smooth muscle cell proliferation. Am. J. Respir. Cell Mol. Biol. 27, 732–738. doi: 10.1165/rcmb.4768
Milne, G. R., and Palmer, T. M. (2011). Anti-inflammatory and immunosuppressive effects of the A2A adenosine receptor. ScientificWorldJournal 11, 320–339. doi: 10.1100/tsw.2011.22
Minamino, T., Christou, H., Hsieh, C. M., Liu, Y., Dhawan, V., Abraham, N. G., et al. (2001). Targeted expression of heme oxygenase-1 prevents the pulmonary inflammatory and vascular responses to hypoxia. Proc. Natl. Acad. Sci. U.S.A. 98, 8798–8803. doi: 10.1073/pnas.161272598.
Miura, T., Liu, Y., Kita, H., Ogawa, T., and Shimamoto, K. (2000). Roles of mitochondrial ATP-sensitive K channels and PKC in anti-infarct tolerance afforded by adenosine A1 receptor activation. J. Am. Coll. Cardiol. 35, 238–245. doi: 10.1016/S0735-1097(99)00493-3
Monahan, T. S., Sawmiller, D. R., Fenton, R. A., and Dobson, J. G. Jr. (2000). Adenosine A(2a)-receptor activation increases contractility in isolated perfused hearts. Am. J. Physiol. Heart Circ. Physiol. 279, H1472–H1481.
Morrison, R. R., Talukder, M. A., Ledent, C., and Mustafa, S. J. (2002). Cardiac effects of adenosine in A(2A) receptor knockout hearts: uncovering A(2B) receptors. Am. J. Physiol. Heart Circ. Physiol. 282, H437–H444. doi: 10.1152/ajpheart.00723.2001
Mouchaers, K. T., Schalij, I., de Boer, M. A., Postmus, P. E., van Hinsbergh, V. W., van Nieuw Amerongen, G. P., et al. (2010). Fasudil reduces monocrotaline-induced pulmonary arterial hypertension: comparison with bosentan and sildenafil. Eur. Respir. J. 36, 800–807. doi: 10.1183/09031936.00130209
Mubagwa, K., and Flameng, W. (2001). Adenosine, adenosine receptors and myocardial protection: an updated overview. Cardiovasc. Res. 52, 25–39. doi: 10.1016/S0008-6363(01)00358-3
Muller, C. E., and Jacobson, K. A. (2011). Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim. Biophys. Acta 1808, 1290–1308. doi: 10.1016/j.bbamem.2010.12.017
Mustafa, S. J., Morrison, R. R., Teng, B., and Pelleg, A. (2009). Adenosine receptors and the heart: role in regulation of coronary blood flow and cardiac electrophysiology. Handb. Exp. Pharmacol. 193, 161–188. doi: 10.1007/978-3-540-89615-9_6
Nagaoka, T., Gebb, S. A., Karoor, V., Homma, N., Morris, K. G., McMurtry, I. F., et al. (2006). Involvement of RhoA/Rho kinase signaling in pulmonary hypertension of the fawn-hooded rat. J. Appl. Physiol. 100, 996–1002. doi: 10.1152/japplphysiol.01028.2005
Neely, C. F., Jin, J., and Keith, I. M. (1997). A1-adenosine receptor antagonists block endotoxin-induced lung injury. Am. J. Physiol. 272(2 Pt 1), L353–L361.
Newby, A. C., and Holmquist, C. A. (1981). Adenosine production inside rat polymorphonuclear leucocytes. Biochem. J. 200, 399–403. doi: 10.1042/bj2000399
Newby, A. C., Worku, Y., and Holmquist, C. A. (1985). Adenosine formation. Evidence for a direct biochemical link with energy metabolism. Adv. Myocardiol. 6, 273–284.
Nootens, M., Schrader, B., Kaufmann, E., Vestal, R., Long, W., and Rich, S. (1995). Comparative acute effects of adenosine and prostacyclin in primary pulmonary hypertension. Chest 107, 54–57. doi: 10.1378/chest.107.1.54
Olafsson, B., Forman, M. B., Puett, D. W., Pou, A., Cates, C. U., Friesinger, G. C., et al. (1987). Reduction of reperfusion injury in the canine preparation by intracoronary adenosine: importance of the endothelium and the no-reflow phenomenon. Circulation 76, 1135–1145. doi: 10.1161/01.CIR.76.5.1135
Pak, M. A., Haas, H. L., Decking, U. K., and Schrader, J. (1994). Inhibition of adenosine kinase increases endogenous adenosine and depresses neuronal activity in hippocampal slices. Neuropharmacology 33, 1049–1053. doi: 10.1016/0028-3908(94)90142-2
Pearl, R. G. (1994). Adenosine produces pulmonary vasodilation in the perfused rabbit lung via an adenosine A2 receptor. Anesth. Analg. 79, 46–51. doi: 10.1213/00000539-199407000-00010
Peart, J. N., and Headrick, J. P. (2007). Adenosinergic cardioprotection: multiple receptors, multiple pathways. Pharmacol. Ther. 114, 208–221. doi: 10.1016/j.pharmthera.2007.02.004
Periasamy, M., and Huke, S. (2001). SERCA pump level is a critical determinant of Ca(2+)homeostasis and cardiac contractility. J. Mol. Cell Cardiol. 33, 1053–1063. doi: 10.1006/jmcc.2001.1366
Playa, H., Lewis, T. A., Ting, A., Suh, B. C., Munoz, B., Matuza, R., et al. (2014). Dilazep analogues for the study of equilibrative nucleoside transporters 1 and 2 (ENT1 and ENT2). Bioorg. Med. Chem. Lett. 24, 5801–5804. doi: 10.1016/j.bmcl.2014.10.026
Ponnoth, D. S., Nadeem, A., Tilley, S., and Mustafa, S. J. (2010). Involvement of A1 adenosine receptors in altered vascular responses and inflammation in an allergic mouse model of asthma. Am. J. Physiol. Heart Circ. Physiol. 299, H81–H87. doi: 10.1152/ajpheart.01090.2009
Ponnoth, D. S., Sanjani, M. S., Ledent, C., Roush, K., Krahn, T., and Mustafa, S. J. (2009). Absence of adenosine-mediated aortic relaxation in A(2A) adenosine receptor knockout mice. Am. J. Physiol. Heart Circ. Physiol. 297, H1655–H1660. doi: 10.1152/ajpheart.00192.2009
Prentice, D. J., and Hourani, S. M. (1996). Activation of multiple sites by adenosine analogues in the rat isolated aorta. Br. J. Pharmacol. 118, 1509–1517. doi: 10.1111/j.1476-5381.1996.tb15567.x
Prentice, D. J., and Hourani, S. M. (2000). Characterisation of adenosine receptors mediating relaxation in hamster isolated aorta. Naunyn Schmiedebergs Arch. Pharmacol. 362, 427–434. doi: 10.1007/s002100000292
Qian, G., Cao, J., Chen, C., Wang, L., Huang, X., Ding, C., et al. (2013). Paeoniflorin inhibits pulmonary artery smooth muscle cells proliferation via upregulating A2B adenosine receptor in rat. PLOS ONE 8:e69141. doi: 10.1371/journal.pone.0069141
Rabinovitch, M., Guignabert, C., Humbert, M., and Nicolls, M. R. (2014). Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 115, 165–175. doi: 10.1161/CIRCRESAHA.113.301141
Raja, S. G. (2012). Evaluation of clinical efficacy of fasudil for the treatment of pulmonary arterial hypertension. Recent Pat. Cardiovasc. Drug Discov. 7, 100–104. doi: 10.2174/157489012801227238
Ralevic, V., and Burnstock, G. (1998). Receptors for purines and pyrimidines. Pharmacol. Rev. 50, 413–492.
Ramakers, B. P., Pickkers, P., Deussen, A., Rongen, G. A., van den Broek, P., van der Hoeven, J. G., et al. (2008). Measurement of the endogenous adenosine concentration in humans in vivo: methodological considerations. Curr. Drug Metab. 9, 679–685. doi: 10.2174/138920008786049249
Ray, C. J., and Marshall, J. M. (2006). The cellular mechanisms by which adenosine evokes release of nitric oxide from rat aortic endothelium. J. Physiol. 570(Pt 1), 85–96. doi: 10.1113/jphysiol.2005.099390
Reeves, J. T., Groves, B. M., and Weir, E. K. (1991). Adenosine and selective reduction of pulmonary vascular resistance in primary pulmonary hypertension. Circulation 84, 1437–1439. doi: 10.1161/01.CIR.84.3.1437
Rump, L. C., Jabbari, T. J., von Kugelgen, I., and Oberhauser, V. (1999). Adenosine mediates nitric-oxide-independent renal vasodilation by activation of A2A receptors. J. Hypertens. 17(12 Pt 2), 1987–1993. doi: 10.1097/00004872-199917121-00032
Ryan, J. J., and Archer, S. L. (2014). The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ. Res. 115, 176–188. doi: 10.1161/CIRCRESAHA.113.301129
Saadjian, A. Y., Paganelli, F., Gaubert, M. L., Levy, S., and Guieu, R. P. (1999). Adenosine plasma concentration in pulmonary hypertension. Cardiovasc. Res. 43, 228–236. doi: 10.1016/S0008-6363(99)00059-0
Sakao, S., Tatsumi, K., and Voelkel, N. F. (2009). Endothelial cells and pulmonary arterial hypertension: apoptosis, proliferation, interaction and transdifferentiation. Respir. Res. 10:95. doi: 10.1186/1465-9921-10-95
Sartore, S., Chiavegato, A., Faggin, E., Franch, R., Puato, M., Ausoni, S., et al. (2001). Contribution of adventitial fibroblasts to neointima formation and vascular remodeling: from innocent bystander to active participant. Circ. Res. 89, 1111–1121. doi: 10.1161/hh2401.100844
Sassi, Y., Ahles, A., Truong, D. J., Baqi, Y., Lee, S. Y., Husse, B., et al. (2014). Cardiac myocyte-secreted cAMP exerts paracrine action via adenosine receptor activation. J. Clin. Invest. 124, 5385–5397. doi: 10.1172/JCI74349
Sawada, N., Itoh, H., Ueyama, K., Yamashita, J., Doi, K., Chun, T. H., et al. (2000). Inhibition of rho-associated kinase results in suppression of neointimal formation of balloon-injured arteries. Circulation 101, 2030–2033. doi: 10.1161/01.CIR.101.17.2030
Schermuly, R. T., Ghofrani, H. A., Wilkins, M. R., and Grimminger, F. (2011). Mechanisms of disease: pulmonary arterial hypertension. Nat. Rev. Cardiol. 8, 443–455. doi: 10.1038/nrcardio.2011.87
Shang, P., He, Z. Y., Chen, J. F., Huang, S. Y., Liu, B. H., Liu, H. X., et al. (2015). Absence of the adenosine A2A receptor confers pulmonary arterial hypertension through RhoA/ROCK signaling pathway in mice. J. Cardiovasc. Pharmacol. 66, 569–575. doi: 10.1097/FJC.0000000000000305
Shryock, J. C., and Belardinelli, L. (1997). Adenosine and adenosine receptors in the cardiovascular system: biochemistry, physiology, and pharmacology. Am. J. Cardiol. 79, 2–10. doi: 10.1016/S0002-9149(97)00256-7
Silva, A. G., Zapata-Sudo, G., Kummerle, A. E., Fraga, C. A., Barreiro, E. J., and Sudo, R. T. (2005). Synthesis and vasodilatory activity of new N-acylhydrazone derivatives, designed as LASSBio-294 analogues. Bioorg. Med. Chem. 13, 3431–3437. doi: 10.1016/j.bmc.2005.03.003
Simonneau, G., Gatzoulis, M. A., Adatia, I., Celermajer, D., Denton, C., Ghofrani, A., et al. (2013). Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 62(25 Suppl), D34–D41. doi: 10.1016/j.jacc.2013.10.029
Singh, L. S., and Sharma, R. (2000). Purification and characterization of intestinal adenosine deaminase from mice. Mol. Cell. Biochem. 204, 127–134. doi: 10.1023/A:1007087905192
Sisbarro, L., Ihida-Stansbury, K., Stevens, T., Bauer, N., McMurtry, I., and Jones, P. L. (2005). The extracellular matrix microenvironment specifies pulmonary endothelial cell identity: roles of tenascin-C and RhoA. Chest 128(6 Suppl), 564S. doi: 10.1378/chest.128.6_suppl.564S
Soon, E., Holmes, A. M., Treacy, C. M., Doughty, N. J., Southgate, L., Machado, R. D., et al. (2010). Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 122, 920–927. doi: 10.1161/CIRCULATIONAHA.109.933762
Spychala, J. (2000). Tumor-promoting functions of adenosine. Pharmacol. Ther. 87, 161–173. doi: 10.1016/S0163-7258(00)00053-X
St Croix, C. M., and Steinhorn, R. H. (2016). New Thoughts about the Origin of Plexiform Lesions. Am. J. Respir. Crit. Care Med. 193, 484–485. doi: 10.1164/rccm.201510-1959ED
St Hilaire, C., Yang, D., Schreiber, B. M., and Ravid, K. (2008). B-Myb regulates the A(2B) adenosine receptor in vascular smooth muscle cells. J. Cell. Biochem. 103, 1962–1974. doi: 10.1002/jcb.21586
Stacher, E., Graham, B. B., Hunt, J. M., Gandjeva, A., Groshong, S. D., McLaughlin, V. V., et al. (2012). Modern age pathology of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 186, 261–272. doi: 10.1164/rccm.201201-0164OC
Stoggall, S. M., and Shaw, J. S. (1990). The coexistence of adenosine A1 and A2 receptors in guinea-pig aorta. Eur. J. Pharmacol. 190, 329–335. doi: 10.1016/0014-2999(90)94197-6
Sutendra, G., Dromparis, P., Bonnet, S., Haromy, A., McMurtry, M. S., Bleackley, R. C., et al. (2011). Pyruvate dehydrogenase inhibition by the inflammatory cytokine TNFalpha contributes to the pathogenesis of pulmonary arterial hypertension. J. Mol. Med. 89, 771–783. doi: 10.1007/s00109-011-0762-2
Sztrymf, B., Souza, R., Bertoletti, L., Jais, X., Sitbon, O., Price, L. C., et al. (2010). Prognostic factors of acute heart failure in patients with pulmonary arterial hypertension. Eur. Respir. J. 35, 1286–1293. doi: 10.1183/09031936.00070209
Tabrizchi, R., and Bedi, S. (2001). Pharmacology of adenosine receptors in the vasculature. Pharmacol. Ther. 91, 133–147. doi: 10.1016/S0163-7258(01)00152-8
Takeshima, H., Kobayashi, N., Koguchi, W., Ishikawa, M., Sugiyama, F., and Ishimitsu, T. (2012). Cardioprotective effect of a combination of Rho-kinase inhibitor and p38 MAPK inhibitor on cardiovascular remodeling and oxidative stress in Dahl rats. J. Atheroscler. Thromb. 19, 326–336. doi: 10.5551/jat.11114
Talukder, M. A., Morrison, R. R., Jacobson, M. A., Jacobson, K. A., Ledent, C., and Mustafa, S. J. (2002). Targeted deletion of adenosine A(3) receptors augments adenosine-induced coronary flow in isolated mouse heart. Am. J. Physiol. Heart Circ. Physiol. 282, H2183–H2189. doi: 10.1152/ajpheart.00964.2001.
Teichert-Kuliszewska, K., Kutryk, M. J., Kuliszewski, M. A., Karoubi, G., Courtman, D. W., Zucco, L., et al. (2006). Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ. Res. 98, 209–217. doi: 10.1161/01.RES.0000200180.01710.e6
Teng, B., Qin, W., Ansari, H. R., and Mustafa, S. J. (2005). Involvement of p38-mitogen-activated protein kinase in adenosine receptor-mediated relaxation of coronary artery. Am. J. Physiol. Heart Circ. Physiol. 288, H2574–H2580. doi: 10.1152/ajpheart.00912.2004
Tofovic, S. P., Jackson, E. K., and Rafikova, O. (2009). Adenosine deaminase-adenosine pathway in hemolysis-associated pulmonary hypertension. Med. Hypotheses 72, 713–719. doi: 10.1016/j.mehy.2008.12.043
Tsutsui, S., Schnermann, J., Noorbakhsh, F., Henry, S., Yong, V. W., Winston, B. W., et al. (2004). A1 adenosine receptor upregulation and activation attenuates neuroinflammation and demyelination in a model of multiple sclerosis. J. Neurosci. 24, 1521–1529. doi: 10.1523/JNEUROSCI.4271-03.2004
Tuder, R. M., Marecki, J. C., Richter, A., Fijalkowska, I., and Flores, S. (2007). Pathology of pulmonary hypertension. Clin. Chest. Med. 28, 23–42, vii. doi: 10.1016/j.ccm.2006.11.010.
Umapathy, S. N., Kaczmarek, E., Fatteh, N., Burns, N., Lucas, R., Stenmark, K. R., et al. (2013). Adenosine A1 receptors promote vasa vasorum endothelial cell barrier integrity via Gi and Akt-dependent actin cytoskeleton remodeling. PLOS ONE 8:e59733. doi: 10.1371/journal.pone.0059733
Upton, P. D., Long, L., Trembath, R. C., and Morrell, N. W. (2008). Functional characterization of bone morphogenetic protein binding sites and Smad1/5 activation in human vascular cells. Mol. Pharmacol. 73, 539–552. doi: 10.1124/mol.107.041673
Varani, K., Caramori, G., Vincenzi, F., Adcock, I., Casolari, P., Leung, E., et al. (2006). Alteration of adenosine receptors in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 173, 398–406. doi: 10.1164/rccm.200506-869OC
Varani, K., Padovan, M., Govoni, M., Vincenzi, F., Trotta, F., and Borea, P. A. (2010a). The role of adenosine receptors in rheumatoid arthritis. Autoimmun. Rev. 10, 61–64. doi: 10.1016/j.autrev.2010.07.019
Varani, K., Vincenzi, F., Tosi, A., Targa, M., Masieri, F. F., Ongaro, A., et al. (2010b). Expression and functional role of adenosine receptors in regulating inflammatory responses in human synoviocytes. Br. J. Pharmacol. 160, 101–115. doi: 10.1111/j.1476-5381.2010.00667.x
Vecchio, E. A., White, P. J., and May, L. T. (2017). Targeting adenosine receptors for the treatment of cardiac fibrosis. Front. Pharmacol. 8:243. doi: 10.3389/fphar.2017.00243
Voelkel, N. F., Cool, C., Taraceviene-Stewart, L., Geraci, M. W., Yeager, M., Bull, T., et al. (2002). Janus face of vascular endothelial growth factor: the obligatory survival factor for lung vascular endothelium controls precapillary artery remodeling in severe pulmonary hypertension. Crit. Care Med. 30(5 Suppl), S251–S256. doi: 10.1097/00003246-200205001-00013
Voelkel, N. F., Quaife, R. A., Leinwand, L. A., Barst, R. J., McGoon, M. D., Meldrum, D. R., et al. (2006). Right ventricular function and failure: report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation 114, 1883–1891. doi: 10.1161/CIRCULATIONAHA.106.632208
Wagner, D. R., McTiernan, C., Sanders, V. J., and Feldman, A. M. (1998). Adenosine inhibits lipopolysaccharide-induced secretion of tumor necrosis factor-alpha in the failing human heart. Circulation 97, 521–524. doi: 10.1161/01.CIR.97.6.521
Woodiwiss, A. J., Honeyman, T. W., Fenton, R. A., and Dobson, J. G. Jr. (1999). Adenosine A2a-receptor activation enhances cardiomyocyte shortening via Ca2+-independent and -dependent mechanisms. Am. J. Physiol. 276(5 Pt 2), H1434–H1441.
Woolfson, R. G., Patel, V. C., and Yellon, D. M. (1996). Pre-conditioning with adenosine leads to concentration-dependent infarct size reduction in the isolated rabbit heart. Cardiovasc. Res. 31, 148–151. doi: 10.1016/S0008-6363(95)00185-9
Xiao, J. W., Zhu, X. Y., Wang, Q. G., Zhang, D. Z., Cui, C. S., Zhang, P., et al. (2015). Acute effects of Rho-kinase inhibitor fasudil on pulmonary arterial hypertension in patients with congenital heart defects. Circ. J. 79, 1342–1348. doi: 10.1253/circj.CJ-14-1015
Xu, H., Stein, B., and Liang, B. (1996). Characterization of a stimulatory adenosine A2a receptor in adult rat ventricular myocyte. Am. J. Physiol. 270(5 Pt 2), H1655–H1661.
Xu, J., Bian, X., Liu, Y., Hong, L., Teng, T., Sun, Y., et al. (2017). Adenosine A2 receptor activation ameliorates mitochondrial oxidative stress upon reperfusion through the posttranslational modification of NDUFV2 subunit of complex I in the heart. Free Radic. Biol. Med. 106, 208–218. doi: 10.1016/j.freeradbiomed.2017.02.036.
Xu, M. H., Gong, Y. S., Su, M. S., Dai, Z. Y., Dai, S. S., Bao, S. Z., et al. (2011). Absence of the adenosine A2A receptor confers pulmonary arterial hypertension and increased pulmonary vascular remodeling in mice. J. Vasc. Res. 48, 171–183. doi: 10.1159/000316935
Xu, R., and Mao, J. H. (2011). Gene transcriptional networks integrate microenvironmental signals in human breast cancer. Integr. Biol. 3, 368–374. doi: 10.1039/c0ib00087f
Yang, Z., Day, Y. J., Toufektsian, M. C., Ramos, S. I., Marshall, M., Wang, X. Q., et al. (2005). Infarct-sparing effect of A2A-adenosine receptor activation is due primarily to its action on lymphocytes. Circulation 111, 2190–2197. doi: 10.1161/01.CIR.0000163586.62253.A5
Yang, Z., Day, Y. J., Toufektsian, M. C., Xu, Y., Ramos, S. I., Marshall, M. A., et al. (2006). Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4+ T lymphocytes. Circulation 114, 2056–2064. doi: 10.1161/CIRCULATIONAHA.106.649244
Zhang, S., Fantozzi, I., Tigno, D. D., Yi, E. S., Platoshyn, O., Thistlethwaite, P. A., et al. (2003). Bone morphogenetic proteins induce apoptosis in human pulmonary vascular smooth muscle cells. Am. J. Physiol. Lung. Cell Mol. Physiol. 285, L740–L754. doi: 10.1152/ajplung.00284.2002
Zhao, Y. D., Courtman, D. W., Deng, Y., Kugathasan, L., Zhang, Q., and Stewart, D. J. (2005). Rescue of monocrotaline-induced pulmonary arterial hypertension using bone marrow-derived endothelial-like progenitor cells: efficacy of combined cell and eNOS gene therapy in established disease. Circ. Res. 96, 442–450. doi: 10.1161/01.RES.0000157672.70560.7b
Zhong, H., Belardinelli, L., Maa, T., Feoktistov, I., Biaggioni, I., and Zeng, D. (2004). A(2B) adenosine receptors increase cytokine release by bronchial smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 30, 118–125. doi: 10.1165/rcmb.2003-0118OC
Keywords: pulmonary arterial hypertension, cardiopulmonary system, adenosine receptors, cell proliferation, right ventricle dysfunction, pleiotropic effects
Citation: Alencar AKN, Montes GC, Barreiro EJ, Sudo RT and Zapata-Sudo G (2017) Adenosine Receptors As Drug Targets for Treatment of Pulmonary Arterial Hypertension. Front. Pharmacol. 8:858. doi: 10.3389/fphar.2017.00858
Received: 12 July 2017; Accepted: 09 November 2017;
Published: 04 December 2017.
Edited by:
Kenneth A. Jacobson, National Institutes of Health (NIH), United StatesReviewed by:
Katia Varani, University of Ferrara, ItalyCopyright © 2017 Alencar, Montes, Barreiro, Sudo and Zapata-Sudo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gisele Zapata-Sudo, Z3pzdWRvQG9pLmNvbS5icg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.