 Martha A. Clark
Martha A. Clark Morgan M. Goheen
Morgan M. Goheen Carla Cerami
Carla Cerami
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 06 May 2014
Sec. Drug Metabolism and Transport
Volume 5 - 2014 | https://doi.org/10.3389/fphar.2014.00084
This article is part of the Research Topic The Importance Of Iron In Pathophysiologic Conditions View all 44 articles
Iron deficiency affects one quarter of the world's population and causes significant morbidity, including detrimental effects on immune function and cognitive development. Accordingly, the World Health Organization (WHO) recommends routine iron supplementation in children and adults in areas with a high prevalence of iron deficiency. However, a large body of clinical and epidemiological evidence has accumulated which clearly demonstrates that host iron deficiency is protective against falciparum malaria and that host iron supplementation may increase the risk of malaria. Although many effective antimalarial treatments and preventive measures are available, malaria remains a significant public health problem, in part because the mechanisms of malaria pathogenesis remain obscured by the complexity of the relationships that exist between parasite virulence factors, host susceptibility traits, and the immune responses that modulate disease. Here we review (i) the clinical and epidemiological data that describes the relationship between host iron status and malaria infection and (ii) the current understanding of the biological basis for these clinical and epidemiological observations.
Iron deficiency and malaria are significant co-morbidities in large portions of the developing world, and both maladies disproportionately affect pregnant women and children. Malaria causes an estimated 250 million infections and 500,000 deaths annually. Iron deficiency is estimated to affect one quarter of the world's populations causing substantial morbidity. Fortunately, iron deficiency is easily treated with iron supplementation (Okebe et al., 2011). Accordingly the World Health Organization (WHO) recommends routine iron supplementation for children and adults in areas with high prevalence of iron deficiency (Haider et al., 2013; Low et al., 2013). However, the wisdom of universal iron supplementation campaigns in malaria endemic regions has recently been questioned due to clinical evidence that suggests iron deficiency protects against malaria, and that iron supplementation of women and children may increase the incidence of malaria when given without malaria prophylaxis or access to adequate health care (Nyakeriga et al., 2004; Sazawal et al., 2006; Tielsch et al., 2006; Kabyemela et al., 2008; Senga et al., 2011; Veenemans et al., 2011; Gwamaka et al., 2012; Jonker et al., 2012; Esan et al., 2013; Zlotkin et al., 2013). This situation has created a dilemma for health policy makers and health care workers in malaria endemic regions of the world (Prentice et al., 2013).
Despite these clinical and epidemiological studies, the extent to which the human host's iron status affects risk to and severity of malaria infection is unknown. Differences in study design and confounding factors (such as acquired immunity to malaria and hemoglobinopathies) have made the clinical and epidemiological studies difficult to interpret (Prentice et al., 2007). Furthermore, though iron and malaria have been and continue to be studied the exact biological relationship between host iron and malaria virulence remains largely unclear.
Iron deficiency is a condition in which there is insufficient iron in the body to maintain normal physiologic functions. Iron deficiency can be categorized into three stages: iron deficiency without anemia, iron deficiency with mild anemia, and iron deficiency with severe anemia. Iron deficiency anemia occurs when iron stores are exhausted and the supply of iron to tissue is compromised; this condition is defined as anemia with biochemical evidence of iron deficiency. Iron deficiency is most prevalent and severe in young children and women of reproductive age, but can also occur in older children, adolescents, adult men, and the elderly. It is estimated that 50% of pregnant women and 40% of preschool children in the developing world are iron deficient (WHO | Assessing the iron status of populations, 2007; Kassebaum et al., 2014). Often, iron deficiency develops slowly and is not clinically diagnosed until severe anemia is apparent (Stoltzfus, 2003).
Studies suggest that iron deficiency impairs the growth, cognition, and neurological development of children from infancy through adolescence, impairs immune function, and is associated with increased morbidity rates (De-Regil et al., 2011, 2013; Wang et al., 2013). Iron deficiency during pregnancy is associated with multiple adverse outcomes for both mother and infant, including increased risk of hemorrhage, sepsis, maternal mortality, perinatal mortality, and low birth weight (Peña-Rosas et al., 2012a,b). Iron deficiency anemia can be a direct cause of death or contribute indirectly. For example, during child birth an anemic mother cannot afford to lose more than 150 mL of blood, compared with a healthy mother who can lose up to 1 liter of blood and still survive. Thus, the WHO recommends iron supplementation for all men, women, and children in areas where malnutrition is prevalent (WHO | Guidelines on food fortification with micronutrients, 2006).
Host iron metabolism is intimately linked to the host response to infection and inflammation. In the face of infection and inflammation, the human host protein hepcidin becomes elevated and initiates signaling which results in reduced iron absorption into the body along with the redistribution of body iron stores. As a consequence many of the biomarkers utilized to assess host iron status are sensitive to both iron as well as infection. For example, low serum ferritin (serum ferritin reflects total body iron reservoirs) is indicative of iron deficiency. However, ferritin is also an acute phase protein which is elevated in the context of infection, and as a result is not a reliable marker of human iron status in the presence of infection or inflammation. Like serum ferritin, transferrin saturation and transferrin receptor levels are biochemical markers of human iron status that are also sensitive to infection and inflammation. As a result evaluating an individual's iron status during an infection has proven difficult (Aguilar et al., 2012), and the scientific community has struggled to establish formal guidelines.
In 2012 malaria caused an estimated 207 million infections and over 600,000 deaths; 90% of these deaths occurred in sub-Saharan Africa, and 77% occurred in children under five (WHO | World Malaria Report, 2013). At least five species of the eukaryotic Apicomplexan parasite from the genus Plasmodium cause malaria in humans with Plasmodium falciparum being the most common and deadly. Following the bite of a malaria parasite infected mosquito, the sporozoite stage of the parasite enters the bloodstream and travels to the liver, where it subsequently infects liver hepatocytes. Malaria replication in the liver is asymptomatic. Next, the merozoite form of the parasite leaves the liver and enters into circulation to infect host red blood cells (RBCs). During the erythrocytic stage of infection, the parasite repeatedly invades, replicates within, and egresses from host RBCs. This erythrocytic stage of infection is responsible for all symptoms of disease (Miller et al., 2013), and the severity of disease is directly associated with parasite burden (Chotivanich et al., 2000; Dondorp et al., 2005).
A wide range of symptoms can be observed in malaria patients. Clinically however, malaria is categorized as either uncomplicated or complicated. Complicated malaria is further divided into three overlapping syndromes: cerebral malaria, severe anemia, and metabolic acidosis. The clinical syndrome observed in each individual patient is influenced by multiple variables: parasite species, host immune status, and genetic background, as well as the use and timing of antimalarial drugs (Taylor et al., 2010).
Host iron has received significant attention at the clinical level as a major factor that may regulate malaria virulence. The results of clinical studies conducted prior to 2002 which examined the relationship between host iron status and malaria risk are reviewed in three meta-analyses (Shankar, 2000; Oppenheimer, 2001; Gera and Sachdev, 2002). In the interim, two large iron supplementation trials as well as several smaller clinical studies have shed further light on the relationship between host iron status and malaria infection (Table 1). Clinical trials that have examined the relationship between host iron and malaria fall into two basic categories: those that observe the rate of malaria in individuals with iron deficiency anemia, and those that look at the rate of malaria infection in individuals given iron supplementation. Differences in study design exist within both study types, and include: the definition of study participant iron status, the administration of iron alone or with folate, and access to health care. Despite these differences, assessment of the outcome of the clinical studies has led to the general consensus that iron deficiency is protective against malaria, and iron supplementation increases malaria risk in the absence of access to adequate health care (Prentice and Cox, 2012; Spottiswoode et al., 2012; Stoltzfus, 2012).
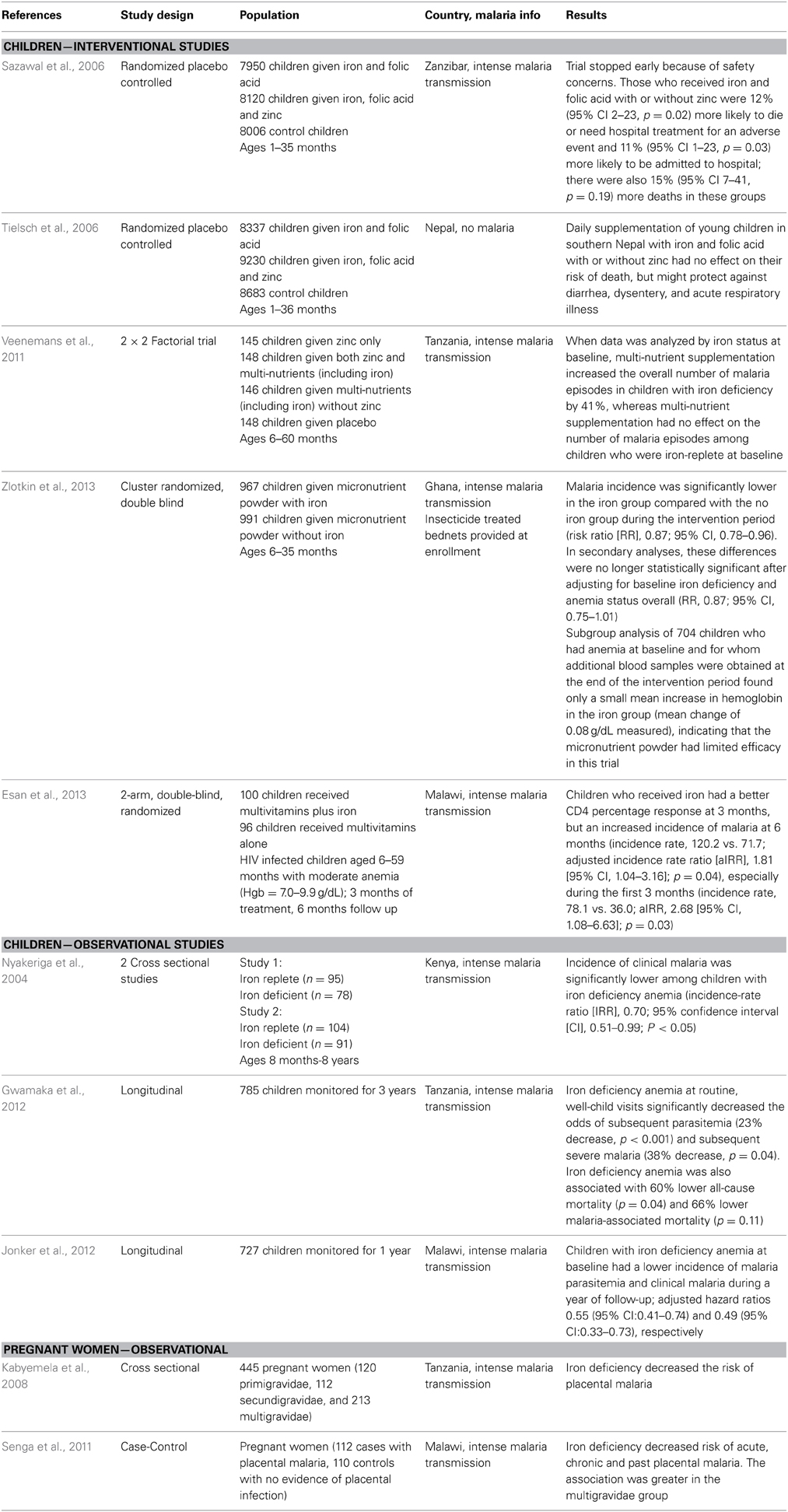
Table 1. Summary of clinical studies on iron deficiency, iron supplementation, and malaria.
While these clinical studies and meta-analyses have been indispensable for determining the relationship between host iron status and malaria risk, it is not clear how iron deficiency protects and why iron supplementation increases risk. Immunity to malaria and high prevalence of genetic traits such as G6PD deficiency and hemoglobinopathies in the study populations limit the capacity of clinical studies to parse out causation. Furthermore, relatively little is known with regards to the role host iron plays in malaria pathogenesis. Iron impacts a broad range of biological processes that have the potential to shape malaria pathogenesis. As a result, even with the most ideal of clinical study designs; the prerequisite knowledge of which aspects of malaria pathogenesis should be studied is largely absent. A better grasp on the underlying biological principals that govern (i) the protection of iron deficiency against malaria and (ii) the increased risk of malaria associated with iron supplementation is critical for managing iron supplementation campaigns in malaria endemic regions.
Iron is an essential nutrient for nearly every living organism including humans and the malaria parasite. Iron impacts a broad range of biological processes; including host and parasite cellular function, erythropoiesis and immune function. The capacity of iron to fluctuate between two oxidation states, ferrous (Fe2+) and ferric (Fe3+), makes it indispensable for many critical biological processes, including DNA replication, cellular respiration, and oxygen transport. However, the same useful biphasic properties of iron which make it indispensable also contribute to its high cytotoxicity. As a result the human host tightly regulates iron availability and usage.
Access to iron is particularly important in the context of host-pathogen interactions. When confronted with infection and inflammation the human host reallocates its iron reservoirs in an effort to deprive invading pathogens of iron. The human protein hepcidin—a rheostat of systemic iron homeostasis—signals the body to decrease absorption of iron in the proximal duodenum and orchestrates the movement of iron from serum into storage within the liver and macrophages (Roy, 2013). As a result of reduced serum iron, erythropoiesis—a process exquisitely sensitive to iron levels—slows in the face of infection as well as inflammation. The human host's active reduction in bioavailable iron protects against a wide range of pathogens (Armitage et al., 2011). Not surprisingly, as many pathogens require access to host iron sources to survive and grow, pathogens have evolved sophisticated iron acquisition systems, and the iron acquisition systems of many bacterial and fungal species have been well described (Skaar, 2010). By comparison how the malaria parasite acquires, regulates, and utilizes iron remains a mystery.
Iron is essential for the survival of the malaria parasite. The parasite multiplies 8–32 times in the course of a single intra-erythrocytic lifecycle. Iron is an essential cofactor for the DNA replication enzyme ribonucleotide reductase, and as a result iron is required to fuel this rapid intra-erythrocytic proliferation (Rubin et al., 1993). Iron is also utilized by the parasite for pyrimidine (Krungkrai et al., 1990; Van Dooren et al., 2006) and heme biosynthesis (Sato and Wilson, 2002; Dhanasekaran et al., 2004; Sato et al., 2004; Nagaraj et al., 2008, 2009, 2010, 2013). As with the human host, the malaria parasite must also balance its need for iron against the cytotoxicity of iron.
The malaria parasite metabolizes host hemoglobin in its acidic digestive vacuole in order to acquire necessary amino acids; however, as discussed below, the parasite does not utilize the iron in host heme. Plasmodium aspartic and cysteine proteases degrade host hemoglobin and release large quantities of toxic iron-laden heme (Goldberg et al., 1990; Subramanian et al., 2009). Apicoblast parasites neutralize the cytotoxic heme produced during hemoglobin metabolism by sequestering the heme in an inert crystal, hemozoin (Rudzinska et al., 1965; Chugh et al., 2013). Despite neutralizing a substantial portion of host heme into hemozoin, some residual heme remains free and becomes oxidized, generating free oxygen radicals (Francis et al., 1997). The parasite possesses powerful thioredoxin and glutathione systems to maintain intracellular redox equilibrium (Jortzik and Becker, 2012). However, even when these redox systems are functioning at full capacity, oxidative stress significantly increases as the parasite matures and replicates within host erythrocytes (Fu et al., 2010). In fact, many antimalarials, including artemisinin, appear to target the parasite's ability to detoxify reactive oxygen species (ROS) (Rosenthal and Meshnick, 1996; Klonis et al., 2013; Ariey et al., 2014). For example, it was recently found that mutations in PF3D7_1343700 (Kelch) can confer resistance to artemisinin. The authors speculate that these mutations cause a disruption of the parasite's ability to detoxify ROS because the efficacy of artemisinin depends on its ability to generate oxygen radicals and some kelch-containing proteins in other organisms have been shown to be involved in the regulation of cytoprotection (Ariey et al., 2014).
Given the relationship between iron, heme, and ROS, it is possible that perturbations in host iron regulation might also affect the malaria parasite's redox equilibrium. Iron responsive proteins (IRPs) and their accompanying iron responsive elements are critical for maintaining cellular iron homeostasis in the human host. IRPs and iron responsive elements are responsible for mobilizing iron when demands are high and moving iron into storage when excess iron may promote ROS formation (Hentze et al., 2010). Loyevsky et al. identified and characterized a P. falciparum IRP, the expression of which was affected by iron starvation as well as iron supplementation (Loyevsky et al., 2001, 2003; Hodges et al., 2005). However, a search of gene databases failed to identify Plasmodium homologs of ferritin, ferroportin, metallothione, a ferrioxamine-based transport system or ferredoxin or siderophore biosynthesis pathways—all proteins and processes utilized by other organisms to acquire, regulate, and store iron (Scholl et al., 2005). Clearly, much remains unknown regarding parasite iron biology.
Realizing the importance of iron for the malaria parasite, researchers have invested extensive time and effort into the investigation of the antimalarial activity of iron chelating agents. These studies have also provided insight into malaria parasite iron biology. In contrast to mammalian cells, which are sensitive to millimolar concentrations of iron chelators, erythrocytic stage malaria parasites are sensitive to micromolar concentrations of iron chelators in vitro and in animal models (Cabantchik et al., 1996). The cytotoxicity of iron chelators is dependent upon the stage of intra-erythrocytic maturation of the malaria parasite and the hydrophobicity of the iron chelator (Lytton et al., 1994). For example, the hydrophilic chelator hydroxamate-based deferoxamine (DFO) has cytostatic activity against the ring stage and cytotoxic activity against the late trophozoite and schizont erythrocytic stages of the parasite (Whitehead and Peto, 1990; Lytton et al., 1994; Cabantchik et al., 1999).
The cytotoxicity of iron chelators against the malaria parasite suggests that the mechanism of action of iron chelators is more complex than simple iron deprivation. Alternative mechanisms have been suggested for some chelators, including the direct inhibition of parasite ribonucleotide reductase activity (Lederman et al., 1984; Lytton et al., 1994). Furthermore, as iron chelators can modulate host immune function, iron chelator antimalarial activity may be a result of modification of the host immune response (Golenser et al., 2006; Li et al., 2012).
Caution must be taken when considering the use of iron chelators to inform our understanding of the biological relationship between iron deficiency and malaria infection. The evidence that iron chelators do more than merely deprive the parasite of iron introduces potential confounding factors into studies that utilize iron chelators as a model for iron deficiency. Furthermore, most iron chelators cannot chelate iron associated with heme, ferritin, or transferrin. Because the iron saturation of each of these host iron reservoirs are reduced in iron deficiency, iron chelators are not suitable for studying the effect of host iron reduction on the malaria parasite.
That said, evidence that chelation of chelatable extracellular and intra-erythrocyte iron does not impact erythrocytic stage P. falciparum growth, suggests that chelatable host iron is not necessary for the erythrocyte stage of infection (Scott et al., 1990). Furthermore, work by Moormann et al. shows that parasite nuclear and mitochondrial transcripts decrease in the presence of the iron chelator DFO (Moormann et al., 1999). These results are consistent with a normal cellular response to iron deprivation. In conclusion, iron chelators are obviously indispensable in the study of iron biology. However, in the case of malaria caution must be taken.
It is inarguable that iron is essential to erythrocytic stage malaria and therefore possible that alterations in host iron levels may tip the balance between inhibiting or promoting parasite growth and virulence. Consequently, the question of how the parasite acquires host iron becomes central. A healthy iron-replete human has 3–4 total grams of iron, which is distributed in hemoglobin contained within circulating RBCs (2.5 g), in iron containing proteins (400 mg), in serum bound to transferrin (3–7 mg), and in storage proteins such as ferritin (1 g). Host iron reservoirs available to erythrocytic stage malaria parasite include: (1) transferrin and non-transferrin bound iron (NTBI) in the serum and (2) intra-erythrocytic iron contained within hemoglobin, ferritin, as well as trace amounts freely bioavailable iron in the RBC cytosol (Figure 1).
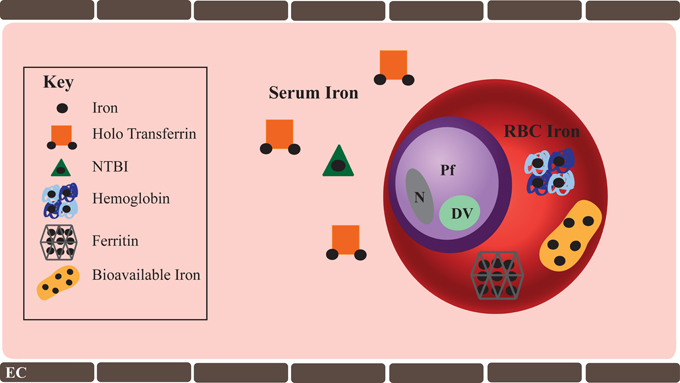
Figure 1. Host Iron available to erythrocytic stage P. falciparum. Host iron immediately available to the erythrocytic stage of P. falciparum include serum and intra-erythrocytic iron. Serum iron ranges from 10 to 27 μM. Transferrin bound iron is the predominant form of iron in the serum, though trace amounts of non-transferrin bound iron (NTBI) are present. In some pathologic conditions such as hemochromatosis, NTBI may be significantly greater. While iron deficiency anemia is characterized by a significant decline in serum iron. RBC iron is found within hemoglobin (20 mM), ferritin (0.7 nM), and as bioavailable iron (1–10 μM). Iron deficiency anemia significantly reduces RBC iron, specifically hemoglobin iron. Shown in the figure are: Pf, P. falciparum; DV, digestive vacuole; N, parasite nucleus; and EC, endothelial cell.
Iron deficiency affects these host iron reservoirs by significantly reducing the availability of both serum iron and intra-erythrocytic iron. Iron supplementation results in brief spikes in serum iron levels (Schümann et al., 2012, 2013), but has little immediate effect on intra-erythrocyte iron. However, approximately 2 weeks following iron supplementation, average intra-erythrocyte iron levels slowly begin improving as new iron-replete RBCs enter into circulation. It is well-documented that virulence of many bacteria is directly associated with the availability of host iron, and as a result iron supplementation can exacerbate infections (Doherty, 2007). Whether described changes in serum and intra-erythrocyte iron stores affect erythrocytic stage malaria infection remains unknown.
The relationship between host serum iron and parasitized RBCs (pRBCs) is especially intriguing (Table 2). Because transferrin has an extremely high affinity for iron (1023M−1 at pH 7.4), NTBI is scarce in healthy individuals. There is strong evidence that transferrin associates with pRBCs but not uninfected RBCs. Work by Pollack et al. shows that pRBCs take up Fe59 bound to human transferrin, and a recent publication by our own group demonstrates that incubation of pRBCs with transferrin and ferric citrate increases the bioavailable iron in pRBCs (Pollack and Fleming, 1984; Clark et al., 2013). The idea that the parasite is able to acquire transferrin bound iron is further supported Surolia et al. who demonstrated that gelonin toxicity toward P. falciparum is 25 times greater when the gelonin is bound to transferrin (Surolia and Misquith, 1996). Moreover, Fry et al. report transferrin reductase activity associated with pRBCs but not uninfected RBCs (Fry, 1989). Additionally, two groups have reported the identification of a P. falciparum transferrin receptor in the RBC membrane of pRBCs (Haldar et al., 1986; Rodriguez and Jungery, 1986). However, a later study by Pollack et al. concluded that transferrin binding of pRBCs is non-specific (Pollack and Schnelle, 1988), and additional studies were unable to detect any acquisition of transferrin bound iron by pRBCs (Peto and Thompson, 1986; Sanchez-Lopez and Haldar, 1992).

Table 2. Relationship between host serum iron and P. falciparum.
Despite strong evidence that transferrin associates with pRBCs, neither iron depletion nor iron supplementation of malaria culture media has any observable effect on parasite growth (Peto and Thompson, 1986; Scott et al., 1990; Sanchez-Lopez and Haldar, 1992; unpublished data Clark et al.). These results challenge the idea that serum iron, specifically transferrin bound iron, contributes to the protection of iron deficiency from malaria and the increased risk of malaria associated with iron supplementation. Yet, it should be noted that malaria culture media contains tenfold less iron than human sera and all existing studies have utilized culture adapted P. falciparum laboratory lines. It is possible laboratory lines have adapted to an iron-starved extracellular environment. Furthermore, because hemoglobin is an essential nutrient for erythrocytic stage malaria, it is impossible to “starve” the parasite of iron in vitro and this may in turn limit the ability to study the effect of serum iron on P. falciparum.
Much less is known about the ability of the malaria parasite to access intra-erythrocytic iron (Table 3). An individual RBC contains 100 fg (20 mM) of iron, the majority of which is contained within hemoglobin. It is estimated that if the parasite were able to access only 1% of this hemoglobin iron all of its iron demands would be fulfilled (Hershko and Peto, 1988; Gabay and Ginsburg, 1993). However, as discussed above, the parasite incorporates the majority of heme released as a result of hemoglobin digestion into hemozoin (Chugh et al., 2013). Despite identification of a Plasmodium heme oxygenase-like protein, which would facilitate release of iron from host heme (Okada, 2009), the parasite does not exhibit enzymatic heme oxygenase activity nor possess a canonical heme oxygenase pathway (Sigala et al., 2012). Even without inherent heme oxygenase activity, it remains possible that non-enzymatic mechanisms release enough iron from trace heme to meet the iron requirements of the parasite. Possible mechanisms include heme breakdown by glutathione or hydrogen peroxide, the conditions for which are predicted to exist within erythrocytic stage parasites (Ginsburg et al., 1998; Loria et al., 1999). However, as the parasite synthesizes heme de novo, it does not seem likely that the parasite draws iron from host heme (Nagaraj et al., 2013).
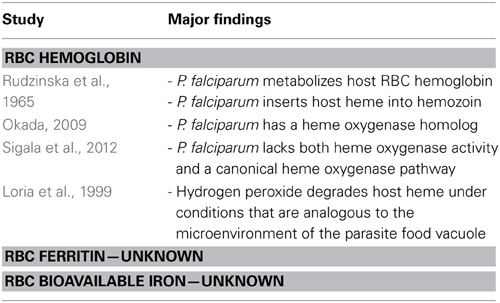
Table 3. Relationship between RBC iron and P. falciparum.
In addition to hemoglobin, RBCs contain residual amounts of biovaialble iron (1–10 μM) as well as iron stored within ferritin (0.7 nM), and it is possible that the parasite is capable of utilizing one or both of these erythrocyte iron reservoirs. Currently, however, there is no reported evidence to either support or refute these possibilities (Scholl et al., 2005). However, despite a lack of evidence that the parasite accesses host intra-erythrocytic iron, recent work by our group has shown that pRBC bioavailable iron content increases as the parasite matures from ring stage to schizont. This observation suggests that iron is released from some form of storage as the parasite develops within host RBCs (Clark et al., 2013). Whether the iron is released from parasite or host storage remains an open question.
Although the precise host iron source(s) the malaria parasite acquires remains unclear, all the potential host iron reservoirs (serum and intra-erythrocyte) available to erythrocytic stage malaria are affected by iron deficiency as well as iron supplementation. Therefore, it is reasonable to hypothesize that iron deprivation and excess iron contribute to the relationship between host iron and malaria risk observed in the clinical studies discussed earlier. That said, even during iron deficiency, the erythrocytic stage of the parasite inhabits the most iron rich environment in the human body. As such it is alternatively possible that neither iron deficiency nor iron supplementation perturb iron reservoirs enough to significantly impact the parasite.
In addition to affecting host iron reservoirs, iron deficiency also induces changes in RBC physiology. One such difference between iron-replete and iron-deficient RBCs is the substitution of zinc for iron in hemogloblin when iron is limiting. This results in zinc protoporphoryin IX levels ten times higher in iron deficient as compared to iron-replete RBCs (Wong et al., 1996). As zinc protoporphoryin IX inhibits hemozoin extension in vitro; it is reasonable to hypothesize that that elevated zinc protoporphoryin IX in iron deficient erythrocytes provides protection against malaria infection by impeding parasite growth (Iyer et al., 2003).
Additional changes to RBC physiology caused by iron deficiency include: microcytosis, greater susceptibility to oxidative stress, reduced ATP content, and decreased deformability (Yip et al., 1983; Acharya et al., 1991; Nagababu et al., 2008; Brandão et al., 2009). Furthermore, iron deficient RBCs experience enhanced eryptotic cell death (Kempe et al., 2006). The altered physiology of microcytic iron deficient RBCs may therefore protect against erythrocytic stage malaria infection. Research by Koka et al. indicates that propagation of the erythrocytic stage of P. falciparum strain BinH is reduced in iron deficient RBCs (Koka et al., 2007). However, earlier work by Luzzie et al. observed abnormal parasite morphology but no difference in the growth of P. falciparum strain UPO in iron deficient as compared to iron-replete RBCs (Luzzi et al., 1990). The differences between these studies may be explained by the use of different P. falciparum isolates which feasibly could have different sensitivities to iron deficient RBCs.
Accelerated host clearance of iron deficient pRBCs is an additional explanation for the protection afforded by iron deficiency against malaria. Results from two studies that examined malaria infection in iron deficient mice both observed a higher clearance rate of pRBCs in iron deficient as compared to iron-replete mice (Koka et al., 2007; Matsuzaki-Moriya et al., 2011). Specifically, Matsuzaki et al. observed elevated phagocytosis of pRBCs in iron deficient as compared to iron-replete mice, and proposed that the increased phagocytosis rate may be attributable to greater phosphatidylserine levels on iron deficient pRBCs as compared to iron-replete pRBCs. Koka et al. similarly observed greater phosphatidylserine levels on P. falciparum human iron deficient pRBCs. Ultimately, these limited data suggests that iron deficiency may provide protection against malaria infection by both impeding erythrocytic stage malaria growth and increasing phagocytosis of iron deficient pRBCs. However, only further investigation will reveal the true relationship between iron deficient RBCs and P. falciparum.
In the absence of sufficient iron for heme synthesis, the human host's erythropoietic rate decreases. Conversely, iron supplementation of individuals with iron deficiency anemia results in a strong erythropoietic response; because the body attempts to recover RBC numbers and replace less viable iron deficient RBCs (Figure 2). It is well-known that P. vivax exclusively infects the very youngest RBCs (reticulocytes). However, P. vivax is not the only Plasmodium species that prefers young RBCs. In fact many species of Plasmodium, including P. falciparum, preferentially infect young RBCs, and furthermore young RBC support greater parasite replication than more mature RBCs (Wilson et al., 1977; Pasvol et al., 1980; Lim et al., 2013). Thus, significant elevation in the erythropoietic rate could put an individual at increased risk of erythrocytic stage P. falciparum infection. Tian et al. have investigated this hypothesis in the context of pregnant women, who are at greater risk of malaria infection than their non-pregnant counterparts and experience increased erythropoietic rates to meet the oxygen demands of the growing fetus. The authors report that P. falciparum growth is significantly greater in the on average younger RBCs taken from pregnant women as compared to the on average older RBCs taken from non-pregnant women (Tian et al., 1998).
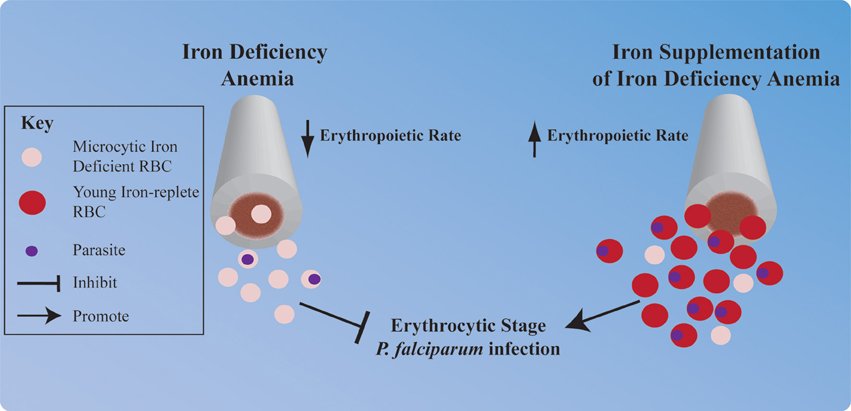
Figure 2. Hypothesized impact of iron deficiency anemia and iron supplementation on P. falciparum erythrocytic infection. Iron deficiency anemia and iron supplementation each profoundly influence human erythropoiesis, and this may influence erythrocytic stage malaria infection. Iron deficiency induced reduction in the erythropoietic rate and synthesis of microcytic iron deficient RBCs may provide protection against P. falciparum infection. Conversely, stimulation of the human host's erythropoietic rate by iron supplementation and subsequent replacement of microcytic iron deficient RBCs with young iron-replete RBCs may increase an individual's risk of erythrocytic stage P. falciparum infection.
Murine models have additionally been used to shed light on the relationship between erythropoiesis and malaria infection. Interestingly, when Chang et al. manipulated the timing of erythropoiesis during the course of a malaria infection it was observed that reticulocytosis early in infection significantly increased infection and morbidity, while reticulocytosis late in infection decreased mortality (Chang et al., 2004). These observations are consistent with recent work by Zhao et al. showing that lipocalin 2, which is elevated during malaria infection, provides protection from malaria infection in mice by limiting reticulocytosis (Zhao et al., 2012).
Furthermore, mathematical modeling by Cromer et al. makes several key predictions that support a role for erythropoiesis in driving the protection from malaria associated with iron deficiency anemia and increased risk associated with iron supplementation. First, their model predicts that low reticulocyte production rate—as would be observed in iron deficiency—in combination with a parasite that prefers reticulocytes, could result in a less severe infection. Second, high reticulocyte production—as would be observed in iron deficient individuals responding to iron supplementation—could increase severity of malaria infection (Cromer et al., 2009). These results indicate that limiting reticulocytosis early in infection is important for limiting erythrocytic stage malaria infection and further support the hypothesis that iron supplementation-induced reticulocytosis significantly increases the risk of erythrocytic stage P. falciparum infection.
Together, these observations provide insight into potential cellular mechanisms contributing to the protection of iron deficiency against malaria, and the increased risk of malaria associated with iron supplementation. With regard to iron deficiency, altered RBC physiology may limit P. falciparum propagation within iron deficient RBCs and increase clearance of iron deficient pRBCs. Furthermore, the reduced erythropoietic rate and subsequent reduction in an iron deficient individual's hematocrit may additionally contribute to protection. Conversely, the increased erythropoietic rate triggered by iron supplementation paired with the preference of P. falciparum for young RBCs may be partially responsible for the increased risk of malaria infection that is associated with iron supplementation.
Overall, the available evidence supports a link between (i) iron deficiency and protection from malaria infection and (ii) iron supplementation and increased risk of malaria. However, there is still much to be learned. Furthermore, study of the competition between the malaria parasite and the human host for iron can serve as a translational model to identify critical molecular mechanisms of P. falciparum pathogenesis (see questions in Table 4). Most importantly, however, such research will help the global health community reach their goal of devising a strategy for safely administering iron supplementation in malaria endemic regions.

Table 4. Questions for future translational research.
Martha A. Clark, Morgan M. Goheen, and Carla Cerami wrote and edited the manuscript. All authors have read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The work was supported by the National Institute of Child Health and Human Development under award number U01HD061235 (to CCH). We thank Dan Raiten, Steven R. Meshnick, and Con Beckers for many useful discussions.
Acharya, J., Punchard, N. A., Taylor, J. A., Thompson, R. P., and Pearson, T. C. (1991). Red cell lipid peroxidation and antioxidant enzymes in iron deficiency. Eur. J. Haematol. 47, 287–291. doi: 10.1111/j.1600-0609.1991.tb01573.x
Aguilar, R., Moraleda, C., Quintó, L., Renom, M., Mussacate, L., Macete, E., et al. (2012). Challenges in the diagnosis of iron deficiency in children exposed to high prevalence of infections. PLoS ONE 7:e50584. doi: 10.1371/journal.pone.0050584
Ariey, F., Witkowski, B., Amaratunga, C., Beghain, J., Langlois, A.-C., Khim, N., et al. (2014). A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 505, 50–55. doi: 10.1038/nature12876
Armitage, A. E., Eddowes, L. A., Gileadi, U., Cole, S., Spottiswoode, N., Selvakumar, T. A., et al. (2011). Hepcidin regulation by innate immune and infectious stimuli. Blood 118, 4129–4139. doi: 10.1182/blood-2011-04-351957
Brandão, M. M., Castro Mde, L., Fontes, A., Cesar, C. L., Costa, F. F., and Saad, S. T. (2009). Impaired red cell deformability in iron deficient subjects. Clin. Hemorheol. Microcirc. 43, 217–221. doi: 10.3233/CH-2009-1211
Cabantchik, Z. I., Glickstein, H., Golenser, J., Loyevsky, M., and Tsafack, A. (1996). Iron chelators: mode of action as antimalarials. Acta Haematol. 95, 70–77. doi: 10.1159/000203952
Cabantchik, Z. I., Moody-Haupt, S., and Gordeuk, V. R. (1999). Iron chelators as anti-infectives; malaria as a paradigm. FEMS Immunol. Med. Microbiol. 26, 289–298. doi: 10.1111/j.1574-695X.1999.tb01401.x
Chang, K.-H., Tam, M., and Stevenson, M. M. (2004). Modulation of the course and outcome of blood-stage malaria by erythropoietin-induced reticulocytosis. J. Infect. Dis. 189, 735–743. doi: 10.1086/381458
Chotivanich, K., Udomsangpetch, R., Simpson, J. A., Newton, P., Pukrittayakamee, S., Looareesuwan, S., et al. (2000). Parasite multiplication potential and the severity of Falciparum malaria. J. Infect. Dis. 181, 1206–1209. doi: 10.1086/315353
Chugh, M., Sundararaman, V., Kumar, S., Reddy, V. S., Siddiqui, W. A., Stuart, K. D., et al. (2013). Protein complex directs hemoglobin-to-hemozoin formation in Plasmodium falciparum. Proc. Natl. Acad. Sci. U.S.A. 110, 5392–5397. doi: 10.1073/pnas.1218412110
Clark, M., Fisher, N. C., Kasthuri, R., and Cerami Hand, C. (2013). Parasite maturation and host serum iron influence the labile iron pool of erythrocyte stage Plasmodium falciparum. Br. J. Haematol. 161, 262–269. doi: 10.1111/bjh.12234
Cromer, D., Stark, J., and Davenport, M. P. (2009). Low red cell production may protect against severe anemia during a malaria infection–insights from modeling. J. Theor. Biol. 257, 533–542. doi: 10.1016/j.jtbi.2008.12.019
De-Regil, L. M., Jefferds, M. E. D., Sylvetsky, A. C., and Dowswell, T. (2011). Intermittent iron supplementation for improving nutrition and development in children under 12 years of age. Cochrane Database Syst. Rev. CD009085. doi: 10.1002/14651858.CD009085.pub2
De-Regil, L. M., Suchdev, P. S., Vist, G. E., Walleser, S., and Peña-Rosas, J. P. (2013). Home fortification of foods with multiple micronutrient powders for health and nutrition in children under two years of age (Review). Evid. Based Child Health 8, 112–201. doi: 10.1002/ebch.1895
Dhanasekaran, S., Chandra, N. R., Sagar, B. K. C., Rangarajan, P. N., and Padmanaban, G. (2004). δ-Aminolevulinic acid dehydratase from Plasmodium falciparum; indigenous versus imported. J. Biol. Chem. 279, 6934–6942. doi: 10.1074/jbc.M311409200
Dondorp, A. M., Desakorn, V., Pongtavornpinyo, W., Sahassananda, D., Silamut, K., Chotivanich, K., et al. (2005). Estimation of the total parasite biomass in acute falciparum malaria from plasma PfHRP2. PLoS Med. 2:e204. doi: 10.1371/journal.pmed.0020204
Esan, M. O., van Hensbroek, M. B., Nkhoma, E., Musicha, C., White, S. A., Ter Kuile, F. O., et al. (2013). Iron supplementation in HIV-infected Malawian children with anemia: a double-blind, randomized controlled trial. Clin. Infect. Dis. 57, 1626–1634. doi: 10.1093/cid/cit528
Francis, S. E., Sullivan, D. J. Jr., and Goldberg, D. E. (1997). Hemoglobin metabolism in the malaria parasite Plasmodium falciparum. Annu. Rev. Microbiol. 51, 97–123. doi: 10.1146/annurev.micro.51.1.97
Fry, M. (1989). Diferric transferrin reductase in Plasmodium falciparum-infected erythrocytes. Biochem. Biophys. Res. Commun. 158, 469–473. doi: 10.1016/S0006-291X(89)80071-3
Fu, Y., Tilley, L., Kenny, S., and Klonis, N. (2010). Dual labeling with a far red probe permits analysis of growth and oxidative stress in P. falciparum-infected erythrocytes. Cytom. Part J. Int. Soc. Anal. Cytol. 77, 253–263. doi: 10.1002/cyto.a.20856
Gabay, T., and Ginsburg, H. (1993). Hemoglobin denaturation and iron release in acidified red blood cell lysate–a possible source of iron for intraerythrocytic malaria parasites. Exp. Parasitol. 77, 261–272. doi: 10.1006/expr.1993.1084
Gera, T., and Sachdev, H. P. S. (2002). Effect of iron supplementation on incidence of infectious illness in children: systematic review. BMJ 325, 1142. doi: 10.1136/bmj.325.7373.1142
Ginsburg, H., Famin, O., Zhang, J., and Krugliak, M. (1998). Inhibition of glutathione-dependent degradation of heme by chloroquine and amodiaquine as a possible basis for their antimalarial mode of action. Biochem. Pharmacol. 56, 1305–1313. doi: 10.1016/S0006-2952(98)00184-1
Goldberg, D. E., Slater, A. F., Cerami, A., and Henderson, G. B. (1990). Hemoglobin degradation in the malaria parasite Plasmodium falciparum: an ordered process in a unique organelle. Proc. Natl. Acad. Sci. U.S.A. 87, 2931–2935. doi: 10.1073/pnas.87.8.2931
Golenser, J., Domb, A., Mordechai-Daniel, T., Leshem, B., Luty, A., and Kremsner, P. (2006). Iron chelators: correlation between effects on Plasmodium spp. and immune functions. J. Parasitol. 92, 170–177. doi: 10.1645/GE-3517.1
Gwamaka, M., Kurtis, J. D., Sorensen, B. E., Holte, S., Morrison, R., Mutabingwa, T. K., et al. (2012). Iron deficiency protects against severe Plasmodium falciparum malaria and death in young children. Clin. Infect. Dis. 54, 1137–1144. doi: 10.1093/cid/cis010
Haider, B. A., Olofin, I., Wang, M., Spiegelman, D., Ezzati, M., Fawzi, W. W., et al. (2013). Anaemia, prenatal iron use, and risk of adverse pregnancy outcomes: systematic review and meta-analysis. BMJ 346, f3443. doi: 10.1136/bmj.f3443
Haldar, K., Henderson, C. L., and Cross, G. A. (1986). Identification of the parasite transferrin receptor of Plasmodium falciparum-infected erythrocytes and its acylation via 1,2-diacyl-sn-glycerol. Proc. Natl. Acad. Sci. U.S.A. 83, 8565–8569. doi: 10.1073/pnas.83.22.8565
Hentze, M. W., Muckenthaler, M. U., Galy, B., and Camaschella, C. (2010). Two to Tango: regulation of mammalian iron metabolism. Cell 142, 24–38. doi: 10.1016/j.cell.2010.06.028
Hershko, C., and Peto, T. E. (1988). Deferoxamine inhibition of malaria is independent of host iron status. J. Exp. Med. 168, 375–387. doi: 10.1084/jem.168.1.375
Hodges, M., Yikilmaz, E., Patterson, G., Kasvosve, I., Rouault, T. A., Gordeuk, V. R., et al. (2005). An iron regulatory-like protein expressed in Plasmodium falciparum displays aconitase activity. Mol. Biochem. Parasitol. 143, 29–38. doi: 10.1016/j.molbiopara.2005.05.004
Iyer, J. K., Shi, L., Shankar, A. H., and Sullivan, D. J. Jr. (2003). Zinc protoporphyrin IX binds heme crystals to inhibit the process of crystallization in Plasmodium falciparum. Mol. Med. Camb. Mass 9, 175–182. doi: 10.2119/2003-00010.Sullivan
Jonker, F. A. M., Calis, J. C. J., van Hensbroek, M. B., Phiri, K., Geskus, R. B., Brabin, B. J., et al. (2012). Iron status predicts malaria risk in Malawian preschool children. PLoS ONE 7:e42670. doi: 10.1371/journal.pone.0042670
Jortzik, E., and Becker, K. (2012). Thioredoxin and glutathione systems in Plasmodium falciparum. Int. J. Med. Microbiol. 302, 187–194. doi: 10.1016/j.ijmm.2012.07.007
Kabyemela, E. R., Fried, M., Kurtis, J. D., Mutabingwa, T. K., and Duffy, P. E. (2008). Decreased susceptibility to Plasmodium falciparum infection in pregnant women with iron deficiency. J. Infect. Dis. 198, 163–166. doi: 10.1086/589512
Kassebaum, N. J., Jasrasaria, R., Naghavi, M., Wulf, S. K., Johns, N., Lozano, R., et al. (2014). A systematic analysis of global anemia burden from 1990 to 2010. Blood 123, 615–624. doi: 10.1182/blood-2013-06-508325
Kempe, D. S., Lang, P. A., Duranton, C., Akel, A., Lang, K. S., Huber, S. M., et al. (2006). Enhanced programmed cell death of iron-deficient erythrocytes. FASEB J. 20, 368–370. doi: 10.1096/fj.05-4872fje
Klonis, N., Creek, D. J., and Tilley, L. (2013). Iron and heme metabolism in Plasmodium falciparum and the mechanism of action of artemisinins. Curr. Opin. Microbiol. 16, 722–727. doi: 10.1016/j.mib.2013.07.005
Koka, S., Föller, M., Lamprecht, G., Boini, K. M., Lang, C., Huber, S. M., et al. (2007). Iron deficiency influences the course of malaria in Plasmodium berghei infected mice. Biochem. Biophys. Res. Commun. 357, 608–614. doi: 10.1016/j.bbrc.2007.03.175
Krungkrai, J., Cerami, A., and Henderson, G. B. (1990). Pyrimidine biosynthesis in parasitic protozoa: purification of a monofunctional dihydroorotase from Plasmodium berghei and Crithidia fasciculata. Biochemistry (Mosc.) 29, 6270–6275. doi: 10.1021/bi00478a023
Lederman, H. M., Cohen, A., Lee, J. W., Freedman, M. H., and Gelfand, E. W. (1984). Deferoxamine: a reversible S-phase inhibitor of human lymphocyte proliferation. Blood 64, 748–753.
Li, G., Pone, E. J., Tran, D. C., Patel, P. J., Dao, L., Xu, Z., et al. (2012). Iron inhibits activation-induced cytidine deaminase enzymatic activity and modulates immunoglobulin class switch DNA recombination. J. Biol. Chem. 287, 21520–21529. doi: 10.1074/jbc.M112.366732
Lim, C., Hansen, E., Desimone, T. M., Moreno, Y., Junker, K., Bei, A., et al. (2013). Expansion of host cellular niche can drive adaptation of a zoonotic malaria parasite to humans. Nat. Commun. 4, 1638. doi: 10.1038/ncomms2612
Loria, P., Miller, S., Foley, M., and Tilley, L. (1999). Inhibition of the peroxidative degradation of haem as the basis of action of chloroquine and other quinoline antimalarials. Biochem. J. 339(pt 2), 363–370. doi: 10.1042/0264-6021:3390363
Low, M., Farrell, A., Biggs, B.-A., and Pasricha, S.-R. (2013). Effects of daily iron supplementation in primary-school-aged children: systematic review and meta-analysis of randomized controlled trials. CMAJ 185, E791–E802. doi: 10.1503/cmaj.130628
Loyevsky, M., LaVaute, T., Allerson, C. R., Stearman, R., Kassim, O. O., Cooperman, S., et al. (2001). An IRP-like protein from Plasmodium falciparum binds to a mammalian iron-responsive element. Blood 98, 2555–2562. doi: 10.1182/blood.V98.8.2555
Loyevsky, M., Mompoint, F., Yikilmaz, E., Altschul, S. F., Madden, T., Wootton, J. C., et al. (2003). Expression of a recombinant IRP-like Plasmodium falciparum protein that specifically binds putative plasmodial IREs. Mol. Biochem. Parasitol. 126, 231–238. doi: 10.1016/S0166-6851(02)00278-5
Luzzi, G. A., Torii, M., Aikawa, M., and Pasvol, G. (1990). Unrestricted growth of Plasmodium falciparum in microcytic erythrocytes in iron deficiency and thalassaemia. Br. J. Haematol. 74, 519–524. doi: 10.1111/j.1365-2141.1990.tb06344.x
Lytton, S. D., Mester, B., Libman, J., Shanzer, A., and Cabantchik, Z. I. (1994). Mode of action of iron (III) chelators as antimalarials: II. Evidence for differential effects on parasite iron-dependent nucleic acid synthesis. Blood 84, 910–915.
Matsuzaki-Moriya, C., Tu, L., Ishida, H., Imai, T., Suzue, K., Hirai, M., et al. (2011). A critical role for phagocytosis in resistance to malaria in iron-deficient mice. Eur. J. Immunol. 41, 1365–1375. doi: 10.1002/eji.201040942
Miller, L. H., Ackerman, H. C., Su, X., and Wellems, T. E. (2013). Malaria biology and disease pathogenesis: insights for new treatments. Nat. Med. 19, 156–167. doi: 10.1038/nm.3073
Moormann, A. M., Hossler, P. A., and Meshnick, S. R. (1999). Deferoxamine effects on Plasmodium falciparum gene expression. Mol. Biochem. Parasitol. 98, 279–283. doi: 10.1016/S0166-6851(98)00163-7
Nagababu, E., Gulyani, S., Earley, C. J., Cutler, R. G., Mattson, M. P., and Rifkind, J. M. (2008). Iron-deficiency anemia enhances red blood cell oxidative stress. Free Radic. Res. 42, 824–829. doi: 10.1080/10715760802459879
Nagaraj, V. A., Arumugam, R., Gopalakrishnan, B., Jyothsna, Y. S., Rangarajan, P. N., and Padmanaban, G. (2008). Unique properties of Plasmodium falciparum porphobilinogen deaminase. J. Biol. Chem. 283, 437–444. doi: 10.1074/jbc.M706861200
Nagaraj, V. A., Arumugam, R., Prasad, D., Rangarajan, P. N., and Padmanaban, G. (2010). Protoporphyrinogen IX oxidase from Plasmodium falciparum is anaerobic and is localized to the mitochondrion. Mol. Biochem. Parasitol. 174, 44–52. doi: 10.1016/j.molbiopara.2010.06.012
Nagaraj, V. A., Prasad, D., Rangarajan, P. N., and Padmanaban, G. (2009). Mitochondrial localization of functional ferrochelatase from Plasmodium falciparum. Mol. Biochem. Parasitol. 168, 109–112. doi: 10.1016/j.molbiopara.2009.05.008
Nagaraj, V. A., Sundaram, B., Varadarajan, N. M., Subramani, P. A., Kalappa, D. M., Ghosh, S. K., et al. (2013). Malaria parasite-synthesized heme is essential in the mosquito and liver stages and complements host heme in the blood stages of infection. PLoS Pathog. 9:e1003522. doi: 10.1371/journal.ppat.1003522
Nyakeriga, A. M., Troye-Blomberg, M., Dorfman, J. R., Alexander, N. D., Bäck, R., Kortok, M., et al. (2004). Iron deficiency and malaria among children living on the coast of Kenya. J. Infect. Dis. 190, 439–447. doi: 10.1086/422331
Okada, K. (2009). The novel heme oxygenase-like protein from Plasmodiumfalciparum converts heme to bilirubin IXalpha in the apicoplast. FEBS Lett. 583, 313–319. doi: 10.1016/j.febslet.2008.12.015
Okebe, J. U., Yahav, D., Shbita, R., and Paul, M. (2011). Oral iron supplements for children in malaria-endemic areas. Cochrane Database Syst. Rev. CD006589. doi: 10.1002/14651858.CD006589.pub3
Oppenheimer, S. J. (2001). Iron and its relation to immunity and infectious disease. J. Nutr. 131, 616S–633S. discussion: 633S–635S.
Pasvol, G., Weatherall, D. J., and Wilson, R. J. (1980). The increased susceptibility of young red cells to invasion by the malarial parasite Plasmodium falciparum. Br. J. Haematol. 45, 285–295. doi: 10.1111/j.1365-2141.1980.tb07148.x
Peña-Rosas, J. P., De-Regil, L. M., Dowswell, T., and Viteri, F. E. (2012a). Daily oral iron supplementation during pregnancy. Cochrane Database Syst. Rev. 12, CD004736. doi: 10.1002/14651858.CD004736.pub4
Peña-Rosas, J. P., De-Regil, L. M., Dowswell, T., and Viteri, F. E. (2012b). Intermittent oral iron supplementation during pregnancy. Cochrane Database Syst. Rev. 7, CD009997. doi: 10.1002/14651858.CD009997
Peto, T. E., and Thompson, J. L. (1986). A reappraisal of the effects of iron and desferrioxamine on the growth of Plasmodium falciparum in vitro: the unimportance of serum iron. Br. J. Haematol. 63, 273–280. doi: 10.1111/j.1365-2141.1986.tb05550.x
Pollack, S., and Fleming, J. (1984). Plasmodium falciparum takes up iron from transferrin. Br. J. Haematol. 58, 289–293. doi: 10.1111/j.1365-2141.1984.tb06087.x
Pollack, S., and Schnelle, V. (1988). Inability to detect transferrin receptors on P. falciparum parasitized red cells. Br. J. Haematol. 68, 125–129. doi: 10.1111/j.1365-2141.1988.tb04190.x
Prentice, A. M., and Cox, S. E. (2012). Iron and malaria interactions: research needs from basic science to global policy. Adv. Nutr. Bethesda Md 3, 583–591. doi: 10.3945/an.111.001230
Prentice, A. M., Ghattas, H., Doherty, C., and Cox, S. E. (2007). Iron metabolism and malaria. Food Nutr. Bull. 28, S524–539.
Prentice, A. M., Verhoef, H., and Cerami, C. (2013). Iron fortification and malaria risk in children. JAMA 310, 914–915. doi: 10.1001/jama.2013.6771
Rodriguez, M. H., and Jungery, M. (1986). A protein on Plasmodium falciparum-infected erythrocytes functions as a transferrin receptor. Nature 324, 388–391. doi: 10.1038/324388a0
Rosenthal, P. J., and Meshnick, S. R. (1996). Hemoglobin catabolism and iron utilization by malaria parasites. Mol. Biochem. Parasitol. 83, 131–139. doi: 10.1016/S0166-6851(96)02763-6
Roy, C. N. (2013). An update on iron homeostasis: make new friends, but keep the old. Am. J. Med. Sci. 346, 413–419. doi: 10.1097/MAJ.0000000000000190
Rubin, H., Salem, J. S., Li, L. S., Yang, F. D., Mama, S., Wang, Z. M., et al. (1993). Cloning, sequence determination, and regulation of the ribonucleotide reductase subunits from Plasmodium falciparum: a target for antimalarial therapy. Proc. Natl. Acad. Sci. U.S.A. 90, 9280–9284. doi: 10.1073/pnas.90.20.9280
Rudzinska, M. A., Trager, W., and Bray, R. S. (1965). Pinocytotic uptake and the digestion of hemoglobin in malaria parasites. J. Protozool. 12, 563–576. doi: 10.1111/j.1550-7408.1965.tb03256.x
Sanchez-Lopez, R., and Haldar, K. (1992). A transferrin-independent iron uptake activity in Plasmodium falciparum-infected and uninfected erythrocytes. Mol. Biochem. Parasitol. 55, 9–20. doi: 10.1016/0166-6851(92)90122-Z
Sato, S., Clough, B., Coates, L., and Wilson, R. J. M. (2004). Enzymes for heme biosynthesis are found in both the mitochondrion and plastid of the malaria parasite Plasmodium falciparum. Protist 155, 117–125. doi: 10.1078/1434461000169
Sato, S., and Wilson, R. J. M. (2002). The genome of Plasmodium falciparum encodes an active delta-aminolevulinic acid dehydratase. Curr. Genet. 40, 391–398. doi: 10.1007/s00294-002-0273-3
Sazawal, S., Black, R. E., Ramsan, M., Chwaya, H. M., Stoltzfus, R. J., Dutta, A., et al. (2006). Effects of routine prophylactic supplementation with iron and folic acid on admission to hospital and mortality in preschool children in a high malaria transmission setting: community-based, randomised, placebo-controlled trial. Lancet 367, 133–143. doi: 10.1016/S0140-6736(06)67962-2
Scholl, P. F., Tripathi, A. K., and Sullivan, D. J. (2005). Bioavailable iron and heme metabolism in Plasmodium falciparum. Curr. Top. Microbiol. Immunol. 295, 293–324. doi: 10.1007/3-540-29088-5_12
Schümann, K., Kroll, S., Romero-Abal, M.-E., Georgiou, N. A., Marx, J. J. M., Weiss, G., et al. (2012). Impact of oral iron challenges on circulating non-transferrin-bound iron in healthy Guatemalan males. Ann. Nutr. Metab. 60, 98–107. doi: 10.1159/000336177
Schümann, K., Solomons, N. W., Orozco, M., Romero-Abal, M. E., and Weiss, G. (2013). Differences in circulating non-transferrin-bound iron after oral administration of ferrous sulfate, sodium iron EDTA, or iron polymaltose in women with marginal iron stores. Food Nutr. Bull. 34, 185–193.
Scott, M. D., Ranz, A., Kuypers, F. A., Lubin, B. H., and Meshnick, S. R. (1990). Parasite uptake of desferroxamine: a prerequisite for antimalarial activity. Br. J. Haematol. 75, 598–602. doi: 10.1111/j.1365-2141.1990.tb07805.x
Senga, E. L., Harper, G., Koshy, G., Kazembe, P. N., and Brabin, B. J. (2011). Reduced risk for placental malaria in iron deficient women. Malar. J. 10, 47. doi: 10.1186/1475-2875-10-47
Shankar, A. H. (2000). Nutritional modulation of malaria morbidity and mortality. J. Infect. Dis. 182(Suppl. 1), S37–S53. doi: 10.1086/315906
Sigala, P. A., Crowley, J. R., Hsieh, S., Henderson, J. P., and Goldberg, D. E. (2012). Direct tests of enzymatic heme degradation by the malaria parasite Plasmodium falciparum. J. Biol. Chem. 287, 37793–37807. doi: 10.1074/jbc.M112.414078
Skaar, E. P. (2010). The battle for iron between bacterial pathogens and their vertebrate hosts. PLoS Pathog. 6:e1000949. doi: 10.1371/journal.ppat.1000949
Spottiswoode, N., Fried, M., Drakesmith, H., and Duffy, P. E. (2012). Implications of malaria on iron deficiency control strategies. Adv. Nutr. Bethesda Md 3, 570–578. doi: 10.3945/an.111.001156
Stoltzfus, R. J. (2003). Iron deficiency: global prevalence and consequences. Food Nutr. Bull. 24, S99–S103.
Stoltzfus, R. J. (2012). Iron and malaria interactions: programmatic ways forward. Adv. Nutr. Bethesda Md 3, 579–582. doi: 10.3945/an.111.000885
Subramanian, S., Hardt, M., Choe, Y., Niles, R. K., Johansen, E. B., Legac, J., et al. (2009). Hemoglobin cleavage site-specificity of the Plasmodium falciparum cysteine proteases falcipain-2 and falcipain-3. PLoS ONE 4:e5156. doi: 10.1371/journal.pone.0005156
Surolia, N., and Misquith, S. (1996). Cell surface receptor directed targeting of toxin to human malaria parasite, Plasmodium falciparum. FEBS Lett. 396, 57–61. doi: 10.1016/0014-5793(96)01065-4
Taylor, S. M., Molyneux, M. E., Simel, D. L., Meshnick, S. R., and Juliano, J. J. (2010). Does this patient have malaria? JAMA 304, 2048–2056. doi: 10.1001/jama.2010.1578
Tian, L. P., Nelson, E. A., Senok, A. C., Yu, L. M., Oppenheimer, S. J., and Li, K. (1998). Red cell age and susceptibility to malaria during pregnancy. Acta Obstet. Gynecol. Scand. 77, 717–721. doi: 10.1080/j.1600-0412.1998.770704.x
Tielsch, J. M., Khatry, S. K., Stoltzfus, R. J., Katz, J., LeClerq, S. C., Adhikari, R., et al. (2006). Effect of routine prophylactic supplementation with iron and folic acid on preschool child mortality in southern Nepal: community-based, cluster-randomised, placebo-controlled trial. Lancet 367, 144–152. doi: 10.1016/S0140-6736(06)67963-4
Van Dooren, G. G., Stimmler, L. M., and McFadden, G. I. (2006). Metabolic maps and functions of the Plasmodium mitochondrion. FEMS Microbiol. Rev. 30, 596–630. doi: 10.1111/j.1574-6976.2006.00027.x
Veenemans, J., Milligan, P., Prentice, A. M., Schouten, L. R. A., Inja, N., van der Heijden, A. C., et al. (2011). Effect of supplementation with zinc and other micronutrients on malaria in Tanzanian children: a randomised trial. PLoS Med. 8:e1001125. doi: 10.1371/journal.pmed.1001125
Wang, B., Zhan, S., Gong, T., and Lee, L. (2013). Iron therapy for improving psychomotor development and cognitive function in children under the age of three with iron deficiency anaemia. Cochrane Database Syst. Rev. 6, CD001444. doi: 10.1002/14651858.CD001444.pub2
Whitehead, S., and Peto, T. E. (1990). Stage-dependent effect of deferoxamine on growth of Plasmodium falciparum in vitro. Blood 76, 1250–1255.
WHO | Assessing the iron status of populations. (2007). WHO. Available online at: http://www.who.int/nutrition/publications/micronutrients/anaemia_iron_deficiency/9789241596107/en/ (Accessed January 10, 2014).
WHO | Guidelines on food fortification with micronutrients. (2006). WHO. Available online at: http://www.who.int/nutrition/publications/micronutrients/9241594012/en/index.html (Accessed January 10, 2014).
WHO | World Malaria Report. (2013). WHO. Available online at: http://www.who.int/malaria/publications/world_malaria_report_2013/report/en/index.html (Accessed January 13, 2014).
Wilson, R. J., Pasvol, G., and Weatherall, D. J. (1977). Invasion and growth of Plasmodium falciparum in different types of human erythrocyte. Bull. World Health Organ. 55, 179–186.
Wong, S. S., Qutishat, A. S., Lange, J., Gornet, T. G., and Buja, L. M. (1996). Detection of iron-deficiency anemia in hospitalized patients by zinc protoporphyrin. Clin. Chim. Acta Int. J. Clin. Chem. 244, 91–101. doi: 10.1016/0009-8981(95)06200-9
Yip, R., Mohandas, N., Clark, M. R., Jain, S., Shohet, S. B., and Dallman, P. R. (1983). Red cell membrane stiffness in iron deficiency. Blood 62, 99–106.
Zhao, H., Konishi, A., Fujita, Y., Yagi, M., Ohata, K., Aoshi, T., et al. (2012). Lipocalin 2 bolsters innate and adaptive immune responses to blood-stage malaria infection by reinforcing host iron metabolism. Cell Host Microbe 12, 705–716. doi: 10.1016/j.chom.2012.10.010
Keywords: malaria, iron, iron deficiency anemia, Plasmodium falciparum, iron supplementation
Citation: Clark MA, Goheen MM and Cerami C (2014) Influence of host iron status on Plasmodium falciparum infection. Front. Pharmacol. 5:84. doi: 10.3389/fphar.2014.00084
Received: 15 January 2014; Paper pending published: 12 March 2014;
Accepted: 04 April 2014; Published online: 06 May 2014.
Edited by:
Raffaella Gozzelino, Instituto Gulbenkian de Ciência, PortugalReviewed by:
Leann Tilley, Melbourne University, AustraliaCopyright © 2014 Clark, Goheen and Cerami. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carla Cerami, Epidemiology, Gillings School of Global Public Health, University of North Carolina at Chapel Hill, CB# 7435, Chapel Hill, NC 27599, USA e-mail:Y2NlcmFtaUB1bmMuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.