 Li-Long Pan
Li-Long Pan Ming Qin
Ming Qin Xin-Hua Liu
Xin-Hua Liu Yi-Zhun Zhu
Yi-Zhun Zhu- 1Department of Pharmacology, School of Pharmacy, Fudan University, Shanghai, China
- 2State Key Laboratory of Quality Research in Chinese Medicine and School of Pharmacy, Macau University of Science and Technology, Macau, China
Hydrogen sulfide (H2S), the third endogenous gaseous signaling molecule alongside nitric oxide (NO) and carbon monoxide, is synthesized by multiple enzymes in cardiovascular system. Similar to other gaseous mediators, H2S has demonstrated a variety of biological activities, including anti-oxidative, anti-apoptotic, pro-angiogenic, vasodilating capacities and endothelial NO synthase modulating activity, and regulates a wide range of pathophysiological processes in cardiovascular disorders. However, the underlying mechanisms by which H2S mediates cardiovascular homeostasis are not fully understood. This review focuses on the recent progress on functional and mechanistic aspects of H2S in the inflammatory and immunoregulatory processes of cardiovascular disorders, importantly myocardial ischemia, heart failure, and atherosclerosis. Moreover, we highlight the challenges for developing H2S-based therapy to modulate the pathological processes in cardiovascular diseases. A better understanding of the immunomodulatory and biochemical functions of H2S might provide new therapeutic strategies for these cardiovascular diseases.
Introduction
Cardiovascular diseases, the leading cause of death worldwide, are multifactorial resulting from disorders of the heart and circulation (Murphy et al., 2015), which cause immense health and economic burdens in all countries (Benjamin et al., 2017). The main risk factors associated with cardiovascular diseases are unhealthy lifestyle and lack of physical activity (American Heart Association Nutrition Committe et al., 2006). Accumulating evidence has demonstrated that the excess risk of cardiovascular outcomes is associated with changing endogenous H2S levels (Yang et al., 2008; Kondo et al., 2013; Mani et al., 2013; Wallace and Wang, 2015).
H2S, a colorless gas with characteristic rotten egg smell, is recognized an environmental hazard and a toxic agent for long. In addition, before the discovery that H2S is present in most organ systems in mammals including humans, it was believed that the gas was a byproduct of metabolic processes by microbes in the atmosphere. The pioneering study by the neuroscientist Hideo Kimura demonstrated that H2S at the physiological concentration facilitated hippocampal long-term potentiation in the nervous system, proposing that this gasotransmitter acts as a neuromodulator (Abe and Kimura, 1996). Later, H2S has become recognized widely as the third endogenous gaseous mediator alongside NO and CO for its modulatory effects on many signaling molecules, including kinases, phosphatases, and transcription factors (Abe and Kimura, 1996; Wang, 2002; Sun et al., 2011). Subsequently, it has been found to regulate both physiological and pathophysiological processes but at specific concentrations. Over the past decade, H2S has been found to be synthesized primarily through metabolic processes from cysteine and homocysteine in a variety of tissues where it functions as a signaling molecule (Szabo, 2007). H2S exerts its cellular effects by directly transport across cell membranes by simple diffusion without the need of specific membrane receptors and it is also involved in the modulation of many pathophysiological processes in cardiovascular system (Chunyu et al., 2003; Mathai et al., 2009; Wang et al., 2009; Liu et al., 2012; Pan et al., 2012). So far, a plethora of investigations have been performed on therapeutic values of H2S in cardiovascular diseases, which reveal that H2S at physiological levels has an important role in cardiovascular homeostasis, and inhibitors of endogenous H2S production or H2S donors exert significant effects in cardiovascular diseases, including heart failure, ischemic myocardium, atherosclerosis, and hypertension (Yang et al., 2008; Wang et al., 2009; Calvert et al., 2010; Miao et al., 2016a). Furthermore, empirical studies have elucidated several mechanisms of H2S-mediated cardiovascular protective activities, which include, but are not restricted to, anti-oxidation (Chang et al., 2008; Wang et al., 2009; Huang et al., 2017), anti-apoptosis (Yan et al., 2017), ion channel regulation (Pan et al., 2008; Ma et al., 2015), pro-angiogenesis (Cai et al., 2007), and anti-inflammation (Pan et al., 2011; Wallace and Wang, 2015). The realization of biological importance of H2S in numerous cells, tissues and organs is now shedding light on the pathogenesis of various cardiovascular diseases, and paving the way for innovative therapeutic interventions (Wallace and Wang, 2015; Zheng et al., 2017). Meanwhile, regulation of H2S functions during cardiovascular diseases remains to be better understood.
The Modulation of Endogenous H2S Biosynthesis in Cardiovascular System
Endogenous H2S is produced in mammalian tissues by primarily enzymatic or non-enzymatic pathways (Li et al., 2011; Martelli et al., 2012; Pan et al., 2012; Mani et al., 2014). Current understanding of H2S biology has arisen mostly from research work focused on enzymatic pathways (Liu et al., 2012; Wallace and Wang, 2015). The majority of endogenous H2S is synthesized by CBS (Szabo, 2007), CSE (Zhao et al., 2001), and 3-MST (Shibuya et al., 2009a,b). CBS and CSE may produce H2S from cysteine alone or from cysteine with homocysteine (Kimura, 2015). 3-MST produces endogenous H2S from one of the following substrates: 3-MP by CAT, thioredoxin, dihydrolipoic acid and D-cysteine along with DAO (Mikami et al., 2011; Yadav et al., 2013). All three H2S-synthesizing enzymes have been reported to be expressed by cardiovascular cells (Yang and Wang, 2015) (Figure 1). The distribution of H2S-producing enzymes in mammalian tissues is tissue-specific (Pan et al., 2012). CBS expression predominates in the brain, nervous system, liver, and kidneys, while CSE is a major H2S-synthesizing enzyme present in cardiovascular system under normal physiological conditions (Yang and Wang, 2015). 3-MST, along with CAT, accounts for H2S production in vascular endothelium in cardiovascular system (Shibuya et al., 2009a). In addition to their tissue-specific distribution, the intracellular localization of the three enzymes is different (Pan et al., 2012; Donnarumma et al., 2017). While CBS and CSE are cytosolic enzymes (Pan et al., 2012), 3-MST is present in both the mitochondrial and the cytosol with approximately two thirds of 3-MST found in the mitochondria (Li et al., 2009). Despite these findings, there is no definitive information regarding the relative contributions of each of the three enzymes on circulating and tissue H2S levels (Donnarumma et al., 2017). To maintain an appropriate physiological balance of H2S metabolism, endogenous H2S is inactivated in the biological systems by the enzymes ETHE1, SQR, and CDO, or by means of mitochondrial oxidation, cytosolic methylation, scavenging by glutathione disulfide or other metallo- or disulfide-containing molecules, as well as by release from the lungs (Mani et al., 2014; Donnarumma et al., 2017; Rose et al., 2017).
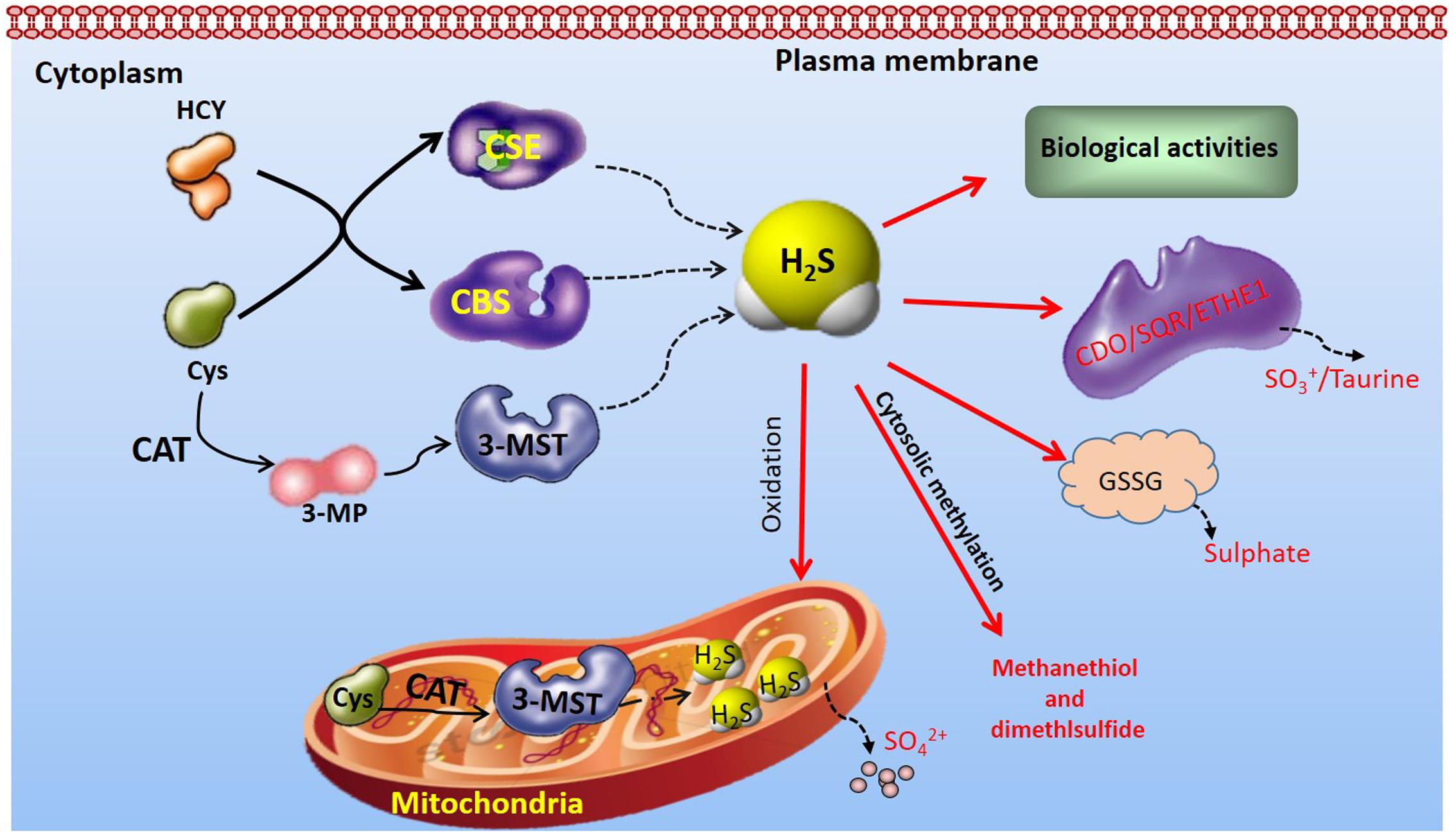
FIGURE 1. Overview of H2S-producting enzymes distribution and endogenous H2S metabolism CBS and CSE, distributed in the cytoplasm, produce H2S from Cys alone or from cysteine with HCY. In contrast, endogenous H2S production by 3-MST is primarily from 3-MP, which is produced from cysteine and α-ketoglutarate by CAT. 3-MST is distributed in both mitochondria and cytoplasm. The endogenous H2S is inactivated in the biological systems by the enzymes ETHE1, SQR, and CDO, or by means of mitochondrial oxidation, cytosolic methylation, scavenging by GSSG or other metallo- or disulfide-containing molecules. 3-MP, 3-mercaptopyruvate; 3-MST, 3-mercaptopyruvate sulfurtransferase; CAT, cysteine aminotransferase; CBS, Cystathionine β-synthase; CDO, cysteine dioxygenase; CSE, cystathionine γ-lyase; Cys, cysteine; HCY, homocysteine; ETHE1, ethylmalonic encephalopathy protein 1; GSSG, glutathione disulfide; SQR, sulfur:quinone oxidoreductase.
The critical roles of H2S at its physiologically relevant concentrations on the cardiovascular homeostasis have been well documented (Sun et al., 2011; Liu et al., 2012; Pan et al., 2012; Wang R. et al., 2015; Nagpure and Bian, 2016). The H2S levels in plasma and in organ tissues are regulated strictly by its generation and consumption under physiologic conditions (Liu et al., 2012). The H2S concentrations vary in different cells, tissues and organs and maintained within a certain range (Pan et al., 2012). Physiological levels of H2S range from 15 nM to 300 μM in vivo and the wide range of H2S levels may be due to variable detection methods used and the tissues analyzed (Liu et al., 2012; Hackfort and Mishra, 2016). Notably, significant changes of endogenous H2S levels (change of H2S-producing enzyme expression or its activity) have been clearly correlated to the pathogenesis of cardiovascular diseases, including heart failure, myocardial ischemia and atherosclerosis as indicated by both experimental and clinical evidence (Jiang et al., 2005; Feng et al., 2015; Gao et al., 2015; Wang W. et al., 2015; Yang and Wang, 2015; Kanagy et al., 2017; Li et al., 2017). Supplementation with exogenous H2S or modulation of endogenous H2S production markedly attenuates myocardial injury and improves cardiac function. For example, administration of SG1002 (a novel H2S prodrug, 400 mg) is reported to result in increased H2S levels and circulating NO bioavailability, and decreased circulating natriuretic peptide levels in patients with heart failure (Polhemus et al., 2015).
Immuneregulatory Effects of H2S in Cardiovascular Diseases
The physiological and biomedical importance of H2S has been recognized in the cardiovascular homeostasis. Accumulating evidence has demonstrated the beneficial effects of H2S-based therapies in cardiovascular disorders, including atherosclerosis, ischemic and heart diseases, which have been well addressed earlier by high-quality reviews (Szabo, 2007; Jin et al., 2010; Liu et al., 2012; Wang, 2012; Polhemus and Lefer, 2014). The roles of H2S in modulating inflammatory and immune processes during cardiovascular diseases have been emerging. H2S has been shown to regulate various immune cell functions, such as T-cell activation and proliferation, monocyte and polymorphonuclear cell apoptosis, leukocyte adhesion and infiltration, and inflammatory cytokine release by immune cells. Recent evidence has highlighted that H2S also actively regulates immuno-inflammatory processes in cardiovascular diseases. The current review focuses on immune-inflammatory modulation in H2S-mediated cardiovascular homeostasis in conditions including myocardial ischemia, heart failure, and atherosclerosis.
Myocardial Ischemia
Myocardial infarction is the leading cause of death worldwide with a yearly incidence of 1 million cases (Mozaffarian et al., 2015). During the deprivation of oxygen-carrying blood, combined with nutrient starvation, cardiomyocytes become insensitive to oxygen, leading to MI (Ghaderi et al., 2017). For those patients who undergo acute MI, the most effective therapeutic intervention is timely and effective myocardial reperfusion via revascularization combined with routine medical therapy (Hausenloy and Yellon, 2013). However, myocardial reperfusion can itself trigger cardiomyocyte death, an important complication of reperfusion therapy for MI, known as myocardial reperfusion injury (Boag et al., 2017). To date, no effective treatment has been identified. Among the pathological mechanisms underlying MI/R injury, inflammation and inflammatory cell infiltration, together with the activation of innate and adaptive immune responses, are the hallmarks of MI and reperfusion injury (Yellon and Hausenloy, 2007; Nahrendorf et al., 2010; Coggins and Rosenzweig, 2012; Frangogiannis, 2012; Swirski and Nahrendorf, 2013; Liu et al., 2016). Accumulating evidence suggests that modulation of excessive inflammation activation by negative regulation of toll-like receptor signaling and recruitment of inflammatory cells represents a promising therapeutic approach for MI and reperfusion injury (Yellon and Hausenloy, 2007; Coggins and Rosenzweig, 2012).
As the third endogenous gasotransmitter, H2S has emerged as an important mediator in maintaining cardiovascular homeostasis. Our group demonstrated for the first time that decreased plasma H2S levels were associated with increased infarct size and mortality. Administration of sodium hydrogen sulfide (NaHS, an exogenous H2S donor) decreased the infarct size of the left ventricle and MI-associated mortality in rats (Zhu et al., 2007). However, the changes of H2S levels during myocardial ischemia are still controversial. Ali et al. (2016) observed that serum H2S was significantly increased in ST-elevation acute MI patients. Li et al. (2016) have shown that plasma H2S levels decreased after acute MI surgery in rats. This observation is consistent with the previous study that either exogenous H2S administration or modulation of endogenous H2S production reduced myocardial ischemia-reperfusion injury in experimental models through restoration of H2S levels after ischemia (Elrod et al., 2007). The controversial results on serum H2S levels in ischemic myocardium cannot be readily explained by the different detection methods or the different species tested. However, the inconsistent results of serum H2S indeed merit further investigations.
In myocardium, enhanced H2S levels, whether by H2S supplementation or increased endogenous H2S production, have been found to protect the heart against ischemic injury. The exact cardioprotective mechanism of H2S has yet to be clarified but a number of molecular mechanisms have been identified, including vasodilation, anti-inflammation, antioxidation, anti-apoptosis, and modulation of cellular metabolism (Pan et al., 2012). MI and reperfusion injury trigger a complex immune-inflammatory responses in the injured myocardium, including inflammatory leukocyte infiltration and release of cytokines, such as IL-6, IL-8 and TNF-α. Recently, several studies, including our own, have demonstrated that H2S plays an important role in immune-inflammatory processes during MI and reperfusion injury (Elrod et al., 2007; Sodha et al., 2009; Miao et al., 2016a,b) (Figure 2). Neutrophils and leukocytes migrate into the infarcted myocardium during the first few hours after the onset of ischemia and peak after 1 day (Yellon and Hausenloy, 2007). Exogenous H2S administration or overexpression of the H2S-producing enzyme CSE significantly decreased leukocytes and neutrophil infiltration within the ischemic zone and markedly reduced myocardial inflammatory cytokine production during MI and reperfusion injury (Elrod et al., 2007). Additionally, exogenous H2S therapy was shown to attenuate cardiomyocyte apoptosis in the AAR of heart in a rat model of MI/R, whereas it decreased polymorphonuclear leukocyte accumulation and inflammatory mediators in the AAR from rat hearts subjected to regional MI/R (Sivarajah et al., 2009). Meanwhile, recruited monocytes/macrophages persist for days in the infarct area and contribute to inflammation, phagocytosis, proteolysis, angiogenesis, and collagen deposition (Nahrendorf et al., 2010). Modulated macrophage infiltration decreased inflammation, diminished interstitial fibrosis and improved cardiac remodeling and dysfunction (Nahrendorf et al., 2010). In a murine MI model subjected to pre- and post-coronary artery occlusion, exogenous H2S treatment reduced the recruitment of CD11b+ Gr-1+ myeloid cells to the myocardium, inhibited their migration from the splenic reservoir, and decreased serum TNF-α and IL-1β levels, thereby protecting against ischemic myocardial injury (Zhang et al., 2014). Recently, our study demonstrated that exogenous H2S treatment increased macrophage infiltration into the infarcted myocardium at the early stage of MI in both wild type and CSE-deficient mice (Miao et al., 2016b). In this study, exogenous H2S treatment promoted the migration of macrophages in vitro. Meanwhile, exogenous H2S treatment induced the activation of phosphor-Src, -Pyk2, -FAK397, and -FAK925. Moreover, exogenous H2S treatment induced internalization of integrin β1 on macrophage surface and promoted migration of macrophages and activation of Src signaling (Miao et al., 2016b). In our very recent study, we further demonstrated that exogenous H2S treatment ameliorated post-MI pathological cardiac remodeling and dysfunction in wild-type and CSE-deficient mice, decreased infarct size and mortality, and promoted M2 polarization of macrophages at the early stage of MI (Miao et al., 2016a). Notably, adoptive transfer of exogenous H2S-treated bone marrow-derived macrophages into wild-type and CSE-deficient mice with depleted macrophages also improved MI-induced cardiac dysfunction. A similar profile was also observed by Ji et al. (2017) that exogenous H2S treatment promoted microglia switch from a pro-inflammatory M1 phenotype to the modulatory M2 phenotype in ischemic stroke mice. Further mechanistic investigations demonstrated that exogenous H2S-induced M2 polarization of macrophages was achieved by enhanced mitochondrial biogenesis and fatty acid oxidation (Miao et al., 2016a).
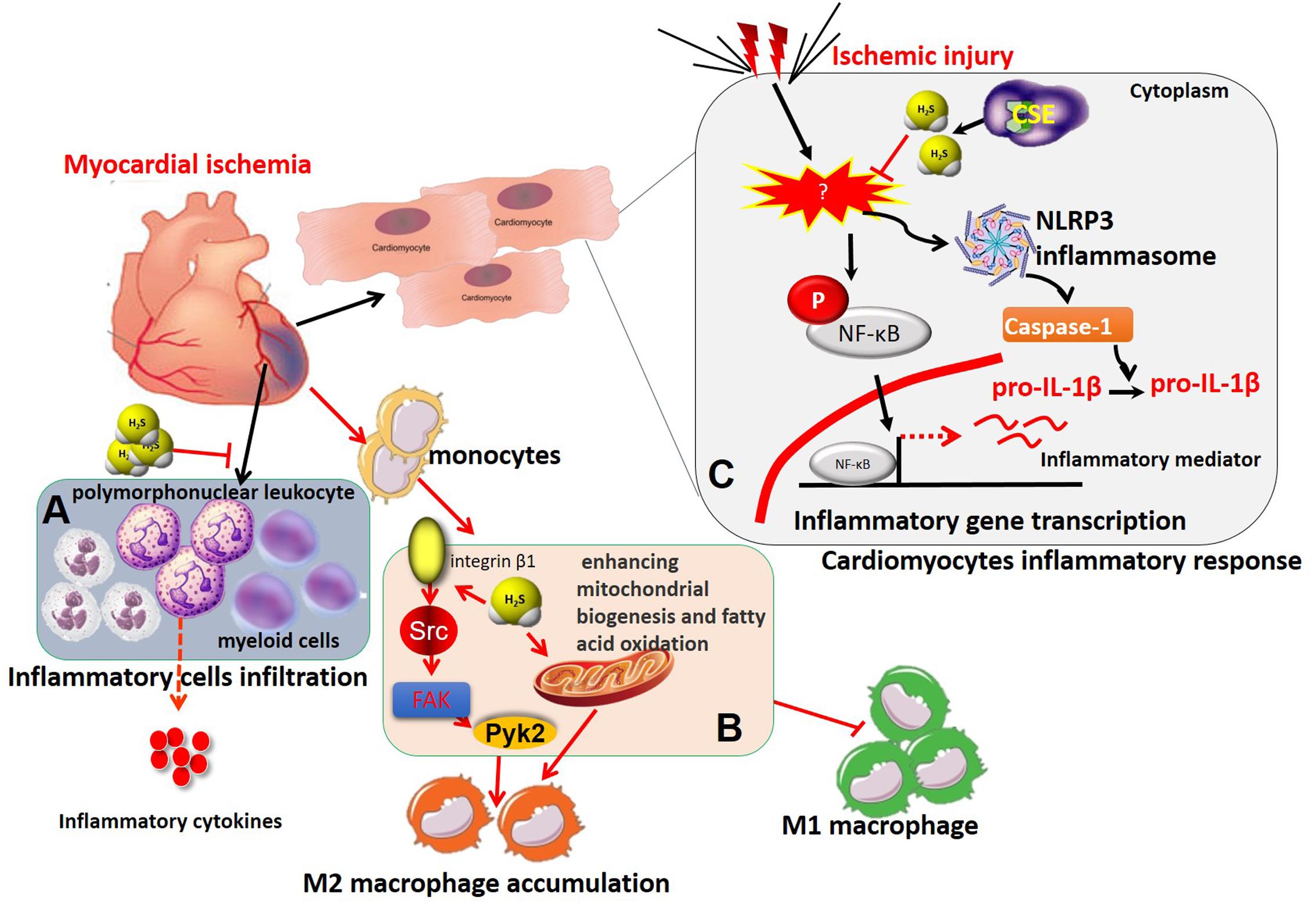
FIGURE 2. The immunomodulatory role of H2S in myocardial ischemia. (A) H2S treatment protected against ischemic myocardial injury by suppressing the recruitment of CD11b+ Gr-1+ myeloid cells and polymorphonuclear leukocytes to the ischemic myocardium and subsequent release of inflammatory cytokines, including TNF-α, IL-1β, and so on. (B) H2S recruited macrophages and induces M2 macrophage polarization in myocardial infarction by integrin β1-Src-FAK/Pyk2-Rac pathway and enhancing mitochondrial biogenesis and fatty acid oxidation. (C) H2S inhibited activation of NF-κB and NLRP3 inflammasome, subsequent inflammatory mediator expression and inflammatory responses in ischemia-stimulated cardiomyocytes. IL-1β, interleukin-1β; NLRP3, nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3; TNF-α, tumor necrosis factor-α.
In addition to regulatory effects of H2S on immune cell infiltration and phenotype switch, it also directly inhibits inflammatory responses in ischemic myocardium. For example, we found that S-propargyl-cysteine (SPRC, a novel endogenous H2S modulator) markedly attenuated LPS-induced TNF-α, ICAM-1, and iNOS expression in cardiomyocytes through modulation of CSE/H2S pathway by impairing inhibitory κBα (IκBα)/nuclear factor-κB (NF-κB inflammatory signaling and by activating PI3K/Akt signaling pathway (Pan et al., 2011). In addition, Toldo et al. (2014) revealed that Na2S (a H2S donor) administration during MI/R in vivo or in vitro prevented the activation of nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 (NLRP3) inflammasome and caspase-1, a macromolecular complex responsible for sensing tissue injury or ‘danger’ and amplifying the inflammatory responses. Moreover, the NLRP3 inflammasome-inhibiting effects of H2S were completely abolished with deletion of microRNA-21, demonstrating that H2S suppressed myocardial inflammatory responses by inhibition of NLRP3 inflammasome activation and was dependent on microRNA-21 (Toldo et al., 2014) (Figure 2).
Heart Failure
Heart failure is an inability of the heart to adequately meet the metabolic needs of the body, a clinical disease causing significant morbidity and mortality. Heart failure is the final outcome of conditions with varying etiologies. Atherosclerosis, risk factors, and comorbidities such as diabetes and obesity, many of which have an inflammatory component, typically precede MI injury (Odegaard and Chawla, 2013). The smoldering immune-inflammatory response impedes infarct healing by interfering with resolution of local inflammation and delaying the reparative phase (Swirski and Nahrendorf, 2013). Although the prognosis of patients with acute MI is largely determined by the extent of myocardial tissue loss, immune-inflammation also plays a critical role in the evolution of MI-induced cardiac remodeling and may tip the balance in favor of heart failure. Kondo et al. (2013) reported that the myocardial and circulating H2S levels were markedly reduced in experimental models of heart failure. In addition, they found that CSE-deficient mice exhibited greater cardiac dilatation and dysfunction compared to wild-type mice after transverse aortic constriction. In contrast, cardiac-specific CSE transgenic mice maintained cardiac structure and function after transverse aortic constriction. Recently, we also demonstrated that SPRC (a novel endogenous H2S modulator) therapy prevented doxorubicin-induced heart failure partially via regulation of gp130/STAT3 pathways. All these data suggest that both exogenous and endogenous H2S exhibit cardioprotective effects in heart failure.
In heart failure, accumulating experimental and clinical evidence points to a gradual state of immune-inflammatory activation accompanied by the progression of ventricular dysfunction with leukocyte activation and release of inflammatory mediators. For example, the inflammatory biomarker C-reactive protein, and inflammatory cytokines, such as TNF-α and IL-6, increase systemically in heart failure, and leukocytosis is associated with disease progression (Shirazi et al., 2017). Our previous study demonstrated that exogenous H2S administration markedly inhibited inflammatory cytokine expression in an in vivo model of heart failure associated with improving cardiac function and attenuating myocardial fibrosis (Pan et al., 2013). This potent beneficial pharmacological effects of H2S, at least partially, was associated with decreased Nox4/ROS/ERK1/2 signaling and increased HO-1 expression (Pan et al., 2013). In addition, H2S also inhibited chronic inflammatory responses and attenuated myocardial hypertrophy in experimental models of myocardial infarction and pressure overload induced via transverse aortic constriction (Nishida et al., 2012). Furthermore, exogenous H2S treatment also reduced recruitment of CD11b+Gr-1+ cells in infarct myocardium and peripheral blood and attenuated cardiac dilation in chronic ischemia-mediated infarcted myocardium in mice (Wu et al., 2017). These findings support the emerging view that H2S has potent immuno-inflammatory regulatory activities in ischemia-induced heart failure, resulted in reduced interstitial fibrosis, cardiac hypertrophy as well as improved overall survival (Figure 3).
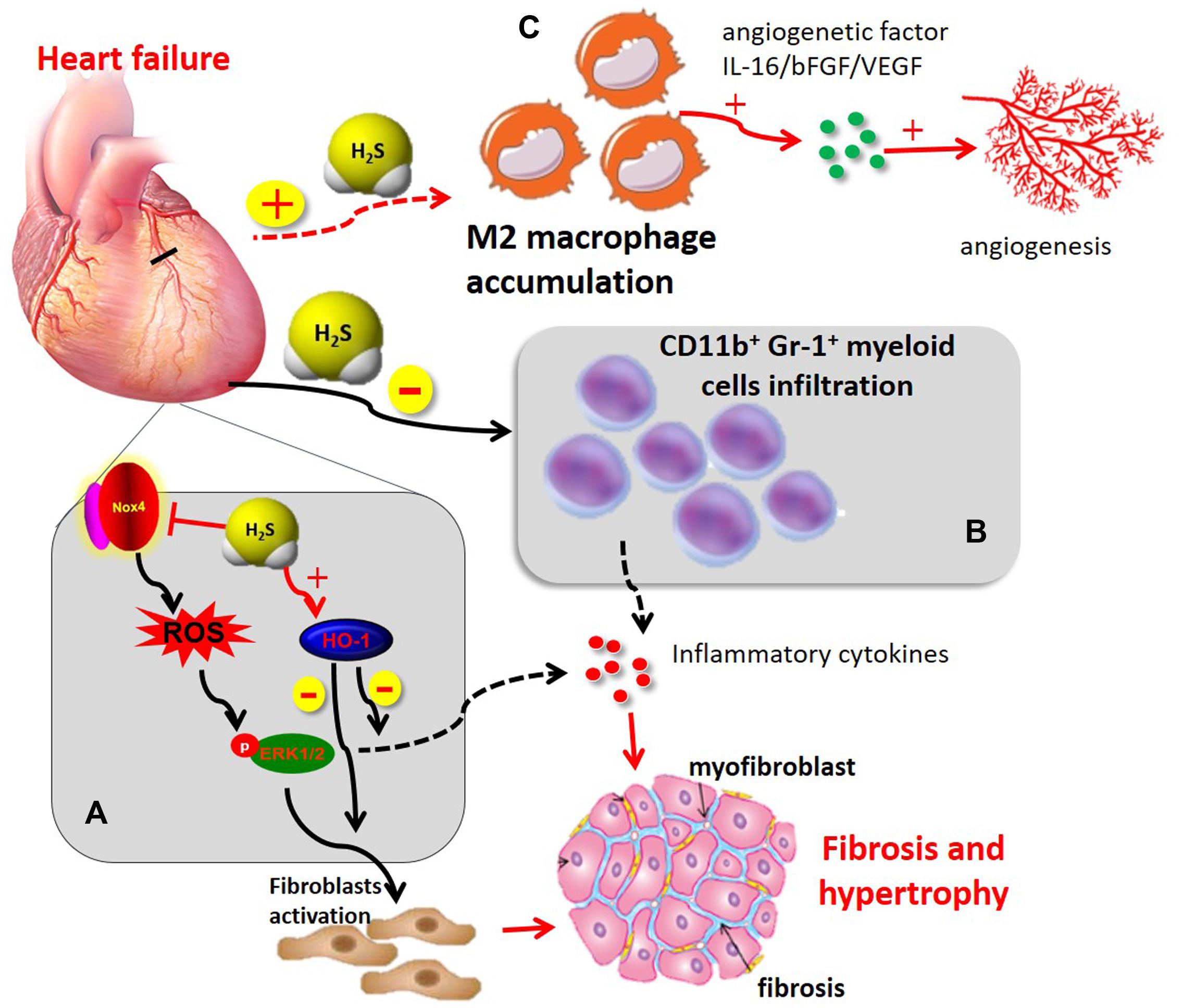
FIGURE 3. The immunomodulatory role of H2S in heart failure (A) H2S administration induced cardioprotection in chronic MI by improving cardiac function, attenuating myocardial fibrosis, and inhibiting chronic inflammatory mediators. This beneficial effect of H2S, at least in part, was associated with a decrease of Nox4/ROS/ERK1/2 signaling axis and an increase in HO-1 expression (B) H2S attenuated cardiac dilation through inhibiting recruitment of CD11b+Gr-1+ cells in infarct myocardium and inflammatory mediator release. (C) H2S induced M2 macrophage polarization and recruitment in myocardial infarction, thereby contributing to angiogenetic factor release and subsequent angiogenesis. ERK1/2, extracellular signal-regulated kinase 1/2; HO-1, heme oxygenase-1; Nox4, NADPH oxidase 4; ROS, reactive oxygen species.
Angiogenesis is a complex biological process that leads to increased blood flow and promotes cardiac repair and myocardium survival during heart failure. Therefore, promoting myocardial angiogenesis is a novel therapeutic strategy for the treatment of heart failure (Bao et al., 2013; Wang and Cai, 2016). In recent years, the gasotransmitter H2S has become apparent that it is capable of mediating angiogenesis and improving cardiac function after heart failure (Givvimani et al., 2011; Polhemus et al., 2013). In a mouse model of transverse aortic constriction-induced heart failure, chronic H2S treatment with diallyl trisulfide improved left ventricular remodeling and function by inducing angiogenesis via upregulation of VEGF and endothelial NO synthase. Additionally, the study also indicated that H2S upregulated the endogenous antioxidants, GPx1 and HO-1 (Polhemus et al., 2013). These results were further confirmed by our previous study that chronic H2S therapy with SPRC induced angiogenesis by a mechanism involving STAT-3 interacting with VEGF receptor 2 in a rat model of chronic myocardial ischemia (Kan et al., 2014). Recruitment of monocytes/macrophages to the site of injury not only promote inflammatory responses and pathological tissue remodeling but is also required for the resolution of inflammation and regenerative activities, such as angiogenesis (Lavine et al., 2014; Howangyin et al., 2016). One proposed explanation for these observations is that distinct macrophage subpopulations may mediate inflammatory (M1) and reparative (M2) macrophage activities (Lavine et al., 2014; Kolluru et al., 2015; Miao et al., 2016a). H2S modulates monocytes/macrophages phenotypes, which produces beneficial effects in angiogenesis in chronic ischemic diseases. Although there is no direct evidence that H2S modulates macrophage phenotype in heart failure, Kolluru et al. (2015) using femoral artery ligation model has demonstrated that CSE dysregulation or deficiency as well as endogenous H2S production have a significant effect on monocytes/macrophage recruitment and subsequent expression of angiogenetic factors (bFGF and VEGF) under ischemic conditions. Yet exogenously administering H2S or modulation of endogenous H2S production promoted monocyte/macrophage recruitment and angiogenetic factor expression, leading to angiogenesis and restored blood flow (Kolluru et al., 2015). However, the contribution of M1 vs. M2 macrophages in H2S-mediated angiogenetic responses in heart failure has yet to be clarified in future investigation. In addition, CD4+T lymphocyte deficiency delays the transition from M1 to M2 macrophages and impairs healing of the heart (Hofmann et al., 2012). Likewise, depletion of dendritic cells disturbs resolution of inflammation (Hofmann et al., 2012; Swirski and Nahrendorf, 2013). There is still lack of direct evidence for an immunoregulatory role of H2S on immune cell phenotypes in heart failure. Given that leukocytes play a key role in heart failure, the immunoregulatory function of H2S on different immune cell subsets merits further investigations.
Taken together, preclinical evidence suggests that H2S significantly improve cardiac function in the setting of heart failure via immunoregulatory activities, including modulating immune cell phenotypes, suppressing inflammatory responses and inflammatory cell infiltration, which represents a therapeutic strategy for heart failure (Figure 3).
Atherosclerosis
Atherosclerosis, a vascular disease at the susceptible sites in medium and large-sized arteries, is the pathological basis of coronary heart disease and the major cause of death in developed countries. The development of atherosclerosis is a complex multifactorial process that involves vascular inflammation, VSMC proliferation and migration, thrombus formation, as well as abnormal immune responses including monocyte infiltration and differentiation, and lesion-resident macrophage conversion into foam cells (Pan et al., 2012). Through the factors that initiate plaque formation and are currently being debated, it is no longer news that atherosclerosis is more than a mere cholesterol storage disease. Immune inflammation in the pathogenesis of atherosclerosis has now gained widespread recognition (Libby and Hansson, 2015).
Recent studies have suggested that dysfunctional CSE and reduced endogenous H2S levels are linked to the pathogenesis of atherosclerosis (Wang et al., 2009; Mani et al., 2014). Exogenous H2S treatment protects rat aortic VSMC from hyperhomocysteine- or ROS-induced cytotoxicity, which is considered independent of atherogenic risk factors (Yan et al., 2006). In a genetic model of hyperhomocysteinemia, CBS-/-ApoE-/- mice exhibited accelerated aortic atherosclerosis compared with ApoE-/- mice after 6 months of age in the absence of dietary manipulation (Wang et al., 2003). Since only 2% of CBS-/-ApoE-/- mice survived up to 6 months of age, the pathophysiological relevance of CBS-/- to H2S metabolism in atherosclerosis is not clear (Wang et al., 2003). At the same time, CSE expression and H2S production were reduced during neointimal hyperplasia in carotid artery in rats, and that exogenous H2S treatment markedly reduced neointimal formation (Meng et al., 2007). Furthermore, Wang et al. (2009) reported that plasma H2S level and H2S production in atherosclerotic aortic tissues were decreased in ApoE-/- atherosclerotic mice. Exogenous H2S treatment resulted in elevated plasma H2S level and reduced the atherosclerotic plaque size in the aortic root of ApoE-/- mice, whereas DL-propargylglycine (PPG, a potent CSE inhibitor) reduced plasma H2S level and enlarged plaque size in the aorta (Wang et al., 2009). Furthermore, atherogenic diet feeding to ApoE-/- CSE-/- mice exacerbated the development of atherosclerosis compared to mice with only ApoE or CSE deficiency. Treatment of CSE-/- mice with exogenous H2S inhibited the progression of atherosclerosis (Mani et al., 2013), which provided clear evidence that supports a protective role of H2S against atherosclerosis.
Initially, exploration of the immune-inflammatory aspects of atherogenesis focused on the intima, the site where atheromata take root. As probing has deepened, researchers have come to recognize that influence arising from all three layers of arteries can affect the pathophysiology of this disease (Libby and Hansson, 2015; Gistera and Hansson, 2017). Indeed, immune-inflammatory responses participate in atherosclerosis by modifying the arterial tree at various levels (Libby and Hansson, 2015). Accumulating evidence has indicated that H2S is involved in the immune-inflammatory processes in atherosclerosis in a number of preclinical models of atherosclerosis (Figure 4). The vascular endothelial dysfunction, characterized by the loss or dysregulation of the homeostasis, is considered an important early event in the development of atherosclerosis (Mani et al., 2014). Endothelial dysfunction is associated with increased oxidative stress, adhesion molecules expression, synthesis of inflammatory and pro-thrombotic factors, and abnormal modulation of vascular tone (Mani et al., 2014). Wang et al. (2009) demonstrated that ICAM-1 levels were significantly increased, accompanied by increased size of the atherosclerotic plaque in ApoE-/- atherosclerotic mice. The ICAM-1 levels and atherosclerotic plaque were reduced in aortas of ApoE-/- mice following treatment with exogenous H2S. The inhibitory mechanism of H2S on ICAM-1 expression was addressed in endothelial cells in vitro, where exogenous H2S was shown to inhibit NF-κB activation (Wang et al., 2009). We also studied the protective effects of exogenous H2S on TNF-α-induced dysfunction in human umbilical vein endothelial cells in vitro (Pan et al., 2012). Mechanically, exogenous H2S inhibited TNF-α-induced ICAM-1 and VCAM-1 protein expression, P-selectin and E-selectin mRNA expression, as well as monocyte adhesion to endothelial cells (Pan et al., 2012). Similarly, Feng et al. (2017) found that CSE/H2S pathway was significantly downregulated in the development of pulmonary vascular endothelial inflammation. H2S treatment could reduce pulmonary vascular pressure, relieve pulmonary vascular remodeling, inhibit pulmonary vascular endothelial cellular inflammation, and attenuate the NF-κB signaling pathway in pulmonary arterial endothelial cells. In contrast, Zanardo et al. (2006) demonstrated that suppression of endogenous H2S production, by CSE blockade using β-cyano-L-alanine, led to enhanced leukocyte adhesion, leukocyte infiltration, and edema formation while H2S donors produced opposite effects.
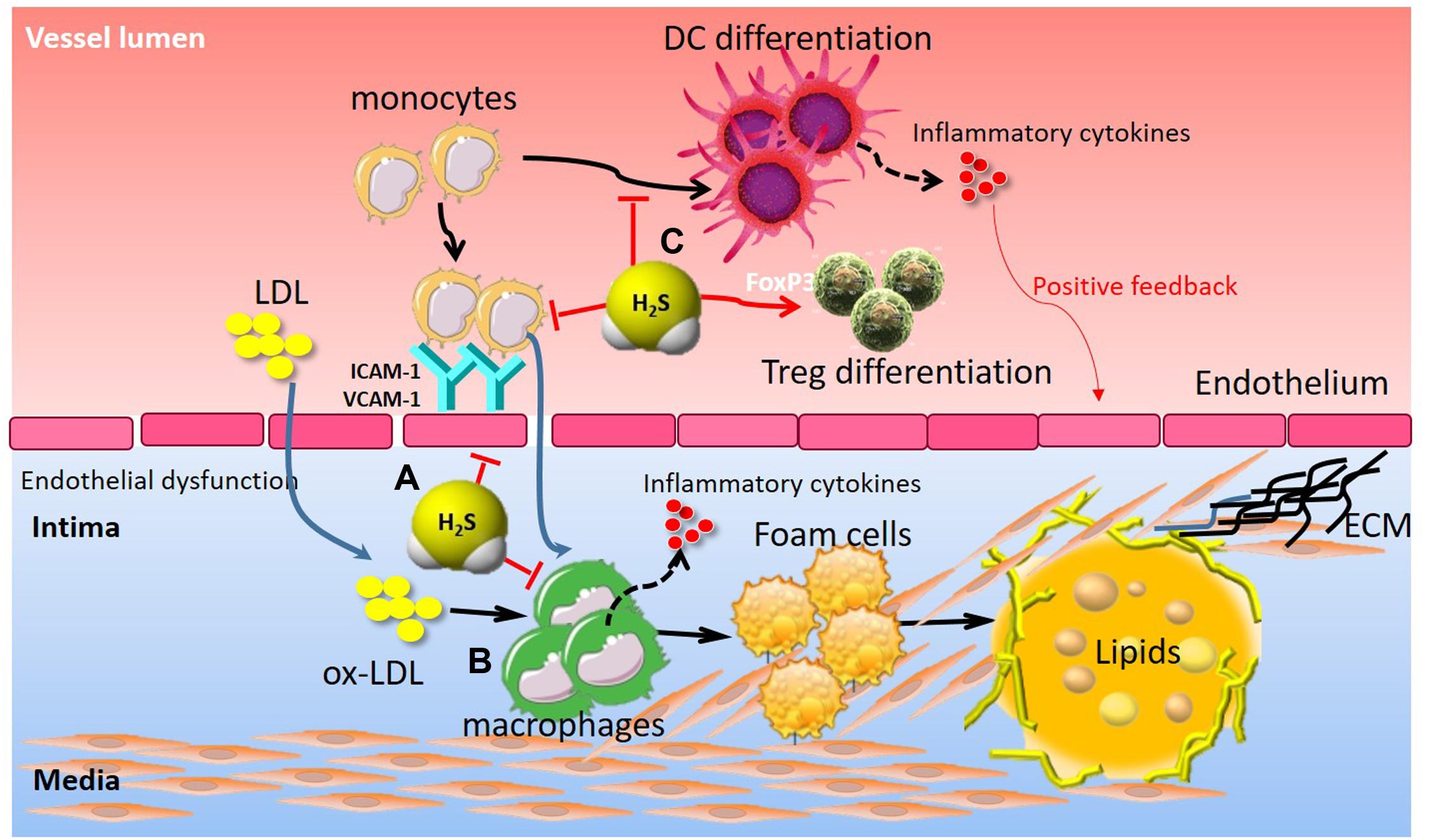
FIGURE 4. The immunomodulatory role of H2S in atherosclerosis. (A) H2S attenuated endothelial dysfunction, adhesion molecules (ICAM-1, VCAM-1) expression, and leukocyte adhesion. (B) H2S inhibited monocyte activation and foam cell formation, thereby contributing to inflammatory cytokine release, VSMC proliferation and subsequent atherosclerotic plaque formation. (C) H2S suppressed monocyte differentiation into inflammatory dendritic cells and induced CD4+Foxp3+ Treg cell differentiation, thereby contributing to vascular immune homeostasis. Treg, regulatory T cell; ICAM-1, intercellular adhesion molecule-1; VCAM-1, vascular cell adhesion molecule-1.
Macrophage is thought to play an important role in atherosclerosis by generating lipid-laden foam cells and by secreting inflammatory mediators (Moore and Tabas, 2011). Macrophage uptake of oxidized-low density lipoproteins (oxLDL) contributes to formation of lipid-laden “foam cells,” the primary component of atherosclerotic lesions (Moore and Tabas, 2011). However, H2S plays an inhibitory role in macrophage-derived foam cell formation. In vitro, Wang et al. (2013) demonstrated that oxLDL may down-regulate the CSE/H2S pathway, which exerts an anti-inflammatory effect on oxLDL-stimulated macrophage by suppressing JNK/NF-κB signaling. Treatment with exogenous H2S or upregulation of endogenous H2S by CSE overexpression markedly attenuated oxLDL-mediated inflammatory responses and JNK/NF-κB signaling activation (Wang et al., 2013). Du et al. (2014) further elucidated that the sulfhydration of free thiol group on cysteine 38 in NF-κB p65 served as a molecular mechanism by which H2S inhibited NF-κB activation in oxLDL-induced macrophage inflammation. In addition, H2S also abrogated oxLDL-induced macrophage foam cell formation. Mechanistically, H2S inhibited oxLDL-induced intracellular lipid accumulation, reduced TC, EC, and EC/TC ratio in macrophages by down-regulating expressions of CD36, scavenger receptor A and acyl-coenzyme A: cholesterol acyltransferase-1 (Zhao et al., 2011). In agreement with these findings, H2S treatment attenuated high glucose + oxLDL-induced foam cell formation (Xie et al., 2016). The protective effect of H2S can be, at least in part, attributed to Nrf2 activation via Keap1 S-sulfhydration at Cys151. Furthermore, H2S significantly inhibited macrophage accumulation and reduced the aortic atherosclerotic lesion in ApoE-/- mice. However, PPG demonstrated the opposite effect: enlarging the lesion area and macrophage accumulation in the lesions of ApoE-/- mice (Lin et al., 2016). H2S supplementation also reduced lesion zone and macrophage infiltration in diabetic LDLr-/- mice. The expression of adhesion molecules and macrophages/monocyte recruitment in sub-endothelial space are critical for the initiation and development of atherosclerotic lesions. The preferential accumulation of Ly-6Chigh monocytes in the growing atheromata relied on the C-C chemokine receptor type 2 (CCR2)-monocyte chemotactic protein-1 (CCL2), CX3CR1-CX3CL1, and CCR5-CCL5 (Gao et al., 2015), and neutralizing these axes in mice almost abolished atherosclerosis via reduced macrophage infiltration and increased plaque stability (Moore and Tabas, 2011). In vivo, Zhang et al. (2012) observed that H2S treatment downregulated CX3CR1 and CX3CL1 expression on macrophages through modulation of the transcription factors PPAR-γ and NF-κB. They also demonstrated that, by interfering with the CX3CL1/CX3CR1 dyad, supplementation of mice with the exogenous H2S reduced the development of atherosclerotic plaques (Zhang et al., 2012). They further found in a clinical study that plasma H2S level was markedly reduced, whereas plasma CCL2 and CX3CL1 levels were substantially increased in patients with ACS compared to patients with SAP or non-CAD patients (Gao et al., 2015). Furthermore, patients with ACS exhibited significantly higher proportions of CD14+CCR2+CX3CR1+ (intermediate monocytes, Mon2) and CD14+CCR2-CX3CR1+ monocytes (non-classical monocytes, Mon3) but a lower percentage of CD14+CCR2+CX3CR1- monocytes (classical monocytes, Mon1) than patients with SAP or non-CAD did Gao et al. (2015). Lastly, they identified that plasma H2S level was negatively correlated with the proportion of Mon2 monocyte subsets, suggesting that impaired endogenous H2S synthesis in ACS may facilitate monocyte subset conversion from Mon1 to Mon2 or Mon3, leading to atherosclerotic plaque instability, and the development of ACS (Gao et al., 2015). However, the precise mechanism by which H2S regulates monocyte phenotypes in CAD remains to be better understood. Similarly, in methionine/choline-deficient diet-induced experimental steatohepatitis in mice, H2S treatment significantly prevented CX3CR1+CD11b+/F4-80+ cell accumulation and decreased circulating and hepatic TNF-α levels (Sutti et al., 2015). These CX3CR1+ cells were further characterized by the co-expression of inflammatory monocyte (Ly6C, CD11b) and dendritic cell (CD11c, MHCII) markers as well as by a sustained TNF-α production, suggesting that H2S could prevent monocyte differentiation into inflammatory monocyte-derived inflammatory dendritic cells and limit their M1 polarization (Sutti et al., 2015).
T-helper (Th) cells play a critical role in mediating adaptive immunity. During TCR activation in a particular cytokine milieu, naive CD4+T cells may differentiate into different lineages of Th cells, including Th1, Th2, Th17, and Treg cells. Accumulating evidence has shown that peripheral activation of Treg and subsequent recruitment to atherosclerotic plaque limit the lesion progression in experimental models by down-regulating inflammatory responses which include multiple mechanisms (Libby et al., 2013; Delgado-Maroto et al., 2017). However, minor populations of Foxp3+ Treg cells were found in human atherosclerotic plaques at all stages of the disease (Gistera and Hansson, 2017). Transfer of Foxp3+ Treg cells decreased atherosclerosis in hypercholesterolaemic mice (Gistera and Hansson, 2017). More recently, Yang et al. (2015) found that reduced H2S levels were responsible for impaired CD4+Foxp3+ Treg cell differentiation and function as well as immune dysfunction in mice. Treatment of H2S donor rescued Treg-cell-deficient phenotypes of immune dysfunction in CBS-/- mice and WT Treg cell infusion could partially rescue autoimmunity in CBS-/- mice (Yang et al., 2015). The immune regulatory mechanisms by H2S are that H2S affected sulfhydration of nuclear transcription factor Y subunit beta (NFYB) to control NFYB complex binding to the Tet1 and Tet2 promoters, forming a H2S-NFYB-Tet axis to regulate Treg differentiation and immune homeostasis (Yang et al., 2015). Furthermore, H2S can enhance TCR-dependent T cell activation and IL-2 expression. H2S also enhances T cell proliferation and lineage determination via altering cytoskeletal actin dynamics and increasing the reorientation of the microtubule-organizing center (Miller et al., 2012), suggesting that H2S represents a novel immunomodulatory molecule for T cell responses. Therefore, it may be a novel therapeutic approach for chronic immune-inflammatory responses in atherosclerosis via targeting H2S metabolism.
Challenges for H2S Research and Future Perspectives
Since the first demonstration of the expression of H2S-producing enzymes in the mammalian system, there have been numerous experimental studies conducted on the role of H2S modulation, by ways of overexpression/inhibition of H2S-synthesizing enzymes or H2S donor, on cardiovascular homeostasis (Salloum, 2015). Mechanisms underlying H2S signaling have been uncovered; however, a lot of unknowns on how H2S influences cardiovascular homeostasis remain to be further investigated. Immune-inflammatory responses play a decisive role in different phases of cardiovascular diseases (Gistera and Hansson, 2017; Jones et al., 2017). Data from basic studies support immunoregulatory functions of H2S and therefore the potential of H2S to modulate the immune-inflammatory response to prevent cardiovascular disorders, including ischemic heart disease, atherosclerosis, heart failure, and so on. In other cardiovascular diseases such as hypertension, H2S has been demonstrated to play an important role (Yang et al., 2008). As arterial inflammation and immune dysregulation are involved in the pathogenesis of the disease (Smith and Ferguson, 2016) and H2S has been shown to maintain immune homeostasis, it could be postulated that H2S may play a positive role in such condition. So far literature has been limited to provide further evidence and hence is not covered in the current review, which merit future investigation and verification.
Both pro- and anti-inflammatory effects of H2S have been reported. In numerous studies including our studies, H2S has been characterized for its anti-inflammatory role (Cao and Bian, 2016; Zhou et al., 2016; Feng et al., 2017). In contrast, recent work from different groups has shown a key role of H2S as an inflammatory mediator (Li et al., 2011; Bhatia, 2015). These contradictory observations may result from different experimental settings and approaches, such as cell culture and/or in vivo disease models. However, a number of elegant studies suggest that H2S is a potent anti-inflammatory molecule, specifically in the cardiovascular diseases (Polhemus and Lefer, 2014; Miao et al., 2016a). Although the data are debatable, they suggest that H2S may be a double-edged sword and controversies are warranted to encourage future studies to better understand the biological significance of this gaseous molecule in cardiovascular homeostasis. We believe that resolving these issues would drastically advance H2S research.
H2S has a number of biological effects on cardiovascular systems. However, the molecular targets of H2S remains to be fully uncovered. KATP channels in many cellular systems are accountable for the effects of H2S. In other cases, H2S seems not to act on the same channels. Until now, the potential molecular targets for H2S are likely to include intracellular proteins or enzymes (such as p66Shc, phospholamban, protein tyrosine phosphatase 1B, mitogen-activated extracellular signal-regulated kinase 1, ATP synthase subunit α, etc.), and transcription factors (such as NF-κB, kelch-like ECH-associating protein 1, specific protein-1 and interferon regulatory factor-1, etc.) as well as membrane receptors (vascular endothelial growth factor receptor 2, insulin receptor, and epidermal growth factor receptor) in cardiovascular system (Li et al., 2011; Ge et al., 2014; Wang et al., 2016). Underlying these functions are the atomic biology, interaction between sulfur atoms and target molecules. In this regard, Tao et al. found a molecular switch in H2S-targeting receptor, the cysteine1024 (Cys1024)-S-S-Cys1045 disulfhide bond, in the intracellular kinase domain of vascular endothelial growth factor receptor 2 (Tao et al., 2013), which has prompted the field of H2S biology to a new landmark. Although remarkable progress has been made in delineating the role of the potential targets by H2S in cardiovascular homeostasis, one challenging question that remains in this field is the identification of more precise protein targets that mediate numerous physiological functions. These proteins propose novel targets for therapeutic intervention and drug design in cardiovascular homeostasis, which may accelerate the development and application of H2S related drugs in the future.
Modulation of endogenous H2S levels as a novel potential therapeutic strategy for cardioprotection in patients undergoing cardiovascular disorders and the changed H2S-producing enzymes expression/activities have been directly related to the endogenous H2S generation. Meanwhile, the three H2S-generating enzymes have been broadly localized in cardiovascular system (Pan et al., 2012). Downregulation of the three H2S-producing enzymes is associated with chronic cardiovascular pathologies (Kanagy et al., 2017; Merz et al., 2017). Unfortunately, the H2S-producing enzymes responsible for H2S production and biological function in cardiovascular homeostasis are not clear and consistent in current literature (Kuo et al., 2016; Li et al., 2016). Meanwhile, the roles of H2S degradative enzymes ETHE1, SQR and CDO in cardiovascular immune homeostasis remain largely unexplored and merit further investigation (Rose et al., 2017).
Over the last decade, considerable evidence has been collected, which points to a functional role for H2S in cardiovascular homeostasis, representing a novel promising therapeutic strategy for cardiovascular diseases. However, the majority of the cardiovascular studies involving H2S have been investigated and established in healthy, juvenile, and small animals, making them far removed from the clinical setting of the typical cardiovascular diseases. Meanwhile, improved understanding on the protective actions of H2S, together with rapid development of novel H2S donors (Polhemus and Lefer, 2014; Hackfort and Mishra, 2016; Zheng et al., 2017), has raised heightened enthusiasm for the translational studies. Currently, there are three cardiovascular H2S trials on clinicaltrials.gov. Therefore, it will be expedient to move one step forward to confirm rigorously the therapeutic effects of H2S in larger animal models before making a complete transition to the clinic.
As with the development of small molecule H2S donors, there are organ-specific issues that need to be considered. Given its ubiquitous nature, it is not surprising that H2S has important functions in a wide range of physiological and pathophysiological processes. H2S delivery will produce a wide range of biological activities, including unwanted side effects. Therefore, the speed and amount of H2S release from different donors should be controllable. Otherwise, a novel H2S donor that could specifically target an organ system would alleviate undesirable effects. Because the cardiovascular system is a circulatory system, the cardiovascular-specific delivery of H2S maintained cardiovascular homeostasis is one of the key challenges being explored in the field.
Conclusion
H2S is a ubiquitous gasotransmitter and plays a critical role in immune homeostasis in cardiovascular disorders. Significant changes of endogenous H2S levels (change of H2S-producing enzyme expression or its activity) have been clearly correlated to immune-inflammatory responses in a variety of cardiovascular diseases. We have summarized the latest knowledge on the immune-inflammatory modulatory functions of H2S in cardiovascular diseases and discussed the possible cellular and molecular mechanisms by which it exerts cardiovascular protective actions as well as its therapeutic potential for cardiovascular diseases. Although the molecular targets of H2S remain to be fully elucidated, considerable evidence has demonstrated that H2S is a novel immune-modulator in cardiovascular homeostasis. Insights into the molecular targets of H2S in immune-inflammatory processes may help better understanding of the pathophysiology of these diseases.
Author Contributions
X-HL and Y-ZZ designed the subject content of the review article. L-LP, MQ, and X-HL conducted initial search of literature, drafted the manuscript, and prepared the figures and tables. X-HL and Y-ZZ had primary responsibility for final content. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from National Natural Science Foundation of China (Nos. 81573420, 81673428, 81330080), “Zhuo Xue” Talent Plan of Fudan University, Faculty Research Grant of MUST (FRG-17-006-SP) and Macau FDCT grants (055/2016/A2 and 039/2016/A).
Abbreviations
3-MP, 3-mercaptopyruvate; 3-MST, 3-mercaptopyruvate sulfurtransferase; AAR, area at risk; ACS, acute coronary syndrome; CAD, coronary artery disease; CAT, cysteine aminotransferase; CBS, cystathionine-β-synthase; CDO, cysteine dioxygenase; CO, carbon monoxide; CSE, cystathionine-γ-lyase; DAO, D-amino acid oxidase; EC, esterified cholesterol; ETHE1, ethylmalonic encephalopathy protein 1; ERK1/2, extracellular signal-regulated kinase 1/2; Foxp3, forkhead box protein P3; H2S, hydrogen sulfide; HO-1, heme oxygenase-1; ICAM-1, intercellular adhesion molecule-1; iNOS, inducible nitric oxide synthase; IL, interleukin; MI, myocardial infarction; MI/R, myocardial ischemia/reperfusion; NF-κB, nuclear factor-κB; NLRP3, nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3; NO, nitric oxide; Nox4, NADPH oxidase 4; oxLDL, oxidized LDL; PPG, DL-propargylglycine; PPAR-γ, proliferators-activated receptor-γ; ROS, reactive oxygen species; SAP, stable angina pectoris; SQR, sulfur:quinone oxidoreductase; SPRC, S-propargyl-cysteine; STAT3, signal transducer and activator of transcription 3; TC, total cholesterol; TCR, T-cell receptor; TNF-α, tumor necrosis factor-α; Tregs, regulatory T cells; VCAM-1, vascular cell adhesion molecule-1; VEGF, vascular endothelial growth factor; VSMC, vascular smooth muscle cell.
References
Abe, K., and Kimura, H. (1996). The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 16, 1066–1071.
Ali, S. E., Farag, M. A., Holvoet, P., Hanafi, R. S., and Gad, M. Z. (2016). A comparative metabolomics approach reveals early biomarkers for metabolic response to acute myocardial infarction. Sci. Rep. 6:36359. doi: 10.1038/srep36359
American Heart Association Nutrition Committe, Lichtenstein, A. H., Appel, L. J., Brands, M., Carnethon, M., Daniels, S., et al. (2006). Diet and lifestyle recommendations revision 2006: a scientific statement from the American Heart Association Nutrition Committee. Circulation 114, 82–96.
Bao, W., Ballard, V. L., Needle, S., Hoang, B., Lenhard, S. C., Tunstead, J. R., et al. (2013). Cardioprotection by systemic dosing of thymosin beta four following ischemic myocardial injury. Front. Pharmacol. 4:149. doi: 10.3389/fphar.2013.00149
Benjamin, E. J., Blaha, M. J., Chiuve, S. E., Cushman, M., Das, S. R., Deo, R., et al. (2017). Heart disease and stroke statistics-2017 update: a report from the American Heart Association. Circulation 135, e146–e603. doi: 10.1161/CIR.0000000000000485
Bhatia, M. (2015). H2S and inflammation: an overview. Handb. Exp. Pharmacol. 230, 165–180. doi: 10.1007/978-3-319-18144-8_8
Boag, S. E., Andreano, E., and Spyridopoulos, I. (2017). Lymphocyte communication in myocardial ischemia/reperfusion injury. Antioxid. Redox Signal. 26, 660–675. doi: 10.1089/ars.2016.6940
Cai, W. J., Wang, M. J., Moore, P. K., Jin, H. M., Yao, T., and Zhu, Y. C. (2007). The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc. Res. 76, 29–40. doi: 10.1016/j.cardiores.2007.05.026
Calvert, J. W., Elston, M., Nicholson, C. K., Gundewar, S., Jha, S., Elrod, J. W., et al. (2010). Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia-induced heart failure in mice. Circulation 122, 11–19. doi: 10.1161/CIRCULATIONAHA.109.920991
Cao, X., and Bian, J. S. (2016). The role of hydrogen sulfide in renal system. Front. Pharmacol. 7:385. doi: 10.3389/fphar.2016.00385
Chang, L., Geng, B., Yu, F., Zhao, J., Jiang, H., Du, J., et al. (2008). Hydrogen sulfide inhibits myocardial injury induced by homocysteine in rats. Amino Acids 34, 573–585. doi: 10.1007/s00726-007-0011-8
Chunyu, Z., Junbao, D., Dingfang, B., Hui, Y., Xiuying, T., and Chaoshu, T. (2003). The regulatory effect of hydrogen sulfide on hypoxic pulmonary hypertension in rats. Biochem. Biophys. Res. Commun. 302, 810–816. doi: 10.1016/S0006-291X(03)00256-0
Coggins, M., and Rosenzweig, A. (2012). The fire within: cardiac inflammatory signaling in health and disease. Circ. Res. 110, 116–125. doi: 10.1161/CIRCRESAHA.111.243196
Delgado-Maroto, V., Benitez, R., Forte-Lago, I., Morell, M., Maganto-Garcia, E., Souza-Moreira, L., et al. (2017). Cortistatin reduces atherosclerosis in hyperlipidemic ApoE-deficient mice and the formation of foam cells. Sci. Rep. 7:46444. doi: 10.1038/srep46444
Donnarumma, E., Trivedi, R. K., and Lefer, D. J. (2017). Protective actions of H2S in acute myocardial infarction and heart failure. Compr. Physiol. 7, 583–602. doi: 10.1002/cphy.c160023
Du, J., Huang, Y., Yan, H., Zhang, Q., Zhao, M., Zhu, M., et al. (2014). Hydrogen sulfide suppresses oxidized low-density lipoprotein (ox-LDL)-stimulated monocyte chemoattractant protein 1 generation from macrophages via the nuclear factor kappaB (NF-kappaB) pathway. J. Biol. Chem. 289, 9741–9753. doi: 10.1074/jbc.M113.517995
Elrod, J. W., Calvert, J. W., Morrison, J., Doeller, J. E., Kraus, D. W., Tao, L., et al. (2007). Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. U.S.A. 104, 15560–15565. doi: 10.1073/pnas.0705891104
Feng, S., Chen, S., Yu, W., Zhang, D., Zhang, C., Tang, C., et al. (2017). H2S inhibits pulmonary arterial endothelial cell inflammation in rats with monocrotaline-induced pulmonary hypertension. Lab. Invest. 97, 268–278. doi: 10.1038/labinvest.2016.129
Feng, S. J., Li, H., and Wang, S. X. (2015). Lower hydrogen sulfide is associated with cardiovascular mortality, which involves cPKCbetaII/Akt pathway in chronic hemodialysis patients. Blood Purif. 40, 260–269. doi: 10.1159/000439580
Frangogiannis, N. G. (2012). Regulation of the inflammatory response in cardiac repair. Circ. Res. 110, 159–173. doi: 10.1161/CIRCRESAHA.111.243162
Gao, L., Xu, Z., Yin, Z., Chen, K., Wang, C., and Zhang, H. (2015). Association of hydrogen sulfide with alterations of monocyte chemokine receptors, CCR2 and CX3CR1 in patients with coronary artery disease. Inflamm. Res. 64, 627–635. doi: 10.1007/s00011-015-0844-7
Ge, S. N., Zhao, M. M., Wu, D. D., Chen, Y., Wang, Y., Zhu, J. H., et al. (2014). Hydrogen sulfide targets EGFR Cys797/Cys798 residues to induce Na(+)/K(+)-ATPase endocytosis and inhibition in renal tubular epithelial cells and increase sodium excretion in chronic salt-loaded rats. Antioxid. Redox Signal. 21, 2061–2082. doi: 10.1089/ars.2013.5304
Ghaderi, S., Alidadiani, N., Dilaver, N., Heidari, H. R., Parvizi, R., Rahbarghazi, R., et al. (2017). Role of glycogen synthase kinase following myocardial infarction and ischemia-reperfusion. Apoptosis 22, 887–897. doi: 10.1007/s10495-017-1376-0
Gistera, A., and Hansson, G. K. (2017). The immunology of atherosclerosis. Nat. Rev. Nephrol. 13, 368–380. doi: 10.1038/nrneph.2017.51
Givvimani, S., Munjal, C., Gargoum, R., Sen, U., Tyagi, N., Vacek, J. C., et al. (2011). Hydrogen sulfide mitigates transition from compensatory hypertrophy to heart failure. J. Appl. Physiol. 110, 1093–1100. doi: 10.1152/japplphysiol.01064.2010
Hackfort, B. T., and Mishra, P. K. (2016). Emerging role of hydrogen sulfide-microRNA crosstalk in cardiovascular diseases. Am. J. Physiol. Heart Circ. Physiol. 310, H802–H812. doi: 10.1152/ajpheart.00660.2015
Hausenloy, D. J., and Yellon, D. M. (2013). Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J. Clin. Invest. 123, 92–100. doi: 10.1172/JCI62874
Hofmann, U., Beyersdorf, N., Weirather, J., Podolskaya, A., Bauersachs, J., Ertl, G., et al. (2012). Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation 125, 1652–1663. doi: 10.1161/CIRCULATIONAHA.111.044164
Howangyin, K. Y., Zlatanova, I., Pinto, C., Ngkelo, A., Cochain, C., Rouanet, M., et al. (2016). Myeloid-epithelial-reproductive receptor tyrosine kinase and milk fat globule epidermal growth factor 8 coordinately improve remodeling after myocardial infarction via local delivery of vascular endothelial growth factor. Circulation 133, 826–839. doi: 10.1161/CIRCULATIONAHA.115.020857
Huang, P., Shen, Z., Yu, W., Huang, Y., Tang, C., Du, J., et al. (2017). Hydrogen sulfide inhibits high-salt diet-induced myocardial oxidative stress and myocardial hypertrophy in dahl rats. Front. Pharmacol. 8:128. doi: 10.3389/fphar.2017.00128
Ji, J., Xiang, P., Li, T., Lan, L., Xu, X., Lu, G., et al. (2017). NOSH-NBP, a novel nitric oxide and hydrogen sulfide- releasing hybrid, attenuates ischemic stroke-induced neuroinflammatory injury by modulating microglia polarization. Front. Cell Neurosci. 11:154. doi: 10.3389/fncel.2017.00154
Jiang, H. L., Wu, H. C., Li, Z. L., Geng, B., and Tang, C. S. (2005). [Changes of the new gaseous transmitter H2S in patients with coronary heart disease]. Di Yi Jun Yi Da Xue Xue Bao 25, 951–954.
Jin, H. F., Du, J. B., and Tang, C. S. (2010). [“Waste gas is not waste”: advance in the research of hydrogen sulfide]. Sheng Li Xue Bao 62, 495–504.
Jones, D. P., True, H. D., and Patel, J. (2017). Leukocyte trafficking in cardiovascular disease: insights from experimental models. Mediators Inflamm. 2017:9746169. doi: 10.1155/2017/9746169
Kan, J., Guo, W., Huang, C., Bao, G., Zhu, Y., and Zhu, Y. Z. (2014). S-propargyl-cysteine, a novel water-soluble modulator of endogenous hydrogen sulfide, promotes angiogenesis through activation of signal transducer and activator of transcription 3. Antioxid. Redox Signal. 20, 2303–2316. doi: 10.1089/ars.2013.5449
Kanagy, N. L., Szabo, C., and Papapetropoulos, A. (2017). Vascular biology of hydrogen sulfide. Am. J. Physiol. Cell Physiol. 312, C537–C549. doi: 10.1152/ajpcell.00329.2016
Kimura, H. (2015). Signaling molecules: hydrogen sulfide and polysulfide. Antioxid. Redox Signal. 22, 362–376. doi: 10.1089/ars.2014.5869
Kolluru, G. K., Bir, S. C., Yuan, S., Shen, X., Pardue, S., Wang, R., et al. (2015). Cystathionine gamma-lyase regulates arteriogenesis through NO-dependent monocyte recruitment. Cardiovasc. Res. 107, 590–600. doi: 10.1093/cvr/cvv198
Kondo, K., Bhushan, S., King, A. L., Prabhu, S. D., Hamid, T., Koenig, S., et al. (2013). H2S protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation 127, 1116–1127. doi: 10.1161/CIRCULATIONAHA.112.000855
Kuo, M. M., Kim, D. H., Jandu, S., Bergman, Y., Tan, S., Wang, H., et al. (2016). MPST but not CSE is the primary regulator of hydrogen sulfide production and function in the coronary artery. Am. J. Physiol. Heart Circ. Physiol. 310, H71–H79. doi: 10.1152/ajpheart.00574.2014
Lavine, K. J., Epelman, S., Uchida, K., Weber, K. J., Nichols, C. G., Schilling, J. D., et al. (2014). Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. U.S.A. 111, 16029–16034. doi: 10.1073/pnas.1406508111
Li, H., Mani, S., Wu, L., Fu, M., Shuang, T., Xu, C., et al. (2017). The interaction of estrogen and CSE/H2S pathway in the development of atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 312, H406–H414. doi: 10.1152/ajpheart.00245.2016
Li, L., Hsu, A., and Moore, P. K. (2009). Actions and interactions of nitric oxide, carbon monoxide and hydrogen sulphide in the cardiovascular system and in inflammation–a tale of three gases! Pharmacol. Ther. 123, 386–400. doi: 10.1016/j.pharmthera.2009.05.005
Li, L., Rose, P., and Moore, P. K. (2011). Hydrogen sulfide and cell signaling. Annu. Rev. Pharmacol. Toxicol. 51, 169–187. doi: 10.1146/annurev-pharmtox-010510-100505
Li, N., Wang, M. J., Jin, S., Bai, Y. D., Hou, C. L., Ma, F. F., et al. (2016). The H2S donor NaHS changes the expression pattern of H2S-Producing Enzymes after Myocardial Infarction. Oxid. Med. Cell. Longev. 2016:6492469. doi: 10.1155/2016/6492469
Libby, P., and Hansson, G. K. (2015). Inflammation and immunity in diseases of the arterial tree: players and layers. Circ. Res. 116, 307–311. doi: 10.1161/CIRCRESAHA.116.301313
Libby, P., Lichtman, A. H., and Hansson, G. K. (2013). Immune effector mechanisms implicated in atherosclerosis: from mice to humans. Immunity 38, 1092–1104. doi: 10.1016/j.immuni.2013.06.009
Lin, Y., Chen, Y., Zhu, N., Zhao, S., Fan, J., and Liu, E. (2016). Hydrogen sulfide inhibits development of atherosclerosis through up-regulating protein S-nitrosylation. Biomed. Pharmacother. 83, 466–476. doi: 10.1016/j.biopha.2016.07.003
Liu, J., Wang, H., and Li, J. (2016). Inflammation and inflammatory cells in myocardial infarction and reperfusion injury: a double-edged sword. Clin. Med. Insights Cardiol. 10, 79–84. doi: 10.4137/CMC.S33164
Liu, Y. H., Lu, M., Hu, L. F., Wong, P. T., Webb, G. D., and Bian, J. S. (2012). Hydrogen sulfide in the mammalian cardiovascular system. Antioxid. Redox Signal. 17, 141–185. doi: 10.1089/ars.2011.4005
Ma, S. F., Luo, Y., Ding, Y. J., Chen, Y., Pu, S. X., Wu, H. J., et al. (2015). Hydrogen sulfide targets the Cys320/Cys529 Motif in Kv4.2 to inhibit the ito potassium channels in cardiomyocytes and regularizes fatal arrhythmia in myocardial infarction. Antioxid. Redox Signal. 23, 129–147. doi: 10.1089/ars.2014.6094
Mani, S., Li, H., Untereiner, A., Wu, L., Yang, G., Austin, R. C., et al. (2013). Decreased endogenous production of hydrogen sulfide accelerates atherosclerosis. Circulation 127, 2523–2534. doi: 10.1161/CIRCULATIONAHA.113.002208
Mani, S., Untereiner, A., Wu, L., and Wang, R. (2014). Hydrogen sulfide and the pathogenesis of atherosclerosis. Antioxid. Redox Signal. 20, 805–817. doi: 10.1089/ars.2013.5324
Martelli, A., Testai, L., Breschi, M. C., Blandizzi, C., Virdis, A., Taddei, S., et al. (2012). Hydrogen sulphide: novel opportunity for drug discovery. Med. Res. Rev. 32, 1093–1130. doi: 10.1002/med.20234
Mathai, J. C., Missner, A., Kugler, P., Saparov, S. M., Zeidel, M. L., Lee, J. K., et al. (2009). No facilitator required for membrane transport of hydrogen sulfide. Proc. Natl. Acad. Sci. U.S.A. 106, 16633–16638. doi: 10.1073/pnas.0902952106
Meng, Q. H., Yang, G., Yang, W., Jiang, B., Wu, L., and Wang, R. (2007). Protective effect of hydrogen sulfide on balloon injury-induced neointima hyperplasia in rat carotid arteries. Am. J. Pathol. 170, 1406–1414. doi: 10.2353/ajpath.2007.060939
Merz, T., Stenzel, T., Nussbaum, B., Wepler, M., Szabo, C., Wang, R., et al. (2017). Cardiovascular disease and resuscitated septic shock lead to the downregulation of the H2S-producing enzyme cystathionine-gamma-lyase in the porcine coronary artery. Intensive Care Med. Exp. 5, 17. doi: 10.1186/s40635-017-0131-8
Miao, L., Shen, X., Whiteman, M., Xin, H., Shen, Y., Xin, X., et al. (2016a). Hydrogen sulfide mitigates myocardial infarction via promotion of mitochondrial biogenesis-dependent M2 polarization of macrophages. Antioxid. Redox Signal. 25, 268–281. doi: 10.1089/ars.2015.6577
Miao, L., Xin, X., Xin, H., Shen, X., and Zhu, Y. Z. (2016b). Hydrogen sulfide recruits macrophage migration by integrin beta1-Src-FAK/Pyk2-Rac pathway in myocardial infarction. Sci. Rep. 6:22363. doi: 10.1038/srep22363
Mikami, Y., Shibuya, N., Kimura, Y., Nagahara, N., Ogasawara, Y., and Kimura, H. (2011). Thioredoxin and dihydrolipoic acid are required for 3-mercaptopyruvate sulfurtransferase to produce hydrogen sulfide. Biochem. J. 439, 479–485. doi: 10.1042/BJ20110841
Miller, T. W., Wang, E. A., Gould, S., Stein, E. V., Kaur, S., Lim, L., et al. (2012). Hydrogen sulfide is an endogenous potentiator of T cell activation. J. Biol. Chem. 287, 4211–4221. doi: 10.1074/jbc.M111.307819
Moore, K. J., and Tabas, I. (2011). Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355. doi: 10.1016/j.cell.2011.04.005
Mozaffarian, D., Benjamin, E. J., Go, A. S., Arnett, D. K., Blaha, M. J., Cushman, M., et al. (2015). Heart disease and stroke statistics–2015 update: a report from the American Heart Association. Circulation 131, e29–322. doi: 10.1161/CIR.0000000000000157
Murphy, S. L., Kochanek, K. D., Xu, J., and Heron, M. (2015). Deaths: final data for 2012. Natl. Vital Stat. Rep. 63, 1–117. doi: 10.1136/vr.h753
Nagpure, B. V., and Bian, J. S. (2016). Interaction of hydrogen sulfide with nitric oxide in the cardiovascular system. Oxid. Med. Cell Longev. 2016:6904327. doi: 10.1155/2016/6904327
Nahrendorf, M., Pittet, M. J., and Swirski, F. K. (2010). Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation 121, 2437–2445. doi: 10.1161/CIRCULATIONAHA.109.916346
Nishida, M., Sawa, T., Kitajima, N., Ono, K., Inoue, H., Ihara, H., et al. (2012). Hydrogen sulfide anion regulates redox signaling via electrophile sulfhydration. Nat. Chem. Biol. 8, 714–724. doi: 10.1038/nchembio.1018
Odegaard, J. I., and Chawla, A. (2013). Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science 339, 172–177. doi: 10.1126/science.1230721
Pan, L. L., Liu, X. H., Gong, Q. H., Yang, H. B., and Zhu, Y. Z. (2012). Role of cystathionine gamma-lyase/hydrogen sulfide pathway in cardiovascular disease: a novel therapeutic strategy? Antioxid. Redox Signal. 17, 106–118. doi: 10.1089/ars.2011.4349
Pan, L. L., Liu, X. H., Gong, Q. H., and Zhu, Y. Z. (2011). S-Propargyl-cysteine (SPRC) attenuated lipopolysaccharide-induced inflammatory response in H9c2 cells involved in a hydrogen sulfide-dependent mechanism. Amino Acids 41, 205–215. doi: 10.1007/s00726-011-0834-1
Pan, L. L., Liu, X. H., Shen, Y. Q., Wang, N. Z., Xu, J., Wu, D., et al. (2013). Inhibition of NADPH oxidase 4-related signaling by sodium hydrosulfide attenuates myocardial fibrotic response. Int. J. Cardiol. 168, 3770–3778. doi: 10.1016/j.ijcard.2013.06.007
Pan, T. T., Neo, K. L., Hu, L. F., Yong, Q. C., and Bian, J. S. (2008). H2S preconditioning-induced PKC activation regulates intracellular calcium handling in rat cardiomyocytes. Am. J. Physiol. Cell Physiol. 294, C169–C177.
Polhemus, D. J., Kondo, K., Bhushan, S., Bir, S. C., Kevil, C. G., Murohara, T., et al. (2013). Hydrogen sulfide attenuates cardiac dysfunction after heart failure via induction of angiogenesis. Circ. Heart Fail. 6, 1077–1086. doi: 10.1161/CIRCHEARTFAILURE.113.000299
Polhemus, D. J., and Lefer, D. J. (2014). Emergence of hydrogen sulfide as an endogenous gaseous signaling molecule in cardiovascular disease. Circ. Res. 114, 730–737. doi: 10.1161/CIRCRESAHA.114.300505
Polhemus, D. J., Li, Z., Pattillo, C. B., Gojon, G. Sr., Gojon, G. Jr., Giordano, T., et al. (2015). A novel hydrogen sulfide prodrug, SG1002, promotes hydrogen sulfide and nitric oxide bioavailability in heart failure patients. Cardiovasc. Ther. 33, 216–226. doi: 10.1111/1755-5922.12128
Rose, P., Moore, P. K., and Zhu, Y. Z. (2017). H2S biosynthesis and catabolism: new insights from molecular studies. Cell Mol. Life Sci. 74, 1391–1412. doi: 10.1007/s00018-016-2406-8
Salloum, F. N. (2015). Hydrogen sulfide and cardioprotection–Mechanistic insights and clinical translatability. Pharmacol. Ther. 152, 11–17. doi: 10.1016/j.pharmthera.2015.04.004
Shibuya, N., Mikami, Y., Kimura, Y., Nagahara, N., and Kimura, H. (2009a). Vascular endothelium expresses 3-mercaptopyruvate sulfurtransferase and produces hydrogen sulfide. J. Biochem. 146, 623–626. doi: 10.1093/jb/mvp111
Shibuya, N., Tanaka, M., Yoshida, M., Ogasawara, Y., Togawa, T., Ishii, K., et al. (2009b). 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid. Redox Signal. 11, 703–714. doi: 10.1089/ars.2008.2253
Shirazi, L. F., Bissett, J., Romeo, F., and Mehta, J. L. (2017). Role of inflammation in heart failure. Curr. Atheroscler. Rep. 19, 27. doi: 10.1007/s11883-017-0660-3
Sivarajah, A., Collino, M., Yasin, M., Benetti, E., Gallicchio, M., Mazzon, E., et al. (2009). Anti-apoptotic and anti-inflammatory effects of hydrogen sulfide in a rat model of regional myocardial I/R. Shock 31, 267–274. doi: 10.1097/SHK.0b013e318180ff89
Smith, P. M., and Ferguson, A. V. (2016). Recent advances in central cardiovascular control: sex, ROS, gas and inflammation. F1000Res. doi: 10.12688/f1000research.7987.1
Sodha, N. R., Clements, R. T., Feng, J., Liu, Y., Bianchi, C., Horvath, E. M., et al. (2009). Hydrogen sulfide therapy attenuates the inflammatory response in a porcine model of myocardial ischemia/reperfusion injury. J. Thorac. Cardiovasc. Surg. 138, 977–984. doi: 10.1016/j.jtcvs.2008.08.074
Sun, Y., Tang, C. S., Du, J. B., and Jin, H. F. (2011). Hydrogen sulfide and vascular relaxation. Chin. Med. J. (Engl) 124, 3816–3819.
Sutti, S., Locatelli, I., Bruzzi, S., Jindal, A., Vacchiano, M., Bozzola, C., et al. (2015). CX3CR1-expressing inflammatory dendritic cells contribute to the progression of steatohepatitis. Clin. Sci. (Lond.) 129, 797–808. doi: 10.1042/CS20150053
Swirski, F. K., and Nahrendorf, M. (2013). Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science 339, 161–166. doi: 10.1126/science.1230719
Szabo, C. (2007). Hydrogen sulphide and its therapeutic potential. Nat. Rev. Drug Discov. 6, 917–935. doi: 10.1038/nrd2425
Tao, B. B., Liu, S. Y., Zhang, C. C., Fu, W., Cai, W. J., Wang, Y., et al. (2013). VEGFR2 functions as an H2S-targeting receptor protein kinase with its novel Cys1045-Cys1024 disulfide bond serving as a specific molecular switch for hydrogen sulfide actions in vascular endothelial cells. Antioxid. Redox Signal. 19, 448–464. doi: 10.1089/ars.2012.4565
Toldo, S., Das, A., Mezzaroma, E., Chau, V. Q., Marchetti, C., Durrant, D., et al. (2014). Induction of microRNA-21 with exogenous hydrogen sulfide attenuates myocardial ischemic and inflammatory injury in mice. Circ. Cardiovasc. Genet. 7, 311–320. doi: 10.1161/CIRCGENETICS.113.000381
Wallace, J. L., and Wang, R. (2015). Hydrogen sulfide-based therapeutics: exploiting a unique but ubiquitous gasotransmitter. Nat. Rev. Drug Discov. 14, 329–345. doi: 10.1038/nrd4433
Wang, H., and Cai, J. (2016). The role of microRNAs in heart failure. Biochim. Biophys. Acta 1863, 2019–2030. doi: 10.1016/j.bbadis.2016.11.034
Wang, H., Jiang, X., Yang, F., Gaubatz, J. W., Ma, L., Magera, M. J., et al. (2003). Hyperhomocysteinemia accelerates atherosclerosis in cystathionine beta-synthase and apolipoprotein E double knock-out mice with and without dietary perturbation. Blood 101, 3901–3907. doi: 10.1182/blood-2002-08-2606
Wang, M. J., Cai, W. J., and Zhu, Y. C. (2016). Hydrogen sulphide in cardiovascular system: a cascade from interaction between sulphur atoms and signalling molecules. Life Sci. 153, 188–197. doi: 10.1016/j.lfs.2016.03.057
Wang, R. (2002). Two’s company, three’s a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J. 16, 1792–1798. doi: 10.1096/fj.02-0211hyp
Wang, R. (2012). Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol. Rev. 92, 791–896. doi: 10.1152/physrev.00017.2011
Wang, R., Szabo, C., Ichinose, F., Ahmed, A., Whiteman, M., and Papapetropoulos, A. (2015). The role of H2S bioavailability in endothelial dysfunction. Trends Pharmacol. Sci. 36, 568–578. doi: 10.1016/j.tips.2015.05.007
Wang, W., Feng, S. J., Li, H., Zhang, X. D., and Wang, S. X. (2015). Correlation of lower concentrations of hydrogen sulfide with activation of protein kinase CbetaII in uremic accelerated atherosclerosis patients. Chin. Med. J. (Engl.) 128, 1465–1470. doi: 10.4103/0366-6999.157653
Wang, X. H., Wang, F., You, S. J., Cao, Y. J., Cao, L. D., Han, Q., et al. (2013). Dysregulation of cystathionine gamma-lyase (CSE)/hydrogen sulfide pathway contributes to ox-LDL-induced inflammation in macrophage. Cell. Signal. 25, 2255–2262. doi: 10.1016/j.cellsig.2013.07.010
Wang, Y., Zhao, X., Jin, H., Wei, H., Li, W., Bu, D., et al. (2009). Role of hydrogen sulfide in the development of atherosclerotic lesions in apolipoprotein E knockout mice. Arterioscler. Thromb. Vasc. Biol. 29, 173–179. doi: 10.1161/ATVBAHA.108.179333
Wu, T., Li, H., Wu, B., Zhang, L., Wu, S. W., Wang, J. N., et al. (2017). Hydrogen sulfide reduces recruitment of CD11b+Gr-1+ cells in mice with myocardial infarction. Cell Transplant 26, 753–764. doi: 10.3727/096368917X695029
Xie, L., Gu, Y., Wen, M., Zhao, S., Wang, W., Ma, Y., et al. (2016). Hydrogen sulfide induces keap1 S-sulfhydration and suppresses diabetes-accelerated atherosclerosis via Nrf2 activation. Diabetes Metab. Res. Rev. 65, 3171–3184. doi: 10.2337/db16-0020
Yadav, P. K., Yamada, K., Chiku, T., Koutmos, M., and Banerjee, R. (2013). Structure and kinetic analysis of H2S production by human mercaptopyruvate sulfurtransferase. J. Biol. Chem. 288, 20002–20013. doi: 10.1074/jbc.M113.466177
Yan, S. K., Chang, T., Wang, H., Wu, L., Wang, R., and Meng, Q. H. (2006). Effects of hydrogen sulfide on homocysteine-induced oxidative stress in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 351, 485–491. doi: 10.1016/j.bbrc.2006.10.058
Yan, X., Wu, H., Wu, Z., Hua, F., Liang, D., Sun, H., et al. (2017). The new synthetic H2S-releasing SDSS protects MC3T3-E1 osteoblasts against H2O2-induced apoptosis by suppressing oxidative stress, inhibiting MAPKs, and activating the PI3K/Akt pathway. Front. Pharmacol. 8:07. doi: 10.3389/fphar.2017.00007
Yang, G., and Wang, R. (2015). H2S and blood vessels: an overview. Handb. Exp. Pharmacol. 230, 85–110. doi: 10.1007/978-3-319-18144-8_4
Yang, G., Wu, L., Jiang, B., Yang, W., Qi, J., Cao, K., et al. (2008). H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science 322, 587–590. doi: 10.1126/science.1162667
Yang, R., Qu, C., Zhou, Y., Konkel, J. E., Shi, S., Liu, Y., et al. (2015). Hydrogen sulfide promotes Tet1- and Tet2-mediated Foxp3 demethylation to drive regulatory T cell differentiation and maintain immune homeostasis. Immunity 43, 251–263. doi: 10.1016/j.immuni.2015.07.017
Yellon, D. M., and Hausenloy, D. J. (2007). Myocardial reperfusion injury. N. Engl. J. Med. 357, 1121–1135. doi: 10.1056/NEJMra071667
Zanardo, R. C., Brancaleone, V., Distrutti, E., Fiorucci, S., Cirino, G., and Wallace, J. L. (2006). Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 20, 2118–2120. doi: 10.1096/fj.06-6270fje
Zhang, H., Guo, C., Wu, D., Zhang, A., Gu, T., Wang, L., et al. (2012). Hydrogen sulfide inhibits the development of atherosclerosis with suppressing CX3CR1 and CX3CL1 expression. PLOS ONE 7:e41147. doi: 10.1371/journal.pone.0041147
Zhang, Y., Li, H., Zhao, G., Sun, A., Zong, N. C., Li, Z., et al. (2014). Hydrogen sulfide attenuates the recruitment of CD11b(+)Gr-1(+) myeloid cells and regulates Bax/Bcl-2 signaling in myocardial ischemia injury. Sci. Rep. 4:4774. doi: 10.1038/srep04774
Zhao, W., Zhang, J., Lu, Y., and Wang, R. (2001). The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 20, 6008–6016. doi: 10.1093/emboj/20.21.6008
Zhao, Z. Z., Wang, Z., Li, G. H., Wang, R., Tan, J. M., Cao, X., et al. (2011). Hydrogen sulfide inhibits macrophage-derived foam cell formation. Exp. Biol. Med. (Maywood) 236, 169–176. doi: 10.1258/ebm.2010.010308
Zheng, Y., Yu, B., De La Cruz, L. K., Roy Choudhury, M., Anifowose, A., and Wang, B. (2017). Toward hydrogen sulfide based therapeutics: critical drug delivery and developability issues. Med. Res. Rev. doi: 10.1002/med.21433 [Epub ahead of print].
Zhou, Y., Wu, Z., Cao, X., Ding, L., Wen, Z., and Bian, J. S. (2016). HNO suppresses LPS-induced inflammation in BV-2 microglial cells via inhibition of NF-kappaB and p38 MAPK pathways. Pharmacol. Res. 111, 885–895. doi: 10.1016/j.phrs.2016.08.007
Keywords: myocardial ischemia, heart failure, atherosclerosis, inflammation, hydrogen sulfide
Citation: Pan L-L, Qin M, Liu X-H and Zhu Y-Z (2017) The Role of Hydrogen Sulfide on Cardiovascular Homeostasis: An Overview with Update on Immunomodulation. Front. Pharmacol. 8:686. doi: 10.3389/fphar.2017.00686
Received: 21 May 2017; Accepted: 13 September 2017;
Published: 26 September 2017.
Edited by:
Jinsong Bian, National University of Singapore, SingaporeReviewed by:
Hongfang Jin, Peking University First Hospital, ChinaYong Ji, Nanjing Medical University, China
Copyright © 2017 Pan, Qin, Liu and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin-Hua Liu, bGl1eGluaHVhQGZ1ZGFuLmVkdS5jbg== Yi-Zhun Zhu, eXp6aHVAbXVzdC5lZHUubW8=