
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 01 May 2014
Sec. Pharmacology of Ion Channels and Channelopathies
Volume 5 - 2014 | https://doi.org/10.3389/fphar.2014.00067
This article is part of the Research Topic CaMKII in Cardiac Health and Disease View all 19 articles
 Mei-ling A. Joiner*
Mei-ling A. Joiner* Olha M. Koval
Olha M. KovalCaMKII is a newly discovered resident of mitochondria in the heart. Mitochondrial CaMKII promotes poor outcomes after heart injury from a number of pathological conditions, including myocardial infarction (MI), ischemia reperfusion (IR), and stress from catecholamine stimulation. A study using the inhibitor of CaMKII, CaMKIIN, with expression delimited to myocardial mitochondria, indicates that an underlying cause of heart disease results from the opening of the mitochondrial permeability transition pore (mPTP). Evidence from electrophysiological and other experiments show that CaMKII inhibition likely suppresses mPTP opening by reducing Ca2+ entry into mitochondria. However, we expect other proteins involved in Ca2+ signaling in the mitochondria are affected with CaMKII inhibition. Several outstanding questions remain for CaMKII signaling in heart mitochondria. Most importantly, how does CaMKII, without the recognized N-terminal mitochondrial targeting sequence transfer to mitochondria?
CaMKII activity promotes heart failure by mediating pathological effects of ischemia reperfusion (IR) through induction of both apoptosis and necrosis (Salas et al., 2009). Cytosolic inhibition of CaMKII attenuates cell death in the heart that results from catecholamine stress, myocardial infarction (MI) or IR (Yang et al., 2006). The increase in cell death via CaMKII activity involves mitochondrial pro-death pathways (Salas et al., 2009; Joiner et al., 2012). Further, either membrane partitioned or mitochondrial matrix expression of a specific and potent inhibitor of CaMKII, CaMKIIN, reduces cell death from MI, catecholamine stress, and IR (Joiner et al., 2012). Therefore, inhibiting CaMKII either in the cytosol or in mitochondria can block CaMKII activity leading to cell death. CaMKII protein targets in cytosol are well studied and include Ca2+ entry pathways and proteins involved with Ca2+ handling at the ER (Salas et al., 2009; Koval et al., 2010; Ozcan and Tabas, 2010; Zhang et al., 2010). Mitochondrial-triggered cell death occurs from Ca2+ overload or excess reactive oxygen species (ROS) production in the mitochondria (Crompton and Costi, 1988; Gunter and Pfeiffer, 1990; Lemasters et al., 2009). Inhibiting or eliminating mitochondrial CaMKII activity reduced cell death in a number of cellular models of pathology (Timmins et al., 2009; Joiner et al., 2012; Yun et al., 2013). Reducing cell death by mitochondria Ca2+ overload may occur by either decreasing Ca2+ uptake or reducing mitochondrial permeability transition pore (mPTP) formation (Griffiths and Halestrap, 1993; Elrod et al., 2010; Pan et al., 2013) (Figure 1). The predominant mitochondrial Ca2+ uptake and efflux are via the mitochondrial calcium uniporter (MCU) and Na+/Ca2+ antiporters (NCLX), respectively. However, a number of other channels have been described for these processes (Figure 1 and described in recent reviews, including Ryu et al., 2010). Regulation of these ion channel complexes by post-translational modification and auxiliary proteins is best described for the MCU. In addition to Ca2+ exchange through channel proteins, Ca2+ can be sequestered in the matrix by forming phosphate complexes. These Ca2+-phosphate complexes allow accumulation of Ca2+ in the matrix during periods of high levels cytosolic Ca2+ (Wei et al., 2012). In this review we focus on effects of CaMKII in mitochondrial Ca2+ uptake and permeability transition.
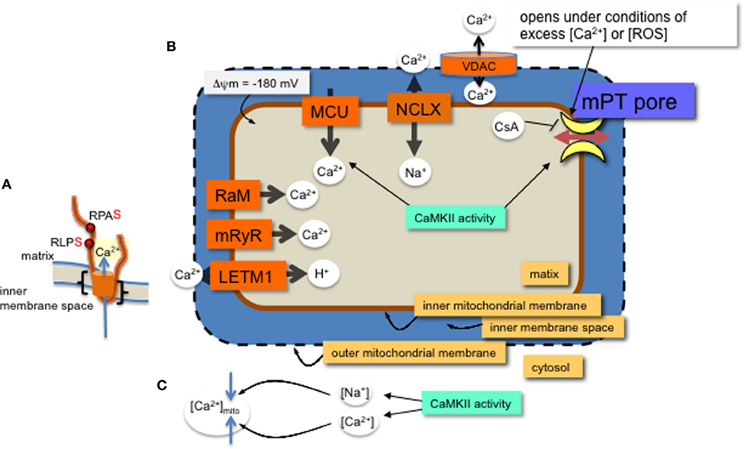
Figure 1. MCU channel in the inner mitochondrial membrane. (A) A single monomer of MCU is shown (orange) with two phospho-serine residues (red dots, with consensus amino acids adjacent) on the N-terminal region in the matrix. The two transmembrane domains are indicated (black brackets). Layers of regulation include accessory proteins and a protein similar to MCU, MCUb (not shown). (B) Mitochondrial Ca2+ channels and exchangers on the inner membrane. Ca2+ predominantly enters the matrix through the MCU channel and efflux is via the Na+-Ca2+ exchanger (NCLX). Other channels and an exchanger that were found to regulate Ca2+ across the inner membrane are the rapid mode of uptake (RaM), the ryanodine receptor (mRyR) and the Ca2+-H+ exchanger (LETM1). Voltage dependent anion channels (VDAC) allow ions and metabolites across the outer membrane. The proton (H+) gradient, a major component of the membrane potential (ΔΨ), is generated from the electron transport chain and drives the flow of H+ through ATP synthase in a reaction coupled to the generation of ATP from ADP and inorganic phosphate. The membrane potential produces a driving force for matrix Ca2+ accumulation. Excess Ca2+ or ROS will open the permeability transition pore, which can be inhibited with CsA or CaMKIIN. Mitochondrial CaMKII activity regulates 1. Ca2+ entry through the MCU and 2. transition pore opening. (C) CaMKII activity in the cytosol can increase both Ca2+ and Na+ ion levels in the cytosol with opposite effects on mitochondrial matrix Ca2+ accumulation.
Mitochondria take up Ca2+ primarily via the MCU (Kirichok et al., 2004; Baughman et al., 2011; De Stefani et al., 2011). The MCU pore-forming channel, a 350 amino acid protein, has two predicted transmembrane helices (Figure 1, from amino acids Lys233 to Trp255 and Thr266 to Met 283), each spanning the inner mitochondrial membrane with the N- and C-terminal ends extending into the matrix. The MCU channel is composed of two pore forming proteins, MCUa and MCUb, as well as at least three regulatory proteins, MICU1, MICU2, and EMRE (Perocchi et al., 2010; Sancak et al., 2013). Although a number of post-translational modifications were identified for MICU1 (Hornbeck et al., 2012), it is not known how these affect Ca2+ current or whether CaMKII phosphorylates any of the accessory proteins or MCUb.
A number of research groups use patch clamp onto mitoplasts (exposed inner membrane of mitochondrial) to measure ion currents, such as the MCU current, across the inner mitochondrial membrane. In order for the patch pipette to access the mitochondrial inner membrane, the mitochondria must be swollen to rupture the outer mitochondrial membrane. These manipulations raise the issue of whether, or how closely, the channel activity observed with patch-clamp corresponds to the in vivo state. Technical aspects of mitoplast patch-clamping remain highly non-standardized, as evidenced by inter-study variation in mitoplast capacitance (0.8–5 pF), (Kirichok et al., 2004; Fieni et al., 2012; Chaudhuri et al., 2013; Dolga et al., 2013; Hoffman et al., 2013) and patch-electrode resistance (4–40 MΩ) (Kirichok et al., 2004; Dolga et al., 2013). In combination with these technical aspects is the broader uncertainty in basic physiological characteristics of mitochondria, such as the ionic composition of the matrix. Reduced experimental conditions along with technical issues for patch-clamp studies of mitochondrial inner membrane ion currents can complicate data interpretation. For example, theoretical estimates of ionic currents are orders of magnitude lower than the currents through mitochondrial channels directly measured by patch-clamp (Kane and Pavlov, 2013). Accordingly, to understand channel modifications and regulation, studies should include a variety of techniques, not only patch clamp. Despite these acknowledged variations, overall effects of manipulating CaMKII function or its target sites in MCU have revealed effects of CaMKII activation on mitochondrial Ca2+ uptake. That is, phosphorylation of two serine sites on the N-terminus of MCU present a phenotype when mutated. Significantly, the CaMKII-induced larger current can be prevented by serine to alanine mutations of these two residues on MCU (Joiner et al., 2012) (Figure 1).
In order to identify proteins in the mitochondrial CaMKII pathway, we deduced that MCU may be a target for CaMKII because well-established CaMKII targets in heart cells are located near Ca2+ sources (Koval et al., 2010; Purohit et al., 2013). Furthermore, recent publications that showed either CaMKII inhibition (Yang et al., 2006) or the MCU inhibitor, Ru360 (García-Rivas Gde et al., 2006) are protective from IR damage in heart. An immunoprecipitation assay indicated that mitochondrial CaMKII and the MCU interact in a complex (Joiner et al., 2012) and others showed that accumulation of mitochondrial Ca2+ is activated through CaMKII signaling (Timmins et al., 2009). Ca2+ current through the MCU is increased with CaMKII activation as shown with patch-clamp measurements onto prepared mitoplasts and expression of CaMKIIN in the matrix reduced mitochondrial Ca2+ uptake (Joiner et al., 2012). Others found that CaMKII inhibitors block A23187-stimulated arachidonic acid release, LDH release and the decrease in the subsequent mPTP formation is attributed to reduced MCU current (Yun et al., 2013). Taken together, these studies indicate that CaMKII activation in mitochondria is responsible for excess Ca2+ uptake under pathological conditions, which ultimately leads to increased levels of cell death. Conversely, in the absence of MCU, using a knock out mouse lacking the MCU channel, Pan et al showed no protective effect preventing necrosis in the heart after IR (Pan et al., 2013). This study suggests that phosphorylation by CaMKII of a protein(s), other than on the MCU, underlies the transition to cell death by CaMKII activation.
Inhibiting mitochondrial CaMKII with CaMKIIN expression decreased cell death following MI, catecholamine stress and IR. As suggested above, phosphorylation of the MCU by CaMKII may promote cell death under stress conditions. However, reducing MCU current does not appear effective in reducing apoptosis (Pan et al., 2013), therefore, an alternative pathway for CaMKII inhibition may be to delay mPTP opening. The mitochondrial permeability transition allows flow of molecules of up to 1500 Daltons to pass across the inner mitochondrial membrane, leading to mitochondrial swelling, and cell death through apoptosis or necrosis. Opening of the transition pore on the inner membrane occurs under pathophysiological conditions and is triggered by either excess Ca2+ or ROS (Kim et al., 2006; Lemasters et al., 2009). The ATP synthase complex on the inner membrane is a leading contender for molecular identity of the transition pore (Giorgio et al., 2013). In addition, the phosphate carrier and auxiliary regulatory proteins are possible components (Halestrap, 2009). Blocking the opening of the mPTP with the inhibitor, cyclosporin A (CsA) (Nicolli et al., 1996; Halestrap and Brenner, 2003) can reduce cell death from stress and reduce infarct size in hearts after IR in patients (Piot et al., 2008). A number of mitochondrial kinases appear to regulate mPTP opening (Miura et al., 2010; Azarashvili et al., 2014). Furthermore, like CsA, CaMKII inhibition increases matrix Ca2+ capacity because expressing CaMKIIN in the mitochondrial matrix allowed as much or more Ca2+ retention as did CsA (Joiner et al., 2012), suggesting a level of regulation by CaMKII and other kinases in transition pore formation.
Protective effects of CaMKII inhibition may occur via auxiliary proteins to the transition pore. CaMKII regulates the interaction of carnitine palmitoyltransferase I with its inhibitor, malonyl CoA, to affect fatty acid metabolism in mitochondria (Sharma et al., 2010) with possible consequences for mPTP opening (Moon et al., 2012). A study using a knockdown approach to decrease a number of kinases, including CaMKII, showed decreases in ATP synthase activity correlated with a reduction in kinase activity (Sugawara et al., 2013). As mentioned above, components of the ATP synthase complex may form the transition pore under stress conditions (Giorgio et al., 2013). Taken together, excess CaMKII activation may promote mitochondrial cell death by its link to energy production and transition pore formation.
Many post-translational modifications are being discovered in the mitochondrial proteome that are yet to be established as functionally significant. Using phosphoproteomics is one way to identify potential CaMKII targets in the mitochondria. However, it has been argued that few transient protein phosphorylation events are physiologically relevant (Clarke et al., 2008; Covian and Balaban, 2012), citing few phospho-sites revealed with a phospho-protein fluorescent dye on mitochondrial lysate before and after treatment to induce mPTP inhibition (Clarke et al., 2008), and also reasoning that phosphorylation can occur spontaneously, without a kinase. In contrast, numerous sensitive phosphoproteomic studies have identified hundreds of phospho-sites on mitochondrial proteins under different treatment regimes (Lee et al., 2007; Witze et al., 2007; Zhao et al., 2011; Koc and Koc, 2012), some of which are indeed functionally relevant as described in the previous two sections. The spontaneous reaction argument is reminiscent of the early days of assigning a role to superoxide dismutase. Arguments that an enzyme for the superoxide radical dismutation to H2O2 was not necessary, as the reaction could occur rapidly without an enzyme (Fridovich, 1983), were eventually overruled by findings that superoxide dismutase over expression or reduced expression can lead to drastic physiological changes in vivo (Antonarakis et al., 2004). Ultimately, uncovering functionally relevant phospho-sites, for CaMKII and other kinases, in the mitochondrial proteome will require extensive study of the individual sites under different phosphorylation conditions along with mutation analysis.
Protein phosphorylation sites identified by phosphoproteomics described above may be attributed to kinases other than CaMKII. The consensus phosphorylation site, a serine or threonine, three amino acids down stream of an arginine (RxxS/T, x represents any amino acid) for CaMKII phosphorylation is shared by a number of other kinases, for example, PKCdelta (www.kinexus.ca). However, specificity of the inhibitor CaMKIIN occurs even in overexpression systems, where for example both CaMKII and PKC could phosphorylate the same consensus site. When each kinase was co-expressed with CaMKIIN, only phosphorylation by CaMKII was inhibited (Chang et al., 1998). Therefore, using the CaMKII inhibitor, CaMKIIN, endogenous to brain (Chang et al., 1998) raises confidence that CaMKII, rather than a different kinase, is responsible for promoting cell death under stress conditions in heart.
CaMKII activity outside of mitochondria contributes to mitochondrial Ca2+ homeostasis. CaMKII activity elevates diastolic sarcoplasmic reticulum (SR) Ca2+ leak (Curran et al., 2007), which was later shown to contribute to mitochondrial Ca2+ overload (Zhang et al., 2010) specifically, under pathophysiological conditions such as rapid cardiomyocyte pacing (Sepúlveda et al., 2013) and diabetes (Luo et al., 2013), but also with the extreme physiological condition of endurance exercise (Rose et al., 2007). Disruption of cytosolic Ca2+ homeostasis promotes mitochondrial Ca2+ overload (Lemasters et al., 2009). Using genetic tools to overexpress CaMKII or the inhibitor of CaMKII, CaMKIIN, in different cell compartments will lead to a better understanding of where CaMKII activity is required for promoting disease with particular models of stress, including MI, IR, excess catecholamine stimulation, and metabolic diseases. Conversely, CaMKII is a major contributor to myocyte Na+ homeostasis in heart failure (Wagner et al., 2006) and Na+ accumulation in heart failure was shown to influence mitochondrial Ca2+ load via enhanced NCLX -mediated Ca2+ removal (Maack et al., 2006). Thus, indirectly, cytosolic CaMKII can regulate mitochondrial Ca2+ levels.
The role of CaMKII functioning in the mitochondria in physiology and disease is in the early stages of research and discovery. CaMKII may be central to regulating mitochondrial homeostasis as its activity is regulated by both Ca2+ (Miller and Kennedy, 1986) and ROS (Erickson et al., 2008) signaling pathways. CaMKII effects in the mitochondria are likely to be numerous and uncovering target sites promises to reveal regulation of mitochondrial signaling pathways that tune cellular responses for cardiac output.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Antonarakis, S. E., Lyle, R., Dermitzakis, E. T., Reymond, A., and Deutsch, S. (2004). Chromosome 21 and down syndrome: from genomics to pathophysiology. Nat. Rev. Genet. 5, 725–738. doi: 10.1038/nrg1448
Azarashvili, T., Odinokova, I., Bakunts, A., Ternovsky, V., Krestinina, O., Tyynelä, J., et al. (2014). Potential role of subunit c of F0F1-ATPase and subunit c of storage body in the mitochondrial permeability transition. Effect of the phosphorylation status of subunit c on pore opening. Cell Calcium 55, 69–77. doi: 10.1016/j.ceca.2013.12.002
Baughman, J. M., Perocchi, F., Girgis, H. S., Plovanich, M., Belcher-Timme, C. A., Sancak, Y., et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. doi: 10.1038/nature10234
Chang, B. H., Mukherji, S., and Soderling, T. R. (1998). Characterization of a calmodulin kinase II inhibitor protein in brain. Proc. Natl. Acad. Sci. U.S.A. 95, 10890–10895. doi: 10.1073/pnas.95.18.10890
Chaudhuri, D., Sancak, Y., Mootha, V. K., and Clapham, D. E. (2013). MCU encodes the pore conducting mitochondrial calcium currents. Elife 2:e00704. doi: 10.7554/eLife.00704
Clarke, S. J., Khaliulin, I., Das, M., Parker, J. E., Heesom, K. J., and Halestrap, A. P. (2008). Inhibition of mitochondrial permeability transition pore opening by ischemic preconditioning is probably mediated by reduction of oxidative stress rather than mitochondrial protein phosphorylation. Circ. Res. 102, 1082–1090. doi: 10.1161/CIRCRESAHA.107.167072
Covian, R., and Balaban, R. S. (2012). Cardiac mitochondrial matrix and respiratory complex protein phosphorylation. Am. J. Physiol. Heart Circ. Physiol. 303, H940–H966. doi: 10.1152/ajpheart.00077.2012
Crompton, M., and Costi, A. (1988). Kinetic evidence for a heart mitochondrial pore activated by Ca2+, inorganic phosphate and oxidative stress. Eur. J. Biochem. 178, 489–501. doi: 10.1111/j.1432-1033.1988.tb14475.x
Curran, J., Hinton, M. J., Ríos, E., Bers, D. M., and Shannon, T. R. (2007). Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ. Res. 100, 391–398. doi: 10.1161/01.RES.0000258172.74570.e6
De Stefani, D., Raffaello, A., Teardo, E., Szabo, I., and Rizzuto, R. (2011). A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340. doi: 10.1038/nature10230
Dolga, A. M., Netter, M. F., Perocchi, F., Doti, N., Meissner, L., Tobaben, S., et al. (2013). Mitochondrial small conductance SK2 channels prevent glutamate-induced oxytosis and mitochondrial dysfunction. J. Biol. Chem. 288, 10792–10804. doi: 10.1074/jbc.M113.453522
Elrod, J. W., Wong, R., Mishra, S., Vagnozzi, R. J., Sakthievel, B., Goonasekera, S. A., et al. (2010). Cyclophilin D controls mitochondrial pore-dependent Ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J. Clin. Invest. 120, 3680–3687. doi: 10.1172/JCI43171
Erickson, J. R., Joiner, M. A., Guan, X., Kutschke, W., Yang, J., Oddis, C. V., et al. (2008). A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133, 462–474. doi: 10.1016/j.cell.2008.02.048
Fieni, F., Bae Lee, S., Jan, Y. N., and Kirichok, Y. (2012). Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat. Commun. 3, 1317. doi: 10.1038/ncomms2325
Fridovich, I. (1983). Superoxide radical: an endogenous toxicant. Annu. Rev. Pharmacol. Toxicol. 23, 239–257. doi: 10.1146/annurev.pa.23.040183.001323
García-Rivas Gde, J., Carvajal, K., Correa, F., and Zazueta, C. (2006). Ru360, a specific mitochondrial calcium uptake inhibitor, improves cardiac post-ischaemic functional recovery in rats in vivo. Br. J. Pharmacol. 149, 829–837. doi: 10.1038/sj.bjp.0706932
Giorgio, V., von Stockum, S., Antoniel, M., Fabbro, A., Fogolari, F., Forte, M., et al. (2013). Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. U.S.A. 110, 5887–5892. doi: 10.1073/pnas.1217823110
Griffiths, E. J., and Halestrap, A. P. (1993). Protection by cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. J. Mol. Cell. Cardiol. 25, 1461–1469. doi: 10.1006/jmcc.1993.1162
Gunter, T. E., and Pfeiffer, D. R. (1990). Mechanisms by which mitochondria transport calcium. Am. Cell Physiol. 258, C755–C786.
Halestrap, A., and Brenner, C. (2003). The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr. Med. Chem. 10, 1507–1525. doi: 10.2174/0929867033457278
Halestrap, A. P. (2009). What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 46, 821–831. doi: 10.1016/j.yjmcc.2009.02.021
Hoffman, N. E., Chandramoorthy, H. C., Shamugapriya, S., Zhang, X., Rajan, S., Mallilankaraman, K., et al. (2013). MICU1 motifs define mitochondrial calcium uniporter binding and activity. Cell Rep. 5, 1576–1588. doi: 10.1016/j.celrep.2013.11.026
Hornbeck, P. V., Kornhauser, J. M., Tkachev, S., Zhang, B., Skrzypek, E., Murray, B., et al. (2012). PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 40, D261–D270. doi: 10.1093/nar/gkr1122
Joiner, M. A., Koval, O., Li, J., He, B. J., Allamargot, C., Gao, Z., et al. (2012). CaMKII determines mitochondrial stress responses in heart. Nature 491, 269–273. doi: 10.1038/nature11444
Kane, D. A., and Pavlov, E. V. (2013). Calculation of ion currents across the inner membrane of functionally intact mitochondria. Channels 7, 426–431. doi: 10.4161/chan.26290
Kim, J.-S., Jin, Y., and Lemasters, J. J. (2006). Reactive oxygen species, but not Ca2+ overloading, trigger pH- and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 290, H2024–H2034. doi: 10.1152/ajpheart.00683.2005
Kirichok, Y., Krapivinsky, G., and Clapham, D. E. (2004). The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427, 360–364. doi: 10.1038/nature02246
Koc, E. C., and Koc, H. (2012). Regulation of mammalian mitochondrial translation by post-translational modifications. Biochim. Biophys. Acta 1819, 1055–1066. doi: 10.1016/j.bbagrm.2012.03.003
Koval, O. M., Guan, X., Wu, Y., Joiner, M. A., Gao, Z., Chen, B., et al. (2010). CaV1.2 beta-subunit coordinates CaMKII-triggered cardiomyocyte death and afterdepolarizations. Proc. Natl. Acad. Sci. U.S.A. 107, 4996–5000. doi: 10.1073/pnas.0913760107
Lee, J., Xu, Y., Chen, Y., Sprung, R., Kim, S. C., Xie, S., et al. (2007). Mitochondrial phosphoproteome revealed by an improved IMAC method and MS/MS/MS. Mol. Cell. Proteomics 6, 669–676. doi: 10.1074/mcp.M600218-MCP200
Lemasters, J. J., Theruvath, T. P., Zhong, Z., and Nieminen, A.-L. (2009). Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta 1787, 1395–1401. doi: 10.1016/j.bbabio.2009.06.009
Luo, M., Guan, X., Luczak, E. D., Lang, D., Kutschke, W., Gao, Z., et al. (2013). Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J. Clin. Invest. 123, 1262–1274. doi: 10.1172/JCI65268
Maack, C., Cortassa, S., Aon, M. A., Ganesan, A. N., Liu, T., and O'Rourke, B. (2006). Elevated cytosolic Na+ decreases mitochondrial Ca2+ Uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ. Res. 99, 172–182. doi: 10.1161/01.RES.0000232546.92777.05
Miller, S. G., and Kennedy, M. B. (1986). Regulation of brain Type II Ca2+calmodulin-dependent protein kinase by autophosphorylation: a Ca2+-triggered molecular switch. Cell 44, 861–870. doi: 10.1016/0092-8674(86)90008-5
Miura, T., Tanno, M., and Sato, T. (2010). Mitochondrial kinase signalling pathways in myocardial protection from ischaemia/reperfusion-induced necrosis. Cardiovasc. Res. 88, 7–15. doi: 10.1093/cvr/cvq206
Moon, S. H., Jenkins, C. M., Kiebish, M. A., Sims, H. F., Mancuso, D. J., and Gross, R. W. (2012). Genetic ablation of calcium-independent phospholipase A2gamma (iPLA2gamma) attenuates calcium-induced opening of the mitochondrial permeability transition pore and resultant cytochrome c release. J. Biol. Chem. 287, 29837–29850. doi: 10.1074/jbc.M112.373654
Nicolli, A., Basso, E., Petronilli, V., Wenger, R. M., and Bernardi, P. (1996). Interactions of cyclophilin with the mitochondrial inner membrane and regulation of the permeability transition pore, a cyclosporin a-sensitive channel. J. Biol. Chem. 271, 2185–2192.
Ozcan, L., and Tabas, I. (2010). Pivotal role of calcium/calmodulin-dependent protein kinase II in ER stress-induced apoptosis. Cell Cycle 9, 223–224. doi: 10.4161/cc.9.2.10596
Pan, X., Liu, J., Nguyen, T., Liu, C., Sun, J., Teng, Y., et al. (2013). The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 15, 1464–1472. doi: 10.1038/ncb2868
Perocchi, F., Gohil, V. M., Girgis, H. S., Bao, X. R., McCombs, J. E., Palmer, A. E., et al. (2010). MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 467, 291–296. doi: 10.1038/nature09358
Piot, C., Croisille, P., Staat, P., Thibault, H., Rioufol, G., Mewton, N., et al. (2008). Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 359, 473–481. doi: 10.1056/NEJMoa071142
Purohit, A., Rokita, A. G., Guan, X., Chen, B., Koval, O. M., Voigt, N., et al. (2013). Oxidized ca2+/calmodulin-dependent protein kinase II triggers atrial fibrillation. Circulation 128, 1748–1757. doi: 10.1161/CIRCULATIONAHA.113.003313
Rose, A. J., Frøsig, C., Kiens, B., Wojtaszewski, F. P. Jr., and Richter, E. A. (2007). Effect of endurance exercise training on Ca2+ calmodulin-dependent protein kinase II expression and signalling in skeletal muscle of humans. J. Physiol. 583, 785–795. doi: 10.1113/jphysiol.2007.138529
Ryu, S.-Y., Beutner, G., Dirksen, R. T., Kinnally, K. W., and Sheu, S.-S. (2010). Mitochondrial ryanodine receptors and other mitochondrial Ca2+ permeable channels. FEBS Lett. 584, 1948–1955. doi: 10.1016/j.febslet.2010.01.032
Salas, M. A., Valverde, C. A., Sanchez, G., Said, M., Rodriguez, J. S., Portiansky, E. L., et al. (2009). The signalling pathway of CaMKII-mediated apoptosis and necrosis in the ischemia/reperfusion injury. J. Mol. Cell. Cardiol. 48, 1298–1306. doi: 10.1016/j.yjmcc.2009.12.015
Sancak, Y., Markhard, A. L., Kitami, T., Kovács-Bogdán, E., Kamer, K. J., Udeshi, N. D., et al. (2013). EMRE Is an essential component of the mitochondrial calcium uniporter complex. Science 342, 1379–1382. doi: 10.1126/science.1242993
Sepúlveda, M., Gonano, L. A., Back, T. G., Chen, S. R. W., and Vila Petroff, M. (2013). Role of CaMKII and ROS in rapid pacing-induced apoptosis. J. Mol. Cell. Cardiol. 63, 135–145. doi: 10.1016/j.yjmcc.2013.07.013
Sharma, V., Abraham, T., So, A., Allard, M., and McNeill, J. (2010). Functional effects of protein kinases and peroxynitrite on cardiac carnitine palmitoyltransferase-1 in isolated mitochondria. Mol. Cell. Biochem. 337, 223–237. doi: 10.1007/s11010-009-0303-2
Sugawara, K., Fujikawa, M., and Yoshida, M. (2013). Screening of protein kinase inhibitors and knockdown experiments identified four kinases that affect mitochondrial ATP synthesis activity. FEBS Lett. 587, 3843–3847. doi: 10.1016/j.febslet.2013.10.012
Timmins, J. M., Ozcan, L., Seimon, T. A., Li, G., Malagelada, C., Backs, J., et al. (2009). Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J. Clin. Invest. 119, 2925–2941. doi: 10.1172/JCI38857
Wagner, S., Dybkova, N., Rasenack, E. C., Jacobshagen, C., Fabritz, L., Kirchhof, P., et al. (2006). Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Invest. 116, 3127–3138. doi: 10.1172/JCI26620
Wei, A.-C., Liu, T., Winslow, R. L., and O'Rourke, B. (2012). Dynamics of matrix-free Ca2+ in cardiac mitochondria: two components of Ca2+ uptake and role of phosphate buffering. J. Gen. Physiol. 139, 465–478. doi: 10.1085/jgp.201210784
Witze, E. S., Old, W. M., Resing, K. A., and Ahn, N. G. (2007). Mapping protein post-translational modifications with mass spectrometry. Nat. Methods 4, 798–806. doi: 10.1038/nmeth1100
Yang, Y., Zhu, W.-Z., Joiner, M., Zhang, R., Oddis, C. V., Hou, Y., et al. (2006). Calmodulin kinase II inhibition protects against myocardial cell apoptosis in vivo. Am. J. Physiol. Heart Circ. Physiol. 291, H3065–H3075. doi: 10.1152/ajpheart.00353.2006
Yun, B., Lee, H., Ghosh, M., Cravatt, B. F., Hsu, K.-L., Bonventre, J. V., et al. (2013). Serine hydrolase inhibitors block necrotic cell death by preventing calcium overload of the mitochondria and permeability transition pore formation. J. Biol. Chem. 289, 1491–1504. doi: 10.1074/jbc.M113.497651
Zhang, T., Guo, T., Mishra, S., Dalton, N. D., Kranias, E. G., Peterson, K. L., et al. (2010). Phospholamban ablation rescues sarcoplasmic reticulum Ca2+ handling but exacerbates cardiac dysfunction in CaMKII{delta}C transgenic mice. Circ. Res. 106, 354–362. doi: 10.1161/CIRCRESAHA.109.207423
Zhao, X., León, I. R., Bak, S., Mogensen, M., Wrzesinski, K., Højlund, K., et al. (2011). Phosphoproteome analysis of functional mitochondria isolated from resting human muscle reveals extensive phosphorylation of inner membrane protein complexes and enzymes. Mol. Cell. Proteomics 10:M110.000299. doi: 10.1074/mcp.M110.000299
Keywords: mitochondria, cell death, mitochondrial calcium uniporter, CaMKII, CaMKIIN
Citation: Joiner MA and Koval OM (2014) CaMKII and stress mix it up in mitochondria. Front. Pharmacol. 5:67. doi: 10.3389/fphar.2014.00067
Received: 09 January 2014; Accepted: 24 March 2014;
Published online: 01 May 2014.
Edited by:
Andrew G. Edwards, Oslo University Hospital and Simula Research Laboratory, NorwayReviewed by:
David A. Brown, East Carolina University, USACopyright © 2014 Joiner and Koval. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mei-ling A. Joiner, University of Iowa, 258 Newton Rd., 2256 CBRB, Iowa City, IA 52242, USA e-mail:bWVpLWxpbmctam9pbmVyQHVpb3dhLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.