 Xiaoou Hou1,2†Yuqing Yuan1,3†Yulan Sheng1,3†Baoshi Yuan1Yali Wang1,2Jiyue Zheng4
Xiaoou Hou1,2†Yuqing Yuan1,3†Yulan Sheng1,3†Baoshi Yuan1Yali Wang1,2Jiyue Zheng4 Chun-Feng Liu1,2Xiaohu Zhang4*
Chun-Feng Liu1,2Xiaohu Zhang4* Li-Fang Hu1,2,4*
Li-Fang Hu1,2,4*- 1Institute of Neuroscience, Soochow University, Suzhou, China
- 2Department of Neurology and Suzhou Clinical Research Center of Neurological Disease, The Second Affiliated Hospital of Soochow University, Suzhou, China
- 3Department of Pharmacology, School of Pharmacy, Soochow University, Suzhou, China
- 4Jiangsu Key Laboratory of Translational Research and Therapy for Neuro-Psychiatric-Diseases and College of Pharmaceutical Sciences, Soochow University, Suzhou, China
The neuromodulator hydrogen sulfide (H2S) was shown to exert neuroprotection in different models of Parkinson’s disease (PD) via its anti-inflammatory and anti-apoptotic properties. In this study, we evaluated the effect of an H2S slow-releasing compound GYY4137 (GYY) on a mouse PD model induced by acute injection with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). GYY was intraperitoneally (i.p.) injected once daily into male C57BL/6J mice 3 days before and 2 weeks after MPTP (14 mg/kg, four times at 2-h intervals, i.p.) administration. Saline was given as a control. Behavioral tests (rotarod, balance beam, and grid walking) showed that 50 mg/kg GYY significantly ameliorated MPTP-caused motor impairments. At lower doses (12.5 and 25 mg/kg) GYY exhibited a less obvious effect. Consistent with this, immunohistochemistry and western blot analysis demonstrated that 50 mg/kg GYY attenuated the loss of tyrosine hydroxylase (TH) positive neurons in the substantia nigra and the decrease of TH expression in the striatum of MPTP-treated mice. Moreover, at this regimen GYY relieved the nitrative stress, as indicated by the decreases in nitric oxide (NO) generation and neuronal NO synthase (nNOS) upregulation elicited by MPTP in the striatum. The suppression of GYY on nNOS expression was verified in vitro, and the results further revealed that Akt activation may participate in the inhibition by GYY on nNOS upregulation. More important, GYY reduced the nitrated modification of α-synuclein, a PD-related protein, in MPTP-induced mice. Overall, our findings suggest that GYY attenuated dopaminergic neuron degeneration and reduced α-synuclein nitration in the midbrain, thus exerting neuroprotection in MPTP-induced mouse model of PD.
Introduction
Hydrogen sulfide is the third gasotransmitter next to NO and carbon monoxide. It regulates a variety of physiologic and pathologic processes in a wide range of biological systems. For example, the cse (a gene encoding the H2S synthase CSE) deficient mice developed hypertension early at 7 weeks after birth (Yang et al., 2008). H2S also exerted a beneficial role in atherosclerosis and related disorders (Wang et al., 2009). We previously demonstrated its anti-fibrotic property in chronic kidney disease (Song et al., 2014). Indeed, H2S was first proposed as an endogenous neuromodulator by Abe and Kimura (1996). However, it was until recently that its function in the central nervous system gains the attention of scientists. Increasing studies identify a potential role of H2S in neurodegeneration. For example, sodium hydrosulfide (NaHS, an H2S fast-releasing salt) reduced amyloid beta-peptide-induced neuronal injury and ameliorated the learning memory impairment in APP/PS1 transgenic mice (Fan et al., 2013; Liu et al., 2016). In addition, cse deficiency contributed to the neurodegeneration in Huntington’s disease (Paul et al., 2014).
Parkinson’s disease is the second most common neurodegenerative disorder, affecting approximately 1.7% of the population over 65 years old. Pathologically, it is featured by dopaminergic neuron losses in the SN and formation of inclusion bodies that are composed of α-synuclein (α-syn). Its etiology remains elusive. Several pathogenic factors such as oxidative stress, mitochondrial dysfunction, protein misfolding and neuroinflammation, have been reported to be involved. In addition, higher level expressions of NO and its synthases were detected in the brains of PD patients and animal models (Eve et al., 1998; Dehmer et al., 2000). Inhibitors or depletion of NOS protected against dopaminergic neuron degeneration in MPTP-induced mouse models (Schulz et al., 1995; Liberatore et al., 1999). These studies strongly suggest a role of nitrative stress in PD progression.
Inhaled H2S or NaHS has been shown to exhibit neuroprotection in neurotoxins-induced rodent models of PD (Hu et al., 2010; Kida et al., 2011; Lu et al., 2012). Of note, manipulation of H2S gas deals with a lot of safety issues in practice. NaHS is unstable in water solution. Moreover, NaHS or Na2S delivers a rapid bolus of H2S in aqueous solution within a quite short period (seconds) that does not accurately mimic the biological process of H2S release in vivo. These limit our understanding to the role of H2S in PD pathogenesis. As such, in this study we evaluated the effect of GYY, which slowly releases low but consistent amounts of H2S within several hours in aqueous solution at physiologic pH and temperature (Li et al., 2008), in an MPTP-induced mouse model of PD, and the underlying mechanism involved. Our findings clearly demonstrate a protective action of GYY against dopaminergic neuron losses caused by MPTP via its anti-nitrative stress property and inhibition on α-syn nitration, and thus consolidate a favorable role of H2S in PD development.
Materials and Methods
Animals and MPTP Treatment
Male C57BL/6J mice (8–10 weeks old; 23–28 g weight) were purchased from the SLRC Laboratory (Shanghai, China). The experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Soochow University. All mice were housed in a SPF animal facility with a temperature at 21–25°C and a controlled light–dark cycle.
Parkinson’s disease animal models were established by intraperitoneally (i.p.) injecting the mice with MPTP (14 mg/kg, Sigma-Aldrich, St. Louis, MO, United States) four times at 2-h intervals. Saline was given as controls. To evaluate the effect of GYY (Cayman Chemical, United States), the mice were administered with GYY at individual doses (12.5, 25, and 50 mg/kg, i.p.) once daily for 3 days prior to MPTP treatment, and continued throughout the experimental period. The animals were sacrificed 2 weeks after MPTP administration for biochemical studies.
Behavioral Assessment
Behavioral assessments were performed at 2 weeks after MPTP injection. The data were analyzed by independent observers blinded to the treatments.
Rotarod test was performed to estimate motor balance and coordination. Before test, each mouse was trained three times on the rod with an accelerating speed at 5–20 rpm/min. During the test session, the rotational speed remained constant at 18 rpm/min. A trial was stopped if the mouse fell down or the latency to fall reached 5 min. Each mouse was subjected to three trials with an interval of 30 min during the test session, and the average value of the latency to stay on the rod was calculated and included for further analysis.
Balance Beam Walking
The mice were placed on a batten and tempted with fodder to cross a wooded balance beam (64 cm long, 1.5 cm wide, 15 cm high). The test was stopped and repeated again if the mouse fell off. The time that a mouse successfully transversed the balance beam was recorded. The test was repeated three times at 5 min intervals, and the mean value of the walking time was presented as the data for analysis. A longer latency to cross the beam indicated a poorer motor coordination.
Grid walking test was applied to evaluate the sensorimotor coordination of hindlimbs in mice. The mice were placed on a 50 ∗ 40 cm wire grid with 3 ∗ 3 cm grid squares (iron wire at a diameter of 4 mm) in a quiet room with dim lighting, and allowed to walk for 30 s. The number of hindlimb slips was counted by an independent experimenter when the paw completely failed to hold a rung. The animal was put on the grid twice for habituation before test without pre-training. Each trial was repeated three times and the average of foot slips was used for analysis.
Immunohistochemical Staining
Following the behavioral tests, the animals were deeply anesthetized and perfused with 0.1 M phosphate buffer followed by 4% paraformaldehyde (PFA). Mouse brains were immediately removed from the skull and post-fixed in PFA solution overnight. After that, brains were dehydrated in a serial of alcohol solution and dimethylbenzene was applied to make the brains transparent. Brains were then embedded in paraffin wax. Next, 4 μm thickness sections were cut through SN compacta (SNpc), and every fourth slice was reserved. For staining, sections were deparaffinized and rehydrated, followed by epitope retrieval by boiling the sections in 0.01 M citrate buffer (pH 6.0) for 10–15 min and cooling at room temperature for 20–30 min. After that, sections were thoroughly washed in 0.01 M PBS three times. Endogenous peroxidase activity was blocked with 3% H2O2 in 0.01 M PBS for 30 min. Next, the sections were incubated in 5% BSA with 0.1% Triton X-100 in PBS for 30 min, followed by incubation with mouse monoclonal anti-TH (1:1000, Sigma, St. Louis, MO, United States) at 4°C overnight. Subsequently, the sections were washed in PBS, incubated with goat anti-mouse secondary antibody at room temperature for 1 h and stained using a DAB kit (Gene Tech, GK500705, China). The sections were observed and photographed using the Zeiss microscope (Carl Zeiss, 37081, Germany).
Striatal MPP+ Level Analysis
Striatal MPP+ levels were determined as previously described (Jackson-Lewis and Przedborski, 2007) with minor modifications. In brief, striatum and whole blood were collected 90 min after four injections of MPTP (14 mg/kg, i.p.) at 2-h intervals. The tissue samples were homogenized by sonication in 0.2 M chilled perchloric acid and then centrifuged at 15,000 g for 30 min at 4°C. The whole blood samples were kept at room temperature for 30 min and subjected to centrifuged at 1000 g, 30 min. Serum was harvested from the resulting supernatant, and mixed with 0.2 M perchloric acid before subjected to the second centrifuged at 15,000 g for 30 min at 4°C. The supernatants were transferred into a clean eppendorf tube for MPTP and MPP+ analysis.
MPTP/MPP+ was analyzed by HPLC equipped with UV detectors (MPTP at 295 nm; MPP+ at 245 nm). An aliquot (20 μl) of each supernatant was eluted through a C18 reverse phase column (5 μm, 250 mm × 4.6 mm, SHIMADZU VP-ODS) attached to the HPLC system (Waters, Milford, MA, United States) at a flow rate of 1.0 ml/min. The mobile phase consists of 15% acetonitrile, 50 mM potassium phosphate adjusted to pH 3.2 with ultrapure 18 M phosphoric acid. The detection limit is 3 ng/ml and this method provides good reproducibility.
NO Production Determination
The striatal NO content was evaluated by measuring the accumulation of nitrite and nitrate using the kit from Jiancheng Bioengineering Institute (Nanjing, Jiangsu, China). Briefly, striatum tissues were homogenized (1:10 m/v) in lysis buffer and centrifuged at 15,000 g for 30 min at 4°C. Next, an aliquote (50 μl) of the resulting supernatant was mixed with the dilution buffer at an equal volume, followed by addition with 5 μl nicotinamide adenine dinucleotide phosphate (NADPH), 10 μl flavin adenine dinucleotide (FAD) and 5 μl nitrate reductase and incubation at 37°C for 15 min. After that, 10 μl lactate dehydrogenase (LDH) was added and incubated for another 5 min. Subsequently, 50 μl Griess reagent I (0.1% naphthylethylene diaminedihydrochloride in 1% sulfanilamide) and 50 μl Griess reagent II (2.5% H3PO4) were added and incubated for 10 min. The optical density of the mixture was measured at 540 nm using a microplate reader (Tecan Infinite M200 PRO, Switzerland). The results were calculated by a standard curve with sodium nitrite, and expressed as μmol per gram protein. The protein concentrations of striatal homogenates were determined using the BCA protein assay kit (Thermoscientific, United States).
Western Blot Analysis
Whole striatum tissues were homogenized with lysis buffer and centrifuged at 13,200 rpm for 20 min at 4°C. Protein concentrations of the homogenates were determined using the BCA protein assay kit. Equal amounts of total proteins were loaded in 8–12% sodium dodecyl sulfate polyacrylamide gels and then transferred onto PVDF membranes. Membranes were incubated with specific antibodies: anti-TH (1:2000, Sigma, T1299, United States), anti-nNOS (1:1000, BD, 610308, New York, United States), anti-nitrated α-syn (1:750, Thermo Scientific, MA5-16142, United States), anti-α-syn (1:500, CST, 2642, United States), anti-p-Akt (1:500, CST, 4060s, United States), anti-Akt (1:1000, CST, 4691s, United States), and anti-β-actin (1:5000, Sigma, A3584, United States) at 4°C overnight. Blots were then briefly washed with PBS followed by incubation with the corresponding secondary antibodies (Jackson Lab, United States) for 1 h at room temperature. Protein densities were detected using an enhanced chemiluminescence kit and analyzed by ImageJ software.
Cell Culture and Treatment
PC12 cells were purchased from the Institute of Cell Biology, Chinese Academy of Sciences (China), and cultured in RPMI1640 medium with 10% fetal bovine serum and 1% penicillin/streptomycin. The cultures were maintained in a incubator with 5% CO2/95% air at 37°C. For experimentation, cells were seeded into a 12-well plate and cultured overnight before treatment.
Statistical Analysis
All data were presented as mean ± SEM of at least three independent experiments. All statistical analyses were performed with GraphPad Prism 5.0 software. The differences among groups were analyzed using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc analysis. Statistical significance was set at P < 0.05.
Results
GYY Improves Motor Deficits Induced by MPTP in Mouse
To study the ability of GYY to affect MPTP-induced impairment in motor function, we assessed the animals’ motor coordination using rotarod, balance beam walking, and grid walking test at 2 weeks after MPTP injection. The results are shown in Figure 1. Compared to saline-treated group (control), the latency to fall off the rod significantly decreased in MPTP-injected mice (Figure 1A). Co-treatment with GYY at 25 and 50 mg/kg prolonged the time of staying on the rod compared to MPTP-only group. Balance beam walking test also showed that 50 mg/kg GYY co-treatment improved the MPTP-induced motor deficits, as indicated by a shorter time period to cross the beam compared to MPTP only group (Figure 1B). Similarly, in grid walking test we found that MPTP injection increased the number of foot slips in mice, implying that the sensorimotor function was also impaired by MPTP. This was markedly attenuated by 50 mg/kg GYY co-treatment (Figure 1C). And the treatment with GYY alone at 50 mg/kg, the maximal tested dose in this study, did not have any impact on the motor behavior.
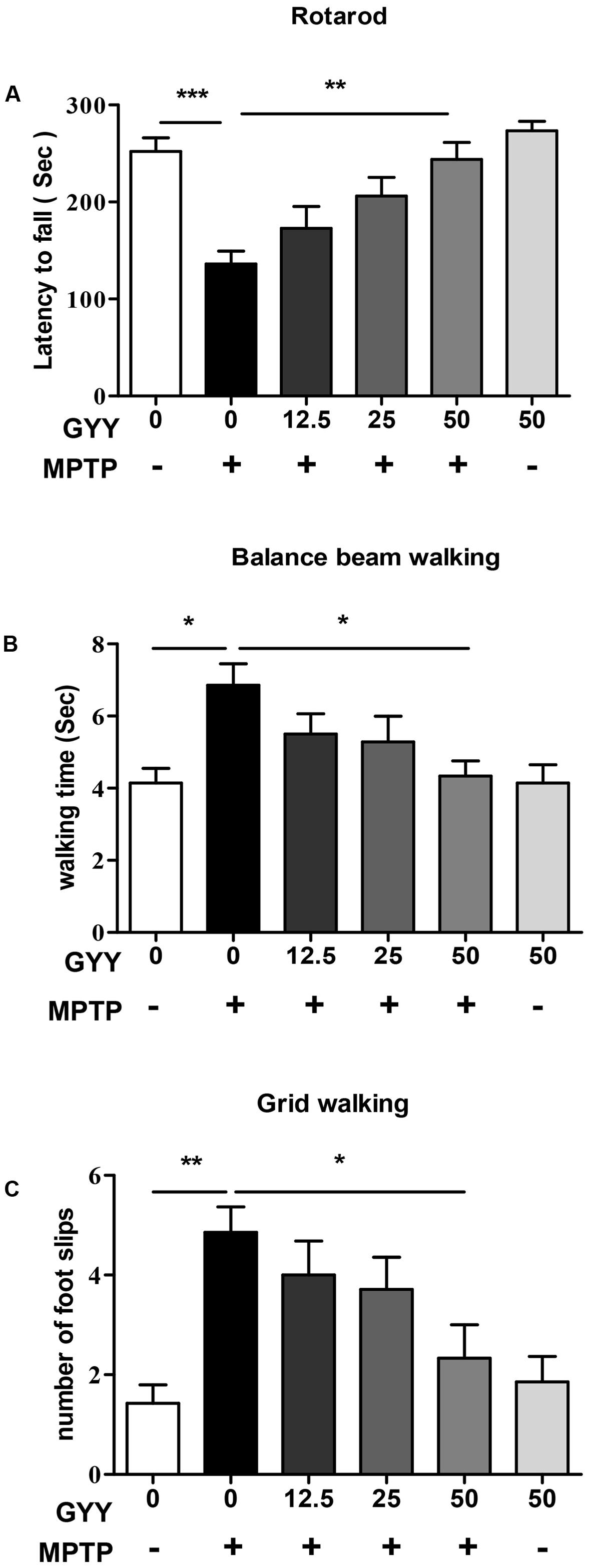
FIGURE 1. GYY ameliorated motor deficits in MPTP-induced mouse model of PD. Performance on the rotarod (A) and balance beam (B) was impaired by MPTP administration, as indicated by a shorter latency to stay on the rod (A) and a longer period to cross the beam (B) in MPTP-injected mice. The number of foot slips in the hindpaw (C) was also increased by MPTP injection compared to controls. GYY (12.5, 25, and 50 mg/kg/d, i.p.), administered 3 days before and 2 weeks after MPTP injection, alleviated the MPTP-induced motor dysfunction to a different extent, and the effect of 50 mg/kg GYY was consistently significant. Treatment with 50 mg/kg GYY or saline alone did not affect the motor behavior. Results are presented as mean ± SEM from 6 to 7 mice per group. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001; one-way ANOVA.
Striatal MPP+ Levels Are not Affected by GYY Preventive Treatment
As MPTP-caused neurotoxicity to dopaminergic neurons is dependent on the metabolism of MPTP to its active metabolite MPP+ via monoamine oxidase (MAO) in the nigrostriatal system, the effect of GYY on striatal MPP+ levels was determined in a parallel study. In this part, 10 male C57BL/6J mice (10 weeks old) were applied and randomly divided into two groups: MPTP and GYY + MPTP group (n = 5 per group). For MPTP group, the mice received four injections with MPTP (14 mg/kg, i.p., at 2 h intervals). For GYY + MPTP group, the mice were treated with GYY (50 mg/kg, i.p.) once daily for 3 days followed by MPTP administration at 1 h after the last injection with GYY. No significant changes were observed in the striatal MPP+ level in MPTP-treated mice with and without GYY (50 mg/kg, i.p.) pretreatment (MPTP 3.90 ± 1.98 nmol/mg tissue; GYY + MPTP group, 3.96 ± 1.46 nmol/mg tissue; P > 0.05; Figure 2), which was measured 90 min after the last MPTP injection. Measurement of serum MPP+ levels revealed no significant difference between the two tested groups either (MPTP 3.25 ± 0.39 μg/ml; GYY + MPTP group, 3.14 ± 0.33 μg/ml, P > 0.05). This indicates that the observed effects of GYY on MPTP-induced neurotoxicity are not caused by the reduction of MPTP conversion into MPP+.
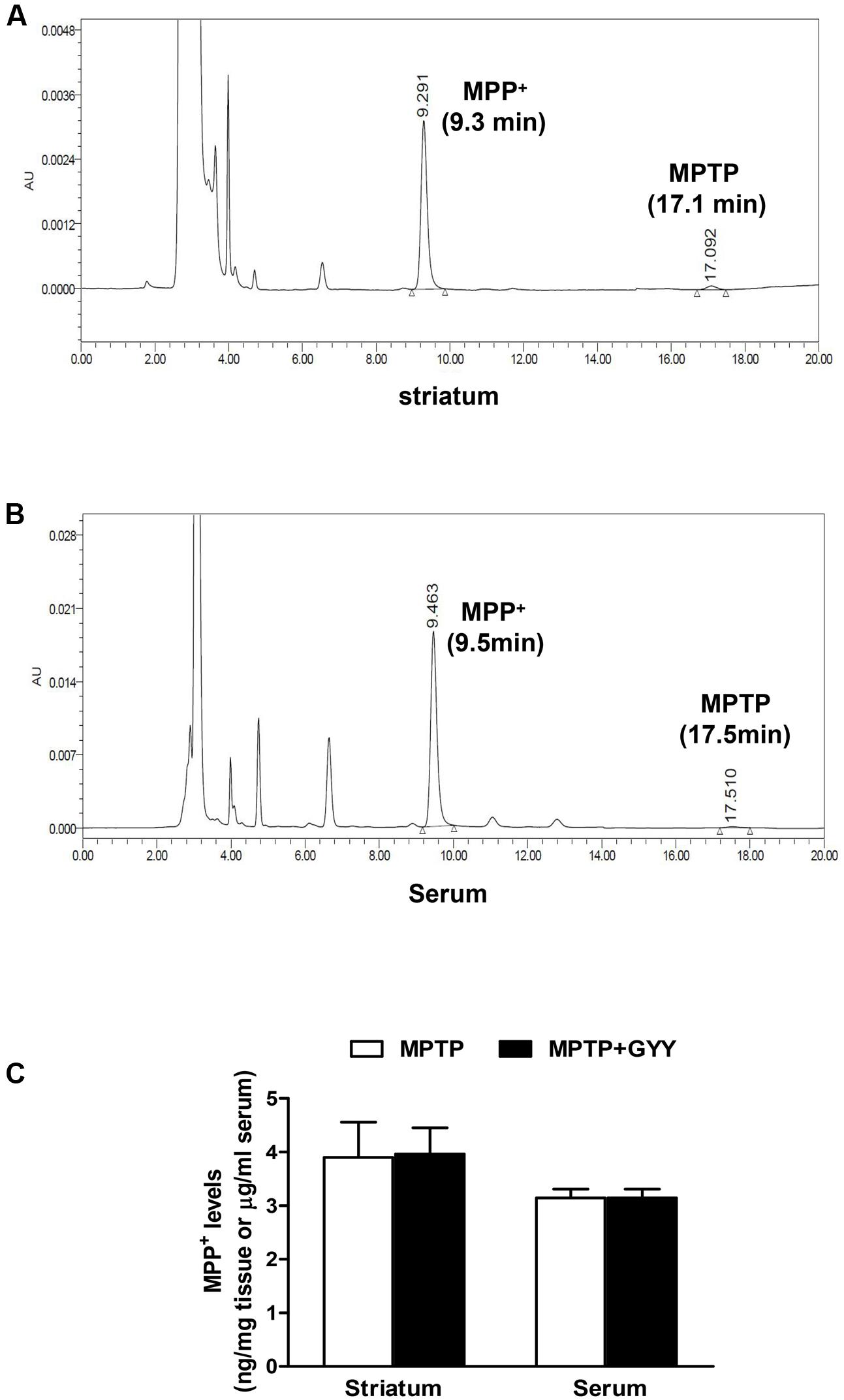
FIGURE 2. GYY did not affect MPTP metabolism. Striatal and serum MPP+ levels quantification by HPLC in combination with UV detection. Panels show typical chromatographs of striatal (A) and serum (B) MPP+ and MPTP. Typical retention times for MPP+ at 9.3 min and MPTP at 17.5 min. MPTP levels, almost near the detection limit (3 ng/ml), were much lower than those of MPP+ and thus were not included for analysis. Mice received four injections of MPTP (14 mg/kg, i.p., at 2 h intervals), with or without GYY (50 mg/kg, i.p.) pretreatment for 3 days (five mice per group). MPTP was given at 1 h after the last GYY injection. Striatum and whole blood were harvested 90 min after the last MPTP administration. GYY pretreatment did not affect the striatal and serum MPP+ levels resulting from MPTP metabolism (C). Student’s t-test. N = 5 for serum sample; n = 10 for striatal homogenates (both sides of the striatum from five mice per group).
GYY Attenuates Dopaminergic Neuron Losses in the Substantia Nigra
The dopaminergic neuron degeneration was examined by quantifying the number of TH immunoreactive neurons in the SNpc using immunohistochemistry study. The result showed that MPTP caused an approximate 40% decrease in the number of dopaminergic neurons in the SN compared to saline, and this was attenuated in the mice co-treated with 50 mg/kg GYY (Figures 3A,B). Treatment with GYY alone did not have any impact on TH-positive staining in the SN. Western blot analysis showed that 50 mg/kg GYY co-treatment prevented the reduction of striatal TH protein level caused by MPTP (Figure 3C). These results indicate that co-treatment with GYY at a suitable dose was able to prevent dopaminergic neuron losses in the SN of MPTP-treated mice.
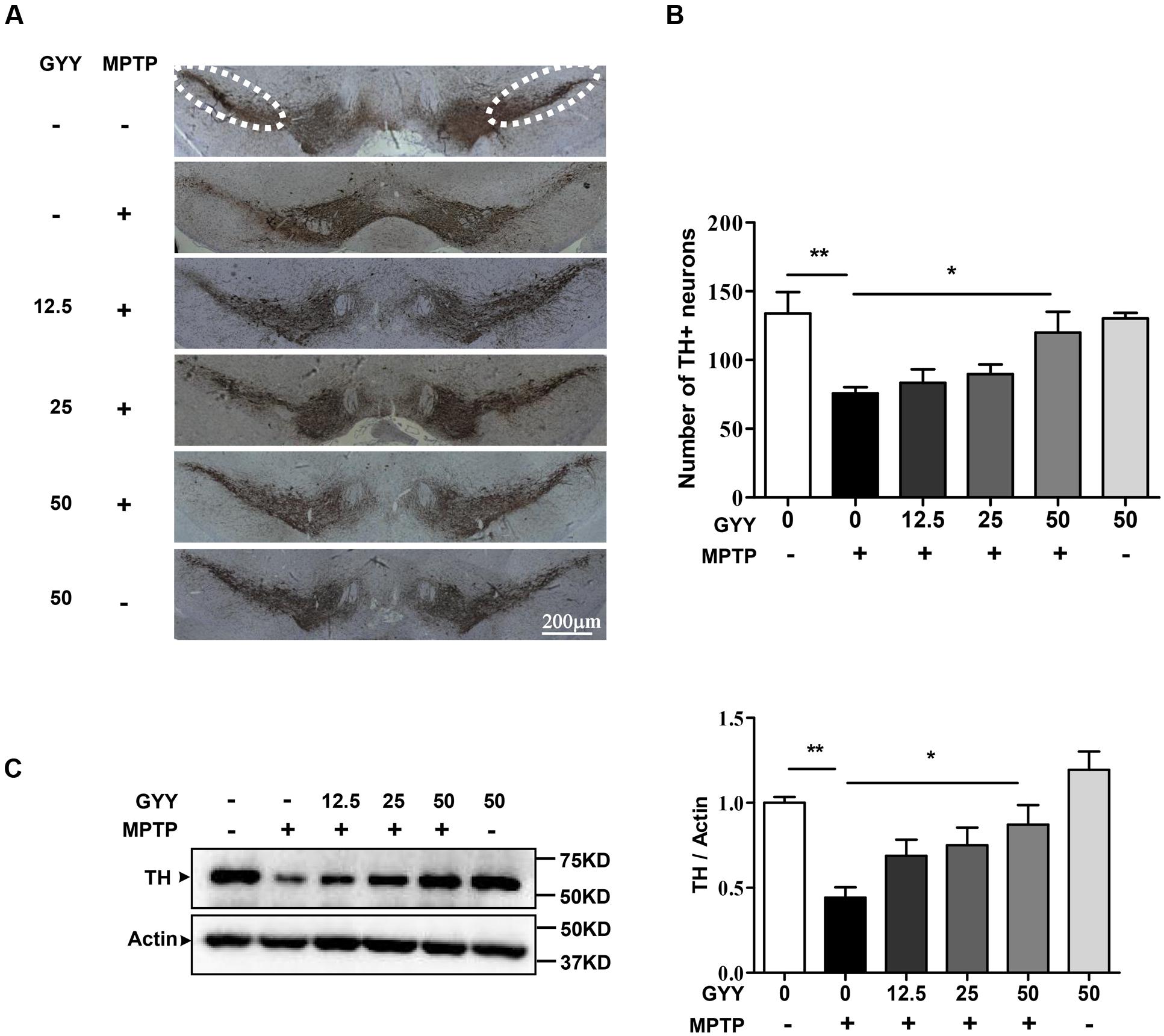
FIGURE 3. GYY attenuated dopaminergic neuron losses in MPTP-treated mice. (A) MPTP injection caused a reduction in the number of TH positive neurons in the SNpc (the circled area in dotted line). Group data was shown in (B). The number of TH positive neurons in both sides was counted from six brain slices for each mouse, with 3–5 mouse brains per group. The counting was performed by the experimenters blind to the treatments. (C) Striatal TH expression in MPTP-intoxicated mice was markedly reduced. N = 6 per group. Co-treatment with 50 mg/kg GYY was able to attenuate the loss of TH neuron in the SNpc (A,B) and the decrease of TH expression (C) in the striatum. The molecular masses of the protein markers were indicated to the right of the blots. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001; one-way ANOVA.
GYY Mitigates MPTP-Induced NO Overproduction and Nitrated Modification of α-Synuclein in the Striatum
Overproduction of NO and its associated nitrative stress participate in PD pathology (Jimenez-Jimenez et al., 2016). In line with previous studies (Dehmer et al., 2000), we observed an accumulation of nitrite, a metabolite of NO, in the striatum of the mice receiving MPTP administration (Figure 4A). This increase of NO generation was abated in the mice co-treated with GYY at the dose of 25 and 50 mg/kg, respectively. Likewise, co-treatment with GYY at this regimen inhibited the MPTP-induced upregulation of nNOS expression in the striatum (Figure 4B). Treatment with GYY alone did not modulate NO or nNOS expression level. The result was verified by an in vitro study, which demonstrated that pre-treatment with 100 μM GYY prevented MPP+ (100 μM, 24 h) elicited increase of nNOS expression in PC12 cells (Figure 4C).
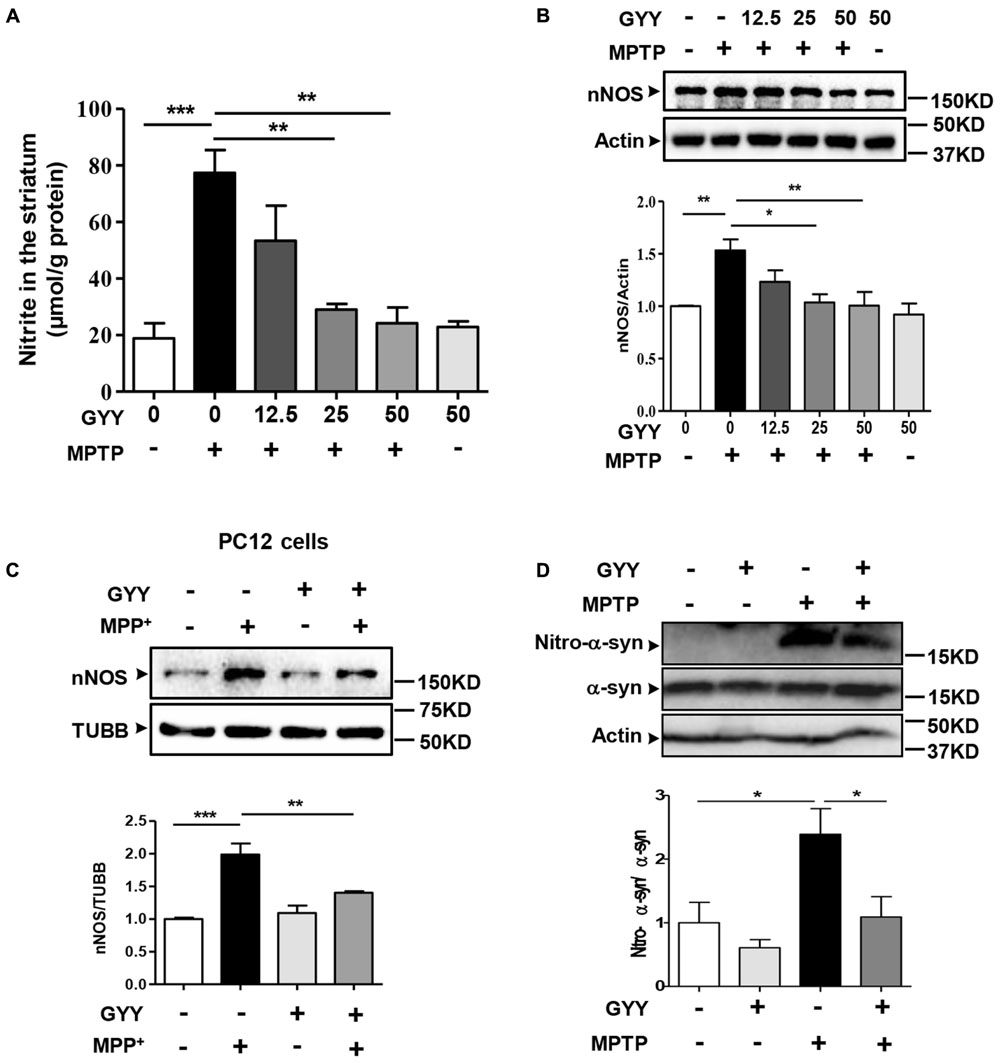
FIGURE 4. GYY mitigated MPTP-induced nitrative stress and α-syn nitration in the striatum. (A) Co-treatment with 50 mg/kg GYY abolished NO overproduction in the striatum of MPTP-injected mice. The nitrite level was normalized by the protein concentration of striatal homogenates, n = 3. (B) Western blot study showed an upregulation of striatal nNOS induced by MPTP compared to controls, and this was attenuated by GYY (25, 50 mg/kg) co-treatment. N = 5 per group. Treatment with 50 mg/kg GYY alone did not affect NO generation or nNOS expression. (C) Effect of GYY on nNOS expressions in PC12 cells treated with 100 μM MPP+ or vehicle for 24 h. Cells were pretreated with 100 μM GYY for 1 h before MPP+ being added. Relative values were obtained after normalization to loading controls (Tubulin/TUBB), n = 4. (D) Co-treatment with 50 mg/kg GYY led to a reduced nitrated modification of α-syn, but not the total α-syn in the striatum, indicated by a lower ratio of nitro-α-syn over total α-syn compared to MPTP-treated mice. Actin served as loading controls. Mean ± SEM, n = 5. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001; one-way ANOVA.
Excessive NO generation often results in peroxynitrite formation and tyrosine nitration in proteins. Human α-syn contains four tyrosine residues (Tyr39, Tyr125, Tyr133, and Tyr136) that can be nitrated (Schildknecht et al., 2013), which may play a role in protein aggregation in PD. Therefore, we further examined the effect of GYY on α-syn nitration by western blot using the specific antibody against nitrated α-syn (Tyr125, Tyr133). The result showed that although the total α-syn level was not altered in the striatum following MPTP injection, the nitrated α-syn level was markedly elevated in MPTP-treated mice, and this elevation was abolished by GYY co-treatment (Figure 4D).
Akt Signaling Mediates GYY-Induced Inhibition on nNOS Upregulation
Last, we studied the signaling pathway that underlies the inhibition by GYY on nNOS expression. Treatment with 100 μM MPP+ was found to reduce the phosphorylation of Akt Ser 473 in a time-dependent manner, which was detected early at 0.25 h and remained to be dramatically declined at 1 h after treatment in PC12 cells (Figure 5A). Pretreatment with 100 μM GYY almost reversed the decline of Akt phosphorylation caused by MPP+ exposure (Figure 5B). However, co-treatment with Akt inhibitor perifosine (5 μM) was able to attenuate the GYY-mediated inhibition on nNOS expression in MPP+-treated cells (Figure 5C). These results indicate that Akt signaling may be activated and thus contribute to the inhibition on nNOS expression by GYY.
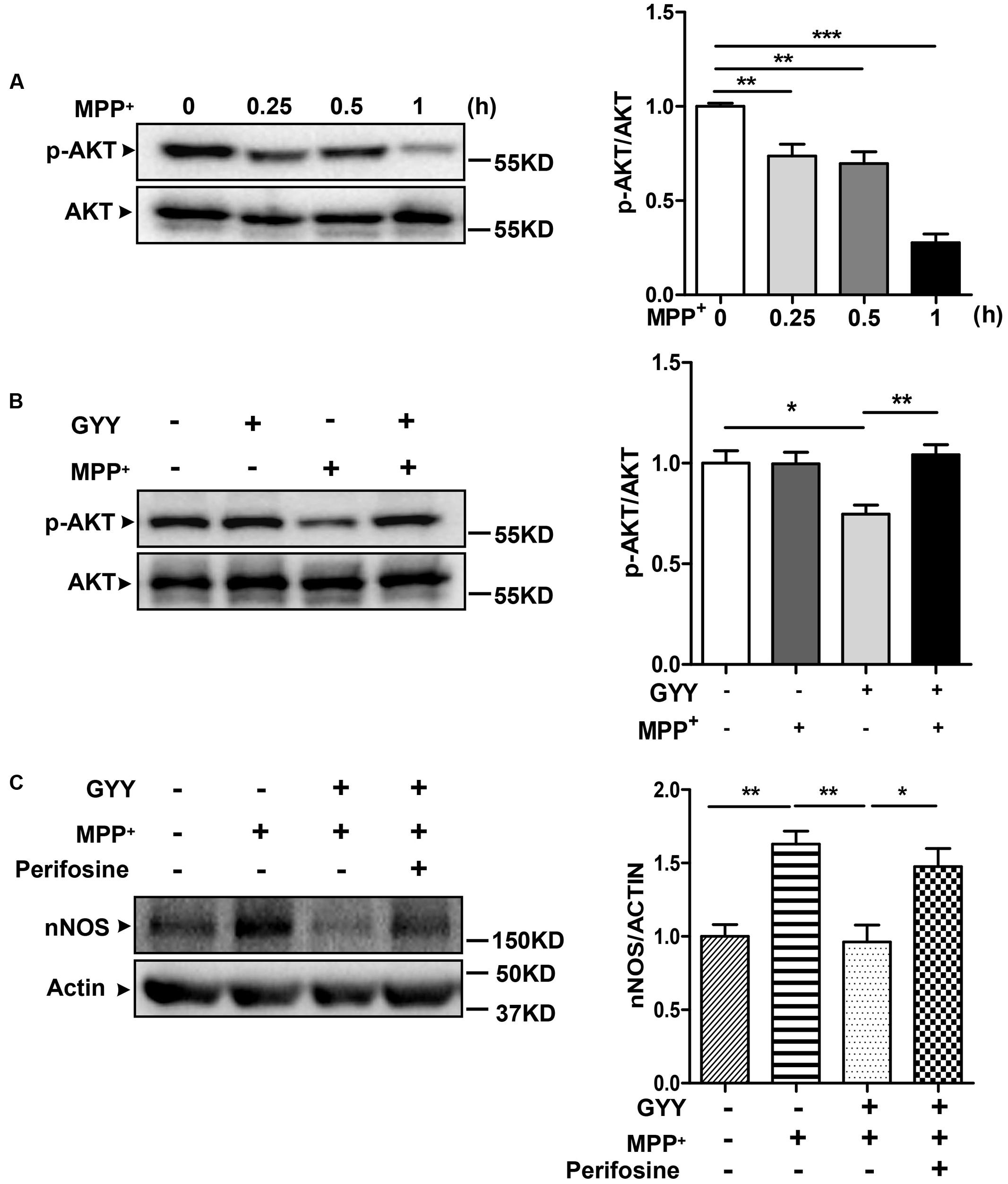
FIGURE 5. Akt activation was involved in the inhibition by GYY on nNOS expression. (A) Decreases of AKT phosphorylation at different time points after 100 μM MPP+ treatment in PC12 cells, n = 3. (B) Pretreatment with 100 μM GYY for 1 h was found to reverse the decrease of AKT phosphorylation caused by MPP+ in cells. N = 6. (C) The inhibition of GYY pretreatment on nNOS expression was abolished in the presence of AKT inhibitor perifosine. PC12 cells were pretreated with perifosine (5 μM) for 1 h, followed by incubation with GYY for 1 h and subsequent exposure to MPP+ for 24 h. N = 5. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001; one-way ANOVA.
Discussion
In this study we demonstrate the potential of GYY as a neuroprotectant against dopaminergic neuron losses in a mouse model of PD based on the following findings. First, intraperitoneal injection with GYY at an appropriate dose (50 mg/kg) alleviated MPTP-induced motor impairments in mice. Second, this H2S slow-releasing compound attenuated dopaminergic neuron losses in the SNpc caused by MPTP. Last, GYY mitigated MPTP-induced nitrative stress and α-syn nitration in vivo. In addition, GYY preventive treatment did not affect MPTP metabolism, as serum and striatal MPP+ levels in GYY + MPTP group did not differ from those in MPTP group. To our knowledge, this is the first study demonstrating the neuroprotection of an H2S slow-releasing compound in the MPTP-induced mouse model of PD.
Hydrogen sulfide is linked to the pathogenesis of PD. Hu et al. (2010) first reported that NaHS exerted neuroprotection against dopaminergic neuron losses in 6-hydroxydopamine and rotenone-induced rat models of PD. This beneficial role of H2S in PD was subsequently confirmed by other studies. Two independent groups demonstrated that systemic administration with NaHS or inhaled H2S prevented neurodegeneration and movement dysfunction in the MPTP-induced mouse model of PD (Kida et al., 2011; Paul et al., 2014). Vandiver et al. (2013) demonstrated that sulfhydration, a post-translational modification afforded by H2S, mediated the neuroprotective actions of parkin, and this study again highlighted a potential role of H2S in PD. However, it should be noted that most studies utilized NaHS as an H2S donor, which often leads to a burst of H2S release in solution. This does not mimick the biological synthesis of H2S, which is tightly regulated in vivo. To fully understand the role of H2S in PD, it is essential to assess the effect of H2S slow-releasing compounds in PD models. GYY is a water-soluable compound that releases H2S slowly over a period of hours in vitro and in vivo, producing H2S-related beneficial effects in several pathological conditions (Lin et al., 2016; Weber et al., 2017). Thus, in this study we evaluated the impact of GYY on a classic mouse model of PD induced by acute MPTP injection. Functionally, administration of GYY improved the motor impairments. Pathologically, GYY co-treatment attenuated the loss of dopaminergic neurons in the SN. Moreover, GYY pretreatment did not alter the striatal MPP+ level, which excluded the possibility that GYY inhibited the MPTP metabolism via MAO although sulfides was reported to be MAO inhibitors (Warenycia et al., 1989; Stewart et al., 2014). These findings suggest the protective effects of GYY in this PD model. In support of this, two recent articles published in Movement Disorders proposed that the action of coffee consumption and smoking in lowering the risk of PD may be linked to H2S. Coffee intake was shown to improve the Prevotella population, which served as an important source of H2S generation and was decreased in the gut of PD patients (Cakmak, 2015, 2016; Scheperjans et al., 2015). But, it remains to delineate whether the role of H2S in PD relates to the gut microbiota.
Human α-syn contains four tyrosine residues, which can be nitrated by peroxynitrite when excessive NO is generated. 3-nitrotyrosine, as a footprint of tyrosine nitration in proteins, was detected in both PD patients and neurotoxins-induced animal models of PD (Schulz et al., 1995; Good et al., 1998). Moreover, several studies demonstrate that lewy bodies, the pathologic hallmark of PD and other synucleinopathies, largely contain α-syn that is modified by nitration on tyrosine residues (Giasson et al., 2000). So far, the pathophysiological function of nitrated α-syn remains elusive. Some studies report that α-syn nitration may facilitate protein aggregates formation and thus contribute to the pathogenic processes of PD and other synucleinopathies. Nitrated α-syn was suggested to serve as a clinical biomarker for PD diagnosis (Fernandez et al., 2013). In this study, we found that MPTP caused an increase of nitrated but not the total α-syn level in the striatum, and this increase was attenuated by GYY co-treatment. This also indicates the inhibition by GYY on nitrative stress induced by MPTP.
As a sibling of NO, H2S has been demonstrated to directly and indirectly interact with NO in biological systems. Moreover, H2S signaling, mediated by S-sulfhydration of its targeting proteins, often functions in an opposite manner to NO (Mustafa et al., 2009; Lu et al., 2013). In this study, GYY not only reduced the excessive generation of NO but also blocked the increase of nNOS expression induced by MPTP in the striatum. However, treatment with GYY did not have an obvious impact on NO or nNOS expression in control mice. This may be due to the relative lower level of NO under physiological conditions. Alternatively, the slower release of H2S from GYY which results in a consistent but lower level of H2S may also explain the lack of a direct effect of GYY on NO and nNOS expression, as previously reported (Li et al., 2008). Here, we demonstrated an indirect effect of GYY on nNOS expression, which may, at least in part, be mediated by Akt signaling, because GYY was found to enhance Akt activation. And in the presence of the Akt inhibitor perifosine, GYY-induced inhibition on nNOS expression was obviously abated. Therefore, activation of Akt signaling may participate in the suppression on nitrative stress and the neuroprotection by GYY on this mouse model. However, it should be cautious that a number of treatments have been shown to be efficacious in the animal models but finally failed in human clinical trials. Therefore, much more preclinical investigations need to be carried out to fully understand the role of H2S and its related strategies for treatment in PD.
In summary, our data demonstrated that GYY, a novel H2S slow-releasing compound, was protective in an in vivo mouse model of PD via its inhibition on nitrative stress. The findings may have implications for the research and development of H2S-based therapies for PD.
Ethics Statement
This study was carried out in accordance with the recommendations of the Laboratory Animal Care Guidelines of the Institutional Animal Care and Use Committee (IACUC) of Soochow University. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of Soochow University.
Author Contributions
XH: study design, data acquisition, data analysis and manuscript preparation. YY and YS: experiments design, sample collect and data analysis. BY, YW, and JZ: sample acquisition, data collection, statistical analysis. C-FL and XZ: study design and manuscript revision. L-FH determined the study theme, designed the experiments, ensured all the data and approved the final version to be published. All authors have contributed to this article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the National Key Plan for Scientific Research and Development of China (No. 2016YFC1306002), National Natural Science Foundation of China (Nos. 81171212, 81571233), and the Natural Science Foundation of Jiangsu Province of China (No. BK2011294). This was also partly supported by Suzhou Clinical Research Center of Neurological Disease (Szzx201503) and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Abbreviations
CSE, cystathionine-γ-lyase; GYY, GYY4137; HPLC, high-performance liquid chromatography; H2S, hydrogen sulfide; i.p., intraperitoneal; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; NO, nitric oxide; NOS, NO synthase; PD, Parkinson’s disease; SN, substantia nigra; α-syn, α-synuclein; TH, tyrosine hydroxylase.
References
Abe, K., and Kimura, H. (1996). The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 16, 1066–1071.
Cakmak, Y. O. (2015). Provotella-derived hydrogen sulfide, constipation, and neuroprotection in Parkinson’s disease. Mov. Disord. 30, 1151. doi: 10.1002/mds.26258
Cakmak, Y. O. (2016). Coffee consumption, smoking, and Parkinson’s disease? The beneficial role of hydrogen sulfide. Mov. Disord. 31, 429. doi: 10.1002/mds.26526
Dehmer, T., Lindenau, J., Haid, S., Dichgans, J., and Schulz, J. B. (2000). Deficiency of inducible nitric oxide synthase protects against MPTP toxicity in vivo. J. Neurochem. 74, 2213–2216. doi: 10.1046/j.1471-4159.2000.0742213.x
Eve, D. J., Nisbet, A. P., Kingsbury, A. E., Hewson, E. L., Daniel, S. E., Lees, A. J., et al. (1998). Basal ganglia neuronal nitric oxide synthase mRNA expression in Parkinson’s disease. Brain Res. Mol. Brain Res. 63, 62–71. doi: 10.1016/S0169-328X(98)00259-9
Fan, H., Guo, Y., Liang, X., Yuan, Y., Qi, X., Wang, M., et al. (2013). Hydrogen sulfide protects against amyloid beta-peptide induced neuronal injury via attenuating inflammatory responses in a rat model. J. Biomed. Res. 27, 296–304. doi: 10.7555/JBR.27.20120100
Fernandez, E., Garcia-Moreno, J. M., Martin De Pablos, A., and Chacon, J. (2013). May the evaluation of nitrosative stress through selective increase of 3-nitrotyrosine proteins other than nitroalbumin and dominant tyrosine-125/136 nitrosylation of serum alpha-synuclein serve for diagnosis of sporadic Parkinson’s disease? Antioxid. Redox Signal. 19, 912–918. doi: 10.1089/ars.2013.5250
Giasson, B. I., Duda, J. E., Murray, I. V., Chen, Q., Souza, J. M., Hurtig, H. I., et al. (2000). Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 290, 985–989. doi: 10.1126/science.290.5493.985
Good, P. F., Hsu, A., Werner, P., Perl, D. P., and Olanow, C. W. (1998). Protein nitration in Parkinson’s disease. J. Neuropathol. Exp. Neurol. 57, 338–342. doi: 10.1097/00005072-199804000-00006
Hu, L. F., Lu, M., Tiong, C. X., Dawe, G. S., Hu, G., and Bian, J. S. (2010). Neuroprotective effects of hydrogen sulfide on Parkinson’s disease rat models. Aging Cell 9, 135–146. doi: 10.1111/j.1474-9726.2009.00543.x
Jackson-Lewis, V., and Przedborski, S. (2007). Protocol for the MPTP mouse model of Parkinson’s disease. Nat. Protoc. 2, 141–151. doi: 10.1038/nprot.2006.342
Jimenez-Jimenez, F. J., Alonso-Navarro, H., Herrero, M. T., Garcia-Martin, E., and Agundez, J. A. (2016). An update on the role of nitric oxide in the neurodegenerative processes of Parkinson’s disease. Curr. Med. Chem. 23, 2666–2679. doi: 10.2174/0929867323666160812151356
Kida, K., Yamada, M., Tokuda, K., Marutani, E., Kakinohana, M., Kaneki, M., et al. (2011). Inhaled hydrogen sulfide prevents neurodegeneration and movement disorder in a mouse model of Parkinson’s disease. Antioxid. Redox Signal. 15, 343–352. doi: 10.1089/ars.2010.3671
Li, L., Whiteman, M., Guan, Y. Y., Neo, K. L., Cheng, Y., Lee, S. W., et al. (2008). Characterization of a novel, water-soluble hydrogen sulfide-releasing molecule (GYY4137): new insights into the biology of hydrogen sulfide. Circulation 117, 2351–2360. doi: 10.1161/CIRCULATIONAHA.107.753467
Liberatore, G. T., Jackson-Lewis, V., Vukosavic, S., Mandir, A. S., Vila, M., Mcauliffe, W. G., et al. (1999). Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat. Med. 5, 1403–1409. doi: 10.1038/70978
Lin, S., Visram, F., Liu, W., Haig, A., Jiang, J., Mok, A., et al. (2016). GYY4137, a slow-releasing hydrogen sulfide donor, ameliorates renal damage associated with chronic obstructive uropathy. J. Urol. 196, 1778–1787. doi: 10.1016/j.juro.2016.05.029
Liu, Y., Deng, Y., Liu, H., Yin, C., Li, X., and Gong, Q. (2016). Hydrogen sulfide ameliorates learning memory impairment in APP/PS1 transgenic mice: a novel mechanism mediated by the activation of Nrf2. Pharmacol. Biochem. Behav. 15, 207–216. doi: 10.1016/j.pbb.2016.11.002
Lu, C., Kavalier, A., Lukyanov, E., and Gross, S. S. (2013). S-sulfhydration/desulfhydration and S-nitrosylation/denitrosylation: a common paradigm for gasotransmitter signaling by H2S and NO. Methods 62, 177–181. doi: 10.1016/j.ymeth.2013.05.020
Lu, M., Zhao, F. F., Tang, J. J., Su, C. J., Fan, Y., Ding, J. H., et al. (2012). The neuroprotection of hydrogen sulfide against MPTP-induced dopaminergic neuron degeneration involves uncoupling protein 2 rather than ATP-sensitive potassium channels. Antioxid. Redox Signal. 17, 849–859. doi: 10.1089/ars.2011.4507
Mustafa, A. K., Gadalla, M. M., Sen, N., Kim, S., Mu, W., Gazi, S. K., et al. (2009). H2S signals through protein S-sulfhydration. Sci. Signal. 2:ra72. doi: 10.1126/scisignal.2000464
Paul, B. D., Sbodio, J. I., Xu, R., Vandiver, M. S., Cha, J. Y., Snowman, A. M., et al. (2014). Cystathionine gamma-lyase deficiency mediates neurodegeneration in Huntington’s disease. Nature 509, 96–100. doi: 10.1038/nature13136
Scheperjans, F., Aho, V., Pereira, P. A., Koskinen, K., Paulin, L., Pekkonen, E., et al. (2015). Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 30, 350–358. doi: 10.1002/mds.26069
Schildknecht, S., Gerding, H. R., Karreman, C., Drescher, M., Lashuel, H. A., Outeiro, T. F., et al. (2013). Oxidative and nitrative alpha-synuclein modifications and proteostatic stress: implications for disease mechanisms and interventions in synucleinopathies. J. Neurochem. 125, 491–511. doi: 10.1111/jnc.12226
Schulz, J. B., Matthews, R. T., Muqit, M. M., Browne, S. E., and Beal, M. F. (1995). Inhibition of neuronal nitric oxide synthase by 7-nitroindazole protects against MPTP-induced neurotoxicity in mice. J. Neurochem. 64, 936–939. doi: 10.1046/j.1471-4159.1995.64020936.x
Song, K., Wang, F., Li, Q., Shi, Y. B., Zheng, H. F., Peng, H., et al. (2014). Hydrogen sulfide inhibits the renal fibrosis of obstructive nephropathy. Kidney Int. 85, 1318–1329. doi: 10.1038/ki.2013.449
Stewart, J., Drexler, D. M., Leet, J. E., Mcnaney, C. A., and Herbst, J. J. (2014). Labware additives identified to be selective monoamine oxidase-B inhibitors. J. Biomol. Screen. 19, 1409–1414. doi: 10.1177/1087057114551523
Vandiver, M. S., Paul, B. D., Xu, R., Karuppagounder, S., Rao, F., Snowman, A. M., et al. (2013). Sulfhydration mediates neuroprotective actions of parkin. Nat. Commun. 4, 1626. doi: 10.1038/ncomms2623
Wang, Y., Zhao, X., Jin, H., Wei, H., Li, W., Bu, D., et al. (2009). Role of hydrogen sulfide in the development of atherosclerotic lesions in apolipoprotein E knockout mice. Arterioscler. Thromb. Vasc. Biol. 29, 173–179. doi: 10.1161/ATVBAHA.108.179333
Warenycia, M. W., Smith, K. A., Blashko, C. S., Kombian, S. B., and Reiffenstein, R. J. (1989). Monoamine oxidase inhibition as a sequel of hydrogen sulfide intoxication: increases in brain catecholamine and 5-hydroxytryptamine levels. Arch. Toxicol. 63, 131–136. doi: 10.1007/BF00316435
Weber, G., Pushpakumar, S., and Sen, U. (2017). Hydrogen sulfide alleviates hypertensive kidney dysfunction through an epigenetic mechanism. Am. J. Physiol. Heart Circ. Physiol. 312, H874–H885. doi: 10.1152/ajpheart.00637.2016
Keywords: GYY4137, hydrogen sulfide, Parkinson’s disease, nitric oxide, α-synuclein nitration
Citation: Hou X, Yuan Y, Sheng Y, Yuan B, Wang Y, Zheng J, Liu C-F, Zhang X and Hu L-F (2017) GYY4137, an H2S Slow-Releasing Donor, Prevents Nitrative Stress and α-Synuclein Nitration in an MPTP Mouse Model of Parkinson’s Disease. Front. Pharmacol. 8:741. doi: 10.3389/fphar.2017.00741
Received: 02 July 2017; Accepted: 02 October 2017;
Published: 30 October 2017.
Edited by:
Junbao Du, Peking University First Hospital, ChinaReviewed by:
Guangdong Yang, Laurentian University, CanadaPatrick Lewis, University of Reading, United Kingdom
Copyright © 2017 Hou, Yuan, Sheng, Yuan, Wang, Zheng, Liu, Zhang and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li-Fang Hu, aHVsaWZhbmdAc3VkYS5lZHUuY24= Xiaohu Zhang, bGl0aWdlcl85OUB5YWhvby5jb20=
†These authors have contributed equally to this work.