 Zvi Ioav Cabantchik
Zvi Ioav Cabantchik- Department of Biological Chemistry, Institute of Life Sciences, Hebrew University of Jerusalem, Jerusalem, Israel
In living systems iron appears predominantly associated with proteins, but can also be detected in forms referred as labile iron, which denotes the combined redox properties of iron and its amenability to exchange between ligands, including chelators. The labile cell iron (LCI) composition varies with metal concentration and substances with chelating groups but also with pH and the medium redox potential. Although physiologically in the lower μM range, LCI plays a key role in cell iron economy as cross-roads of metabolic pathways. LCI levels are continually regulated by an iron-responsive machinery that balances iron uptake versus deposition into ferritin. However, LCI rises aberrantly in some cell types due to faulty cell utilization pathways or infiltration by pathological iron forms that are found in hemosiderotic plasma. As LCI attains pathological levels, it can catalyze reactive O species (ROS) formation that, at particular threshold, can surpass cellular anti-oxidant capacities and seriously damage its constituents. While in normal plasma and interstitial fluids, virtually all iron is securely carried by circulating transferrin (Tf; that renders iron essentially non-labile), in systemic iron overload (IO), the total plasma iron binding capacity is often surpassed by a massive iron influx from hyperabsorptive gut or from erythrocyte overburdened spleen and/or liver. As plasma Tf approaches iron saturation, labile plasma iron (LPI) emerges in forms that can infiltrate cells by unregulated routes and raise LCI to toxic levels. Despite the limited knowledge available on LPI speciation in different types and degrees of IO, LPI measurements can be and are in fact used for identifying systemic IO and for initiating/adjusting chelation regimens to attain full-day LPI protection. A recent application of labile iron assay is the detection of labile components in intravenous iron formulations per se as well as in plasma (LPI) following parenteral iron administration.
Introduction
The various forms of iron present in biological fluids are largely determined by the chemical composition of the medium, particularly its reductive power (commonly dictated by the GSH-NADPH/NADH levels) and the repertoire of substances with metal complexing groups (e.g., carboxylates, phosphates, amides, thiolates, and hydroxylates). In extracellular fluids, essentially all the iron is safely carried by the protein transferrin (Tf) that shields the Fe(III) from the environment and renders it virtually redox-inactive as well as non-exchangeable with, or displaceable by, physiological substances or metals. Thus Tf-bound iron (TBI) is a tightly bound form of ferric iron that by definition is non-labile and it remains as such, until conformational changes triggered by binding of Tf to its cognitive Tf receptor (TfR), in conjunction with medium acidification, jointly lead to its release and ensuing reduction to ferrous iron (Crichton, 2001; Cairo and Recalcati, 2007). That complex series of reactions is mechanistically integrated in the physiological process of receptor mediated endocytosis (RME) of TBI that operates in all mammalian cells and serves as the key route of regulated iron uptake (Figure 1).
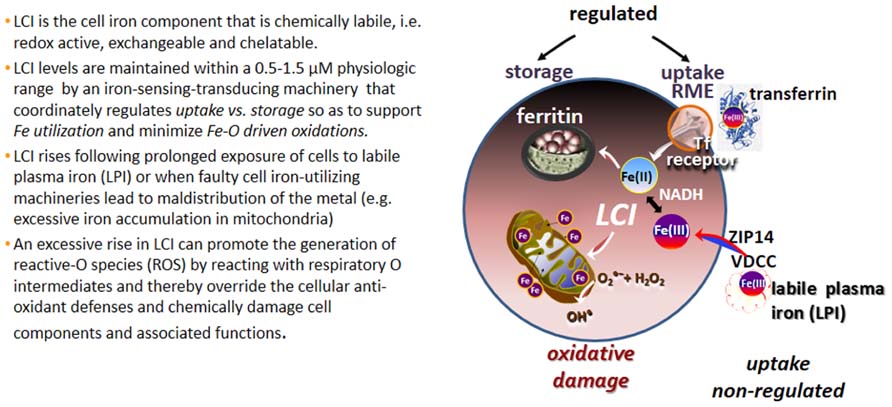
FIGURE 1. Labile cell iron (LCI) and labile plasma iron (LPI). Proposed routes of labile iron ingress into cells comprise voltage dependent Ca channels (VDCC; Oudit et al., 2006), the Zn transporter ZIP14 (Liuzzi et al., 2006) and others (Sohn et al., 2012).
In cellular compartments almost all (>95%) of the iron is also protein-bound, either directly to protein residues or via iron-containing prosthetic groups such as heme or iron sulfur clusters (ISC; Crichton, 2001). Although most of the iron-containing proteins are endowed with catalytic metal centers, for this exposé we consider the redox-active metal as stricto senso labile if it is also physiologically exchangeable and/or pharmacologically chelatable. This fits with the more traditional definition of labile cell iron (LCI) that was introduced in order to describe transitory (i.e., exchangeable) forms of cell iron that are important for cell iron metabolism and homeostasis (Jacobs, 1977; Crichton, 2001). We describe here LCI in physiological and pathophysiological states and its potential relevance to pharmacology and therapeutics. Methodological aspects related to the analysis of LCI and its counterpart in plasma, labile plasma iron (LPI), are the subject of other reviews (Esposito et al., 2002; Kakhlon and Cabantchik, 2002; Petrat et al., 2002; Kruszewski, 2003; Breuer et al., 2007; Brissot et al., 2012).
Labile Cell Iron
Many attempts made to chemically define LCI in extracts from lysed cells led biochemists to implicate various Fe3+ complexes as potential LCI candidates [e.g., nucleotides and even glutathione (GSH); Weaver and Pollack, 1989; Hider and Kong, 2011]. However, those implications should be interpreted with caution as changes in environments (including pH and redox potential) that result from cell de-compartmentalization are likely to result in metal redistribution among potential metal binders. Therefore, in this review we regard LCI as a dynamic parameter that is relevant only for living cells and that prevails under defined conditions, spatial, temporal and environmental (Cabantchik et al., 2009).
Physiology
The most compelling evidence that LCI prevails in physiological conditions in most mammalian cells, is the demonstrable ability of permeating iron chelators to inhibit the in situ (cell or organelle) catalytic contribution of labile iron to basal as well as peroxide-stimulated cell reactive O species (ROS) formation and to inhibit iron-dependent cell functions (Glickstein et al., 2005). LCI is per se a generic term used to describe labile iron in the cell as a whole, or in particular cell compartments. The cytosolic and organellar components of LCI are comprised of chelatable complexes of both Fe2+and Fe3+, whose relative proportions largely reflect (or are dictated by) the reductive properties and chemical composition of the compartment in question and those in turn depend on cell metabolism (Petrat et al., 2002; Breuer et al., 2007).
The cytosolic LCI, that is “strategically” located at the cross-roads of cell iron metabolism, serves both as metabolic source of metal but also as indicator of cell iron levels. Thus, cells sense and regulate LCI by balancing the uptake of circulating TBI with the storage of unutilized (surplus?) cell iron in shells of ferritin molecules (Figure 1). As expected for a dynamic cell parameter, the various LCI pools are likely to vary over time in response to chemical or biological stimuli as well as to metabolic demands/responses. For example, in human K562 or murine erythroleukemia cells, exposure to increasing TBI concentration results in a commensurate rise in cytosolic and mitochondrial LI indicating increased iron uptake by RME and ensuing release into cytosol (and almost concurrent delivery to mitochondria; Shvartsman et al., 2007, 2010; Levi and Rovidac, 2009). Similar results are obtained by modulation of cell ferritin expression (Picard et al., 1998; Kakhlon et al., 2001). Conversely, LCI levels fall in iron starvation, following stable or transient overexpression of cytosolic or mitochondrial ferritin as well as in some mitochondrial disorders that involve aberrant mitochondrial iron accumulation (Rouault, 2006, 2012; Levi and Rovidac, 2009).
The search for putative LCI components have led also to complementary searches for LCI counterparts, namely putative iron chaperons or carriers that, in analogy with copper chaperons, might mediate “safe” iron traffic within cells via formation of non-labile or occluded metal complexes. Accordingly, PBP proteins were implicated as facilitators of iron incorporation into ferritin (Shi et al., 2008) and 2,4 dihydroxybenzoate (2,4-DHB) as universal cytosol-mitochondrial iron chaperon (Devireddy et al., 2010). It must be stressed however, that: (a) neither the putative presence of nM concentrations of 2,4-DHB, seemingly detected in mammalian cells, nor (b) the almost undetectable chelating ability of 2,4-DHB per se in buffered medium, let alone (c) in a cellular milieu that is comprised of mM concentrations of organo-phosphates and -carboxylates, could possibly confer upon DHB a putative role as cell iron chaperon, carrier, or the like (Shvartsman and Cabantchik, 2012). For hemoglobin synthesizing reticulocytes, it was hypothesized that iron derived from uptake of TBI by RME is delivered to mitochondria by a mechanism referred as “kiss and run” (Zhang et al., 2005; Sheftel et al., 2007). In that mechanism, vesicle-mitochondria interactions were proposed to provide bridges or channels for trans-vesicular transfer of Fe2+ to cytosol.
In live-cell fluorescence studies done on human K562 erythroleukemia or murine erythroleukemia (MEL) cells, changes in cytosolic LCI levels occurred in response to increasing concentrations of TBI (but also non-TBI (NTBI) compounds), and almost in parallel to changes in mitochondrial LCI (Shvartsman et al., 2007, 2010). Those studies indicated the operation of various routes of cell iron traffic, most of which are manifested as transient changes in the levels of the cytosolic LCI pool. Observations describing a rapid delivery of iron from the point of entry into cells to mitochondria have also been observed in cells exposed to various NTBI substances (e.g., organic iron salts; Shvartsman et al., 2007). Moreover, in the intestinal CACO-2 cell model, iron trafficked intracellularly from mucosal to serosal cell phases appeared to be shielded in dimetal transporter 1 (DMT1)-containing endocytic vesicles (Núñez et al., 1994; Núñez et al., 1999; Linder et al., 2006). Hitherto, the demonstration of “cell iron passages” as “safe and efficient” routes of iron delivery mediated by cell chaperons or vesicles, awaits experimental support. However, their possible contribution to cell iron physiology does not exclude cellular iron traffic also (or primarily) via LCI pools, whose levels are dynamically monitored live with metal-sensing or redox-sensing probes in response to physiological challenges.
Pathology
It is generally accepted that a major and persistent rise in LCI levels can compromise cell integrity, since excessively accumulated labile iron is prone to engage in the catalytic generation of noxious ROS from reactive O intermediates (ROIs) and those can override the cell antioxidant defenses (Figure 1; Kruszewski, 2003). These reactions occur primarily in mitochondria, which not only tend to accumulate excessive LI in various disorders but also suffer the consequences of local ROS formation and ensuing damage. In the various types of systemic siderosis (primary or transfusional), the etiopathology of iron overload (IO) is classically associated with the ability of components of the hemosiderotic plasma to infiltrate cells and raise the LCI levels in both cytosol and mitochondria (Kakhlon and Cabantchik, 2002; Cabantchik et al., 2009). However, a unique feature in inherited mitochondrial disorders caused by faulty biosynthesis/assembly of heme or ISC is the under-utilization of iron that results in mitochondrial iron accumulation/deposition and ensuing cytosolic iron deprivation (Rouault, 2006, 2012; Breuer and Cabantchik, 2009; Camaschella, 2009; Li-Hsuan Huang et al., 2011). That scenario of iron maldistribution (that might be causatively inter-related) is promoted by a vicious circle of increased cell iron uptake induced by a reduction in cytosolic LCI (Li-Hsuan Huang et al., 2011). The latter in turn induces TfR expression via activation of the iron-regulatory IRP-IRE system (Cairo and Recalcati, 2007), leading to increased TfFe uptake and ensuing iron deposition in mitochondria. The resulting phenotypes of regional siderosis are demonstrated in sideroblastic anemia (SA) caused by the mutated gene alas2 (for XLSA), or glrx5 (for SA) or slc25a38 (for SA) but also in neuro-siderosis caused by the mutated gene fxn (for FRDA) or abcb7 (for x-linked SA with ataxia, xlsa/a; Rouault, 2006, 2012; Camaschella, 2009; Li-Hsuan Huang et al., 2011).
Pharmacology
To the extent that a persistently elevated LCI is a risk factor for cell survival (Li-Hsuan Huang et al., 2011; Fleming and Ponka, 2012), metal detoxification by chelation should be regarded as the most direct mode of pharmacological intervention (Hershko et al., 2005; Pietrangelo, 2007; Porter, 2009; Ma et al., 2012; Camaschella, 2013). However, such an approach should be endowed with specificity for labile forms of iron and designed with an adequate regimen so as not to generate long term metal deficiency, local or systemic. In principle, the major goal of chelation is the neutralization or attenuation of iron propensity for catalyzing radical formation. In order for a chelator to meet that goal in an iron overloaded tissue, it must be permeant to cells and endowed with an effective binding affinity for the labile metal (to ensure specificity) and a mode of coordinating complexation that should render the metal essentially non-labile. Those are exemplified by: (a) the tridentate chelator deferasirox (DFR) that binds Fe3+ with 2:1 stoichiometry and (b) the bidentate deferiprone (DFP) that binds it with 3:1 stoichiometry. As in systemic siderosis, the targets of chelation are both within cells and in extracellular fluids, the goal of chelation is twofold: (a) metal detoxification by means of reducing the iron burden in overloaded tissues and (b) prevention of tissue iron accumulation, by maintaining a low level of LPI, namely the labile components of plasma NTBI (Figure 2).
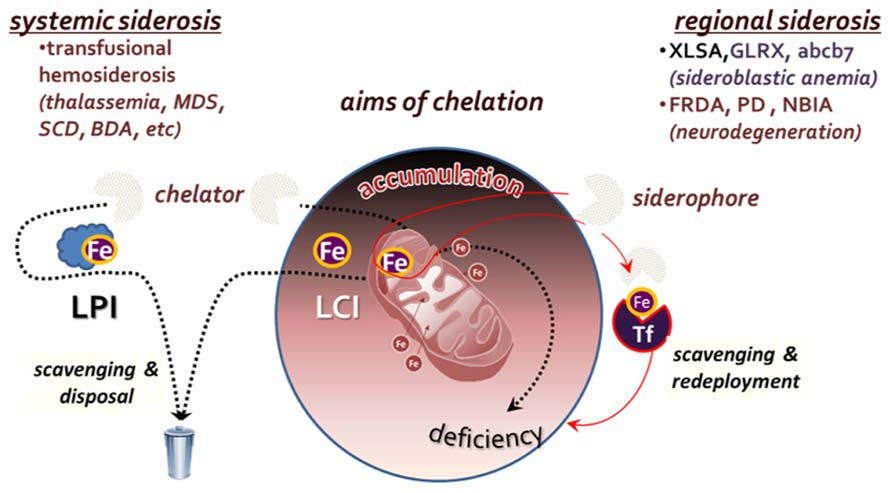
FIGURE 2. Aims of chelation therapy for systemic versus regional siderosis. In systemic siderosis, primarily transfusional hemosiderosis (thalassemia major, myelodysplastic syndrome-MDS, sickle cell disease-SCD or Blackfan Diamond anemia-BDA), the major aim is to reduce body iron burden by scavenging and disposing the iron that accumulated in tissues and fluids. In regional siderosis, iron accumulation in particular cell compartments is accompanied by a deficiency in another cell compartment (e.g., in various types of sideroblastic anemia or the neurodegenerative disorders Friedreich ataxia-FRDA), or the maldistribution of iron is between different organs. For this cases, chelation is designed to redistribute the accumulated iron within the same cells by direct scavenging and redeployment or via transfer to extracellular transferrin (Tf) and recapture by receptor-mediated endocytosis-RME.
Unlike in systemic siderosis, in regional siderosis, iron is generally maldistributed among cells or cell organelles, often accumulating in some at the expense of iron-sufficient ones. Scavenging of accumulated iron by chelation might accomplish regional detoxification but also concurrently generate deprivation in iron-sufficient or iron-deficient regions (e.g., tissues, cells, or organelles; Breuer and Cabantchik, 2009). Thus, for regional siderosis, treatment should not be limited to scavenging of surplus iron but should be followed by its redeployment, either within or across cells, as depicted in Figure 2. Such mode of metal redistribution can be accomplished by chelators with siderophore properties, namely membrane permeant chelators that have a combined accessibility and iron affinity for scavenging LCI in cell and organelles but also the ability to transfer the chelated metal to cell acceptors or the plasma iron acceptor Tf (Kakhlon and Cabantchik, 2002; Breuer and Cabantchik, 2009). A paradigm for that modus operandi is given by DFP, that in addition of scavenging and redeploying iron between cells and extracellular Tf, it has also the ability to correct iron maldistribution per se (Sohn et al., 2008, 2011; Breuer and Cabantchik, 2009; Kakhlon et al., 2010; e.g., between mitochondria and cytosol, as found in cells rendered frataxin deficient by shRNA suppression technology) as well as most of the affected cell properties (Kakhlon et al., 2008, 2010).
Determination of LCI in Living Cells
The determination of LCI in living cells has been based on spectroscopic probes (herewith referred as fluorescence metal sensors FMS) that detect labile iron in situ and in real time by one of the following mechanisms (Petrat et al., 2002; Glickstein et al., 2006; Ma et al., 2006; Breuer et al., 2007): (a) reversible quenching of fluorescence upon binding of iron to a probe endowed with a fluorescent-tagged chelating unit or (b) generation of fluorescence by metal-catalyzed oxidation of a fluorogenic probe (Glickstein et al., 2006; Ma et al., 2006). Changes in fluorescence signal are attributed to labile iron if they are prevented or reversed by a strong and permeant iron chelator that can swiftly gain access to cell compartments and scavenge LCI as well as FMS-chelated iron (!). FMSs like calcein blue (CALB as shown in Figure 3) or calcein green (CALG as shown in Figure 4) are comprised of one or two metal chelating arms (aminodiacetate or iminodiacetate) linked to fluorescent probes (e.g., fluorescein or methylumbelliferone) that undergo signal quenching upon binding of transition metals like Fe or Cu. The probes can be easily loaded into cells via membrane permeant precursors that are non-fluorescent and non-chelating, e.g., CALG-AM or CALB-AM; AM-acetomethoxy. These probes are hydrolyzed intracellularly, releasing the fluorescent, impermeant chelating CALG (binds iron 1:1) or CALB (binds iron 2:1) that interact (reversibly) with resident labile iron and undergo quenching commensurate with LCI concentrations. That mode of iron sensing is depicted schematically at the bottom of Figures 3 and 4 for CALB and CALG, respectively, whereby iron added to the free CALB or CALG quenches the FMS and addition of a relatively high concentration of a strong iron chelator restores the quenched fluorescence. In CALB-laden human hepatic HepG2 cells (Figure 3), the FMS fills the cytosolic space and binds a fraction of LCI that can be revealed upon addition of excess permeant chelator. Thus addition of DFR elicits a rise in fluorescence ΔF that corresponds to Fe released form the quenched CALB-Fe the chelator (ΔF can be converted to actual concentration of LCI with the aid of appropriate calibration curves as demonstrated earlier for CALG (Epsztejn et al., 1997; Cabantchik et al., 2002; Petrat et al., 2002; Breuer et al., 2007).
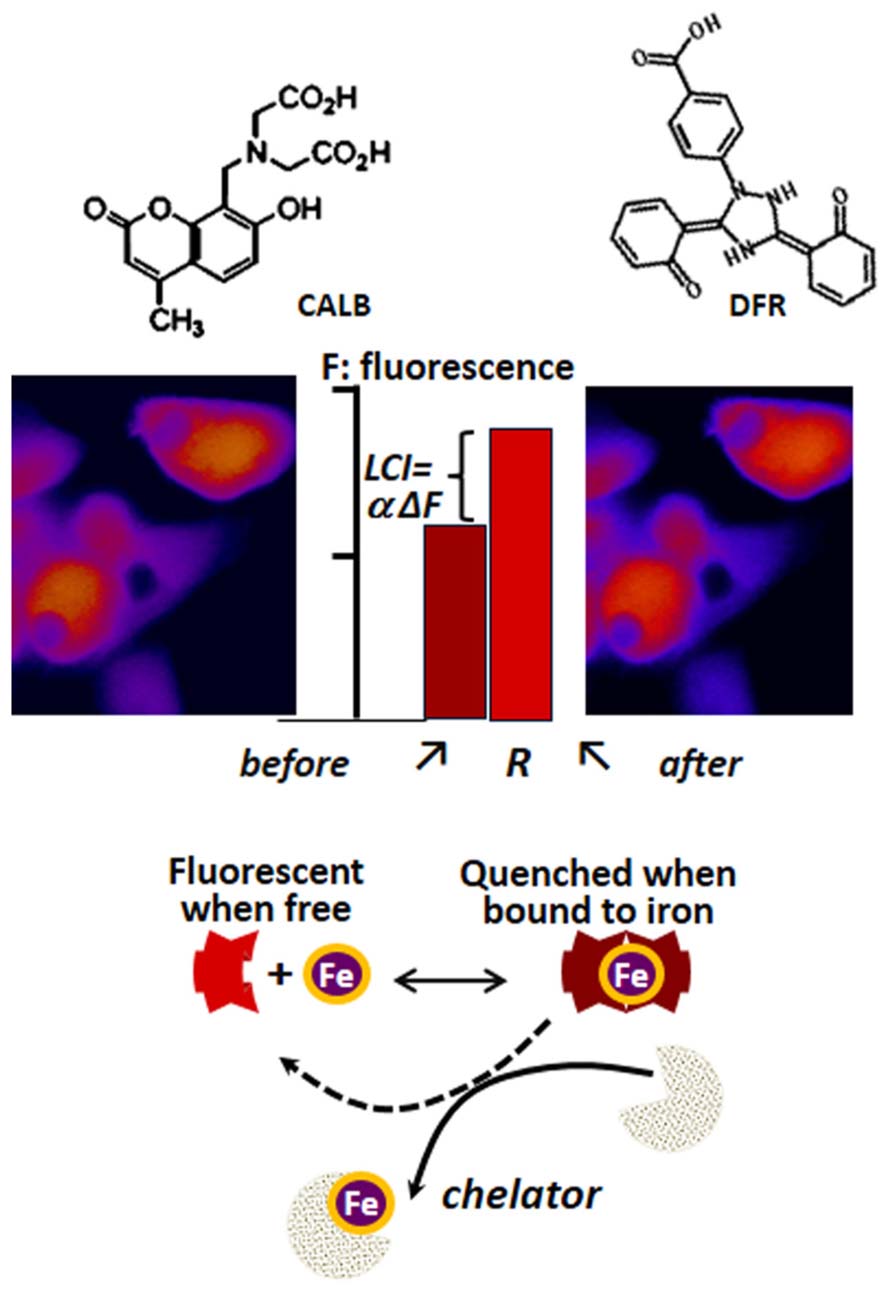
FIGURE 3. Principle of labile iron detection with fluorescent metal sensors (FMS) using fluorescence microscopy imaging. Calcein blue (CALB) as FMS paradigm. The blue fluorescent umbelliferyl iminodiacetic acid binds Fe(III) with a 2:1 or 3:1 stoichiometry and undergoes fluorescence (F) quenching which can be reversed by addition of excess amounts of the chelator deferasirox (DFR). That process, schematically depicted in the lower part of the figure, can be applied to cells (e.g., human hepatic hepG2 cells) by loading them with the acetomethoxy ester (CALB-AM) precursor that generates the fluorescent CALB in the cytosol (fluorescence microscopy imaging, pseudcolor: blue represents relatively low F signal and red high F signal). The addition of an excess amount of a strong permeant chelator (e.g., deferasirox, DFR) leads to a rise in intracellular fluorescence ΔF that denotes the amount of CALB-Fe generated by binding of CALB to LCI. The observed ΔF, which is proportional to LCI, can be converted into actual concentration of iron (Epsztejn et al., 1997; Esposito et al., 2002) that represents the cytosolic LCI pool.
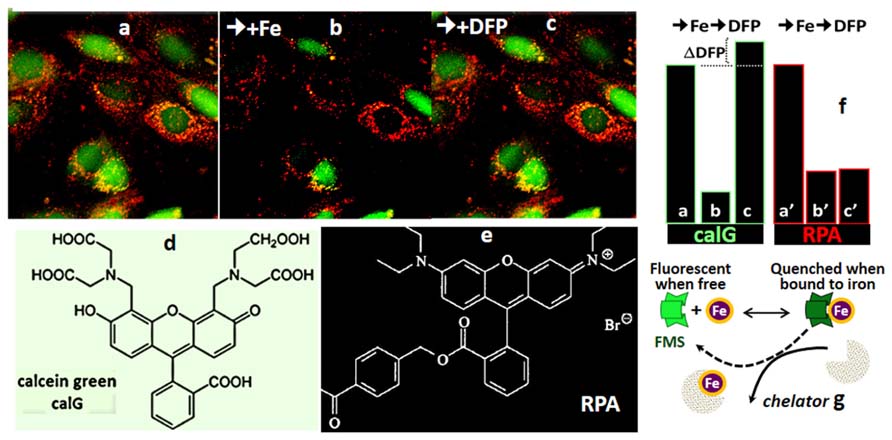
FIGURE 4. Calcein green (CALG) and RPA as FMS for simultaneous assessment of Fe ingress into cytosol and mitochondria (fluorescence microscopy imaging). The indicated fluorescent FMSs CALG (d) (loaded into the cell cytosol via the CALG-AM precursor) and red RPA (f) (loaded into cell mitochondria by potentiometric distribution) (a) were monitored with time by fluorescence microscopy imaging. Following addition of 5 μM ferric-hydroxyquinoline (1:1) for 10 min (b) the fluorescence intensity (in relative units) in both cytosol and mitochondria dropped substantially (from a to b and a′ to b′ for CALG and RPA, respectively). The reduction in the fluorescence signal elicited by the added Fe provides a measure for the amount of labile metal that gained access to the cytosol in 10 min, until Fe ingress was stopped by addition of 20 μM of the impermeant chelator DTPA-diethylenetriamine pentaacetic acid (to chelate all extracellular Fe). Subsequent addition of 50 μM of the permeant chelator DFP, led to the recovery CALG fluorescence quenched by the added Fe as well the original LCI, denoted by ΔDFP. On the other hand, the quenched RPA signal was not recovered by addition of DFP, due to the relatively poor reversibility of phenanthroline-complexed metal in physiological conditions. g depicts the principle of CALG use as FMS.
The principle of LCI measurement demonstrated with the FMS CALB can also be used to assess dynamic changes in various LCI pools with organelle targeted FMS (Glickstein et al., 2005; Shvartsman et al., 2007, 2010; Sohn et al., 2008; Porter, 2009; Cabantchik et al., 2013). Figure 4 depicts h9c2 cardiac cells double labeled with CALG (cytosol and nucleus) and RPA, a red rhodamine-phenanthroline iron-sensitive probe that targets potentiometrically (binds iron 3:1) to mitochondria. Addition of a permeant iron source reduces the fluorescent intensity of both CALG and RPA (indicating ingress of labile iron to the respective compartment) and subsequent addition of the permeant chelator DFP restores the quenched fluorescence signal of CALG, but not that of RPA (that binds iron tightly and demands extreme conditions for scavenging the bound metal from the phenanthroline:metal 3:1 complex). Parallel time-dependent changes in cytosolic and mitochondrial LCI compartments can be monitored by flow cytometry or fluorescence microscopy as means to assess iron transport into the respective compartments in response to addition of TBI or NTBI (Shvartsman et al., 2007, 2010). As demonstrated in Figure 5 for TBI, the relative time-dependent changes in fluorescence intensity obtained for either CALG or RPA by flow cytometry match those obtained independently by fluorescence microscopy imaging. Figure 5 depicts both the resident cytosolic LCI prior to and after addition of TBI (labeled LCI and LCI+, respectively) that are revealed as changes in CALG fluorescence after addition of the permeant tridentate chelator (salicylaldehyde isonicotinoyl hydrazone SIH). When time-dependent profiles are done on probe labeled cells exposed to different substrate concentrations, it is also possible to obtain the kinetic parameters of iron ingress into the respective LCI pools (cytosolic and mitochondrial), by converting ΔF to LCI concentrations, as described elsewhere (Shvartsman et al., 2007, 2010).
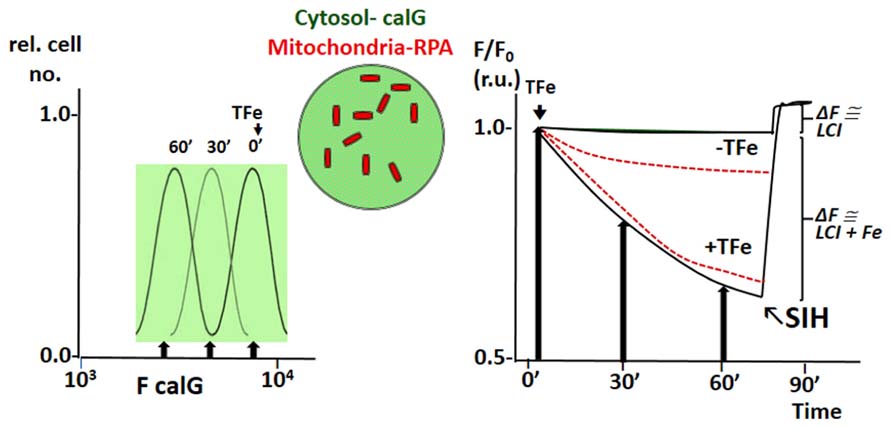
FIGURE 5. Example of simultaneous tracing of Fe ingress (from TfFe) into cytosol and mitochondria of human K562 cells by flow cytometry (FC) and front-face fluorescence (plate reader; modified from references Shvartsman et al., 2010; Shvartsman and Cabantchik, 2012). The fluorescent FMSs calcein green (CALG) and RPA are loaded into K562 cells as described in previous figures, the first in the cytosol and the second in mitochondria (depicted schematically in the center of the figure). Fluorescence intensity (F) is followed with time at different time points by FC or continuously by spectrofluorimetry in a fluorescence plate reader using wavelength settings for fluorescein (CALG) and rhodamine (RPA). The left panel depicts schematically the FC profiles of cell samples taken at different times following addition TfFe (only F for green fluorescent calG is shown; arrow indicated median F values). The right panel shows values of F at different time points (following addition of TfFe or none, as indicated) normalized for values of F at t time 0 (F/F0) for both CALG (black lines) and RPA (red broken lines) as obtained in a fluorescence plate reader. The arrows indicate the normalized (to time 0) median fluorescence values obtained in the left panel (indicated by arrows). The relatively flat lines represent the mean values in cells not exposed to Tf, to indicate the extent of probe leakage during the measurements. In order to assess the basal LCI level (no TfFe added) and that raised by Fe (from TfFe) after 75′ exposure to TfFe, we obtain a measure of CALG-Fe both in basal conditions (no TfFe added) and at a given time point following addition of TfFe (75′). The measure of CALG-Fe is obtained by addition of a strong permeant iron chelator SIH (salicyl-isonicotinoyl-hydrazone) that scavenges Fe from CALF-Fe raising CALG fluorescence intensity commensurately. The ΔF obtained by addition of SIH provides a measure of LCI.
Various types of FMS have been applied to cells as sensors for monitoring changes in LCI by following stoichiometry quenching of fluorescence (Rauen et al., 2007; Prus and Fibach, 2008a, b; Ma et al., 2012). Among those are probes based on phenanthroline, deferrioxamine, or hydroxypyridinones chelating moieties and others originally designed for monitoring intracellular Ca. The major shortcoming of most those probes is the difficulty in reversing the quenched signal in physiological conditions and thereby the difficulty in unequivocally assigning the quenched signal to in situ LCI.
Targeted FMSs have also been used to demonstrate intracellular redistribution of iron (scavengery and redeployment) by addition of the chelator siderophore DFP in a cell model of iron maldistribution (Kakhlon et al., 2008; Sohn et al., 2008, 2011). As depicted in Figure 6, h9c2 cells, were pre-labeled with the FMSs RPA (red, for mitochondria) and the fluorescence quenched histone-CALG-Fe (green, for the nucleus) and incubated with the bidentate chelator DFP. As schematized in the lower panel, an increase in nuclear fluorescence elicited by added DFP denotes iron scavengery from the CALG-Fe moiety, whereas a decrease in RPA fluorescence denoted DFP-mediated redeployment of Fe to mitochondria. As some DFP-Fe might also egress from cells into a medium containing apo-Tf, iron can redeployed to Tf, that in turn can also deliver the metal to other cells. This is depicted in the right panel, whereby the medium from the h9c2 cells treated with DFP offered to iron-deficient erythroid MEL cells supported hemoglobin Hb synthesis.
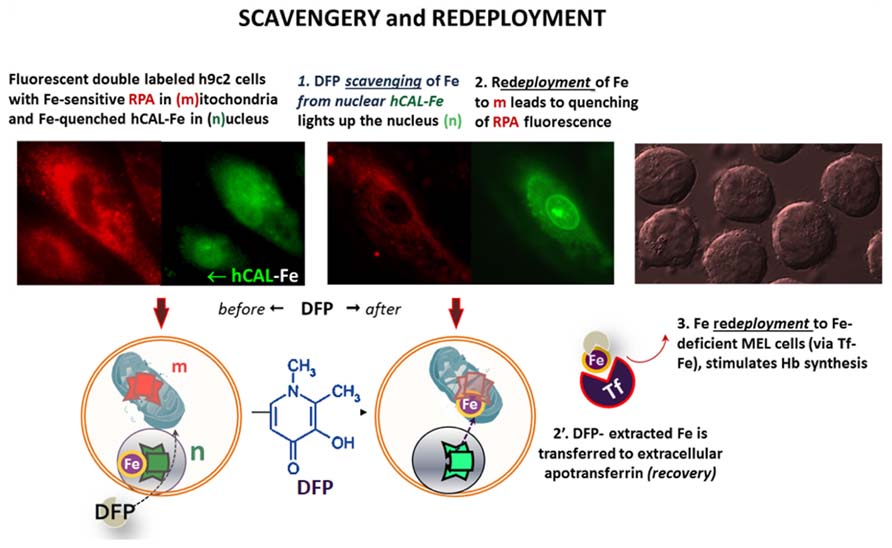
FIGURE 6. Intracellular and transcellular redistribution of iron. The proof of principle of intracellular scavengery and redeployment is depicted schematically in the lower part of the figure, whereas the upper part depicts the experimental results as visualized by fluorescence microscopy imaging. Murine h9c2 cardiomyocytes were double labeled with fluorescent metal sensors: red RPA in mitochondria and green histone-CALG precomplexed to Fe (that partially quenches the probe fluorescence) in the nucleus. Addition of the chelator-siderophore deferiprone (DFP), leads to (1) the scavenging of the Fe from hCAL-Fe, manifested as an increase in nuclear fluorescence and (2) a concomitant quenching of mitochondrial-RPA red fluorescence, that denotes the redeployment of Fe from the intracellular generated DFP-Fe chelate. In addition, DFP-Fe chelates that egress from cells (2′) can transfer the Fe to extracellular apotransferrin (Tf) and the latter (3) in turn, can furnish the metal to Fe-deficient murine erythroleukemia cells (MEL) and thereby support hemoglobinization. All the described steps were experimentally demonstrated in references (Sohn et al., 2008, 2011).
An additional mode of monitoring LCI in living cells is by assessing the ability of labile iron to catalyze oxidation of fluorogenic analogs of dihydrofluorescein or dihydrorhodamine loaded into cells. In the presence of ROIs (spontaneously generated in the cell or prompted by addition of oxidants like H2O2 or other peroxides) the labile metal converts in situ the non-fluorescent dihydro-probes into the respective fluorescent probes. As with the other FMS, the contribution of LCI to the time-dependent generation of fluorescence is assessed by the degree of inhibition attained by addition of a permeant chelator (Glickstein et al., 2006; Ma et al., 2006).
LPI and NTBI
Pathology and Pharmacology
In systemic IO, plasma is the compartment that harbors the pathophysiological source of uncontrolled iron ingress into cells and ensuing tissue IO (Hershko et al., 1978, 2005; Graham et al., 1979; Pietrangelo, 2007). The most implicated component of that source is LPI (Graham et al., 1979), the plasma counterpart of LCI that represents the labile fraction of plasma NTBI (Breuer et al., 2000; Esposito et al., 2003). As LPI comprises the redox-active and exchangeable forms of iron in native plasma that are also direct pharmacological targets of chelation (Pootrakul et al., 2004), it represents a parameter with potential diagnostic as well as therapeutic (i.e., theragnostic) value (Cabantchik et al., 2013). The generic term NTBI was originally introduced to denote the non-physiological, low molecular weight forms of iron that is not tightly associated with Tf and appears in plasma of IO patients (Hershko et al., 1978; Graham et al., 1979). Those forms were postulated on the basis that iron levels in plasma of IO patients often supersede total iron binding capacity (TIBC). Since a substantial fraction of NTBI is apparently adsorbed to plasma proteins, its chemical detection has posed technical difficulties in native plasma/serum unless pretreated with metal mobilizing agents like the polycarboxylate nitrilotriacetate (NTA) or diethylenetriamine pentaacetic acid (DTPA; Hershko et al., 1978; Graham et al., 1979; Singh et al., 1990; Gosriwatana et al., 1999; Kolb et al., 2009). Although several tests have been performed to ascertain that the applied extraction measures do not generally lead to some mobilization of Tf iron, those claims have been challenged leading to the application of more gentle mobilization conditions (Kolb et al., 2009). However, in general, most mobilizing maneuvers applied to plasma/sera that contains any of the chelators used in clinical practice, can lead to mobilization from highly saturated Tf (Kolb et al., 2009). A more serious difficulty pertains to the use of the generic term NTBI to denote plasma iron forms that are unique to IO and presumed to be potentially toxic. Defining “something that is by what it is not” (classically known as an apophasis, from Greek άπóφασις), can be confusing and in practice oxymoronic. For example, plasma that does not contain pathological species contains in fact NTBI in the form of ferritin or iron-chelates generated during iron chelation therapy or polymeric iron that is given supplementary via parenteral routes and detected in plasma by NTBI assays. On the other hand LPI is a parameter that denotes, operationally, the level of labile iron species in native plasma/serum irrespective of treatment or medium composition, an asset that can also become a liability if plasma/serum is not properly stored or some components of serum that affect its redox properties undergo extreme modifications (Breuer et al., 2012).
Attempts were done to identify the membrane transport mechanisms involved in NTBI uptake into cells that cause IO and oxidative damage. However, few studies took into consideration : (a) the actual repertoire of iron permeant species (i.e., the iron complexes that comprise the iron overloaded plasma containing NTBI) and their relative concentrations at different degrees of IO, (b) the composition of the medium milieu (e.g., the presence of high protein that limits substrate availability and redox-active substances that affect substrate composition) and (c) the different transport agencies present in different cell types. Those agencies comprise a variety of transporters or carriers of metals or metal-complexes, ionic channels or even the means for non-specific adsorptive endocytosis. As most NTBI transport studies have only been done with model simulating substrates in protein-free media and with model cells, their relevance to the etiopathology of tissue iron accumulation in systemic IO remains to be experimentally demonstrated (Figure 1). Recent studies indicated dimeric and oligomeric iron-citrate complexes as potential NTBI candidates (Evans et al., 2008), although the chemical speciation is likely to vary with the level of IO and the natural history of the disease in a given patient (Silva and Hider, 2009). Likewise, NTBI association with plasma proteins is exacerbated in oxidative conditions, such as those that prevail under inflammation or in chronic diabetes (Arezes et al., 2013). A single study with T lymphocytes demonstrated that oligomeric ferric citrate is taken up by T lymphocytes (Arezes et al., 2013) and another study indicated that a substantial fraction of iron that accumulated from exposure of cells to NTBI-containing human plasma was by adsorptive endocytosis (Sohn et al., 2012).
Determination of LPI/plasma NTBI
The measurement of a few μM of plasma NTBI in the presence of 70 to >100% saturated TBI (30–50 μM) poses some methodological problems. Two different approaches have tried to overcome them (schematically depicted in Figure 7): (1) by extraction of plasma NTBI with iron mobilizing/extraction/chelating agents like NTA or DTPA followed by size filtration (to separate NTBI from TBI) and detection of the iron by iron complex formation and direct measurement of colored complexes or following HPLC analysis (Singh et al., 1990; Gosriwatana et al., 1999; Kolb et al., 2009) and (2) by the detection of LPI in native plasma/serum by: (a) prompting resident LPI (with physiological concentrations of ascorbate) to either catalytically convert the non-fluorescent probe dihydrorhodamine (DHR) into the fluorescent rhodamine (R; Esposito et al., 2003) or co-catalyze with bleomycin chemical changes in chromophoric substrates (Halliwell et al., 1988; Evans and Halliwell, 1994) or (b) binding the labile iron with a fluorescent-chelator that undergoes a commensurate quenching of fluorescence, as exemplified in Figure 7 with fluoresceinated DFO (FDFO; Breuer et al., 2001). For either modality (a or b) of method 2, a specific iron chelator (DFO or DFP in excess) is added to a parallel sample in order to assess the specific contribution of labile iron to the observed change in fluorescence elicited in the test sample.
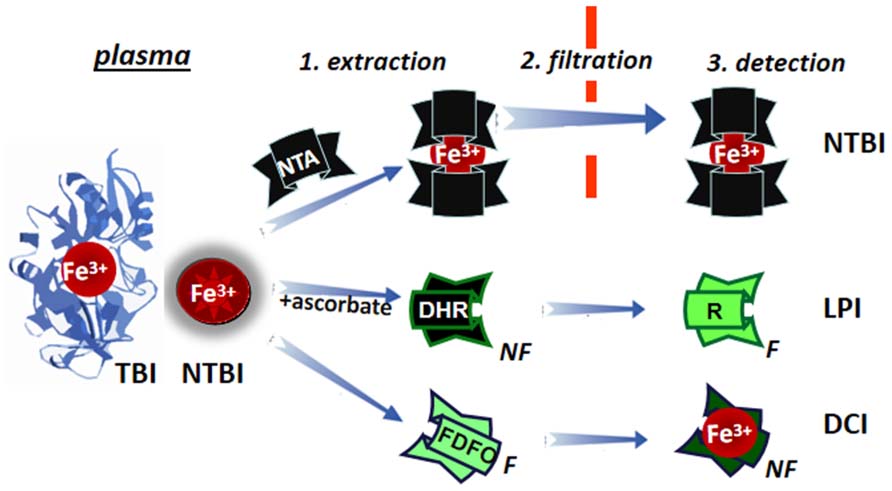
FIGURE 7. Principles of determination of LPI and of plasma NTBI. Plasma/serum non-TBI (transferrin bound iron) indicated as NTBI is measured by the three indicated methodologies. NTBI is measured by its extraction (1) from serum with relatively high (10–80 mM) concentrations of nitrilotriacetic acid (NTA), size filtration (2) and iron detection by colorimetric methods or by addition of iron complexing agents and analysis by HPLC. The labile plasma iron (LPI) component of NTBI is analyzed by exposing native plasma or serum to physiological concentration of ascorbate in the presence of the fluorogenic dihydrorhodamine (DHR), whose time-dependent conversion from non-fluorescent (NF) to green (F) fluorescent 1,2,3, rhodamine (R) is followed in a plate reader (or in a fluorimeter) and the fraction of the signal change inhibited by deferrioxamine (or another strong and specific iron chelator) is attributed to labile iron. Performance of LPI measurements in the presence of 0.1–1.0 mM NTA, provides an equivalent estimate of NTBI (Breuer et al., 2012). The directly chelatable iron (DCI) component of NTBI is assessed with a strong fluorescent (F) chelator such as fluoresceinated-deferrioxamine (FDFO) that upon binding of Fe(III) undergoes stoichiometric quenching to non-fluorescent (NF) chelate FDFO-Fe.
The LPI/NTBI assays have been used extensively to monitor chelation efficacy in transfusional siderotic patients treated with different chelators (Cabantchik et al., 2005; Daar et al., 2009; Zanninelli et al., 2009; Greenberg et al., 2010; Danjou et al., 2014) or hemochromatosis patients following phleboptomy (Le Lan et al., 2005), particularly for establishing: (a) the attainment of sufficiently low LPI (<0.1 μM) at trough levels of chelator as a goal of a particular therapeutic regimen (Cabantchik et al., 2005; Zanninelli et al., 2009) an the appearance of LPI as a criterion for initiating chelation therapy in polytransfused children (Danjou et al., 2014). An attempt has also been done to compare different assays for assessing NTBI (not LPI; Jacobs et al., 2005) but a newer completed one that included also LPI should provide more comprehensive and updated information regarding the usefulness of the LPI/NTBI assays in the clinical setting.
Conclusion and Perspectives
With presently available methodologies based on FMS, it has become possible to obtain real time measures of LCI and their compartments in both physiological and pathological conditions (i.e., systemic and regional siderosis) and of LPI/NTBI in body fluids as early marker of systemic IO and treatment efficacy. Of particular importance is the ability to assess biological responses to pharmacological measures (e.g., intravenous iron supplementation or iron chelation) and correlate them with clinical outcomes. That area still needs further strengthening, particularly in establishing the thresholds of changes in LPI and LCI that define a given pathological state, either overt or impending.
Unfortunately, hitherto, it has been difficult to define the chemical components of LCI and LPI in biological systems, largely because of their heterogeneous character and variable nature of the media they dwell in. The same pertains to hypothesized iron chaperons that might be operative in mammalian cells and play some physiological role in intracellular iron traffic and metabolism. Although LCI is the parameter closely associated with tissue iron accumulation and ensuing iron-dependent damage, from the clinical perspective, only LPI has thus far proven diagnostically therapeutically relevant for assessing impending/emerging IO and therapeutically, for initiating chelation and monitoring its short term and long term efficacy.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Zvi Ioav Cabantchik was supported by the A&M Della Pergola Chair in Life Sciences, served on the Scientific Advisory Board of Novartis ESB, received in the past research grants from NIH, EEC (5 and 6 framework), Israel Science Foundation, AFIRNE (Paris), Novartis, Shire and Apopharma (also lecture honoraria from the last two) and is presently a consultant for Aferrix Ltd and Hinoman Ltd, Israel.
References
Arezes, J., Costa, M., Vieira, I., Dias, V., Kong, X. L., Fernandes, R., et al. (2013). Non-transferrin-bound iron (NTBI) uptake by T lymphocytes: evidence for the selective acquisition of oligomeric ferric citrate species. PLoS ONE 8:e79870. doi: 10.1371/journal.pone.0079870
Breuer, W., and Cabantchik, Z. I. (2009). Disorders affecting iron distribution: causes, consequences and possible treatments. Available at: http://www.bloodmed.com/800000/mini-reviews1.asp?id=253&p=1&v=1 (accessed January 6, 2009).
Breuer, W., Ermers, M. J., Pootrakul, P., Abramov, A., Hershko, C., and Cabantchik, Z. I. (2001). Deferrioxamine-chelatable iron a component of serum non-transferrin-bound iron used for assessing chelation therapy. Blood 97, 792–798. doi: 10.1182/blood.V97.3.792
Breuer, W., Ghoti, H., Shattat, A., Goldfarb, A., Koren, A., Levin, C., et al. (2012). Non-transferrin bound iron in Thalassemia: differential detection of redox active forms in children and older patients. Am. J. Hematol. 87, 55–61. doi: 10.1002/ajh.22203
Breuer, W., Hershko, C., and Cabantchik, Z. I. (2000). The importance of non-transferrin bound iron in disorders of iron metabolism. Transfus. Sci. 23, 185–191. doi: 10.1016/S0955-3886(00)00087-4
Breuer, W., Shvartsman, M., and Cabantchik, Z. I. (2007). Intracellular labile iron. a review. Int. J. Biochem. Cell Biol. 40, 350–354. doi: 10.1016/j.biocel.2007.03.010
Brissot, P., Ropert, M., Le Lan, C., and Loreal, O. (2012). Non-transferrin bound iron: a key role in iron overload and iron toxicity. Biochim. Biophys. Acta 1820, 403–410. doi: 10.1016/j.bbagen.2011.07.014
Cabantchik, Z. I., Breuer, W., Zanninelli, G., and Cianciulli, P. (2005). LPI-labile plasma iron in iron overload. Best Pract. Res. Clin. Haematol. 18, 277–287. doi: 10.1016/j.beha.2004.10.003
Cabantchik, Z. I., Fibach, E., and Breuer, W. (2009). Can Labile Plasma Iron (LPI) and Labile Cell Iron (LCI) Levels Serve as Early Indicators of Chelation Efficacy in Iron Overload? Available at: http://www.bloodmed.com/800000/mini-reviews1.asp?id=254 (accessed July 14, 2009).
Cabantchik, Z. I., Kakhlon, O., Epsztejn, S., Zanninelli, G., and Breuer, W. (2002). Intracellular and extracellular labile iron pools. Adv. Exp. Med. Biol. 509, 55–75. doi: 10.1007/978-1-4615-0593-8_4
Cabantchik, Z. I., Sohn, Y. S., Breuer, W., and Espósito, B. P. (2013). The Molecular and Cellular Basis of Iron Toxicity in Iron Overload Disorders. Diagnostic and Therapeutic Approaches. Thalassemia Reports, 3, 7–13.
Cairo, G., and Recalcati, S. (2007). Iron-regulatory proteins: molecular biology and patho-physiological implications. Expert Rev. Mol. Med. 9, 1–13. doi: 10.1017/S1462399407000531
Camaschella, C. (2009). Hereditary sideroblastic anemias: pathophysiology, diagnosis, and treatment. Semin. Hematol. 46, 371–377. doi: 10.1053/j.seminhematol.2009.07.001
Camaschella, C. (2013). Treating iron overload. N. Engl. J. Med. 368, 2325–2327. doi: 10.1056/NEJMcibr1304338
Crichton, R. (2001). Inorganic Biochemistry of Iron Metabolism From Molecular Mechanisms to Clinical Consequences, 2nd Edn. London: Wiley.
Daar, S., Pathare, A., Nick, H., Kriemler-Krahn, U., Hmissi, A., Habr, D., et al. (2009). Reduction in labile plasma iron during treatment with deferasirox a once-daily oral iron chelator in heavily iron-overloaded patients with beta-thalassaemia. Eur. J. Haematol. 82, 454–457. doi: 10.1111/j.1600-0609.2008.01204.x
Danjou, F., Cabantchik, Z. I., Origa, R., Moi, P., Marcias, M., Barella, S., et al. (2014). A decisional algorithm to start iron chelation in patients with beta thalassemia. Haematologica 99, e38–e40. doi: 10.3324/haematol.2013.098202
Devireddy, L. R., Hart, D. O., Goetz, D. H., and Green, M. R. (2010). A mammalian siderophore synthesized by an enzyme with a bacterial homolog involved in enterobactin production. Cell 141, 1006–1017. doi: 10.1016/j.cell.2010.04.040
Epsztejn, S., Kakhlon, O., Breuer, W., Glickstein, H., and Cabantchik, Z. I. (1997). A fluorescence assay for the labile iron pool (LIP) of mammalian cells. Anal. Biochem. 248, 31–40. doi: 10.1006/abio.1997.2126
Esposito, B. P., Breuer, W., Sirankapracha, P., Pootrakul, P., Hershko, C., and Cabantchik, Z. I. (2003). Labile plasma iron in iron overload: redox activity and susceptibility to chelation. Blood 102, 2670–2677. doi: 10.1182/blood-2003-03-0807
Esposito, B. P., Epsztejn, S., Breuer, W., and Cabantchik, Z. I. (2002). A review of fluorescence methods for assessing labile iron in cells and biological fluids. Anal. Biochem. 304, 1–18. doi: 10.1006/abio.2002.5611
Evans, P. J., and Halliwell, B. (1994). Measurement of iron and copper in biological systems: bleomycin and copper-phenanthroline assays. Methods Enzymol. 233, 82–89. doi: 10.1016/S0076-6879(94)33010-7
Evans, R. W., Rafique, R., Zarea, A., Rapisarda, C., Cammack, R., Evans, P. J., et al. (2008). Nature of non-transferrin-bound iron: studies on iron citrate complexes and thalassemic sera. J. Biol. Inorg. Chem. 13, 57–74. doi: 10.1007/s00775-007-0297-8
Fleming, R. E., and Ponka, P. (2012). Iron overload in human disease. N. Engl. J. Med. 366, 348–359. doi: 10.1056/NEJMra1004967
Glickstein, H., Ben-El, R. B., Link, G., Breuer, W., Konijn, A. M., Hershko, C., et al. (2006). Action of chelators in iron-loaded cardiac cells: accessibility to intracellular labile iron and functional consequences. Blood 108, 3195–3203. doi: 10.1182/blood-2006-05-020867
Glickstein, H., El, R. B., Shvartsman, M., and Cabantchik, Z. I. (2005). Intracellular labile iron pools as direct targets of iron chelators: a fluorescence study of chelator action in living cells. Blood 106, 3242–3250. doi: 10.1182/blood-2005-02-0460
Gosriwatana, I., Loreal, O., Lu, S., Brissot, P., Porter, J., and Hider, R. C. (1999). Quantification of non-transferrin-bound iron in the presence of unsaturated transferrin. Anal. Biochem. 273, 212–220. doi: 10.1006/abio.1999.4216
Graham, G., Bates, G. W., Rachmilewitz, E. A., and Hershko, C. (1979). Nonspecific serum iron in thalassemia: quantitation and chemical reactivity. Am. J. Hematol. 6, 207–217. doi: 10.1002/ajh.2830060305
Greenberg, P. L., Koller, C. A., Cabantchik, Z. I., Warsi, G., Glynos, T., Paley, C., et al. (2010). Prospective assessment of effects on iron overload parameters of deferasirox therapy in patients with myelodysplastic syndromes. Leuk. Res. 34, 1560–1565. doi: 10.1016/j.leukres.2010.06.013
Halliwell, B., Aruoma, O. I., Mufti, G., and Bomford, A. (1988). Bleomycin-detectable iron in serum from leukemic patients before and after chemotherapy. Therapeutic implications for treatment with oxidant-generating drugs. FEBS Lett. 241, 202–204. doi: 10.1016/0014-5793(88)81061-5
Hershko, C., Graham, G., Bates, G. W., and Rachmilewitz, E. A. (1978). Non-specific serum iron in thalassaemia: an abnormal serum iron fraction of potential toxicity. Br. J. Haematol. 40, 255–263. doi: 10.1111/j.1365-2141.1978.tb03662.x
Hershko, C., Link, G., Konijn, A. M., and Cabantchik, Z. I. (2005). Iron Chelation therapy. Curr. Hematol. Rep. 4, 110–116.
Hider, R. C., and Kong, X. L. (2011). Glutathione: a key component of the cytoplasmic labile iron pool. Biometals 24, 1179–1187. doi: 10.1007/s10534-011-9476-8
Jacobs, A. (1977). An intracellular transit iron pool. Blood 50, 4331–4336. doi: 10.1002/9780470720325.ch5
Jacobs, E. M., Hendriks, J. C., van Tits, B. L., Evans, P. J., Breuer, W., Liu, D. Y., et al. (2005). Results of an international round robin for the quantification of serum non-transferrin-bound iron: need for defining standardization and a clinically relevant isoform. Anal. Biochem. 341, 241–250. doi: 10.1016/j.ab.2005.03.008
Kakhlon, O., Breuer, W., Munnich, A., and Cabantchik, Z. I. (2010). Iron relocation therapy for the treatment of diseases of regional iron accumulation. Rev. Can. J. Physiol. Pharmacol. 88, 187–196. doi: 10.1139/Y09-128
Kakhlon, O., and Cabantchik, Z. I. (2002). The labile iron pool: characterization, measurement, and participation in cellular processes. Free Radic. Biol. Med. 33, 1037–1046. doi: 10.1016/S0891-5849(02)01006-7
Kakhlon, O., Gruenbaum, Y., and Cabantchik, Z. I. (2001). Repression of ferritin expression increases the labile iron pool and oxidative stress and short-term growth of human erythroleukemia cells. Blood 97, 2863–2871. doi: 10.1182/blood.V97.9.2863
Kakhlon, O., Manning, H., Breuer, W., Melamed-Book, N., Lu, C., Cortopassi, G., et al. (2008). Cell functions impaired by frataxin deficiency are restored by drug-mediated iron relocation. Blood 112, 5219–5227. doi: 10.1182/blood-2008-06-161919
Kolb, A. M., Smit, N. P., Lentz-Ljuboje, R., Osanto, S., and van Pelt, J. (2009). Non-transferrin bound iron measurement is influenced by chelator concentration. Anal. Biochem. 385, 13–19. doi: 10.1016/j.ab.2008.11.003
Kruszewski, M. (2003). Labile iron pool: the main determinant of cellular response to oxidative stress. Mutat. Res. 29, 81–92. doi: 10.1016/j.mrfmmm.2003.08.004
Le Lan, C., Loréal, O., Cohen, T., Ropert, M., Glickstein, H., and Lainé, F. (2005). Redox active plasma iron in C282Y/C282Y hemochromatosis. Blood 105, 4527–4531. doi: 10.1182/blood-2004-09-3468
Levi, S., and Rovidac, E. (2009). The role of iron in mitochondrial function. Biochim. Biophys. Acta 1790, 629–636. doi: 10.1016/j.bbagen.2008.09.008
Li-Hsuan Huang, M., Lane, D. J. R., and Richardson, D. R. (2011). The mitochondrion as a modulator of iron metabolism and its role in disease. Antioxid. Redox Signal. 15, 3003–3019. doi: 10.1089/ars.2011.3921
Linder, M. C., Moriya, M., Whon, A. M., Kassa, A., and Gilley, C. (2006). Vesicular transport of Fe and interaction with other metal ions in polarized Caco2 Cell monolayers. Biol. Res. 39, 143–156. doi: 10.4067/S0716-97602006000100016
Liuzzi, J. P., Aydemir, F., Nam, H., Knutson, M. D., and Cousins, R. J. (2006). Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc. Natl. Acad. Sci. U.S.A. 103, 13612–13617. doi: 10.1073/pnas.0606424103
Ma, Y., DeGroot, H., Liu, Z., Hider, R., and Petrat, F. (2006). Chelation and determination of labile iron in primary hepatocytes by pyridinone fluorescent probes. Biochem. J. 395, 49–55. doi: 10.1042/BJ20051496
Ma, Y., Zhou, T., Kong, X., and Hider, R. C. (2012). Chelating agents for the treatment of systemic iron overload. Rev. Curr. Med. Chem. 19, 2816–2827. doi: 10.2174/092986712800609724
Núñez, M. T., Álvarez-Hernández, X., Smith, M., Tapia, V., and Glass, J. (1994). Role of redox systems on Fe3+ uptake by transformed human intestinal epithelial (Caco-2) cells. Am. J. Physiol. 267, C1582–C1588.
Núñez, M. T., Núñez-Millacura, C., Beltrán, M., Tapia, V., and Álvarez-Hernández, X. (1999). Apotransferrin and holotransferrin undergo different endocytic cycles in intestinal epithelia (Caco-2) cells. J. Biol. Chem. 272, 19425–19428. doi: 10.1074/jbc.272.31.19425
Oudit, G. Y., Trivieri, M. G., Khaper, N., Liu, P. P., and Backx, P. H. (2006). Role of L-type Ca2+ channels in iron transport and iron-overload cardiomyopathy. J. Mol. Med. 84, 349–364. doi: 10.1007/s00109-005-0029-x
Petrat, F., de Groot, H., Sustmann, R., and Rauen, U. (2002). The chelatable iron pool in living cells: a methodically defined quantity. Biol. Chem. 383, 489–502. doi: 10.1515/BC.2002.051
Picard, V., Epsztejn, S., Santambrogio, P., Cabantchik, Z. I., and Beaumont, C. (1998). Role of ferritin in the control of the labile iron pool in murine erythroleukemia cells. J. Biol. Chem. 273, 15382–15386. doi: 10.1074/jbc.273.25.15382
Pietrangelo, A. (2007). Iron chelation beyond transfusion iron overload. Am. J. Hematol. 82, 1142–1146. doi: 10.1002/ajh.21101
Pootrakul, P., Breuer, W., Sametband, M., Sirankapracha, P., Hershko, C., and Cabantchik, Z. I. (2004). Labile plasma iron (LPI) as an indicator of chelatable plasma redox activity in iron overloaded beta-thalassaemia/HbE patients treated with an oral chelator. Blood 104, 1504–1510. doi: 10.1182/blood-2004-02-0630
Porter, J. B. (2009). Pathophysiology of transfusional iron overload: contrasting patterns in thalassemia major and sickle cell disease Hemoglobin 33, S37–S45. doi: 10.3109/03630260903346627
Prus, E., and Fibach, E. (2008a). Flow cytometry measurement of the labile iron pool in human hematopoietic cells. Cytometry 73, 22–27. doi: 10.1002/cyto.a.20491
Prus, E., and Fibach, E. (2008b). The labile iron pool in human erythroid cells. Br. J. Haematol. 142, 301–307. doi: 10.1111/j.1365-2141.2008.07192.x
Rauen, U., Springer, A., Weisheit, D., Petrat, F., Korth, H. G., de Groot, H., et al. (2007). Assessment of chelatable mitochondrial iron by using mitochondrion-selective fluorescent iron indicators with different iron-binding affinities. Chem. Biochem. 8, 341–352. doi: 10.1002/cbic.200600311
Rouault, T. A. (2006). The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 8, 406–414. doi: 10.1038/nchembio807
Rouault, T. A. (2012). Biogenesis of iron-sulfur clusters in mammalian cells: new insights and relevance to human disease. Dis. Model. Mech. 5, 155–164. doi: 10.1242/dmm.009019
Sheftel, A. D., Zhang, A. S., Brown, C., Shirihai, O. S., and Ponka, P. (2007). Direct interorganellar transfer of iron from endosome to mitochondrion. Blood 110, 125–132. doi: 10.1182/blood-2007-01-068148
Shi, H., Bencze, K. Z., Stemmler, T. L., and Philpott, C. C. (2008). A cytosolic iron chaperone that delivers iron to ferritin. Science 32, 1207–1210. doi: 10.1126/science.1157643
Shvartsman, M., and Cabantchik, Z. I. (2012). Intracellular iron trafficking: role of cytosolic ligands. Biometals 25, 711–723. doi: 10.1007/s10534-012-9529-7
Shvartsman, M., Fibach, E., and Cabantchik, Z. I. (2010). Transferrin-iron routing to the cytosol and mitochondria as studied by live and real-time fluorescence. Biochem. J. 429, 185–193. doi: 10.1042/BJ20100213
Shvartsman, M., Kikkeri, A., Shanzer, A., and Cabantchik, Z. I. (2007). Iron accesses mitochondria from a cytosolic pool of non-labile iron. Biological and clinical implications. Am. J. Physiol. Cell Physiol. 293, C1383–C1394. doi: 10.1152/ajpcell.00054.2007
Silva, A. M., and Hider, R. C. (2009). Influence of non-enzymatic post-translation modifications on the ability of human serum albumin to bind iron Implications for non-transferrin-bound iron speciation. Biochim. Biophys. Acta 1794, 1449–1458. doi: 10.1016/j.bbapap.2009.06.003
Singh, S., Hider, R. C., and Porter, J. B. (1990). A direct method for quantification of non-transferrin-bound iron. Anal. Biochem. 86, 320–323. doi: 10.1016/0003-2697(90)90088-Q
Sohn, Y. S., Breuer, W., Munnich, A., and Cabantchik, Z. I. (2008). Redistribution of accumulated cell iron: a modality of chelation with therapeutic implications. Blood 111, 1690–1699. doi: 10.1182/blood-2007-07-102335
Sohn, Y. S., Ghoti, H., Breuer, W., Rachmilewitz, E. A., Attar, S., Weiss, G., et al. (2012). The role of endocytic pathways in cellular uptake of plasma non-transferrin iron. Haematologica 97, 670–678. doi: 10.3324/haematol.2011.054858
Sohn, Y. S., Mitterstiller, A. M., Breuer, W., Weiss, G., and Cabantchik, Z. I. (2011). Rescuing iron overloaded macrophages by conservative relocation of accumulated metal. Br. J. Pharmacol. 164, 406–418. doi: 10.1111/j.1476-5381.2010.01120.x
Weaver, J., and Pollack, S. (1989). Low-Mr iron isolated from guinea pig reticulocytes as AMP-Fe and ATP-Fe complexes. Biochem. J. 26, 787–790.
Zanninelli, G., Breuer, W., and Cabantchik, Z. I. (2009). Daily labile plasma iron as an indicator of chelator activity in Thalassaemia major patients. Br. J. Haematol. 147, 744–751. doi: 10.1111/j.1365-2141.2009.07907.x
Keywords: iron, iron metabolism, chelator, siderophore, mitochondria, iron overload, oxidative stress, fluorescence
Citation: Cabantchik ZI (2014) Labile iron in cells and body fluids: physiology, pathology, and pharmacology. Front. Pharmacol. 5:45. doi: 10.3389/fphar.2014.00045
Received: 12 January 2014; Paper pending published: 19 February 2014;
Accepted: 26 February 2014; Published online: 13 March 2014.
Edited by:
Paolo Arosio, University of Brescia, ItalyReviewed by:
Stanislav Yanev, Bulgarian Academy of Sciences, BulgariaDario Finazzi, University of Brescia, Italy
Copyright © 2014 Cabantchik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zvi Ioav Cabantchik, Department of Biological Chemistry, Institute of Life Sciences, Hebrew University of Jerusalem, Safra Campus at Givat Ram, Jerusalem 91904, Israel e-mail:aW9hdkBjYy5odWppLmFjLmls