 Yan Wang1
Yan Wang1 Mark Del Borgo2Huey W. Lee1Dhaniel Baraldi1Baydaa Hirmiz2
Mark Del Borgo2Huey W. Lee1Dhaniel Baraldi1Baydaa Hirmiz2 Tracey A. Gaspari1
Tracey A. Gaspari1 Kate M. Denton3
Kate M. Denton3 Marie-Isabel Aguilar2
Marie-Isabel Aguilar2 Chrishan S. Samuel1
Chrishan S. Samuel1 Robert E. Widdop1*
Robert E. Widdop1*- 1Department of Pharmacology, Cardiovascular Disease Program, Biomedicine Discovery Institute, Monash University, Clayton, VIC, Australia
- 2Department of Biochemistry and Molecular Biology, Cardiovascular Disease Program, Biomedicine Discovery Institute, Monash University, Clayton, VIC, Australia
- 3Department of Physiology, Cardiovascular Disease Program, Biomedicine Discovery Institute, Monash University, Clayton, VIC, Australia
There are a number of therapeutic targets to treat organ fibrosis that are under investigation in preclinical models. There is increasing evidence that stimulation of the angiotensin II type 2 receptor (AT2R) is a novel anti-fibrotic strategy and we have reviewed the published in vivo preclinical data relating to the effects of compound 21 (C21), which is the only nonpeptide AT2R agonist that is currently available for use in chronic preclinical studies. In particular, the differential influence of AT2R on extracellular matrix status in various preclinical fibrotic models is discussed. Collectively, these studies demonstrate that pharmacological AT2R stimulation using C21 decreases organ fibrosis, which has been most studied in the setting of cardiovascular and renal disease. In addition, AT2R-mediated anti-inflammatory effects may contribute to the beneficial AT2R-mediated anti-fibrotic effects seen in preclinical models.
Introduction
The renin angiotensin system (RAS) is one of the most important systems to regulate hemodynamics, blood pressure, and tissue remodeling processes. The RAS is a circulating as well as a local hormonal system (Campbell, 1987) such that local generation of angiotensin occurs in many tissues including the brain, heart, kidney, and vasculature (Te Riet et al., 2015). Various components of circulating and tissue RAS that are important for regulating vascular and cardiac contractility, fluid and electrolyte homeostasis, as well as extracellular matrix (ECM) production have been reviewed elsewhere (Campbell, 1987; Te Riet et al., 2015).
There are two major subtypes of angiotensin receptors, the angiotensin II subtype 1 receptor (AT1R) and angiotensin II subtype 2 receptor (AT2R) (Herichova and Szantoova, 2013; Karnik et al., 2015). It is well established that activation of AT1R by angiotensin II (Ang II) mediates pathophysiological effects such as vasoconstriction, proliferation, fibrosis, oxidative stress, and inflammation (Sadoshima and Izumo, 1993; Ferrario and Strawn, 2006; Whaley-Connell et al., 2013), which occurs in multiple organs including heart, kidney, liver, lungs, vascular smooth muscle, and brain (Mehta and Griendling, 2007; Karnik et al., 2015). On the other hand, activation of AT2R is thought to counter-regulate the pathophysiological effects induced by AT1R and exert vasodilator, anti-fibrotic, anti-proliferative, and anti-inflammatory effects (Widdop et al., 2003; Jones et al., 2008) as well as natriuretic and antihypertensive effects in renal disease (Carey, 2017). The AT2R is also very topical in the context of neuropathic pain as it has recently been reported that an old AT2R antagonist has been repurposed to treat neuropathic pain. For general interest, an historical account of this recent discovery is also noted (see Keppel Hesselink and Schatman, 2017). However, the current review will focus on recent evidence for an anti-fibrotic effect due to the pharmacological stimulation of AT2R in the context of cardiovascular disease.
Cardiovascular Disease (CVD)
Cardiovascular disease is the leading cause of morbidity and mortality globally (Moran et al., 2014). While progress is being made in addressing CVD risk factors such as high blood pressure, diabetes, obesity, and high cholesterol (Moran et al., 2014), less therapeutic intervention has been directed at some of the underlying pathological changes occurring in relevant organs. In particular, the ECM is now considered an important site for therapeutic intervention (Rockey et al., 2015).
In the heart, for example, hypertensive heart disease is characterized by myocardial ECM expansion due to excess collagen accumulation (Weber, 1989). Injurious stimuli such as myocardial inflammation, cardiac overload, or cardiomyocyte death may activate pro-fibrotic pathways. Several cell types are involved in this process directly by producing matrix proteins (fibroblasts) or indirectly by secreting fibrogenic mediators (macrophages, mast cells, lymphocytes, cardiomyocytes) that in turn promote fibroblast-mediated ECM production. Transforming growth factor (TGF)-β1 is considered the main pro-fibrogenic mediator and promotes the transdifferentiation of fibroblasts into myofibroblasts that contribute to myofibroblast-mediated collagen synthesis leading to excess collagen complex deposition in the ECM (Tomasek et al., 2002; Rockey et al., 2015). This is one of the key cellular events that drive cardiac fibrosis. The accumulation of collagen (scar tissue) replaces cardiomyocytes that leads to the loss of structural integrity of the myocardium (Weber, 1989; Weber et al., 2013). The distinction between reactive interstititial fibrosis and reparative fibrosis, as occurs following myocardial infraction (MI), is not always well defined (Schelbert et al., 2014). In any case, the consequences of ECM expansion such as increased myocardial collagen deposition in patients results in heart dysfunction (Anderson et al., 1993; Brilla et al., 2000; Diez et al., 2002; Weber et al., 2013; Schelbert et al., 2014). Indeed, it has been estimated that fibrotic diseases contributed to about 45% of mortality in Western countries and may be higher in developing countries (Rosenbloom et al., 2013).
Targeting RAS as Therapeutic Treatment for CVDs
Angiotensin II subtype 1 receptor blockers (ARBs) and angiotensin converting enzyme (ACE) inhibitors (ACEi) are effective treatments for hypertension based on the concept of blocking the AngII–AT1R-axis mediated pathological effects (Karnik et al., 2015). Both treatments are effective in hypertensive patients and their antihypertensive effects appear equivalent (Vijan, 2009; Bavishi et al., 2016), although both ARBs and ACEi exhibit only limited capacity to improve cardiovascular outcome in hypertensive patients beyond blood pressure reductions (van Vark et al., 2012; Bavishi et al., 2016). By contrast, anti-fibrotic effects of ACEi and ARBs were clearly demonstrated in tissue biopsies in small well-controlled trials (Brilla et al., 2000; Diez et al., 2002; Querejeta et al., 2004), which were designed to measure cardiac ECM status (although this is clearly not possible in large outcome trials). Therefore, it was not surprising that many studies subsequently combined ACEi and ARBs (dual RAS inhibition) in the hope that this strategy would maximize any potential cardiovascular remodeling (such as fibrosis reduction) to improve clinical outcomes. However, the impact was in fact the opposite: there was an increased risk of adverse renal events such as hyperkalemia and acute renal failure together with symptomatic hypotension (Yusuf et al., 2008; Messerli et al., 2010; Makani et al., 2013). Indeed, dual RAS inhibition is now contraindicated in most cardiovascular guidelines. Clearly, novel treatments are needed that can exert anti-fibrotic effects alone or in combination with individual RAS inhibitors.
At2R Knock Out and Over-Expression Studies
Initially, there were conflicting reports on the anti-fibrotic effects of AT2R deletion on cardiac remodeling evoked by pressure overload, Ang II infusion, or myocardial infarction (MI) that were most likely due to the background mouse strains [see Widdop et al. (2003) for review]. Generally, there is strong evidence demonstrating the protective role of AT2R activation since AT2R knock out mice exhibited enhanced cardiac perivascular (Akishita et al., 2000), renal (Ma et al., 1998; Chow et al., 2014), and liver (Nabeshima et al., 2006) fibrosis following pro-fibrotic stimuli. Furthermore, cardiac overexpression of AT2R was protective against Ang II-induced fibrosis (Kurisu et al., 2003), cardiac hypertrophy in spontaneously hypertensive rats (Metcalfe et al., 2004), and during post-infarct remodeling (Yang et al., 2002; Bove et al., 2004; Isbell et al., 2007; Qi et al., 2012). While detrimental effects of cardiac AT2R overexpression have been reported (Nakayama et al., 2005), recent evidence suggests that there is an optimal AT2R transgene copy number required to protect against MI-induced cardiac hypertrophy and fibrosis (Xu et al., 2014).
Direct Pharmacological At2R Stimulation is Anti-Fibrotic
The development of the selective nonpeptide AT2R agonist compound 21 (C21) (Wan et al., 2004) provided another approach for the understanding of AT2R function. Compound 21 is highly AT2R-selective (Wan et al., 2004; Bosnyak et al., 2011) although some off target effects such as interference with cellular calcium transport have been reported (Verdonk et al., 2012), albeit at concentrations orders of magnitude greater than its AT2R binding affinity, as we have previously discussed (McCarthy et al., 2013). Therefore, C21 can generally be considered as a selective AT2R agonist and has been used in this context by many research groups. In a seminal study, Kaschina et al. (2008) reported that following 7 days of treatment with C21, the scarring associated with post-MI remodeling was reduced, which correlated with significantly improved cardiac function (Kaschina et al., 2008). In addition, Gelosa et al. (2009) reported that chronic treatment with C21 reduced kidney inflammation and fibrosis in stroke-prone spontaneously hypertensive rats (SHRSP), although the main focus of this study was on stroke protection (Gelosa et al., 2009). Subsequently, there have been a handful of studies published that clearly show that AT2R stimulation using C21 exerts anti-fibrotic effects in hearts of SHRSP (Rehman et al., 2012), following chronic MI in rats (Lauer et al., 2014), in vasculature of rats treated with the nitric oxide synthase inhibitor L-NAME (Paulis et al., 2012), and in lungs during pulmonary hypertension (Bruce et al., 2015). While C21 is generally considered to be AT2R selective (Bosnyak et al., 2011), not all these studies used the AT2R antagonist to confirm an AT2R effect. In addition, renal anti-fibrotic effects of C21 have also been reported in kidneys insulted by doxorubicin (Hrenak et al., 2013) or different forms of diabetic nephropathy (Castoldi et al., 2014; Koulis et al., 2015). Details of all aforementioned studies are provided in Table 1. Collectively, these studies document a protective role of the C21–AT2R axis against organ fibrosis. Intriguingly, it was recently reported that the AT2R may form heterodimers with other class A G-protein-coupled receptors, such as relaxin family peptide receptor (RXFP1) to regulate fibrosis progression, as the anti-fibrotic effects of relaxin in the kidney were actually prevented by genetic or pharmacological inhibition of AT2Rs (Chow et al., 2014).

TABLE 1. Summary of anti-fibrotic and related protective effects evoked by chronic treatment with C21.
Potential Anti-Fibrotic Mechanisms of At2R
A number of anti-fibrotic mechanisms are likely to be associated with the changes evoked by C21 (Table 1). The activation of the pro-inflammatory nuclear factor-κBα (NFκB) pathway is a central transcriptional effector of inflammatory signaling. Nuclear factor-κB activation triggers gene transcription of many inflammatory cytokines, chemokines, and vascular adhesion molecules such as TNF-α, IL-1β, and IL-6 in fibrotic hearts (Torre-Amione et al., 1996; Francis et al., 1998; Plenz et al., 1998).
Rompe et al. (2010) were the first to show that C21 could exert a direct anti-inflammatory effect as C21 inhibited NFκB activation leading to reduced TNF-α-mediated IL-6 release from human dermal fibroblasts. The anti-fibrotic effect caused by C21 was consistently associated with reduced inflammatory responses and inflammatory cell infiltration in a variety of animal models/organs (Table 1) and in other studies not directly assessing fibrosis (Matavelli et al., 2011, 2015; Sampson et al., 2016). In particular, C21-mediated renal anti-inflammatory effects occurred within 4 days in hypertensive rats (Matavelli et al., 2011) and modestly protected against diabetic nephropathy in a short-term (4 week) model in rats (Matavelli et al., 2015) whereas C21 consistently evoked renal anti-inflammatory and anti-fibrotic effects in a longer term model of diabetic nephropathy in rats (Castoldi et al., 2014) and mice (Koulis et al., 2015). Taken together, these studies suggest that C21 inhibits inflammatory responses during the development of fibrosis via activation of AT2R.
TGF-β1 is a major pro-fibrotic factor that plays a key role in the development of tissue fibrosis (Lijnen et al., 2000). TGF-β stimulates fibroblasts to differentiate into pro-secretory myofibroblasts that in turn enhance ECM protein synthesis (Desmouliere et al., 1993; Tomasek et al., 2002; Berk et al., 2007). At the same time, matrix metalloproteinases (MMPs) degrade ECM proteins and this process is tightly controlled by tissue inhibitors of metalloproteinases (TIMPs) (Weber et al., 2013). However, in an injured organ, TGF-β1 upregulates the expression of protease inhibitors such as plasminogen activator inhibitor (PAI)-1 and TIMPs which contribute to ECM preservation (Schiller et al., 2004).
Given that macrophages are a source of TGF-β1, the inhibition of macrophage infiltration via AT2R activation could contribute to reduced TGF-β1 stimulation of fibrotic pathways. In addition, direct stimulation of AT2R is well known to increase nitric oxide and cyclic guanosine monophosphate (cGMP) levels, particularly in the kidneys (Siragy and Carey, 1996, 1997) and vasculature (see Widdop et al., 2003), noting that decreased cGMP levels following AT2R stimulation have also been reported (Karnik et al., 2015). Importantly, in vivo treatment with C21 increased NO/cGMP levels in kidneys (Matavelli et al., 2011, 2015) in keeping with a predominant AT2R-cGMP stimulatory effect. Interestingly, cGMP was reported to inhibit TGF-β signaling (Gong et al., 2011), thereby providing another mechanism for AT2R stimulation to modify fibrosis production. Indeed, a number of the anti-fibrotic effects of C21 already described were associated with marked reductions in TGF-β1 in heart (Lauer et al., 2014), lung (Bruce et al., 2015), and kidney (Matavelli et al., 2011; Koulis et al., 2015), suggesting that the inhibition of the TGF-β1 cascade is a common mechanism of the anti-fibrotic effect caused by AT2R activation. As TGF-β1 acutely increased AT2R expression in skeletal muscle (Painemal et al., 2013), it is possible that a similar compensatory response to cardiovascular injury contributes to increased AT2R expression in CVD, although the role of such interactions on AT2R expression during chronic AT2R stimulation is not known.
In terms of collagen metabolism affecting ECM turnover, the effect of AT2R activation on collagen degradation and the regulation of the MMP/TIMP balance is likely to depend on the experimental conditions studied, such as whether the main driver for fibrosis is reparative (in the case of MI) or persistent reactive fibrosis (in the case of hypertensive heart disease). Associated with the anti-fibrotic effect of C21, MMP2/9 levels were either unchanged in SHRSP hearts (Rehman et al., 2012), increased in diabetic murine kidneys (Koulis et al., 2015), or decreased in MI-injured rat hearts (Lauer et al., 2014). These discrepant results are likely to reflect the different requirements of ECM in such models. For example, following MI, cardiac TGF-β1 and MMP levels were elevated whereas cardiac TIMP levels were reduced (Lauer et al., 2014). These somewhat opposing changes caused by MI itself, i.e., pro-fibrotic TGF-β1 activity together with increased proteolytic activity seen by raised MMP-9/TIMP-1 ratio, reflects the need to repair and remodel the heart following MI. In this instance, C21 appears to protect the heart by reducing widespread collagen production (decreased TGF-β1) and attenuating volume expansion (decreased MMP-9/TIMP-1 ratio). By contrast, the ability of C21 to reduce fibrosis in persistent reactive fibrotic models of CVD probably reflects both impaired collagen production (decreased TGF-β1 and collagen), as well as increased degradation due to raised MMP levels (Koulis et al., 2015), which is clearly different to abruptly developing MI-induced cardiac remodeling (Figure 1).
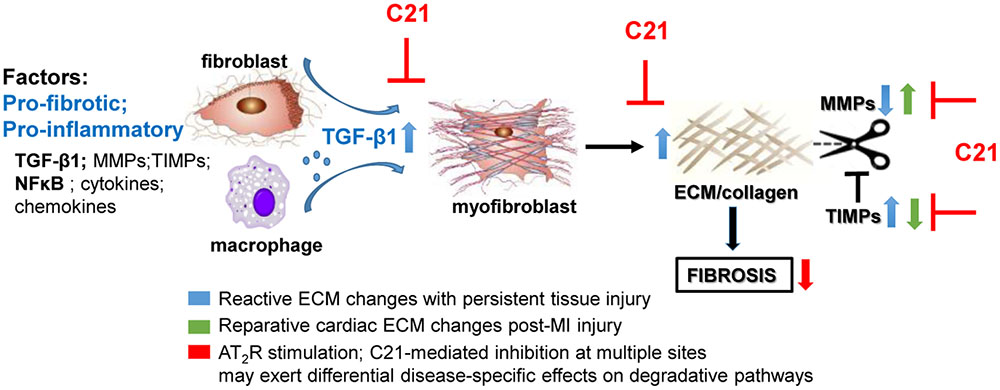
FIGURE 1. Potential mechanisms involved in the anti-fibrotic actions of AT2R stimulation based on the effects of C21 (inhibitory sites in red). AT2R stimulation consistently reduces inflammatory and pro-fibrotic factors such as TGF-β1 thereby inhibiting myofibroblast differentiation and ECM production. However, the effects of AT2R stimulation on ECM turnover may differ depending on the type of fibrosis/disease model studied. C21 inhibited the proteolytic left ventricular expansion associated with MI-induced injury (green arrows) whereas AT2R stimulation is more likely to inhibit ECM preservation (blue arrows) associated with persistent injury (e.g., hypertension), thus facilitating ECM degradation.
Conclusion and Future Directions
Collectively, these studies demonstrate that pharmacological AT2R stimulation evokes decreases in organ fibrosis, most studied in the heart and kidneys to date. The effects of C21 on cardiac ECM remodeling may differ depending on the preclinical fibrotic model studied (Figure 1), which is likely to reflect the prevailing circumstances in response to injury, i.e., replacement fibrosis following MI versus persistent reactive interstitial fibrosis seen in hypertensive heart disease. However, AT2R stimulation also usually involves an anti-inflammatory effect that may contribute to the beneficial AT2R-mediated anti-fibrotic effects. Most data related to chronic AT2R stimulation have been obtained using C21, although there are a number of other AT2R agonists beginning to emerge in the literature (Jones et al., 2011; Guimond et al., 2014; Del Borgo et al., 2015; Mahmood and Pulakat, 2015) that require rigourous in vivo testing in a similar manner to C21. Such studies will shed further light on the clinical potential of AT2R agonists in CVD.
Author Contributions
RW and CS conceived the review; YW wrote the first draft; MDB and DB provided literature searches and contributed to draft. YW, HL, BH, TG, and RW contributed and performed experiments in Figure 1. RW, CS, M-IA, and KD did major revisions to the draft manuscript and approved final submission.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work from the authors’ laboratories was supported in part by grants from the National Health and Medical Research Council (NHMRC) of Australia (GNT1045848, GNT1101552, and GNT1127792), and NHMRC Senior Research Fellowships to KD (GNT1041844) and CS (GNT1041766).
References
Akishita, M., Iwai, M., Wu, L., Zhang, L., Ouchi, Y., Dzau, V. J., et al. (2000). Inhibitory effect of angiotensin II type 2 receptor on coronary arterial remodeling after aortic banding in mice. Circulation 102, 1684–1689. doi: 10.1161/01.CIR.102.14.1684
Anderson, K. P., Walker, R., Urie, P., Ershler, P. R., Lux, R. L., and Karwandee, S. V. (1993). Myocardial electrical propagation in patients with idiopathic dilated cardiomyopathy. J. Clin. Invest. 92, 122–140. doi: 10.1172/JCI116540
Bavishi, C., Bangalore, S., and Messerli, F. H. (2016). Renin angiotensin aldosterone system inhibitors in hypertension: is there evidence for benefit independent of blood pressure reduction? Prog. Cardiovasc. Dis. 59, 253–261. doi: 10.1016/j.pcad.2016.10.002
Berk, B. C., Fujiwara, K., and Lehoux, S. (2007). ECM remodeling in hypertensive heart disease. J. Clin. Invest. 117, 568–575. doi: 10.1172/JCI31044
Bosnyak, S., Jones, E. S., Christopoulos, A., Aguilar, M. I., Thomas, W. G., and Widdop, R. E. (2011). Relative affinity of angiotensin peptides and novel ligands at AT1 and AT2 receptors. Clin. Sci. 121, 297–303. doi: 10.1042/CS20110036
Bove, C. M., Yang, Z., Gilson, W. D., Epstein, F. H., French, B. A., Berr, S. S., et al. (2004). Nitric oxide mediates benefits of angiotensin II type 2 receptor overexpression during post-infarct remodeling. Hypertension 43, 680–685. doi: 10.1161/01.HYP.0000115924.94236.91
Brilla, C. G., Funck, R. C., and Rupp, H. (2000). Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation 102, 1388–1393. doi: 10.1161/01.CIR.102.12.1388
Bruce, E., Shenoy, V., Rathinasabapathy, A., Espejo, A., Horowitz, A., Oswalt, A., et al. (2015). Selective activation of angiotensin AT2 receptors attenuates progression of pulmonary hypertension and inhibits cardiopulmonary fibrosis. Br. J. Pharmacol. 172, 2219–2231. doi: 10.1111/bph.13044
Campbell, D. J. (1987). Circulating and tissue angiotensin systems. J. Clin. Invest. 79, 1–6. doi: 10.1172/JCI112768
Carey, R. M. (2017). Update on angiotensin AT2 receptors. Curr. Opin. Nephrol. Hypertens 26, 91–96. doi: 10.1097/MNH.0000000000000304
Castoldi, G., di Gioia, C. R., Bombardi, C., Maestroni, S., Carletti, R., Steckelings, U. M., et al. (2014). Prevention of diabetic nephropathy by compound 21, selective agonist of angiotensin type 2 receptors, in Zucker diabetic fatty rats. Am. J. Physiol. Renal Physiol. 307, F1123–F1131. doi: 10.1152/ajprenal.00247.2014
Chow, B. S., Kocan, M., Bosnyak, S., Sarwar, M., Wigg, B., Jones, E. S., et al. (2014). Relaxin requires the angiotensin II type 2 receptor to abrogate renal interstitial fibrosis. Kidney Int. 86, 75–85. doi: 10.1038/ki.2013.518
Del Borgo, M. P., Wang, Y., Bosnyak, S., Khan, M., Walters, P., Spizzo, I., et al. (2015). β-Pro7Ang III is a novel highly selective angiotensin II type 2 receptor (AT2R) agonist, which acts as a vasodepressor agent via the AT2R in conscious spontaneously hypertensive rats. Clin. Sci. 129, 505–513. doi: 10.1042/CS20150077
Desmouliere, A., Geinoz, A., Gabbiani, F., and Gabbiani, G. (1993). Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 122, 103–111. doi: 10.1083/jcb.122.1.103
Diez, J., Querejeta, R., Lopez, B., Gonzalez, A., Larman, M., and Martinez Ubago, J. L. (2002). Losartan-dependent regression of myocardial fibrosis is associated with reduction of left ventricular chamber stiffness in hypertensive patients. Circulation 105, 2512–2517. doi: 10.1161/01.CIR.0000017264.66561.3D
Ferrario, C. M., and Strawn, W. B. (2006). Role of the renin-angiotensin-aldosterone system and proinflammatory mediators in cardiovascular disease. Am. J. Cardiol. 98, 121–128. doi: 10.1016/j.amjcard.2006.01.059
Francis, S. E., Holden, H., Holt, C. M., and Duff, G. W. (1998). Interleukin-1 in myocardium and coronary arteries of patients with dilated cardiomyopathy. J. Mol. Cell Cardiol. 30, 215–223. doi: 10.1006/jmcc.1997.0592
Gelosa, P., Pignieri, A., Fandriks, L., de Gasparo, M., Hallberg, A., Banfi, C., et al. (2009). Stimulation of AT2 receptor exerts beneficial effects in stroke-prone rats: focus on renal damage. J. Hypertens. 27, 2444–2451. doi: 10.1097/HJH.0b013e3283311ba1
Gong, K., Xing, D., Li, P., Hilgers, R. H., Hage, F. G., Oparil, S., et al. (2011). cGMP inhibits TGF-beta signaling by sequestering Smad3 with cytosolic beta2-tubulin in pulmonary artery smooth muscle cells. Mol. Endocrinol. 25, 1794–1803. doi: 10.1210/me.2011-1009
Guimond, M. O., Hallberg, M., Gallo-Payet, N., and Wallinder, C. (2014). Saralasin and sarile are AT2 receptor agonists. ACS Med. Chem. Lett. 5, 1129–1132. doi: 10.1021/ml500278g
Herichova, I., and Szantoova, K. (2013). Renin-angiotensin system: upgrade of recent knowledge and perspectives. Endocr. Regul. 47, 39–52. doi: 10.4149/endo_2013_01_39
Hrenak, J., Arendasova, K., Rajkovicova, R., Aziriova, S., Repova, K., Krajcirovicova, K., et al. (2013). Protective effect of captopril, olmesartan, melatonin and compound 21 on doxorubicin-induced nephrotoxicity in rats. Physiol. Res. 62(Suppl. 1), S181–S189.
Isbell, D. C., Voros, S., Yang, Z., DiMaria, J. M., Berr, S. S., French, B. A., et al. (2007). Interaction between bradykinin subtype 2 and angiotensin II type 2 receptors during post-MI left ventricular remodeling. Am. J. Physiol. Heart Circ. Physiol. 293, H3372–H3378. doi: 10.1152/ajpheart.00997.2007
Jones, E. S., Del Borgo, M. P., Kirsch, J. F., Clayton, D., Bosnyak, S., Welungoda, I., et al. (2011). A single β-amino acid substitution to angiotensin II confers AT2 receptor selectivity and vascular function. Hypertension 57, 570–576. doi: 10.1161/HYPERTENSIONAHA.110.164301
Jones, E. S., Vinh, A., McCarthy, C. A., Gaspari, T. A., and Widdop, R. E. (2008). AT2 receptors: functional relevance in cardiovascular disease. Pharmacol. Ther. 120, 292–316. doi: 10.1016/j.pharmthera.2008.08.009
Karnik, S. S., Unal, H., Kemp, J. R., Tirupula, K. C., Eguchi, S., Vanderheyden, P. M., et al. (2015). International union of basic and clinical pharmacology. XCIX. Angiotensin receptors: interpreters of pathophysiological angiotensinergic stimuli [corrected]. Pharmacol. Rev. 67, 754–819. doi: 10.1124/pr.114.010454
Kaschina, E., Grzesiak, A., Li, J., Foryst-Ludwig, A., Timm, M., Rompe, F., et al. (2008). Angiotensin II type 2 receptor stimulation: a novel option of therapeutic interference with the renin-angiotensin system in myocardial infarction? Circulation 118, 2523–2532. doi: 10.1161/CIRCULATIONAHA.108.784868
Keppel Hesselink, J. M., and Schatman, M. E. (2017). EMA401: an old antagonist of the AT2R for a new indication in neuropathic pain. J. Pain Res. 10, 439–443. doi: 10.2147/JPR.S128520
Koulis, C., Chow, B. S., McKelvey, M., Steckelings, U. M., Unger, T., Thallas-Bonke, V., et al. (2015). AT2R agonist, compound 21, is reno-protective against type 1 diabetic nephropathy. Hypertension 65, 1073–1081. doi: 10.1161/HYPERTENSIONAHA.115.05204
Kurisu, S., Ozono, R., Oshima, T., Kambe, M., Ishida, T., Sugino, H., et al. (2003). Cardiac angiotensin II type 2 receptor activates the kinin/NO system and inhibits fibrosis. Hypertension 41, 99–107. doi: 10.1161/01.HYP.0000050101.90932.14
Lauer, D., Slavic, S., Sommerfeld, M., Thone-Reineke, C., Sharkovska, Y., Hallberg, A., et al. (2014). Angiotensin type 2 receptor stimulation ameliorates left ventricular fibrosis and dysfunction via regulation of tissue inhibitor of matrix metalloproteinase 1/matrix metalloproteinase 9 axis and transforming growth factor beta1 in the rat heart. Hypertension 63, e60–e67. doi: 10.1161/HYPERTENSIONAHA.113.02522
Lijnen, P. J., Petrov, V. V., and Fagard, R. H. (2000). Induction of cardiac fibrosis by transforming growth factor-beta(1). Mol. Genet. Metab. 71, 418–435. doi: 10.1006/mgme.2000.3032
Ma, J., Nishimura, H., Fogo, A., Kon, V., Inagami, T., and Ichikawa, I. (1998). Accelerated fibrosis and collagen deposition develop in the renal interstitium of angiotensin type 2 receptor null mutant mice during ureteral obstruction. Kidney Int. 53, 937–944. doi: 10.1111/j.1523-1755.1998.00893.x
Mahmood, A., and Pulakat, L. (2015). Differential effects of β-blockers, angiotensin II receptor blockers and a novel AT2R agonist NP-6A4 on stress response of nutrient-starved cardiovascular cells. PLoS ONE 10:e0144824. doi: 10.1371/journal.pone.0144824
Makani, H., Bangalore, S., Desouza, K. A., Shah, A., and Messerli, F. H. (2013). Efficacy and safety of dual blockade of the renin-angiotensin system: meta-analysis of randomised trials. BMJ 346:f360. doi: 10.1136/bmj.f360
Matavelli, L. C., Huang, J., and Siragy, H. M. (2011). Angiotensin AT(2) receptor stimulation inhibits early renal inflammation in renovascular hypertension. Hypertension 57, 308–313. doi: 10.1161/HYPERTENSIONAHA.110.164202
Matavelli, L. C., Zatz, R., and Siragy, H. M. (2015). A nonpeptide angiotensin II type 2 receptor agonist prevents renal inflammation in early diabetes. J. Cardiovasc. Pharmacol. 65, 371–376. doi: 10.1097/FJC.0000000000000207
McCarthy, C. A., Widdop, R. E., Denton, K. M., and Jones, E. S. (2013). Update on the angiotensin AT2 receptor. Curr. Hypertens Rep. 15, 25–30. doi: 10.1007/s11906-012-0321-4
Mehta, P. K., and Griendling, K. K. (2007). Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 292, C82–C97.
Messerli, F. H., Staessen, J. A., and Zannad, F. (2010). Of fads, fashion, surrogate endpoints and dual RAS blockade. Eur. Heart J. 31, 2205–2208. doi: 10.1093/eurheartj/ehq255
Metcalfe, B. L., Huentelman, M. J., Parilak, L. D., Taylor, D. G., Katovich, M. J., Knot, H. J., et al. (2004). Prevention of cardiac hypertrophy by angiotensin II type-2 receptor gene transfer. Hypertension 43, 1233–1238. doi: 10.1161/01.HYP.0000127563.14064.fd
Moran, A. E., Roth, G. A., Narula, J., and Mensah, G. A. (2014). 1990-2010 global cardiovascular disease atlas. Glob. Heart 9, 3–16. doi: 10.1016/j.gheart.2014.03.1220
Nabeshima, Y., Tazuma, S., Kanno, K., Hyogo, H., Iwai, M., Horiuchi, M., et al. (2006). Anti-fibrogenic function of angiotensin II type 2 receptor in CCl4-induced liver fibrosis. Biochem. Biophys. Res. Commun. 346, 658–664. doi: 10.1016/j.bbrc.2006.05.183
Nakayama, M., Yan, X., Price, R. L., Borg, T. K., Ito, K., Sanbe, A., et al. (2005). Chronic ventricular myocyte-specific overexpression of angiotensin II type 2 receptor results in intrinsic myocyte contractile dysfunction. Am. J. Physiol. Heart Circ. Physiol. 288, H317–H327.
Painemal, P., Acuña, M. J., Riquelme, C., Brandan, E., and Cabello-Verrugio, C. (2013). Transforming growth factor type beta 1 increases the expression of angiotensin II receptor type 2 by a SMAD- and p38 MAPK-dependent mechanism in skeletal muscle. Biofactors 39, 467–475. doi: 10.1002/biof.1087
Paulis, L., Becker, S. T., Lucht, K., Schwengel, K., Slavic, S., Kaschina, E., et al. (2012). Direct angiotensin II type 2 receptor stimulation in Nomega-nitro-L-arginine-methyl ester-induced hypertension: the effect on pulse wave velocity and aortic remodeling. Hypertension 59, 485–492. doi: 10.1161/HYPERTENSIONAHA.111.185496
Plenz, G., Song, Z. F., Reichenberg, S., Tjan, T. D., Robenek, H., and Deng, M. C. (1998). Left-ventricular expression of interleukin-6 messenger-RNA higher in idiopathic dilated than in ischemic cardiomyopathy. Thorac. Cardiovasc. Surg. 46, 213–216. doi: 10.1055/s-2007-1010227
Qi, Y., Li, H., Shenoy, V., Li, Q., Wong, F., Zhang, L., et al. (2012). Moderate cardiac-selective overexpression of angiotensin II type 2 receptor protects cardiac functions from ischaemic injury. Exp. Physiol. 97, 89–101. doi: 10.1113/expphysiol.2011.060673
Querejeta, R., Lopez, B., Gonzalez, A., Sanchez, E., Larman, M., Martinez Ubago, J. L., et al. (2004). Increased collagen type I synthesis in patients with heart failure of hypertensive origin: relation to myocardial fibrosis. Circulation 110, 1263–1268. doi: 10.1161/01.CIR.0000140973.60992.9A
Rehman, A., Leibowitz, A., Yamamoto, N., Rautureau, Y., Paradis, P., and Schiffrin, E. L. (2012). Angiotensin type 2 receptor agonist compound 21 reduces vascular injury and myocardial fibrosis in stroke-prone spontaneously hypertensive rats. Hypertension 59, 291–299. doi: 10.1161/HYPERTENSIONAHA.111.180158
Rockey, D. C., Bell, P. D., and Hill, J. A. (2015). Fibrosis–A common pathway to organ injury and failure. N. Engl. J. Med. 372, 1138–1149. doi: 10.1056/NEJMra1300575
Rompe, F., Artuc, M., Hallberg, A., Alterman, M., Stroder, K., Thone-Reineke, C., et al. (2010). Direct angiotensin II type 2 receptor stimulation acts anti-inflammatory through epoxyeicosatrienoic acid and inhibition of nuclear factor kappaB. Hypertension 55, 924–931. doi: 10.1161/HYPERTENSIONAHA.109.147843
Rosenbloom, J., Mendoza, F. A., and Jimenez, S. A. (2013). Strategies for anti-fibrotic therapies. Biochim. Biophys. Acta 1832, 1088–1103. doi: 10.1016/j.bbadis.2012.12.007
Sadoshima, J., and Izumo, S. (1993). Molecular characterization of angiotensin II–induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ. Res. 73, 413–423. doi: 10.1161/01.RES.73.3.413
Sampson, A. K., Irvine, J. C., Shihata, W. A., Dragoljevic, D., Lumsden, N., Huet, O., et al. (2016). Compound 21, a selective agonist of angiotensin AT2 receptors, prevents endothelial inflammation and leukocyte adhesion in vitro and in vivo. Br. J. Pharmacol. 173, 729–740. doi: 10.1111/bph.13063
Schelbert, E. B., Fonarow, G. C., Bonow, R. O., Butler, J., and Gheorghiade, M. (2014). Therapeutic targets in heart failure: refocusing on the myocardial interstitium. J. Am. Coll. Cardiol. 63, 2188–2198. doi: 10.1016/j.jacc.2014.01.068
Schiller, M., Javelaud, D., and Mauviel, A. (2004). TGF-beta-induced SMAD signaling and gene regulation: consequences for extracellular matrix remodeling and wound healing. J. Dermatol. Sci. 35, 83–92. doi: 10.1016/j.jdermsci.2003.12.006
Siragy, H. M., and Carey, R. M. (1996). The subtype-2 (AT2) angiotensin receptor regulates renal cyclic guanosine 3′, 5′-monophosphate and AT1 receptor-mediated prostaglandin E2 production in conscious rats. J. Clin. Invest. 97, 1978–1982. doi: 10.1172/JCI118630
Siragy, H. M., and Carey, R. M. (1997). The subtype 2 (AT2) angiotensin receptor mediates renal production of nitric oxide in conscious rats. J. Clin. Invest. 100, 264–269. doi: 10.1172/JCI119531
Te Riet, L., van Esch, J. H., Roks, A. J., van den Meiracker, A. H., and Danser, A. H. (2015). Hypertension: renin-angiotensin-aldosterone system alterations. Circ. Res. 116, 960–975. doi: 10.1161/CIRCRESAHA.116.303587
Tomasek, J. J., Gabbiani, G., Hinz, B., Chaponnier, C., and Brown, R. A. (2002). Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 3, 349–363. doi: 10.1038/nrm809
Torre-Amione, G., Kapadia, S., Lee, J., Durand, J. B., Bies, R. D., Young, J. B., et al. (1996). Tumor necrosis factor-alpha and tumor necrosis factor receptors in the failing human heart. Circulation 93, 704–711. doi: 10.1161/01.CIR.93.4.704
van Vark, L. C., Bertrand, M., Akkerhuis, K. M., Brugts, J. J., Fox, K., Mourad, J. J., et al. (2012). Angiotensin-converting enzyme inhibitors reduce mortality in hypertension: a meta-analysis of randomized clinical trials of renin-angiotensin-aldosterone system inhibitors involving 158,998 patients. Eur. Heart J. 33, 2088–2097. doi: 10.1093/eurheartj/ehs075
Verdonk, K., Durik, M., Abd-Alla, N., Batenburg, W. W., van den Bogaerdt, A. J., van Veghel, R., et al. (2012). Compound 21 induces vasorelaxation via an endothelium- and angiotensin II type 2 receptor-independent mechanism. Hypertension 60, 722–729. doi: 10.1161/HYPERTENSIONAHA.112.196022
Vijan, S. G. (2009). Angiotensin-converting enzyme inhibitors (ACEIs), not angiotensin receptor blockers (ARBs), are preferred and effective mode of therapy in high cardiovascular risk patients. J. Indian Med. Assoc. 107, 178–182.
Wan, Y., Wallinder, C., Plouffe, B., Beaudry, H., Mahalingam, A. K., Wu, X., et al. (2004). Design, synthesis, and biological evaluation of the first selective nonpeptide AT2 receptor agonist. J. Med. Chem. 47, 5995–6008. doi: 10.1021/jm049715t
Weber, K. T. (1989). Cardiac interstitium in health and disease: the fibrillar collagen network. J. Am. Coll. Cardiol. 13, 1637–1652. doi: 10.1016/0735-1097(89)90360-4
Weber, K. T., Sun, Y., Bhattacharya, S. K., Ahokas, R. A., and Gerling, I. C. (2013). Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat. Rev. Cardiol. 10, 15–26. doi: 10.1038/nrcardio.2012.158
Whaley-Connell, A., Habibi, J., Rehmer, N., Ardhanari, S., Hayden, M. R., Pulakat, L., et al. (2013). Renin Inhibition and AT1R blockade improve metabolic signaling, oxidant stress and myocardial tissue remodeling. Metab. Clin. Exp. 62, 861–872. doi: 10.1016/j.metabol.2012.12.012
Widdop, R. E., Jones, E. S., Hannan, R. E., and Gaspari, T. A. (2003). Angiotensin AT2 receptors: cardiovascular hope or hype? Br. J. Pharmacol. 140, 809–824. doi: 10.1038/sj.bjp.0705448
Xu, J., Sun, Y., Carretero, O. A., Zhu, L., Harding, P., Shesely, E. G., et al. (2014). Effects of cardiac overexpression of the angiotensin II type 2 receptor on remodeling and dysfunction in mice post-myocardial infarction. Hypertension 63, 1251–1259. doi: 10.1161/HYPERTENSIONAHA.114.03247
Yang, Z., Bove, C. M., French, B. A., Epstein, F. H., Berr, S. S., DiMaria, J. M., et al. (2002). Angiotensin II type 2 receptor overexpression preserves left ventricular function after myocardial infarction. Circulation 106, 106–111. doi: 10.1161/01.CIR.0000020014.14176.6D
Keywords: AT2 receptor, compound 21, cardiac fibrosis, renal fibrosis, inflammation
Citation: Wang Y, Del Borgo M, Lee HW, Baraldi D, Hirmiz B, Gaspari TA, Denton KM, Aguilar M-I, Samuel CS and Widdop RE (2017) Anti-fibrotic Potential of AT2 Receptor Agonists. Front. Pharmacol. 8:564. doi: 10.3389/fphar.2017.00564
Received: 30 May 2017; Accepted: 09 August 2017;
Published: 31 August 2017.
Edited by:
Nicolau Beckmann, Novartis Institutes for BioMedical Research, United StatesReviewed by:
Anthony Dart, The Alfred Hospital, AustraliaIan Dixon, University of Manitoba, Canada
Lakshmidevi Pulakat, University of Missouri, United States
Copyright © 2017 Wang, Del Borgo, Lee, Baraldi, Hirmiz, Gaspari, Denton, Aguilar, Samuel and Widdop. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert E. Widdop, cm9iZXJ0LndpZGRvcEBtb25hc2guZWR1