
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 08 May 2014
Sec. Pharmacology of Ion Channels and Channelopathies
Volume 5 - 2014 | https://doi.org/10.3389/fphar.2014.00101
This article is part of the Research Topic CaMKII in Cardiac Health and Disease View all 19 articles
 Emmanuel Camors
Emmanuel Camors Héctor H. Valdivia*
Héctor H. Valdivia*Ryanodine receptors (RyRs) and inositol triphosphate receptors (InsP3Rs) are structurally related intracellular calcium release channels that participate in multiple primary or secondary amplified Ca2+ signals, triggering muscle contraction and oscillatory Ca2+ waves, or activating transcription factors. In the heart, RyRs play an indisputable role in the process of excitation–contraction coupling as the main pathway for Ca2+ release from sarcoplasmic reticulum (SR), and a less prominent role in the process of excitation–transcription coupling. Conversely, InsP3Rs are believed to contribute in subtle ways, only, to contraction of the heart, and in more important ways to regulation of transcription factors. Because uncontrolled activity of either RyRs or InsP3Rs may elicit life-threatening arrhythmogenic and/or remodeling Ca2+ signals, regulation of their activity is of paramount importance for normal cardiac function. Due to their structural similarity, many regulatory factors, accessory proteins, and post-translational processes are equivalent for RyRs and InsP3Rs. Here we discuss regulation of RyRs and InsP3Rs by CaMKII phosphorylation, but touch on other kinases whenever appropriate. CaMKII is emerging as a powerful modulator of RyR and InsP3R activity but interestingly, some of the complexities and controversies surrounding phosphorylation of RyRs also apply to InsP3Rs, and a clear-cut effect of CaMKII on either channel eludes investigators for now. Nevertheless, some effects of CaMKII on global cellular activity, such as SR Ca2+ leak or force-frequency potentiation, appear clear now, and this constrains the limits of the controversies and permits a more tractable approach to elucidate the effects of phosphorylation at the single channel level.
Ryanodine receptors (RyRs) are the calcium release channels of sarcoplasmic reticulum (SR) that provide the majority of the calcium ions (Ca2+) that are needed for contraction of cardiac and skeletal muscle. In the heart, these intracellular Ca2+ release channels are essential for normal cell development and indispensable for life, as demonstrated by the fact that genetic ablation of RYR2, the gene encoding for the cardiac isoform of the RyR channel (RyR2), causes death at early embryonic stage in mice (Takeshima et al., 1998). In humans, single amino acid mutations in RyR2 lead to life-threatening cardiac arrhythmias, also as demonstrated by the ~160 mutations that have been linked so far to catecholaminergic polymorphic ventricular tachycardia (CPVT; Priori and Chen, 2011). Thus, in their intracellular environment, RyR2 channels must be finely regulated so that their output signal (Ca2+) may induce finely graded cell contraction without igniting cellular processes that may lead to aberrant electrical activity or pathological cellular remodeling. This review will concentrate on regulation of RyR2 by phosphorylation, a common post-translational process that confers dynamic functional modulation to a myriad of ion channels and transporters. In the majority of cases, phosphorylation of ion channels or transporters provides an additional layer of regulation without actually being the effector or trigger of activity, i.e., in the case of RyR channels, phosphorylation modulates the effect of Ca2+ on cardiac RyR (RyR2) or depolarization on skeletal RyR (RyR1) without having the inherent ability to open or close the channel per se. In a useful analogy, phosphorylation and Ca2+/depolarization would be similar to the “volume” and “on/off” switches, respectively, of the radio in which RyR songs are played. However, as we will see on several sections of this review, this mode of regulation is so controversial that a definitive role for RyR2 phosphorylation has not been reached yet, despite the paramount importance of this process in physiological and pathophysiological conditions. RyR2 may be phosphorylated in vitro and in vivo by several kinases, but here we will concentrate on CaMKIIδ (the most abundant CaMKII expressed in the heart and referred to here simply as “CaMKII”), and incorporate protein kinase A (PKA) whenever relevant, as these two kinases share some common transduction pathways, bear the most relevance in cardiac diseases and have been studied the most. Similarly, at least two splice variants of CaMKIIδ are expressed in cardiomyocytes, CaMKIIδC (cytosolic) and CaMKIIδB (nuclear), and we will identify their differential effect whenever appropriate.
The RyR2 channel is a homotetrameric protein of ~2 million Da that is often complexed with several accessory proteins (see below), forming an intricate multi-protein array (Marx et al., 2000; Bers, 2004; Valdivia, 2013). The RyR2 monomer (almost 5,000 amino acids) is organized as a series of discrete domains, with the carboxy-terminal segment crossing the SR membrane as few as four and as many as ten times (depending on the model) and forming the Ca2+-permeable pore, whereas the bulk of the protein (~90%) protrudes into the cytosol to bridge a ~15–20 nm gap between the SR and t-tubule membranes (Hamilton and Serysheva, 2009; Capes et al., 2011; Van Petegem, 2012). The cytosolic portion of the channel contains multiple regulatory domains, most prominently Ca2+ activation and inactivation sites, but also binding sites for energy sensors such as nucleotides (ATP, ADP, and AMP) and inorganic phosphate, metabolites such as pyruvate, fatty acids and polyamines, and ions such as Mg2+, H+, and Cl- (Zucchi and Ronca-Testoni, 1997; Fill and Copello, 2002; Meissner, 2004). The cytoplasmic site harbors also multiple phosphorylation epitopes (see below). Therefore, the RyR2 channel may act as a molecular switchboard that integrates a multitude of cytosolic signals such as dynamic and steady Ca2+ fluctuations, oxidation, metabolic states, β- adrenergic stimulation (phosphorylation), etc. and transduces these cytosolic signals to the channel pore to release appropriate amounts of Ca2+. Furthermore, Ca2+ release is critically influenced by luminal (intra-SR) factors such as Ca2+ content and protein interactions, thus conferring RyR2 channels an additional role as integrative switch-valves that restrict luminal Ca2+ overload, e.g., during sympathetic stimulation. Most of the signal-decoding structures are integral domains of the RyR2 protein, but as if this huge structural assembly were not sufficiently complex, RyR2 channels are also capable of protein–protein interactions that allow them to bind, in some cases steadily and in other cases in a time- and Ca2+-dependent manner, to smaller and independently regulated accessory proteins that add another layer of versatility (and complexity) to regulation of Ca2+ release in vivo. The best known RyR2-interacting proteins are calmodulin (CaM), which tonically inhibits Ca2+ release (Balshaw et al., 2002; Yang et al., 2014) FKBP12.6, which stabilizes RyR2 closures (Marx et al., 2000; Kushnir and Marks, 2010; but see also Timerman et al., 1996; Xiao et al., 2007), sorcin, which inhibits Ca2+ release in a Ca2+-dependent manner (Farrell et al., 2003), and the ternary complex triadin-junctin-calsequestrin, which “senses” luminal Ca2+ content and modulates RyR2 activity, probably by acting as direct channel ligands (Gyorke et al., 2004). More recently, RyR2 has been found to hold anchoring sites for PKA, protein phosphatase 1 (PP1) and 2A (PP1 and PP2A), phosphodiesterase 4D3 (PDE4D3) and CaMKII (Marx et al., 2000; Currie et al., 2004; Lehnart et al., 2005), underscoring the importance of RyR2 regulation by phosphorylation.
The multi-protein complex described above that includes kinases and phosphatases associated to the RyR2 strongly suggest that this channel is an avid target of downstream signaling effectors of the β-adrenergic system. PKA is a classical effector of the β-adrenergic pathway and, although the extent of activation of CaMKII by β-adrenergic stimulation and the precise signaling pathways involved are still incompletely understood, it is also accepted that CaMKII is involved in the inotropic and lusitropic effects of sympathetic stimulation (Grimm and Brown, 2010). Therefore, RyR2 proteins are natural targets of PKA and CaMKII and indeed, stimulation of beating hearts with β-adrenergic agonists readily triggers phosphorylation of RyR2 (Takasago et al., 1989; Witcher et al., 1991; Benkusky et al., 2007). Which kinase phosphorylates RyR2 to a greater extent? As early as 1989 (and before the controversy surrounding the stoichiometry of CaMKII and PKA phosphorylation of RyR2 that started in 2000, see below), Takasago et al. (1989) reported that the extent of RyR2 phosphorylation by CaMKII was ~4 times greater than that of PKA in dog hearts. This was confirmed by Witcher et al. (1991) in the same animal species. Later, using phospho-specific antibodies and autoradiograms, Rodriguez et al. (2003) confirmed that CaMKII phosphorylated at least four sites per each PKA-phosphorylated site. Hence, there is significant evidence indicating that RyR2 are far better substrates for CaMKII than they are for PKA, which is not entirely surprising given that the phosphorylation consensus for CaMKII: R/K-X-X-S/T (where X is any amino acid residue) is less stringent than that for PKA: R-R-X-S/T (Pinna and Ruzzene, 1996; however, as we will see below, a CaMKII or PKA phosphorylation consensus does not necessarily result in CaMKII or PKA phosphorylation).
Despite the presence of multiple phosphorylation consensuses in each RyR2 subunit (George, 2008) and the fact that RyR2 channels are eager substrates for several kinases (see above), only three phospho-sites have been discovered to date. Let’s discuss the most salient features of each of these sites.
Serine 2808 (S2808, mouse and human nomenclature) was first discovered by Witcher et al. (1991) as a CaMKII site but later Marx et al. (2000), Wehrens et al. (2006) labeled it as an exclusive PKA site, despite the fact that RyR2 channels with specific ablation of this phospho-epitope (RyR2-S2808A) were still phosphorylated by PKA (Benkusky et al., 2007). So, a major issue with this site is whether it’s a preferred substrate for CaMKII or PKA. Since the study by Marx et al. (2000) reporting that S2808 was hyperphosphorylated in heart failure patients and that its phosphorylation enhanced dramatically the activity of RyR2 channels, several groups have studied this phospho-site in detail (Jiang et al., 2002; Rodriguez et al., 2003; Stange et al., 2003; Currie et al., 2004; Ai et al., 2005; Xiao et al., 2005; Carter et al., 2006; Kohlhaas et al., 2006; Huke and Bers, 2008; MacDonnell et al., 2008; Fischer et al., 2013), with the majority of evidence pointing to S2808 being a target for PKA, CaMKII and possibly PKG. Also, most studies find S2808 constitutively phosphorylated [basal phosphorylation ~50–75% in several animal species and humans (Jiang et al., 2002; Rodriguez et al., 2003; Carter et al., 2006; Huke and Bers, 2008)], thus raising doubts that this site may be a reliable index of abnormal PKA-phosphorylation in heart failure patients. Indeed, Benkusky et al. (2007) found that mice with genetic ablation of the S2808 phospho-epitope (RyR2-S2808A) do not alter their β-adrenergic response, have cardiomyocyte function almost unchanged, and are not significantly protected against the maladaptive cardiac remodeling induced by chronic stress. Further, although PKA phosphorylation of RyR2 modified single-channel activity, its effect was modest and occurred at activating (systolic) [Ca2+], only, not at the expected low (diastolic) [Ca2+], where it would cause significant Ca2+ leak (Bers et al., 2003). The lack of protection against heart failure dysfunction as well as the normal β-adrenergic response of the RyR2-S2808A mice were confirmed by MacDonnell et al. (2008), Zhang et al. (2012) using the same transgenic mouse line. In the scheme of Marx et al. (2000), which is continuously validated and extended by additional reports by the same group (for example, Wehrens et al., 2006; Shan et al., 2010a, b), PKA phosphorylation of S2808 led to dissociation of FKBP12.6, which in turn destabilized the closed state of the channel and induced multiple subconductance states, overall increasing RyR2 Ca2+ fluxes, especially at diastolic [Ca2+]. However, most of the central tenets of this scheme have not been confirmed by others (reviewed by George, 2008; Currie, 2009; Eschenhagen, 2010; Bers, 2012; Valdivia, 2012). Specifically, Stange et al. (2003) found no effect of ablating the S2808 phospho-site (RyR2-S2808A) or simulating constitutive activation (RyR2-S2808D) in the activity of the channel or its affinity for FKBP12.6. That phosphorylation of RyR2 does not appear to dissociate FKBP12.6 was also found by Xiao et al. (2004), Guo et al. (2010). Also, there are no reports, except by the Marks’ group, indicating that RyR2 phosphorylation induces subconductance states, presumably the hallmark of FKBP12.6 dissociation. Lastly, several groups have independently reported that FKBP12.6 does not affect RyR2 activity at all (Timerman et al., 1996; Xiao et al., 2007) or that it has modest effects, only (Guo et al., 2010). Thus, although it is indisputable that S2808 is phosphorylated by PKA, CaMKII and possibly other kinases, the functional output of such reaction has been difficult to pin down and is the subject of intense debate. Further studies are needed and should continue to provide insights as drugs designed to prevent FKBP12.6 dissociation enter clinical trials and testing in humans.
Serine 2814 (S2814) was discovered by Wehrens et al. (2004) as a CaMKII site and, although there is consensus that CaMKII is the preferential kinase for this site, there is less agreement that it is the only CaMKII site in RyR2. As mentioned above, S2808 is also a target for CaMKII, and since there are at least ~4 CaMKII sites for each PKA site, this suggests that there are still other CaMKII sites yet to be discovered. Although S2814 is only six amino acid residues downstream of S2808 and forms part of the same RyR2 “phosphorylation hot spot” (Valdivia, 2012; Yuchi et al., 2012) the role of S2814 has been less controversial because so far it appears to be a more specific substrate for CaMKII, despite the fact that it forms part of a non-canonical CaMKII phosphorylation consensus: 2805RRISQTSQVSV2815 (S2808 and S2814 are underlined). Interestingly, another serine residue within that hot spot, S2811, is also part of a non-canonical CaMKII consensus and yet, it appears to be phosphorylated in vitro by PKA and CaMKII (Yuchi et al., 2012) and in vivo in mice stimulated by β-adrenergic agonists (Huttlin et al., 2010). Whether S2811 contributes to the effect of CaMKII phosphorylation of RyR2, or distorts the signal of phospho-specific antibodies pS2808 and pS2814 (Huke and Bers, 2008), making it difficult to discern the specificity of kinases for these phospho-epitopes, is still unclear. In quiescent cardiomyocytes, S2814 is barely phosphorylated (unlike S2808), and although its activity-dependent phosphorylation may be prevented by CaMKII inhibitors, the basal phosphorylation at rest is maintained by a Ca2+-dependent kinase other than CaMKII (Huke and Bers, 2008). Thus, at least two Ca2+-dependent kinases phosphorylate S2814, possibly leading to the same functional output (discussed below). Whether S2814 phosphorylation contributes to the inotropic effects induced by β-adrenergic stimulation has not been firmly established. However, mice with germline ablation of the S2814 phospho-epitope (RyR2-S2814A) appear more resilient than WT against a variety of cardiac insults. van Oort et al. (2010) found that RyR2-S2814A mice were protected from catecholaminergic- and pacing-induced tachyarrhythmias, whereas Ather et al. (2013) reported that arrhythmogenic spontaneous Ca2+ waves that were prevalent in the mdx mice (Duchenne muscular dystrophy model), were suppressed by crossbreeding with the RyR2-S2814A mice. In a protocol of ischemia-reperfusion injury, the RyR2-S2814A mice exhibited significantly fewer premature beats (that could be ascribed to delayed afterdepolarizations) than WT, a protection that was not seen in mice with ablation of two phospholamban phospho-sites (PLB-DM; Said et al., 2011). Respress et al. (2012) reported by RyR2-S2814 mice fared much better than WT after TAC (transverse aortic constriction)-induced heart failure, but interestingly, were not protected against MI (myocardial infarction)-induced heart failure, proposing that CaMKII phosphorylation of RyR2 plays a role in non-ischemic forms of heart failure, only. Purohit et al. (2013) reported that CaMKII activation and phosphorylation of S2814 were required to induce atrial fibrillation in angiotensin-infused mice. Thus, several laboratories (albeit all of them using the same mouse line) have independently bestowed on S2814 a preponderant role in cardiac protection, wherein inhibition of S2814 phosphorylation averts the functional and structural damage to the heart induced by heart failure, atrial fibrillation, and other insults. Since it appears illogical that S2814 phosphorylation, a seemingly common reaction, was naturally designed to wreak havoc in the heart’s function and structure, it is therefore important to demarcate the limits in which S2814 phosphorylation turns pathogenic. Interestingly, mice with constitutive activation of S2814 (S2814D), have structurally and functionally normal hearts without arrhythmias (van Oort et al., 2010) which is surprising in the context of the presumably malicious role played by S2814 phosphorylation described above.
Finally, Serine 2030 (S2030) was found by Xiao et al. (2005) using classical phospho-epitope mapping. Although S2030 is squarely within a CaMKII phosphorylation consensus (2027R-L-L-S2030), oddly this site is preferentially phosphorylated by PKA, at least in vitro. This site, therefore, like the other sites discussed above, does not follow in silico predictions of kinase specificity and conforms instead to the cryptic rules of steric hindrance, topological association to specific kinases, substrate availability, etc. that operate in vivo and separate predicted from actual phosphorylation sites. Experiments in vitro revealed a major role for S2030 in the control of RyR2 activity, and in heart failure patients, it appeared as a reliable marker of RyR2 dysfunction (Xiao et al., 2006), presumably even superseding S2808 (Marx et al., 2000). The RyR2 channel appears to be completely unphosphorylated at S2030 in quiescent cardiac myocytes, and phosphorylation is promoted by β-adrenergic stimulation (Huke and Bers, 2008), in line with in vitro experiments that indicate PKA phosphorylation of this site. However, the precise role of S2030 in intracellular Ca2+ homeostasis and cardiac performance, and its involvement in pathological states of the heart are not well understood yet. Mouse lines with genetic ablation of this site (RyR2-S2030A) have just been generated (Camors et al., 2014) and should shed light on the functional role of this novel phopho-site.
Many lines of evidence demonstrate that RyR2 channels are phosphorylated in vitro and in vivo, but a fundamental question still pervades the field: what is the functional effect of RyR2 phosphorylation? This appears as a simple question, set for a straightforward answer, but examining the diverse and apparently opposite effects that have been published in the topic in the last ~2 decades, the only safe response seems to be that phosphorylation does something to RyR2 activity. All potential functional outcomes for RyR2 phosphorylation (increase, decrease, and no effect on activity) have been reported, with tantalizing hints toward, but no clear factors responsible for, the radical differences. Also, the question needs to be framed in a specific integrative context for a defined response to hold some truth. For example, whereas there is compelling evidence that PKA can alter RyR2 activity at the single channel level (Hain et al., 1995; Valdivia et al., 1995; Marx et al., 2000; Jiang et al., 2002; Uehara et al., 2002; Carter et al., 2006; Wehrens et al., 2006; Benkusky et al., 2007; Li et al., 2013), multiple self-regulatory systems operating in intact cells (not to mention the whole heart) may bring down this response to undetectable levels so that the answer from the cellular viewpoint would appear to be “no effect.”
Although not always sure foretellers of a defined outcome, there are some factors that modify the activity of RyR2s and are likely to complicate their response to phosphorylation. First, as stated above, RyR2 channels contain multiple phosphorylation sites that, depending on their phospho-state, may attenuate or synergize the effect of the other sites, or may require prior phosphorylation to activate the whole protein. This has become evident in experiments in which it has been possible to link variable levels of phosphorylation with defined single-channel activity (Carter et al., 2006) and also where it is clear that phosphatases activate RyR2 channels to higher levels than either PKA or CaMKII alone (Lokuta et al., 1995; Terentyev et al., 2003), suggesting that dephosphorylation uncovers a set of phospho-sites that modulate RyR2 activity but are not affected by either kinase. Second, RyR2 activity has long been established to be dependent on the speed of Ca2+ application [as inferred by Fabiato in his classical experiments that characterized CICR (Fabiato, 1985) and demonstrated in single channel experiments (Gyorke and Fill, 1993)], and this in turn may greatly influence the overall effect of phosphorylation (Valdivia et al., 1995; Jiang et al., 2002). For example, PKA phosphorylation of RyR2 channels increases their transient component of activity (peak activation) and accelerates their rate of adaptation to a steady level of activity. In cellular settings, this effect would translate into faster rates of Ca2+ release in response to a fast and transient Ca2+ entry (such as ICa) and into little effects on steady RyR2 activity. In fact, experiments in which SR Ca2+ load and ICa were kept constant showed that β-adrenergic stimulation of ventricular myocytes accelerated the rate of Ca2+ release (Ginsburg and Bers, 2004), and in permeabilized cells with constant [Ca2+], PKA had little effect on RyR2 activity (Li et al., 2002). Thus, not all effects of RyR2 phosphorylation are detectable at constant [Ca2+], the preferred method of testing for such effects; fast and transient Ca2+ stimuli are required to unveil some of its most critical effects. Third, there exist self-correcting mechanisms that preclude persistent activation (or inhibition) of RyR2s in intact cells and can mitigate effects of phosphorylation in a few beats (Eisner et al., 1998). These mechanisms invoke restoration of steady-state Ca2+ fluxes when a single component of the excitation–contraction coupling machinery is perturbed or malfunctions. Thus, in its simplest terms, if the effect of PKA phosphorylation of RyR2 channels was to cause a persistent Ca2+ leak (as postulated by Marx et al., 2000), then the persistent leak of Ca2+ would necessarily cause at least partial SR Ca2+ depletion, which in turn would re-tune Ca2+ release and stop the leak. Thus, again, RyR2 responses to phosphorylation that are discretely detected at the single channel level for a relatively long period of time, may be short-lived or undetectable in cellular settings due to self-correcting mechanisms. Lastly, other factors such as Mg2+ (Li et al., 2013) and luminal [Ca2+] (Xiao et al., 2005) seem to be required in just about the right quantity for phosphorylation to exert maximal effects. Overall then, the response of RyRs to phosphorylation is neither simple nor monotonous; it is complicated by factors intrinsic and extrinsic to the channel protein and depends critically on the context (molecular, cellular, whole heart) in which it is examined.
Despite the recognized difficulty in linking RyR2 phosphorylation with a defined functional output, the current model of RyR2 phosphorylation is extremely simple, and needs revision. In its simplest version, PKA phosphorylation of S2808 dissociates FKBP12.6 and activates the channel by inducing long-lived subconducting states, whereas CaMKII phosphorylation of S2814 activates the channel by a different mechanism (Marx et al., 2000; Wehrens et al., 2004). However, this model disregards the S2030 phospho-site, a bona fide PKA site (Xiao et al., 2006), and does not give weight to the potential contribution of other phospho-sites, hitherto uncovered but convincingly supported by several lines of evidence. This model also does not bode well with recent structural data, wherein S2808 and S2814, by virtue of its immediacy (only six residues apart in a ~5000 amino acid subunit), form part of a larger “phosphorylation hotspot” that encompasses S2811 and possibly T2810 (2805RRISQTSQVSV2815; Yuchi et al., 2012). This phosphorylation hotspot is harbored in a flexible loop connecting two symmetrical repeats interacting with one another through β strand interactions (Yuchi et al., 2012). Because the flexible loop is exposed to solvent and protrudes prominently on the RyR2 surface, it is not surprising that the whole phosphorylation hotspot may be easily accessed by several kinases. Because of the tight clustering of phospho-sites in this hotspot, there emerges the interesting possibility that they all provide functional redundancy, i.e., phosphorylation of S2808 may lead to the same downstream effects as phosphorylation of S2811, or S2814. In this scenario, the sequential addition of phosphate groups to the hotspot would lead to a graded (instead of all-or-none) response of RyR2 to kinases, which could explain the widely variable response detected by investigators to a specific phosphorylation maneuver. Of course, this alternative model based on the phosphorylation hotspot alone does not explain the distinct effect of CaMKII and PKA on RyR2 activity (see below), and to fill this void it is necessary to invoke the participation of S2030 and likely other as-yet-unrecognized phospho-sites. Thus, in an obligatorily more complex model (Figure 1) that we call “multi-phosphosite model” (Valdivia, 2012), the differential effect of PKA and CaMKII on RyR2 activity is dictated by the integrated response of the phosphorylation hotspot and of additional phosphorylation sites. For example, phosphorylation of S2808 and S2030 by PKA could coordinate channel openings in response to fast calcium stimuli (Valdivia et al., 1995; Ginsburg and Bers, 2004), and phosphorylation of S2814 and other CaMKII site(s) could open RyR2s at diastolic [Ca2+], which would translate in Ca2+ leak. Certainly, until we understand the molecular basis by which the phosphorylation hotspot and other phospho-sites talk to the channel’s gating domains, this structurally-based model will remain speculative and incomplete. Nonetheless, it takes into consideration compelling evidence on the existence of various phosphorylation sites and departs substantially from the simplified notion that one kinase phosphorylates one site and produces one effect.
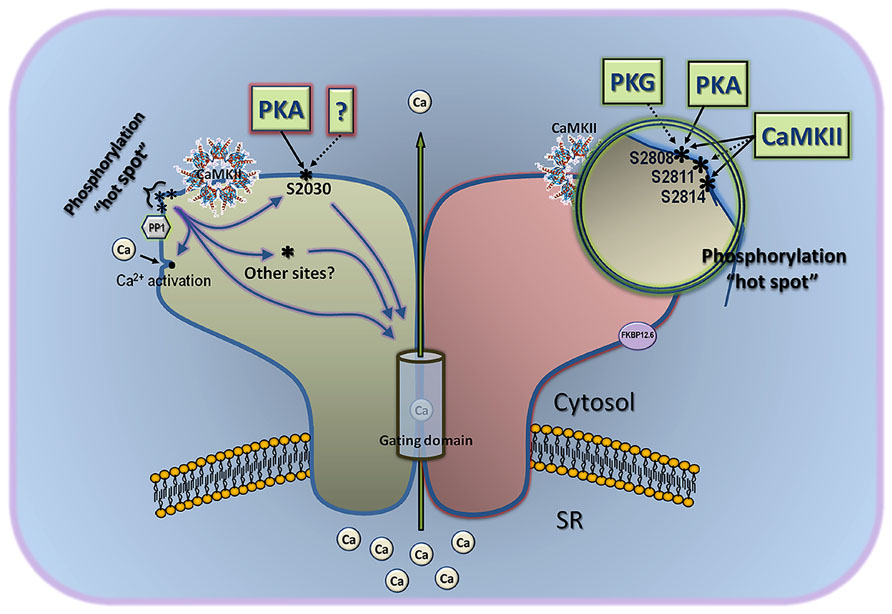
FIGURE 1. Multi-site model of RyR2 phosphorylation. This model considers the three phospho-sites known to date, and gives also significant weight to other as-yet-uncovered sites. The classical sites S2808 and S2814 are part of a “phosphorylation hotspot” that is located in a protruding part of the channel, is targeted by several kinases, and may contain other phospho-epitopes not yet characterized (for example, S2811). Phosphorylation of individual residues within this hotspot may be undistinguishable by the channel’s gating domain, but gradual addition of phosphate groups here may contribute to a tunable effect instead of an all-or-none response. Assuming that the phosphorylation hotspot works collectively toward a single effect, the differential regulation of PKA and CaMKII on channel gating may come about by the combined effect of each kinase on phospho-residues of the hotspot and other phosphorylation sites, such as S2030. This model also accommodates solo effects of S2030 or the phosphorylation hotspot on gating domains of the channel, as well as indirect effect on gating via interaction with classical Ca2+ activation sites. CaMKII and protein phosphatase 1 (PP1) are depicted close to the phosphorylation hotspot because the latter is readily phosphorylated/dephosphorylated by endogenous CaMKII and PP1. Demonstrated effect of kinases on S2808, S2814, and S2030 in intact cells or hearts is shown with a solid line, and in vitro effect is shown with a broken line.
Ryanodine receptor 2 channels have long been known to be a suitable substrate for CaMKII. In fact, in 1984, long before the controversies surrounding RyR2 phosphorylation and before the purification of RyR2, Seiler et al. (1984) observed that “high molecular weight proteins,” later identified as RyR2 channels, were excellent substrates for CaMKII. In the experiments of Marx et al. (2000), where RyR2 was initially presented as a structural scaffold for multiple accessory proteins, CaMKII was not identified as part of that macromolecular complex. However, Currie et al. (2004) and J.H. Brown’s group (reviewed in Grimm and Brown, 2010), have provided ample evidence that CaMKII is intimately associated to RyR2 (although the actual biding site is not known), logically portending an important effect of CaMKII on modulation of RyR2 activity. At the single channel level, the preponderance of results suggests that CaMKII activates RyR2 channels, but again, the results are not unanimous. Let’s briefly review the reports.
Although indirect evidence of RyR2 modulation by CaMKII was initially obtained by [3H]ryanodine binding assays (Takasago et al., 1989), the first direct demonstration was provided by Witcher et al. (1991) using single channel recordings of canine RyR2 channels reconstituted in lipid bilayers. These authors observed activation of RyR2 channels by both, endogenous (cardiac SR-resident) and exogenous (purified from brain homogenates) CaMKII, and correlated their results with biochemical assays showing additive levels of phosphorylation by endogenous and exogenous CaMKII. This was also the report that unveiled S2808 (S2809 in dogs) as a major CaMKII site, although the proportion of the total RyR2 phosphorylation for which S2808 was solely responsible could not be determined. Thus, if the endogenous CaMKII of Witcher et al. (1991) is the same CaMKII intimately attached to RyR2 (Currie et al., 2004; Grimm and Brown, 2010), it appears that this CaMKII phosphorylates a subset of RyR2 phospho-sites only, fewer than what is actually possible by exogenous CaMKII. Thus, the recurring point that emerges is that multiple CaMKII sites concur in the RyR2 protein, each potentially exerting a defined level of control in the channel and not always phosphorylated by a given experimental condition. This might have been why Lokuta et al. (1995) detected inhibition of single RyR2 channel activity by exogenous CaMKII, similar to Takasago et al. (1989), who used exogenous CaMKII in [3H]ryanodine binding experiments. Nevertheless, multiple CaMKII sites as justification for apparent discrepancies may not be universally applied, as Hain et al. (1995) found activation of RyR2 channels by CaMKII, using methods and animal species (dog) similar to Lokuta et al. (1995). The four studies mentioned above [Witcher et al. (1991), Lokuta et al. (1995), Takasago et al. (1989), Hain et al. (1995)] all used high (μM) [Ca2+] to keep the RyR2 channels open, and none of them frontally addressed the question of whether CaMKII activates RyR2 at low (diastolic) [Ca2+], a question of paramount importance given the current thinking that CaMKII increases SR Ca2+ leak (see below). This was technically difficult at the time because CaMKII itself was known to require Ca2+ as a cofactor for activation, and the Ca2+-free, auto-phosphorylated active form of the enzyme was not widely known. Hence, more studies are needed to clarify the Ca2+-dependence of CaMKII effect on isolated RyR2 channels. Lastly, in an oversimplified scheme that largely ignored the overwhelming evidence for the aforementioned multiple CaMKII sites, Wehrens et al. (2004) found that CaMKII activated single RyR2 channels and postulated that ablation of a single phospho-site (S2815) was sufficient to inhibit CaMKII phosphorylation completely. In summary, CaMKII phosphorylation of isolated RyR2 channels is readily detected at the biochemical level, and the preponderance of results indicate that CaMKII activates RyR2 channels by targeting multiple sites; however, some studies find that CaMKII may inhibit RyR2 channel activity and the nature of this apparent discrepancy is not easily explained. Furthermore, whether CaMKII phosphorylation activates individual RyR2 channels at low (diastolic) [Ca2+] has not been firmly established yet and needs more refined studies.
Many studies have addressed the role of CaMKII phosphorylation in SR Ca2+ release and excitation–contraction coupling of intact ventricular myocytes, and most of them are detailed in excellent reviews (George, 2008; Currie, 2009; Grimm and Brown, 2010; Currie et al., 2011; Luo and Anderson, 2013; Bers, 2014). Here we will simplify the discussion by concentrating on the studies that have addressed the role of CaMKII phosphorylation on the most direct indicators of RyR2 function, namely, SR Ca2+ leak, spontaneous Ca2+ waves, and Ca2+ sparks. This circumvents the problem of interpreting CaMKII effects on RyR2 based on whole cell results or global Ca2+ transients, which are the product of multiple nodes of activity interacting in complex ways.
Most studies find that β-adrenergic stimulation increases SR Ca2+ leak, and that chronic adrenergic stimulation of ventricular myocytes such as that occurring in heart failure produces a cellular substrate favorable for generation of Ca2+-triggered arrhythmias. To what extent are CaMKII and RyR2 channels responsible for these effects? We examined in preceding paragraphs that, although the precise transduction pathways have not been completely elucidated yet, it is clear that CaMKII is activated upon β-adrenergic stimulation of the heart (Grimm and Brown, 2010). In addition, a significant number of studies [but not all (Yang et al., 2007)] find that CaMKII activation increases SR Ca2+ leak (Maier et al., 2003; Currie et al., 2004; Guo et al., 2006; Curran et al., 2007) and that the SR Ca2+ leak that is characteristically increased in heart failure (Kirchhefer et al., 1999; Marx et al., 2000; Ai et al., 2005) may be prevented by specific CaMKII inhibition (Wu et al., 2002; Ai et al., 2005; Curran et al., 2010; Sossalla et al., 2010; Respress et al., 2012), but not by PKA inhibition (Curran et al., 2010). Hence, making the reasonable assumption that RyR2 channels are the main pathway for SR Ca2+ leak, and deriving from the reports above that CaMKII activation evokes arrhythmogenic SR Ca2+ leak, then it is fair to conclude that CaMKII phosphorylation activates RyR2 at diastolic [Ca2+] to produce unchecked SR Ca2+ release that is capable of bringing membrane potential to threshold (delayed afterdepolarizations) and quite possibly ignite cellular pathways that lead to cardiac remodeling. From this perspective, CaMKII is presented as an arrhythmogenic, deleterious kinase, and RyR2 its main instrument of deraignment. Obviously, the seemingly belittled positive effect of CaMKII in normal cell function cannot be discounted, and a balance between physiological and pathological effects of CaMKII activation must exist in vivo. Thus, an emerging notion is that normally, acute CaMKII and PKA activation result in an increased magnitude and rate of Ca2+ release, respectively (Ginsburg and Bers, 2004), which account in no small part for the inotropic effects of β-adrenergic stimulation. This hypothesis is supported by studies that find that CaMKII increases fractional Ca2+ release (reviewed in Anderson et al., 2011) and that PKA increases the rate of Ca2+ release, only, in cells with controlled L-type Ca2+ channel trigger and SR Ca2+ content (Ginsburg and Bers, 2004). The role of CaMKII in force-frequency stimulation is also well known (Krishna et al., 2013). On the other hand, it appears that most of the deleterious effects of CaMKII are exerted under conditions that allow its chronic activation. It has become evident that CaMKII is not only a sensor of Ca2+ signals, but it is also exquisitely sensitive to oxidative stress (Luczak and Anderson, 2014). Oxidative stress is an important ingredient of the pathogenic recipe that deranges cardiomyocytes in atrial fibrillation, heart failure, sinus node dysfunction, and other cardiomyopathies. Thus, persistent activation of CaMKII by reactive oxygen species (ROS), is an expected (and demonstrated) side effect of many cardiac insults, and a constitutively activated CaMKII has an ample range of action (hence the name multi-functional), including several ion channels and transporters that control membrane excitability and excitation–contraction coupling (Bers and Grandi, 2009), and others that control excitation–transcription coupling (Bers, 2011). Since, as we noted above, RyR2 channels are natural and avid substrates of CaMKII and are themselves affected by oxidative stress (Donoso et al., 2011), the contribution of RyR2 to altered Ca2+ homeostasis in these cardiac pathologies is almost assured. Overall, then, CaMKII walks a fine line separating “good” from “evil,” and a great part of this dichotomy is dictated by its effect on RyR2 channels and its capacity to induce (potentially excessive) SR Ca2+ leak. Studies aimed at demarcating the pivotal point in which CaMKII contributes to health or disease continues at great strides. Until then, inhibiting CaMKII phosphorylation of RyR2 channels as targeted approach to prevent the excessive Ca2+ leak and the spontaneous Ca2+ waves that undergird several cardiomyopathies appears enticing, but needs further studies.
In the majority of mammalian cells, complex intracellular Ca2+ signals elicited by neurohormonal stimuli are mediated through the generation of inositol 1,4,5-triphosphate (InsP3) and the activation of its receptor, the InsP3R. InsP3 originates from the hydrolysis of the membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) by phospholipase C (PLC; reviewed in Foskett et al., 2007; Taylor and Tovey, 2010). Concomitant binding of InsP3 and Ca2+ is necessary for InsP3R activation, and this in turn leads to Ca2+ release within the cytoplasm. Multiple stimuli by a given agonist result in repetitive InsP3R-mediated Ca2+ releases (Ca2+ oscillations) that propagate as Ca2+ waves through the entire cell first and may ultimately stimulate neighboring cells to form inter-cellular Ca2+ waves. In cardiac myocytes, localization of InsP3Rs in the nuclear envelop underlies their role in excitation–transcription coupling, but recent evidence (mostly from atrial cells) suggest that although their abundance is low in the t-tubules and the sarcolemma, InsP3Rs can also rev up excitation–contraction coupling and eventually trigger cellular arrhythmias (Mackenzie et al., 2002; Zima and Blatter, 2004; Li et al., 2005). Similar to RyR2 channels, InsP3Rs are scaffolding proteins highly regulated by ions (Ca2+, H+), nucleotides (ATP), accessory proteins (FKBP12, calmodulin), and also undergo major post-translational modifications such as phosphorylation (Taylor and Tovey, 2010). The first evidence for InsP3Rs phosphorylation was obtained in the late 80s by Greengard’s and Sydner’s groups from rat cerebella (Supattapone et al., 1988; Walaas et al., 1988). Today, at least 15 different kinases and phosphatases, including CaMKII, are postulated to target the InsP3Rs and regulate their activity in an isoform- and tissue-specific manner. This second part of this review will focus on the newly discovered CaMKII site (Ser150), on the physiological consequences of CaMKII phosphorylation of InsP3R, and on the mechanisms by which this event may participate in cardiac Ca2+ signaling. Further details on the general properties of InsP3R and their role in cardiac myocytes may be found in excellent reviews (Foskett et al., 2007; Kockskämper et al., 2008; Vanderheyden et al., 2009; Taylor and Tovey, 2010).
In mammals, three different genes (ITPR1, ITPR2, and IPTR3) encode for ~300 kDa subunits that assemble as homo- or hetero-tetramers to form a functional InsP3R channel. Similar to RyR2, the low resolution of InsP3Rs crystals shows a mushroom-like structure with a large cytosolic cap constituted by the N-terminal and central regions of the protein and a short stem, inserted into the membrane by the C-terminal domain (Da Fonseca et al., 2003). The InsP3-binding domain (or “core”) is located within the first amino acids of the channel, while up to eight Ca2+ binding sites are distributed throughout its entire sequence (including two sites within the InsP3-binding core; Mignery et al., 1990; Mignery and Sudhof, 1990; Pietri et al., 1990; Sienaert et al., 1996, 1997). The Ca2+ pore is formed by a classic P-loop between the membrane-spanning segments 5 and 6. Altogether the organization of InsP3Rs suggests that the opening of the channel depends on large allosteric movements that, by analogy with the RyR2s, may be finely tuned by phosphorylation (Van Petegem, 2012).
Depending on the subtype, the apparent Kd of the receptors for InsP3 varies from 10 to 80 nM (with InsP3R-2>>InsP3R-1>InsP3R-3; Newton et al., 1994). Remarkably, the binding of InsP3 to the receptor channel is stoichiometric and regulates the properties of the InsP3R-mediated Ca2+ release. At low intracellular concentrations, InsP3-binding induces the opening of single InsP3R channel promoting a unitary Ca2+ release (dubbed “Ca2+ blip”). Increasing InsP3 concentrations facilitate the opening of clusters of InsP3Rs, allowing CICR within a cluster. At high concentration, InsP3 promotes CICR between the clusters and intracellular Ca2+ waves (Bootman et al., 1997). A noteworthy property of the InsP3Rs is their high affinity for Ca2+, causing maximal activity at diastolic [Ca2+]i (100 nM) while in similar conditions RyR2 are closed (Ramos-Franco et al., 1998). Therefore in myocytes stimulated by Gq–protein coupled receptor (GqPCR) agonists, InsP3Rs-mediated Ca2+ release may occur during diastole and “prime” the RyR2 to open, thus potentially increasing RyR2-mediated SR Ca2+ leak and triggering of arrhythmias (Mackenzie et al., 2002; Zima and Blatter, 2004; Li et al., 2005).
Since the first reports of InsP3R phosphorylation more than two decades ago (Supattapone et al., 1988; Walaas et al., 1988), the growing number of kinases that target InsP3Rs and either decrease or increase their activity have uncovered the complexity of InsP3Rs regulation. Among those enzymes, recent observations demonstrate that CaMKII plays a critical role for InsP3R function.
The consequences of CaMKII phosphorylation of InsP3R were first ascertained in permeabilized embryonic mouse fibroblasts (Zhang et al., 1993). In these cells, CaMKII phosphorylation permits the activation of the InsP3R channel upon addition of InsP3 or an agonist cocktail (bradykinin + GTPγS). This activation takes place at intracellular [Ca2+] ranging from 30 to 100 nM. At higher [Ca2+]i, calcineurin activity prevails and dephosphorylation of the channel occurs. In agreement with these results, Cameron et al. (1995) observed that CaMKII enhanced the InsP3-sensitive Ca2+ flux in rat cerebellum microsomes. However, these outcomes were challenged by observations made in intact HeLa cells stimulated by histamine (Zhu et al., 1996). In this model, it is the Ca2+ released by the InsP3R that activates CaMKII, which phosphorylates back InsP3R to terminate the release. In a second step, the activation of a phosphatase (likely PP1/PP2A) dephosphorylates InsP3Rs to restore its activity. This model is particularly elegant since the authors also demonstrated that activated CaMKII facilitates the ER Ca2+ refilling by increasing SERCA activity (likely through PLN phosphorylation). As a consequence, the alternation between phosphorylated and dephosphorylated states of InsP3Rs seemed to be the basic mechanism for histamine-induced intracellular Ca2+ oscillations (Figure 2, left panel). Later, similar inhibitory effects of InsP3Rs phosphorylation by CaMKII were reported in channels reconstituted in lipid bilayers (Bare et al., 2005) as well as in Xenopus oocytes (Matifat et al., 2001) and bovine endothelial cells (Aromolaran and Blatter, 2005). Today, the origin of these conflicting observations remains unclear but a fair approximation is that it is linked to the high variability of the InsP3Rs sequence induced by alternative splicing, or to tissue-specific expression of accessory proteins (Foskett et al., 2007).
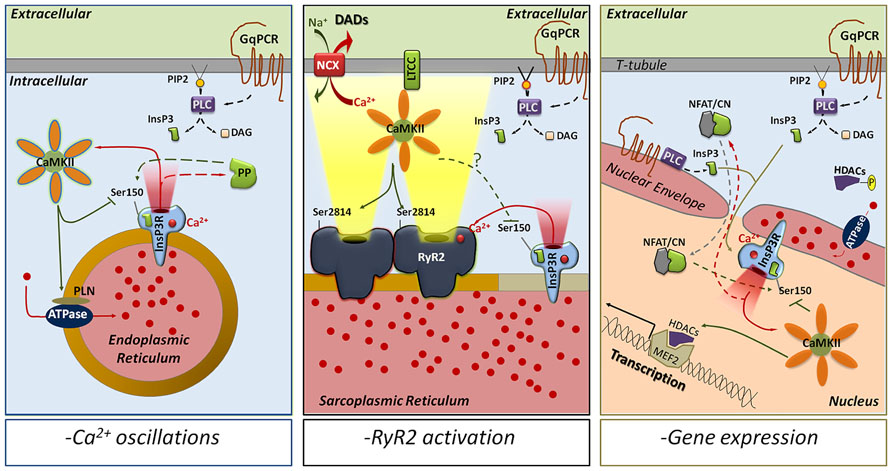
FIGURE 2. CaMKII phosphorylation of Ser150 as a modulator of InsP3R-mediated Ca2+ release. The activation of GqPCRs leads to the production of InsP3 by PLC and initiates InsP3R-mediated Ca2+ release. The released Ca2+ activates CaMKII, which in turn phosphorylates InsP3R-S150 to inhibit channel function. Depending on the intracellular location, CaMKII phosphorylation of InsP3Rs could be implicated in multiple responses. In the endoplasmic reticulum of most of the mammalian cells (left panel), CaMKII phosphorylation of S150 alternates with protein phosphatases dephosphorylation to trigger intracellular Ca2+ oscillations and mediate complex Ca2+ signals. In the SR of cardiac myocytes (middle panel), InsP3Rs are able to activate RyR2s by CICR even though they seem to be excluded from the dyadic cleft. This increases Ca2+ transients and might trigger delayed afterdepolarizations (via NCX activation). CaMKII phosphorylation of InsP3R-S150, which has not been determined yet, could then prevent the uncontrolled activation of RyR2s and decrease the incidence of arrhythmias. In the nucleus (right panel), InsP3R Ca2+ release activates CaMKII, which phosphorylates HDAC proteins to promote transcription. S150- phosphorylation by CaMKII could limit InsP3Rs activation in time and space. However, if associated with calcineurin-dephosphorylation of S150, CaMKII could favor InsP3R-dependent Ca2+ oscillations as secondary mechanism to activate gene expression. ATPase, sarco/endoplasmic reticulum Ca2+ ATPase; NFAT/CN, NFAT-calcineurin complex; DAD, delayed after depolarization; DAG, diacylglycerol; GqPCR, Gq–protein coupled receptor; HDAC, histone deacetylase; LTCC, L-type Ca2+ channel; MEF2, myocyte enhancer factor-2; NCX, Na+/Ca2+ exchanger; PLC, phospholipase C; PIP2, phosphatidylinositol 4,5-bisphosphate; PP, protein phosphatase (1, 2A, and/or 2B).
Recently, Mignery et al. (1990), Maxwell et al. (2012) identified Ser150 (S150) as a distinct CaMKII phosphorylation site on InsP3R-2s. They demonstrated in lipid bilayers that the replacement of S150 by the non-phosphorylatable residue alanine (InsP3R-2S150A) abrogates the inhibitory effects of CaMKII phosphorylation. Conversely, the phospho-mimetic mutant InsP3R-2S150E reproduced the blunted channel activity of phosphorylated WT receptors. Noticeably, S150 belongs to a suppressor domain located prior to the InsP3-binding core. The ablation of that domain increases InsP3R affinity for InsP3 but additionally blocks Ca2+ release by the channel (Uchida et al., 2003). The proposed mechanism is that the suppressor domain participates in the transduction of the allosteric movements that are necessary for channel opening (Uchida et al., 2003). Therefore, it is possible that phosphorylation of S150 by CaMKII also blocks the transmission of the activation signal to the pore domain rather than decreasing the affinity of the InsP3Rs for the InsP3. This hypothesis is reinforced by the fact that high [InsP3]i is not able to reverse the inhibition of InsP3Rs activity after CaMKII phosphorylation (Zhu et al., 1996).
The mammalian heart expresses all three InsP3R isoforms. InsP3R-1 is abundant in endothelial cells and Purkinje fibers. InsP3R-2 is the main isoform of cardiac myocytes and pacemaker cells. Finally, InsP3R-3s are present in all cell types but at a lower level (5–15% total InsP3R expression; Perez et al., 1997; Lipp et al., 2000; Ju et al., 2011). Although the roles of InsP3Rs in the cardiac function have been overlooked, recent evidence assigns to InsP3R-2s an important role in pathologic hypertrophy and Ca2+-triggered arrhythmias.
In both atrial and ventricular myocytes, InsP3R-2s are mainly present in the nuclear envelope where their implication in pathological excitation–transcription coupling has been recently uncovered (reviewed in Kockskämper et al., 2008). During cardiac hypertrophy induced by pressure overload or chronic activation of the GqPCRs, the InsP3R-2s activation leads to a subsequent release of Ca2+ in the nucleoplasm, which activates the nuclear isoform of CaMKIIδ (CaMKIIδB). CaMKIIδB phosphorylates the histone deacetylases (HDACs) to induce their nuclear export and relieve their inhibition on the transcription factor MEF-2 (myocyte enhancer factor-2; Zhu et al., 2000; Wu et al., 2006; Ago et al., 2010). In this context the inhibition of the InsP3Rs by a CaMKIIδB feedback phosphorylation would limit in time and space the InsP3R-mediated Ca2+ release and consequently be anti-hypertrophic. Conversely, a possible pro-hypertrophic role for CaMKIIδB phosphorylation of InsP3R is borne out from recent observations. First, InsP3R-mediated Ca2+ release is able to activate two targets of calmodulin: CaMKIIδB and calcineurin, a perinuclear phosphatase that translocates to the nucleus complexed with the transcription factor NFAT (reviewed in Molkentin, 2000; Bootman et al., 2009). The inhibition of either CaMKII or calcineurin prevents the InsP3R-mediated cardiac hypertrophy (Molkentin et al., 1998; Taigen et al., 2000; Zhu et al., 2000). Second, in addition to their classical localization in the sarcolemma, GqPCRs have been recently identified in the nuclear envelope as well as in t-tubules that spread near the nucleus of cardiomyocyte (Tadevosyan et al., 2012; Ibarra et al., 2013). This allows for a rapid and local production of InsP3 that diffuses and signals into the nucleus independently from the cytosolic InsP3 concentration (Ibarra et al., 2013). Finally, Luo et al. (2008) showed that neonatal rat cardiac myocytes stimulated by a GqPCR agonist exhibit repetitive InsP3R-mediated Ca2+ waves (or Ca2+ oscillations) that take place in the nucleus autonomously from the cytosolic [Ca2+]. Interestingly, frequency-dependent Ca2+ oscillations have been shown to increase gene expression via the activation of the calcineurin/NF-AT pathway (Dolmetsch et al., 1998). Altogether, these observations suggest that hypertrophic signals may be generated by intra-nuclear Ca2+ oscillations independently (frequency, amplitude, and duration) from the excitation–contraction coupling. InsP3-dependent oscillations would be created through InsP3Rs activation–inhibition cycles mediated by calcineurin dephosphorylation and CaMKIIδB phosphorylation of S150, respectively (Figure 2, right panel).
In cardiac myocytes, the expression of InsP3R-2s is remarkably low compared to RyR2s (for an InsP3R-2:RyR2 ratio of 1:50 to 1:100 in ventricular myocytes). In addition, most of the immunofluorescence studies that have observed partial localization of InsP3R-2s in the sub-sarcolemmal space of atrial myocytes, failed to detect InsP3R-2s in the surface sarcolemma or t-tubules of ventricular myocytes (Bare et al., 2005; Escobar et al., 2011; except Mohler et al., 2003). Activation of InsP3R-2s by a GqPCR agonist produces, in the myocyte, a small and slow cytosolic Ca2+ spark-like release dubbed “Ca2+ puffs” (Kockskämper et al., 2008). Ca2+ puffs can activate the neighboring RyR2s to increase the Ca2+ spark frequency and the Ca2+ transient amplitude. In pathological conditions, InsP3R-2 mediated Ca2+ release can ultimately cause, in atrial cells, Ca2+ alternans and increased susceptibility to arrhythmias (Mackenzie et al., 2002; Zima and Blatter, 2004; Li et al., 2005). Moreover, although their expression is not detected in t-tubules, some reports support a similar role for InsP3R-mediated release in ventricular myocyte (Domeier et al., 2008; Signore et al., 2013 but not Mackenzie et al., 2002; Zima and Blatter, 2004; Li et al., 2005). All together, these data suggest that InsP3R-2s are not directly involved in excitation–contraction coupling but participate as modulators that “prime” the RyR2s to increase their sensitivity to diastolic [Ca2+] and LTCC current. In that context, the inhibition of InsP3R-dependent Ca2+ release following CaMKIIδC phosphorylation of InsP3R-Ser150 would block RyR2 potentiation and exert an anti-arrhythmic effect. However, in an extensive phospho-proteome analysis that identified the vast majority of proteins phosphorylated during β-adrenergic stimulation in mice, Huttlin et al. (2010) failed to detect phosphorylation of the cardiac InsP3R-2s, including S150, while InsP3R-1 and -3 were targeted by PKA at Ser1588 and Ser934, respectively. This suggests that the InsP3R-2 channels expressed in the SR are not located in the vicinity of the CaMKIIδC/PKA microdomains, and therefore they may be excluded from the dyad containing the RyR2s (Figure 2, central panel). In agreement with that result, Signore et al. (2013) described in mouse ventricular myocytes that the InsP3R-2 effects are mediated through the activation of the NCX and the increase of the action potential duration rather than a direct effect on RyR2s. Overall, these observations suggest an absence of a direct cross-talk between RyR2s and InsP3Rs and advocate the hypothesis that InsP3Rs mediate their effects on RyR2s by limited Ca2+ diffusion from the InsP3R release sites to the RyR2 sites.
Interestingly, Huttlin et al. (2010) also identified two cardiac-specific epitopes on InsP3R-3 (Ser930 and Ser2189) for which the targeting kinase(s) remain(s) undetermined. Noticeably, S2189 of InsP3R-3 contains the CaMKII consensus sequence (Pinna and Ruzzene, 1996) and is absent from InsP3R-1 or -2. More studies will be necessary to determine whether this InsP3R-3 phosphorylation occurs in cardiac myocytes and the mechanisms by which it affects cardiac function.
CaMKII is a pleiotropic kinase that targets several ion channels and transporters in the heart, and RyR2 and InsP3R channels are among its principal substrates. Interestingly, despite the relatively strong structural similarity between these two intracellular Ca2+ release channels, the majority (but not all) of the studies indicate that CaMKII phosphorylation of RyR2s and InsP3Rs leads to antithetical outcomes. On one hand, CaMKII phosphorylation is presumed to increase RyR2 activity and promote SR Ca2+ leak that, when excessive, may trigger cardiac arrhythmias. On the other hand, CaMKII phosphorylation of nuclear InsP3Rs is presumed to inhibit InsP3Rs-mediated Ca2+ release and to prevent intra-nuclear Ca2+ oscillations (although cytosolic InsP3R-mediated Ca2+ release seems to promote arrhythmias, too). The magnitude of the effect of phosphorylation on these channels is also purportedly different since CaMKII modulates RyR2 channel activity, only, while it appears to play an on/off function on InsP3Rs. The bases of both of these differences are not immediately apparent and in fact, there is no universal agreement that such differences exist. As can be derived from the preceding paragraphs, a unified scheme on the effect of phosphorylation on both, RyR2 and InsP3Rs is yet to be forged. Nonetheless, independent of its precise mechanism of action on these channels, an emerging notion is that excessive CaMKII activity is detrimental for cardiac performance, and the potential salutary effect of blocking its chronic effects appears worth pursuing since its action on RyR2s and InsP3Rs might prevent both Ca2+-mediated arrhythmias and Ca2+-dependent activation of hypertrophic gene programs.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Randall Loaiza and Francisco Alvarado for carefully reading this manuscript. This work was supported by National Institutes of Health grants RO1-HL055438, PO1-HL094291, and RO1-HL108175 to Héctor H. Valdivia.
Ago, T., Yang, Y., Zhai, P., and Sadoshima, J. (2010). Nifedipine inhibits cardiac hypertrophy and left ventricular dysfunction in response to pressure overload. J. Cardiovasc. Transl. Res. 3, 304–313. doi: 10.1007/s12265-010-9182-x
Ai, X., Curran, J. W., Shannon, T. R., Bers, D. M., and Pogwizd, S. M. (2005). Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 97, 1314–1322. doi: 10.1161/01.RES.0000194329.41863.89
Anderson, M. E., Brown, J. H., and Bers, D. M. (2011). CaMKII in myocardial hypertrophy and heart failure. J. Mol. Cell. Cardiol. 51, 468–473. doi: 10.1016/j.yjmcc.2011.01.012
Aromolaran, A. A., and Blatter, L. A. (2005). Modulation of intracellular Ca2+ release and capacitative Ca2+ entry by CaMKII inhibitors in bovine vascular endothelial cells. Am. J. Physiol. Cell Physiol. 289, C1426–C1436. doi: 10.1152/ajpcell.00262.2005
Ather, S., Wang, W., Wang, Q., Li, N., Anderson, M. E., and Wehrens, X. H. (2013). Inhibition of CaMKII phosphorylation of RyR2 prevents inducible ventricular arrhythmias in mice with Duchenne muscular dystrophy. Heart Rhythm 10, 592–599. doi: 10.1016/j.hrthm.2012.12.016
Balshaw, D. M., Yamaguchi, N., and Meissner, G. (2002). Modulation of intracellular calcium-release channels by calmodulin. J. Membr. Biol. 185, 1–8. doi: 10.1007/s00232-001-0111-4
Bare, D. J., Kettlun, C. S., Liang, M., Bers, D. M., and Mignery, G. A. (2005). Cardiac type 2 inositol 1,4,5-trisphosphate receptor: interaction and modulation by calcium/calmodulin-dependent protein kinase II. J. Biol. Chem. 280, 15912–15920. doi: 10.1074/jbc.M414212200
Benkusky, N. A., Weber, C. S., Scherman, J. A., Farrell, E. F., Hacker, T. A., John, M. C., et al. (2007). Intact beta-adrenergic response and unmodified progression toward heart failure in mice with genetic ablation of a major protein kinase A phosphorylation site in the cardiac ryanodine receptor. Circ. Res. 101, 819–829. doi: 10.1161/CIRCRESAHA.107.153007
Bers, D. M. (2004). Macromolecular complexes regulating cardiac ryanodine receptor function. J. Mol. Cell. Cardiol. 37, 417–429. doi: 10.1016/j.yjmcc.2004.05.026
Bers, D. M. (2011). Ca(2)(+)-calmodulin-dependent protein kinase II regulation of cardiac excitation-transcription coupling. Heart Rhythm 8, 1101–1104. doi: 10.1016/j.hrthm.2011.01.030
Bers, D. M. (2012). Ryanodine receptor S2808 phosphorylation in heart failure: smoking gun or red herring. Circ. Res. 110, 796–799. doi: 10.1161/CIRCRESAHA.112.265579
Bers, D. M. (2014). Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annu. Rev. Physiol. 76, 107–127. doi: 10.1146/annurev-physiol-020911-153308
Bers, D. M., Eisner, D. A., and Valdivia, H. H. (2003). Sarcoplasmic reticulum Ca2+ and heart failure: roles of diastolic leak and Ca2+ transport. Circ. Res. 93, 487–490. doi: 10.1161/01.RES.0000091871.54907.6B
Bers, D. M., and Grandi, E. (2009). Calcium/calmodulin-dependent kinase II regulation of cardiac ion channels. J. Cardiovasc. Pharmacol. 54, 180–187. doi: 10.1097/FJC.0b013e3181a25078
Bootman, M. D., Berridge, M. J., and Lipp, P. (1997). Cooking with calcium: the recipes for composing global signals from elementary events. Cell 91, 367–373. doi: 10.1016/S0092-8674(00)80420-1
Bootman, M. D., Fearnley, C., Smyrnias, I., Macdonald, F., and Roderick, H. L. (2009). An update on nuclear calcium signalling. J. Cell Sci. 122, 2337–2350. doi: 10.1242/jcs.028100
Cameron, A. M., Steiner, J. P., Roskams, A. J., Ali, S. M., Ronnett, G. V., and Snyder, S. H. (1995). Calcineurin associated with the inositol 1,4,5-trisphosphate receptor-FKBP12 complex modulates Ca2+ flux. Cell 83, 463–472. doi: 10.1016/0092-8674(95)90124-8
Camors, E., Loaiza, R., Alvarado, F., Zhao, Y., Powers, P., and Valdivia, H. H. (2014). Preventing RyR2-S2808 and RyR2-S2814 phosphorylation does not alter the β-adrenergic response of mouse hearts. Biophys. J. 106:108a.
Capes, E. M., Loaiza, R., and Valdivia, H. H. (2011). Ryanodine receptors. Skelet. Muscle 1:18. doi: 10.1186/2044-5040-1-18
Carter, S., Colyer, J., and Sitsapesan, R. (2006). Maximum phosphorylation of the cardiac ryanodine receptor at serine-2809 by protein kinase a produces unique modifications to channel gating and conductance not observed at lower levels of phosphorylation. Circ. Res. 98, 1506–1513. doi: 10.1161/01.RES.0000227506.43292.df
Curran, J., Brown, K. H., Santiago, D. J., Pogwizd, S., Bers, D. M., and Shannon, T. R. (2010). Spontaneous Ca waves in ventricular myocytes from failing hearts depend on Ca(2+)-calmodulin-dependent protein kinase II. J. Mol. Cell. Cardiol. 49, 25–32. doi: 10.1016/j.yjmcc.2010.03.013
Curran, J., Hinton, M. J., Rios, E., Bers, D. M., and Shannon, T. R. (2007). Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ. Res. 100, 391–398. doi: 10.1161/01.RES.0000258172.74570.e6
Currie, S. (2009). Cardiac ryanodine receptor phosphorylation by CaM Kinase II: keeping the balance right. Front. Biosci. 14:5134–5156. doi: 10.2741/3591
Currie, S., Elliott, E. B., Smith, G. L., and Loughrey, C. M. (2011). Two candidates at the heart of dysfunction: the ryanodine receptor and calcium/calmodulin protein kinase II as potential targets for therapeutic intervention-An in vivo perspective. Pharmacol. Ther. 131, 204–220. doi: 10.1016/j.pharmthera.2011.02.006
Currie, S., Loughrey, C. M., Craig, M. A., and Smith, G. L. (2004). Calcium/calmodulin-dependent protein kinase IIdelta associates with the ryanodine receptor complex and regulates channel function in rabbit heart. Biochem. J. 377, 357–366. doi: 10.1042/BJ20031043
Da Fonseca, P. C., Morris, S. A., Nerou, E. P., Taylor, C. W., and Morris, E. P. (2003). Domain organization of the type 1 inositol 1,4,5-trisphosphate receptor as revealed by single-particle analysis. Proc. Natl. Acad. Sci. U.S.A. 100, 3936–3941. doi: 10.1073/pnas.0536251100
Dolmetsch, R. E., Xu, K., and Lewis, R. S. (1998). Calcium oscillations increase the efficiency and specificity of gene expression. Nature 392, 933–936. doi: 10.1038/31960
Domeier, T. L., Zima, A. V., Maxwell, J. T., Huke, S., Mignery, G. A., and Blatter, L. A. (2008). IP3 receptor-dependent Ca2+ release modulates excitation-contraction coupling in rabbit ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 294, H596–H604. doi: 10.1152/ajpheart.01155.2007
Donoso, P., Sanchez, G., Bull, R., and Hidalgo, C. (2011). Modulation of cardiac ryanodine receptor activity by ROS and RNS. Front. Biosci. 16:553–567. doi: 10.2741/3705
Eisner, D. A., Trafford, A. W., Diaz, M. E., Overend, C. L., and O’Neill, S. C. (1998). The control of Ca release from the cardiac sarcoplasmic reticulum: regulation versus autoregulation. Cardiovasc. Res. 38, 589–604. doi: 10.1016/S0008-6363(98)00062-5
Eschenhagen, T. (2010). Is ryanodine receptor phosphorylation key to the fight or flight response and heart failure? J. Clin. Invest. 120, 4197–4203. doi: 10.1172/JCI45251
Escobar, M., Cardenas, C., Colavita, K., Petrenko, N. B., and Franzini-Armstrong, C. (2011). Structural evidence for perinuclear calcium microdomains in cardiac myocytes. J. Mol. Cell. Cardiol. 50, 451–459. doi: 10.1016/j.yjmcc.2010.11.021
Fabiato, A. (1985). Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J. Gen. Physiol. 85, 247–289. doi: 10.1085/jgp.85.2.247
Farrell, E. F., Antaramian, A., Rueda, A., Gomez, A. M., and Valdivia, H. H. (2003). Sorcin inhibits calcium release and modulates excitation-contraction coupling in the heart. J. Biol. Chem. 278, 34660–34666. doi: 10.1074/jbc.M305931200
Fill, M., and Copello, J. A. (2002). Ryanodine receptor calcium release channels. Physiol. Rev. 82, 893–922. doi: 10.1152/physrev.00013.2002
Fischer, T. H., Herting, J., Tirilomis, T., Renner, A., Neef, S., Toischer, K., et al. (2013). Ca2+/calmodulin-dependent protein kinase II and protein kinase A differentially regulate sarcoplasmic reticulum Ca2+ leak in human cardiac pathology. Circulation 128, 970–981. doi: 10.1161/CIRCULATIONAHA.113.001746
Foskett, J. K., White, C., Cheung, K. H., and Mak, D. O. (2007). Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 87, 593–658. doi: 10.1152/physrev.00035.2006
George, C. H. (2008). Sarcoplasmic reticulum Ca2+ leak in heart failure: mere observation or functional relevance? Cardiovasc. Res. 77, 302–314. doi: 10.1093/cvr/cvm006
Ginsburg, K. S., and Bers, D. M. (2004). Modulation of excitation-contraction coupling by isoproterenol in cardiomyocytes with controlled SR Ca2+ load and Ca2+ current trigger. J. Physiol. 556, 463–480. doi: 10.1113/jphysiol.2003.055384
Grimm, M., and Brown, J. H. (2010). Beta-adrenergic receptor signaling in the heart: role of CaMKII. J. Mol. Cell. Cardiol. 48, 322–330. doi: 10.1016/j.yjmcc.2009.10.016
Guo, T., Cornea, R. L., Huke, S., Camors, E., Yang, Y., Picht, E., et al. (2010). Kinetics of FKBP12.6 binding to ryanodine receptors in permeabilized cardiac myocytes and effects on Ca sparks. Circ. Res. 106, 1743–1752. doi: 10.1161/CIRCRESAHA.110.219816
Guo, T., Zhang, T., Mestril, R., and Bers, D. M. (2006). Ca2+/Calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ. Res. 99, 398–406. doi: 10.1161/01.RES.0000236756.06252.13
Gyorke, I., Hester, N., Jones, L. R., and Gyorke, S. (2004). The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys. J. 86, 2121–2128. doi: 10.1016/S0006-3495(04)74271-X
Gyorke, S., and Fill, M. (1993). Ryanodine receptor adaptation: control mechanism of Ca(2+)-induced Ca2+ release in heart. Science 260, 807–809. doi: 10.1126/science.8387229
Hain, J., Onoue, H., Mayrleitner, M., Fleischer, S., and Schindler, H. (1995). Phosphorylation modulates the function of the calcium release channel of sarcoplasmic reticulum from cardiac muscle. J. Biol. Chem. 270, 2074–2081. doi: 10.1074/jbc.270.5.2074
Hamilton, S. L., and Serysheva, I. I. (2009). Ryanodine receptor structure: progress and challenges. J. Biol. Chem. 284, 4047–4051. doi: 10.1074/jbc.R800054200
Huke, S., and Bers, D. M. (2008). Ryanodine receptor phosphorylation at Serine 2030, 2808 and 2814 in rat cardiomyocytes. Biochem. Biophys. Res. Commun. 376, 80–85. doi: 10.1016/j.bbrc.2008.08.084
Huttlin, E. L., Jedrychowski, M. P., Elias, J. E., Goswami, T., Rad, R., Beausoleil, S. A., et al. (2010). A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189. doi: 10.1016/j.cell.2010.12.001
Ibarra, C., Vicencio, J. M., Estrada, M., Lin, Y., Rocco, P., Rebellato, P., et al. (2013). Local control of nuclear calcium signaling in cardiac myocytes by perinuclear microdomains of sarcolemmal insulin-like growth factor 1 receptors. Circ. Res. 112, 236–245. doi: 10.1161/CIRCRESAHA.112.273839
Jiang, M. T., Lokuta, A. J., Farrell, E. F., Wolff, M. R., Haworth, R. A., and Valdivia, H. H. (2002). Abnormal Ca2+ release, but normal ryanodine receptors, in canine and human heart failure. Circ. Res. 91, 1015–1022. doi: 10.1161/01.RES.0000043663.08689.05
Ju, Y. K., Liu, J., Lee, B. H., Lai, D., Woodcock, E. A., Lei, M., et al. (2011). Distribution and functional role of inositol 1,4,5-trisphosphate receptors in mouse sinoatrial node. Circ. Res. 109, 848–857. doi: 10.1161/CIRCRESAHA.111.243824
Kirchhefer, U., Schmitz, W., Scholz, H., and Neumann, J. (1999). Activity of cAMP-dependent protein kinase and Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human hearts. Cardiovasc. Res. 42, 254–261. doi: 10.1016/S0008-6363(98)00296-X
Kockskämper, J., Zima, A. V., Roderick, H. L., Pieske, B., Blatter, L. A., and Bootman, M. D. (2008). Emerging roles of inositol 1,4,5-trisphosphate signaling in cardiac myocytes. J. Mol. Cell. Cardiol. 45, 128–147. doi: 10.1016/j.yjmcc.2008.05.014
Kohlhaas, M., Zhang, T., Seidler, T., Zibrova, D., Dybkova, N., Steen, A., et al. (2006). Increased sarcoplasmic reticulum calcium leak but unaltered contractility by acute CaMKII overexpression in isolated rabbit cardiac myocytes. Circ. Res. 98, 235–244. doi: 10.1161/01.RES.0000200739.90811.9f
Krishna, A., Valderrabano, M., Palade, P. T., and Clark, J. W. Jr. (2013). Rate-dependent Ca2+ signalling underlying the force-frequency response in rat ventricular myocytes: a coupled electromechanical modeling study. Theor. Biol. Med. Model. 10:54. doi: 10.1186/1742-4682-10-54
Kushnir, A., and Marks, A. R. (2010). The ryanodine receptor in cardiac physiology and disease. Adv. Pharmacol. 59, 1–30. doi: 10.1016/S1054-3589(10)59001-X
Lehnart, S. E., Wehrens, X. H., Reiken, S., Warrier, S., Belevych, A. E., Harvey, R. D., et al. (2005). Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell 123, 25–35. doi: 10.1016/j.cell.2005.07.030
Li, J., Imtiaz, M. S., Beard, N. A., Dulhunty, A. F., Thorne, R., Vanhelden, D. F., et al. (2013). ss-Adrenergic stimulation increases RyR2 activity via intracellular Ca2+ and Mg2+ regulation. PLoS ONE 8:e58334. doi: 10.1371/journal.pone.0058334
Li, X., Zima, A. V., Sheikh, F., Blatter, L. A., and Chen, J. (2005). Endothelin-1-induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol-1,4,5-trisphosphate(IP3)-receptor type 2-deficient mice. Circ. Res. 96, 1274–1281. doi: 10.1161/01.RES.0000172556.05576.4c
Li, Y., Kranias, E. G., Mignery, G. A., and Bers, D. M. (2002). Protein kinase A phosphorylation of the ryanodine receptor does not affect calcium sparks in mouse ventricular myocytes. Circ. Res. 90, 309–316. doi: 10.1161/hh0302.105660
Lipp, P., Laine, M., Tovey, S. C., Burrell, K. M., Berridge, M. J., Li, W., et al. (2000). Functional InsP3 receptors that may modulate excitation-contraction coupling in the heart. Curr. Biol. 10, 939–942. doi: 10.1016/S0960-9822(00)00624-2
Lokuta, A. J., Rogers, T. B., Lederer, W. J., and Valdivia, H. H. (1995). Modulation of cardiac ryanodine receptors of swine and rabbit by a phosphorylation-dephosphorylation mechanism. J. Physiol. 487, 609–622.
Luczak, E. D., and Anderson, M. E. (2014). CaMKII oxidative activation and the pathogenesis of cardiac disease. J. Mol. Cell. Cardiol. doi: 10.1016/j.yjmcc.2014.02.004 [Epub ahead of print].
Luo, D., Yang, D., Lan, X., Li, K., Li, X., Chen, J., et al. (2008). Nuclear Ca2+ sparks and waves mediated by inositol 1,4,5-trisphosphate receptors in neonatal rat cardiomyocytes. Cell Calcium 43, 165–174. doi: 10.1016/j.ceca.2007.04.017
Luo, M., and Anderson, M. E. (2013). Mechanisms of altered Ca(2)(+) handling in heart failure. Circ. Res. 113, 690–708. doi: 10.1161/CIRCRESAHA.113.301651
MacDonnell, S. M., Garcia-Rivas, G., Scherman, J. A., Kubo, H., Chen, X., Valdivia, H., et al. (2008). Adrenergic regulation of cardiac contractility does not involve phosphorylation of the cardiac ryanodine receptor at serine 2808. Circ. Res. 102, e65–e72. doi: 10.1161/CIRCRESAHA.108.174722
Mackenzie, L., Bootman, M. D., Laine, M., Berridge, M. J., Thuring, J., Holmes, A., et al. (2002). The role of inositol 1,4,5-trisphosphate receptors in Ca(2+) signalling and the generation of arrhythmias in rat atrial myocytes. J. Physiol. 541, 395–409. doi: 10.1113/jphysiol.2001.013411
Maier, L. S., Zhang, T., Chen, L., Desantiago, J., Brown, J. H., and Bers, D. M. (2003). Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ. Res. 92, 904–911. doi: 10.1161/01.RES.0000069685.20258.F1
Marx, S. O., Reiken, S., Hisamatsu, Y., Jayaraman, T., Burkhoff, D., Rosemblit, N., et al. (2000). PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101, 365–376. doi: 10.1016/S0092-8674(00)80847-8
Matifat, F., Hague, F., Brule, G., and Collin, T. (2001). Regulation of InsP3-mediated Ca2+ release by CaMKII in Xenopus oocytes. Pflugers Arch. 441, 796–801. doi: 10.1007/s004240000479
Maxwell, J. T., Natesan, S., and Mignery, G. A. (2012). Modulation of inositol 1,4,5-trisphosphate receptor type 2 channel activity by Ca2+/calmodulin-dependent protein kinase II (CaMKII)-mediated phosphorylation. J. Biol. Chem. 287, 39419–39428. doi: 10.1074/jbc.M112.374058
Meissner, G. (2004). Molecular regulation of cardiac ryanodine receptor ion channel. Cell Calcium 35, 621–628. doi: 10.1016/j.ceca.2004.01.015
Mignery, G. A., Newton, C. L., Archer, B. T. III, and Sudhof, T. C. (1990). Structure and expression of the rat inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 265, 12679–12685.
Mignery, G. A., and Sudhof, T. C. (1990). The ligand binding site and transduction mechanism in the inositol-1,4,5-triphosphate receptor. EMBO J. 9, 3893–3898.
Mohler, P. J., Schott, J. J., Gramolini, A. O., Dilly, K. W., Guatimosim, S., Dubell, W. H., et al. (2003). Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 421, 634–639. doi: 10.1038/nature01335
Molkentin, J. D. (2000). Calcineurin and beyond: cardiac hypertrophic signaling. Circ. Res. 87, 731–738. doi: 10.1161/01.RES.87.9.731
Molkentin, J. D., Lu, J. R., Antos, C. L., Markham, B., Richardson, J., Robbins, J., et al. (1998). A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 93, 215–228. doi: 10.1016/S0092-8674(00)81573-1
Newton, C. L., Mignery, G. A., and Sudhof, T. C. (1994). Co-expression in vertebrate tissues and cell lines of multiple inositol 1,4,5-trisphosphate (InsP3) receptors with distinct affinities for InsP3. J. Biol. Chem. 269, 28613–28619.
Perez, P. J., Ramos-Franco, J., Fill, M., and Mignery, G. A. (1997). Identification and functional reconstitution of the type 2 inositol 1,4,5-trisphosphate receptor from ventricular cardiac myocytes. J. Biol. Chem. 272, 23961–23969. doi: 10.1074/jbc.272.38.23961
Pietri, F., Hilly, M., and Mauger, J. P. (1990). Calcium mediates the interconversion between two states of the liver inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 265, 17478–17485.
Pinna, L. A., and Ruzzene, M. (1996). How do protein kinases recognize their substrates? Biochim. Biophys. Acta 1314, 191–225. doi: 10.1016/S0167-4889(96)00083-3
Priori, S. G., and Chen, S. R. (2011). Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ. Res. 108, 871–883. doi: 10.1161/CIRCRESAHA.110.226845
Purohit, A., Rokita, A. G., Guan, X., Chen, B., Koval, O. M., Voigt, N., et al. (2013). Oxidized Ca(2+)/calmodulin-dependent protein kinase II triggers atrial fibrillation. Circulation 128, 1748–1757. doi: 10.1161/CIRCULATIONAHA.113.003313
Ramos-Franco, J., Fill, M., and Mignery, G. A. (1998). Isoform-specific function of single inositol 1,4,5-trisphosphate receptor channels. Biophys. J. 75, 834–839. doi: 10.1016/S0006-3495(98)77572-1
Respress, J. L., Van Oort, R. J., Li, N., Rolim, N., Dixit, S. S., Dealmeida, A., et al. (2012). Role of RyR2 phosphorylation at S2814 during heart failure progression. Circ. Res. 110, 1474–1483. doi: 10.1161/CIRCRESAHA.112.268094
Rodriguez, P., Bhogal, M. S., and Colyer, J. (2003). Stoichiometric phosphorylation of cardiac ryanodine receptor on serine 2809 by calmodulin-dependent kinase II and protein kinase A. J. Biol. Chem. 278, 38593–38600. doi: 10.1074/jbc.C301180200
Said, M., Becerra, R., Valverde, C. A., Kaetzel, M. A., Dedman, J. R., Mundina-Weilenmann, C., et al. (2011). Calcium-calmodulin dependent protein kinase II (CaMKII): a main signal responsible for early reperfusion arrhythmias. J. Mol. Cell. Cardiol. 51, 936–944. doi: 10.1016/j.yjmcc.2011.08.010
Seiler, S., Wegener, A. D., Whang, D. D., Hathaway, D. R., and Jones, L. R. (1984). High molecular weight proteins in cardiac and skeletal muscle junctional sarcoplasmic reticulum vesicles bind calmodulin, are phosphorylated, and are degraded by Ca2+-activated protease. J. Biol. Chem. 259, 8550–8557.
Shan, J., Betzenhauser, M. J., Kushnir, A., Reiken, S., Meli, A. C., Wronska, A., et al. (2010a). Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J. Clin. Invest. 120, 4375–4387. doi: 10.1172/JCI37649
Shan, J., Kushnir, A., Betzenhauser, M. J., Reiken, S., Li, J., Lehnart, S. E., et al. (2010b). Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice. J. Clin. Invest. 120, 4388–4398. doi: 10.1172/JCI32726
Sienaert, I., De Smedt, H., Parys, J. B., Missiaen, L., Vanlingen, S., Sipma, H., et al. (1996). Characterization of a cytosolic and a luminal Ca2+ binding site in the type I inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 271, 27005–27012. doi: 10.1074/jbc.271.43.27005
Sienaert, I., Missiaen, L., De Smedt, H., Parys, J. B., Sipma, H., and Casteels, R. (1997). Molecular and functional evidence for multiple Ca2+-binding domains in the type 1 inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 272, 25899–25906. doi: 10.1074/jbc.272.41.25899
Signore, S., Sorrentino, A., Ferreira-Martins, J., Kannappan, R., Shafaie, M., Del Ben, F., et al. (2013). Inositol 1, 4, 5-trisphosphate receptors and human left ventricular myocytes. Circulation 128, 1286–1297. doi: 10.1161/CIRCULATIONAHA.113.002764
Sossalla, S., Fluschnik, N., Schotola, H., Ort, K. R., Neef, S., Schulte, T., et al. (2010). Inhibition of elevated Ca2+/calmodulin-dependent protein kinase II improves contractility in human failing myocardium. Circ. Res. 107, 1150–1161. doi: 10.1161/CIRCRESAHA.110.220418
Stange, M., Xu, L., Balshaw, D., Yamaguchi, N., and Meissner, G. (2003). Characterization of recombinant skeletal muscle (Ser-2843) and cardiac muscle (Ser-2809) ryanodine receptor phosphorylation mutants. J. Biol. Chem. 278, 51693–51702. doi: 10.1074/jbc.M310406200
Supattapone, S., Worley, P. F., Baraban, J. M., and Snyder, S. H. (1988). Solubilization, purification, and characterization of an inositol trisphosphate receptor. J. Biol. Chem. 263, 1530–1534.
Tadevosyan, A., Vaniotis, G., Allen, B. G., Hebert, T. E., and Nattel, S. (2012). G protein-coupled receptor signalling in the cardiac nuclear membrane: evidence and possible roles in physiological and pathophysiological function. J. Physiol. 590, 1313–1330. doi: 10.1113/jphysiol.2011.222794
Taigen, T., De Windt, L. J., Lim, H. W., and Molkentin, J. D. (2000). Targeted inhibition of calcineurin prevents agonist-induced cardiomyocyte hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 97, 1196–1201. doi: 10.1073/pnas.97.3.1196
Takasago, T., Imagawa, T., and Shigekawa, M. (1989). Phosphorylation of the cardiac ryanodine receptor by cAMP-dependent protein kinase. J. Biochem. 106, 872–877.
Takeshima, H., Komazaki, S., Hirose, K., Nishi, M., Noda, T., and Iino, M. (1998). Embryonic lethality and abnormal cardiac myocytes in mice lacking ryanodine receptor type 2. EMBO J. 17, 3309–3316. doi: 10.1093/emboj/17.12.3309
Taylor, C. W., and Tovey, S. C. (2010). IP(3) receptors: toward understanding their activation. Cold Spring Harb. Perspect. Biol. 2:a004010. doi: 10.1101/cshperspect.a004010
Terentyev, D., Viatchenko-Karpinski, S., Gyorke, I., Terentyeva, R., and Gyorke, S. (2003). Protein phosphatases decrease sarcoplasmic reticulum calcium content by stimulating calcium release in cardiac myocytes. J. Physiol. 552, 109–118. doi: 10.1113/jphysiol.2003.046367
Timerman, A. P., Onoue, H., Xin, H. B., Barg, S., Copello, J., Wiederrecht, G., et al. (1996). Selective binding of FKBP12.6 by the cardiac ryanodine receptor. J. Biol. Chem. 271, 20385–20391. doi: 10.1074/jbc.271.34.20385
Uchida, K., Miyauchi, H., Furuichi, T., Michikawa, T., and Mikoshiba, K. (2003). Critical regions for activation gating of the inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 278, 16551–16560. doi: 10.1074/jbc.M300646200
Uehara, A., Yasukochi, M., Mejia-Alvarez, R., Fill, M., and Imanaga, I. (2002). Gating kinetics and ligand sensitivity modified by phosphorylation of cardiac ryanodine receptors. Pflugers Arch. 444, 202–212. doi: 10.1007/s00424-002-0791-3
Valdivia, H. H. (2012). Ryanodine receptor phosphorylation and heart failure: phasing out s2808 and “criminalizing” s2814. Circ. Res. 110, 1398–1402. doi: 10.1161/CIRCRESAHA.112.270876
Valdivia, H. H. (2013). “Ionic basis of sarcoplasmic reticulum ion fluxes in heart muscle,” in Cardiac Electrophysiology: From Cell to Bedside, 5th Edn, eds D. Zipes and J. Jalife (New York: Saunders).
Valdivia, H. H., Kaplan, J. H., Ellis-Davies, G. C., and Lederer, W. J. (1995). Rapid adaptation of cardiac ryanodine receptors: modulation by Mg2+ and phosphorylation. Science 267, 1997–2000. doi: 10.1126/science.7701323
Vanderheyden, V., Devogelaere, B., Missiaen, L., De Smedt, H., Bultynck, G., and Parys, J. B. (2009). Regulation of inositol 1,4,5-trisphosphate-induced Ca2+ release by reversible phosphorylation and dephosphorylation. Biochim. Biophys. Acta 1793, 959–970. doi: 10.1016/j.bbamcr.2008.12.003
van Oort, R. J., Mccauley, M. D., Dixit, S. S., Pereira, L., Yang, Y., Respress, J. L., et al. (2010). Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation 122, 2669–2679. doi: 10.1161/CIRCULATIONAHA.110.982298
Van Petegem, F. (2012). Ryanodine receptors: structure and function. J. Biol. Chem. 287, 31624–31632. doi: 10.1074/jbc.R112.349068
Walaas, S. I., Horn, R. S., Albert, K. A., Adler, A., and Walaas, O. (1988). Phosphorylation of multiple sites in a 15,000 dalton proteolipid from rat skeletal muscle sarcolemma, catalyzed by adenosine 3′, 5′-monophosphate-dependent and calcium/phospholipid-dependent protein kinases. Biochim. Biophys. Acta 968, 127–137. doi: 10.1016/0167-4889(88)90052-3
Wehrens, X. H., Lehnart, S. E., Reiken, S. R., and Marks, A. R. (2004). Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ. Res. 94, e61–e70. doi: 10.1161/01.RES.0000125626.33738.E2
Wehrens, X. H., Lehnart, S. E., Reiken, S., Vest, J. A., Wronska, A., and Marks, A. R. (2006). Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc. Natl. Acad. Sci. U.S.A. 103, 511–518. doi: 10.1073/pnas.0510113103
Witcher, D. R., Kovacs, R. J., Schulman, H., Cefali, D. C., and Jones, L. R. (1991). Unique phosphorylation site on the cardiac ryanodine receptor regulates calcium channel activity. J. Biol. Chem. 266, 11144–11152.
Wu, X., Zhang, T., Bossuyt, J., Li, X., Mckinsey, T. A., Dedman, J. R., et al. (2006). Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J. Clin. Invest. 116, 675–682. doi: 10.1172/JCI27374
Wu, Y., Temple, J., Zhang, R., Dzhura, I., Zhang, W., Trimble, R., et al. (2002). Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation 106, 1288–1293. doi: 10.1161/01.CIR.0000027583.73268.E7
Xiao, B., Jiang, M. T., Zhao, M., Yang, D., Sutherland, C., Lai, F. A., et al. (2005). Characterization of a novel PKA phosphorylation site, serine-2030, reveals no PKA hyperphosphorylation of the cardiac ryanodine receptor in canine heart failure. Circ. Res. 96, 847–855. doi: 10.1161/01.RES.0000163276.26083.e8
Xiao, B., Sutherland, C., Walsh, M. P., and Chen, S. R. (2004). Protein kinase A phosphorylation at serine-2808 of the cardiac Ca2+-release channel (ryanodine receptor) does not dissociate 12.6-kDa FK506-binding protein (FKBP12.6). Circ. Res. 94, 487–495. doi: 10.1161/01.RES.0000115945.89741.22
Xiao, B., Zhong, G., Obayashi, M., Yang, D., Chen, K., Walsh, M. P., et al. (2006). Ser-2030, but not Ser-2808, is the major phosphorylation site in cardiac ryanodine receptors responding to protein kinase A activation upon beta-adrenergic stimulation in normal and failing hearts. Biochem. J. 396, 7–16. doi: 10.1042/BJ20060116
Xiao, J., Tian, X., Jones, P. P., Bolstad, J., Kong, H., Wang, R., et al. (2007). Removal of FKBP12.6 does not alter the conductance and activation of the cardiac ryanodine receptor or the susceptibility to stress-induced ventricular arrhythmias. J. Biol. Chem. 282, 34828–34838. doi: 10.1074/jbc.M707423200
Yang, D., Zhu, W. Z., Xiao, B., Brochet, D. X., Chen, S. R., Lakatta, E. G., et al. (2007). Ca2+/calmodulin kinase II-dependent phosphorylation of ryanodine receptors suppresses Ca2+ sparks and Ca2+ waves in cardiac myocytes. Circ. Res. 100, 399–407. doi: 10.1161/01.RES.0000258022.13090.55
Yang, Y., Guo, T., Oda, T., Chakraborty, A., Chen, L., Uchinoumi, H., et al. (2014). Cardiac myocyte Z-line calmodulin is mainly RyR2-bound, and reduction is arrhythmogenic and occurs in heart failure. Circ. Res. 114, 295–306. doi: 10.1161/CIRCRESAHA.114.302857
Yuchi, Z., Lau, K., and Van Petegem, F. (2012). Disease mutations in the ryanodine receptor central region: crystal structures of a phosphorylation hot spot domain. Structure 20, 1201–1211. doi: 10.1016/j.str.2012.04.015
Zhang, B. X., Zhao, H., and Muallem, S. (1993). Ca(2+)-dependent kinase and phosphatase control inositol 1,4,5-trisphosphate-mediated Ca2+ release. Modification by agonist stimulation. J. Biol. Chem. 268, 10997–11001.
Zhang, H., Makarewich, C. A., Kubo, H., Wang, W., Duran, J. M., Li, Y., et al. (2012). Hyperphosphorylation of the cardiac ryanodine receptor at serine 2808 is not involved in cardiac dysfunction after myocardial infarction. Circ. Res. 110, 831–840. doi: 10.1161/CIRCRESAHA.111.255158
Zhu, D. M., Tekle, E., Chock, P. B., and Huang, C. Y. (1996). Reversible phosphorylation as a controlling factor for sustaining calcium oscillations in HeLa cells: involvement of calmodulin-dependent kinase II and a calyculin A-inhibitable phosphatase. Biochemistry 35, 7214–7223. doi: 10.1021/bi952471h
Zhu, W., Zou, Y., Shiojima, I., Kudoh, S., Aikawa, R., Hayashi, D., et al. (2000). Ca2+/calmodulin-dependent kinase II and calcineurin play critical roles in endothelin-1-induced cardiomyocyte hypertrophy. J. Biol. Chem. 275, 15239–15245. doi: 10.1074/jbc.275.20.15239
Zima, A. V., and Blatter, L. A. (2004). Inositol-1,4,5-trisphosphate-dependent Ca(2+) signalling in cat atrial excitation-contraction coupling and arrhythmias. J. Physiol. 555, 607–615. doi: 10.1113/jphysiol.2003.058529
Keywords: ryanodine receptors, inositol triphosphate receptor, CaMKII, sarcoplasmic reticulum, Ca2+ leak, myocardium
Citation: Camors E and Valdivia HH (2014) CaMKII regulation of cardiac ryanodine receptors and inositol triphosphate receptors. Front. Pharmacol. 5:101. doi: 10.3389/fphar.2014.00101
Received: 13 March 2014; Accepted: 17 April 2014;
Published online: 08 May 2014.
Edited by:
Donald M. Bers, University of California at Davis, USACopyright © 2014 Camors and Valdivia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Héctor H. Valdivia, Division of Cardiovascular Medicine, Department of Internal Medicine, Center for Arrhythmia Research, University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48109, USA email:aHZhbGRpdkB1bWljaC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.