 Chen-Ming Zeng
Chen-Ming Zeng Lin-Lin Chang
Lin-Lin Chang Hong Zhu
Hong Zhu Bo Yang
Bo Yang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 14 March 2017
Sec. Cancer Molecular Targets and Therapeutics
Volume 8 - 2017 | https://doi.org/10.3389/fphar.2017.00119
This article is part of the Research Topic Cancer Metabolism: Molecular Targeting and Implications for Therapy View all 11 articles
Aldo–keto reductases comprise of AKR1C1–AKR1C4, four enzymes that catalyze NADPH dependent reductions and have been implicated in biosynthesis, intermediary metabolism, and detoxification. Recent studies have provided evidences of strong correlation between the expression levels of these family members and the malignant transformation as well as the resistance to cancer therapy. Mechanistically, most studies focus on the catalytic-dependent function of AKR1C isoforms, like their impeccable roles in prostate cancer, breast cancer, and drug resistance due to the broad substrates specificity. However, accumulating clues showed that catalytic-independent functions also played critical roles in regulating biological events. This review summarizes the catalytic-dependent and -independent roles of AKR1Cs, as well as the small molecule inhibitors targeting these family members.
The AKR1C1–AKR1C4 genes are located on chromosome 10 p15-p14 and comprise of 12 exons. And the average molecular weight of enzymes is estimated to be 34–42 kDa. These enzymes share a high percentage of amino-acid sequence identity that ranges from 84 to 98%. In particular, AKR1C1 and AKR1C2, differ by only seven amino-acid residues (Jez et al., 1997).
The AKR1C isoforms play pivotal roles in NADPH dependent reductions. Therefore, the enzymes are highly related to malignant cancer involve NADPH reductive progress like PCa, breast cancer, and etc. Whereas, discoveries about the catalytic-independent role of the AKR1C isoforms, has also been revealed, including their function as a coactivator, regulation in E3-ligase-ubiquirin system, cell sensitivity, apoptosis, and metastasis.
AKR1C isoforms catalyze NADPH dependent reductions at the C3, C5, C17, and C20 positions on the steroid nucleus and side-chain and act as 3-keto-, 17-keto-, and 20- ketosteroid reductases to varying extents in humans (Rižner and Penning, 2014).
AKR1C4 is mainly liver-specific (Deyashiki et al., 1994) and recently it has been proved to be related to manic/hypomanic irritability in males (Johansson et al., 2011, 2012). AKR1C4 efficiently catalyzes the reduction of 5α-pregnane-3,20-dione to yield 3α-hydroxy-5α-pregnan-20-one (allopregnanolone) which is the precursor of and rosterone (Higaki et al., 2003).
The AKR1C3 protein is also known as PGF synthase that catalyzes the conversion of prostaglandins H2 and D2 into PGF2α and 9α,11β-PGF2α respectively (Suzuki-Yamamoto et al., 1999). It has the highest catalytic efficiency of the AKR1C enzymes to interconvert testosterone with Δ4-androstene-3,17-dione (Sharma et al., 2006). The enzyme will also reversibly reduce 5α-DHT, estrogen and progesterone to produce 3α-androstanediol, 17β-estradiol and 20α-hydroxprogesterone, respectively (Penning et al., 2001). There are also significant correlations between the expression levels of AKR1C3 and CRPC. And AKR1C3 overexpression is proved to be a promising biomarker for PCa progression (Tian et al., 2014; Hagberg Thulin et al., 2016). Positive AKR1C3 immunoreactivity was also extensively present in both adenocarcinoma and squamous cell carcinoma arising from the lung and the gastroesophageal junction (Miller et al., 2012). Strong correlations between AKR1C3 and tumors were also demonstrated in human colorectal cancer (Hanada et al., 2012; Nakarai et al., 2015), columnar epithelium (Miller et al., 2012), and endometriosis (Sinreih et al., 2015a; Gibson et al., 2016).
While AKR1C4 and AKR1C3 are almost exclusively in the liver and prostate respectively, AKR1C1 and AKR1C2 are most prominent in the mammary glands includes breast cancer, endometrial cancer, colorectal cancer (Hanada et al., 2012; Hofman et al., 2015; Sinreih et al., 2015b; Wang et al., 2016; Wenners et al., 2016). AKR1C2, is also known as bile-acid binding protein and DD2, has lower catalytic efficiencies but preferentially reduces 3-ketosteroids. AKR1C2 preferentially reduces DHT to the weak metabolite 5α-androstane-3α,17β-diol (3α-diol) without conversion of 3α-diol to DHT in the PC-3 cell line (Ji et al., 2003). Progesterone is found to be essential for maintenance of early pregnancy (Agrawal and Hirsch, 2012) and blunting estrogen signaling in endometrial cancer (Ham et al., 1975). And AKR1C1 is the predominant 20-ketosteroid reductase in man and play an important role in reductive inactivation of progesterone into 20α-DHP (Rižner et al., 2006).
Prostate cancer is the most commonly diagnosed solid tumor and the second cause of cancer-related mortality (Tanaka et al., 1993). Androgens drive PCa cell growth via the AR. Accordingly, ADT has been the mainstay in the treatment of advanced PCa patients. However, patients eventually relapse and develop into the lethal form of the disease, termed CRPC (Zong and Goldstein, 2013; Zhang et al., 2016a).
Recent evidence suggests that CRPC may be caused by augmented androgen/AR signaling, generally involving AR overexpression (Yuan and Balk, 2009; Taylor et al., 2010; Shiota et al., 2011). Therefore, newer therapies that target androgen metabolizing AR are being developed and have shown clinical efficacy, indicating the continued importance of the androgen signaling axis in advanced PCa (Higano and Crawford, 2011).
AKR1C3 plays an important role for the biosynthesis of testosterone and estradiol. Elevated levels of AKR1C3 expression in CRPC over PCa have been reported (Tian et al., 2014). The differential distribution of AKR1C isoforms includes AKR1C1 and AKR1C2 has been implicated in the maintenance of a pro-estrogenic or a pro-androgenic state which contributes to development of CRPC as well (Hofland et al., 2010).
High affinity binding of DHT to the AR initiates androgen-dependent gene activation and contributes to PCa development and progression. DHT is synthesized predominantly by 5α-reduction of testosterone (5α-DHT) (Mohler et al., 2011).
In the prostate, 5α-DHT can be reduced to 3α-diol through the action of reductive 3α-HSDs. Between the two major 3α-HSD isozymes, AKR1C2 and AKR1C3, in human prostate, both isozymes catalyze the reversible reduction of 5α-DHT activity toward the weakly androgenic metabolite 3α-diol, which is recognized as a weak androgen with low affinity toward the AR. AKR1C1, which is associated with the HSD3B pathway of DHT metabolism, expressed at higher levels than AKR1C2, catalyzes the irreversible conversion of DHT to 3β-diol (Zhang et al., 2016a). Therefore, the 3α-HSD regulate the occupancy of the AR (Ji et al., 2003; Yepuru et al., 2013).
Recent study has found a first-in-class orally available inhibitor of AKR1C3, ASP9521, which demonstrated anti-tumor activity in vitro and in vivo preclinical models (Loriot et al., 2014). SN33638, a selective inhibitor of AKR1C3, can prevent the conversion of PGD2 to11β-PGF2α. However, due to the involvement of additional enzymes in testosterone and 17β-estradiol synthesis, its activity at preventing steroid hormone reduction and resultant CRPC and ER-positive breast cancer growth is limited to small subpopulation of CRPC patients with tumors that have upregulated AKR1C3 expression and are dependent on AKR1C3 for producing the testosterone required for their growth (Yin et al., 2014).
Breast cancer is the most frequently diagnosed cancer in women worldwide. The ovarian steroid hormone, progesterone, and its nuclear receptor, the progesterone receptor, are implicated in the progression of breast cancer (Ross et al., 2000). Progesterone binding to its receptor supports an increased progesterone-responsive gene expression and therewith tumor growth and progression (Ji et al., 2004).
AKR1C3 is known to be abundantly expressed in breast cancer tissues, and high levels are often associated with adverse clinical outcome. AKR1C3 is capable to produce intratumorally testosterone and 17β-estradiol by reducing the androgen precursors and estrogen, respectively. The local conversion of less potent hormones to more potent ones will lead to nuclear receptor activation and tumor progression. Therefore, AKR1C3 has recently been identified as a potential therapeutic target in both CRPC and ER-positive breast cancer. AKR1C3 is responsible for the reduction of PGD2 to11β-PGF2α, both of which were reported to demonstrate similar affinities toward their cognate receptor, Prostaglandin receptor (FP receptor). And the action of FP receptor ligands results in carcinoma cell survival in breast cancer (Yoda et al., 2015). AKR1C3 is also associated with doxorubicin resistance in human breast cancer (Zhong et al., 2015).
However, a large proportion (about 30–60%) of breast tumors are PR negative (McGuire et al., 1982; Taucher et al., 2003; Rexhepaj et al., 2008), and about 90% of normal proliferating breast epithelial cells are receptor negative (Robinson et al., 2000). Patients with receptor-negative tumors do not respond to current steroid hormone-based therapies and generally have significantly higher risk of recurrence and mortality compared with patients with tumors that are ER- and/or PR-positive (Wiebe et al., 2013). Overall, this means that for receptor-negative breast cancers, current explanations based on estrogen and progesterone actions and receptors are inadequate, and the related hormone-based therapies are ineffective. Therefore, it is critical to reveal the potential mechanism in regulating breast cancer.
The expression of AKR1C1 and AKR1C2 was found reduced in tumorous breast tissue (Lewis et al., 2004). Then in vitro studies had shown that progesterone metabolites can regulate PR-negative breast cell tumor formation and growth as well as tumor regression and maintenance of normalcy. Progesterone is degraded to its metabolite 20α-DHP by AKR1C1 and to 3α-HP by AKR1C2. These metabolites promote suppression of cell proliferation and adhesion. These 20α-DHP and 3α-HP bind to specific plasma membrane receptors, separate from classical HRs, and influence anti-proliferative functions on mitosis, apoptosis, and cytoskeletal and adhesion molecules (Lewis et al., 2004). Evidence has also been presented that progesterone metabolites, 5αP exhibits pro-cancer effects.
Resistance to anticancer drugs and organ specific toxicity are two of the major problems in chemotherapy. Although this phenomenon has been repeatedly observed in the experimental setting, to our knowledge it has not been clinically exploited. An emerging theme is the role of AKRs in cancer chemotherapeutic drug resistance (Barski et al., 2008). And the induction of AKRs was found to be correlated with changes in drug’s properties.
Among the mechanisms of resistance, metabolic inactivation by carbonyl reduction is a major cause of chemotherapy failure that applies to drugs bearing a carbonyl moiety. Oracin is a promising potential cytostatic drug which is presently in phase II clinical trials. Continuously studies found that AKR1C1, AKR1C2, and AKR1C4 mediate the carbonyl reduction of the novel anticancer drug oracin (6-[2-(2-hydroxyethyl)-aminoethyl]-5,11-dioxo-5,6-dihydro-11H-indeno [1,2-c]isoquinoline) to its inactive metabolite DHO (Wsol et al., 2007; Novotna et al., 2008).
AKR1C3 does also catalyze the inactivation of the anticancer drug doxorubicin. Doxorubicin undergoes metabolic detoxification by carbonyl reduction to the corresponding C13 alcohol metabolite, doxorubicinol (Minotti et al., 2004). In comparison to doxorubicin, doxorubicinol exhibited dramatically reduced cytotoxicity, reduced DNA-binding activity, and strong localization to extra nuclear lysosomes (Heibein et al., 2012). Induction of AKR1C1 and AKR1C3 has been shown to efficiently abolish the efficacy of daunorubicin chemotherapy for leukemic U937 cells by metabolizing both DNR and cytotoxic aldehydes derived from ROS-linked lipid peroxidation (Matsunaga et al., 2014). Aldo–keto reductase 1C3 (AKR1C3) is also linked to doxorubicin resistance in human breast cancer which resulted from activation of anti-apoptosis PTEN/Akt pathway via PTEN loss (Zhong et al., 2015). And the reduction of daunorubicin and idarubicin, which is catalyzed by AKR1C3, also contributes to the resistance of cancer cells to anthracycline treatment (Hofman et al., 2014).
The biochemical basis for resistance to cisplatin in a human ovarian cancer cell line has also been reported to be due to overexpression of the AKR1C1 though the underlying mechanism has not been revealed yet (Deng et al., 2002). Knockdown of both AKR1C1 and AKR1C3 in the resistant cells or treatment of the cells with specific inhibitors of the AKRs increased the sensitivity to cisplatin toxicity (Matsunaga et al., 2013).
Previous studies about AKR1C isoforms mostly revealed their biological function in an catalytic-dependent role. However, their non-catalytic functions have remained elusive until Yepuru M. found that AKR1C3 can function as an AR-selective coactivator.
Early studies presented that AKR1C3 catalyzes the adrenal androgens into testosterone, which binds to AR or get converted to DHT, resulting in ligand occupancy of AR. Therefore, AKR1C3 is proposed to play a vital role in the emergence of CRPC by activation of its enzyme activity.
Notably, it was recently reported that AKR1C3 can regulate AR activity in a catalytically independent role. Yepuru M. and his co-workers found that as an enzyme converts androstenedione to testosterone, AKR1C3 also acts as a selective coactivator for the AR to promote CRPC growth. AR can interact with AKR1C3 and get recruited to the ARE on the promoter of androgen responsive genes. Thus, recruits related cofactors leading to activation of transcription on reduction of target genes. And while the full-length of proteins is necessary to mediate AKR1C3’s enzymatic functions, amino acids 171–237 were sufficient to mediate the AR activation. These observations identify AKR1C3 a high priority target in PCa progression, considering its dual role as a coactivator and androgen biosynthetic enzyme (Figure 1).
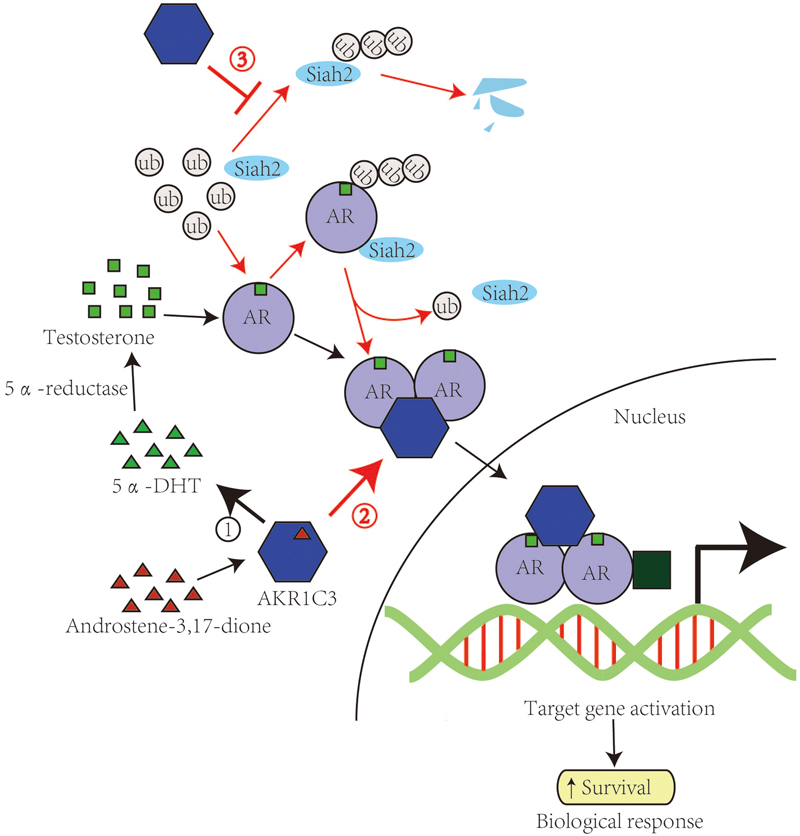
FIGURE 1. AKR1C3 promotes PCa via catalytic-dependent and independent roles. The catalytic and catalytic-independent functions of AKR1C3 in the progression of PCa are shown in black arrows and red arrows respectively. AKR1C3 catalyzes androstene-3,17-dione into 5α-DHT, which is reduced into testosterone by 5α-reductase and binds to androgen recptor. AKR1C3 can also bind to dimerizated and phosphorylated androgen receptors and function as a coactivator of AR. AKR1C3 is able to stabilize Siah2 and thus enhance AR transcriptional activity.
Another example of a catalytically independent role of AKR1C3 on AR activity was found in regulating Siah2 stability. Ubiquitin ligase Siah2 was reported to enhance AR transcriptional activity and PCa cell growth (Qi et al., 2013). Further study found that AKR1C3 shows the ability to bind and stabilize Siah2 by blocking Siah2 self-ubiquitination and degradation (Fan et al., 2015).
Interactions between steroid biosynthetic enzymes and steroid receptors may be exceedingly complex and involved in a variety of hormone-dependent cancers (Yepuru et al., 2013). Future clinical trials with AKR1C3 inhibitors will be needed to show their potential to be the next generation of tissue-specific therapeutics for CRPC. Therefore, identification of mechanisms underlying the non-catalytic function of AKR1C3 may provide new targets for development of novel AKR1C3 inhibitors that complement inhibitors targeting AKR1C3 catalytic activity as potential CRPC therapy (Figure 1).
The AKRs were also found to be implicated in cell sensitivity, growth, metastasis, and apoptosis in a catalytic independent role, though the underlying mechanisms are still not revealed yet.
Firstly, there was evidence that after short-term and long-term cadmium exposure, the expression of AKR1C1 was elevated which implies the role of ARKs in cell sensitivity (Garrett et al., 2013). Then studies found that AKR1C3 siRNA significantly enhanced cell radio sensitivity (Xie et al., 2013). Consistently with this, overexpression of AKR1C3 enhances resistance of cancer cells to radiation (Xiong et al., 2014; Sun et al., 2016). Participation of AKR1C3 in cancer development is also well proven. Down-regulation of AKR1C3significantly decreases PCa and MCF7 breast cancer cell growth (Downs et al., 2011; Zhang et al., 2016b). Besides, silencing of AKR1C3 increases LCN2 expression and inhibits metastasis in cervical cancer (Wu et al., 2014). AKR1C2 is mostly involved in the process of metastasis. Li et al. (2016) identified two powerful genes in the liver cancer metastasis process, AEG-1 and AKR1C2. And then AEG-1 was proved to promote metastasis through downstream AKR1C2 and NF1 in liver cancer (Li et al., 2014b, 2016). Since AEG-1 and AKR1C2 promote metastasis, inhibiting those two genes would effectively control metastasis.
These findings may provide novel potential clinical targets against metastasis in liver cancer patients. Notably, AKR1C2 is also involved in apoptosis induced by Panax ginseng polysaccharide (Li et al., 2014a).
Several types of AKR1C1 inhibitors have been identified, including, benzodiazepines, steroid carboxylates, phytoestrogens, derivatives of pyrimidine, phthalimide, anthranilic acid and cyclopentane, flavones and ruthenium complexes (Usami et al., 2002; Bauman et al., 2005; Brozic et al., 2006b, 2009; Stefane et al., 2009; Liu et al., 2011; Traven et al., 2015). Notably, 3-bromo-5-phenylsalicylic acid, an inhibitor designed based on the structure of AKR1C1 in ternary complex with NADP+ and DCL, its phenyl group targets a non-conserved hydrophobic pocket in the active site of the enzyme lined by residues Leu54, Leu308 and Phe311, resulting in a 21-fold improved potency (Ki = 4 nM) over the structurally similar AKR1C2 (Carbone et al., 2009). Moreover, compound 3-bromo-5-phenylsalicylic acid significantly decreased the metabolism of progesterone in the cells with an IC50 value of 460 nM.
Structure between AKR1C1 and AKR1C2 is rather similar, only differs by one active-site residue (Leu54 versus Val54). Therefore, the selectivity of inhibitors targeting AKR1C1 and AKR1C2 is rather low, and newly designed inhibitors that mostly interact with Leu54 in AKR1C1 are needed as to improve the selectivity over AKR1C2. Derivatives of BPSA, 3-chloro-5-phenylsalicylic acid (Ki = 0.86 nM), is 24-fold more selective for AKR1C1 over AKR1C2. Furthermore, the compound potently inhibited the metabolism of progesterone by AKR1C1 in the cells with an IC50 value of 100 nM (El-Kabbani et al., 2010).
AKR1C3 is inhibited by several classes of AKR1C3 inhibitors, including cinnamic acid (Brozic et al., 2006a), non-steroidal anti-inflammatory drugs (NSAIDs) and their derivatives (Gobec et al., 2005; Byrns et al., 2008; Liedtke et al., 2013), steroid hormone analoges (Bydal et al., 2009), flavonoids (Skarydova et al., 2009), cyclopentanes (Stefane et al., 2009), benzoic acids (Adeniji et al., 2011; Jamieson et al., 2012), progestins (Beranic et al., 2011), baccharin analogs (Zang et al., 2015), ruthenium complexes (Kljun et al., 2016), and the most widely used anti-diabetes drugs, sulfonylureas (Zhao et al., 2015). Most inhibitors of AKR1C3 are carboxylic acids, whose transport into cells is likely dominated by carrier-mediated processes. Therefore, development of non-carboxylate inhibitors of AKR1C3 like 1-(4-(piperidin-1-ylsulfonyl)phenyl)pyrrolidin-2-ones (Heinrich et al., 2013) and morpholylureasis essential (Flanagan et al., 2014).
Critical concern in exploiting AKR1C3 inhibitors is the cross inhibition of AKR1C subfamily members, as they have high amino acid sequence identity and structural similarity. This prompts us to find new inhibitors with new molecular skeleton or binding domains (Table 1).
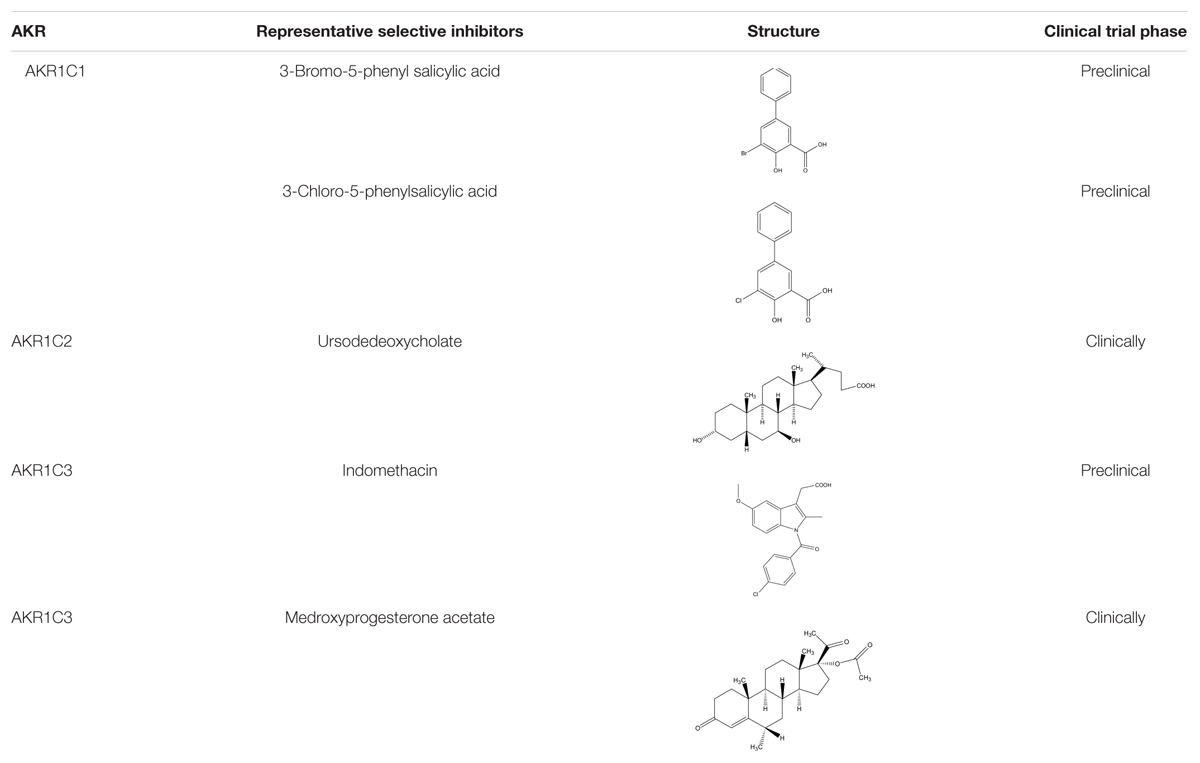
TABLE 1. Small molecular inhibitors.
The aldo–keto reductases AKR1C1–AKR1C4 is a series of four proteins with a multitude of functions. Recent advances have been made in terms of the roles played by this family among a variety of diseases, particularly, those functions related to their catalytic activities. However, some clues showed that the catalytic-independent functions of these proteins are totally arousing as well, and aiding in highlight the AKR1C family as promising anti-cancer targets for cancer treatment. Nonetheless, exploration of more potent AKR1C-targeting strategies to interrupt their catalytic activities or the other critical functions is still in urgent need. We rest assured that future developments in this area will absolutely enrich our understanding of the AKR1C isoforms and provide new avenues for using this knowledge to improve cancer therapy (Table 2).

TABLE 2. Role of human AKRs in health and disease.
BY and HZ conceived, designed the conception of review article, and made the amendments of the paper. C-MZ conducted the paper. L-LC, M-DY, JC, and Q-JH collected the related research articles.
This study was supported by National Natural Science Foundation of China (81673458).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
3α-HP, 3α-hydroxyprogesterone; 5αP, 5α-dihydroprogesterone; 20α-DHP, 20α-dihydroprogesterone; ADT, androgen deprivation therapy; AKR1C isoforms, Aldo–keto reductases; AR, androgen receptor; CRPC, castration-resistant prostate cancer; DHO, dihydrooracin; DHT, dihydrotestosterone; ER, estrogen receptor; HR, hormone receptors; PCa, prostate cancer; PGF, prostaglandin factor; PR, prostaglandin receptor.
Adeniji, A. O., Twenter, B. M., Byrns, M. C., Jin, Y., Winkler, J. D., and Penning, T. M. (2011). Discovery of substituted 3-(phenylamino)benzoic acids as potent and selective inhibitors of type 5 17beta-hydroxysteroid dehydrogenase (AKR1C3). Bioorg. Med. Chem. Lett. 21, 1464–1468. doi: 10.1016/j.bmcl.2011.01.010
Agrawal, V., and Hirsch, E. (2012). Intrauterine infection and preterm labor. Paper Presented at: Seminars in Fetal and Neonatal Medicine. Amsterdam: Elsevier.
Barski, O. A., Tipparaju, S. M., and Bhatnagar, A. (2008). The aldo-keto reductase superfamily and its role in drug metabolism and detoxification. Drug Metab. Rev. 40, 553–624. doi: 10.1080/03602530802431439
Bauman, D. R., Rudnick, S. I., Szewczuk, L. M., Jin, Y., Gopishetty, S., and Penning, T. M. (2005). Development of nonsteroidal anti-inflammatory drug analogs and steroid carboxylates selective for human aldo-keto reductase isoforms: potential antineoplastic agents that work independently of cyclooxygenase isozymes. Mol. Pharmacol. 67, 60–68. doi: 10.1124/mol.104.006569
Beranic, N., Gobec, S., and Rizner, T. L. (2011). Progestins as inhibitors of the human 20-ketosteroid reductases, AKR1C1 and AKR1C3. Chem. Biol. Interact 191, 227–233. doi: 10.1016/j.cbi.2010.12.012
Brozic, P., Cesar, J., Kovac, A., Davies, M., Johnson, A. P., Fishwick, C. W., et al. (2009). Derivatives of pyrimidine, phthalimide and anthranilic acid as inhibitors of human hydroxysteroid dehydrogenase AKR1C1. Chem. Biol. Interact 178, 158–164. doi: 10.1016/j.cbi.2008.10.019
Lewis, M. J., Wiebe, J. P., and Heathcote, J. G. (2004). Expression of progesterone metabolizing enzyme genes (AKR1C1, AKR1C2, AKR1C3, SRD5A1, SRD5A2) is altered in human breast carcinoma. BMC Cancer 4:27. doi: 10.1186/1471-2407-4-27
Brozic, P., Golob, B., Gomboc, N., Rizner, T. L., and Gobec, S. (2006a). Cinnamic acids as new inhibitors of 17beta-hydroxysteroid dehydrogenase type 5 (AKR1C3). Mol. Cell. Endocrinol. 248, 233–235.
Brozic, P., Smuc, T., Gobec, S., and Rizner, T. L. (2006b). Phytoestrogens as inhibitors of the human progesterone metabolizing enzyme AKR1C1. Mol. Cell. Endocrinol. 259, 30–42.
Bydal, P., Luu-The, V., Labrie, F., and Poirier, D. (2009). Steroidal lactones as inhibitors of 17β-hydroxysteroid dehydrogenase type 5: chemical synthesis, enzyme inhibitory activity, and assessment of estrogenic and androgenic activities. Eur. J. Med. Chem. 44, 632–644. doi: 10.1016/j.ejmech.2008.03.020
Byrns, M. C., Steckelbroeck, S., and Penning, T. M. (2008). An indomethacin analogue, N-(4-chlorobenzoyl)-melatonin, is a selective inhibitor of aldo-keto reductase 1C3 (type 2 3α-HSD, type 5 17β-HSD, and prostaglandin F synthase), a potential target for the treatment of hormone dependent and hormone independent malignancies. Biochem. Pharmacol. 75, 484–493. doi: 10.1016/j.bcp.2007.09.008
Carbone, V., Zhao, H. T., Chung, R., Endo, S., Hara, A., and El-Kabbani, O. (2009). Correlation of binding constants and molecular modelling of inhibitors in the active sites of aldose reductase and aldehyde reductase. Bioorg. Med. Chem. 17, 1244–1250. doi: 10.1016/j.bmc.2008.12.024
Deng, H. B., Parekh, H. K., Chow, K. C., and Simpkins, H. (2002). Increased expression of dihydrodiol dehydrogenase induces resistance to cisplatin in human ovarian carcinoma cells. J. Biol. Chem. 277, 15035–15043. doi: 10.1074/jbc.M112028200
Deyashiki, Y., Ogasawara, A., Nakayama, T., Nakanishi, M., Miyabe, Y., Sato, K., et al. (1994). Molecular cloning of two human liver 3 α-hydroxysteroid/dihydrodiol dehydrogenase isoenzymes that are identical with chlordecone reductase and bile-acid binder. Biochem. J. 299, 545–552. doi: 10.1042/bj2990545
Downs, T. M., Burton, D. W., Araiza, F. L., Hastings, R. H., and Deftos, L. J. (2011). PTHrP stimulates prostate cancer cell growth and upregulates aldo-keto reductase 1C3. Cancer Lett. 306, 52–59. doi: 10.1016/j.canlet.2011.02.027
El-Kabbani, O., Scammells, P. J., Day, T., Dhagat, U., Endo, S., Matsunaga, T., et al. (2010). Structure-based optimization and biological evaluation of human 20alpha-hydroxysteroid dehydrogenase (AKR1C1) salicylic acid-based inhibitors. Eur. J. Med. Chem. 45, 5309–5317. doi: 10.1016/j.ejmech.2010.08.052
Fan, L., Peng, G., Hussain, A., Fazli, L., Guns, E., Gleave, M., et al. (2015). The steroidogenic enzyme AKR1C3 regulates stability of the ubiquitin ligase Siah2 in prostate cancer cells. J. Biol. Chem. 290, 20865–20879. doi: 10.1074/jbc.M115.662155
Flanagan, J. U., Atwell, G. J., Heinrich, D. M., Brooke, D. G., Silva, S., Rigoreau, L. J., et al. (2014). Morpholylureas are a new class of potent and selective inhibitors of the type 5 17-beta-hydroxysteroid dehydrogenase (AKR1C3). Bioorg. Med. Chem. 22, 967–977. doi: 10.1016/j.bmc.2013.12.050
Garrett, S. H., Clarke, K., Sens, D. A., Deng, Y., Somji, S., and Zhang, K. K. (2013). Short and long term gene expression variation and networking in human proximal tubule cells when exposed to cadmium. BMC Med. Genomics 6(Suppl. 1):S2. doi: 10.1186/1755-8794-6-S1-S2
Gibson, D. A., Simitsidellis, I., Cousins, F. L., Critchley, H. O., and Saunders, P. T. (2016). Intracrine androgens enhance decidualization and modulate expression of human endometrial receptivity genes. Sci. Rep. 6:19970. doi: 10.1038/srep19970
Gobec, S., Brožiè, P., and Rižner, T. L. (2005). Nonsteroidal anti-inflammatory drugs and their analogues as inhibitors of aldo-keto reductase AKR1C3: new lead compounds for the development of anticancer agents. Bioorg. Med. Chem. Lett. 15, 5170–5175. doi: 10.1016/j.bmcl.2005.08.063
Hagberg Thulin, M., Nilsson, M. E., Thulin, P., Ceraline, J., Ohlsson, C., Damber, J. E., et al. (2016). Osteoblasts promote castration-resistant prostate cancer by altering intratumoral steroidogenesis. Mol. Cell. Endocrinol. 422, 182–191. doi: 10.1016/j.mce.2015.11.013
Ham, E., Cirillo, V., Zanetti, M. E., and Kuehl, F. (1975). Estrogen-directed synthesis of specific prostaglandins in uterus. Proc. Natl. Acad. Sci. U.S.A. 72, 1420–1424. doi: 10.1073/pnas.72.4.1420
Hanada, N., Takahata, T., Zhou, Q., Ye, X., Sun, R., Itoh, J., et al. (2012). Methylation of the KEAP1 gene promoter region in human colorectal cancer. BMC Cancer 12:66. doi: 10.1186/1471-2407-12-66
Heibein, A. D., Guo, B., Sprowl, J. A., Maclean, D. A., and Parissenti, A. M. (2012). Role of aldo-keto reductases and other doxorubicin pharmacokinetic genes in doxorubicin resistance, DNA binding, and subcellular localization. BMC Cancer 12:381. doi: 10.1186/1471-2407-12-381
Heinrich, D. M., Flanagan, J. U., Jamieson, S. M., Silva, S., Rigoreau, L. J., Trivier, E., et al. (2013). Synthesis and structure-activity relationships for 1-(4-(piperidin-1-ylsulfonyl)phenyl)pyrrolidin-2-ones as novel non-carboxylate inhibitors of the aldo-keto reductase enzyme AKR1C3. Eur. J. Med. Chem. 62, 738–744. doi: 10.1016/j.ejmech.2013.01.047
Higaki, Y., Usami, N., Shintani, S., Ishikura, S., El-Kabbani, O., and Hara, A. (2003). Selective and potent inhibitors of human 20α-hydroxysteroid dehydrogenase (AKR1C1) that metabolizes neurosteroids derived from progesterone. Chem. Biol. Interact. 143, 503–513. doi: 10.1016/S0009-2797(02)00206-5
Higano, C. S., and Crawford, E. D. (2011). New and emerging agents for the treatment of castration-resistant prostate cancer. Urol. Oncol. 29, S1–S8. doi: 10.1016/j.urolonc.2011.08.013
Hofland, J., van Weerden, W. M., Dits, N. F., Steenbergen, J., van Leenders, G. J., Jenster, G., et al. (2010). Evidence of limited contributions for intratumoral steroidogenesis in prostate cancer. Cancer Res. 70, 1256–1264. doi: 10.1158/0008-5472.CAN-09-2092
Hofman, J., Malcekova, B., Skarka, A., Novotna, E., and Wsol, V. (2014). Anthracycline resistance mediated by reductive metabolism in cancer cells: the role of aldo-keto reductase 1C3. Toxicol. Appl. Pharmacol. 278, 238–248. doi: 10.1016/j.taap.2014.04.027
Hofman, J., Skarka, A., Havrankova, J., and Wsol, V. (2015). Pharmacokinetic interactions of breast cancer chemotherapeutics with human doxorubicin reductases. Biochem. Pharmacol. 96, 168–178. doi: 10.1016/j.bcp.2015.05.005
Jamieson, S. M. F., Brooke, D. G., Heinrich, D., Atwell, G. J., Silva, S., Hamilton, E. J., et al. (2012). 3-(3,4-dihydroisoquinolin-2(1 H)-ylsulfonyl)benzoic acids: highly potent and selective inhibitors of the type 5 17-β-hydroxysteroid dehydrogenase AKR1C3. J. Med. Chem. 55, 7746–7758. doi: 10.1021/jm3007867
Jez, J. M., Bennett, M. J., Schlegel, B. P., Lewis, M., and Penning, T. M. (1997). Comparative anatomy of the aldo-keto reductase superfamily. Biochem. J. 326, 625–636. doi: 10.1042/bj3260625
Ji, Q., Aoyama, C., Nien, Y. D., Liu, P. I., Chen, P. K., Chang, L., et al. (2004). Selective loss of AKR1C1 and AKR1C2 in breast cancer and their potential effect on progesterone signaling. Cancer Res. 64, 7610–7617. doi: 10.1158/0008-5472.CAN-04-1608
Ji, Q., Chang, L., VanDenBerg, D., Stanczyk, F. Z., and Stolz, A. (2003). Selective reduction of AKR1C2 in prostate cancer and its role in DHT metabolism. Prostate 54, 275–289.
Johansson, A. G., Nikamo, P., Schalling, M., and Landen, M. (2011). AKR1C4 gene variant associated with low euthymic serum progesterone and a history of mood irritability in males with bipolar disorder. J. Affect. Disord. 133, 346–351. doi: 10.1016/j.jad.2011.04.009
Johansson, A. G., Nikamo, P., Schalling, M., and Landen, M. (2012). Polymorphisms in AKR1C4 and HSD3B2 and differences in serum DHEAS and progesterone are associated with paranoid ideation during mania or hypomania in bipolar disorder. Eur. Neuropsychopharmacol. 22, 632–640. doi: 10.1016/j.euroneuro.2012.01.007
Kljun, J., Anko, M., Traven, K., Sinreih, M., Pavlič, R., Peršič, Š., et al. (2016). Pyrithione-based ruthenium complexes as inhibitors of aldo-keto reductase 1C enzymes and anticancer agents. Dalton Trans. 45, 11791–11800. doi: 10.1039/c6dt00668j
Li, C., Tian, Z. N., Cai, J. P., Chen, K. X., Zhang, B., Feng, M. Y., et al. (2014a). Panax ginseng polysaccharide induces apoptosis by targeting Twist/AKR1C2/NF-1 pathway in human gastric cancer. Carbohydr. Polym. 102, 103–109. doi: 10.1016/j.carbpol.2013.11.016
Li, C., Wu, X., Zhang, W., Li, J., Liu, H., Hao, M., et al. (2014b). AEG-1 promotes metastasis through downstream AKR1C2 and NF1 in liver cancer. Oncol. Res. 22, 203–211. doi: 10.3727/096504015X14386062091352
Li, C., Wu, X., Zhang, W., Li, J., Liu, H., Hao, M., et al. (2016). High-content functional screening of AEG-1 and AKR1C2 for the promotion of metastasis in liver cancer. J. Biomol. Screen. 21, 101–107. doi: 10.1177/1087057115603310
Liedtke, A. J., Adeniji, A. O., Chen, M., Byrns, M. C., Jin, Y., Christianson, D. W., et al. (2013). Development of potent and selective indomethacin analogues for the inhibition of AKR1C3 (Type 5 17beta-hydroxysteroid dehydrogenase/prostaglandin F synthase) in castrate-resistant prostate cancer. J. Med. Chem. 56, 2429–2446. doi: 10.1021/jm3017656
Liu, X., Piao, Y. S., and Arnold, J. T. (2011). Transforming growth factor beta1 increase of hydroxysteroid dehydrogenase proteins is partly suppressed by red clover isoflavones in human primary prostate cancer-derived stromal cells. Carcinogenesis 32, 1648–1654. doi: 10.1093/carcin/bgr206
Loriot, Y., Fizazi, K., Jones, R. J., Van Den Brande, J., Molife, R. L., Omlin, A., et al. (2014). Safety, tolerability and anti-tumour activity of the androgen biosynthesis inhibitor ASP9521 in patients with metastatic castration-resistant prostate cancer: multi-centre phase I/II study. Invest. New Drugs 32, 995–1004. doi: 10.1007/s10637-014-0101-x
Matsunaga, T., Hojo, A., Yamane, Y., Endo, S., El-Kabbani, O., and Hara, A. (2013). Pathophysiological roles of aldo-keto reductases (AKR1C1 and AKR1C3) in development of cisplatin resistance in human colon cancers. Chem. Biol. Interact. 202, 234–242. doi: 10.1016/j.cbi.2012.09.024
Matsunaga, T., Yamaguchi, A., Morikawa, Y., Kezuka, C., Takazawa, H., Endo, S., et al. (2014). Induction of aldo-keto reductases (AKR1C1 and AKR1C3) abolishes the efficacy of daunorubicin chemotherapy for leukemic U937 cells. Anticancer Drugs 25, 868–877. doi: 10.1097/CAD.0000000000000112
McGuire, W. L., Osborne, C. K., Clark, G. M., and Knight, W. A. III (1982). Steroid hormone receptors and carcinoma of the breast. Am. J. Physiol. 243, E99–E102.
Miller, V. L., Lin, H. K., Murugan, P., Fan, M., Penning, T. M., Brame, L. S., et al. (2012). Aldo-keto reductase family 1 member C3 (AKR1C3) is expressed in adenocarcinoma and squamous cell carcinoma but not small cell carcinoma. Int. J. Clin. Exp. Pathol. 5, 278–289.
Minotti, G., Menna, P., Salvatorelli, E., Cairo, G., and Gianni, L. (2004). Anthracyclines: molecular advances and pharmacologie developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 56, 185–229. doi: 10.1124/pr.56.2.6
Mohler, J. L., Titus, M. A., and Wilson, E. M. (2011). Potential prostate cancer drug target: bioactivation of androstanediol by conversion to dihydrotestosterone. Clin. Cancer Res. 17, 5844–5849. doi: 10.1158/1078-0432.CCR-11-0644
Nakarai, C., Osawa, K., Akiyama, M., Matsubara, N., Ikeuchi, H., Yamano, T., et al. (2015). Expression of AKR1C3 and CNN3 as markers for detection of lymph node metastases in colorectal cancer. Clin. Exp. Med. 15, 333–341. doi: 10.1007/s10238-014-0298-1
Novotna, R., Wsol, V., Xiong, G., and Maser, E. (2008). Inactivation of the anticancer drugs doxorubicin and oracin by aldo-keto reductase (AKR) 1C3. Toxicol. Lett. 181, 1–6. doi: 10.1016/j.toxlet.2008.06.858
Penning, T. M., Burczynski, M. E., Jez, J. M., Lin, H.-K., Ma, H., Moore, M., et al. (2001). Structure-function aspects and inhibitor design of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3). Mol. Cell. Endocrinol. 171, 137–149. doi: 10.1016/S0303-7207(00)00426-3
Qi, J., Tripathi, M., Mishra, R., Sahgal, N., Fazli, L., Ettinger, S., et al. (2013). The E3 ubiquitin ligase Siah2 contributes to castration-resistant prostate cancer by regulation of androgen receptor transcriptional activity. Cancer Cell 23, 332–346. doi: 10.1016/j.ccr.2013.02.016
Rexhepaj, E., Brennan, D. J., Holloway, P., Kay, E. W., McCann, A. H., Landberg, G., et al. (2008). Novel image analysis approach for quantifying expression of nuclear proteins assessed by immunohistochemistry: application to measurement of oestrogen and progesterone receptor levels in breast cancer. Breast Cancer Res. 10, R89. doi: 10.1186/bcr2187
Rižner, T. L., and Penning, T. M. (2014). Role of aldo–keto reductase family 1 (AKR1) enzymes in human steroid metabolism. Steroids 79, 49–63. doi: 10.1016/j.steroids.2013.10.012
Rižner, T. L., Šmuc, T., Rupreht, R., Šinkovec, J., and Penning, T. M. (2006). AKR1C1 and AKR1C3 may determine progesterone and estrogen ratios in endometrial cancer. Mol. Cell. Endocrinol. 248, 126–135. doi: 10.1016/j.mce.2005.10.009
Robinson, G. W., Hennighausen, L., and Johnson, P. F. (2000). Side-branching in the mammary gland: the progesterone-Wnt connection. Genes Dev. 14, 889–894.
Ross, R. K., Paganini-Hill, A., Wan, P. C., and Pike, M. C. (2000). Effect of hormone replacement therapy on breast cancer risk: estrogen versus estrogen plus progestin. J. Natl. Cancer Inst. 92, 328–332. doi: 10.1093/jnci/92.4.328
Sharma, K. K., Lindqvist, A., Zhou, X. J., Auchus, R. J., Penning, T. M., and Andersson, S. (2006). Deoxycorticosterone inactivation by AKR1C3 in human mineralocorticoid target tissues. Mol. Cell. Endocrinol. 248, 79–86. doi: 10.1016/j.mce.2005.10.024
Shiota, M., Yokomizo, A., and Naito, S. (2011). Increased androgen receptor transcription: a cause of castration-resistant prostate cancer and a possible therapeutic target. J. Mol. Endocrinol. 47, R25–R41. doi: 10.1530/JME-11-0018
Sinreih, M., Anko, M., Kene, N. H., Kocbek, V., and Rizner, T. L. (2015a). Expression of AKR1B1, AKR1C3 and other genes of prostaglandin F2alpha biosynthesis and action in ovarian endometriosis tissue and in model cell lines. Chem. Biol. Interact. 234, 320–331. doi: 10.1016/j.cbi.2014.11.009
Sinreih, M., Anko, M., Zukunft, S., Adamski, J., and Rizner, T. L. (2015b). Important roles of the AKR1C2 and SRD5A1 enzymes in progesterone metabolism in endometrial cancer model cell lines. Chem. Biol. Interact. 234, 297–308. doi: 10.1016/j.cbi.2014.11.012
Skarydova, L., Zivna, L., Xiong, G., Maser, E., and Wsol, V. (2009). AKR1C3 as a potential target for the inhibitory effect of dietary flavonoids. Chem. Biol. Interact. 178, 138–144. doi: 10.1016/j.cbi.2008.10.015
Stefane, B., Brozic, P., Vehovc, M., Rizner, T. L., and Gobec, S. (2009). New cyclopentane derivatives as inhibitors of steroid metabolizing enzymes AKR1C1 and AKR1C3. Eur. J. Med. Chem. 44, 2563–2571. doi: 10.1016/j.ejmech.2009.01.028
Sun, S. Q., Gu, X., Gao, X. S., Li, Y., Yu, H., Xiong, W., et al. (2016). Overexpression of AKR1C3 significantly enhances human prostate cancer cells resistance to radiation. Oncotarget 7, 48050–48058. doi: 10.18632/oncotarget.10347
Suzuki-Yamamoto, T., Nishizawa, M., Fukui, M., Okuda-Ashitaka, E., Nakajima, T., Ito, S., et al. (1999). cDNA cloning, expression and characterization of human prostaglandin F synthase. FEBS Lett. 462, 335–340. doi: 10.1016/S0014-5793(99)01551-3
Tanaka, H., Hiyama, T., Hanai, A., and Fujimoto, I. (1993). The research group for population-based cacner registration in japan: interhospital differences in cancer survivals in Japan. Jpn. J. Clin. Oncol. 23, 191–198.
Taucher, S., Rudas, M., Gnant, M., Thomanek, K., Dubsky, P., Roka, S., et al. (2003). Sequential steroid hormone receptor measurements in primary breast cancer with and without intervening primary chemotherapy. Endocr. Relat. Cancer 10, 91–98. doi: 10.1677/erc.0.0100091
Taylor, B. S., Schultz, N., Hieronymus, H., Gopalan, A., Xiao, Y., Carver, B. S., et al. (2010). Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22. doi: 10.1016/j.ccr.2010.05.026
Tian, Y., Zhao, L., Zhang, H., Liu, X., Zhao, L., Zhao, X., et al. (2014). AKR1C3 overexpression may serve as a promising biomarker for prostate cancer progression. Diagn. Pathol. 9:42. doi: 10.1186/1746-1596-9-42
Traven, K., Sinreih, M., Stojan, J., Sersen, S., Kljun, J., Bezensek, J., et al. (2015). Ruthenium complexes as inhibitors of the aldo-keto reductases AKR1C1-1C3. Chem. Biol. Interact. 234, 349–359. doi: 10.1016/j.cbi.2014.11.005
Usami, N., Yamamoto, T., Shintani, S., Higaki, Y., Ishikura, S., Katagiri, Y., et al. (2002). Substrate specificity of human 3(20)α-hydroxysteroid dehydrogenase for neurosteroids and its inhibition by benzodiazepines. Biol. Pharm. Bull. 25, 441–445. doi: 10.1248/bpb.25.441
Wang, Y., Wang, Y., Zhang, Z., Park, J. Y., Guo, D., Liao, H., et al. (2016). Mechanism of progestin resistance in endometrial precancer/cancer through Nrf2-AKR1C1 pathway. Oncotarget 7, 10363–10372. doi: 10.18632/oncotarget.7004
Wenners, A., Hartmann, F., Jochens, A., Roemer, A. M., Alkatout, I., Klapper, W., et al. (2016). Stromal markers AKR1C1 and AKR1C2 are prognostic factors in primary human breast cancer. Int. J. Clin. Oncol. 21, 548–556. doi: 10.1007/s10147-015-0924-2
Wiebe, J. P., Zhang, G., Welch, I., and Cadieux-Pitre, H. A. (2013). Progesterone metabolites regulate induction, growth, and suppression of estrogen- and progesterone receptor-negative human breast cell tumors. Breast Cancer Res. 15:R38. doi: 10.1186/bcr3422
Wsol, V., Szotakova, B., Martin, H. J., and Maser, E. (2007). Aldo-keto reductases (AKR) from the AKR1C subfamily catalyze the carbonyl reduction of the novel anticancer drug oracin in man. Toxicology 238, 111–118. doi: 10.1016/j.tox.2007.05.021
Wu, C. H., Ko, J. L., Chen, S. C., Lin, Y. W., Han, C. P., Yang, T. Y., et al. (2014). Clinical implications of aldo-keto reductase family 1 member C3 and its relationship with lipocalin 2 in cancer of the uterine cervix. Gynecol. Oncol. 132, 474–482. doi: 10.1016/j.ygyno.2013.11.032
Xie, L., Yu, J., Guo, W., Wei, L., Liu, Y., Wang, X., et al. (2013). Aldo-keto reductase 1C3 may be a new radioresistance marker in non-small-cell lung cancer. Cancer Gene Ther. 20, 260–266. doi: 10.1038/cgt.2013.15
Xiong, W., Zhao, J., Yu, H., Li, X., Sun, S., Li, Y., et al. (2014). Elevated expression of AKR1C3 increases resistance of cancer cells to ionizing radiation via modulation of oxidative stress. PLoS ONE 9:e111911. doi: 10.1371/journal.pone.0111911
Yepuru, M., Wu, Z., Kulkarni, A., Yin, F., Barrett, C. M., Kim, J., et al. (2013). Steroidogenic enzyme AKR1C3 is a novel androgen receptor-selective coactivator that promotes prostate cancer growth. Clin. Cancer Res. 19, 5613–5625. doi: 10.1158/1078-0432.CCR-13-1151
Yin, Y. D., Fu, M., Brooke, D. G., Heinrich, D. M., Denny, W. A., and Jamieson, S. M. (2014). The activity of SN33638, an inhibitor of AKR1C3, on testosterone and 17beta-estradiol production and function in castration-resistant prostate cancer and ER-positive breast cancer. Front. Oncol. 4:159. doi: 10.3389/fonc.2014.00159
Yoda, T., Kikuchi, K., Miki, Y., Onodera, Y., Hata, S., Takagi, K., et al. (2015). 11beta-Prostaglandin F2alpha, a bioactive metabolite catalyzed by AKR1C3, stimulates prostaglandin F receptor and induces slug expression in breast cancer. Mol. Cell. Endocrinol. 413, 236–247. doi: 10.1016/j.mce.2015.07.008
Yuan, X., and Balk, S. P. (2009). Mechanisms mediating androgen receptor reactivation after castration. Urol. Oncol. 27, 36–41. doi: 10.1016/j.urolonc.2008.03.021
Zang, T., Verma, K., Chen, M., Jin, Y., Trippier, P. C., and Penning, T. M. (2015). Screening baccharin analogs as selective inhibitors against type 5 17beta-hydroxysteroid dehydrogenase (AKR1C3). Chem. Biol. Interact. 234, 339–348. doi: 10.1016/j.cbi.2014.12.015
Zhang, A., Zhang, J., Plymate, S., and Mostaghel, E. A. (2016a). Classical and non-classical roles for pre-receptor control of DHT metabolism in prostate cancer progression. Horm. Cancer 7, 104–113. doi: 10.1007/s12672-016-0250-9
Zhang, B., Hu, X. J., Wang, X. Q., Theriault, J. F., Zhu, D. W., Shang, P., et al. (2016b). Human 3alpha-hydroxysteroid dehydrogenase type 3: structural clues of 5alpha-DHT reverse binding and enzyme down-regulation decreasing MCF7 cell growth. Biochem. J. 473, 1037–1046. doi: 10.1042/BCJ20160083
Zhao, Y., Zheng, X., Zhang, H., Zhai, J., Zhang, L., Li, C., et al. (2015). In vitro inhibition of AKR1Cs by sulphonylureas and the structural basis. Chem. Biol. Interact. 240, 310–315. doi: 10.1016/j.cbi.2015.09.006
Zhong, T., Xu, F., Xu, J., Liu, L., and Chen, Y. (2015). Aldo-keto reductase 1C3 (AKR1C3) is associated with the doxorubicin resistance in human breast cancer via PTEN loss. Biomed. Pharmacother. 69, 317–325. doi: 10.1016/j.biopha.2014.12.022
Keywords: aldo–keto reductases, catalytic-dependent, catalytic-independent, inhibitor, therapy
Citation: Zeng C-M, Chang L-L, Ying M-D, Cao J, He Q-J, Zhu H and Yang B (2017) Aldo–Keto Reductase AKR1C1–AKR1C4: Functions, Regulation, and Intervention for Anti-cancer Therapy. Front. Pharmacol. 8:119. doi: 10.3389/fphar.2017.00119
Received: 12 October 2016; Accepted: 27 February 2017;
Published: 14 March 2017.
Edited by:
Shanmugasundaram Ganapathy-Kanniappan, Johns Hopkins School of Medicine, USAReviewed by:
Massimiliano Agostini, University of Rome Tor Vergata, ItalyCopyright © 2017 Zeng, Chang, Ying, Cao, He, Zhu and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong Zhu, aG9uZ3podUB6anUuZWR1LmNu Bo Yang, eWFuZzkyNEB6anUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.