 Charles B. B. Gray
Charles B. B. Gray Joan Heller Brown
Joan Heller Brown
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 11 February 2014
Sec. Pharmacology of Ion Channels and Channelopathies
Volume 5 - 2014 | https://doi.org/10.3389/fphar.2014.00015
This article is part of the Research Topic CaMKII in Cardiac Health and Disease View all 19 articles
In this review we discuss the localization and function of the known subtypes of calcium/calmodulin dependent protein kinase IIδ (CaMKIIδ) and their role in cardiac physiology and pathophysiology. The CaMKII holoenzyme is comprised of multiple subunits that are encoded by four different genes called CaMKIIα, β, γ, and δ. While these four genes have a high degree of sequence homology, they are expressed in different tissues. CaMKIIα and β are expressed in neuronal tissue while γ and δ are present throughout the body, including in the heart. Both CaMKIIγ and δ are alternatively spliced in the heart to generate multiple subtypes. CaMKIIδ is the predominant cardiac isoform and is alternatively spliced in the heart to generate the CaMKIIδB subtype or the slightly less abundant δC subtype. The CaMKIIδB mRNA sequence contains a 33bp insert not present in δC that codes for an 11-amino acid nuclear localization sequence. This review focuses on the localization and function of the CaMKIIδ subtypes δB and δC and the role of these subtypes in arrhythmias, contractile dysfunction, gene transcription, and the regulation of Ca2+ handling.
Calcium/calmodulin dependent protein kinase II (CaMKII) is a multimeric enzyme consisting of distinct subunits encoded by four different genes known as CaMKIIα, β, γ, and δ. These genes have a high degree of sequence homology but show differential tissue expression. CaMKIIα and β are predominantly expressed in neuronal tissue while γ and δ are present throughout the body, including the heart (Bennett et al., 1983; Tobimatsu and Fujisawa, 1989). CaMKIIδ is the predominant cardiac isoform and is alternatively spliced to generate multiple subtypes (Edman and Schulman, 1994).
Schworer et al. (1993) were the first to demonstrate that there are different subtypes of CaMKIIδ expressed in various tissues. The authors reported four distinct proteins with differential expression patterns and named them CaMKIIδ1-4. CaMKIIδ2, and CaMKIIδ3 were shown to be identical except for the insertion of an 11-amino acid sequence in the variable domain of CaMKIIδ3, the more abundant of the two subtypes in the heart (Schworer et al., 1993). Around the same time, Edman and Schulman (1994) identified these same CaMKIIδ subtypes in rat heart and characterized their catalytic activity and regulation by calcium-liganded calmodulin (Ca2+/CaM). They refer to the predominant cardiac subtypes as CaMKIIδB and CaMKIIδC (the convention that will be used in this review), which correspond to the δ3 and δ2 subtypes, respectively. The structure of these proteins is shown in Figure 1. CaMKIIδB and δC possess similar catalytic activity and sensitivity to Ca2+/CaM. Furthermore, both subtypes can undergo autophosphorylation and acquire a similar degree of Ca2+-independent or autonomous activity (Edman and Schulman, 1994). In the years that followed, seven additional splice variants of the CaMKIIδ gene, referred to as CaMKIIδ5-11, were identified. Only one of these, CaMKIIδ9, is expressed in the adult heart (Figure 1; Mayer et al., 1994, 1995; Hoch et al., 1998, 1999).
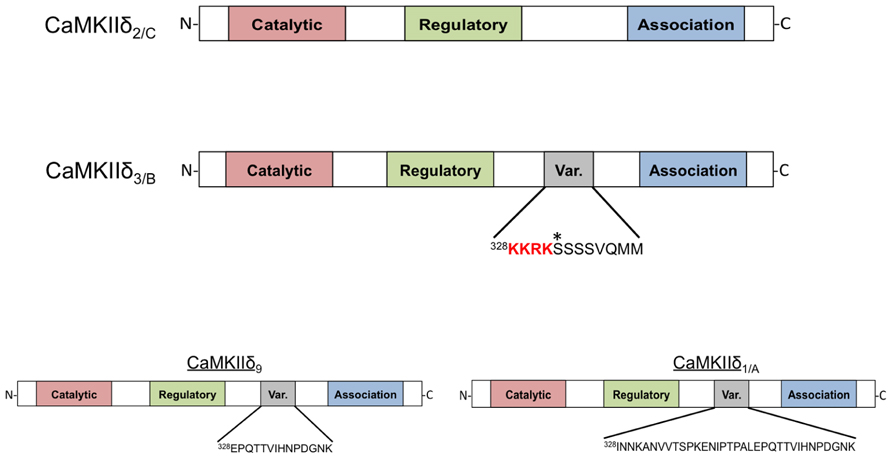
FIGURE 1. Domain scheme of CaMKIIδ monomers. The four subtypes shown are distinguished by the amino acid sequence of a variable region between the regulatory and association domains. CaMKIIδ2/C lacks this variable domain while CaMKIIδ3/B contains an 11-amino acid insertion that contains the canonical nuclear localization signal (NLS) sequence KKRK. An asterisk denotes a phosphorylation site (Ser332) in this domain that affects NLS accessibility. CaMKIIδ9 contains a different variable domain sequence that is also present in the longer variable region of CaMKIIδ1/A. All of these subtypes can be detected in adult murine heart with the exception of CaMKIIδ1/A, which is normally only present in neonatal mouse heart.
The 11-amino acid insert in CaMKIIδB (328KKRKSSSSVQMM) is also present in some splice variants of CaMKIIα and γ; this conservation suggests an important function (Schworer et al., 1993). Srinivasan et al. (1994) showed that when constructs of CaMKIIδB are transfected into fibroblasts the expressed protein is localized to the nucleus. This is not the case for constructs of CaMKIIδC, implying that that the additional amino acid sequence present in CaMKIIδB is responsible for nuclear localization (Srinivasan et al., 1994). A similar differential localization pattern was also observed when CaMKIIδ subtypes were expressed neonatal rat ventricular myocytes (NRVMs; Ramirez et al., 1997). Further studies showed that the 11-amino acid insert in CaMKIIδB can confer nuclear localization when inserted into the variable domain of CaMKIIα and that mutagenesis of the first two lysines in the insert abrogates the nuclear localization of these constructs. Thus it is widely accepted that the CaMKIIδB variable domain contains a nuclear localization signal (NLS).
CaMKII heteromultimerization is permissive in that the CaMKII holoenzyme can include subunits from multiple CaMKII genes and multiple splice variants of those genes (Bennett et al., 1983; Yamauchi et al., 1989). It seems likely that more than a single CaMKIIδ subtype is present in a single CaMKIIδ multimer and accordingly the ratio of δB to δC in a multimer could regulate the localization of the holoenzyme. This has been demonstrated experimentally. When CaMKIIδB and δC are cotransfected into fibroblasts or NRVMs, the localization of the expressed protein can be shifted in accordance with the ratio of the expressed CaMKIIδ subtypes, i.e., highly expressed δC sequesters δB in the cytosol and blocks its nuclear localization (Srinivasan et al., 1994; Ramirez et al., 1997). The opposite is also true: high relative expression of δB can localize δC to the nucleus. While not well appreciated, CaMKIIδB localization can also be regulated by phosphorylation. A serine (Ser332) immediately adjacent to the NLS of CaMKIIδB was shown to be a site of phosphorylation by CaMKI and CaMKIV in vitro. Phosphorylation prevents association of δB with the NLS receptor m-pendulin and thus limits localization of CaMKIIδB to the nucleus (Heist et al., 1998). Remarkably this mode of regulation is also seen when the NLS is moved from the middle of the protein to the N-terminus, suggesting that conformational changes are not required for phosphorylation to block the NLS.
Relative expression of CaMKIIδ subtypes is altered during cardiomyocyte differentiation and maturation and in association with the development of heart failure and ischemia/reperfusion (I/R) injury (Hoch et al., 1998, 1999; Colomer et al., 2003; Peng et al., 2010). In humans CaMKIIδB mRNA is selectively upregulated during heart failure (Hoch et al., 1999). The altered expression of particular subtypes suggests the possibility of a regulated process governing CaMKIIδ mRNA splicing because transcriptional regulation would not be expected to alter the ratio of CaMKIIδ subtypes. Alternative splicing factor/pre-mRNA-splicing factor SF2 (ASF/SF2) was initially described by Krainer and Maniatis (1985) and subsequently mice lacking ASF/SF2 expression were demonstrated to have incomplete processing of CaMKIIδ mRNA (Krainer and Maniatis, 1985; Xu et al., 2005). Specifically, enhanced expression of the δA subtype [δ1 in the nomenclature of Schworer et al. (1993)] was observed while expression of CaMKIIδB and δC was diminished. Figure 1 also depicts the structure of the δA subtype, which is expressed in the fetal heart. ASF/SF2 can be regulated by phosphorylation. Protein kinase A (PKA)-mediated ASF/SF2 phosphorylation has been correlated with alternative splicing of CaMKIIδ in heart and brain (Gu et al., 2011). Additionally, regulation of ASF/SF2 by Protein phosphatase 1 γ (PP1γ) has been demonstrated to affect CaMKIIδ splicing (Huang et al., 2013). CaMKIIδA expression is increased in models of isoproterenol-induced cardiac hypertrophy and thus regulation of CaMKIIδ splicing by PKA and PP1γ may be relevant in the context of chronic β-adrenergic stimulation (Li et al., 2011). The RNA binding proteins Fox 1 (RBFOX1) and 2 (RBFOX2) collaborate with ASF/SF2 to induce proper CaMKIIδ splicing (Han et al., 2011) and factors that regulate these proteins could also influence the expression of CaMKIIδ subtypes. Thus, CaMKIIδ splicing is a dynamic and regulated process. The role of this system in the heart has not been extensively explored but could be of major importance since regulation of CaMKIIδ splicing may account for altered subtype expression and CaMKIIδ signaling in physiological and pathophysiological settings.
The differential localization and function of CaMKIIδ subtypes could be of considerable importance to understanding the role of this enzyme in normal physiology and disease states. Early studies demonstrated that expression of CaMKIIδB in NRVMs induced atrial natriuretic factor (ANF) expression and led to increased myofilament organization, both hallmarks of cardiac hypertrophy, while expression of CaMKIIδC did not (Ramirez et al., 1997). This finding suggested that nuclear CaMKIIδ localization is required to regulate gene expression. Consistent with this notion are data indicating that CaMKIIδB signaling activates several transcription factors including myocyte enhancer factor 2 (MEF2), GATA4, and heat shock factor 1 (HSF1; Little et al., 2009; Lu et al., 2010; Peng et al., 2010). The significance of the hypertrophic responses elicited by δB in vitro was explored further by generation of CaMKIIδB transgenic (TG) mice (Zhang et al., 2002). These animals, which overexpress δB under the control of the cardiac-specific α-myosin heavy chain (α-MHC) promoter, demonstrate the enhanced expression of hypertrophic markers observed in NRVMs expressing CaMKIIδB. CaMKIIδBTG animals develop hypertrophy and moderate cardiac dysfunction by 4 months of age. Thus, CaMKIIδB expression appears to be sufficient to induce cardiac hypertrophy. Surprisingly, despite the increased CaMKII activity in the CaMKIIδBTG mouse heart, phosphorylation of the canonical cardiac CaMKII substrate phospholamban (PLN) at its CaMKII site (Thr17) was not increased but rather was decreased relative to WT mice. PLN phosphorylation at the PKA site (Ser16) was similarly reduced. These data were related to increases in phosphatase activity (Zhang et al., 2002), but also implied that CaMKIIδB did not lead to robust phosphorylation of PLN. A subsequent paper that examined CaMKIIδBTG animals at a younger age to avoid changes in phosphatase activity confirmed that phosphorylation of PLN and another cardiac CaMKII substrate, the cardiac ryanodine receptor (RyR2), was not increased by cardiac CaMKIIδB expression (Zhang et al., 2007). This finding is consistent with a predominantly nuclear localization and function of the δB subtype.
CaMKIIδB has also been suggested to regulate expression of the Na+/Ca2+ exchanger (NCX1) during the development of cardiac dysfunction following trans-aortic constriction (TAC; Lu et al., 2011). The conclusion that δB was the subtype involved in NCX1 regulation relied on the use of a constitutively active construct of CaMKIIδB in which a Thr to Asp mutation (T287D) simulates autophosphorylation. Interestingly, the authors found that this construct was excluded from the nucleus (Lu et al., 2010). This differs from the localization pattern described above (Srinivasan et al., 1994; Ramirez et al., 1997) and can be explained as a result of phosphorylation of Ser332 in the 11-amino acid insert of δB (Figure 1). The observation that mutation of Ser332 to Ala restores nuclear localization of constitutively active CaMKIIδB (Backs et al., 2006) confirms the role of this site in the cytosolic localization of the active construct. The possibility that phosphorylation of Ser332 might regulate CaMKIIδB localization in the intact heart has not been evaluated, but such a mechanism could contribute to the observation that CaMKIIδB is found outside the nucleus even in the absence of multimerization with δC (Mishra et al., 2011).
CaMKIIδC transgenic mice have also been generated and demonstrate a strikingly different phenotype from mice that express CaMKIIδB. While cardiac dysfunction is relatively moderate and takes months to develop in CaMKIIδBTG animals, mice expressing δC rapidly progress to heart failure and premature death (Zhang et al., 2003). By 6 weeks of age CaMKIIδCTG animals display marked changes in cardiac morphology and by 12 weeks these animals display severe cardiac dysfunction and upregulation of hypertrophic genes.
Expression of the cardiac sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) is diminished in δCTG mice as occurs in other models of heart failure. Since SERCA regulates Ca2+ reuptake into the sarcoplasmic reticulum (SR), this decrease would diminish SR Ca2+ loading. On the other hand, the CaMKIIδCTG mice show hyperphosphorylation of PLN at Thr17, which should improve SERCA function. In addition δCTG animals display marked increases in phosphorylation of the RyR2, the channel through which Ca2+ exits the SR. Taken together, these changes would predict dysregulation of SR Ca2+ cycling and excitation–contraction coupling. This was substantiated in an accompanying paper that systematically analyzed and demonstrated dysregulation of cardiac Ca2+ handling in mice expressing δC (Maier et al., 2003). Specifically it was shown that SR Ca2+ stores were depleted in myocytes from these animals, explaining the observation that isolated myocytes displayed diminished twitch shortening amplitude. Furthermore, Maier et al. (2003) showed that the frequency and duration of Ca2+ sparks, or spontaneous intracellular Ca2+-release events, was markedly increased in myocytes from animals expressing δC. Hyperphosphorylation of RyR2 by CaMKIIδC was hypothesized to underly the enhanced leak of Ca2+ from the SR, and this was verified by the demonstration that acute inhibition of CaMKII in δCTG myocytes rescues the altered Ca2+ handling (Maier et al., 2003). In other experiments, acute expression of δC in rabbit cardiomyocytes was shown to be sufficient to induce SR Ca2+ sparks and diminished SR Ca2+ loading (Kohlhaas et al., 2006). These findings imply that direct regulation of Ca2+ handling targets including RyR2 by CaMKIIδC can account for the dysregulation of Ca2+ and contractile function seen in myocytes from δCTG animals (Figure 2).
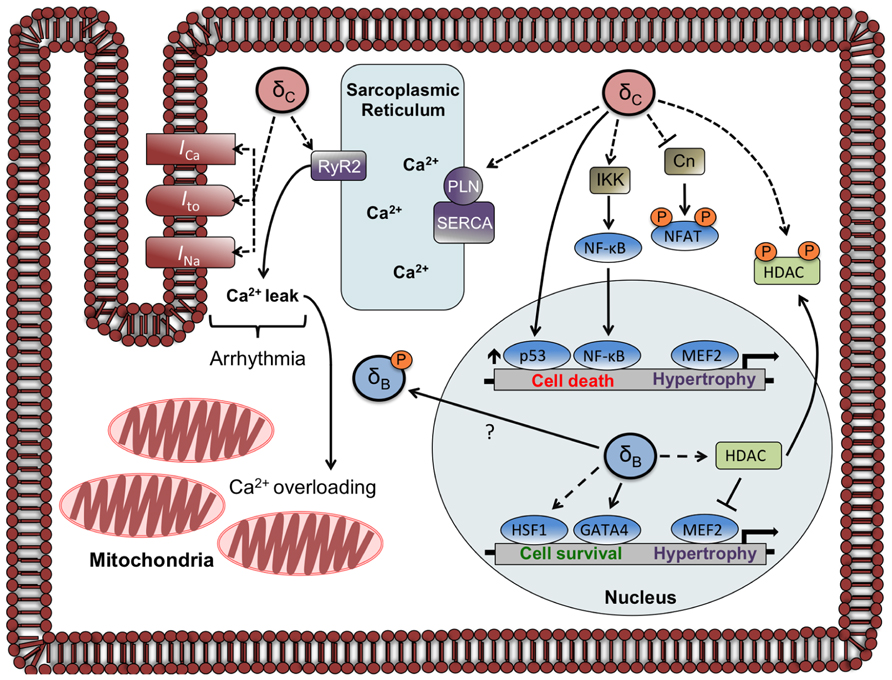
FIGURE 2. Localization and function of CaMKIIδ subtypes in the adult cardiomyocyte. The circles labeled δC and δB represent CaMKIIδ multimers that are composed primarily of δC and δB subunits, respectively. Documented phosphorylation events are indicated by dashed lines. CaMKIIδC regulates Ca2+ homeostasis and currents involved in arrhythmogenesis through phosphorylation of Ca2+ handling proteins and channels. CaMKIIδC can also affect gene transcription through direct and indirect mechanisms including phosphorylation of NFAT and HDAC (sequestering them in the cytosol), increases in p53, and increased nuclear import of NF-κB. The CaMKIIδB subtype has little effect on phosphorylation of Ca2+ handling proteins but increases gene expression through HDAC phosphorylation and nuclear export and activation of HSF1 and GATA4. A putative mechanism for δBredistribution is depicted, showing δBexiting or being excluded from the nucleus due to phosphorylation at a site (Ser332) adjacent to its NLS.
Dysregulation of excitation–contraction coupling by CaMKII is thought to contribute to arrythmogenesis in a variety of contexts, as supported by the increased incidence of arrhythmogenic events in CaMKIIδCTG mice (Anderson et al., 1998; Wu et al., 2002; Wagner et al., 2006). Overexpression of CaMKIIδC not only induces more spontaneous arrhythmias but also enhances the susceptibility of mice to arrhythmogenic challenge by β-adrenergic stimulation. Sag et al. (2009) found that much of the proarrhythmogenic effects of β-adrenergic stimulation on SR Ca2+ leak were significantly inhibited by treatment of myocytes with KN-93, an inhibitor of CaMKII. Furthermore the SR Ca2+ leak induced by isoproterenol did not occur in myocytes from mice lacking CaMKIIδ. These findings collectively implicate SR Ca2+ leak as one of the key mechanisms in δC-mediated arrhythmias (Sag et al., 2009). The notion that hyperphosphorylation of RyR2 at the CaMKII site (Ser2814) contributes to arrhythmias and SR Ca2+ leak is supported by the finding that mutation of Ser2814 to Ala (S2814A) blocks the ability of CaMKII to induce Ca2+ sparks (van Oort et al., 2010). The autosomal dominant form of catecholaminergic polymorphic ventricular tachycardia (CPVT) can be caused by the RyR2 mutation R4496C and mice carrying this mutation are predisposed to arrhythmia and ventricular fibrillation. Enhanced CaMKIIδC expression and activity are implicated in the etiology of premature death in CPVT as expression of CaMKIIδC exacerbates the effects of the R4496C mutation (Dybkova et al., 2011). As mentioned earlier RyR2 Ser2814 phosphorylation is increased by expression of CaMKIIδC (but not by δB) in vivo (Zhang et al., 2007) and the effects of mutating this site (van Oort et al., 2010) emphasize the importance of RyR2 phosphorylation by CaMKII in SR Ca2+ leak and arrhythmia.
Other targets besides those at the SR may contribute to the arrhythmogenic phenotype of CaMKIIδC mice. The cardiac sodium channel NaV1.5 is physically associated with CaMKIIδC based on coimmunoprecipitation of these proteins from CaMKIIδCTG animals and NaV1.5 is phosphorylated in mice expressing δC (Wagner et al., 2006). CaMKIIδC phosphorylates NaV1.5 at multiple sites and phosphorylation appears to elicit the loss-of-function changes in NaV1.5 gating that are observed in the context of CaMKIIδC expression in vitro (Ashpole et al., 2012; Koval et al., 2012). Incomplete inactivation of NaV1.5 generates a late Na+ current (INa), which can prolong the duration of the action potential and contribute to arrhythmias. Additionally, increased INa can lead to Na+-overloading of the cardiomyocyte, which contributes to diminished diastolic contractile performance (Maltsev et al., 1998). Late INa is observed in CaMKIIδCTG mice and inhibition of this current ameliorates arrhythmia and diastolic dysfunction in these animals (Sossalla et al., 2011). Modulation of INa therefore appears to contribute to the phenotype of CaMKIIδC mice with respect to arrhythmia development; additionally the CaMKIIδC subtype likely regulates the L-type Ca2+ channel (LTCC) and repolarizing potassium currents (Ito and IK1; McCarron et al., 1992; Wagner et al., 2009). Thus, a multitude of mechanisms link CaMKIIδC to arrhythmogenesis.
Arrhythmias may contribute to the premature death of CaMKIIδCTG mice but there are also marked decreases in contractile function in these animals. Since alterations to cardiomyocyte Ca2+ handling are seen in relatively young CaMKIIδCTG mice and precede the development of heart failure, it is possible that dysregulated Ca2+ homeostasis (specifically SR Ca2+ leak) is an initiating event in δC-induced heart failure. Specifically, as a consequence of SR Ca2+ leak and SERCA downregulation, the SR Ca2+ load is diminished which would compromise contractile function. To determine whether diminished SR Ca2+ load is the primary causal event leading to contractile dysfunction and premature death in response to δC overexpression, we crossed the δCTG mice with mice in which the SERCA regulatory protein PLN was deleted (PLN-KO). Deletion of PLN in the context of δC overexpression normalized SR Ca2+ levels and the contractile function of isolated myocytes was restored (Zhang et al., 2010). Remarkably the development of cardiac dysfunction in vivo was not rescued but instead was accelerated in the δCTG/PLN-KO mice. In addition SR Ca2+ leak was enhanced. It was hypothesized that the increased SR Ca2+ load, in the context of RyR2 hyperphosphorylation, precipitated greater Ca2+ leak and further suggested that the accelerated development of cardiac dysfunction was due to mitochondrial Ca2+ overloading (Zhang et al., 2010). These observations and their interpretation places central importance on the Ca2+ leak elicited by δC-mediated phosphorylation of RyR2 in the development of heart failure. Further support for this hypothesis comes from the finding that CaMKIIδ knockout mice have attenuated contractile dysfunction in response to pressure overload induced by TAC and myocytes from these animals show diminished SR Ca2+ leak in response to TAC (Ling et al., 2009). Additionally, mice expressing the RyR2 S2814A mutation are protected from the development of heart failure in response to pressure overload (Respress et al., 2012) consistent with a critical role for CaMKII-mediated RyR2 phosphorylation. We recently crossed CaMKIIδC mice with those expressing RyR2 S2814A; if the hypothesis is correct these mice will show diminished SR Ca2+ leak and improved contractile function when compared to CaMKIIδCTG mice.
Another approach used to determine the role of RyR2 phosphorylation and SR Ca2+ leak in the phenotype of CaMKIIδCTG mice was to cross the CaMKIIδCTG mice with mice expressing SR-targeted autocamtide-2-related inhibitory peptide (SR-AIP; Huke et al., 2011). AIP simulates the regulatory domain of CaMKII and inhibits the kinase, and SR-AIP mice have been shown to display diminished phosphorylation of CaMKII substrates at the SR (Ji et al., 2003). A reduction in the extent of PLN and RyR2 hyperphosphorylation observed in CaMKIIδCTG mice was conferred by SR-AIP. There were associated changes in Ca2+ handling that indicated a modest improvement in SR Ca2+ leak. Despite the salutary effects of SR-AIP in cells from δCTG mice, in vivo cardiac function was not improved. One possible explanation for these findings is that the degree of inhibition of RyR2 phosphorylation conferred by SR-AIP was insufficient to prevent the effects of CaMKIIδC overexpression. Alternatively, while δC-mediated phosphorylation of targets at the SR including RyR2 and PLN is of considerable consequence, targets of CaMKII elsewhere in the cell may also contribute to the pathogenesis of cardiac dysfunction induced by CaMKIIδC.
Mitochondrial Ca2+ is elevated in mice overexpressing δC in the context of intact SR Ca2+ load (Zhang et al., 2010) and increases in mitochondrial Ca2+ are known to induce opening of the mitochondrial permeability transition pore (MPTP) and cell death (Halestrap and Davidson, 1990). Considering the central importance of mitochondria in the regulation of cell death and of cell death in the development of heart failure (Wencker et al., 2003), any pathway by which CaMKIIδC induces mitochondrial Ca2+ overloading and subsequent loss of mitochondrial integrity would be predicted to contribute to the development of contractile dysfunction and heart failure. To test the role of mitochondrial dysregulation in the cardiomyopathy that develops in δCTG animals, CaMKIIδCTG mice were crossed with mice lacking expression of cyclophilin D, a mitochondrial protein required for the formation of the MPTP. The ability of high Ca2+ to induce swelling of isolated mitochondria, an index of MPTP opening, was impaired in the CaMKIIδCTG mice lacking cyclophilin D, but development of dilated cardiomyopathy and premature death of these mice was not diminished. Indeed these responses were exacerbated when compared to δCTG mice with intact cyclophilin D expression. The authors suggest that cyclophilin D may actually play a beneficial role in stress responses, as they observed that TAC-induced heart failure development was also made more severe by genetic deletion of cyclophilin D (Elrod et al., 2010). However, CaMKIIδC is found at mitochondria and a recent seminal study by Joiner et al. (2012) identified the mitochondrial Ca2+ uniporter (MCU) as a potential target of CaMKII (Mishra et al., 2011; Joiner et al., 2012). While phosphorylation of the MCU by CaMKII was not shown to occur in vivo, a CaMKII-dependent change in the function of the MCU was evidenced by data demonstrating that a CaMKII inhibitory peptide targeted to the mitochondria diminished mitochondrial Ca2+ uptake and inhibited apoptosis in mice subjected to myocardial infarction and I/R injury.
The discussion above, and indeed most of the literature, considers the role of CaMKIIδ-mediated phosphorylation and regulation of Ca2+ handling proteins and ion channels. Chronic elevations in CaMKIIδ expression and activity are observed in humans with heart failure (Hoch et al., 1999) and these long-term changes are likely to elicit altered gene expression. As discussed earlier, CaMKIIδB induces the expression of hypertrophic genes in myocytes and transgenic mice, consistent with its primarily nuclear localization (Ramirez et al., 1997; Zhang et al., 2002). Other work showed that the CaMKIIδB subtype is required for GATA-4 binding to the B cell lymphoma 2 (Bcl-2) promoter and subsequent gene expression (Little et al., 2009). Furthermore, CaMKIIδB was shown to phosphorylate the transcription factor HSF1 thereby increasing its transcriptional activity (Peng et al., 2010). Taken together, these observations imply that it is the CaMKIIδB subtype that regulates gene expression as a result of its actions in the nucleus.
It is not necessarily the case, however, that gene regulation requires CaMKIIδ to be localized to the nuclear compartment. Despite its primarily cytosolic localization, CaMKIIδC overexpressed in mouse heart increased phosphorylation of histone deacetylase 4 (HDAC4), resulting in activation of the transcription factor MEF2 (Zhang et al., 2007). CaMKIIδC has also been demonstrated to regulate nuclear localization of nuclear factor of activated T cells (NFATs) in NRVM. The ability of CaMKIIδC to decrease nuclear NFAT was blocked by coexpression of a dominant-negative construct of CaMKIIδC and was shown to be elicited by phosphorylation and inhibition of the Ca2+/CaM dependent phosphatase calcineurin (Cn; MacDonnell et al., 2009), presumably in the cytosol. Alteration of Ca2+ homeostasis by cytosolic CaMKIIδC expression may indirectly affect gene expression and additionally the constitutively active CaMKIIδB utilized in the studies discussed above (Lu et al., 2011) is cytosolic and yet regulates expression of NCX1.
Regulation of gene expression by CaMKIIδB has been demonstrated to promote cardiomyocyte survival while the opposite is true for CaMKIIδC. CaMKIIδB was shown to protect cardiomyocytes from doxorubicin-induced cell death via transcriptional upregulation of Bcl-2 (Little et al., 2009). Along similar lines, CaMKIIδB contributes to cardioprotection from H2O2 by increasing inducible heat shock protein 70 (iHSP70) expression (Peng et al., 2010). Conversely, CaMKIIδC activation is implicated in cell death elicited by a variety of stimuli (Zhu et al., 2007). It has been suggested that CaMKIIδC (but not δB) upregulates the proapoptotic transcription factor p53 (Toko et al., 2010), and recent work from our laboratory demonstrates that CaMKIIδC expression in NRVMs activates the proinflammatory transcription factor nuclear factor κB (NF-κB; Ling et al., 2013). We demonstrated that CaMKIIδC increased phosphorylation of IκB Kinase (IKK) and since IKK activation can also upregulate p53(Jia et al., 2013), this pathway may contribute to the proapoptotic response reported by Toko et al. (2010).
There is compelling evidence that the CaMKIIδB and δC subtypes differentially regulate cardiomyocyte Ca2+ handling and survival in vitro. Whether this occurs in vivo under physiological or pathophysiological conditions, and whether δB and δC subserve different functions based on their localization or selective activation, remains to be determined.
It seems likely that the relative levels of endogenous δB and δC determine localization and could therefore impact CaMKIIδ signaling. Hypothetically, a selective increase in CaMKIIδC would result in accumulation of cytosolic CaMKIIδ and depletion of nuclear CaMKIIδ while a selective increase in CaMKIIδB would have the opposite effect. CaMKIIδ redistribution in this manner may contribute to the phenotype of mice that overexpress δB and δC and importantly there are changes in the relative expression of δB and δC in models of heart failure and I/R injury. In both models δC expression is enhanced relative to that of δB (Zhang et al., 2003; Peng et al., 2010). It is not known how this occurs but it is of interest to postulate that in heart failure and during I/R regulation of CaMKIIδ splicing is altered. ASF/SF2 and RBFOX1/2 regulate the splicing of the CaMKIIδ gene and thus expression of δB and δC, but whether changes in splicing occur in and contribute to the development of heart failure or I/R injury remains to be determined. It is likely that the increased δC expression observed in these models is pathogenic.
While CaMKIIδB contains an NLS, this subtype is not completely sequestered in the nucleus (Mishra et al., 2011). As mentioned previously the NLS within the variable domain of δB can be regulated by phosphorylation, which prevents nuclear localization. This type of regulation could be of considerable importance since the nuclear localization of δB appears to correlate with enhanced expression of protective genes and cell survival while cytosolic localization does not (Little et al., 2009; Peng et al., 2010; Lu et al., 2011).
Of additional interest is the neglected CaMKIIδ9. The pioneering work of (Hoch et al., 1998; Mayer et al., 1995) identified δ9 as one of the three subtypes of CaMKIIδ in the adult heart and showed that it is expressed at similar levels to those of CaMKIIδB. δ9 contains a sequence (328EPQTTVIHNPDGNK) not present in δB or δC and thus may possess unique properties that merit further investigation, as the function and localization of δ9 in vivo has not been explored. Along similar lines, CaMKIIδA expression is increased in a model of cardiac hypertrophy (Li et al., 2011), but the possibility that this splice variant is upregulated in and contributes to cardiovascular disease has not been investigated.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Stephanie Dusaban B.A. for her help regarding the figures.
Anderson, M. E., Braun, A. P., Wu, Y., Lu, T., Schulman, H., and Sung, R. J. (1998). KN-93, an inhibitor of multifunctional Ca++/calmodulin-dependent protein kinase, decreases early afterdepolarizations in rabbit heart. J. Pharmacol. Exp. Ther. 287, 996–1006.
Ashpole, N. M., Herren, A. W., Ginsburg, K. S., Brogan, J. D., Johnson, D. E., Cummins, T. R., et al. (2012). Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1.5 gating by multiple phosphorylation sites. J. Biol. Chem. 287, 19856–19869. doi: 10.1074/jbc.M111.322537
Backs, J., Song, K., Bezprozvannaya, S., Chang, S., and Olson, E. N. (2006). CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J. Clin. Invest. 116, 1853–1864. doi: 10.1172/jci27438
Bennett, M. K., Erondu, N. E., and Kennedy, M. B. (1983). Purification and characterization of a calmodulin-dependent protein kinase that is highly concentrated in brain. J. Biol. Chem. 258, 12735–12744.
Colomer, J. M., Mao, L., Rockman, H. A., and Means, A. R. (2003). Pressure overload selectively up- regulates Ca2+/calmodulin-dependent protein kinase II in vivo. Mol. Endocrinol. 17, 183–192. doi: 10.1210/me.2002-0350
Dybkova, N., Sedej, S., Napolitano, C., Neef, S., Rokita, A. G., Hunlich, M., et al. (2011). Overexpression of CaMKIIdeltac in RyR2R4496C+/- knock-in mice leads to altered intracellular Ca2+ handling and increased mortality. J. Am. Coll. Cardiol. 57, 469–479. doi: 10.1016/j.jacc.2010.08.639
Edman, C. F., and Schulman, H. (1994). Identification and characterization of delta B-CaM kinase and delta C-CaM kinase from rat heart, two new multifunctional Ca2+/calmodulin-dependent protein kinase isoforms. Biochim. Biophys. Acta 1221, 89–101. doi: 10.1016/0167-4889(94)90221-6
Elrod, J. W., Wong, R., Mishra, S., Vagnozzi, R. J., Sakthievel, B., Goonasekera, S. A., et al. (2010). Cyclophilin D controls mitochondrial pore-dependent Ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J. Clin. Invest. 120, 3680–3687. doi: 10.1172/jci43171
Gu, Q., Jin, N., Sheng, H., Yin, X., and Zhu, J. (2011). Cyclic AMP-dependent protein kinase A regulates the alternative splicing of CaMKIIdelta. PLoS ONE 6:e25745. doi: 10.1371/journal.pone.0025745
Halestrap, A. P., and Davidson, A. M. (1990). Inhibition of Ca2(+)-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem. J. 268, 153–160.
Han, J., Ding, J. H., Byeon, C. W., Kim, J. H., Hertel, K. J., Jeong, S., et al. (2011). SR proteins induce alternative exon skipping through their activities on the flanking constitutive exons. Mol. Cell. Biol. 31, 793–802. doi: 10.1128/mcb.01117-10
Heist, E. K., Srinivasan, M., and Schulman, H. (1998). Phosphorylation at the nuclear localization signal of Ca2+/calmodulin-dependent protein kinase II blocks its nuclear targeting. J. Biol. Chem. 273, 19763–19771. doi: 10.1074/jbc.273.31.19763
Hoch, B., Haase, H., Schulze, W., Hagemann, D., Morano, I., Krause, E. G., et al. (1998). Differentiation-dependent expression of cardiac delta-CaMKII isoforms. J. Cell. Biochem. 68, 259–268. doi: 10.1002/(SICI)1097-4644(19980201)68:2<259::AID-JCB12>3.0.CO;2-A
Hoch, B., Meyer, R., Hetzer, R., Krause, E. G., and Karczewski, P. (1999). Identification and expression of delta-isoforms of the multifunctional Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circ. Res. 84, 713–721. doi: 10.1161/01.RES.84.6.713
Huang, C., Cao, W., Liao, R., Wang, J., Wang, Y., Tong, L., et al. (2013). PP1gamma functionally augments the alternative splicing of CaMKIIdelta through interacting with ASF. Am. J. Physiol. Cell Physiol. 306, C167–C177. doi: 10.1152/ajpcell.00145.2013
Huke, S., Desantiago, J., Kaetzel, M. A., Mishra, S., Brown, J. H., Dedman, J. R., et al. (2011). SR-targeted CaMKII inhibition improves SR Ca(2)+ handling, but accelerates cardiac remodeling in mice overexpressing CaMKIIdeltaC. J. Mol. Cell. Cardiol. 50, 230–238. doi: 10.1016/j.yjmcc.2010.10.014
Ji, Y., Li, B., Reed, T. D., Lorenz, J. N., Kaetzel, M. A., and Dedman, J. R. (2003). Targeted inhibition of Ca2+/calmodulin-dependent protein kinase II in cardiac longitudinal sarcoplasmic reticulum results in decreased phospholamban phosphorylation at threonine 17. J. Biol. Chem. 278, 25063–25071. doi: 10.1074/jbc.M302193200
Jia, C. H., Li, M., Liu, J., Zhao, L., Lin, J., Lai, P. L., et al. (2013). IKK-beta mediates hydrogen peroxide induced cell death through p85 S6K1. Cell Death Differ. 20, 248–258. doi: 10.1038/cdd.2012.115
Joiner, M. L., Koval, O. M., Li, J., He, B. J., Allamargot, C., Gao, Z., et al. (2012). CaMKII determines mitochondrial stress responses in heart. Nature 491, 269–273. doi: 10.1038/nature11444
Kohlhaas, M., Zhang, T., Seidler, T., Zibrova, D., Dybkova, N., Steen, A., et al. (2006). Increased sarcoplasmic reticulum calcium leak but unaltered contractility by acute CaMKII overexpression in isolated rabbit cardiac myocytes. Circ. Res. 98, 235–244. doi: 10.1161/01.RES.0000200739.90811.9f
Koval, O. M., Snyder, J. S., Wolf, R. M., Pavlovicz, R. E., Glynn, P., Curran, J., et al. (2012). Ca2+/calmodulin-dependent protein kinase II-based regulation of voltage-gated Na+ channel in cardiac disease. Circulation 126, 2084–2094. doi: 10.1161/circulationaha.112.105320
Krainer, A. R., and Maniatis, T. (1985). Multiple factors including the small nuclear ribonucleoproteins U1 and U2 are necessary for pre-mRNA splicing in vitro. Cell 42, 725–736. doi: 10.1016/0092-8674(85)90269-7
Li, C., Cai, X., Sun, H., Bai, T., Zheng, X., Zhou, X. W., et al. (2011). The deltaA isoform of calmodulin kinase II mediates pathological cardiac hypertrophy by interfering with the HDAC4-MEF2 signaling pathway. Biochem. Biophys. Res. Commun. 409, 125–130. doi: 10.1016/j.bbrc.2011.04.128
Ling, H., Gray, C. B., Zambon, A. C., Grimm, M., Gu, Y., Dalton, N., et al. (2013). Ca2+/Calmodulin-dependent protein kinase II delta mediates myocardial ischemia/reperfusion injury through nuclear factor-kappaB. Circ. Res. 112, 935–944. doi: 10.1161/circresaha.112.276915
Ling, H., Zhang, T., Pereira, L., Means, C. K., Cheng, H., Gu, Y., et al. (2009). Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J. Clin. Invest. 119, 1230–1240. doi: 10.1172/jci38022
Little, G. H., Saw, A., Bai, Y., Dow, J., Marjoram, P., Simkhovich, B., et al. (2009). Critical role of nuclear calcium/calmodulin-dependent protein kinase IIdeltaB in cardiomyocyte survival in cardiomyopathy. J. Biol. Chem. 284, 24857–24868. doi: 10.1074/jbc.M109.003186
Lu, Y. M., Huang, J., Shioda, N., Fukunaga, K., Shirasaki, Y., Li, X. M., et al. (2011). CaMKIIdeltaB mediates aberrant NCX1 expression and the imbalance of NCX1/SERCA in transverse aortic constriction-induced failing heart. PLoS ONE 6:e24724. doi: 10.1371/journal.pone.0024724
Lu, Y. M., Shioda, N., Yamamoto, Y., Han, F., and Fukunaga, K. (2010). Transcriptional upregulation of calcineurin Abeta by endothelin-1 is partially mediated by calcium/calmodulin-dependent protein kinase IIdelta3 in rat cardiomyocytes. Biochim. Biophys. Acta 1799, 429–441. doi: 10.1016/j.bbagrm.2010.02.004
MacDonnell, S. M., Weisser-Thomas, J., Kubo, H., Hanscome, M., Liu, Q., Jaleel, N., et al. (2009). CaMKII negatively regulates calcineurin- NFAT signaling in cardiac myocytes. Circ. Res. 105, 316–325. doi: 10.1161/circresaha.109.194035
Maier, L. S., Zhang, T., Chen, L., DeSantiago, J., Brown, J. H., and Bers, D. M. (2003). Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ. Res. 92, 904–911. doi: 10.1161/01.res.0000069685.20258.f1
Maltsev, V. A., Sabbah, H. N., Higgins, R. S., Silverman, N., Lesch, M., and Undrovinas, A. I. (1998). Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation 98, 2545–2552. doi: 10.1161/01.CIR.98.23.2545
Mayer, P., Mohlig, M., Idlibe, D., and Pfeiffer, A. (1995). Novel and uncommon isoforms of the calcium sensing enzyme calcium/calmodulin dependent protein kinase II in heart tissue. Basic Res. Cardiol. 90, 372–379. doi: 10.1007/BF00788498
Mayer, P., Mohlig, M., Schatz, H., and Pfeiffer, A. (1994). Additional isoforms of multifunctional calcium/calmodulin-dependent protein kinase II in rat heart tissue. Biochem. J. 298(Pt 3), 757–758.
McCarron, J. G., McGeown, J. G., Reardon, S., Ikebe, M., Fay, F. S., and Walsh, J. V. Jr. (1992). Calcium-dependent enhancement of calcium current in smooth muscle by calmodulin-dependent protein kinase II. Nature 357, 74–77. doi: 10.1038/357074a0
Mishra, S., Gray, C. B., Miyamoto, S., Bers, D. M., and Brown, J. H. (2011). Location matters: clarifying the concept of nuclear and cytosolic CaMKII subtypes. Circ. Res. 109, 1354–1362. doi: 10.1161/circresaha.111.248401
Peng, W., Zhang, Y., Zheng, M., Cheng, H., Zhu, W., Cao, C. M., et al. (2010). Cardioprotection by CaMKII-deltaB is mediated by phosphorylation of heat shock factor 1 and subsequent expression of inducible heat shock protein 70. Circ. Res. 106, 102–110. doi: 10.1161/circresaha.109.210914
Ramirez, M. T., Zhao, X. L., Schulman, H., and Brown, J. H. (1997). The nuclear deltaB isoform of Ca2+/calmodulin-dependent protein kinase II regulates atrial natriuretic factor gene expression in ventricular myocytes. J. Biol. Chem. 272, 31203–31208. doi: 10.1074/jbc.272.49.31203
Respress, J. L., van Oort, R. J., Li, N., Rolim, N., Dixit, S. S., deAlmeida, A., et al. (2012). Role of RyR2 phosphorylation at S2814 during heart failure progression. Circ. Res. 110, 1474–1483. doi: 10.1161/circresaha.112.268094
Sag, C. M., Wadsack, D. P., Khabbazzadeh, S., Abesser, M., Grefe, C., Neumann, K., et al. (2009). Calcium/calmodulin-dependent protein kinase II contributes to cardiac arrhythmogenesis in heart failure. Circ. Heart Fail. 2, 664–675. doi: 10.1161/circheartfailure.109.865279
Schworer, C. M., Rothblum, L. I., Thekkumkara, T. J., and Singer, H. A. (1993). Identification of novel isoforms of the delta subunit of Ca2+/calmodulin-dependent protein kinase II. Differential expression in rat brain and aorta. J. Biol. Chem. 268, 14443–14449.
Sossalla, S., Maurer, U., Schotola, H., Hartmann, N., Didie, M., Zimmermann, W. H., et al. (2011). Diastolic dysfunction and arrhythmias caused by overexpression of CaMKIIdelta(C) can be reversed by inhibition of late Na(+) current. Basic Res. Cardiol. 106, 263–272. doi: 10.1007/s00395-010-0136-x
Srinivasan, M., Edman, C. F., and Schulman, H. (1994). Alternative splicing introduces a nuclear localization signal that targets multifunctional CaM kinase to the nucleus. J. Cell Biol. 126, 839–852. doi: 10.1083/jcb.126.4.839
Tobimatsu, T., and Fujisawa, H. (1989). Tissue-specific expression of four types of rat calmodulin- dependent protein kinase II mRNAs. J. Biol. Chem. 264, 17907–17912.
Toko, H., Takahashi, H., Kayama, Y., Oka, T., Minamino, T., Okada, S., et al. (2010). Ca2+/calmodulin-dependent kinase IIdelta causes heart failure by accumulation of p53 in dilated cardiomyopathy. Circulation 122, 891–899. doi: 10.1161/circulationaha.109.935296
van Oort, R. J., McCauley, M. D., Dixit, S. S., Pereira, L., Yang, Y., Respress, J. L., et al. (2010). Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation 122, 2669–2679. doi: 10.1161/circulationaha.110.982298
Wagner, S., Dybkova, N., Rasenack, E. C., Jacobshagen, C., Fabritz, L., Kirchhof, P., et al. (2006). Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Invest. 116, 3127–3138. doi: 10.1172/jci26620
Wagner, S., Hacker, E., Grandi, E., Weber, S. L., Dybkova, N., Sossalla, S., et al. (2009). Ca/calmodulin kinase II differentially modulates potassium currents. Circ. Arrhythm. Electrophysiol. 2, 285–294. doi: 10.1161/circep.108.842799
Wencker, D., Chandra, M., Nguyen, K., Miao, W., Garantziotis, S., Factor, S. M., et al. (2003). A mechanistic role for cardiac myocyte apoptosis in heart failure. J. Clin. Invest. 111, 1497–1504. doi: 10.1172/jci17664
Wu, Y., Temple, J., Zhang, R., Dzhura, I., Zhang, W., Trimble, R., et al. (2002). Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation 106, 1288–1293. doi: 10.1161/01.CIR.0000027583.73268.E7
Xu, X., Yang, D., Ding, J. H., Wang, W., Chu, P. H., Dalton, N. D., et al. (2005). ASF/SF2-regulated CaMKIIdelta alternative splicing temporally reprograms excitation-contraction coupling in cardiac muscle. Cell 120, 59–72. doi: 10.1016/j.cell.2004.11.036
Yamauchi, T., Ohsako, S., and Deguchi, T. (1989). Expression and characterization of calmodulin- dependent protein kinase II from cloned cDNAs in Chinese hamster ovary cells. J. Biol. Chem. 264, 19108–19116.
Zhang, T., Guo, T., Mishra, S., Dalton, N. D., Kranias, E. G., Peterson, K. L., et al. (2010). Phospholamban ablation rescues sarcoplasmic reticulum Ca(2+) handling but exacerbates cardiac dysfunction in CaMKIIdelta(C) transgenic mice. Circ. Res. 106, 354–362. doi: 10.1161/circresaha.109.207423
Zhang, T., Johnson, E. N., Gu, Y., Morissette, M. R., Sah, V. P., Gigena, M. S., et al. (2002). The cardiac-specific nuclear delta(B) isoform of Ca2+/calmodulin-dependent protein kinase II induces hypertrophy and dilated cardiomyopathy associated with increased protein phosphatase 2A activity. J. Biol. Chem. 277, 1261–1267. doi: 10.1074/jbc.M108525200
Zhang, T., Kohlhaas, M., Backs, J., Mishra, S., Phillips, W., Dybkova, N., et al. (2007). CaMKIIdelta isoforms differentially affect calcium handling but similarly regulate HDAC/MEF2 transcriptional responses. J. Biol. Chem. 282, 35078–35087. doi: 10.1074/jbc.M707083200
Zhang, T., Maier, L. S., Dalton, N. D., Miyamoto, S., Ross, J. Jr., Bers, D. M., et al. (2003). The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ. Res. 92, 912–919. doi: 10.1161/01.res.0000069686.31472.c5
Keywords: Ca2+/calmodulin-dependent protein kinase II, heart, splice variants, nuclear localization, transgenic mice
Citation: Gray CBB and Heller Brown J (2014) CaMKIIdelta subtypes: localization and function. Front. Pharmacol. 5:15. doi: 10.3389/fphar.2014.00015
Received: 25 December 2013; Accepted: 25 January 2014;
Published online: 11 February 2014.
Edited by:
Eleonora Grandi, University of California Davis, USACopyright © 2014 Gray and Heller Brown. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joan Heller Brown, Department of Pharmacology, University of California at San Diego, San Diego, 9500 Gilman Dr., La Jolla, CA 92093-0636, USA e-mail:amhicm93bkB1Y3NkLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.