 Lisa Sercombe
Lisa Sercombe Tejaswi Veerati
Tejaswi Veerati Fatemeh Moheimani
Fatemeh Moheimani Sherry Y. Wu
Sherry Y. Wu Anil K. Sood
Anil K. Sood Susan Hua
Susan Hua
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 01 December 2015
Sec. Integrative and Regenerative Pharmacology
Volume 6 - 2015 | https://doi.org/10.3389/fphar.2015.00286
This article is part of the Research Topic Nanotechnology and Regenerative Medicine View all 6 articles
The application of liposomes to assist drug delivery has already had a major impact on many biomedical areas. They have been shown to be beneficial for stabilizing therapeutic compounds, overcoming obstacles to cellular and tissue uptake, and improving biodistribution of compounds to target sites in vivo. This enables effective delivery of encapsulated compounds to target sites while minimizing systemic toxicity. Liposomes present as an attractive delivery system due to their flexible physicochemical and biophysical properties, which allow easy manipulation to address different delivery considerations. Despite considerable research in the last 50 years and the plethora of positive results in preclinical studies, the clinical translation of liposome assisted drug delivery platforms has progressed incrementally. In this review, we will discuss the advances in liposome assisted drug delivery, biological challenges that still remain, and current clinical and experimental use of liposomes for biomedical applications. The translational obstacles of liposomal technology will also be presented.
Liposomes are the most common and well-investigated nanocarriers for targeted drug delivery. They have improved therapies for a range of biomedical applications by stabilizing therapeutic compounds, overcoming obstacles to cellular and tissue uptake, and improving biodistribution of compounds to target sites in vivo (Koning and Storm, 2003; Metselaar and Storm, 2005; Ding et al., 2006; Hua and Wu, 2013). Liposomes are defined as phospholipid vesicles consisting of one or more concentric lipid bilayers enclosing discrete aqueous spaces. The unique ability of liposomal systems to entrap both lipophilic and hydrophilic compounds enables a diverse range of drugs to be encapsulated by these vesicles. Hydrophobic molecules are inserted into the bilayer membrane, and hydrophilic molecules can be entrapped in the aqueous center (Koning and Storm, 2003; Metselaar and Storm, 2005; Ding et al., 2006; Hua and Wu, 2013; Figure 1). Furthermore, the large aqueous center and biocompatible lipid exterior permits the delivery of a variety of macromolecules, such as DNA, proteins and imaging agents (Ulrich, 2002; Monteiro et al., 2014). As a drug delivery system, liposomes offer several advantages including biocompatibility, capacity for self-assembly, ability to carry large drug payloads, and a wide range of physicochemical and biophysical properties that can be modified to control their biological characteristics (Koning and Storm, 2003; Metselaar and Storm, 2005; Ding et al., 2006; Hua and Wu, 2013). Liposomal formulations are characterized by properties such as particle size, charge, number of lamellae, lipid composition, and surface modification with polymers and ligands—these all govern their stability in vitro and in vivo (Hua and Wu, 2013; Monteiro et al., 2014). Encapsulation within liposomes protects compounds from early inactivation, degradation and dilution in the circulation (Ulrich, 2002). Liposomes are generally considered to be pharmacologically inactive with minimal toxicity, as they tend to be composed of natural phospholipids (Koning and Storm, 2003; Metselaar and Storm, 2005; Ding et al., 2006; Hua and Wu, 2013); however increasing number of studies have shown that liposomes are not as immunologically inert as once suggested (Szebeni and Moghimi, 2009). Despite the success of liposomal formulations in vivo, their translation into the clinic has progressed incrementally. This review will address the advances, biological challenges, biomedical applications, and translational obstacles of liposomal technology.
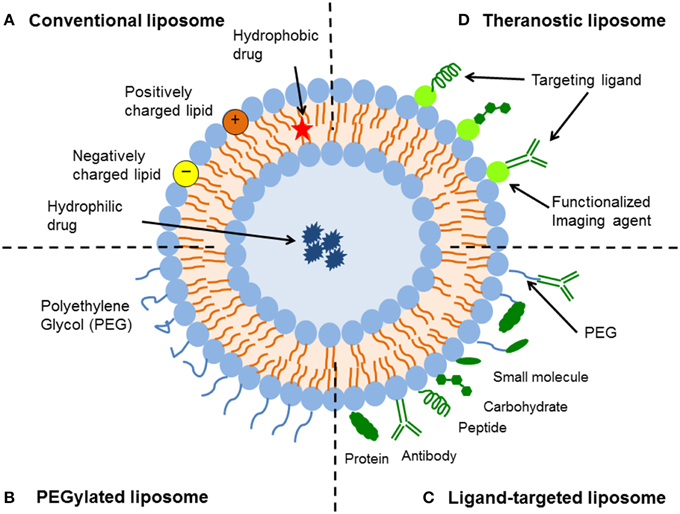
Figure 1. Schematic representation of the different types of liposomal drug delivery systems. (A) Conventional liposome—Liposomes consist of a lipid bilayer that can be composed of cationic, anionic, or neutral (phospho)lipids and cholesterol, which encloses an aqueous core. Both the lipid bilayer and the aqueous space can incorporate hydrophobic or hydrophilic compounds, respectively. (B) PEGylated liposome—Liposome characteristics and behavior in vivo can be modified by addition of a hydrophilic polymer coating, polyethylene glycol (PEG), to the liposome surface to confer steric stabilization. (C) Ligand-targeted liposome—Liposomes can be used for specific targeting by attaching ligands (e.g., antibodies, peptides, and carbohydrates) to its surface or to the terminal end of the attached PEG chains. (D) Theranostic liposome—A single system consist of a nanoparticle, a targeting element, an imaging component, and a therapeutic component.
In general, there are four key types of liposomal delivery systems—conventional liposomes, sterically-stabilized liposomes, ligand-targeted liposomes, and a combination of the above (Figure 1). Conventional liposomes were the first generation of liposomes to be developed. They consist of a lipid bilayer that can be composed of cationic, anionic, or neutral (phospho)lipids and cholesterol, which encloses an aqueous volume (Figure 1). Research on the clinical potential of conventional liposomes began in the 1980s, whereby liposomal delivery proved useful for improving the therapeutic index of encapsulated drugs, such as doxorubicin and amphotericin (Gabizon et al., 1982; Koning and Storm, 2003; Metselaar and Storm, 2005; Ding et al., 2006; Hua and Wu, 2013). Conventional liposomal formulations reduced the toxicity of compounds in vivo, through modifying pharmacokinetics and biodistribution to enhance drug delivery to diseased tissue in comparison to free drug. However, the delivery system was prone to rapid elimination from the bloodstream, therefore limiting its therapeutic efficacy (Gabizon et al., 1991, 1994). This rapid clearance was due to opsonization of plasma components and uptake by fixed macrophages of the reticuloendothelial system (RES), mainly in the liver and spleen (Hua and Wu, 2013).
To improve liposome stability and enhance their circulation times in the blood, sterically-stabilized liposomes were introduced. The hydrophilic polymer, polyethylene glycol (PEG), has been shown to be the optimal choice for obtaining sterically-stabilized liposomes (Figure 1). The establishment of a steric barrier improves the efficacy of encapsulated agents by reducing in vivo opsonization with serum components, and the rapid recognition and uptake by the RES. This not only reduces the elimination of drugs by prolonging blood circulation and providing accumulation at pathological sites, but also attenuates side effects (Torchilin et al., 1992; Northfelt et al., 1996; Ishida et al., 2001b). Steric stabilization strongly influences the pharmacokinetics of liposomes (Gabizon et al., 1993), with reported half-lives varying from 2 to 24 h in rodents (mice and rats) and as high as 45 h in humans, depending on the particle size and the characteristics of the coating polymer (Allen, 1994; Moghimi and Szebeni, 2003). While coating liposomes with PEG results in prolonged circulation times, there can be an offsetting reduction in the ability to interact with the intended targets (Willis and Forssen, 1998; Ulrich, 2002).
Ligand-targeted liposomes offer a vast potential for site-specific delivery of drugs to designated cell types or organs in vivo, which selectively express or over-express specific ligands (e.g., receptors or cell adhesion molecules) at the site of disease (Willis and Forssen, 1998; Hua, 2013). Many types of ligands are available, such as antibodies, peptides/proteins and carbohydrates (Figure 1). The coupling of antibodies, particularly monoclonal antibodies, to create immunoliposomes represents one of the more versatile ligands that can be affixed to liposome surfaces (Bendas, 2001; Puri et al., 2009). One of the advantages of using monoclonal antibodies is their stability and higher binding avidity because of the presence of two binding sites on the molecule. Since lipid assemblies are usually dynamic structures, surface-coupled ligands have a high motional freedom to position themselves for optimal substrate-interactions (Willis and Forssen, 1998). The limited in vivo performance of immunoliposomes, due to poor pharmacokinetics and immunogenicity, has been a major hurdle to achieving their potential as effective site-specific drug carriers (Puri et al., 2009). Therefore, newer generation of liposomes have utilized a combination of the above design platforms to further improve liposomal targeting and associated drug delivery (discussed in Experimental use of liposomes for biomedical applications). For example, integrating target-specific binding of immunoliposomes with the steric stabilization of PEG (thereby creating long-circulating immunoliposomes) has significantly improved the pharmacokinetics of immunoliposomes (Maruyama, 2002). Overall as a drug delivery platform, liposomes offer a dynamic and adaptable technology for enhancing the systemic efficacy of therapeutics in various diseases.
As with any foreign particle that enters the body, liposomes encounter multiple defense systems aimed at recognition, neutralization, and elimination of invading substances. These defenses include the RES, opsonization, and immunogenicity (Willis and Forssen, 1998). While these obstacles must be circumvented for optimal liposome function, other factors such as the enhanced permeability and retention (EPR) effect can be exploited to enhance drug delivery (Sawant and Torchilin, 2012).
The RES is the main site of liposome accumulation following their systemic administration (Poste et al., 1976; Senior, 1987). Primary organs associated with the RES include the liver, spleen, kidney, lungs, bone marrow, and lymph nodes (Senior, 1987). The liver exhibits the largest capacity for liposomal uptake followed by the spleen, which can accumulate liposomes up to 10-fold higher than other RES organs (Chrai et al., 2002). The ability of the RES to sequester liposomes from the circulation is attributed to fenestrations in their microvasculature. Pore diameters in these capillaries can range from 100 to 800 nm, which is large enough for the extravasation and subsequent removal of most drug-loaded liposomes (50–1000 nm in size) (Sapra and Allen, 2003). Liposomes are cleared in the RES by resident macrophages via direct interactions with the phagocytic cells (Chrai et al., 2002). Uptake of liposomes by the RES is typically secondary to vesicle opsonization—that is, the adsorption of plasma proteins such as immunoglobulin, fibronectin, lipoproteins, and/or complement proteins onto the phospholipid membrane (Ishida et al., 2001a; Chrai et al., 2002). However, in vitro studies have demonstrated that liposomal clearance via macrophages can also occur in the absence of plasma proteins (Chrai et al., 2002).
The cells of the RES are also part of the innate immune system, which has raised the question of whether macrophage saturation by liposomes leads to immunosuppression and increases the risk of infections. Excessive liposome deposition in macrophages may impair their phagocytic capacity or modulate other cellular functions; however there have been no reports to date of clinically significant immune suppression at therapeutic doses of non-cytotoxic liposomes (Szebeni and Barenholz, 2009; Szebeni and Moghimi, 2009). The situation is different with anti-cancer liposomes that contain cytotoxic drugs, which are capable of inducing macrophage destruction. Although clinically important blockade of macrophage function in humans have not yet been demonstrated, there have been indirect signs that suggest the possibility of some immune suppression (Szebeni and Barenholz, 2009; Szebeni and Moghimi, 2009). For example, administration of pegylated liposomal doxorubicin (PLD) (Doxil®) in mice showed a dose-dependent clearance saturation effect due to partial blockade of the RES in the liver. This effect was not present after administration of a similar free doxorubicin dose or phospholipid dose in drug-free liposomes (Gabizon et al., 2002). In addition, administration of Doxil® in mice was reported to interfere with the clearance of bacteria from the blood, which was suggested to be due to macrophage suppression (Storm et al., 1998; Szebeni and Barenholz, 2009).
Conjugation of PEG polymers to the liposomal membrane is a key strategy for improving circulation times and preventing removal by the RES through steric stabilization (Oku and Namba, 1994; Ishida et al., 2001a). PEGylation creates a local surface concentration of highly hydrated groups, which sterically inhibits both electrostatic and hydrophobic reactions with plasma proteins and/or cells and thereby reduces liposomal uptake by the RES (Ishida et al., 2001a). The use of PEG significantly minimizes, but does not completely circumvent, liposomal uptake by the RES—with pathways independent of opsonization also possible (Laverman et al., 2001; Sawant and Torchilin, 2012).
The degree of interaction between liposomal drug delivery systems and plasma proteins is important in determining overall nanocarrier biodistribution, efficacy, and toxicity (Hua and Wu, 2013). Plasma proteins have been shown to play a pivotal role in liposomal clearance by the RES via opsonization, as well as in vesicular destabilization (Cullis et al., 1998). Opsonization of liposomes by serum proteins depends on a variety of factors including size, surface charge and stability (Cullis et al., 1998; Ishida et al., 2001a). The extent of this interaction has been shown to decrease with liposome size from 800 to 200 nm in diameter, as small liposomes cannot support opsonic activity (Chrai et al., 2002). This profound effect of liposome size on complement recognition can also affect liver uptake (Chrai et al., 2002). Generally, large unmodified liposomes are eliminated more rapidly than small, neutral, or positively charged liposomes (Oku and Namba, 1994; Laverman et al., 1999; Ulrich, 2002). Nevertheless, the presence of high electrostatic charge can still promote the interaction of liposomes with biomolecules that can serve as opsonins (Laverman et al., 1999; Ishida et al., 2001a). Previous investigation has revealed that large, charged liposomes are cleared within minutes by the liver and less than an hour by the spleen (Senior, 1987; Chrai et al., 2002). The inclusion of cholesterol is an important factor for increasing liposome stability and minimizing phospholipid exchange (Willis and Forssen, 1998). Incorporation of cholesterol into the liposomal membrane abates lipid exchange with other circulating structures (e.g., red blood cells and lipoproteins) that can cause the depletion of high phase transition temperature lipids and their replacement with less physiologically stable components (Willis and Forssen, 1998; Laverman et al., 1999; Ulrich, 2002). Integrating cholesterol into small (approximately 100 nm), electrostatically neutral liposomes has been shown to prolong circulation time in the range of several hours (Geng et al., 2014).
Liposomes that have evaded both the RES and opsonization are subjected to the EPR effect (Sawant and Torchilin, 2012; Nehoff et al., 2014). The EPR effect refers to the increased permeability of the vasculature that supplies pathological tissues (e.g., tumors and conditions involving inflammation). At these sites, deregulations in angiogenesis and/or the increased expression and activation of vascular permeability factors predominates (Nehoff et al., 2014), which leads to fenestrations that can range from 300 to 4700 nm. This allows liposomes to extravasate and accumulate by passive targeting (Hashizume et al., 2000). For example, inflammation results in a dramatic change in blood vessel permeability as the capillary vasculature undergoes structural remodeling to allow leukocyte diapedesis into the peripheral tissue (Klimuk et al., 1999; Hua, 2013). The width of the tight junctional regions between endothelial cells in vivo has been reported to range from 12 to 20 nm (Antohe et al., 2004), however exposure to inflammatory mediators increases permeability of the microvasculature, with the formation of gaps of up to 1 μm (Antohe et al., 2004). Pore sizes ranging from 0.2 to 1.2 μm have been observed, though the size and number of pores are dependent upon the microenvironment of the pathological site (Klimuk et al., 1999; Antohe et al., 2004; Hua, 2013). Importantly, all types of liposomal delivery systems are subjected to the EPR effect, with PEGylated liposomes having an advantage due to having reduced RES clearance and extended circulation time (Sawant and Torchilin, 2012).
The interaction of liposome components with the immune system has contributed to the challenges in translation to clinical use. Synthetic modifications to enhance their utility as drug delivery vehicles can result in antibody production against their various components and/or the encapsulated cargo. For example, repeated injection of PEGylated liposomes has been associated with loss of their long circulating properties and subsequent clearance from the blood (Dams et al., 2000; Ishida et al., 2003, 2006b). This phenomenon is known as the “accelerated blood clearance” (ABC) phenomenon. The ABC phenomenon is a major concern for the clinical application of PEGylated formulations that require multiple dosing regimens. Dams et al. first observed the ABC phenomenon by demonstrating that prior dosing of empty PEGylated liposomes influences the pharmacokinetics and biodistribution of the second dose of liposomes in rats and rhesus monkeys, when the doses were administered with an interval of 7 days (Dams et al., 2000). As a result, the circulation time of the second dose of PEGylated liposomes was significantly reduced, and liposome accumulation in the liver and spleen increased (Dams et al., 2000). Subsequent investigations have verified these findings, with a maximum clearance of liposomes 4–7 days after the initial dose in rats and 10 days in mice (Ishida et al., 2003, 2006b).
The exact mechanism underlying the ABC phenomenon is unclear. This phenomenon is affected by lipid dose, PEG surface density, and the interval between the first and consecutive injections (Ishida and Kiwada, 2008). Repeated injection of empty PEGylated liposomes in rats has been shown to elicit marked anti-PEG IgM production (Ishida et al., 2006b). This immune response is thought to be mediated by the spleen, as the degree of anti-PEG IgM production and the ABC phenomenon is dramatically decreased in splenectomized rats (Ishida et al., 2006a). Interestingly, administration of higher doses of the initial PEGylated liposomes (>1 μmol phospholipids/kg) have been shown to reduce the magnitude of the ABC phenomenon (Ishida et al., 2005). Increasing the phospholipid dose has been suggested to cause PEG-reactive B cells to become apoptotic, reducing anti-PEG IgM production and thus abating the ABC phenomenon (Ishida et al., 2006b; Ishida and Kiwada, 2008). The ABC phenomenon has not been reported to occur in patients receiving PLD, even after multiple-dosing regimens (Laverman et al., 2001). Generally higher doses (15 μmol phospholipid/kg) are administered clinically, which may account for this absence of the ABC phenomenon (Lyass et al., 2000). In addition, this response may also be due to doxorubicin-mediated macrophage death and the inhibition of B-cell proliferation and/or the death of proliferated B-cells (Ishida et al., 2006b; Szebeni and Moghimi, 2009).
Some liposomal systems are able to trigger the innate immune response, with subsequent activation of the complement system to trigger an acute hypersensitivity syndrome known as complement activation–related pseudoallergy (CARPA). The complement system is part of the innate immune response, and is involved in a range of immunological and inflammatory processes (Moghimi and Hunter, 2001). A relatively high percentage of patients (2–45%) have been reported to develop infusion-related hypersensitivity reactions to liposomal drug therapy. In addition, CARPA has been reported with both experimental and clinically approved liposomal formulations (e.g., Doxil®, Ambisome, and DaunoXome®) (Szebeni, 2005; Szebeni and Moghimi, 2009). CARPA is an immediate, non-IgE-mediated hypersensitivity reaction that involves symptoms such as anaphylaxis, facial flushing, facial swelling, headache, chills, and cardiopulmonary distress (Szebeni, 2005—the latter of which may limit the clinical use of potentially reactogenic liposomes in cardiac patients (Szebeni and Barenholz, 2009). General clinical management involves slowing the infusion rate or ceasing therapy, as well as the use of standard allergy medications (e.g., antihistamines, epinephrine, and corticosteroids).
This pseudoallergy is thought to be partly due to the activation of the complement system with the subsequent generation of C3 split-products (e.g., C3d) (Dempsey et al., 1996) and anaphylatoxins C3a and C5a (Szebeni, 2005; Szebeni and Moghimi, 2009). Binding of anaphylatoxins to their specific receptors on immune cells (e.g., mast cells, basophils, and macrophages) elicits the release of a multitude of vasoactive mediators, including histamine, tryptase, platelet-activating factor (PAF), leukotrienes (e.g., LTB2, LTB4, LTC4, LTD4, LTE4), thromboxane A2 (TXA2), and prostaglandins (e.g., PGD2). Initial activation triggers include the binding of IgG, IgM, C reactive protein (CRP), C1q, C3, and potentially, mannose binding lectin (MBL) and ficolin, to the liposomal vesicles (Szebeni and Barenholz, 2009). It should be noted that the sensitivity of different species to liposomal CARPA shows substantial variation, with some species (dogs and pigs) also showing tachyphylaxis (tolerance induction) following additional doses (Szebeni et al., 1999, 2007). Therefore, desensitization protocols using empty liposomes may be used to prevent CARPA, as well as pre-administration of complement inhibitors (e.g., soluble C receptor type 1, anti-C5 antibody, and indomethacin) (Szebeni and Barenholz, 2009).
All types of liposomes can activate the complement system. Liposome size, morphology, charge, lipid composition, bilayer packaging, surface characteristics, and administered lipid dose all regulate complement activation (Szebeni and Moghimi, 2009). Specific liposomal characteristics that enhance the propensity for complement activation include a positive or negative surface charge, increasing size, lack of liposomal homogeneity, endotoxin contamination, presence of aggregates, presence of drugs that can bind to and aggregate liposomes/lipids, presence of cholesterol in the bilayer membrane at ≥70%, and PEGylation with PEG-PE (Szebeni and Barenholz, 2009). Based on these findings, neutral small unilamellar vesicles have been shown to be the least reactogenic of the liposomal platforms (Szebeni and Barenholz, 2009). Formulation strategies to minimize the immunogenicity of liposomes have included methylation of the anionic charge localized on the phosphate oxygen of mPEG-phospholipid conjugate (Moghimi et al., 2006) or the use of other non-ionic lipopolymers and lipid conjugates, such as mPEG-substituted synthetic ceramides (Webb et al., 1998; Szebeni and Moghimi, 2009). The development of immunogenic reactions to liposomal therapies may lead to altered pharmacokinetics, loss of efficacy, and the rise of potentially serious toxicities (e.g., anaphylaxis) (Szebeni and Moghimi, 2009). This should therefore be considered in formulation design and closely monitored for in clinical practice.
The application of liposomes in medicine offers significant prospects for novel and effective treatments in a wide range of pathological conditions. Since the discovery of liposomes over 50 years ago, there has been a significant increase in lipid–based drug delivery research at the experimental in vitro and in vivo phase. Liposomes have been utilized as a drug delivery carrier for a wide range of therapeutic compounds and diagnostic agents, such as drug molecules, gene therapy and bioactive agents (Hua and Wu, 2013). Modifications of these formulations are constantly being investigated in an effort to improve efficacy, reduce RES clearance and minimize toxicity—this includes changes in lipid composition, charge, and the addition of surface coatings and ligands (Hua and Wu, 2013; Monteiro et al., 2014). More recent strategies to improve on conventional or stealth liposomal systems involve active targeting, charged lipids, triggered release, and multi-functional formulations (Puri et al., 2009; Allen and Cullis, 2013; Bozzuto and Molinari, 2015).
Active targeting approaches with the conjugation of targeting ligands to the surface of liposomes have been extensively studied at the experimental level for a variety of biomedical applications, and particularly following parenteral administration (e.g., intravenous and intraperitoneal injection) (Torchilin, 1994; Vingerhoeds et al., 1994; Willis and Forssen, 1998; Noble et al., 2004; Deshpande et al., 2013; Hua and Cabot, 2013; Rip et al., 2014; Hua et al., 2015). Targeting ligands are used to increase the specificity of delivery of encapsulated cargo to and retain it in diseased tissues and cells, with minimal deposition in non-target sites. The notion that ligand-targeted liposomes have a therapeutic advantage over non-targeted liposomes is still subject to debate, with conflicting results in the literature (Ferrari, 2005; Puri et al., 2009; Riehemann et al., 2009). A number of studies have demonstrated enhanced uptake and efficacy of ligand-targeted liposomes in diseased tissue in comparison to non-targeted liposomes in vivo (Vingerhoeds et al., 1994; Puri et al., 2009; Allen and Cullis, 2013; Kraft et al., 2014). For example, attachment of folate to liposomes showed enhanced biodistribution of liposomes in folate-expressing tumors in a murine model (Gabizon et al., 2003). In addition, attachment of ICAM-1 monoclonal antibodies to the surface of loperamide-encapsulated liposomes, demonstrated increased efficacy and localization of the targeted nanoparticles to peripheral inflammatory tissue in a rodent model of musculoskeletal pain (Hua and Cabot, 2013). Conversely, there are studies that have shown no difference in the biodistribution and target tissue accumulation of ligand-targeted liposomes compared with non-targeted liposomes. For example, conjugation of HER2 monoclonal antibody fragments to liposomes did not increase the tumor localization of the nanoparticles, with both targeted and non-targeted liposomes achieving similarly high levels of tumor tissue accumulation (7–8% injected dose/g tumor tissue) in HER2-overexpressing breast cancer xenografts models (Kirpotin et al., 1997, 2006). However, doxorubicin-loaded anti-HER2 immunoliposomes produced significantly superior therapeutic results in comparison to all other control groups, including free doxorubicin, non-targeted liposomal doxorubicin and recombinant anti-HER2 Mab trastuzumab (Park et al., 2002). The mechanism of this enhanced anti-tumor efficacy was clearly not due to enhanced accumulation via antigen binding, but rather the result of the marked difference in pharmacodynamics of the targeted liposomal formulation in vivo, by mediating intracellular drug delivery to HER2-overexpressing cancer cells (Kirpotin et al., 2006). Therefore, it is likely that attachment of targeting moieties enhances therapeutic efficacy by increasing receptor-mediated uptake of drug-encapsulated liposomes into target cells, subsequent to the accumulation of the nanocarriers in the diseased tissues (Kirpotin et al., 2006; Puri et al., 2009).
Despite the improved biodistribution and therapeutic outcomes of ligand-targeted liposomes in a number of preclinical studies, the advantages have so far been negligible in the clinical research phase. Possible reasons for this discrepancy have previously been reviewed (Sawant and Torchilin, 2012; Allen and Cullis, 2013), and include factors such as disease-dependent anatomical and physiological barriers, target accessibility and expression, and formulation stability. The optimal targeting ligand density on the surface of each liposome still remains to be resolved, and will likely depend on characteristics of the molecular target (e.g., location, expression, internalization rate, and immunogenicity) (Puri et al., 2009; Hua and Wu, 2013; Kraft et al., 2014). In addition, detailed analysis of the degree of liposome accumulation, cellular internalization, intracellular functionality and intracellular degradation will be important parameters for clinical validation and translation (Puri et al., 2009). Through extensive experimentation, we are gaining a better understanding of the more appropriate clinical indications for ligand-targeted liposomal formulations.
Furthermore, modifications of the lipid bilayer with charged lipids have also attracted much attention (Bozzuto and Molinari, 2015). Addition of charged lipids to the liposomal bilayer has played an important role in developing bioadhesive, mucoadhesive and nucleic acid-based delivery systems. For example, modifying the surface charge of nano-delivery systems can influence the electrostatic interaction of the nanocarriers with components in the gastrointestinal (GI) tract following oral administration, and theoretically should confer selectivity to diseased tissue (Hua et al., 2015). Cationic nano-delivery systems have been shown to adhere to the mucosal surface within inflamed GI tissue, due to the interaction between the positively charged nanocarrier and the negatively charged intestinal mucosa (Coco et al., 2013). Colonic mucins carry a negative charge since their carbohydrates are substituted with numerous sulfate and sialic acid residues (Larsson et al., 2009; Antoni et al., 2014). Conversely, anionic nano-delivery systems preferentially adhere to inflamed GI tissue via electrostatic interaction with the higher concentration of positively charged proteins in inflamed regions. In particular, high amounts of eosinophil cationic protein and transferrin have been observed in inflamed colon sections in patients with inflammatory bowel disease (IBD) (Carlson et al., 1999; Peterson et al., 2002; Tirosh et al., 2009). Cationic nano-delivery systems are also able to effectively transport large, charged structures, such as DNA and RNA, based on electrostatic interaction between the positively charged phospholipids [e.g., dioleoylphosphatidylethanolamine (DOPE)] and negatively charged nucleic acids (Felgner et al., 1987; Campbell et al., 2002, 2009; Kunstfeld et al., 2003; Wu et al., 2007). Such liposomes have also been demonstrated to have greater interaction with tumor vessels due to the overexpression of negatively charged functional groups on the angiogenic endothelial cell membrane (Ran et al., 2002). It should be noted however, that there is a potential for electrostatic interactions and subsequent binding of these charged nanoparticles with other charge-modifying substances in the circulation or during GI transit (Hua et al., 2015). In addition, recent studies have identified potentially toxic in vitro and in vivo effects with the use of cationic lipids and polymers, including cell shrinking, reduced number of mitoses, vacuolization of the cytoplasm, and detrimental effects on key cellular proteins (e.g., protein kinase C) (Lv et al., 2006). For cationic lipids, the cytotoxic effects are determined by the structure of its hydrophilic group, with quaternary ammonium amphiphiles being more toxic than their tertiary amine counterparts. Inclusion of a heterocyclic ring has been shown to spread the positive charge of the head-group, thus attenuating the toxicity level (Lv et al., 2006).
Another approach to improve therapeutic efficacy of liposomal formulations has been to use triggering modalities for site-specific release of therapeutics from liposomes (Bibi et al., 2012). Strategies that have been utilized include remote triggers (e.g., temperature, ultrasound, magnetic, and light) and local triggers specific to the target site (e.g., enzymes and pH), through the use of specific lipid compositions and coatings (Guo and Szoka, 2003; Andresen et al., 2004; Ponce et al., 2006; Bibi et al., 2012). Of these strategies, the use of an external hyperthermic trigger to release therapeutic compounds from liposomal formulations (e.g., ThermoDox®) appears to be the most promising to date (Needham et al., 2000). Thermosensitive liposomes are modified with temperature-sensitive lipids (e.g., 1,2-distearoyl-sn-glycero-3-phosphocholine, DSPC) and/or polymers [e.g., poly(N-isopropylacrylamide)], which enables the nanocarrier to remain stable and retain their contents at physiologic temperatures. Upon heating, these liposomes undergo a phase change that makes them more permeable, causing the release of their cargo (Kono, 2001). Recent studies have investigated the use of cationic thermosensitive liposomes (CTSL) for tumor targeting (Dicheva et al., 2013, 2014), with promising results showing doxorubicin-encapsulated CTSLs having three-fold higher accumulation at the target site compared to the thermosensitive liposomal formulation (Dicheva et al., 2014). Translation of these drug delivery systems into the clinic has not yet been successful, with issues surrounding therapeutic efficacy (e.g., location of diseased tissue and accessibility for remote triggers) and potential toxicity of particularly synthetic components of the drug delivery system (Bibi et al., 2012; Allen and Cullis, 2013). Overall, this technology is promising and does warrant further investigation to determine which disease states would benefit from this localized treatment platform and how to make such formulations safer for clinical use.
Finally, a number of experimental studies have focussed on complex multi-functional liposomal formulations in an attempt to develop more efficient drug delivery systems. These include liposomal formulations that combine one or more of the following strategies—active targeting with one or more targeting ligands, response to triggers to control drug release, delivery of a combination of therapeutics (e.g., siRNA and small molecule drugs), and biomarker and imaging capabilities (Zhang et al., 2011; Allen and Cullis, 2013; Charron et al., 2015; Cole and Holland, 2015). In particular, theranostic nanoparticles have generated much interest as it is both a therapeutic and diagnostic tool all-in-one (Figure 1). A typical theranostic delivery system would include the nanoparticle, imaging component, targeting ligand, and therapeutic agent. A number of studies have shown effective diagnostic imaging and therapeutic delivery of encapsulated drugs in vivo with theranostic nanosystems, especially in various cancers (Charron et al., 2015; Cole and Holland, 2015). For example, Han et al. (2014) conjugated ECl-GLuc to nickel-chelating liposomes (ECl-GLuc-liposome), and demonstrated significant bioluminescence imaging and targeted drug delivery in both SKOv3 cells in vitro and in ErbB2-overexpressing metastatic ovarian tumors in vivo in a murine model (Han et al., 2014). ECl-GLuc is a recombinant protein generated by fusing the ECl peptide (an artificial ligand of ErbB2) with Gaussia luciferase (GLuc). Although innovative and efficient, translation of multi-functional drug delivery systems to the clinic would need to show significant therapeutic advantage over other therapeutic strategies, due to the added costs and complexities required in the manufacturing process. In addition, multi-functional systems will need to address the potential mismatch between the doses required for the effective use of each component in patients, for example imaging and therapy for theranostic nanosystems (Teli et al., 2010). At this stage, packaging multiple payloads in the same carrier would appear most promising, however sequencing and scale up of this kind of approach would still be challenging (Zhang et al., 2011; Allen and Cullis, 2013).
Many liposomal products are on the market with more in clinical development. Some of the most successful delivery methods rely on PEG conjugated lipids. In fact, the first FDA approved nano-drug, doxorubicin, is delivered using PEGylated liposomes (Ning et al., 2007). Often used in combination with other medicines, PLD treats various types of cancer including AIDS-related Kaposi's sarcoma, leukemia, and ovarian, breast, bone, lung, and brain cancers. PLD has also been found to be an effective alternative to conventional doxorubicin in patients with pre-existing cardiac dysfunction (Schmitt et al., 2012). When doxorubicin is incorporated in PEGylated liposomes, it minimizes the uptake and clearance by the RES, which prolongs the serum and plasma half-life. This allows the PLD to accumulate in the tumor tissue, rather than in non-target healthy tissues (Rahman et al., 2007). Furthermore, the use of PLD ensures that doxorubicin can pass through the myocardium without being released and contributing to cardiac muscle cell toxicity (Rahman et al., 2007). Finally, PLD avoids the high plasma peak levels of free drug, which has been correlated with cardiotoxicity (Lyass et al., 2000). In addition to receiving FDA approval for usage of PLD in 1995, combination therapies of PLD and other drugs (such as bortezomib for the treatment of relapsed or refractory multiple myeloma) have recently received FDA approval (Ning et al., 2007). Another type of PEGylated liposome currently in Phase I trials is PEPO2, an irinotecan-encapsulated liposomal formulation used to treat advanced refractory solid tumors (Chang et al., 2015). Camptothecin, which is formulated in PEGylated stealth liposomes, is also in Phase I trials for ovarian cancer treatment (Zamboni et al., 2009).
In addition to PEG conjugated lipids, conventional and cationic liposomal-based drugs have also been FDA approved. Liposomal amphotericin B for anti-fungal prophylaxis (Chandrasekar, 2008), daunorubicin for the treatment of leukemia and solid tumors (Chang and Yeh, 2012), verteporfin to treat macular degeneration (Chang and Yeh, 2012), cytarabine or cytosine arabinoside to treat neoplastic meningitis and lymphomatous meningitis (Chang and Yeh, 2012; Jahn et al., 2015), and morphine sulfate for pain management are currently on the market (Chang and Yeh, 2012). Regardless, many of these drugs are still undergoing clinical trials to test their effects of dose escalation and therapeutic efficacy (Chang and Yeh, 2012). For example, liposomal amphotericin B is in a prospective Phase II trial to test the safety and tolerability of high doses (Giannella et al., 2015). Advantages of these marketed drugs include a reduced toxicity by increased vasculature permeability/accumulation at the target tissue and an ability to encapsulate drugs of different lipophilicities while protecting them from biodegradation (Immordino et al., 2006; Chang and Yeh, 2012; Allen and Cullis, 2013).
Many more liposomal-based drugs are in various stages of clinical development to test their pharmacokinetics and biodistribution profiles. These include irinotecan SN-38 in Phase I/II to treat colorectal cancer (Zhang et al., 2004; Suenaga et al., 2015) and a liposomal-based all-trans-retinoic acid (ATRA) in Phase II that contains the drug tretinoin to treat acute promyelocytic leukemia and hormone-refractory prostate cancer (Ozpolat et al., 2003). One type of conventional liposomal formation is EndoTAG-1, which carries paclitaxel. It is embedded in a cationic liposome and is in Phase II trials to treat advanced triple-negative breast cancer (Awada et al., 2014) and pancreatic cancer (Löhr et al., 2012). EndoTAG-1 is prepared in a 50:47:3 molar ratio of 1,2-dioleoyl-3-trimethylammonium propane (DOTAP), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), and paclitaxel (Chang and Yeh, 2012). EndoTAG-1 exhibits a greater antivascular effect on tumor vasculature while the usage of DOTAP, a cationic synthetic lipid, in EndoTAG-1 allows for selective affinity to the target tumor (Chang and Yeh, 2012). Another form of paclitaxel, LEP-ETU (liposome-entrapped paclitaxel easy-to-use), is in Phase I/II trials (Zhang et al., 2005; Immordino et al., 2006). LEP-ETU is prepared in a 90:5:5 molar ratio of DOPC, cholesterol, and cardiolipin (Chang and Yeh, 2012). Higher doses of LEP-ET can be safely administered compared to paclitaxel alone (Fetterly et al., 2008). Additionally, the use of cholesterol and cardiolipin allows for greater stability and reduced cardiotoxicity, respectively (Chang and Yeh, 2012). DOPC is a recently developed neutral liposome carrier for siRNA delivery and is currently in clinical testing (Mangala et al., 2009). Another drug in Phase I/II trials is annamycin to treat acute lymphocytic leukemia (Wetzler et al., 2013). With non-cross resistance properties and an enhanced cellular uptake and retention, annamycin's efficacy and anti-tumor activity is enhanced with the use of a targeted liposomal-based delivery system (Zou et al., 1994). These and more liposomal-based drugs that are FDA approved or currently in clinical trials are summarized in Table 1.
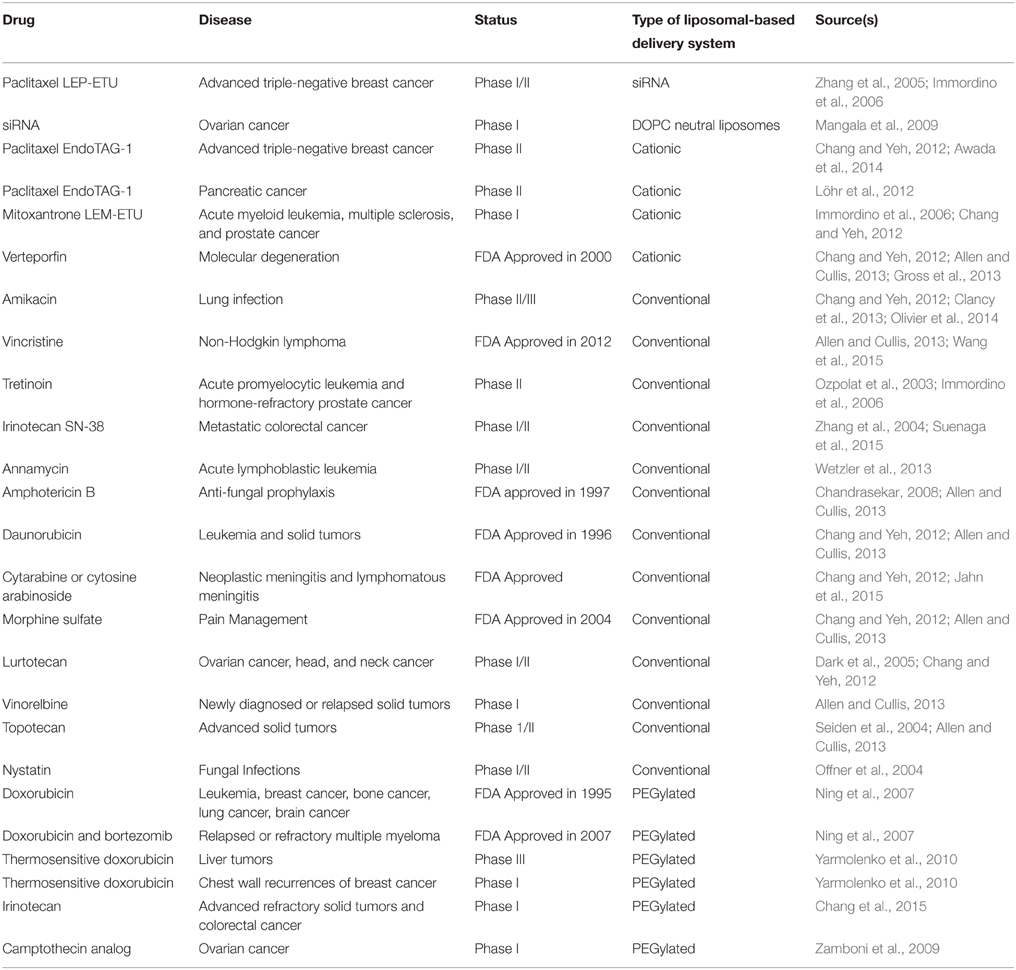
Table 1. Marketed liposomal-based therapeutics and products in clinical development.
Despite considerable research in the last 50 years, the clinical translation of liposome assisted drug delivery platforms has not progressed as quickly as the plethora of positive results would have suggested. Liposomal formulations have demonstrated significant therapeutic advantages for a multitude of biomedical applications, however the major reasons for the bottleneck have been attributed to issues surrounding pharmaceutical manufacturing, government regulations and intellectual property (IP). Similar obstacles are faced by other nano-delivery systems for translation into the clinic (Allen and Cullis, 2004, 2013; Zhang et al., 2008; Sawant and Torchilin, 2012; Narang et al., 2013). Limitations in pharmaceutical development are centered on quality assurance and cost. Quality assurance involves issues surrounding the manufacturing process and stability of the formulation, with nano-delivery systems being affected by (i) scalability of the manufacturing process, (ii) reliability and reproducibility of the final product, (iii) lack of equipment and/or in-house expertise, (iv) chemical instability or denaturation of the encapsulated compound in the manufacturing process, and (v) long term stability issues (Narang et al., 2013). Suitable methods for the industrial scale production of conventional liposomes have been successfully developed, without the need for numerous manufacturing steps or the use of organic solvents (Jaafar-Maalej et al., 2012; Kraft et al., 2014). The challenges arise when the functionality of the liposomal delivery system becomes more complex, such as the addition of surface modification with coatings and/or ligands. Integration of multiple components to a single nanosized carrier requires multiple chemical synthesis steps and formulation processes, which inevitably pose problems for large scale good manufacturing (cGMP) production, increases the cost of production, and makes the evaluation of such products more difficult (Teli et al., 2010; Tinkle et al., 2014). Increasing the number of physicochemical variables in a nanoformulation system also makes it more complicated to assess the pharmacokinetics, pharmacodynamics and toxicology of a formulation following administration (Teli et al., 2010; Tinkle et al., 2014). For example, the use of synthetic coatings and ligands may affect the biocompatibility, biodistribution and toxicology profile of liposomal formulations, and will require further evaluation to understand the interaction of the nanoparticles with biological tissues and cells (Allen and Cullis, 2004, 2013; Zhang et al., 2008; Sawant and Torchilin, 2012; Narang et al., 2013; Tinkle et al., 2014). Improvements in the regulatory framework for the assessment of nanoformulations will require consultation with academia and industry.
IP of liposomal based therapies can be a perplexing issue and is likely to contribute to increasing development costs. IP strategies may vary depending on various factors including non-targeted or targeted liposomal formulations, the design and composition of the liposomes, and drugs that may be encapsulated. Any of these factors may contribute to a weak IP position and reduce the commercial attractiveness of the formulation—this will have implications on further development of the product in the research and development pipeline. Given the complexities of incorporating nanotechnology into biomedical applications, there will likely be multiple patents associated with any given technology and the need for cross-licensing arrangements (Murday et al., 2009). It will be important to simplify the pathway from invention to commercialization through new IP practices and protocols, so as to reduce the time and expense required for negotiating collaboration and licensing agreements (Murday et al., 2009).
Finally, clinical trials of liposomal formulations are generally more complex than conventional formulations, as a number of control groups are required to account for different aspects of the drug delivery system. In order to get to this stage, the formulation has to first pass pharmaceutical and commercial qualities as discussed above. Then we can determine whether therapeutic efficacy in preclinical animal studies translate to success in humans (Allen and Cullis, 2004, 2013; Narang et al., 2013). Even at this stage, the cost-benefit analysis may be a limitation to the clinical translation of some liposomal based therapies when compared to an approved counterpart or existing therapies, in terms of efficacy or drug-related side effects.
The application of liposomes to assist drug delivery has already had a major impact on many biomedical areas. Understanding the advances in liposomal technology to date and the challenges that still need to be overcome, will allow future research to improve on existing platforms and to address the current translational and regulatory limitations. Continued translational success will require communication and collaboration between experts involved in all stages of pharmaceutical development of liposomal technologies, including manufacturing and pharmaceutical design, cellular interactions and toxicology, as well as preclinical and clinical evaluation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by The University of Newcastle, Hunter Medical Research Institute, The Pharmacy Research Trust of New South Wales, and The Rebecca L. Cooper Medical Research Foundation. Portions of this work were supported by the NIH (CA 109298, P50 CA083639, P50 CA098258, CA 177909, UH2 TR000943, CA 16672), the Ovarian Cancer Research Fund, Inc., CPRIT RP110595 and RPI 20214, the RGK Foundation, the Glider Foundation, the Judi A. Rees Ovarian Cancer Research Fund, and the Betty Anne Asche Murray Distinguished Professorship. SW is supported by Foundation for Women's Cancer, Ovarian Cancer Research Funds, and Cancer Prevention Research Institute of Texas training grants (RP101502, RP101489, and RP110595). The authors cordially thank Dr Nagaraj Gopisetty at Newcastle Innovations™ for his assistance on the IP regulations surrounding drug delivery systems.
Allen, T. M. (1994). Long-circulating (sterically stabilized) liposomes for targeted drug delivery. Trends Pharmacol. Sci. 15, 215–220. doi: 10.1016/0165-6147(94)90314-X
Allen, T. M., and Cullis, P. R. (2004). Drug delivery systems: entering the mainstream. Science 303, 1818–1822. doi: 10.1126/science.1095833
Allen, T. M., and Cullis, P. R. (2013). Liposomal drug delivery systems: from concept to clinical applications. Adv. Drug Deliv. Rev. 65, 36–48. doi: 10.1016/j.addr.2012.09.037
Andresen, T. L., Davidsen, J., Begtrup, M., Mouritsen, O. G., and Jørgensen, K. (2004). Enzymatic release of antitumor ether lipids by specific phospholipase A2 activation of liposome-forming prodrugs. J. Med. Chem. 47, 1694–1703. doi: 10.1021/jm031029r
Antohe, F., Lin, L., Kao, G. Y., Poznansky, M. J., and Allen, T. M. (2004). Transendothelial movement of liposomes in vitro mediated by cancer cells, neutrophils or histamine. J. Liposome Res. 14, 1–25. doi: 10.1081/LPR-120039660
Antoni, L., Nuding, S., Wehkamp, J., and Stange, E. F. (2014). Intestinal barrier in inflammatory bowel disease. World J. Gastroenterol. 20, 1165–1179. doi: 10.3748/wjg.v20.i5.1165
Awada, A., Bondarenko, I. N., Bonneterre, J., Nowara, E., Ferrero, J. M., Bakshi, A. V., et al. (2014). A randomized controlled phase II trial of a novel composition of paclitaxel embedded into neutral and cationic lipids targeting tumor endothelial cells in advanced triple-negative breast cancer (TNBC). Ann. Oncol. 25, 824–831. doi: 10.1093/annonc/mdu025
Bendas, G. (2001). Immunoliposomes: a promising approach to targeting cancer therapy. BioDrugs 15, 215–224. doi: 10.2165/00063030-200115040-00002
Bibi, S., Lattmann, E., Mohammed, A. R., and Perrie, Y. (2012). Trigger release liposome systems: local and remote controlled delivery? J. Microencapsul. 29, 262–276. doi: 10.3109/02652048.2011.646330
Bozzuto, G., and Molinari, A. (2015). Liposomes as nanomedical devices. Int. J. Nanomedicine 10, 975–999. doi: 10.2147/IJN.S68861
Campbell, R. B., Fukumura, D., Brown, E. B., Mazzola, L. M., Izumi, Y., Jain, R. K., et al. (2002). Cationic charge determines the distribution of liposomes between the vascular and extravascular compartments of tumors. Cancer Res. 62, 6831–6836.
Campbell, R. B., Ying, B., Kuesters, G. M., and Hemphill, R. (2009). Fighting cancer: from the bench to bedside using second generation cationic liposomal therapeutics. J. Pharm. Sci. 98, 411–429. doi: 10.1002/jps.21458
Carlson, M., Raab, Y., Peterson, C., Hällgren, R., and Venge, P. (1999). Increased intraluminal release of eosinophil granule proteins EPO, ECP, EPX, and cytokines in ulcerative colitis and proctitis in segmental perfusion. Am. J. Gastroenterol. 94, 1876–1883. doi: 10.1111/j.1572-0241.1999.01223.x
Chandrasekar, P. (2008). Amphotericin B lipid complex: treatment of invasive fungal infections in patients refractory to or intolerant of amphotericin B deoxycholate. Ther. Clin. Risk Manag. 4, 1285–1294.
Chang, H. I., and Yeh, M. K. (2012). Clinical development of liposome-based drugs: formulation, characterization, and therapeutic efficacy. Int. J. Nanomedicine 7, 49–60. doi: 10.2147/IJN.S26766
Chang, T. C., Shiah, H. S., Yang, C. H., Yeh, K. H., Cheng, A. L., Shen, B. N., et al. (2015). Phase I study of nanoliposomal irinotecan (PEP02) in advanced solid tumor patients. Cancer Chemother. Pharmacol. 75, 579–586. doi: 10.1007/s00280-014-2671-x
Charron, D. M., Chen, J., and Zheng, G. (2015). Theranostic lipid nanoparticles for cancer medicine. Cancer Treat. Res. 166, 103–127. doi: 10.1007/978-3-319-16555-4_5
Chrai, S. S., Murari, R., and Ahmad, I. (2002). Liposomes (a review) part two: drug delivery systems. BioPharm 17, 40–43.
Clancy, J. P., Dupont, L., Konstan, M. W., Billings, J., Fustik, S., Goss, C. H., et al. (2013). Phase II studies of nebulised Arikace in CF patients with Pseudomonas aeruginosa infection. Thorax 68, 818–825. doi: 10.1136/thoraxjnl-2012-202230
Coco, R., Plapied, L., Pourcelle, V., Jérôme, C., Brayden, D. J., Schneider, Y. J., et al. (2013). Drug delivery to inflamed colon by nanoparticles: comparison of different strategies. Int. J. Pharm. 440, 3–12. doi: 10.1016/j.ijpharm.2012.07.017
Cole, J. T., and Holland, N. B. (2015). Multifunctional nanoparticles for use in theranostic applications. Drug Deliv. Transl. Res. 5, 295–309. doi: 10.1007/s13346-015-0218-2
Cullis, P. R., Chonn, A., and Semple, S. C. (1998). Interactions of liposomes and lipid-based carrier systems with blood proteins: relation to clearance behaviour in vivo. Adv. Drug Deliv. Rev. 32, 3–17.
Dams, E. T., Laverman, P., Oyen, W. J., Storm, G., Scherphof, G. L., van Der Meer, J. W., et al. (2000). Accelerated blood clearance and altered biodistribution of repeated injections of sterically stabilized liposomes. J. Pharmacol. Exp. Ther. 292, 1071–1079.
Dark, G. G., Calvert, A. H., Grimshaw, R., Poole, C., Swenerton, K., Kaye, S., et al. (2005). Randomized trial of two intravenous schedules of the topoisomerase I inhibitor liposomal lurtotecan in women with relapsed epithelial ovarian cancer: a trial of the national cancer institute of Canada clinical trials group. J. Clin. Oncol. 23, 1859–1866. doi: 10.1200/JCO.2005.02.028
Dempsey, P. W., Allison, M. E., Akkaraju, S., Goodnow, C. C., and Fearon, D. T. (1996). C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science 271, 348–350. doi: 10.1126/science.271.5247.348
Deshpande, P. P., Biswas, S., and Torchilin, V. P. (2013). Current trends in the use of liposomes for tumor targeting. Nanomedicine (Lond). 8, 1509–1528. doi: 10.2217/nnm.13.118
Dicheva, B. M., ten Hagen, T. L., Li, L., Schipper, D., Seynhaeve, A. L., van Rhoon, G. C., et al. (2013). Cationic thermosensitive liposomes: a novel dual targeted heat-triggered drug delivery approach for endothelial and tumor cells. Nano Lett. 13, 2324–2331. doi: 10.1021/nl3014154
Dicheva, B. M., ten Hagen, T. L., Schipper, D., Seynhaeve, A. L., van Rhoon, G. C., Eggermont, A. M., et al. (2014). Targeted and heat-triggered doxorubicin delivery to tumors by dual targeted cationic thermosensitive liposomes. J. Control. Release 195, 37–48. doi: 10.1016/j.jconrel.2014.07.058
Ding, B. S., Dziubla, T., Shuvaev, V. V., Muro, S., and Muzykantov, V. R. (2006). Advanced drug delivery systems that target the vascular endothelium. Mol. Interv. 6, 98–112. doi: 10.1124/mi.6.2.7
Felgner, P. L., Gadek, T. R., Holm, M., Roman, R., Chan, H. W., Wenz, M., et al. (1987). Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. U.S.A. 84, 7413–7417. doi: 10.1073/pnas.84.21.7413
Ferrari, M. (2005). Nanovector therapeutics. Curr. Opin. Chem. Biol. 9, 343–346. doi: 10.1016/j.cbpa.2005.06.001
Fetterly, G. J., Grasela, T. H., Sherman, J. W., Dul, J. L., Grahn, A., Lecomte, D., et al. (2008). Pharmacokinetic/pharmacodynamic modeling and simulation of neutropenia during phase I development of liposome-entrapped paclitaxel. Clin. Cancer Res. 14, 5856–5863. doi: 10.1158/1078-0432.CCR-08-1046
Gabizon, A. A., Barenholz, Y., and Bialer, M. (1993). Prolongation of the circulation time of doxorubicin encapsulated in liposomes containing a polyethylene glycol-derivatized phospholipid: pharmacokinetic studies in rodents and dogs. Pharm. Res. 10, 703–708. doi: 10.1023/A:1018907715905
Gabizon, A., Catane, R., Uziely, B., Kaufman, B., Safra, T., Cohen, R., et al. (1994). Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res. 54, 987–992.
Gabizon, A., Chisin, R., Amselem, S., Druckmann, S., Cohen, R., Goren, D., et al. (1991). Pharmacokinetic and imaging studies in patients receiving a formulation of liposome-associated adriamycin. Br. J. Cancer 64, 1125–1132. doi: 10.1038/bjc.1991.476
Gabizon, A., Dagan, A., Goren, D., Barenholz, Y., and Fuks, Z. (1982). Liposomes as in vivo carriers of adriamycin: reduced cardiac uptake and preserved antitumor activity in mice. Cancer Res. 42, 4734–4739.
Gabizon, A., Horowitz, A. T., Goren, D., Tzemach, D., Shmeeda, H., and Zalipsky, S. (2003). In vivo fate of folate-targeted polyethylene-glycol liposomes in tumor-bearing mice. Clin. Cancer Res. 9, 6551–6559.
Gabizon, A., Tzemach, D., Mak, L., Bronstein, M., and Horowitz, A. T. (2002). Dose dependency of pharmacokinetics and therapeutic efficacy of pegylated liposomal doxorubicin (DOXIL) in murine models. J. Drug Target. 10, 539–548. doi: 10.1080/1061186021000072447
Geng, S., Yang, B., Wang, G., Qin, G., Wada, S., and Wang, J. Y. (2014). Two cholesterol derivative-based PEGylated liposomes as drug delivery system, study on pharmacokinetics and drug delivery to retina. Nanotechnology 25:275103. doi: 10.1088/0957-4484/25/27/275103
Giannella, M., Ercolani, G., Cristini, F., Morelli, M., Bartoletti, M., Bertuzzo, V., et al. (2015). High-dose weekly liposomal amphotericin b antifungal prophylaxis in patients undergoing liver transplantation: a prospective phase II trial. Transplantation 99, 848–854. doi: 10.1097/TP.0000000000000393
Gross, N., Ranjbar, M., Evers, C., Hua, J., Martin, G., Schulze, B., et al. (2013). Choroidal neovascularization reduced by targeted drug delivery with cationic liposome-encapsulated paclitaxel or targeted photodynamic therapy with verteporfin encapsulated in cationic liposomes. Mol. Vis. 19, 54–61.
Guo, X., and Szoka, F. C. Jr. (2003). Chemical approaches to triggerable lipid vesicles for drug and gene delivery. Acc. Chem. Res. 36, 335–341. doi: 10.1021/ar9703241
Han, X. J., Wei, Y. F., Wan, Y. Y., Jiang, L. P., Zhang, J. F., and Xin, H. B. (2014). Development of a novel liposomal nanodelivery system for bioluminescence imaging and targeted drug delivery in ErbB2-overexpressing metastatic ovarian carcinoma. Int. J. Mol. Med. 34, 1225–1232. doi: 10.3892/ijmm.2014.1922
Hashizume, H., Baluk, P., Morikawa, S., McLean, J. W., Thurston, G., Roberge, S., et al. (2000). Openings between defective endothelial cells explain tumor vessel leakiness. Am. J. Pathol. 156, 1363–1380. doi: 10.1016/S0002-9440(10)65006-7
Hua, S. (2013). Targeting sites of inflammation: intercellular adhesion molecule-1 as a target for novel inflammatory therapies. Front. Pharmacol. 4:127. doi: 10.3389/fphar.2013.00127
Hua, S., and Cabot, P. J. (2013). Targeted nanoparticles that mimic immune cells in pain control inducing analgesic and anti-inflammatory actions: a potential novel treatment of acute and chronic pain condition. Pain Physician. 16, E199–E216.
Hua, S., Marks, E., Schneider, J. J., and Keely, S. (2015). Advances in oral nano-delivery systems for colon targeted drug delivery in inflammatory bowel disease: selective targeting to diseased versus healthy tissue. Nanomedicine 11, 1117–1132. doi: 10.1016/j.nano.2015.02.018
Hua, S., and Wu, S. Y. (2013). The use of lipid-based nanocarriers for targeted pain therapies. Front. Pharmacol. 4:143. doi: 10.3389/fphar.2013.00143
Immordino, M. L., Dosio, F., and Cattel, L. (2006). Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomedicine 1, 297–315.
Ishida, T., Harada, M., Wang, X. Y., Ichihara, M., Irimura, K., and Kiwada, H. (2005). Accelerated blood clearance of PEGylated liposomes following preceding liposome injection: effects of lipid dose and PEG surface-density and chain length of the first-dose liposomes. J. Control. Release 105, 305–317. doi: 10.1016/j.jconrel.2005.04.003
Ishida, T., Harashima, H., and Kiwada, H. (2001a). Interactions of liposomes with cells in vitro and in vivo: opsonins and receptors. Curr. Drug Metab. 2, 397–409. doi: 10.2174/1389200013338306
Ishida, T., Ichihara, M., Wang, X., and Kiwada, H. (2006a). Spleen plays an important role in the induction of accelerated blood clearance of PEGylated liposomes. J. Control. Release 115, 243–250. doi: 10.1016/j.jconrel.2006.08.001
Ishida, T., Ichihara, M., Wang, X., Yamamoto, K., Kimura, J., Majima, E., et al. (2006b). Injection of PEGylated liposomes in rats elicits PEG-specific IgM, which is responsible for rapid elimination of a second dose of PEGylated liposomes. J. Control. Release 112, 15–25. doi: 10.1016/j.jconrel.2006.01.005
Ishida, T., Kirchmeier, M. J., Moase, E. H., Zalipsky, S., and Allen, T. M. (2001b). Targeted delivery and triggered release of liposomal doxorubicin enhances cytotoxicity against human B lymphoma cells. Biochim. Biophys. Acta 1515, 144–158. doi: 10.1016/S0005-2736(01)00409-6
Ishida, T., and Kiwada, H. (2008). Accelerated blood clearance (ABC) phenomenon upon repeated injection of PEGylated liposomes. Int. J. Pharm. 354, 56–62. doi: 10.1016/j.ijpharm.2007.11.005
Ishida, T., Masuda, K., Ichikawa, T., Ichihara, M., Irimura, K., and Kiwada, H. (2003). Accelerated clearance of a second injection of PEGylated liposomes in mice. Int. J. Pharm. 255, 167–174. doi: 10.1016/S0378-5173(03)00085-1
Jaafar-Maalej, C., Elaissari, A., and Fessi, H. (2012). Lipid-based carriers: manufacturing and applications for pulmonary route. Expert Opin. Drug Deliv. 9, 1111–1127. doi: 10.1517/17425247.2012.702751
Jahn, F., Jordan, K., Behlendorf, T., Globig, C., Schmoll, H. J., Müller-Tidow, C., et al. (2015). Safety and efficacy of liposomal cytarabine in the treatment of neoplastic meningitis. Oncology 89, 137–142. doi: 10.1159/000380913
Kirpotin, D. B., Drummond, D. C., Shao, Y., Shalaby, M. R., Hong, K., Nielsen, U. B., et al. (2006). Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 66, 6732–6740. doi: 10.1158/0008-5472.CAN-05-4199
Kirpotin, D., Park, J. W., Hong, K., Zalipsky, S., Li, W. L., Carter, P., et al. (1997). Sterically stabilized anti-HER2 immunoliposomes: design and targeting to human breast cancer cells in vitro. Biochem. Mosc. 36, 66–75. doi: 10.1021/bi962148u
Klimuk, S. K., Semple, S. C., Scherrer, P., and Hope, M. J. (1999). Contact hypersensitivity: a simple model for the characterization of disease-site targeting by liposomes. Biochim. Biophys. Acta 1417, 191–201. doi: 10.1016/S0005-2736(98)00261-2
Koning, G. A., and Storm, G. (2003). Targeted drug delivery systems for the intracellular delivery of macromolecular drugs. Drug Discov. Today 8, 482–483. doi: 10.1016/S1359-6446(03)02699-0
Kono, K. (2001). Thermosensitive polymer-modified liposomes. Adv. Drug Deliv. Rev. 53, 307–319. doi: 10.1016/S0169-409X(01)00204-6
Kraft, J. C., Freeling, J. P., Wang, Z., and Ho, R. J. (2014). Emerging research and clinical development trends of liposome and lipid nanoparticle drug delivery systems. J. Pharm. Sci. 103, 29–52. doi: 10.1002/jps.23773
Kunstfeld, R., Wickenhauser, G., Michaelis, U., Teifel, M., Umek, W., Naujoks, K., et al. (2003). Paclitaxel encapsulated in cationic liposomes diminishes tumor angiogenesis and melanoma growth in a “humanized” SCID mouse model. J. Invest. Dermatol. 120, 476–482. doi: 10.1046/j.1523-1747.2003.12057.x
Larsson, J. M., Karlsson, H., Sjövall, H., and Hansson, G. C. (2009). A complex, but uniform O-glycosylation of the human MUC2 mucin from colonic biopsies analyzed by nanoLC/MSn. Glycobiology 19, 756–766. doi: 10.1093/glycob/cwp048
Laverman, P., Boerman, O. C., Oyen, W. J., Dams, E. T., Storm, G., and Corstens, F. H. (1999). Liposomes for scintigraphic detection of infection and inflammation. Adv. Drug Deliv. Rev. 37, 225–235.
Laverman, P., Carstens, M. G., Storm, G., and Moghimi, S. M. (2001). Recognition and clearance of methoxypoly(ethyleneglycol)2000-grafted liposomes by macrophages with enhanced phagocytic capacity. Implications in experimental and clinical oncology. Biochim. Biophys. Acta 1526, 227–229. doi: 10.1016/S0304-4165(01)00142-8
Löhr, J. M., Haas, S. L., Bechstein, W. O., Bodoky, G., Cwiertka, K., Fischbach, W., et al. (2012). Cationic liposomal paclitaxel plus gemcitabine or gemcitabine alone in patients with advanced pancreatic cancer: a randomized controlled phase II trial. Ann. Oncol. 23, 1214–1222. doi: 10.1093/annonc/mdr379
Lv, H., Zhang, S., Wang, B., Cui, S., and Yan, J. (2006). Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release 114, 100–109. doi: 10.1016/j.jconrel.2006.04.014
Lyass, O., Uziely, B., Ben-Yosef, R., Tzemach, D., Heshing, N. I., Lotem, M., et al. (2000). Correlation of toxicity with pharmacokinetics of pegylated liposomal doxorubicin (Doxil) in metastatic breast carcinoma. Cancer 89, 1037–1047. doi: 10.1002/1097-0142(20000901)89:5<1037::AID-CNCR13>3.0.CO;2-Z
Mangala, L. S., Han, H. D., Lopez-Berestein, G., and Sood, A. K. (2009). Liposomal siRNA for ovarian cancer. Methods Mol. Biol. 555, 29–42. doi: 10.1007/978-1-60327-295-7_3
Metselaar, J. M., and Storm, G. (2005). Liposomes in the treatment of inflammatory disorders. Expert Opin. Drug Deliv. 465–76. doi: 10.1517/17425247.2.3.465
Moghimi, S. M., Hamad, I., Andresen, T. L., Jørgensen, K., and Szebeni, J. (2006). Methylation of the phosphate oxygen moiety of phospholipid-methoxy(polyethylene glycol) conjugate prevents PEGylated liposome-mediated complement activation and anaphylatoxin production. FASEB J. 20, 2591–2593. doi: 10.1096/fj.06-6186fje
Moghimi, S. M., and Hunter, A. C. (2001). Capture of stealth nanoparticles by the body's defences. Crit. Rev. Ther. Drug Carrier Syst. 18, 527–550. doi: 10.1615/CritRevTherDrugCarrierSyst.v18.i6.30
Moghimi, S. M., and Szebeni, J. (2003). Stealth liposomes and long circulating nanoparticles: critical issues in pharmacokinetics, opsonization and protein-binding properties. Prog. Lipid Res. 42, 463–478. doi: 10.1016/S0163-7827(03)00033-X
Monteiro, N., Martins, A., Reis, R. L., and Neves, N. M. (2014). Liposomes in tissue engineering and regenerative medicine. J. R. Soc. Interface 11:20140459. doi: 10.1098/rsif.2014.0459
Murday, J. S., Siegel, R. W., Stein, J., and Wright, J. F. (2009). Translational nanomedicine: status assessment and opportunities. Nanomedicine 5, 251–273. doi: 10.1016/j.nano.2009.06.001
Narang, A. S., Chang, R. K., and Hussain, M. A. (2013). Pharmaceutical development and regulatory considerations for nanoparticles and nanoparticulate drug delivery systems. J. Pharm. Sci. 102, 3867–3882. doi: 10.1002/jps.23691
Needham, D., Anyarambhatla, G., Kong, G., and Dewhirst, M. W. (2000). A new temperature-sensitive liposome for use with mild hyperthermia: characterization and testing in a human tumor xenograft model. Cancer Res. 60, 1197–1201.
Nehoff, H., Parayath, N. N., Domanovitch, L., Taurin, S., and Greish, K. (2014). Nanomedicine for drug targeting: strategies beyond the enhanced permeability and retention effect. Int. J. Nanomedicine 9, 2539–2555. doi: 10.2147/IJN.S47129
Ning, Y. M., He, K., Dagher, R., Sridhara, R., Farrell, A. T., Justice, R., et al. (2007). Liposomal doxorubicin in combination with bortezomib for relapsed or refractory multiple myeloma. Oncology (Williston Park) 21, 1503–1508. discussion: 11, 13, 16 passim.
Noble, C. O., Kirpotin, D. B., Hayes, M. E., Mamot, C., Hong, K., Park, J. W., et al. (2004). Development of ligand-targeted liposomes for cancer therapy. Expert Opin. Ther. Targets 8, 335–353. doi: 10.1517/14728222.8.4.335
Northfelt, D. W., Martin, F. J., Working, P., Volberding, P. A., Russell, J., Newman, M., et al. (1996). Doxorubicin encapsulated in liposomes containing surface-bound polyethylene glycol: pharmacokinetics, tumor localization, and safety in patients with AIDS-related Kaposi's sarcoma. J. Clin. Pharmacol. 36, 55–63. doi: 10.1002/j.1552-4604.1996.tb04152.x
Offner, F., Krcmery, V., Boogaerts, M., Doyen, C., Engelhard, D., Ribaud, P., et al. (2004). Liposomal nystatin in patients with invasive aspergillosis refractory to or intolerant of amphotericin B. Antimicrob. Agents Chemother. 48, 4808–4812. doi: 10.1128/AAC.48.12.4808-4812.2004
Oku, N., and Namba, Y. (1994). Long-circulating liposomes. Crit. Rev. Ther. Drug Carrier Syst. 11, 231–270.
Olivier, K. N., Shaw, P. A., Glaser, T. S., Bhattacharyya, D., Fleshner, M., Brewer, C. C., et al. (2014). Inhaled amikacin for treatment of refractory pulmonary nontuberculous mycobacterial disease. Ann. Am. Thorac. Soc. 11, 30–35. doi: 10.1513/AnnalsATS.201307-231OC
Ozpolat, B., Lopez-Berestein, G., Adamson, P., Fu, C. J., and Williams, A. H. (2003). Pharmacokinetics of intravenously administered liposomal all-trans-retinoic acid (ATRA) and orally administered ATRA in healthy volunteers. J. Pharm. Pharm. Sci. 6, 292–301.
Park, J. W., Hong, K., Kirpotin, D. B., Colbern, G., Shalaby, R., Baselga, J., et al. (2002). Anti-HER2 immunoliposomes: enhanced efficacy attributable to targeted delivery. Clin. Cancer Res. 8, 1172–1181.
Peterson, C. G., Eklund, E., Taha, Y., Raab, Y., and Carlson, M. (2002). A new method for the quantification of neutrophil and eosinophil cationic proteins in feces: establishment of normal levels and clinical application in patients with inflammatory bowel disease. Am. J. Gastroenterol. 97, 1755–1762. doi: 10.1111/j.1572-0241.2002.05837.x
Ponce, A. M., Vujaskovic, Z., Yuan, F., Needham, D., and Dewhirst, M. W. (2006). Hyperthermia mediated liposomal drug delivery. Int. J. Hyperthermia 22, 205–213. doi: 10.1080/02656730600582956
Poste, G., Papahadjopoulos, D., and Vail, W. J. (1976). Lipid vesicles as carriers for introducing biologically active materials into cells. Methods Cell Biol. 14, 33–71. doi: 10.1016/S0091-679X(08)60468-9
Puri, A., Loomis, K., Smith, B., Lee, J. H., Yavlovich, A., Heldman, E., et al. (2009). Lipid-based nanoparticles as pharmaceutical drug carriers: from concepts to clinic. Crit. Rev. Ther. Drug Carrier Syst. 26, 523–580. doi: 10.1615/CritRevTherDrugCarrierSyst.v26.i6.10
Rahman, A. M., Yusuf, S. W., and Ewer, M. S. (2007). Anthracycline-induced cardiotoxicity and the cardiac-sparing effect of liposomal formulation. Int. J. Nanomedicine 2, 567–583.
Ran, S., Downes, A., and Thorpe, P. E. (2002). Increased exposure of anionic phospholipids on the surface of tumor blood vessels. Cancer Res. 62, 6132–6140.
Riehemann, K., Schneider, S. W., Luger, T. A., Godin, B., Ferrari, M., and Fuchs, H. (2009). Nanomedicine–challenge and perspectives. Angew. Chem. Int. Ed Engl. 48, 872–897. doi: 10.1002/anie.200802585
Rip, J., Chen, L., Hartman, R., van den Heuvel, A., Reijerkerk, A., van Kregten, J., et al. (2014). Glutathione PEGylated liposomes: pharmacokinetics and delivery of cargo across the blood-brain barrier in rats. J. Drug Target. 22, 460–467. doi: 10.3109/1061186X.2014.888070
Sapra, P., and Allen, T. M. (2003). Ligand-targeted liposomal anticancer drugs. Prog. Lipid Res. 42, 439–462. doi: 10.1016/S0163-7827(03)00032-8
Sawant, R. R., and Torchilin, V. P. (2012). Challenges in development of targeted liposomal therapeutics. AAPS J. 14, 303–315. doi: 10.1208/s12248-012-9330-0
Schmitt, C. J., Dietrich, S., Ho, A. D., and Witzens-Harig, M. (2012). Replacement of conventional doxorubicin by pegylated liposomal doxorubicin is a safe and effective alternative in the treatment of non-Hodgkin's lymphoma patients with cardiac risk factors. Ann. Hematol. 91, 391–397. doi: 10.1007/s00277-011-1308-y
Seiden, M. V., Muggia, F., Astrow, A., Matulonis, U., Campos, S., Roche, M., et al. (2004). A phase II study of liposomal lurtotecan (OSI-211) in patients with topotecan resistant ovarian cancer. Gynecol. Oncol. 93, 229–232. doi: 10.1016/j.ygyno.2003.12.037
Senior, J. H. (1987). Fate and behavior of liposomes in vivo: a review of controlling factors. Crit. Rev. Ther. Drug Carrier Syst. 3, 123–193.
Storm, G., ten Kate, M. T., Working, P. K., and Bakker-Woudenberg, I. A. (1998). Doxorubicin entrapped in sterically stabilized liposomes: effects on bacterial blood clearance capacity of the mononuclear phagocyte system. Clin. Cancer Res. 4, 111–115.
Suenaga, M., Mizunuma, N., Matsusaka, S., Shinozaki, E., Ozaka, M., Ogura, M., et al. (2015). Phase II study of reintroduction of oxaliplatin for advanced colorectal cancer in patients previously treated with oxaliplatin and irinotecan: RE-OPEN study. Drug Des. Devel. Ther. 9, 3099–3108. doi: 10.2147/DDDT.S85567
Szebeni, J. (2005). Complement activation-related pseudoallergy: a new class of drug-induced acute immune toxicity. Toxicology 216, 106–121. doi: 10.1016/j.tox.2005.07.023
Szebeni, J., Alving, C. R., Rosivall, L., Bünger, R., Baranyi, L., Bedöcs, P., et al. (2007). Animal models of complement-mediated hypersensitivity reactions to liposomes and other lipid-based nanoparticles. J. Liposome Res. 17, 107–117. doi: 10.1080/08982100701375118
Szebeni, J., and Barenholz, Y. (2009). “Adverse immune effects of liposomes: complement activation, immunogenicity and immune suppression,” in Harnessing Biomaterials for Nanomedicine: Preparation, Toxicity and Applications, ed P. S. Publishing (Singapore: Pan Stanford Publishing), 1–19.
Szebeni, J., Fontana, J. L., Wassef, N. M., Mongan, P. D., Morse, D. S., Dobbins, D. E., et al. (1999). Hemodynamic changes induced by liposomes and liposome-encapsulated hemoglobin in pigs: a model for pseudoallergic cardiopulmonary reactions to liposomes. Role of complement and inhibition by soluble CR1 and anti-C5a antibody. Circulation 99, 2302–2309. doi: 10.1161/01.CIR.99.17.2302
Szebeni, J., and Moghimi, S. M. (2009). Liposome triggering of innate immune responses: a perspective on benefits and adverse reactions. J. Liposome Res. 19, 85–90. doi: 10.1080/08982100902792855
Teli, M. K., Mutalik, S., and Rajanikant, G. K. (2010). Nanotechnology and nanomedicine: going small means aiming big. Curr. Pharm. Des. 16, 1882–1892. doi: 10.2174/138161210791208992
Tinkle, S., McNeil, S. E., Mühlebach, S., Bawa, R., Borchard, G., Barenholz, Y. C., et al. (2014). Nanomedicines: addressing the scientific and regulatory gap. Ann. N.Y. Acad. Sci. 1313, 35–56. doi: 10.1111/nyas.12403
Tirosh, B., Khatib, N., Barenholz, Y., Nissan, A., and Rubinstein, A. (2009). Transferrin as a luminal target for negatively charged liposomes in the inflamed colonic mucosa. Mol. Pharm. 6, 1083–1091. doi: 10.1021/mp9000926
Torchilin, V. P. (1994). Immunoliposomes and PEGylated immunoliposomes: possible use for targeted delivery of imaging agents. Immunomethods 4, 244–258. doi: 10.1006/immu.1994.1027
Torchilin, V. P., Klibanov, A. L., Huang, L., O'Donnell, S., Nossiff, N. D., and Khaw, B. A. (1992). Targeted accumulation of polyethylene glycol-coated immunoliposomes in infarcted rabbit myocardium. FASEB J. 6, 2716–2719.
Ulrich, A. S. (2002). Biophysical aspects of using liposomes as delivery vehicles. Biosci. Rep. 22, 129–150. doi: 10.1023/A:1020178304031
Vingerhoeds, M. H., Storm, G., and Crommelin, D. J. (1994). Immunoliposomes in vivo. Immunomethods 4, 259–272. doi: 10.1006/immu.1994.1028
Wang, X., Song, Y., Su, Y., Tian, Q., Li, B., Quan, J., et al. (2015). Are PEGylated liposomes better than conventional liposomes? A special case for vincristine. Drug Deliv. 29, 1–9. doi: 10.3109/10717544.2015.1027015
Webb, M. S., Saxon, D., Wong, F. M., Lim, H. J., Wang, Z., Bally, M. B., et al. (1998). Comparison of different hydrophobic anchors conjugated to poly(ethylene glycol): effects on the pharmacokinetics of liposomal vincristine. Biomembranes 1372, 272–282. doi: 10.1016/S0005-2736(98)00077-7
Wetzler, M., Thomas, D. A., Wang, E. S., Shepard, R., Ford, L. A., Heffner, T. L., et al. (2013). Phase I/II trial of nanomolecular liposomal annamycin in adult patients with relapsed/refractory acute lymphoblastic leukemia. Clin. Lymphoma Myeloma Leuk. 13, 430–434. doi: 10.1016/j.clml.2013.03.015
Willis, M., and Forssen, E. (1998). Ligand-targeted liposomes. Adv. Drug Deliv. Rev. 29, 249–271. doi: 10.1016/S0169-409X(97)00083-5
Wu, J., Lee, A., Lu, Y., and Lee, R. J. (2007). Vascular targeting of doxorubicin using cationic liposomes. Int. J. Pharm. 337, 329–335. doi: 10.1016/j.ijpharm.2007.01.003
Yarmolenko, P. S., Zhao, Y., Landon, C., Spasojevic, I., Yuan, F., Needham, D., et al. (2010). Comparative effects of thermosensitive doxorubicin-containing liposomes and hyperthermia in human and murine tumours. Int. J. Hyperthermia 26, 485–498. doi: 10.3109/02656731003789284
Zamboni, W. C., Ramalingam, S., Friedland, D. M., Edwards, R. P., Stoller, R. G., Strychor, S., et al. (2009). Phase I and pharmacokinetic study of pegylated liposomal CKD-602 in patients with advanced malignancies. Clin. Cancer Res. 15, 1466–1472. doi: 10.1158/1078-0432.CCR-08-1405
Zhang, H., Wang, G., and Yang, H. (2011). Drug delivery systems for differential release in combination therapy. Expert Opin. Drug Deliv. 8, 171–190. doi: 10.1517/17425247.2011.547470
Zhang, J. A., Anyarambhatla, G., Ma, L., Ugwu, S., Xuan, T., Sardone, T., et al. (2005). Development and characterization of a novel Cremophor EL free liposome-based paclitaxel (LEP-ETU) formulation. Eur. J. Pharm. Biopharm. 59, 177–187. doi: 10.1016/j.ejpb.2004.06.009
Zhang, J. A., Xuan, T., Parmar, M., Ma, L., Ugwu, S., Ali, S., et al. (2004). Development and characterization of a novel liposome-based formulation of SN-38. Int. J. Pharm. 270, 93–107. doi: 10.1016/j.ijpharm.2003.10.015
Zhang, L., Gu, F. X., Chan, J. M., Wang, A. Z., Langer, R. S., and Farokhzad, O. C. (2008). Nanoparticles in medicine: therapeutic applications and developments. Clin. Pharmacol. Ther. 83, 761–769. doi: 10.1038/sj.clpt.6100400
Keywords: liposomes, drug delivery, lipid-based drug delivery system, nanotechnology, biological challenges, translation, accelerated blood clearance, complement activation–related pseudoallergy
Citation: Sercombe L, Veerati T, Moheimani F, Wu SY, Sood AK and Hua S (2015) Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol. 6:286. doi: 10.3389/fphar.2015.00286
Received: 31 July 2015; Accepted: 16 November 2015;
Published: 01 December 2015.
Edited by:
Aaron Tan, University College London, UKReviewed by:
Zejing Wang, Fred Hutchinson Cancer Research Center, USACopyright © 2015 Sercombe, Veerati, Moheimani, Wu, Sood and Hua. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susan Hua, c3VzYW4uaHVhQG5ld2Nhc3RsZS5lZHUuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.