 Jean-Michel Dayer
Jean-Michel Dayer Francesca Oliviero
Francesca Oliviero Leonardo Punzi
Leonardo Punzi- 1Faculty of Medicine, University of Geneva, Geneva, Switzerland
- 2Department of Medicine, University of Padova, Padova, Italy
The history of what, in 1979, was called interleukin-1 (IL-1), orchestrator of leukocyte inter-communication, began many years before then, initially by the observation of fever induction via the endogenous pyrogen (EP) (1974) and then in rheumatology on the role in tissue destruction in rheumatoid diseases via the induction of collagenase and PGE2 in human synovial cells by a mononuclear cell factor (MCF) (1977). Since then, the family has exploded to presently 11 members as well as many membrane-bound and soluble receptor forms. The discovery of a natural Interleukin-1 receptor antagonist (IL-1Ra) in human biological fluids has highlighted the importance of IL-1 and IL-1Ra in human diseases. Evidence delineating its role in autoinflammatory syndromes and the elucidation of the macromolecular complex referred to as “inflammasome” have been instrumental to our understanding of the link with IL-1. At present, the IL-1blockade as therapeutic approach is crucial for many hereditary autoinflammatory diseases, as well as for adult-onset Still’s disease, crystal-induced arthropathies, certain skin diseases including neutrophil-triggered skin diseases, Behçet’s disease and deficiency of IL-1Ra and other rare fever syndromes. Its role is only marginally important in rheumatoid arthritis and is still under debate with regard to osteoarthritis, type 2 diabetes mellitus, cardiovascular diseases and cancer. This brief historical review focuses on some aspects of IL-1, mainly IL-1β and IL-Ra, in rheumatology. There are many excellent reviews focusing on the IL-1 family in general or with regard to specific diseases or biological discoveries.
IL-1 and Its Roots
Historically in rheumatology, the seminal observation of the production of human interstitial collagenase (later called MMP-1) by human synovial cells in patients with rheumatoid arthritis (RA) led to the unraveling of the link between matrix degradation and the biological function of inflammatory molecules produced by immune cells. It was found that the main cellular source of both interstitial collagenase (matrix metalloproteases, MMPs) and prostaglandin E2 (PGE2) in synovial tissue were adherent stellate fibroblast-like cells (ASC), also called type B fibroblastic synovial lining cells (Dayer et al., 1976). Presently, the fibroblast subpopulation in inflamed synovial tissue has proved to be far more complex (Croft et al., 2016).
The question was which are the driving forces for stimulating the synovial cells to produce MMP and PGE2. Seminal studies at the Arthritis Unit, Massachusetts General Hospital, Harvard Medical School) have shown that partial purification of the conditioned medium revealed a protein of approximately 15 kDa, and the factor responsible for this biological activity was called mononuclear cell factor (MCF) (Dayer et al., 1977a,b, 1981) (Figure 1). Interestingly, interactions between T cells and monocyte-macrophages were found to play an important role in the production of IL-1 by monocyte-macrophages, giving rise to the first descriptions of the pathways going from lymphocytes to monocyte-macrophages and synovial fibroblast cells (Dayer et al., 1979a,b).
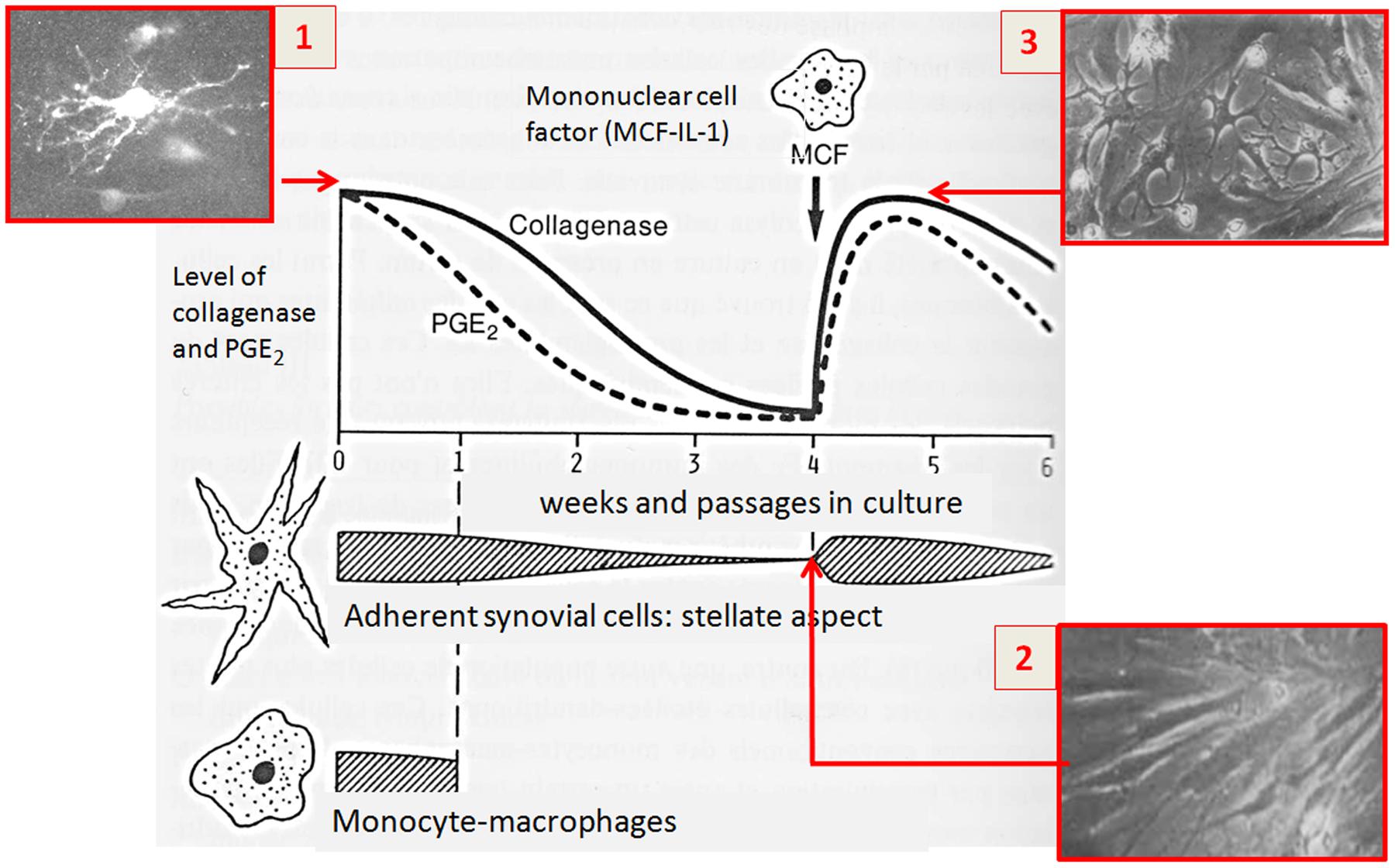
FIGURE 1. Seminal observation about the link between matrix degradation and the biological function of inflammatory molecules produced by immune cells (1976–1977). (1) Cultures of synovial explants from patients with rheumatoid arthritis release large quantities of collagenase and prostaglandins (PGE2) into the media. Secretion of PGE2 declines more rapidly with respect to collagenase whose levels decline after different passages in culture; (2,3) The addition of monocyte-macrophages or their culture supernatants restored collagenase and PGE2 levels. The soluble factor responsible of this effect was later identified in mononuclear cell factor (MCF).
At this early stage of the discovery, before the cloning, different investigators analyzed the soluble factors derived from leucocyte culture supernatants in their respective functional bioassays in order to identify the putative functional specificity. Purified ‘MCF’ proved to have similar properties to the lymphocyte-activating factor (LAF), a factor which had the same molecular weight of approximately 15 kDa and shared chromatographic as well as other biochemical properties (Mizel et al., 1981). It was not until 1979 that the nomenclature and the terminology of interleukin-1 (IL-1), orchestrator of leukocyte communication, was coined at a meeting (Ermatingen, Switzerland) and it referred to products previously identified by several investigators using different bioassays, including lymphocyte activating factor (LAF), MCF, B lymphocyte activating factor (BAFF) and of course endogenous pyrogen (EP) that induced fever. At that time, cytokines were defined only by biochemistry (Aarden et al., 1979). It was defined for the biochemistry properties by a m.w. between 12,000–18,000 with an isoelectric point of 4.5–5.5 in mouse and 6.5–7.5 in human, pH 2 insensitive, does not contain Ia and for the biologic properties as produced by macrophages, with H-2 unrestricted activity, unrestricted species activity, distinguishing bioactivity and does not possess the ability to promote and maintain in vitro long-term cultures of T cells in contrast to IL-2.
When human IL-1 was cloned using an antibody to EP and based on its EP bioactivity, the protein was expressed and the recombinant IL-1 found to have the same biological functions as MCF (Dayer et al., 1986). However, it must be emphasized that in addition to IL-1, natural tumor necrosis factor-cachectin (TNFα) was also demonstrated early on to induce MMP and PGE2 production in synovial cells (Dayer et al., 1985).
As far as LAF is concerned, despite the initial description of its role in the early activation of murine T cells from thymocytes, diseases characterized by an adaptive immune response to specific autoantigens are usually not part of the spectrum of IL-1β-driven disorders.
With regard to the field of rheumatology, other partially purified molecules like catabolin – inducing cartilage degradation – and osteoclast-activating factor (OAF) inducing calcium45 release from bone, to name but a few, were also found to have functions similar to those of IL-1 (Dayer, 2004).
The discovery of the other aspects of the IL-1 and IL-1 receptor families, related to the seminal features of EP and to several other important functions, has been extensively reviewed (Dinarello, 2010, 2015; Dinarello and van der Meer, 2013; Garlanda et al., 2013; Fantuzzi, 2016) as well as the biology of IL-1 alpha (Di Paolo and Shayakhmetov, 2016). The role of IL-1 in rheumatology and its specific interaction with MMPs have also been recently reviewed (Schett et al., 2016; Dayer et al., 2017).
The Story Behind IL-1Ra
As early as 1984, before IL-1 was cloned, the existence of a potential natural inhibitor to IL-1 was suspected. In the bioassay for stimulating collagenase and prostaglandin production, no IL-1 biological activities were detected in the serum or urine of febrile patients (Balavoine et al., 1984). In the light of that finding, Dayer and colleagues (University of Geneva, Switzerland) reported in 1984 that, after biochemical purification, IL-1 was masked by a factor of approximately 17 kDa in the urine of febrile patients with monocytic leukemia and juvenile rheumatoid arthritis (JRA). This seminal observation was presented in 1984 at the Fourth International Lymphokine Workshop (Balavoine et al., 1984). The factor specifically blocked the biological activities of IL-1 (Dayer, 1985; Balavoine et al., 1986).
The first description of the concept underlying the interleukin-1 receptor antagonist (IL-1Ra) was that this natural antagonist could block the binding of a cytokine from the same family to its receptor without affecting those of TNFα (Seckinger et al., 1987a,b). In the field of cytokine, it was the first observation of an inhibitor factor that blocks ligand binding of cytokine and its activities. The discovery of the competitive binding assay of the IL-1 inhibitor (apparent m.w. around 17 kd) to IL-1 at the IL-1 receptor level was crucial for researchers at Synergen (Boulder, CO, United States) who purified and cloned IL-1Ra in 1990. Before IL-1Ra was cloned, the first clinical description of natural IL-Ra was made by following the time course of this specific IL inhibitor in the serum and urine of children with systemic juvenile chronic arthritis, a typical inflammatory syndrome (Prieur et al., 1987). Arend et al. (1985) reported the presence of an inhibitor of IL-1 activity under in vitro conditions, but neither its molecular weight nor the concept of binding to the IL-1 receptor was identified.
IL-1 and Its Extended Family
First described as a secreted product of monocytes and neutrophils ∼35 years ago, interleukin (IL)-1 refers to IL-1α and IL-1β. At present, the IL-1 family comprises a total of 11 members, consisting of the two activating cytokines IL-1α and IL-1β, the IL-1Ra, as well as IL-18, IL-33, four isoforms of IL-36 [IL-36α, IL-36β, IL-36γ and IL-36 receptor antagonist (IL-36Ra)], IL-37 and IL-1 family member 10 (also known as IL-38). IL-1 is processed and activated by a caspase-1-dependent mechanism as well as by caspase-1-independent processes that involve neutrophil proteases (see history in Fantuzzi, 2016).
Interleukin-1 receptor antagonist, IL-36Ra and IL-37 are predominantly anti-inflammatory cytokines. IL-36Ra shares homology with IL-1Ra but is unable to bind to the IL-1R1 (Netea et al., 2015). It has an important role in regulating skin inflammation and its deficiency (linked to mutations in IL-36RN) is associated to a rare form of pustular psoriasis also called deficiency of IL-36-receptor antagonist or DITRA (Marrakchi et al., 2011). Skin lesions, along with multifocal osteomyelitis and periostitis are associated also to IL-1Ra deficiency, termed DIRA, an autosomal recessive autoinflammatory disease caused by mutations affecting IL1RN (Aksentijevich et al., 2009).
Inflammasome, the Macromolecular Complex
The discovery of the macromolecular complex inflammasome resulted from observations made by investigators working with rare diseases (autoinflammatory syndromes), experts in apoptosis, and biochemists analyzing how cells sense the presence of danger. The first to identify the mechanism was a French consortium of scientists in 1997, which was due to the mutation causing familial Mediterranean fever (FMF). The mutated gene encoding an unusual structure of unknown function – named at that time marenostrin (from mare nostrum, referring to the Mediterranean Sea) – a pyrin involved in periodic fevers (French FMF Consortium, 1997). The protease which processes interleukin-1β [IL-1β-converting enzyme (ICE) = caspase-1] was identified in 1989 (Black et al., 1989) and cloned in 1992 (Cerretti et al., 1992; Thornberry et al., 1992). The identification of CARDIAK, a death domain associated with caspase-1, was identified in 1998 (Thome et al., 1998). The mutation in the gene (currently called NLRP3-inflammasome) in recurrent and chronic inflammation was initially detected in a group of rare autoinflammatory conditions, termed cryopyrin-associated periodic syndromes (CAPS) (Hoffman et al., 2001); this protein also belongs to the pyrin family. The link between pyrin, cryopyrin, NLRP3, death domain (CARD) and caspase-1 was established and this macromolecule complex was termed “inflammasome” (Martinon et al., 2002; Srinivasula et al., 2002) (Figure 2).
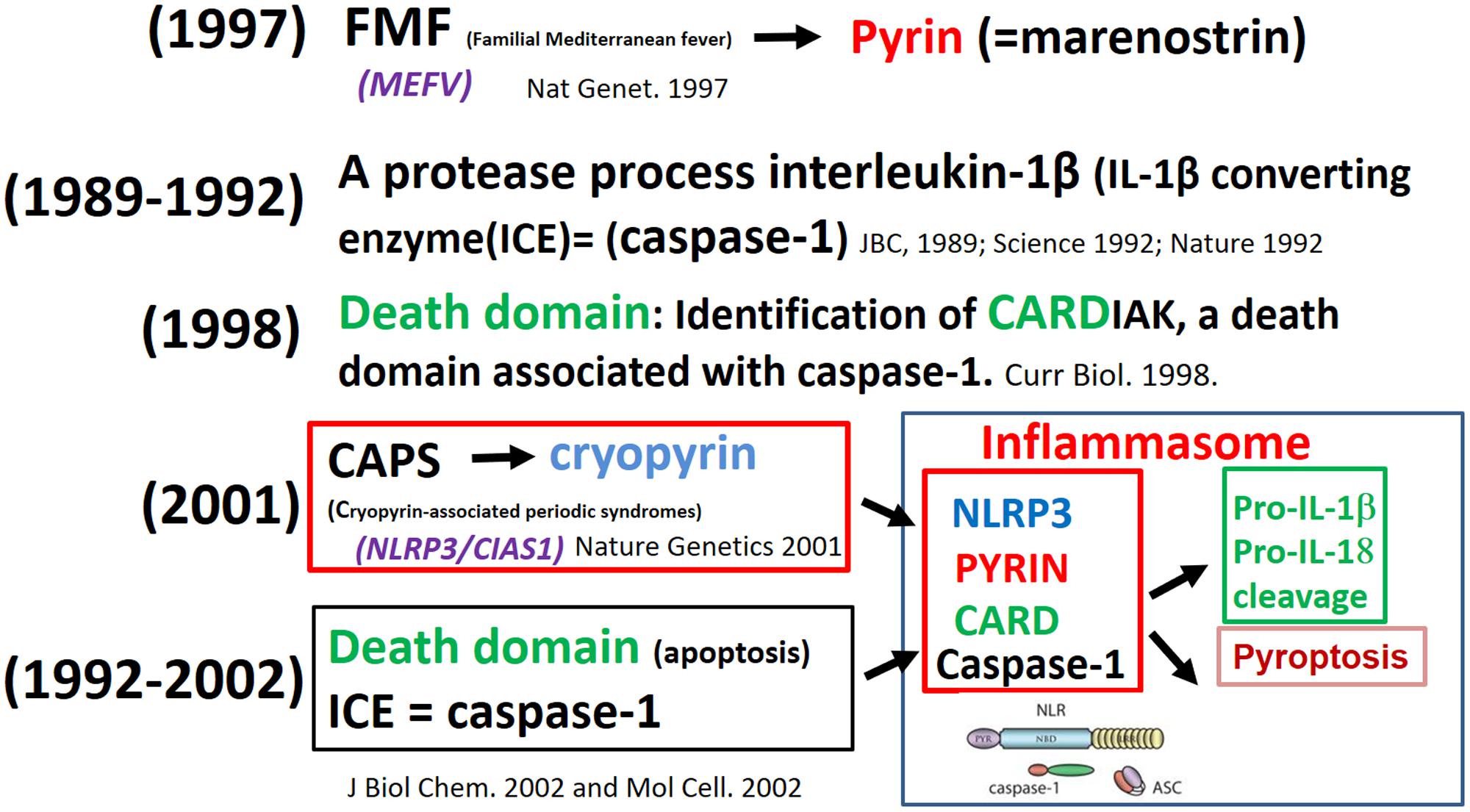
FIGURE 2. From fever of unknown origin to the inflammasome puzzle.
IL-1 Receptors
The search for IL-1 receptors began in 1985 when a high-affinity plasma membrane receptor for human interleukin was identified (Dower et al., 1985); between 1985 and 1988, numerous investigators were working in this field of research (Bird and Saklatvala, 1986; Kilian et al., 1986; Kroggel et al., 1988).
The gene sequence for IL-1β and IL-1Ra receptors was described by Sims et al. (1988). Stylianou et al. (1992) was well established that the type I and not type II receptor led to signal transduction.
Eastgate et al. (1990) demonstrated the existence of an IL-1β binding protein, followed by the findings of Francesco Colotta and Alberto Mantovani in 1993 that the shed molecule of type II receptor was a decoy receptor and probably similar to the IL-1β-binding protein described by Eastgate and Duff.
In a recent book entitled “Body Messages: the Quest for the Proteins of Cellular Communication. Evolution of Message Discovery: The IL Family,” Fantuzzi (2016) elegantly describes the different facets, controversies and surprises characterizing the discovery of the IL-1, IL-1Ra and interleukin receptors families.
Inflammasome, IL-1 and IL-Ra in Crystal-Induced Arthropathies
The first observation that IL-1 could contribute to the pathogenesis of crystal-induced arthritis Duff et al. (1983) demonstrated that monosodium urate (MSU) crystals were able to stimulate mononuclear phagocytes in vitro to produce EP. The authors failed to extend these results to other pathogenic crystals such as hydroxyapatite and calcium pyrophosphate (CPP), hypothesizing that the joint had to be, in some way, “conditioned” to propagate an inflammatory stimulus and that the source of IL-1 could lie outside the joint (Malawista et al., 1985). Subsequently, it became clear that purified crystals alone could not induce IL-1but that an additional stimulus was needed in vitro to generate IL-1 (Giamarellos-Bourboulis et al., 2009). Further ex vivo models identified free fatty acids (Joosten et al., 2010) and synovial fluid (SF) proteins (Scanu et al., 2016) as important triggers of IL-1 release.
An imbalance between the production of IL-1 and IL-1Ra induced by crystals in activated cells was pointed out in 1994 when it became clear that MSU and CPP crystals did not affect IL-1Ra production in neutrophils (Roberge et al., 1994).
Martinon et al. (2006) demonstrated for the first time the involvement of the inflammasome NLRP3 in caspase-1 activation and IL-1 release induced by crystals. IL-1R was also found to play a critical role in amplifying the inflammatory response through a MyD88-dependent mechanism (Chen et al., 2006). These observations, along with the discovery that uric acid was capable to stimulate the innate immune system (Shi et al., 2003), led to the addition of gout to the spectrum of autoinflammatory diseases (Punzi et al., 2012).
The key role that IL-1β plays in gout and pseudo-gout was further confirmed by the effectiveness of blocking agents to IL-1 in that it reduced acute attacks, initially observed after the administration of IL-1Ra anakinra, and then to the anti-IL-1β monoclonal antibody canakinumab (So et al., 2007; Schlesinger et al., 2012; Punzi and So, 2013). Accordingly, the recent EULAR recommendations for the management of gout advocate considering the administration of IL-1 blockers in those patients with frequent flares and contraindications to colchicine, NSAIDs and corticosteroids (Richette et al., 2017). In contrast, Rilonacept, a soluble receptor fusion protein binding both IL-1α and IL-1β, provided no benefit over indomethacin in an RCT (Terkeltaub et al., 2013). Other promising molecules, such as the recombinant human alpha-1-anti-trypsin (AAT)-IgG1 Fc fusion protein, have been shown to mainly target IL-1 in gout (Joosten et al., 2016).
Effect of IL-1 on Cartilage and Bone in OA
The presence of a low molecular weight peptide released from synovial tissue and able to affect cartilage homeostasis was demonstrated at the end of the 1970s (Dingle et al., 1979). This messenger called “catabolin” was able to stimulate chondrocytes to degrade both cartilage proteoglycans and collagen (Saklatvala and Dingle, 1980), and soon its similarity to IL-1 was established (Saklatvala et al., 1984).
Gowen et al. (1983) demonstrated for the first time that IL-1-like factor altered the breakdown of bone by modulating both bone resorption in RA and bone formation in OA. As far as bone resorption is concerned, IL-1Ra was later shown to block the resorptive effect induced by IL-1 in vitro (Seckinger et al., 1990).
Despite the fact that the altered expression of proinflammatory cytokines and chemokines in OA cartilage and synovium is well documented (Kapoor et al., 2011; Raghu et al., 2016), the role of IL-1β has not been fully clarified. Very low levels of IL-1β have been determined in the synovial membrane and SF of patients with early and end-stage OA (Scanzello et al., 2009) although levels were more elevated in the SF cells of OA than in those of controls (Sauerschnig et al., 2014). However, IL-1β levels are significantly lower in OA than in RA (Farahat et al., 1993). In vitro studies have revealed that IL-1β plays a critical role in driving the production of proteolytic enzymes such as MMPs and ADAMTS-4 in OA (Bondeson et al., 2006). But results obtained from different OA animal models showed discrepancies when anti-IL-1β therapy (Fernandes et al., 1999; Zhang et al., 2004) or mice deficient in IL-1 (Clements et al., 2003) were utilized, thus emphasizing that other, IL-1-independent, pathways may be involved in OA pathology (Bondeson et al., 2010).
As far as the association between the IL-1 gene polymorphisms and OA is concerned, the haplotypes IL1A-IL1B-IL1RN and IL1B-IL1RN have, respectively, been shown to confer a higher and a decreased risk of OA (Smith et al., 2004). A study investigating the contribution of gene polymorphisms to dysregulated cytokine expression in OA cartilage revealed significant associations between the TNFα (high) chondrocyte phenotype and IL-1Ra allele 2, and between the TNFα (low) phenotype and IL-1β allele 2 (Moos et al., 2000).
An association between severe hand OA and single nucleotide polymorphisms in the IL-1R1 gene has been observed in family based and case-control analyses (Nakki et al., 2010). Several studies assessing the polymorphism in OA are ongoing, and some have been reviewed in a meta-analysis (Kerkhof et al., 2011).
Toward Clinical Implications
Although IL-1 itself has been used as a therapeutic agent in oncology patients, alone it has little antitumor activity in melanoma, renal cell carcinoma, ovarian carcinoma or other diseases.
Considering that IL-1 seems to protect and restore the bone marrow from lesions induced by radiation or chemotherapy, it has been proposed to use it for treatment, but its modest hematopoietic effects probably do not counterbalance the toxicity necessary to achieve said effects (Veltri and Smith, 1996). IL-1α and IL-1β have also been proposed as vaccine adjuvants (Staats and Ennis, 1999).
The initial attempts to block IL-1 in infectious diseases have been unsuccessful and were accompanied by an increase in infections and side effects (Freeman and Buchman, 2001). Trials monitoring effects of IL-1Ra in patients with RA, which were begun in 1991, have produced interesting results although long-term results have not proved impressive and have demonstrated that this therapy is less effective than anti-TNF therapies. The initial trial targeting IL-1 in RA has been reviewed in detail (Dayer and Bresnihan, 2002).
At present, therapeutic blockade of IL-1 is crucial in many hereditary autoinflammatory diseases, as well as in patients with Still’s disease, crystal-induced arthropathies, certain skin diseases including neutrophil-triggered skin diseases, Behçet’s disease and deficiency of IL-1Ra, and other rare fever syndromes. Its effects have been marginal in RA patients and are still under debate in OA, type 2 diabetes mellitus, cardiovascular diseases and cancer (Larsen et al., 2009; Cantarini et al., 2015; Cavalli and Dinarello, 2015; Sfriso et al., 2016).
Relevant developments are still be expected in future with regard to the IL-1 family and the macromolecular complex which is inflammasome. The complexity of the various forms of the IL-1 receptor family, not discussed here in the history, is far from being fully understood and may lead to new avenues for pharmacological interventions.
Note
We regret that given the brevity of the review with particular focus on rheumatological aspects, it was impossible to acknowledge all the important researchers involved in this field of study and their discoveries and contributions.
Author Contributions
J-MD conceived and drafted the manuscript. FO and LP contributed to the drafting and the revision of the work. All authors approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer SJ and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
References
Aarden, L. A., Brunner, T. K., Cerottini, J. C., Dayer, J.-M., de Weck, A. L., Dinarello, C. A., et al. (1979). Revised nomenclature for antigen nonspecific T cell proliferation and helper factors (letter to the Editor). J. Immunol. 123, 2928–2929.
Aksentijevich, I., Masters, S. L., Ferguson, P. J., Dancey, P., Frenkel, J., van Royen-Kerkhoff, A., et al. (2009). An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N. Engl. J. Med. 360, 2426–2437. doi: 10.1056/NEJMoa0807865
Arend, W. P., Joslin, F. G., and Massoni, R. J. (1985). Effects of immune complexes on production by human monocytes of interleukin 1 or an interleukin 1 inhibitor. J. Immunol. 134, 3868–3875.
Balavoine, J. F., de Rochemonteix, B., Cruchaud, A., and Dayer, J. M. (1984). Identification of interleukin 1-like activity and inhibitor(s) in urine from a patient with acute monoblastic leukemia. Lymphokine Res. 3, 233.
Balavoine, J. F., de Rochemonteix, B., Williamson, K., Seckinger, P., Cruchaud, A., and Dayer, J. M. (1986). Prostaglandin E2 and collagenase production by fibroblasts and synovial cells is regulated by urine-derived human interleukin 1 and inhibitor(s). J. Clin. Invest. 78, 1120–1124. doi: 10.1172/JCI112669
Bird, T. A., and Saklatvala, J. (1986). Identification of a common class of high affinity receptors for both types of porcine interleukin-1 on connective tissue cells. Nature 324, 263–266. doi: 10.1038/324263a0
Black, R. A., Kronheim, S. R., Merriam, J. E., March, C. J., and Hopp, T. P. (1989). A pre-aspartate-specific protease from human leukocytes that cleaves pro-interleukin-1 beta. J. Biol. Chem. 264, 5323–5326.
Bondeson, J., Blom, A. B., Wainwright, S., Hughes, C., Caterson, B., and van den Berg, W. B. (2010). The role of synovial macrophages and macrophage-produced mediators in driving inflammatory and destructive responses in osteoarthritis. Arthritis Rheum. 62, 647–657. doi: 10.1002/art.27290
Bondeson, J., Wainwright, S. D., Lauder, S., Amos, N., and Hughes, C. E. (2006). The role of synovial macrophages and macrophage-produced cytokines in driving aggrecanases, matrix metalloproteinases, and other destructive and inflammatory responses in osteoarthritis. Arthritis Res. Ther. 8, R187. doi: 10.1186/ar2099
Cantarini, L., Lopalco, G., Caso, F., Costa, L., Iannone, F., Lapadula, G., et al. (2015). Effectiveness and tuberculosis-related safety profile of interleukin-1 blocking agents in the management of Behçet’s disease. Autoimmun. Rev. 14, 1–9. doi: 10.1016/j.autrev.2014.08.008
Cavalli, G., and Dinarello, C. A. (2015). Treating rheumatological diseases and co-morbidities with interleukin-1 blocking therapies. Rheumatology 54, 2134–2144. doi: 10.1093/rheumatology/kev269
Cerretti, D. P., Kozlosky, C. J., Mosley, B., Nelson, N., Van Ness, K., Greenstreet, T. A., et al. (1992). Molecular cloning of the interleukin-1 beta-converting enzyme. Science 256, 97–100. doi: 10.1126/science.1373520
Chen, C. J., Shi, Y., Hearn, A., Fitzgerald, K., Golenbock, D., Reed, G., et al. (2006). MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J. Clin. Invest. 116, 2262–2271. doi: 10.1172/JCI28075
Clements, K. M., Price, J. S., Chambers, M. G., Visco, D. M., Poole, A. R., and Mason, R. M. (2003). Gene deletion of either interleukin-1, interleukin-1-converting enzyme, inducible nitric oxide synthase, or stromelysin 1 accelerates the development of knee osteoarthritis in mice after surgical transection of the medial collateral ligament and partial medial meniscectomy. Arthritis Rheum. 48, 3452–3463. doi: 10.1002/art.11355
Croft, A. P., Naylor, A. J., Marshall, J. L., Hardie, D. L., Zimmermann, B., Turner, J., et al. (2016). Rheumatoid synovial fibroblasts differentiate into distinct subsets in the presence of cytokines and cartilage. Arthritis Res. Ther. 18, 270. doi: 10.1186/s13075-016-1156-1
Dayer, J. M. (1985). “Collagenase- and PGE2-stimulating activity (interleukin-1-like) and inhibitor in urine from a patient with monocytic leukemia,” in The Physiologic, Metabolic, and Immunologic Actions of Interleukin-1. Progress in Leukocyte Biology, Vol. 2, eds M. J. Kluger, J. J. Oppenheim, and M. C. Powanda (New York, NY: Alan R. Liss), 429.
Dayer, J. M. (2004). The process of identifying and understanding cytokines: from basic studies to treating rheumatic diseases. Best Pract. Res. Clin. Rheumatol. 18, 31–45. doi: 10.1016/j.berh.2003.09.009
Dayer, J.M., Beutler, B., and Cerami, A. J. (1985). Cachectin/tumor necrosis factor stimulates collagenase and prostaglandin E2 production by human synovial cells and dermal fibroblasts. J. Exp. Med. 162, 2163–2168. doi: 10.1084/jem.162.6.2163
Dayer, J. M., and Bresnihan, B. (2002). Targeting interleukin-1 in the treatment of rheumatoid arthritis. Arthritis Rheum. 46, 574–578. doi: 10.1002/art.10168
Dayer, J. M., Bréard, J., Chess, L., and Krane, S. M. (1979a). Participation of monocyte-macrophages and lymphocytes in the production of a factor that stimulates collagenase and prostaglandin release by rheumatoid synovial cells. J. Clin. Invest. 64, 1386–1392.
Dayer, J. M., de Rochemonteix, B., Burrus, B., Demczuk, S., and Dinarello, C. A. (1986). Human recombinant interleukin 1 stimulates collagenase and prostaglandin E2 production by human synovial cells. J. Clin. Invest. 77, 645–648.
Dayer, J. M., Goldring, S. R., Robinson, D. R., and Krane, S. M. (1979b). Collagenase and prostaglandin in connective tissue destruction: cell-cell and humoral interactions. Bull. Schweiz. Akad. Med. Wiss. 35, 329–343.
Dayer, J. M., Graham, R., Russell, G., and Krane, S. M. (1977a). Collagenase production by rheumatoid synovial cells: stimulation by a human lymphocyte factor. Science 195, 181–183.
Dayer, J. M., Krane, S. M., Russell, R. G., and Robinson, D. R. (1976). Production of collagenase and prostaglandins by isolated adherent rheumatoid synovial cells. Proc. Natl. Acad. Sci. U.S.A. 73, 945–949. doi: 10.1073/pnas.73.3.945
Dayer, J. M., Robinson, D. R., and Krane, S. M. (1977b). Prostaglandin production by rheumatoid synovial cells: stimulation by a factor from human mononuclear cells. J. Exp. Med. 145, 1399–1404.
Dayer, J. M., Stephenson, M. L., Schmidt, E., Karge, W., and Krane, S. M. (1981). Purification of a factor from human blood monocyte-macrophages which stimulates the production of collagenase and prostaglandin E2 by cells cultured from rheumatoid synovial tissues. FEBS Lett. 124, 253. doi: 10.1016/0014-5793(81)80149-4
Dayer, J. M., Williamson, S. J., Croft, A. P., Buckley, C. D., and Chizzolini, C. (2017). “MMPs and cytokines in rheumatology,” in Matrix Metalloproteinases in Health and Disease, ed. C. Brinckerhoff (Singapore: World Scientific Publishing).
Di Paolo, N. C., and Shayakhmetov, D. M. (2016). Interleukin 1α and the inflammatory process. Nat. Immunol. 17, 906–913. doi: 10.1038/ni.3503
Dinarello, C. A. (2010). IL-1: discoveries, controversies and future directions. Eur. J. Immunol. 40, 599–606. doi: 10.1002/eji.201040319
Dinarello, C. A. (2015). The history of fever, leukocytic pyrogen and interleukin-1. Temperature 2, 8–16. doi: 10.1080/23328940.2015.1017086
Dinarello, C. A., and van der Meer, J. W. (2013). Treating inflammation by blocking interleukin-1 in humans. Semin. Immunol. 25, 469–484. doi: 10.1016/j.smim.2013.10.008
Dingle, J. T., Saklatvala, J., Hembry, R., Tyler, J., Fell, H. B., and Jubb, R. (1979). A cartilage catabolic factor from synovium. Biochem. J. 184, 177–180. doi: 10.1042/bj1840177
Dower, S. K., Kronheim, S. R., March, C. J., Conlon, P. J., Hopp, T. P., Gillis, S., et al. (1985). Detection and characterization of high affinity plasma membrane receptors for human interleukin 1. J. Exp. Med. 162, 501–515. doi: 10.1084/jem.162.2.501
Duff, G. W., Atkins, E., and Malawista, S. E. (1983). The fever of gout: urate crystals activate endogenous pyrogen production from human and rabbit mononuclear phagocytes. Trans. Assoc. Am. Physicians 96, 234–245.
Eastgate, J. A., Symons, J. A., and Duff, G. W. (1990). Identification of an interleukin-1 beta binding protein in human plasma. FEBS Lett. 260, 213–216. doi: 10.1016/0014-5793(90)80106-S
Fantuzzi, G. (ed.). (2016). “Evolution of message discovery: the interleukin-1 family,” in Body Messages: The Quest for the Proteins of Cellular Communication, (Cambridge, MA: Harvard University Press), 29–73.
Farahat, M. N., Yanni, G., Poston, R., and Panayi, G. S. (1993). Cytokine expression in synovial membranes of patients with rheumatoid arthritis and osteoarthritis. Ann. Rheum. Dis. 52, 870–875. doi: 10.1136/ard.52.12.870
Fernandes, J., Tardif, G., Martel-Pelletier, J., Lascau-Coman, V., Dupuis, M., Moldovan, F., et al. (1999). In vivo transfer of interleukin-1 receptor antagonist gene in osteoarthritic rabbit knee joints: prevention of osteoarthritis progression. Am. J. Pathol. 154, 1159–1169. doi: 10.1016/S0002-9440(10)65368-0
Freeman, B. D., and Buchman, T. G. (2001). Interleukin-1 receptor antagonist as therapy for inflammatory disorders. Expert Opin. Biol. Ther. 1, 301–308. doi: 10.1517/14712598.1.2.301
French FMF Consortium (1997). A candidate gene for familial Mediterranean fever. Nat. Genet. 17, 25–31. doi: 10.1038/ng0997-25
Garlanda, C., Dinarello, C. A., and Mantovani, A. (2013). The interleukin-1 family: back to the future. Immunity 39, 1003–1018. doi: 10.1016/j.immuni.2013.11.010
Giamarellos-Bourboulis, E. J., Mouktaroudi, M., Bodar, E., van der Ven, J., Kullberg, B. J., Netea, M. G., et al. (2009). Crystals of monosodium urate monohydrate enhance lipopolysaccharide-induced release of interleukin 1 beta by mononuclear cells through a caspase 1-mediated process. Ann. Rheum. Dis. 68, 273–278. doi: 10.1136/ard.2007.082222
Gowen, M., Wood, D. D., Ihrie, E. J., McGuire, M. K., and Russell, R. G. (1983). An interleukin 1 like factor stimulates bone resorption in vitro. Nature 306, 378–380. doi: 10.1038/306378a0
Hoffman, H. M., Mueller, J. L., Broide, D. H., Wanderer, A. A., and Kolodner, R. D. (2001). Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat. Genet. 29, 301–305. doi: 10.1038/ng756
Joosten, L. A., Crian, T. O., Azam, T., Cleophas, M. C., Koenders, M. I., van de Veerdonk, F. L., et al. (2016). Alpha-1-anti-trypsin-Fc fusion protein ameliorates gouty arthritis by reducing release and extracellular processing of IL-1β and by the induction of endogenous IL-1Ra. Ann. Rheum. Dis. 75, 1219–1227. doi: 10.1136/annrheumdis-2014-206966
Joosten, L. A., Netea, M. G., Mylona, E., Koenders, M. I., Malireddi, R. K., Oosting, M., et al. (2010). Engagement of fatty acids with Toll-like receptor 2 drives interleukin-1β production via the ASC/caspase 1 pathway in monosodium urate monohydrate crystal-induced gouty arthritis. Arthritis Rheum. 62, 3237–3248. doi: 10.1002/art.27667
Kapoor, M., Martel-Pelletier, J., Lajeunesse, D., Pelletier, J. P., and Fahmi, H. (2011). Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 7, 33–42. doi: 10.1038/nrrheum.2010.196
Kerkhof, H. J., Doherty, M., Arden, N. K., Abramson, S. B., Attur, M., Bos, S. D., et al. (2011). Large-scale meta-analysis of interleukin-1 beta and interleukin-1 receptor antagonist polymorphisms on risk of radiographic hip and knee osteoarthritis and severity of knee osteoarthritis. Osteoarthritis Cartilage 19, 265–271. doi: 10.1016/j.joca.2010.12.003
Kilian, P. L., Kaffka, K. L., Stern, A. S., Woehle, D., Benjamin, W. R., Dechiara, T. M., et al. (1986). Interleukin 1 alpha and interleukin 1 beta bind to the same receptor on T cells. J. Immunol. 136, 4509–4514.
Kroggel, R., Martin, M., Pingoud, V., Dayer, J. M., and Resch, K. (1988). Two-chain structure of the interleukin 1 receptor. FEBS Lett. 229, 59–62. doi: 10.1016/0014-5793(88)80797-X
Larsen, C. M., Faulenbach, M., Vaag, A., Ehses, J. A., Donath, M. Y., and Mandrup-Poulsen, T. (2009). Sustained effects of interleukin-1 receptor antagonist treatment in type 2 diabetes. Diabetes Care 32, 1663–1668. doi: 10.2337/dc09-0533
Malawista, S. E., Duff, G. W., Atkins, E., Cheung, H. S., and McCarty, D. J. (1985). Crystal-induced endogenous pyrogen production. A further look at gouty inflammation. Arthritis Rheum. 28, 1039–1046. doi: 10.1002/art.1780280911
Marrakchi, S., Guigue, P., Renshaw, B. R., Puel, A., Pei, X. Y., Fraitag, S., et al. (2011). Interleukin-36-receptor antagonist deficiency and generalised pustular psoriasis. N. Engl. J. Med. 365, 620–628. doi: 10.1056/NEJMoa1013068
Martinon, F., Burns, K., and Tschopp, J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426. doi: 10.1016/S1097-2765(02)00599-3
Martinon, F., Pétrilli, V., Mayor, A., Tardivel, A., and Tschopp, J. (2006). Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241. doi: 10.1038/nature04516
Mizel, S. B., Dayer, J. M., Krane, S. M., and Mergenhagen, S. E. (1981). Stimulation of rheumatoid synovial cell collagenase and prostaglandin production by partially purified lymphocyte-activating factor (interleukin 1). Proc. Natl. Acad. Sci. U.S.A. 78, 2474–2477. doi: 10.1073/pnas.78.4.2474
Moos, V., Rudwaleit, M., Herzog, V., Höhlig, K., Sieper, J., and Müller, B. (2000). Association of genotypes affecting the expression of interleukin-1beta or interleukin-1 receptor antagonist with osteoarthritis. Arthritis Rheum. 43, 2417–2422. doi: 10.1002/1529-0131(200011)43:11<2417::AID-ANR7>3.0.CO;2-R
Nakki, A., Kouhia, S. T., Saarela, J., Harilainen, A., Tallroth, K., Videman, T., et al. (2010). Allelic variants of IL1R1 gene associate with severe hand osteoarthritis. BMC Med. Genet. 11:50. doi: 10.1186/1471-2350-11-50
Netea, M. G., van de Veerdonk, F. L., van der Meer, J. W., Dinarello, C. A., and Joosten, L. A. (2015). Inflammasome-independent regulation of IL-1-family cytokines. Annu. Rev. Immunol. 33, 49–77. doi: 10.1146/annurev-immunol-032414-112306
Prieur, A. M., Kaufmann, M. T., Griscelli, C., and Dayer, J. M. (1987). Specific interleukin-1 inhibitor in serum and urine of children with systemic juvenile chronic arthritis. Lancet 2, 1240–1242. doi: 10.1016/j.autrev.2012.07.024
Punzi, L., Scanu, A., Ramonda, R., and Oliviero, F. (2012). Gout as autoinflammatory disease: new mechanisms for more appropriated treatment targets. Autoimmun. Rev. 12, 66–71. doi: 10.1016/j.autrev.2012.07.024
Punzi, L., and So, A. (2013). Serum uric acid and gout: from the past to molecular biology. Curr. Med. Res. Opin. 29(Suppl. 3), 3–8. doi: 10.1185/03007995.2013.790800
Raghu, H., Lepus, C. M., Wang, Q., Wong, H. H., Lingampalli, N., Oliviero, F., et al. (2016). CCL2/CCR2, but not CCL5/CCR5, mediates monocyte recruitment, inflammation and cartilage destruction in osteoarthritis. Ann. Rheum. Dis. 76, 914–922. doi: 10.1136/annrheumdis-2016-210426
Richette, P., Doherty, M., Pascual, E., Barskova, V., Becce, F., Castañeda-Sanabria, J., et al. (2017). 2016 updated EULAR evidence-based recommendations for the management of gout. Ann. Rheum. Dis. 76, 29–42. doi: 10.1136/annrheumdis-2016-209707
Roberge, C. J., de Médicis, R., Dayer, J. M., Rola-Pleszczynski, M., Naccache, P. H., and Poubelle, P. E. (1994). Crystal-induced neutrophil activation. V. Differential production of biologically active IL-1 and IL-1 receptor antagonist. J. Immunol. 152, 5485–5494.
Saklatvala, J., and Dingle, J. T. (1980). Identification of catabolin, a protein from synovium which induces degradation of cartilage in organ culture. Biochem. Biophys. Res. Commun. 96, 1225–1231. doi: 10.1016/0006-291X(80)90082-0
Saklatvala, J., Pilsworth, L. M., Sarsfield, S. J., Gavrilovic, J., and Heath, J. K. (1984). Pig catabolin is a form of interleukin 1. Cartilage and bone resorb, fibroblasts make prostaglandin and collagenase, and thymocyte proliferation is augmented in response to one protein. Biochem. J. 224, 461–466. doi: 10.1042/bj2240461
Sauerschnig, M., Stolberg-Stolberg, J., Schulze, A., Salzmann, G. M., Perka, C., and Dynybil, C. J. (2014). Diverse expression of selected cytokines and proteinases in synovial fluid obtained from osteoarthritic and healthy human knee joints. Eur. J. Med. Res. 19, 65. doi: 10.1186/s40001-014-0065-5
Scanu, A., Oliviero, F., Gruaz, L., Galozzi, P., Luisetto, R., Ramonda, R., et al. (2016). Synovial fluid proteins are required for the induction of interleukin-1β production by monosodium urate crystals. Scand. J. Rheumatol. 45, 384–393. doi: 10.3109/03009742.2015.1124452
Scanzello, C. R., Umoh, E., Pessler, F., Diaz-Torne, C., Miles, T., Dicarlo, E., et al. (2009). Local cytokine profiles in knee osteoarthritis: elevated synovial fluid interleukin-15 differentiates early from end-stage disease. Osteoarthritis Cartilage 17, 1040–1048. doi: 10.1016/j.joca.2009.02.011
Schett, G., Dayer, J. M., and Manger, B. (2016). Interleukin-1 function and role in rheumatic disease. Nat. Rev. Rheumatol. 12, 14–24. doi: 10.1038/nrrheum.2016.166
Schlesinger, N., Alten, R. E., Bardin, T., Schumacher, H. R., Bloch, M., Gimona, A., et al. (2012). Canakinumab for acute gouty arthritis in patients with limited treatment options: results from two randomised, multicentre, active-controlled, double-blind trials and their initial extensions. Ann. Rheum. Dis. 71, 1839–1848. doi: 10.1136/annrheumdis-2011-200908
Seckinger, P., Klein-Nulend, J., Alander, C., Thompson, R. C., Dayer, J. M., and Raisz, L. G. (1990). Natural and recombinant human IL-1 receptor antagonists block the effects of IL-1 on bone resorption and prostaglandin production. J. Immunol. 145, 4181–4184.
Seckinger, P., Lowenthal, J. W., Williamson, K., Dayer, J. M., and MacDonald, H. R. (1987a). A urine inhibitor of interleukin 1 activity that blocks ligand binding. J. Immunol. 139, 1546–1549.
Seckinger, P., Williamson, K., Balavoine, J. F., Mach, B., Mazzei, G., Shaw, A., et al. (1987b). A urine inhibitor of interleukin 1 activity affects both interleukin 1 alpha and 1 beta but not tumor necrosis factor alpha. J. Immunol. 139, 1541–1545.
Sfriso, P., Priori, R., Valesini, G., Rossi, S., Montecucco, C. M., D’Ascanio, A., et al. (2016). Adult-onset Still’s disease: an Italian multicentre retrospective observational study of manifestations and treatments in 245 patients. Clin. Rheumatol. 35, 1683–1689. doi: 10.1007/s10067-016-3308-8
Shi, Y., Evans, J. E., and Rock, K. L. (2003). Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 425, 516–521. doi: 10.1038/nature01991
Sims, J. E., March, C. J., Cosman, D., Widmer, M. B., MacDonald, H. R., McMahan, C. J., et al. (1988). cDNA expression cloning of the IL-1 receptor, a member of the immunoglobulin superfamily. Science 241, 585–589. doi: 10.1126/science.2969618
Smith, A. J., Keen, L. J., Billingham, M. J., Perry, M. J., Elson, C. J., Kirwan, J. R., et al. (2004). Extended haplotypes and linkage disequilibrium in the IL1R1-IL1A-IL1B-IL1RN gene cluster: association with knee osteoarthritis. Genes Immun. 5, 451–460. doi: 10.1038/sj.gene.6364107
So, A., De Smedt, T., Revaz, S., and Tschopp, J. (2007). A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res. Ther. 9, R28. doi: 10.1186/ar2143
Srinivasula, S. M., Poyet, J. L., Razmara, M., Datta, P., Zhang, Z., and Alnemri, E. S. (2002). The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J. Biol. Chem. 277, 21119–21122. doi: 10.1074/jbc.C200179200
Staats, H. F., and Ennis, F. A. Jr. (1999). IL-1 Is an effective adjuvant for mucosal and systemic immune responses when coadministered with protein immunogens. J. Immunol. 162, 6141–6147.
Stylianou, E., O’Neill, L. A., Rawlinson, L., Edbrooke, M. R., Woo, P., and Saklatvala, J. (1992). Interleukin 1 induces NF-kappa B through its type I but not its type II receptor in lymphocytes. J. Biol. Chem. 267, 15836–15841.
Terkeltaub, R. A., Schumacher, H. R., Carter, J. D., Baraf, H. S., Evans, R. R., Wang, J., et al. (2013). Rilonacept in the treatment of acute gouty arthritis: a randomized, controlled clinical trial using indomethacin as the active comparator. Arthritis Res. Ther. 15, R25. doi: 10.1186/ar4159
Thome, M., Hofmann, K., Burns, K., Martinon, F., Bodmer, J. L., Mattmann, C., et al. (1998). Identification of CARDIAK, a RIP-like kinase that associates with caspase-1. Curr. Biol. 8, 885–888. doi: 10.1016/S0960-9822(07)00352-1
Thornberry, N. A., Bull, H. G., Calaycay, J. R., Chapman, K. T., Howard, A. D., Kostura, M. J., et al. (1992). A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 356, 768–774. doi: 10.1038/356768a0
Veltri, S., and Smith, J. W. II. (1996). Interleukin 1 trials in cancer patients: a review of the toxicity, antitumor and hematopoietic effects. Oncologist 1, 190–200. doi: 10.1002/stem.140164
Keywords: interleulin-1, interleukin-1 antagonist, rheumatoid arthritis, inflammasome, autoinflammatory diseases
Citation: Dayer J-M, Oliviero F and Punzi L (2017) A Brief History of IL-1 and IL-1 Ra in Rheumatology. Front. Pharmacol. 8:293. doi: 10.3389/fphar.2017.00293
Received: 21 March 2017; Accepted: 08 May 2017;
Published: 23 May 2017.
Edited by:
Stephan Immenschuh, Hannover Medical School, GermanyReviewed by:
Satish Ramalingam, Chettinad Academy of Research and Education, IndiaSabina Janciauskiene, Hannover Medical School, Germany
Copyright © 2017 Dayer, Oliviero and Punzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean-Michel Dayer, SmVhbi1NaWNoZWwuRGF5ZXJAdW5pZ2UuY2g=