
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 21 September 2017
Sec. Cardiovascular and Smooth Muscle Pharmacology
Volume 8 - 2017 | https://doi.org/10.3389/fphar.2017.00649
 Nils Bögeholz1†
Nils Bögeholz1† Jan S. Schulte2*†
Jan S. Schulte2*† Sven Kaese1†
Sven Kaese1† B. Klemens Bauer1,2Paul Pauls1,2Dirk G. Dechering1Gerrit Frommeyer1Joshua I. Goldhaber3Uwe Kirchhefer2Lars Eckardt1Christian Pott1
B. Klemens Bauer1,2Paul Pauls1,2Dirk G. Dechering1Gerrit Frommeyer1Joshua I. Goldhaber3Uwe Kirchhefer2Lars Eckardt1Christian Pott1 Frank U. Müller2
Frank U. Müller2Background/Objective: The cardiac Na+/Ca2+ exchanger (NCX) has been identified as a promising target to counter arrhythmia in previous studies investigating the benefit of NCX inhibition. However, the consequences of NCX inhibition have not been investigated in the setting of altered NCX expression and function, which is essential, since major cardiac diseases (heart failure/atrial fibrillation) exhibit NCX upregulation. Thus, we here investigated the effects of the NCX inhibitor SEA0400 on the Ca2+ transient amplitude and on proarrhythmia in homozygous NCX overexpressor (OE) and heterozygous NCX knockout (hetKO) mice compared to corresponding wild-types (WTOE/WThetKO).
Methods/Results: Ca2+ transients of field-stimulated isolated ventricular cardiomyocytes were recorded with fluo-4-AM or indo-1-AM. SEA0400 (1 μM) significantly reduced NCX forward mode function in all mouse lines. SEA0400 (1 μM) significantly increased the amplitude of field-stimulated Ca2+ transients in WTOE, WThetKO, and hetKO, but not in OE (% of basal; OE = 98.7 ± 5.0; WTOE = 137.8 ± 5.2*; WThetKO = 126.3 ± 6.0*; hetKO = 140.6 ± 12.8*; *p < 0.05 vs. basal). SEA0400 (1 μM) significantly reduced the number of proarrhythmic spontaneous Ca2+ transients (sCR) in OE, but increased it in WTOE, WThetKO and hetKO (sCR per cell; basal/+SEA0400; OE = 12.5/3.7; WTOE = 0.2/2.4; WThetKO = 1.3/8.8; hetKO = 0.2/5.5) and induced Ca2+ overload with subsequent cell death in hetKO.
Conclusion: The effects of SEA0400 on Ca2+ transient amplitude and the occurrence of spontaneous Ca2+ transients as a proxy measure for inotropy and cellular proarrhythmia depend on the NCX expression level. The antiarrhythmic effect of SEA0400 in conditions of increased NCX expression promotes the therapeutic concept of NCX inhibition in heart failure/atrial fibrillation. Conversely, in conditions of reduced NCX expression, SEA0400 suppressed the NCX function below a critical level leading to adverse Ca2+ accumulation as reflected by an increase in Ca2+ transient amplitude, proarrhythmia and cell death. Thus, the remaining NCX function under inhibition may be a critical factor determining the inotropic and antiarrhythmic efficacy of SEA0400.
Approximately 25% of cardiovascular mortality has been attributed to sudden cardiac death (Priori et al., 2015). Except for betablockers, conventional antiarrhythmic drugs targeting Na+, Ca2+, or K+ channels have not been shown to prevent sudden cardiac death. Due to these unsatisfying results, much effort has been made to identify alternative targets to prevent from life threatening arrhythmias. In this regard, heart failure—the most prevalent precondition for sudden cardiac death—is accompanied by an upregulation of the cardiac Na+/Ca2+ exchanger (NCX) (Menick et al., 2007). NCX is the dominant mechanism for the extrusion of Ca2+ out of the cell. In its electrogenic forward mode NCX extrudes 1 Ca2+ ion in exchange for 3 Na+ ions thus generating a net electrical inward current that depolarizes the cellular membrane potential. In the setting of increased NCX expression—as observed in heart failure or atrial fibrillation—NCX-mediated depolarization is enhanced leading to a prolongation of the action potential duration and favoring the occurrence of early and delayed afterdepolarizations (EADs, DADs). Thus, increased NCX function is regarded as a crucial factor for increased triggered activity and whole heart arrhythmia (Sipido et al., 2006; Pott et al., 2012; Bourgonje et al., 2013).
Conversely, we have recently shown that reduced NCX expression in a murine model with heterozygous knockout of the NCX gene leads to suppression of EADs and DADs (Bögeholz et al., 2015) supporting the above stated hypothesis. Others have evaluated the pharmacological (Nagy et al., 2004; Tanaka et al., 2007; Milberg et al., 2012) inhibition of NCX as an antiarrhythmic strategy. The majority of these studies used the NCX inhibitor SEA0400. Within the group of potent NCX inhibitors, previously represented by the peptide XIP (Li et al., 1991) and the isothiourea derivative KB-R7943 (Iwamoto et al., 1996), SEA0400 was introduced around the millennium. SEA0400 was considered as a major advance, especially due to its improved selectivity and high potency as compared to the mentioned precursors (Nagy et al., 2004). In later studies, the selectivity of SEA0400 has been debated and relativized (Reuter et al., 2002; Birinyi et al., 2005, 2008) due to relevant inhibitory efficacy on the L-type Ca2+ channel on higher concentrations (Birinyi et al., 2005). Other than XIP, SEA0400 is able to permeate the cell membrane. Encouragingly, the majority of studies to investigate the effect of SEA0400 on arrhythmia reported positive results, i.e., antiarrhythmic efficacy (Nagy et al., 2004; Milberg et al., 2012) in absence of negative inotropic effects (Bourgonje et al., 2013) thus potentially qualifying the concept of NCX inhibition as a promising antiarrhythmic strategy. However, there is also data suggesting unbeneficial effects of SEA0400 in the form of increased proarrhythmia (Ozdemir et al., 2008). These studies investigated various animal models and species in the absence or presence of heart failure and partially applied cardiac glycosides, that reduce the NCX forward mode function (O'Neill et al., 1990), to provoke arrhythmia (Nagasawa et al., 2005; Namekata et al., 2009; Nagy et al., 2014). Thus, NCX function was either enhanced due to an increased NCX expression in heart failure models or already reduced by cardiac glycosides which reduce the NCX driving force by increasing intracellular Na+. To the best of our knowledge, there is no study that evaluated the antiarrhythmic and inotropic effects of NCX inhibition by SEA0400 in dependence of the NCX expression and function level. However, this aspect is crucial, since major cardiac diseases like heart failure and atrial fibrillation (Voigt et al., 2012) are accompanied by NCX upregulation, which critically affects cardiac Ca2+ cycling. On the contrary, there are cardiac disease entities that exhibit a reduced NCX function, like the postinfarction (Zhang et al., 1996; Quinn et al., 2003) and diabetic cardiomyopathy (Schaffer et al., 1997; Hattori et al., 2000; Zhao et al., 2014). Thus, we here used a murine model of increased (Adachi-Akahane et al., 1997; Pott et al., 2012) or decreased (Henderson et al., 2004; Bögeholz et al., 2015) NCX expression (and consequently function) and corresponding wild-type mice to evaluate the effects of SEA0400 on Ca2+ transient amplitudes as a proxy measure for inotropy and the occurrence of proarrhythmic Ca2+ releases in dependence of the NCX expression level. Since chronic reduction of NCX expression and function has been demonstrated to prevent arrhythmia (Bögeholz et al., 2015), another more fundamental question to be addressed in this study is, whether acute pharmacological NCX inhibition similarly mediates antiarrhythmic effects. And if so, whether this antiarrhythmic effect does apply exclusively to the setting of increased NCX expression and/or function vs. reduced NCX function, bearing a potential answer toward the question whether there is an optimum of NCX function in the face of inotropy and arrhythmia.
Generation of global homozygous NCX overexpressor (OE) (Adachi-Akahane et al., 1997) and heterozygous knockout (hetKO) (Jordan et al., 2010) mouse model has been described previously. The genetic background of OE mice is C57BL/6 (Pott et al., 2012), whereas hetKO mice are on a combined genetic background of C57BL/6 and CD-1 due to embryonic transfer (Bögeholz et al., 2016). Animals entering experimentation were <12.5 weeks of age, since up to this age there is no evidence for structural remodeling or heart failure in OE (Pott et al., 2012) or hetKO mice (Jordan et al., 2010; Bögeholz et al., 2015). All experiments were approved by the local animal welfare authorities (Landesamt für Natur, Umwelt, und Verbraucherschutz NRW, permission number: 8.84-02.05.20.11.098) and conform to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes.
Ventricular cardiomyocytes were isolated from OE, hetKO and respective wild-type mice (WTOE/WThetKO) as previously described with minor alterations (Schulte et al., 2012; Bögeholz et al., 2015). Mice were sedated and euthanized by carbon dioxide inhalation. Hearts were explanted immediately after decease and perfused retrogradely for 1 min via the aorta with a perfusion solution containing in mM: NaCl 113, KCl 4.7, KH2PO4 0.6, Na2HPO4 0.6, MgSO4 1.2, NaHCO3 12, KHCO3 10, taurine 30, HEPES 10, glucose 11.1, BDM 10, heparin 14.3 IE/ml, pH = 7.4. To isolate rod-shaped viable ventricular myocytes, the extracellular connective tissue was degraded by perfusion for 6 min ± 10 s on 37.1°C with the perfusion solution containing Collagenase type 2 (Worthington Biochemical Corporation) and Protease from streptomyces griseus type XIV (Sigma Aldrich).
Technical and procedural aspects of Ca2+ epifluorescence measurement have been described in detail before (Pott et al., 2012; Bögeholz et al., 2015). Data acquisition was performed using a Ca2+ imaging system, composed of a DeltaRAM monochromator and two Type-710 photomultiplier (Photon Technology International, USA) mounted to an inverse microscope (Diaphot 200, Nikon, Japan). Cardiomyocytes were transferred to a custom-made recording chamber and field-stimulated by platinum wires (Type 223 Stimulator CS, Hugo Sachs Elektronik- Harvard Apparatus, Germany). Fluorescence signals were recorded by FeliX 1.42 software (Photon Technology International, USA). The following external solution (Tyrode's solution) has been applied; in mM: NaCl 140, KCl 5.8, KH2PO4 0.5, Na2HPO4 0.4 MgSO4 0.9, HEPES 10, glucose 11.1, CaCl2 2, pH = 7.3. Experiments were performed at room temperature. The NCX function was assessed by the decay of caffeine (10 mM) induced Ca2+ transients (Bassani et al., 1994; Bögeholz et al., 2015, 2016) using fluo-4-AM (excitation: 488 nm, emission: 522 nm). The ratiometric fluorescent Ca2+ indicator indo-1-AM was employed to record field-stimulated Ca2+ transients and proarrhythmic spontaneous Ca2+ activity (excitation: 344 nm, emission-ratio: 405/495 nm). The amplitude of field-stimulated Ca2+ transients served as a proxy measure for inotropy. The most chosen SEA0400 concentration is 1 μM (Farkas et al., 2008; Namekata et al., 2009; Bourgonje et al., 2013; Nagy et al., 2014). Since previous studies demonstrated a relevant inhibitory effect of SEA0400 on other molecular targets next to NCX, such as the L-type Ca2+ channel, at concentrations higher than 1 μM (Birinyi et al., 2005) we decided not to exceed this threshold. Since we could observe an effect of SEA0400 on Ca2+ transient dynamics in initial dose-response experiments in a range between 0.3 and 1 μM, we decided to apply both concentrations.
Statistical calculations were conducted with SigmaPlot 11.2 (Systat Software; Inc.). Numerical results are expressed as mean value ± standard error of the mean (SEM). Number of replicates are indicated as n = number of independently analyzed cardiomyocytes/number of mice used for cardiomyocyte isolation. Data were analyzed by ANOVA on ranks followed by Dunn's post hoc test or repeated measures ANOVA followed by Holm-Sidak post hoc test. Rates and proportions were quantified using the Fisher's exact test with R.
To test the inhibitory effect of SEA0400 on the NCX forward mode, caffeine application experiments were conducted as reported previously (Bassani et al., 1994; Bögeholz et al., 2015). Before caffeine was applied, cells were field-stimulated in absence or presence of SEA0400, respectively, with 1 Hz for 1 min followed by 10 s of rest. A faster caffeine-induced Ca2+ transient decay indicates a higher NCX function.
As demonstrated previously, in absence of SEA0400 (basal conditions), the time to 50% decay of caffeine-induced Ca2+ transients (T50) was significantly prolonged in heterozygous hetKO and shortened in OE as compared to the corresponding WT confirming a reduced/enhanced NCX-mediated Ca2+ removal rate, respectively (T50 in s: OE: 1.2 ± 0.2+; n = 37/5; WTOE: 2.0 ± 0.1; n = 42/5; hetKO: 3.3 ± 0.2+; n = 26/4; WThetKO: 2.1 ± 0.2; n = 24/4; +p < 0.05 OE vs. WTOE or hetKO vs. WThetKO; ANOVA on ranks test) (Figures 1A–D). When caffeine was applied in the presence of SEA0400 (1 μM), T50 was significantly increased vs. basal in all mouse lines indicating the inhibitory effect of SEA0400 on NCX forward mode (T50 in s; 1 μM SEA0400: OE: 2.9 ± 0.4*; n = 21/3; WTOE: 4.1 ± 0.4*; n = 25/3; hetKO: 5.8 ± 0.4*; n = 24/3; WThetKO: 4.3 ± 0.4*; n = 24/3; *p < 0.05 vs. basal; ANOVA on ranks test) (Figures 1B,D). In presence of SEA0400, T50 still tended to be shortened in OE and prolonged in hetKO compared to WTOE and WThetKO, respectively, however this finding was not statistically significant (p > 0.05 each; ANOVA on ranks test). Notably, the mean T50 value of OE in presence of SEA0400 was larger than in WTOE/WThetKO but still smaller than in hetKO under basal conditions indicating a remaining NCX function in OE ranging between the levels of WTOE/WThetKO and hetKO.
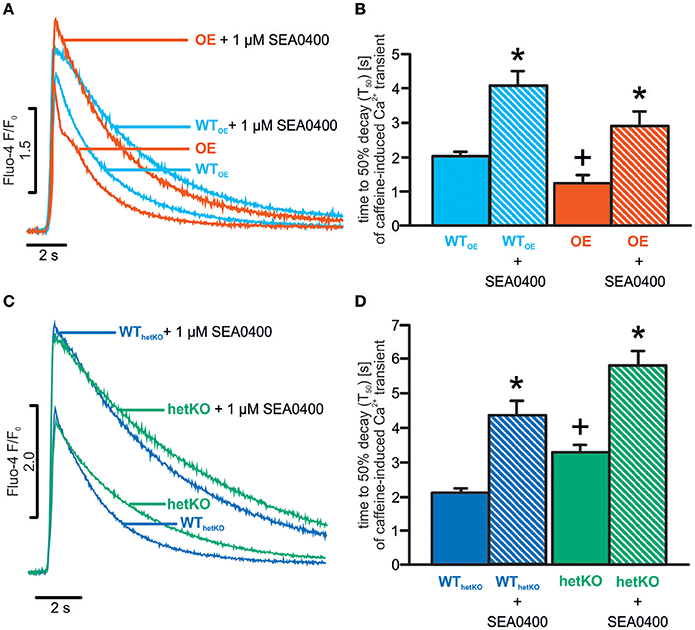
Figure 1. SEA0400 mediated NCX forward mode inhibition in caffeine application experiments. Representative tracings of caffeine-induced Ca2+ transients illustrate the different decay rates in OE (A), hetKO (C) and respective WTs as a direct measure of NCX-mediated Ca2+ removal under basal conditions and during NCX inhibition by SEA0400. Quantification of time to 50% decay (T50) of caffeine-induced Ca2+ transients confirmed increased NCX forward mode function in OE (B) and decreased in hetKO (D), and verified NCX forward mode inhibition by SEA0400 in all investigated groups but to different levels. *p < 0.05 vs. basal; +p < 0.05 OE/hetKO vs. WTOE/WThetKO basal; ANOVA on ranks test.
To test the effects of SEA0400 on the steady-state amplitude of field-stimulated Ca2+ transients, Ca2+ transients were measured in absence and presence of 0.3 and 1 μM SEA0400. Cells were field-stimulated at 1 Hz for 4 min followed by 20 sweeps on 0.125 Hz. This protocol was repeated in presence of 0.3 and 1 μM SEA0400 within the same cell. The amplitude of the Ca2+ transient was quantified at the end of the 1 Hz pacing phase after reaching a steady-state in Ca2+ transient kinetics. The Ca2+ transient amplitude in presence of SEA0400 is given in % of basal conditions.
SEA0400 led to a similar relative increase of the mean Ca2+ transient amplitude in WTOE (n = 27/7), WThetKO (n = 19/4), and hetKO (n = 19/5). However, in OE (n = 27/8) no increase of the Ca2+ transient amplitude was observed under application of SEA0400 (amplitude of field stimulated Ca2+ transients in % of basal; 1 μM SEA0400: OE: 98.7 ± 5.1; n = 23/8; WTOE: 137.8 ± 5.2*; n = 23/7; hetKO: 140.6 ± 12.8*; n = 9/3; WThetKO: 126.3 ± 6.0*; n = 16/4; *p < 0.05 vs. basal; repeated measures ANOVA test) (Figures 2A,B).
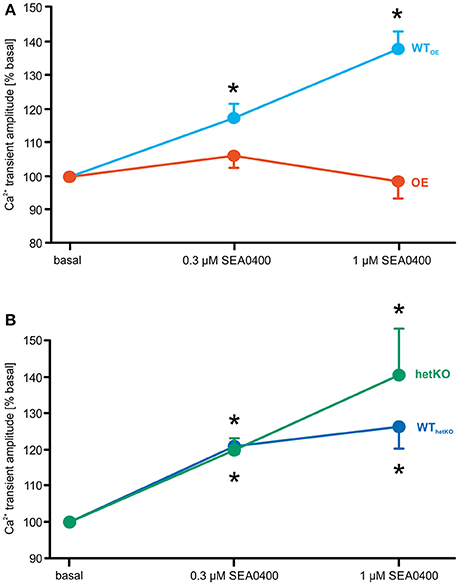
Figure 2. Relative effect of increasing SEA0400 concentrations on the amplitude of field stimulated Ca2+ transients as a measure for inotropy. SEA0400 mediated a significant relative increase of the Ca2+ transient amplitude vs. basal in WTOE (A), WThetKO and hetKO (B), but not in OE (A). Thus, the effect of SEA0400 on the Ca2+ transient amplitude seems to depend on the respective NCX expression level. *p < 0.05 vs. basal; two-way repeated measures ANOVA test.
To investigate the dependence of the antiarrhythmic effects of SEA0400 on NCX expression level, we monitored the occurrence of spontaneous Ca2+ releases (sCR), defined as spontaneously occurring Ca2+ transients between the field stimulated Ca2+ transients, during the 0.125 Hz pacing period in the above stated protocol before and after SEA0400 administration. An increase of sCR suggests an increase of proarrhythmia.
Under basal conditions, the occurrence of sCR (Figure 3) was similar to previously reported findings for OE (Pott et al., 2012) and hetKO (Bögeholz et al., 2015). The fraction of cells exhibiting spontaneous Ca2+ transients was significantly increased in OE compared to WTOE (Figure 3C) (OE: 63%+, WTOE: 7%; +p < 0.05 OE vs. WTOE, Fisher's exact test with R). The average number of sCR per cell (Figure 3B) was likewise increased in OE vs. WTOE (number of sCR/all investigated cells: OE: 12.5 ± 3.8+; n = 27/8; WTOE: 0.2 ± 0.1; n = 27/7; +p < 0.05 OE vs. WTOE; repeated measures ANOVA).
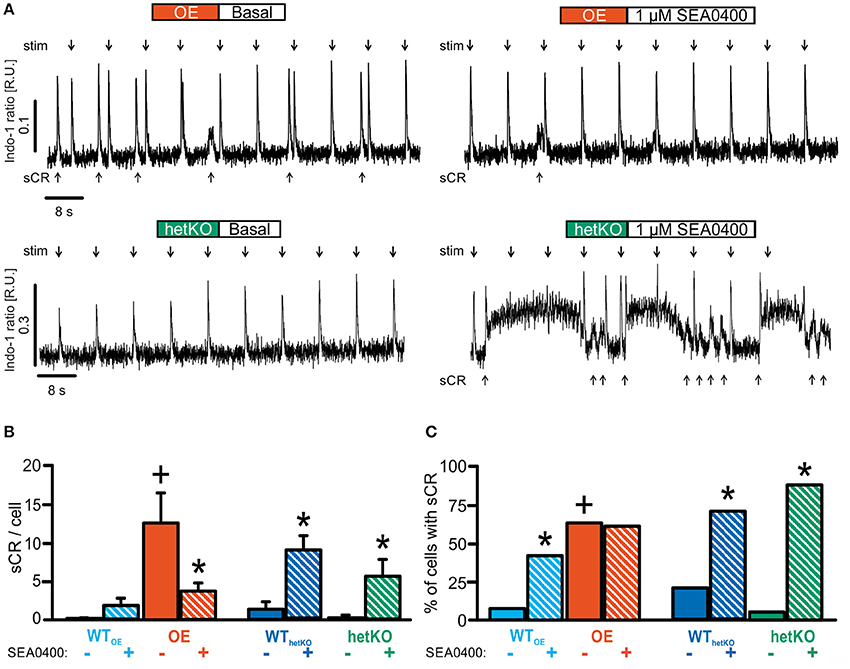
Figure 3. Quantification of spontaneous Ca2+ releases (sCR) as a measure for proarrhythmia. (A) Representative tracings illustrate the occurrence of numerous sCR (arrows below respective tracings) between field-stimulated Ca2+ transients (stim, arrows above respective tracings) in OE but not in hetKO in absence of SEA0400. (B) Quantification of sCR/cell under basal conditions and during exposure with 1 μM SEA0400. (C) Number of sCR-positive cardiomyocytes under basal conditions and during exposure with 1 μM SEA0400. SEA0400 significantly reduced the occurrence of sCR/cell in OE. Conversely, in hetKO SEA0400 increased the occurrence of sCR. Hence, the effect of SEA0400 on the occurrence of proarrhythmic spontaneous Ca2+ transients again depends on the NCX expression level. *p < 0.05 vs. basal; +p < 0.05 vs. corresponding WT basal; two-way repeated measures ANOVA test; z-test.
In hetKO the fraction of cells with sCR (Figure 3C) (hetKO: 5%; n = 19/5 WThetKO: 21%; n = 19/4; p > 0.05 hetKO vs. WThetKO, Fisher's exact test with R) as well as mean number of sCR per cell (Figure 3B) tended to be reduced vs. WThetKO but the results were not statistically significant (number of sCR/all investigated cells: hetKO: 0.2 ± 0.2; WThetKO: 1.3 ± 1.0; p > 0.05 hetKO vs. WThetKO; repeated measures ANOVA).
Application of SEA0400 altered the occurrence of sCR, but the specific effect depended upon the basal NCX expression level. In OE, SEA0400 significantly decreased the mean number of sCR per cell (Figure 3B) (number of sCR/all investigated cells; 1 μM SEA0400; OE: 3.7 ± 1.0*; n = 23/8; *p < 0.05 vs. basal; repeated measures ANOVA) without altering the number of sCR-positive cells (Figure 3C). In contrast, we observed an increase in the occurrence of sCR in groups with normal or reduced NCX expression level. In WTOE and WThetKO SEA0400 significantly increased the number of sCR-positive cells to 41 and 71% (Figure 3C). At the same time the occurrence of sCR per cell was increased significantly in WThetKO and in tendency in WTOE in response to SEA0400 (Figure 3B) (number of sCR/all investigated cells; 1 μM SEA0400: WTOE: 2.4 ± 0.8; n = 23/7; WThetKO: 8.8 ± 1.9*; n = 16/4; *p < 0.05 vs. basal; repeated measures ANOVA). In hetKO application of 1 μM SEA0400 significantly increased both, the occurrence of sCR per cell (Figure 3B) (number of sCR/all investigated cells; 1 μM SEA0400: hetKO: 5.5 ± 2.2*; n = 8/3; *p < 0.05 vs. basal; repeated measures ANOVA) and the number of sCR-positive cells (Figure 3C) (hetKO: 88%*; *p < 0.05 vs. basal, Fisher's exact test with R). Moreover, SEA0400 drastically impaired Ca2+ homeostasis in hetKO as reflected by an abrupt elevation of the diastolic Ca2+ level, incessant Ca2+ oscillations and subsequent cell death, defined as Ca2+ hypercontraction (Figure 4), so that only 42% of all measured hetKO cells could be analyzed. The fraction of cells suffering from Ca2+ hypercontraction was significantly increased in hetKO as compared to WThetKO (Figure 4B) (hetKO: 58%+, WThetKO: 11 %; +p < 0.05 hetKO vs. WThetKO, Fisher's exact test with R) but unaltered between OE and WTOE (OE: 15%, WTOE: 19%; p > 0.05 OE vs. WTOE, Fisher's exact test with R).
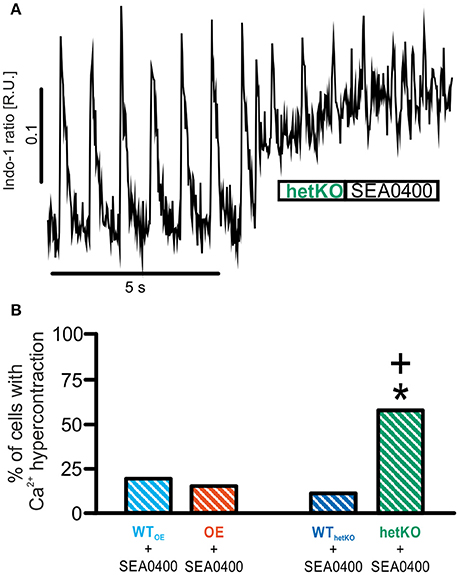
Figure 4. Quantification of Ca2+ hypercontractions as a measure for proarrhythmia. In a fraction of cardiomyocytes application of 1 μM SEA0400 caused a sudden increase of the diastolic Ca2+ level with highly frequent contractions and subsequent cell death (“Ca2+ hypercontraction”) (B) as illustrated by the representative tracing in (A). The proportion of cells with Ca2+ hypercontractions was significantly increased only in the hetKO group. *p < 0.05 vs. basal; +p < 0.05 vs. WT; Fisher's exact test with R.
In the search for novel strategies to counter cardiac arrhythmia, the cardiac Na+/Ca2+ exchanger (NCX) has been identified as a potential target. Due to its electrogenic mode of action (3 Na+: 1 Ca2+) NCX mediates membrane depolarization during Ca2+ extrusion. Hence, NCX may contribute to the development of EADs and facilitate the translation of spontaneous SR Ca2+ releases into DADs, which both may trigger arrhythmia (Sipido et al., 2006; Pott et al., 2012; Bourgonje et al., 2013). Consequently, pharmacological NCX inhibition should have antiarrhythmic effects but may on the other hand increase the cellular Ca2+ content, which could increase inotropy but also adversely promote arrhythmia. Encouragingly, initial work has demonstrated evidence for the antiarrhythmic efficacy of NCX inhibition mediated by SEA0400. However, the present study demonstrates, that the effects of SEA0400 on Ca2+ transient amplitude and proarrhythmia in murine cardiomyocytes are dependent on the NCX expression level.
SEA0400 reduced the NCX forward mode function to a similar extent in all mouse lines thus confirming inhibitory efficacy. At the same time, SEA0400 mediated a significant increase of the Ca2+ transient suggesting a positive inotropic effect in both wild-types (WTOE; WThetKO) and hetKO but interestingly not in OE.
Previous studies found a positive inotropic effect in response to SEA0400 in murine, rat and pig models (Tanaka et al., 2005; Farkas et al., 2008; Ozdemir et al., 2008). Other studies reported rather neutral effects on contractility in rabbit, guinea pig and dog models (Nagy et al., 2004; Tanaka et al., 2007; Bourgonje et al., 2013). These diverse effects may be mostly attributable to differences in Ca2+ handling and basic electrophysiological properties between the various species. Analogous to cardiac glycosides, NCX inhibition can result in a net gain of intracellular Ca2+, which augments the Ca2+ transient amplitude and consequently inotropy. SEA0400 seems to mediate such a positive inotropic effect in smaller rodents (mouse and rat) but not in higher mammals, with the exception for pigs. In smaller rodents, the intracellular Na+ level is elevated compared to higher mammals which results in a reversal potential for NCX (ENa/Ca) close to the diastolic membrane potential (Em) reducing the driving force for the NCX forward mode (Em-ENa/Ca) (Bers, 2002). Moreover, Ca2+ removal from the cytosol in these species largely depends on sarco(endo-)plasmic reticulum Ca2+-ATPase (SERCA) which mediates the re-uptake of Ca2+ ions into the sarcoplasmic reticulum while NCX mediates Ca2+ extrusion to the extracellular compartment. In higher mammals, the competition between SERCA and NCX shifts in favor of NCX, although SERCA still removes the major proportion of cytosolic Ca2+ (Puglisi et al., 1996). To assure a balanced Ca2+ homeostasis, the exact amount of Ca2+ entering the cell mainly via L-type Ca2+ channels has to be extruded to the extracellular compartment via NCX. Thus, SEA0400-mediated reduction of a comparatively low NCX-mediated Ca2+ removal rate as observed in smaller rodents may more likely result in intracellular Ca2+ accumulation than in higher mammals which might explain the observed different inotropic responses in different studies. However, Ozdemir et al. found a positive inotropic response of SEA0400 in pigs as an example for higher mammals (Ozdemir et al., 2008). But they also found a lower NCX current in pig compared to mice, which might explain the positive inotropy via Ca2+ accumulation.
In contrast to both wild-types and hetKO, cardiomyocytes from OE mice did not exhibit a significant increase in the Ca2+ transient amplitude in response to SEA0400. In OE 1 μM SEA0400 reduced the NCX-mediated Ca2+ removal rate on average to a level ranging between the basal hetKO rate and the rate of both wild-types (Figures 1B,D). A NCX-mediated Ca2+ removal rate as low as in hetKO mice under basal conditions was not associated with a measurable increase of the amplitude of Ca2+ transients as reported previously (Bögeholz et al., 2015). Hence, the remaining NCX function in OE in presence of SEA0400 seems to be still high enough to cope with Ca2+ removal and thus to avoid net gain of Ca2+ and consequently an increase in Ca2+ transient amplitude. In this regard, the inhibitory effect of SEA0400 on L-type Ca2+ current has also to be taken into account (Birinyi et al., 2008). It has been shown that SEA0400 inhibits the L-type Ca2+ current either directly via pleiotropic effects, or indirectly by slowing the NCX-mediated Ca2+ removal leading to an increase of subsarcolemmal Ca2+ concentration which in turn promotes Ca2+ dependent inactivation of L-type Ca2+ current (Pott et al., 2005; Bögeholz et al., 2015).
Interestingly, Ozdemir et al. similarly found absence of a positive inotropic effect in MLP-/- mice, a murine model of heart failure (Ozdemir et al., 2008). Due to the heart failing phenotype NCX upregulation is likely, which may prevent from Ca2+ accumulation and positive inotropy. Taken together within the same species the effect of the same concentration of SEA0400 on calcitropy and thus inotropy seems to depend on the NCX expression level and corresponding function.
In previous studies, SEA0400 exhibited antiarrhythmic efficacy on cellular level by suppressing afterdepolarizations (Nagy et al., 2004; Zhao et al., 2012), on whole heart level in vitro (Milberg et al., 2008, 2012) and even in vivo in a canine model (Nagasawa et al., 2005; Bourgonje et al., 2013). On the other hand, proarrhythmic effects mediated by SEA0400 have been reported as well (Ozdemir et al., 2008). The respective NCX function might be considerably heterogeneous in the investigated animal models, i.e., increased in the case of heart failure, or effectively decreased in the setting of cardiac glycoside-induced proarrhythmia. We here demonstrate that the effects of SEA0400 on cellular proarrhythmia again depend on the underlying baseline NCX function within the same species. Considering NCX inhibition as a promising therapeutic strategy, this might be of translational relevance, since major cardiac diseases like heart failure or atrial fibrillation are accompanied by NCX upregulation, whereas other specific cardiac disease entities, like the postinfarction (Zhang et al., 1996; Quinn et al., 2003) and diabetic cardiomyopathy (Schaffer et al., 1997; Hattori et al., 2000) exhibit a reduced NCX function, at least in animal models. There also might be further specific disease entities with altered NCX function that are yet to be further evaluated (Primessnig et al., 2016).
In OE, application of SEA0400 reduced the occurrence of spontaneous Ca2+ transients. In a former study the increased NCX expression and function in OE has been identified as an independent proarrhythmic factor (Pott et al., 2012). Thus, the observed reduction of arrhythmogenic spontaneous Ca2+ transients in OE can be attributed to the SEA0400-mediated inhibition of NCX function.
The underlying molecular mechanism of SEA0400-mediated decrease in the occurrence of spontaneous Ca2+ transients in OE may consist of reduced NCX forward mode with subsequently reduced depolarizing inward current thus avoiding membrane depolarization and the occurrence of EADs and DADs as proarrhythmic triggers. However, in this regard again the inhibitory potency of SEA0400 on L-type Ca2+ current has to be taken into account (Birinyi et al., 2008; Bourgonje et al., 2013), which might contribute to the observed reduction of proarrhythmic spontaneous Ca2+ transients in OE in presence of SEA0400. Supplementary to these molecular mechanisms of arrhythmia, we have previously demonstrated a significant antiarrhythmic effect of SEA0400 on whole-heart arrhythmia (ventricular tachycardia) in a rabbit model of heart failure (Milberg et al., 2012). SEA0400 reduced the occurrence of EADs due to a reduced monophasic action potential duration, dispersion and QT interval. Thus, the antiarrhythmic effects of SEA0400 may be diverse and apply to diverse functional levels.
In contrast to OE, SEA0400 led to an increased occurrence of spontaneous Ca2+ transients in WTOE, WThetKO, and hetKO. Furthermore, Ca2+ homeostasis seemed to be drastically impaired in hetKO treated with SEA0400 as reflected in the increase percentage of cardiomyocytes with hypercontraction and subsequent cell death as a consequence of cytosolic Ca2+ overload. Based on these observations, it seems likely that a critical level of NCX function must remain after SEA0400 treatment to avoid detrimental intracellular Ca2+ accumulation and overload. While the reduced basal NCX function in hetKO led to a phenotype with reduced likelihood of arrhythmia and a balanced Ca2+ homeostasis as reported previously (Bögeholz et al., 2015), lowering NCX function below this level with SEA0400 was clearly detrimental in hetKO, WThetKO, and WTOE. In this regard, one has to distinguish between acute reduction of NCX function via SEA0400 and chronically reduced NCX function via transgenic manipulation. The latter offers the opportunity for long-term adaption beginning from the early stages of development to compensate for the reduced Ca2+ removal rate mediated via NCX. The most important adaptive mechanism in models with reduced (Bögeholz et al., 2015) or absent NCX function (Pott et al., 2007a,b,c) to keep a balanced Ca2+ homeostasis consists of a reduced Ca2+ entry via the L-type Ca2+ channel as mediated by an enhanced Ca2+-dependent inactivation of the L-type Ca2+ current. Thus, if less Ca2+ enters the cell, the reduced NCX function is still sufficient to keep a balanced Ca2+ homeostasis in a chronically adapted system. In contrast, the lack of time for adaptation to acute NCX inhibition by SEA0400 may contribute to the impaired Ca2+ homeostasis in WT and most prominently in hetKO. However, in OE acute application of SEA0400 was not detrimental but reduced the cellular proarrhythmia through acute NCX inhibition. Thus, depending on the baseline NCX expression level, acute SEA0400 application can cause destabilization of intracellular Ca2+ homeostasis leading to arrhythmia and cell death or mediate antiarrhythmic effects.
In conclusion, our results suggest that within the same species beneficial or detrimental effects mediated by SEA0400 depend upon the basal NCX expression and thus function level. A reduced NCX function level as low as that in hetKO has been shown to be antiarrhythmic (Bögeholz et al., 2015). Reducing an already increased proarrhythmic NCX function level, such as that in OE (Pott et al., 2012), by SEA0400 to the baseline hetKO level reduces proarrhythmia without increasing the amplitude of Ca2+ transients. Reducing the NCX function level below this value increases the amplitude of Ca2+ transients due to SR Ca2+ loading but at the same time mediates cellular proarrhythmia as demonstrated in WThetKO, WTOE and most prominently in hetKO where the remaining NCX function is the lowest. Consistent with this observation, a previous study demonstrated an enhanced susceptibility toward arrhythmia in a ventricular specific homozygous NCX knockout mouse model with absence of NCX expression in 80–90% of ventricular cardiomyocytes (Jordan et al., 2010). Taken together, these findings suggest that NCX inhibition may be beneficial, however there seems to be a critical level of the remaining NCX function that is needed for balanced Ca2+ homeostasis. In case of falling below a certain critical threshold, adverse effects occur due to Ca2+ accumulation promoting proarrhythmia (Table 1). Translated into the clinical context, NCX inhibition may be antiarrhythmic in absence of a negative inotropic effect in conditions of increased NCX expression as present in heart failure or atrial fibrillation. Albeit, in conditions of preexisting reduced NCX function, as observed in animal models of postinfarction or diabetic cardiomyopathy, additional NCX inhibition may potentially exert proarrhythmic effects accompanied by increased inotropy due to a reduction of the remaining NCX function below a critical level that is needed for balancing the cellular Ca2+ dynamics. Thus, at-risk groups should be identified in future large-scaled clinical trials investigating specific NCX inhibitors.
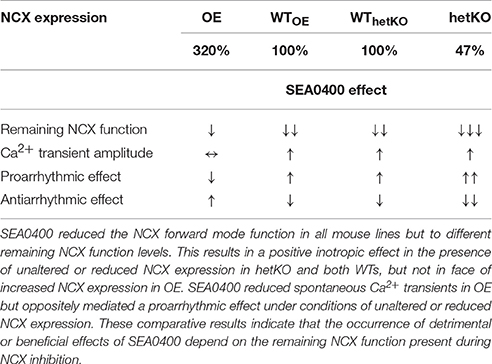
Table 1. Summary of the effects of SEA0400 on Ca2+ handling in dependence on the NCX expression level.
In this study, the Ca2+ transient amplitude served as a proxy measure for inotropy on the cellular level. Though an acute increase of the Ca2+ transient amplitude is usually accompanied by a simultaneous increase in contractility, we did not measure changes in contractility directly. Likewise, although spontaneous Ca2+ releases represent a well-known proarrhythmic substrate, it has to be acknowledged, that clinically relevant whole-heart arrhythmia is a more complex result from a magnitude of involved factors. Thus, future studies should investigate the in vivo-relevance of our findings derived from the cellular level.
Moreover, our findings may not be uncritically transferable into the human (patho-)physiology, since there are substantial differences in cardiac Ca2+ cycling und electrophysiology compared to a murine model. It remains also to be tested whether more specific NCX inhibitors show comparable effects to SEA0400. However, despite these limitations our data suggest that care should be taken when testing NCX inhibitors in clinical studies where we usually do not know the respective NCX expression and function level of individuals, which might determine beneficial or detrimental effects of the treatment approach with an NCX inhibitor.
NB, JS, SK, CP, and FM designed the study. NB, JS, SK, BB, and PP acquired the data. NB, JS, SK, BB, and DD performed the statistical analysis. All authors contributed substantially to the interpretation of the data. NB, JS, and SK wrote the manuscript. BB, PP, DD, GF, JG, UK, LE, CP, and FM critically revised the manuscript. All authors approved the final version of the manuscript.
This work was supported by the Deutsche Stiftung für Herzforschung (F/44/12) to CP, LE, and FM. LE and GF are supported by the Peter-Osypka-Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Adachi-Akahane, S., Lu, L., Li, Z., Frank, J. S., Philipson, K. D., and Morad, M. (1997). Calcium signaling in transgenic mice overexpressing cardiac Na(+)-Ca2+ exchanger. J. Gen. Physiol. 109, 717–729. doi: 10.1085/jgp.109.6.717
Bassani, J. W., Bassani, R. A., and Bers, D. M. (1994). Relaxation in rabbit and rat cardiac cells: species-dependent differences in cellular mechanisms. J. Physiol. 476, 279–293. doi: 10.1113/jphysiol.1994.sp020130
Bers, D. M. (2002). Cardiac Na/Ca exchange function in rabbit, mouse and man: what's the difference? J. Mol. Cell. Cardiol. 34, 369–373. doi: 10.1006/jmcc.2002.1530
Birinyi, P., Acsai, K., Banyasz, T., Toth, A., Horvath, B., Virag, L., et al. (2005). Effects of SEA0400 and KB-R7943 on Na+/Ca2+ exchange current and L-type Ca2+ current in canine ventricular cardiomyocytes. Naunyn Schmiedebergs. Arch. Pharmacol. 372, 63–70. doi: 10.1007/s00210-005-1079-x
Birinyi, P., Toth, A., Jona, I., Acsai, K., Almassy, J., Nagy, N., et al. (2008). The Na+/Ca2+ exchange blocker SEA0400 fails to enhance cytosolic Ca2+ transient and contractility in canine ventricular cardiomyocytes. Cardiovasc. Res. 78, 476–484. doi: 10.1093/cvr/cvn031
Bögeholz, N., Pauls, P., Bauer, B. K., Schulte, J. S., Dechering, D. G., Frommeyer, G., et al. (2015). Suppression of early and late afterdepolarizations by heterozygous knockout of the Na+/Ca2+ exchanger in a murine model. Circ. Arrhythm. Electrophysiol. 8, 1210–1218. doi: 10.1161/CIRCEP.115.002927
Bögeholz, N., Pauls, P., Kaese, S., Schulte, J. S., Lemoine, M. D., Dechering, D. G., et al. (2016). Triggered activity in atrial myocytes is influenced by Na+/Ca2+ exchanger activity in genetically altered mice. J. Mol. Cell. Cardiol. 101, 106–115. doi: 10.1016/j.yjmcc.2016.11.004
Bourgonje, V. J., Vos, M. A., Ozdemir, S., Doisne, N., Acsai, K., Varro, A., et al. (2013). Combined Na(+)/Ca(2+) exchanger and L-type calcium channel block as a potential strategy to suppress arrhythmias and maintain ventricular function. Circ. Arrhythm. Electrophysiol. 6, 371–379. doi: 10.1161/CIRCEP.113.000322
Farkas, A. S., Acsai, K., Nagy, N., Toth, A., Fulop, F., Seprenyi, G., et al. (2008). Na(+)/Ca(2+) exchanger inhibition exerts a positive inotropic effect in the rat heart, but fails to influence the contractility of the rabbit heart. Br. J. Pharmacol. 154, 93–104. doi: 10.1038/bjp.2008.83
Hattori, Y., Matsuda, N., Kimura, J., Ishitani, T., Tamada, A., Gando, S., et al. (2000). Diminished function and expression of the cardiac Na+-Ca2+ exchanger in diabetic rats: implication in Ca2+ overload. J. Physiol. 527(Pt 1), 85–94. doi: 10.1111/j.1469-7793.2000.00085.x
Henderson, S. A., Goldhaber, J. I., So, J. M., Han, T., Motter, C., Ngo, A., et al. (2004). Functional adult myocardium in the absence of Na+-Ca2+ exchange: cardiac-specific knockout of NCX1. Circ. Res. 95, 604–611. doi: 10.1161/01.RES.0000142316.08250.68
Iwamoto, T., Watano, T., and Shigekawa, M. (1996). A novel isothiourea derivative selectively inhibits the reverse mode of Na+/Ca2+ exchange in cells expressing NCX1. J. Biol. Chem. 271, 22391–22397. doi: 10.1074/jbc.271.37.22391
Jordan, M. C., Henderson, S. A., Han, T., Fishbein, M. C., Philipson, K. D., and Roos, K. P. (2010). Myocardial function with reduced expression of the sodium-calcium exchanger. J. Card. Fail. 16, 786–796. doi: 10.1016/j.cardfail.2010.03.012
Li, Z., Nicoll, D. A., Collins, A., Hilgemann, D. W., Filoteo, A. G., Penniston, J. T., et al. (1991). Identification of a peptide inhibitor of the cardiac sarcolemmal Na(+)-Ca2+ exchanger. J. Biol. Chem. 266, 1014–1020.
Menick, D. R., Renaud, L., Buchholz, A., Muller, J. G., Zhou, H., Kappler, C. S., et al. (2007). Regulation of Ncx1 gene expression in the normal and hypertrophic heart. Ann. N. Y. Acad. Sci. 1099, 195–203. doi: 10.1196/annals.1387.058
Milberg, P., Pott, C., Fink, M., Frommeyer, G., Matsuda, T., Baba, A., et al. (2008). Inhibition of the Na+/Ca2+ exchanger suppresses torsades de pointes in an intact heart model of long QT syndrome-2 and long QT syndrome-3. Heart Rhythm 5, 1444–1452. doi: 10.1016/j.hrthm.2008.06.017
Milberg, P., Pott, C., Frommeyer, G., Fink, M., Ruhe, M., Matsuda, T., et al. (2012). Acute inhibition of the Na(+)/Ca(2+) exchanger reduces proarrhythmia in an experimental model of chronic heart failure. Heart Rhythm 9, 570–578. doi: 10.1016/j.hrthm.2011.11.004
Nagasawa, Y., Zhu, B. M., Chen, J., Kamiya, K., Miyamoto, S., and Hashimoto, K. (2005). Effects of SEA0400, a Na+/Ca2+ exchange inhibitor, on ventricular arrhythmias in the in vivo dogs. Eur. J. Pharmacol. 506, 249–255. doi: 10.1016/j.ejphar.2004.11.011
Nagy, N., Kormos, A., Kohajda, Z., Szebeni, A., Szepesi, J., Pollesello, P., et al. (2014). Selective Na(+) /Ca(2+) exchanger inhibition prevents Ca(2+) overload-induced triggered arrhythmias. Br. J. Pharmacol. 171, 5665–5681. doi: 10.1111/bph.12867
Nagy, Z. A., Virag, L., Toth, A., Biliczki, P., Acsai, K., Banyasz, T., et al. (2004). Selective inhibition of sodium-calcium exchanger by SEA-0400 decreases early and delayed after depolarization in canine heart. Br. J. Pharmacol. 143, 827–831. doi: 10.1038/sj.bjp.0706026
Namekata, I., Tsuneoka, Y., Takahara, A., Shimada, H., Sugimoto, T., Takeda, K., et al. (2009). Involvement of the Na(+)/Ca(2+) exchanger in the automaticity of guinea-pig pulmonary vein myocardium as revealed by SEA0400. J. Pharmacol. Sci. 110, 111–116. doi: 10.1254/jphs.08159SC
O'Neill, S. C., Donoso, P., and Eisner, D. A. (1990). The role of [Ca2+]i and [Ca2+] sensitization in the caffeine contracture of rat myocytes: measurement of [Ca2+]i and [caffeine]i. J. Physiol. 425, 55–70. doi: 10.1113/jphysiol.1990.sp018092
Ozdemir, S., Bito, V., Holemans, P., Vinet, L., Mercadier, J. J., Varro, A., et al. (2008). Pharmacological inhibition of na/ca exchange results in increased cellular Ca2+ load attributable to the predominance of forward mode block. Circ. Res. 102, 1398–1405. doi: 10.1161/CIRCRESAHA.108.173922
Pott, C., Henderson, S. A., Goldhaber, J. I., and Philipson, K. D. (2007a). Na+/Ca2+ exchanger knockout mice: plasticity of cardiac excitation-contraction coupling. Ann. N. Y. Acad. Sci. 1099, 270–275. doi: 10.1196/annals.1387.015
Pott, C., Muszynski, A., Ruhe, M., Bogeholz, N., Schulte, J. S., Milberg, P., et al. (2012). Proarrhythmia in a non-failing murine model of cardiac-specific Na+/Ca2+ exchanger overexpression: whole heart and cellular mechanisms. Basic Res. Cardiol. 107, 247. doi: 10.1007/s00395-012-0247-7
Pott, C., Philipson, K. D., and Goldhaber, J. I. (2005). Excitation-contraction coupling in Na+-Ca2+ exchanger knockout mice: reduced transsarcolemmal Ca2+ flux. Circ. Res. 97, 1288–1295. doi: 10.1161/01.RES.0000196563.84231.21
Pott, C., Ren, X., Tran, D. X., Yang, M. J., Henderson, S., Jordan, M. C., et al. (2007b). Mechanism of shortened action potential duration in Na+-Ca2+ exchanger knockout mice. Am. J. Physiol. Cell Physiol. 292, C968–C973. doi: 10.1152/ajpcell.00177.2006
Pott, C., Yip, M., Goldhaber, J. I., and Philipson, K. D. (2007c). Regulation of cardiac L-type Ca2+ current in Na+-Ca2+ exchanger knockout mice: functional coupling of the Ca2+ channel and the Na+-Ca2+ exchanger. Biophys. J. 92, 1431–1437. doi: 10.1529/biophysj.106.091538
Primessnig, U., Schonleitner, P., Holl, A., Pfeiffer, S., Bracic, T., Rau, T., et al. (2016). Novel pathomechanisms of cardiomyocyte dysfunction in a model of heart failure with preserved ejection fraction. Eur. J. Heart Fail. 18, 987–997. doi: 10.1002/ejhf.524
Priori, S. G., Blomstrom-Lundqvist, C., Mazzanti, A., Blom, N., Borggrefe, M., Camm, J., et al. (2015). 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of Cardiology (ESC)endorsed by: association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 36, 2793–2867. doi: 10.1093/eurheartj/ehv316
Puglisi, J. L., Bassani, R. A., Bassani, J. W., Amin, J. N., and Bers, D. M. (1996). Temperature and relative contributions of Ca transport systems in cardiac myocyte relaxation. Am. J. Physiol. 270, H1772–1778.
Quinn, F. R., Currie, S., Duncan, A. M., Miller, S., Sayeed, R., Cobbe, S. M., et al. (2003). Myocardial infarction causes increased expression but decreased activity of the myocardial Na+-Ca2+ exchanger in the rabbit. J. Physiol. 553, 229–242. doi: 10.1113/jphysiol.2003.050716
Reuter, H., Henderson, S. A., Han, T., Matsuda, T., Baba, A., Ross, R. S., et al. (2002). Knockout mice for pharmacological screening: testing the specificity of Na+-Ca2+ exchange inhibitors. Circ. Res. 91, 90–92. doi: 10.1161/01.RES.0000027529.37429.38
Schaffer, S. W., Ballard-Croft, C., Boerth, S., and Allo, S. N. (1997). Mechanisms underlying depressed Na+/Ca2+ exchanger activity in the diabetic heart. Cardiovasc. Res. 34, 129–136. doi: 10.1016/S0008-6363(97)00020-5
Schulte, J. S., Seidl, M. D., Nunes, F., Freese, C., Schneider, M., Schmitz, W., et al. (2012). CREB critically regulates action potential shape and duration in the adult mouse ventricle. Am. J. Physiol. Heart Circ. Physiol. 302, H1998–H2007. doi: 10.1152/ajpheart.00057.2011
Sipido, K. R., Varro, A., and Eisner, D. (2006). Sodium calcium exchange as a target for antiarrhythmic therapy. Handb. Exp. Pharmacol. 171, 159–199. doi: 10.1007/3-540-29715-4_6
Tanaka, H., Namekata, I., Takeda, K., Kazama, A., Shimizu, Y., Moriwaki, R., et al. (2005). Unique excitation-contraction characteristics of mouse myocardium as revealed by SEA0400, a specific inhibitor of Na+-Ca2+ exchanger. Naunyn Schmiedebergs. Arch. Pharmacol. 371, 526–534. doi: 10.1007/s00210-005-1051-9
Tanaka, H., Shimada, H., Namekata, I., Kawanishi, T., Iida-Tanaka, N., and Shigenobu, K. (2007). Involvement of the Na+/Ca2+ exchanger in ouabain-induced inotropy and arrhythmogenesis in guinea-pig myocardium as revealed by SEA0400. J. Pharmacol. Sci. 103, 241–246. doi: 10.1254/jphs.FP0060911
Voigt, N., Li, N., Wang, Q., Wang, W., Trafford, A. W., Abu-Taha, I., et al. (2012). Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 125, 2059–2070. doi: 10.1161/CIRCULATIONAHA.111.067306
Zhang, X. Q., Tillotson, D. L., Moore, R. L., Zelis, R., and Cheung, J. Y. (1996). Na+/Ca2+ exchange currents and SR Ca2+ contents in postinfarction myocytes. Am. J. Physiol. 271, C1800–C1807.
Zhao, S. M., Wang, Y. L., Guo, C. Y., Chen, J. L., and Wu, Y. Q. (2014). Progressive decay of Ca2+ homeostasis in the development of diabetic cardiomyopathy. Cardiovasc. Diabetol. 13:75. doi: 10.1186/1475-2840-13-75
Keywords: Na+/Ca2+ exchanger, arrhythmia, heart failure, novel antiarrhythmic strategies, Ca2+ cycling, transgenic mice
Citation: Bögeholz N, Schulte JS, Kaese S, Bauer BK, Pauls P, Dechering DG, Frommeyer G, Goldhaber JI, Kirchhefer U, Eckardt L, Pott C and Müller FU (2017) The Effects of SEA0400 on Ca2+ Transient Amplitude and Proarrhythmia Depend on the Na+/Ca2+ Exchanger Expression Level in Murine Models. Front. Pharmacol. 8:649. doi: 10.3389/fphar.2017.00649
Received: 20 June 2017; Accepted: 01 September 2017;
Published: 21 September 2017.
Edited by:
Issy Laher, University of British Columbia, CanadaReviewed by:
Nazareno Paolocci, Johns Hopkins University, United StatesCopyright © 2017 Bögeholz, Schulte, Kaese, Bauer, Pauls, Dechering, Frommeyer, Goldhaber, Kirchhefer, Eckardt, Pott and Müller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan S. Schulte, ai5zLnNjaHVsdGVAdW5pLW11ZW5zdGVyLmRl
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.