 Stephen Orena†
Stephen Orena† Tristan S. Maurer
Tristan S. Maurer Li She
Li She Rena Eudy†
Rena Eudy† Vincent BernardoDarla DashPaula LoriaMary E. BankerMeera TugnaitCarlin V. Okerberg
Vincent BernardoDarla DashPaula LoriaMary E. BankerMeera TugnaitCarlin V. Okerberg Jessie Qian
Jessie Qian Carine M. Boustany-Kari*†
Carine M. Boustany-Kari*†- Pfizer Groton Research and Development, Groton, CT, USA
The mineralocorticoid receptor (MR) antagonists PF-03882845 and eplerenone were evaluated for renal protection against aldosterone-mediated renal disease in uninephrectomized Sprague-Dawley (SD) rats maintained on a high salt diet and receiving aldosterone by osmotic mini-pump for 27 days. Serum K+ and the urinary albumin to creatinine ratio (UACR) were assessed following 14 and 27 days of treatment. Aldosterone induced renal fibrosis as evidenced by increases in UACR, collagen IV staining in kidney cortex, and expression of pro-fibrotic genes relative to sham-operated controls not receiving aldosterone. While both PF-03882845 and eplerenone elevated serum K+ levels with similar potencies, PF-03882845 was more potent than eplerenone in suppressing the rise in UACR. PF-03882845 prevented the increase in collagen IV staining at 5, 15 and 50 mg/kg BID while eplerenone was effective only at the highest dose tested (450 mg/kg BID). All doses of PF-03882845 suppressed aldosterone-induced increases in collagen IV, transforming growth factor-β 1 (Tgf-β 1), interleukin-6 (Il-6), intermolecular adhesion molecule-1 (Icam-1) and osteopontin gene expression in kidney while eplerenone was only effective at the highest dose. The therapeutic index (TI), calculated as the ratio of the EC50 for increasing serum K+ to the EC50 for UACR lowering, was 83.8 for PF-03882845 and 1.47 for eplerenone. Thus, the TI of PF-03882845 against hyperkalemia was 57-fold superior to that of eplerenone indicating that PF-03882845 may present significantly less risk for hyperkalemia compared to eplerenone.
Introduction
Mounting clinical evidence suggests a key role for aldosterone in the progression of nephropathy. Inhibitors of the renin-angiotensin-aldosterone system (RAAS), such as angiotensin receptor blockers (ARB) and angiotensin-converting enzyme inhibitors (ACEI) have been demonstrated to delay the onset of macroalbuminuria or frank nephropathy in diabetics (Brenner et al., 2001; Lewis et al., 2001; Parving et al., 2001; Remuzzi et al., 2002; Ruggenenti et al., 2004). Nonetheless, a significant proportion of patients continue to progress to macroalbuminuria and overt nephropathy (Parving et al., 2001; Ruggenenti et al., 2004). Many patients on ACEI or ARB treatment exhibit “aldosterone escape,” whereby the initial response of decreased aldosterone and urinary albumin to creatinine ratio (UACR) is followed by a rebound of aldosterone and reversion of UACR to pre-treatment levels (Lijnen et al., 1982; Pitt, 1995). The mineralocorticoid receptor (MR) antagonist spironolactone effectively reduced UACR in diabetic nephropathy patients exhibiting “aldosterone escape” while on ACEI therapy (Sato et al., 2003). Moreover, eplerenone, another marketed MR antagonist, decreased UACR in type 2 diabetics with mild to moderate nephropathy to a greater extent than that achieved with ACEI treatment alone, and independently of changes in blood pressure (Epstein, 2006; Epstein et al., 2006). However, inhibition of the RAAS with ACEI, ARB, direct renin inhibitors (DRI) or MR antagonists especially in dual or triple combination, increased the risk of hyperkalemia in patients with heart failure (HF), or chronic kidney disease (McMahon, 2001; Sica, 2002; Palmer, 2004; Weir and Rolfe, 2010).
Aldosterone regulates Na+ and K+ balance and hence extracellular fluid volume and blood pressure (Levy et al., 2004; Muldowney et al., 2009). Under conditions of low blood volume or reduced renal perfusion, plasma angiotensin II (angII) and K+ levels increase and synergistically stimulate aldosterone secretion from the adrenals thereby promoting Na+ and water retention in the proximal tubule and driving sodium-dependent K+ secretion in the distal tubule of the kidney (Palmer, 2004). Thus, when renal filtration is severely compromised, as in patients with chronic kidney disease (CKD), cardiometabolic syndrome or HF, Na+ reabsorption in the proximal tubule becomes highly efficient. Under these conditions, urinary volume and Na+ delivery to the distal tubule become rate limiting for Na+-dependent K+ secretion and hyperkalemia may ensue (Palmer, 2004). Plasma aldosterone is increased in some patients with CKD (Berl et al., 1978; Hene et al., 1982), diabetes (Hollenberg et al., 2004), and metabolic syndrome (Bochud et al., 2006), and plasma aldosterone correlates positively with UACR (Bianchi et al., 2005) and negatively with glomerular filtration rates (GFR) in CKD patients (Quinkler et al., 2005). However, and given the pivotal role of aldosterone in regulating K+ excretion, the beneficial UACR lowering effect of MR antagonists may be offset by an increased risk of hyperkalemia. Therefore, a MR antagonist that decreases UACR without increasing the risk of hyperkalemia would be highly desirable for the treatment and prevention of diabetic nephropathy.
PF-03882845 is a selective and potent non-steroidal MR antagonist. To determine whether PF-03882845 differentiates from traditional steroidal MR antagonists, we sought to investigate the effect of PF-03882845 on UACR and electrolyte balance in preclinical studies. The concentration effect curves for the prevention of increases in UACR and for elevation of serum K+ were established for PF-03882845 and eplerenone in a rat model of aldosterone-induced renal damage. Additionally, the effect of the compounds on urinary Na+/K+ ratio, a surrogate biomarker for electrolyte disturbances, was investigated. Our data indicate a larger therapeutic index, calculated as the ratio of the EC50 for increasing serum K+ or urinary Na+/K+ to the EC50 for UACR lowering, for PF-03882845 compared to eplerenone.
Materials and Methods
All procedures were conducted in accordance with Institutional Animal Care and Use Committee (IACUC) guidelines and regulations at Pfizer Inc (Groton, CT).
Selectivity and Potency of PF-03882845 and Eplerenone
Huh7 cells were transiently transfected with a luciferase reporter gene under the control of a Gal4 response element (Gal4-RE-luc) and a plasmid containing the Gal4 DNA binding domain fused to MR ligand binding domain (Gal4-MR-LBD). Transfected cells were treated for 24 h with the EC80 concentration of activating ligand in the presence or absence of PF-03882845 or eplerenone in serum free medium. Luciferase activity was measured and the IC50 determined for each compound.
Effect of Chronic Administration of Eplerenone and PF-03882845 on Aldosterone-Mediated Renal Injury
Study A
Fifty five male uninephrectomized Sprague Dawley (SD) rats (Charles River, MA) were singly housed and provided food and water ad-libitum. Following an acclimation period, baseline 24 h urine samples were collected and analyzed for albumin and creatinine measurement. Baseline blood was taken by tail bleed for measurement of serum K+. Rats were then randomized into 5 groups. Four groups of rats (n = 11 per group) were implanted subcutaneously (SC) with Alzet mini-pumps (Alzet, Cupertino, CA) to deliver 2.5 μ L/h aldosterone (0.75 μ g/h) in 0.01% dimethylsulfoxide (DMSO) under isoflurane anesthesia. One group was sham operated (Veh Sham, n = 11). Immediately post-surgery, all rats were switched to a 6% high salt diet (Teklad TD.90230) and water containing 0.3% KCl ad-libitum. The following day, treatment with eplerenone was initiated in the 4 groups of rats that were implanted with aldosterone mini-pumps. Rats received either vehicle (Veh Aldo), or one of 3 doses of eplerenone: 15, 50, or 450 mg/kg BID by oral gavage. The sham-operated group also received vehicle (Veh Sham). Dosing was performed at 6 am and 4 pm for a duration of 27 days. Eplerenone was dissolved in vehicle comprised of 0.5% methyl cellulose, 0.1% polysorbate 80 in water.
Study B
For study B the same protocol as study A was used, but PF-03882845 was administered in lieu of eplerenone at doses of 5, 15, and 50 mg/kg BID by oral gavage. PF-03882845 was dissolved in vehicle comprised of 0.5% methyl cellulose, 0.1% polysorbate 80 in water.
Urine Collection
Twenty-four hours urine samples were collected at baseline, on Days 14 and 25 of treatment. Urine volume was recorded and samples were utilized for creatinine and albumin concentrations.
Plasma/Serum Collection
Rats were bled at baseline for plasma K+ measurement. Following the first dose of the respective compounds, rats were bled at 1, 2, 4, 7 h for drug levels measurements. On Day 14 of treatment, rats were bled at 7 h post the first dose of the day for plasma drug levels and serum K+ measurements. Additionally on Day 26, rats were bled at 1, 2, 4, 7 h post-dose for plasma drug levels measurement. On Day 27 (at necropsy, ~16 h post final dose), rats were bled for measurement of serum K+ and plasma drug levels measurements.
Tissue Collection
On Day 27 rats were euthanized with CO2, in accordance with IACUC guidelines and regulations at Pfizer Inc (Groton, CT). Kidney was removed, and weighed. A section of kidney was fixed in 10% formalin for histological analysis and a second portion of kidney (200 mg) was stored in a RNA stabilization reagent (RNAlater, Qiagen) for gene expression analysis.
Histology
Kidney sections were stained with a rabbit anti-collagen-IV antibody (Thermo Scientific) and detected with an anti-rabbit antibody conjugated to horseradish peroxidase (HRP). Slides were counterstained with hematoxylin. Data were expressed as percent collagen IV staining of the kidney cortex.
RT-PCR
Kidney samples (~30 mg) were removed from RNAlater and homogenized in 700 μ L of buffer RLT for RNA purification using an RNeasy Mini Kit (Qiagen) with on-column DNase treatment. Purified RNA was quantitated by NanoDrop. Samples (20 ng) were analyzed by RT-PCR to assess relative mRNA expression of Collagen-IV (Col4a1), osteopontin (Spp1), interleukin-6 (Il-6), intercellular adhesion molecule 1 (Icam-1), monocyte chemoattractant protein-1 (Mcp-1) and transforming growth factor β-1 (Tgf-β 1) using gene expression assays (Applied Biosystems), RNA to Ct Kit (Applied Biosystems) and the SDS-7900HT (Applied Biosystems). Expression data were normalized to mRNA expression of the endogenous control hypoxanthine phosphoribosyltransferase 1 (Hprt1). Data were expressed as relative quantitation (RQ) units using Veh Sham animal No. 2 as comparator (ie RQ equal to “1”).
Biomarker Measurements
Serum K+, urinary albumin and creatinine were measured using the Siemens Advia 2400 Chemistry Analyzer and reagents from Siemens Healthcare Diagnostics (Washington, DC). UACR was calculated as the ratio of urinary albumin concentration to creatinine concentration. Plasma osteopontin was measured on day 27 samples using an enzyme immunoadsorbent assay (Enzo Life Sciences, Inc., Farmingdale NY).
Compound Exposure
Plasma was analyzed for eplerenone or PF-03882845 using a non-GLP LC/MS/MS method. All calculations were performed in Watson LIMS (v7.2, Thermo, Inc, Philadelphia, PA) and individual data were used in all pharmacokinetic analyses. The area under the plasma concentration-time curve (AUC(0-last)) was estimated using the linear trapezoidal rule.
Pharmacokinetic/Pharmacodynamic (PK-PD) Modeling for Study A and Study B
Population PK-PD modeling was conducted in NONMEM (version VI 2.0; GloboMax LLC. Hanover, MD). Analysis was conducted in a piece-wise manner whereby individual pharmacokinetic parameters estimated for each animal in step 1 were fixed during estimation of pharmacodynamic parameters in the second step.
The pharmacokinetics of eplerenone and PF-03882845 were characterized with one and two compartmental models, respectively. In both cases first order absorption and elimination was assumed. Decreases in exposure upon multiple dosing for both compounds were characterized assuming that clearance was induced between Days 1 and 7 and remained constant thereafter. Population variance on the rate of absorption, elimination and volume of distribution were assumed in order to provide the best possible description of exposure in individual animals. Individual pharmacokinetic parameter estimates were carried forward in modeling the PK-PD relationship to UACR and serum K+.
Increases in UACR in vehicle controls were modeled with an indirect response model where the initial value (i.e., UACR at the start of the study) represents a non-steady state condition that varies between individual rats. Drug effect was parameterized such that the increase in UACR was inhibited via an indirect response Imax model (Equation 1). Both eplerenone and PF-03882845 PK-PD data were modeled simultaneously. The Imax, Kin, and Kout parameters were assumed to be drug independent and only the IC50 parameter was assumed to be drug dependent.
For modeling purposes, serum K+ data were transformed according to Equation 2. This formula allows for the calculation of drug-induced changes in serum K+ (from baseline), which are above and beyond that measured in the vehicle group. T × T and T × 0 represent the measured serum K+ in each drug treated individual at times equal to “T” and 0, respectively. VehT and Veh0 represent the mean measured serum K+ in the vehicle group at times equal to “T” and 0, respectively.
The relationship between drug exposure and Δ serum K+ was modeled using a modified indirect response model whereby drug creates a concentration-dependent input rate (Equation 3). Both eplerenone and PF-03882845 PK-PD data were modeled simultaneously. The Kin, max, and Kout parameters were assumed to be drug independent and only the EC50 parameter was assumed to be drug dependent.
Acute Effect of MR Antagonists on Urinary Na+/K+ Ratio
Male, carotid artery cannulated, Sprague Dawley (SD) rats between 11 and 12 weeks of age were obtained (Charles River, MA), singly housed and acclimated in metabolic chambers for one week prior to the start of the experiment. Water and food (Purina 5001 pellets) were provided ad-libitum throughout the study. In one study, rats were randomly assigned to receive either vehicle (0.5% methyl cellulose, 0.1% polysorbate 80) alone or eplerenone at one of 3 doses: 5, 30 or 300 mg/kg. In a second study, rats were randomly assigned to receive either vehicle (0.5% methyl cellulose, 0.1% polysorbate 80) or PF-03882845 at one of 3 doses: 3, 10 and 30 mg/kg. On the evening prior to treatment administration, baseline urine was collected from 4 pm until 8 am on the day of treatment. Animals were then orally gavaged with their respective treatments at 8 am and urine collected at intervals of 0–2, 2–4, and 4–7 h post dose. After collection, urine was stored at −80°C until analyzed. Baseline blood was collected from carotid artery cannulas prior to dosing, and at 1, 2, 4, and 7 h post dose. Samples were centrifuged and plasma was collected and stored at −80°C for compound exposure measurements using the methodology described above. Urine sodium and K+ were measured using the Siemens Advia 2400 Chemistry Analyzer with reagents from Siemens Healthcare Diagnostics. Data were expressed as urinary Na+/K+ ratio.
PK Modeling for Eplerenone
A two compartment distribution model with zero order absorption was used (1 h duration = observed Tmax). Clearance (CL) was estimated as a function of dose:
where Theta(1) is CL at 30 mg/kg, and PWR is a power function allowing non-linear change in CL with dose. Conditional estimates of CL, V1, and Q were obtained. A proportional residual error model was used.
PK Modeling for PF-03882845
A one compartment distribution model with first order absorption was used. Conditional estimates of KA, CL, and V1 were obtained. A proportional residual error model was used.
PK/PD Modeling of Urinary Na+/K+
A two compartment linear model with first order absorption was used for both drugs. Conditional PK parameter estimates were read in with the input data. For PF-03882845, drug was dosed into compartment 1 (depot); Q was fixed to 0 and V2 was fixed to 1. For eplerenone, KA was fixed to 1 and drug was “infused” into compartment 2 over 1 h. The PD endpoint was the “double delta” in urinary Na+/K+ ratio (measured at the midpoint of the urine collection interval). Mean vehicle Na+/K+ ratio at time = 0 (Veh0) and at each urine collection interval (VehT) was estimated using vehicle data from both drug studies combined. Individual animal Na+/K+ ratio observed at time = 0 (T × 0) and at the midpoint of collection intervals (T × T) were used to calculate the “double delta” change in Na+/K+ ratio as follows:
Using this approach each animal's time zero response = 0.
Statistical Analyses
UACR and serum K+ data were analyzed using a mixed ANOVA model accounting for repeated measures where treatment group, days post operation and treatment day interaction effect were introduced as a fixed factor whereas the animal factor was considered as a random effect nested in the treatment factor. The baseline value for each animal was introduced as a covariate. The primary comparisons were vehicle sham group vs. the other four groups and the vehicle aldosterone group vs. treatment groups. A two-sided 5% test was conducted.
For Study A and Study B, kidney gene expression data, collagen IV staining data and plasma osteopontin data were analyzed with a One-Way ANOVA and a Tukey post test using GraphPad Prism 5. When unequal variances were observed, data were log-transformed prior to analysis. The primary comparisons were vehicle sham group vs. the other four groups and the vehicle aldosterone group vs. treatment groups.
Results
Potency of PF-03882845 and Eplerenone
PF-03882845 exhibited a greater potency compared to eplerenone in a serum free functional reporter assay utilizing the human MR ligand binding domain [PF-03882845 geometric mean IC50 = 0.755 nM (90% confidence interval: 0.501–1.11 nM; n = 6), eplerenone IC50 = 109 nM (90% confidence interval: 79.3–150 nM; n = 6)].
Effect of Eplerenone and PF-03882845 on UACR and Serum K+
Baseline UACR levels were not different amongst groups. Aldosterone infusion increased UACR levels relative to the vehicle sham group in both Study A and Study B by Day 14 (Figures 1A, 2A, respectively). This effect was more pronounced by Day 26 (Figures 1B, 2B, respectively). The highest dose of eplerenone (450 mg/kg BID) prevented the increase on Days 14 and 26 of treatment (Figures 1A,B) while all doses of PF-03882845 were efficacious in blunting UACR elevations (Figures 2A,B). Aldosterone decreased serum K+ on Days 14 and 26 in Study A (Figures 3A,B) but only on Day 26 in Study B (Figures 4A,B). By Day 26, eplerenone prevented the aldosterone-induced decrease in serum K+ at the doses of 50 mg/kg and 450 mg/kg BID (Figure 3B). A similar effect was observed with PF-03882845 at the highest dose tested (50 mg/kg BID; Figure 4).
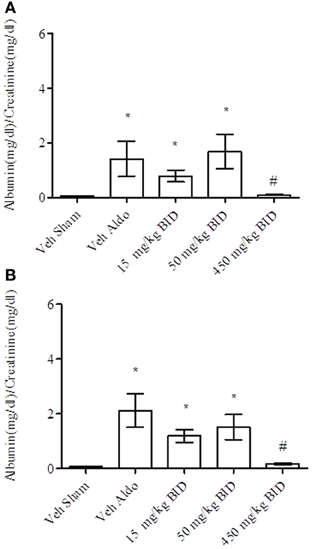
Figure 1. Effect of eplerenone on UACR on day 14 (A) and day 26 (B). Data are represented as mean ± standard error of the mean. See Materials and Methods for statistical analysis. *Indicates a significant difference from the vehicle sham (Veh Sham) group; #Indicates a significant difference from the vehicle + aldosterone (Veh Aldo) group.
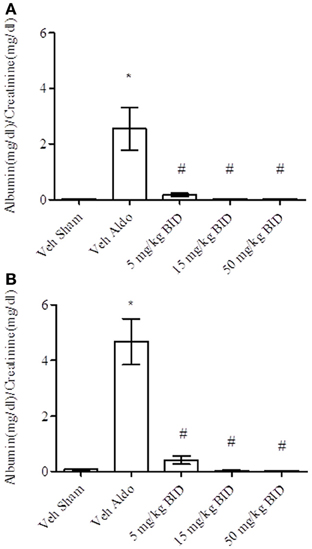
Figure 2. Effect of PF-03882845 on UACR on day 14 (A) and day 26 (B). Data are represented as mean ± standard error of the mean. See Materials and Methods for statistical analysis. *Indicates a significant difference from the vehicle sham (Veh Sham) group; #Indicates a significant difference from the vehicle + aldosterone (Veh Aldo) group.
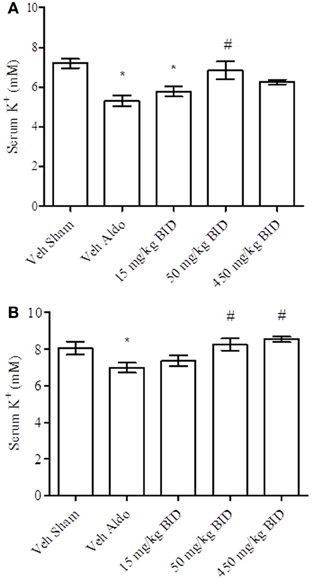
Figure 3. Effect of eplerenone on serum K+ on day 14 (A) and day 26 (B). Data are represented as mean ± standard error of the mean. See Materials and Methods for statistical analysis. *Indicates a significant difference from the vehicle sham (Veh Sham) group; # Indicates a significant difference from the vehicle + aldosterone (Veh Aldo) group.
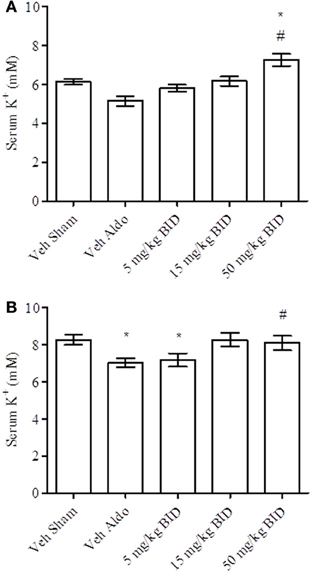
Figure 4. Effect of PF-03882845 on serum K+ on day 14 (A) and day 26 (B). Data are represented as mean ± standard error of the mean. See Materials and Methods for statistical analysis. *Indicates a significant difference from the vehicle sham (Veh Sham) group; #Indicates a significant difference from the vehicle + aldosterone (Veh Aldo) group.
PK-PD Modeling of UACR and Serum K+
Model estimated values of the EC50 for UACR and for serum K+ are depicted in Figure 5 and Table 1. Eplerenone and PF-03882845 resulted in overlapping concentration effect curves for serum K+ (Figure 5B) and the derived EC50 values were not significantly different (Table 1). By contrast, PF-03882845 was more potent than eplerenone in preventing the rise in UACR as observed in the modeled concentration-effect curves (Figure 5A). The resulting EC50 was 64-fold lower compared to eplerenone (Figure 5A and Table 1). The therapeutic index (TI), calculated as the ratio of EC50 for increasing serum K+ to the EC50 for suppressing UACR, was 1.47 for eplerenone and 84 for PF-03882845 (Table 1).
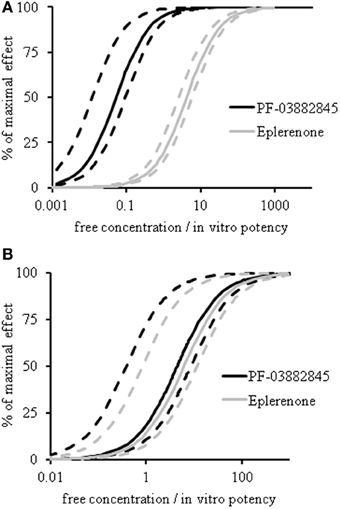
Figure 5. PK-PD modeling of UACR and serum potassium. Concentration-effect curves of eplerenone (gray lines) and PF-03882845 (black lines) for UACR (A) and serum K+ (B) following 27 days of treatment. Dashed lines indicate the 95% upper and lower confidence intervals. Data were modeled using an indirect response model. A double delta equation was used to express the pharmacodynamic response for serum K+.
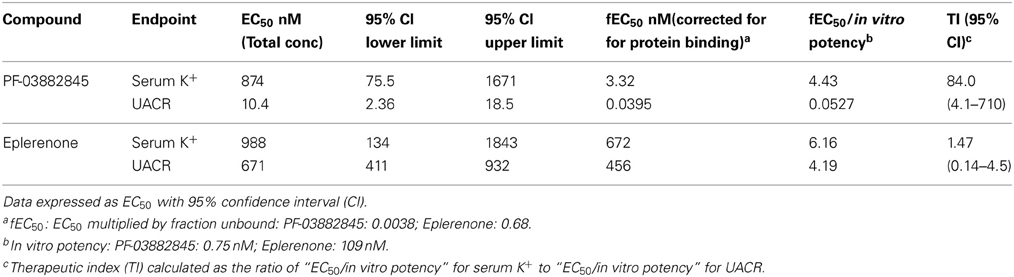
Table 1. Chronic effect of PF-03882845 and eplerenone on UACR and serum K+.
Effect of PF-03882845 and Eplerenone on Collagen IV Staining in Kidney
Continuous infusion with aldosterone significantly increased collagen IV staining in kidney cortex (Figures 6B,F) vs. the vehicle sham group (Figures 6A,E) in Study A and Study B, respectively. Eplerenone prevented the increase in collagen IV staining at the 450 mg/kg BID dose only (Figures 6C,D,J) while all three doses of PF-03882845 were efficacious (Figures 6G–I, 6K).
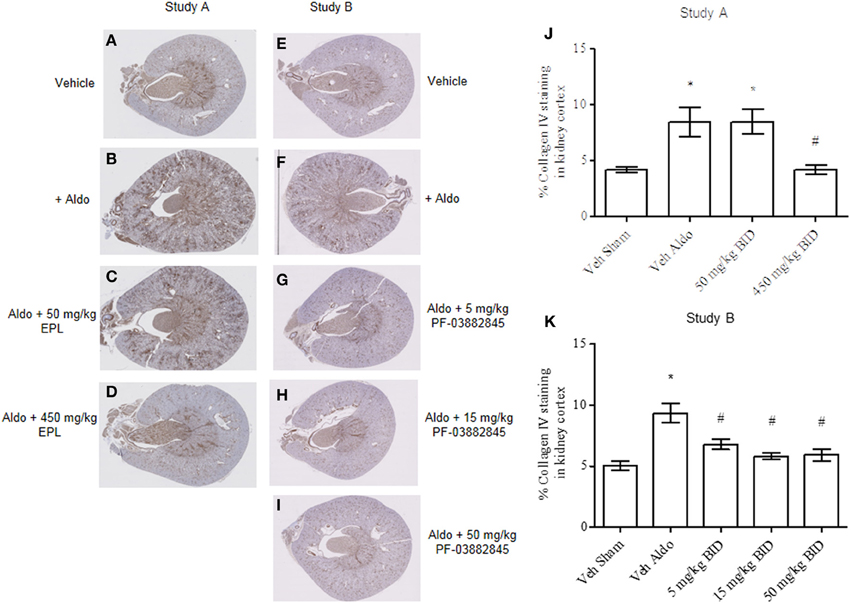
Figure 6. Representative images of collagen IV immunohistochemical localization in kidney cortex. Study A, vehicle sham (A), vehicle + aldosterone (B), 50 mg/kg BID eplerenone (C), and 450 mg/kg BID eplerenone (D), groups following 27 days of treatment; and Study B, kidney cortex from sham vehicle (E), vehicle + aldosterone (F), 5 mg/kg BID PF-03882845 (G), 15 mg/kg BID PF-03882845 (H), and 50 mg/kg BID PF-03882845 (I) following 27 days of treatment. Data expressed as % collagen IV staining in kidney cortex and represented as mean ± standard error of the mean: Study A (J) and Study B (K). See Materials and Methods for statistical analysis. *Indicates a significant difference from the vehicle sham (Veh Sham) group; #Indicates a significant difference from the vehicle + aldosterone (Veh Aldo) group.
Effect of PF-03882845 and Eplerenone on Gene Expression in Kidney
In studies A and B, aldosterone significantly increased mRNA expression of collagen 4a1, transforming growth factor β-1 (Tgf-β 1), interleukin-6 (Il-6), intercellular adhesion molecule-1 (Icam-1), monocyte chemoattractant protein-1 (Mcp-1), and osteopontin (Spp1) in the kidney on Day 27. This was prevented by PF-03882845 at all doses tested (Table 2), while eplerenone prevented the rise in these genes only at the highest dose tested (4.0 mg/kg BID) (Table 3).
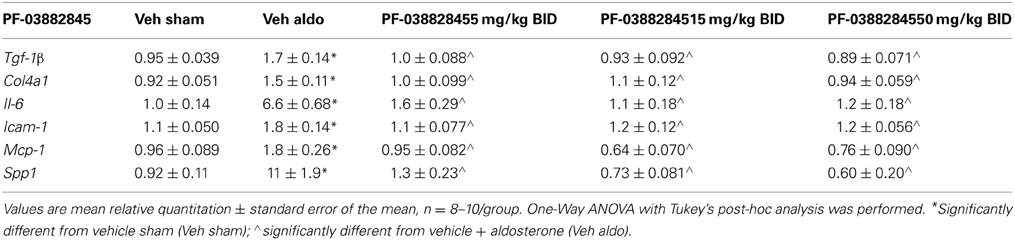
Table 2. Effect of PF-03882845 on relative gene expression in kidney on day 27.
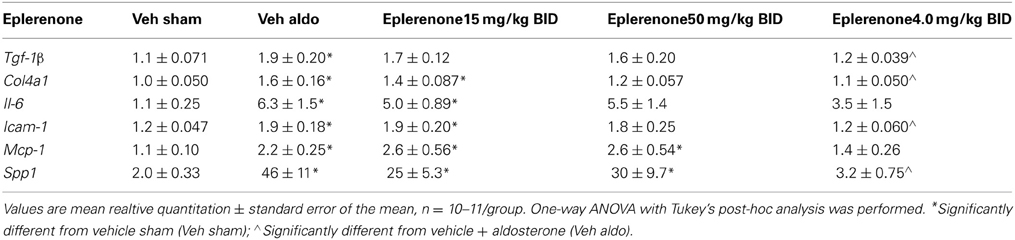
Table 3. Effect of eplerenone on gene expression in kidney on day 27.
Effect of PF-03882845 and Eplerenone on Plasma Osteopontin
Aldosterone increased plasma osteopontin levels in Study A (vehicle: 93.6 ± 16.4 ng/ml; vehicle + aldosterone: 345 ± 205 ng/ml (mean ± SEM)) and this effect was blunted by eplerenone at the 450 mg/kg BID dose (62.2 ± 7.98 ng/ml), while the doses of 15 or 50 mg/kg BID were not different from the vehicle + aldosterone group (eplerenone 15 mg/kg BID: 204 ± 35.1; eplerenone 50 mg/kg BID: 234 ± 85.6 ng/ml). In study B, aldosterone increased plasma osteopontin levels (vehicle: 87.3 ± 13.9 ng/ml; vehicle + aldosterone: 494 ± 90.1 ng/ml). This effect was suppressed at all doses of PF-03882845 (PF-03882845.5 mg/kg BID: 108 ± 13.5, 15 mg/kg BID: 77.1 ± 15.7 and 50 mg/kg BID: 82.4 ± 17.0 ng/ml, respectively).
Acute Effect of PF-03882845 and Eplerenone on Urinary Na+/K+ Ratio
Urinary Na+/K+ ratio was assessed in SD rats following single doses of PF-03882845 or eplerenone. Concentration-effect curves were generated using an indirect response model. In agreement with the chronic effects of these compounds on serum K+ levels, the EC50 values for the acute effect on urinary Na+/K+ ratios were not different when corrected for the respective free fraction and in vitro potency (PF-03882845 (mean [95% confidence interval]: 3.10 [1.58–4.62]) and eplerenone (5.92 [2.22–9.63]) (Figure 7).
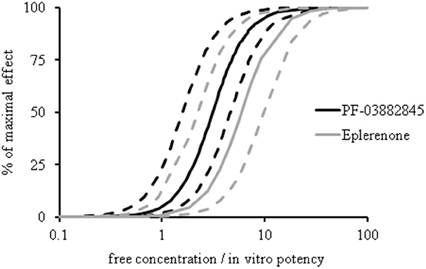
Figure 7. Effect of eplerenone and PF-03882845 on urinary Na+/K+ ratio. SD rats received single doses of 0, 5, 30, or 3.0 mg/kg eplerenone or 0, 3, 10, or 30 mg/kg PF-03882845 and urinary Na+/K+ was assessed at 0–2, 2–4, and 4–7 h post-dose. Both compounds yielded analogous concentration-effect curves for urinary Na+/K+. Data were expressed as free concentration normalized to in vitro IC50 (0.75 nM for PF-03882845 and 109 nM for eplerenone in a serum free assay). Data were modeled using an indirect response model. Dashed lines indicate the 95% lower and upper confidence boundary limits.
Discussion
We investigated whether PF-03882845, a novel non-steroidal MR antagonist, differentiates from traditional steroidal MR antagonists, such as eplerenone. Our findings indicate that PF-03882845 exhibits a greater TI, calculated as the ratio of the EC50 for increasing serum K+ to the EC50 for UACR lowering, compared to eplerenone. Thus, our results support the development of PF-03882845 for the treatment of diabetic nephropathy, a condition in which the patient is particularly at risk for hyperkalemia.
Aldosterone binding to the MR mediates sodium and water retention in exchange for K+ secretion in the renal tubules. These effects appear to be downstream of the serum- and glucocorticoid-inducible kinase-1 (SGK1) (Lang et al., 2006; Terada et al., 2008). Indeed, aldosterone has been demonstrated to stimulate Sgk1 mRNA expression (Naray-Fejes-Toth et al., 1999) and phosphorylation (Flores et al., 2005) in cortical collecting duct cells. Activated SGK1 inhibits turnover of the epithelial sodium channel (ENaC) thereby enhancing ENaC abundance at the plasma membrane (Butterworth et al., 2009) via phosphorylation and inhibition of the ubiquitinase Nedd4-2 which marks ENaC for proteasomal degradation (Flores et al., 2005). Sodium transport is also regulated by the With-No-K (lysine) kinase (WNK) family of kinases amongst which WNK4 has been demonstrated to phosphorylate and inhibit ENaC and the Na+ and Cl− cotransporter (NCC) (Hoorn et al., 2009). In turn, WNK4 is phosphorylated and inhibited by SGK1 and by the “long form”-WNK1 (L-WNK1). Thus, aldosterone increases Na+ retention via SGK1 mediated phosphorylation/inhibition of WNK4, thereby relieving inhibition of ENaC and NCC (Hoorn et al., 2009; Vallon et al., 2009).
Aldosterone modulates K+ secretion via SGK1 upregulation of the renal outer medullary potassium (ROMK) channels in the thick ascending limb of Henle's loop, the distal tubule and in cortical collecting duct cells. L-WNK1 and WNK4 downregulate ROMK by stimulating clathrin-dependent endocytosis and this is abrogated by SGK1 mediated phosphorylation of Wnk4 (Welling and Ho, 2009). In addition to effects on ENaC and ROMK, aldosterone increases K+ secretion in exchange for sodium uptake via the basolateral Na+/K+ ATPase (Djelidi et al., 2001).
In the present study, aldosterone treatment of uninephrectomized rats depressed serum K+ levels as anticipated. In contrast, treatment with the MR antagonists blunted this effect, likely through preventing aldosterone-induction of ENaC, ROMK and the Na+/K+ ATPase. PK-PD modeling demonstrated that PF-03882845 and eplerenone were equipotent for increasing serum K+ levels relative to the vehicle + aldosterone group and for increasing the urinary Na+/K+ ratio following single doses in normal SD rats. Effects of MR antagonists on urinary Na+/K+ ratio have been well-characterized in rodents as well as humans (McInnes et al., 1982). Early on, Kagawa demonstrated blockade of mineralocorticoid activity in adrenalectomized rats by measuring urinary Na+/K+ ratio (Kagawa, 1960). This method was further simplified by Brandish et al. who revealed similar findings in rats with intact adrenal glands (Brandish et al., 2008). We have previously demonstrated translatability of this biomarker from rat to humans using preclinical and clinical data generated with eplerenone (Eudy et al., 2011). Our present findings extend these results by demonstrating similar concentration effect curves for urinary Na+/K+ ratio generated in normal SD rats following single doses of MR antagonists, and for serum K+ in an animal model of nephropathy following chronic exposure to the compounds. Thus, urinary Na+/K+ ratio in a healthy state appears to be a useful biomarker for assessing the risk of hyperkalemia in a renally compromised condition.
Interestingly, PK-PD modeling revealed that PF-03882845 was more potent than eplerenone in lowering UACR despite equipotent effects on serum potassium, thereby yielding a 57-fold greater therapeutic index against hyperkalemia compared to eplerenone. Albuminuria, a widely used surrogate biomarker of renal damage, has been previously demonstrated to be decreased by treatment with MR antagonists in patients with nephropathy (Sato et al., 2003; Epstein et al., 2006) and rodents with renal impairment (Blasi et al., 2003), supporting its modulation by aldosterone. Our results agree with these findings, and further highlight the potential of PF-03882845 in preventing/reducing renal damage, while posing less risk for hyperkalemia compared to a traditional MR antagonist. Concurrent with its effect on UACR, PF-03882845 was more potent than eplerenone in suppressing collagen IV gene and protein expression in kidney cortex. Collagen IV has been associated with glomerulosclerosis in experimental models (Floege et al., 1992; Bergijk et al., 1998; Lee et al., 1998) and humans (Kim et al., 1991; Tamsma et al., 1994; Esposito et al., 1996) with kidney disease. Renal biopsies from patients with diabetic nephropathy indicated a clear increase in α 2 type IV collagen gene and protein in glomeruli compared to controls (Adler et al., 2000). Importantly, aldosterone has been shown to stimulate collagen synthesis in rat renal fibroblasts (Nagai et al., 2005) and aldosterone infusion stimulated collagen III gene expression as assessed by in situ hybridization (Blasi et al., 2003). We expand on these findings by demonstrating increases of collagen IV gene and protein expression in kidney cortex with aldosterone infusion, and its blunting by the novel MR antagonist PF-03882845. Furthermore, our data indicate a greater potency of PF-03882845 compared to eplerenone for suppressing aldosterone-induced expression of proinflammatory and profibrotic genes in the kidney such as Tgf-β, Il-6, Icam-1, Mcp-1 and Spp1. ICAM-1 has been implicated in glomerular inflammation (Riser et al., 2001). Il-6 has been shown to upregulate MCP-1 in human mesangial cells (Coletta et al., 2000). The latter was upregulated in urine of patients with diabetic nephropathy (DN) (Wada et al., 2000) and was decreased by spironolactone treatment (Takebayashi et al., 2006) suggesting regulation by the MR. TGF-β 1 has been demonstrated to reduce collagenase production thereby leading to accumulation of extracellular matrix components (Wolf, 2006). Importantly, Tgf-β 1 is increased in plasma of type 2 diabetics (Pfeiffer et al., 1996) and urine of type 2 diabetics with clinical diabetic nephropathy (Bertoluci et al., 2006) further supporting its role in the pathogenesis of this disease. In accordance with our findings, aldosterone infusion has been shown to increase TGF-β 1 protein expression in rat kidney (Juknevicius et al., 2004) and both TGF-β 1 and aldosterone independently stimulated connective tissue growth factor (CTGF), an inducer of renal fibroblast growth and synthesis of extracellular matrix proteins (Yokoi et al., 2001). In the present studies, aldosterone infusion also resulted in an increased intra-renal gene expression of osteopontin (Spp1), an extracellular matrix protein, chemokine and growth factor recently shown to be regulated by the MR in renal mesangial cells (Gauer et al., 2008). This effect was prevented more potently by PF-03882845 as compared to eplerenone. Importantly, plasma levels of osteopontin mirrored these findings. These results further support a greater potency of PF-03882845 vs. eplerenone in preventing renal damage as plasma levels of osteopontin have been shown to correlate with progression of diabetic nephropathy (Yamaguchi et al., 2004).
While our results clearly indicate a greater potency of PF-03882845 in reducing UACR compared to eplerenone, leading to an increased TI against hyperkalemia, mechanisms underlying these findings are yet to be determined. At least 3 hypotheses may explain the present data. First, PF-03882845 may differentially affect coactivator recruitment in different cell types leading to a greater repression of profibrotic pathways. Second, a greater uptake of PF-03882845 relative to eplerenone in cell types involved in filtration and fibrosis such as mesangial cells and/or podocytes compared to epithelial cells that regulate electrolyte balance could account for these findings. However, to-date, no evidence of active uptake of PF-03882845 into cells has been demonstrated. Third, the increased potency of PF-03882845 against fibrosis may be driven through off-target effects. Recently, aldosterone has been demonstrated to elicit effects on vascular smooth muscle cells at physiological concentrations through the GPR30 receptor (Gros et al., 2011). Further work is warranted to elucidate these findings.
In conclusion, PF-03882845, a novel non-steroidal MR antagonist, appears to present less of a risk for hyperkalemia while maintaining efficacy in preventing renal damage in a rodent model of nephropathy. Should these results translate into humans, PF-03882845 may present a safe and efficacious alternative for the treatment of chronic kidney disease.
Conflict of Interest Statement
The authors are or were previously employed by Pfizer Inc, and possibly own stock options from Pfizer Inc. The authors and editor declare that while the author C. M. Boustany-Kari and the editor R. Fryer are currently employed by Boehringer-Ingelheim Pharmaceuticals, Inc., USA, there has been no conflict of interest during the review and handling of this manuscript.
Acknowledgments
We thank Catherine Ambler and Anthony Mahan in Research Formulations for formulating all the compounds used in this study, Jennifer Stanislawski in Drug Safety Research and Development (DSRD) for assaying urine samples for sodium and potassium concentrations. Jonathan Bauman and Timothy Strelevitz, in Pharmacokinetics, Pharmacodynamics, and Metabolism (PDM) for contribution to preclinical PK analysis of both compounds.
References
Adler, S. G., Feld, S., Striker, L., Striker, G., Lapage, J., Esposito, C., et al. (2000). Glomerular type IV collagen in patients with diabetic nephropathy with and without additional glomerular disease. Kidney Int. 57, 2084–2092. doi: 10.1046/j.1523-1755.2000.00058.x
Bergijk, E. C., Van Alderwegen, I. E., Baelde, H. J., De Heer, E., Funabiki, K., Miyai, H., et al. (1998). Differential expression of collagen IV isoforms in experimental glomerulosclerosis. J. Pathol. 184, 307–315. doi: 10.1002/(SICI)1096-9896(199803)184:3<307::AID-PATH5>3.3.CO;2-N
Berl, T., Katz, F. H., Henrich, W. L., De Torrente, A., and Schrier, R. W. (1978). Role of aldosterone in the control of sodium excretion in patients with advanced chronic renal failure. Kidney Int. 14, 228–235. doi: 10.1038/ki.1978.114
Bertoluci, M. C., Uebel, D., Schmidt, A., Thomazelli, F. C., Oliveira, F. R., and Schmid, H. (2006). Urinary TGF-beta1 reduction related to a decrease of systolic blood pressure in patients with type 2 diabetes and clinical diabetic nephropathy. Diabetes Res. Clin. Pract. 72, 258–264. doi: 10.1016/j.diabres.2005.10.027
Bianchi, S., Bigazzi, R., and Campese, V. M. (2005). Antagonists of aldosterone and proteinuria in patients with CKD: an uncontrolled pilot study. Am. J. Kidney Dis. 46, 45–51. doi: 10.1053/j.ajkd.2005.03.007
Blasi, E. R., Rocha, R., Rudolph, A. E., Blomme, E. A., Polly, M. L., and Mcmahon, E. G. (2003). Aldosterone/salt induces renal inflammation and fibrosis in hypertensive rats. Kidney Int. 63, 1791–1800. doi: 10.1046/j.1523-1755.2003.00929.x
Bochud, M., Nussberger, J., Bovet, P., Maillard, M. R., Elston, R. C., Paccaud, F., et al. (2006). Plasma aldosterone is independently associated with the metabolic syndrome. Hypertension 48, 239–245. doi: 10.1161/01.HYP.0000231338.41548.fc
Brandish, P. E., Chen, H., Szczerba, P., and Hershey, J. C. (2008). Development of a simplified assay for determination of the antimineralocorticoid activity of compounds dosed in rats. J. Pharmacol. Toxicol. Methods 57, 155–160. doi: 10.1016/j.vascn.2008.01.002
Brenner, B. M., Cooper, M. E., De Zeeuw, D., Keane, W. F., Mitch, W. E., Parving, H. H., et al. (2001). Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N. Engl. J. Med. 345, 861–869. doi: 10.1056/NEJMoa011161
Butterworth, M. B., Edinger, R. S., Frizzell, R. A., and Johnson, J. P. (2009). Regulation of the epithelial sodium channel by membrane trafficking. Am. J. Physiol. Renal Physiol. 296, F10–F24. doi: 10.1152/ajprenal.90248.2008
Coletta, I., Soldo, L., Polentarutti, N., Mancini, F., Guglielmotti, A., Pinza, M., et al. (2000). Selective induction of MCP-1 in human mesangial cells by the IL-6/sIL-6R complex. Exp. Nephrol. 8, 37–43. doi: 10.1159/000059327
Djelidi, S., Beggah, A., Courtois-Coutry, N., Fay, M., Cluzeaud, F., Viengchareun, S., et al. (2001). Basolateral translocation by vasopressin of the aldosterone-induced pool of latent Na-K-ATPases is accompanied by alpha1 subunit dephosphorylation: study in a new aldosterone-sensitive rat cortical collecting duct cell line. J. Am. Soc. Nephrol. 12, 1805–1818.
Epstein, M. (2006). Aldosterone blockade: an emerging strategy for abrogating progressive renal disease. Am. J. Med. 119, 912–919. doi: 10.1016/j.amjmed.2006.03.038
Epstein, M., Williams, G. H., Weinberger, M., Lewin, A., Krause, S., Mukherjee, R., et al. (2006). Selective aldosterone blockade with eplerenone reduces albuminuria in patients with type 2 diabetes. Clin. J. Am. Soc. Nephrol. 1, 940–951. doi: 10.2215/CJN.00240106
Esposito, C., Striker, L. J., Patel, A., Peten, E., Liu, Z. H., Sakai, H., et al. (1996). Molecular analysis of glomerular diseases in renal biopsies: initial results of a collaborative international study. The international study group for molecular study of kidney biopsies. Proc. Assoc. Am. Physicians 108, 209–217.
Eudy, R. J., Sahasrabudhe, V., Sweeney, K., Tugnait, M., King-Ahmad, A., Near, K., et al. (2011). The use of plasma aldosterone and urinary sodium to potassium ratio as translatable quantitative biomarkers of mineralocorticoid receptor antagonism. J. Transl. Med. 9, 180. doi: 10.1186/1479-5876-9-180
Floege, J., Alpers, C. E., Burns, M. W., Pritzl, P., Gordon, K., Couser, W. G., et al. (1992). Glomerular cells, extracellular matrix accumulation, and the development of glomerulosclerosis in the remnant kidney model. Lab. Invest. 66, 485–497.
Flores, S. Y., Loffing-Cueni, D., Kamynina, E., Daidie, D., Gerbex, C., Chabanel, S., et al. (2005). Aldosterone-induced serum and glucocorticoid-induced kinase 1 expression is accompanied by Nedd phosphorylation and increased Na+ transport in cortical collecting duct cells. J. Am. Soc. Nephrol. 16, 2279–2287. doi: 10.1681/ASN.2004100828
Gauer, S., Hauser, I. A., Obermuller, N., Holzmann, Y., Geiger, H., and Goppelt-Struebe, M. (2008). Synergistic induction of osteopontin by aldosterone and inflammatory cytokines in mesangial cells. J. Cell. Biochem. 103, 615–623. doi: 10.1002/jcb.21433
Gros, R., Ding, Q., Sklar, L. A., Prossnitz, E. E., Arterburn, J. B., Chorazyczewski, J., et al. (2011). GPR30 expression is required for the mineralocorticoid receptor-independent rapid vascular effects of aldosterone. Hypertension 57, 442–451. doi: 10.1161/HYPERTENSIONAHA.110.161653
Hene, R. J., Boer, P., Koomans, H. A., and Mees, E. J. (1982). Plasma aldosterone concentrations in chronic renal disease. Kidney Int. 21, 98–101. doi: 10.1038/ki.1982.14
Hollenberg, N. K., Stevanovic, R., Agarwal, A., Lansang, M. C., Price, D. A., Laffel, L. M., et al. (2004). Plasma aldosterone concentration in the patient with diabetes mellitus. Kidney Int. 65, 1435–1439. doi: 10.1111/j.1523-1755.2004.00524.x
Hoorn, E. J., Van Der Lubbe, N., and Zietse, R. (2009). The renal WNK kinase pathway: a new link to hypertension. Nephrol. Dial. Transplant. 24, 1074–1077. doi: 10.1093/ndt/gfp013
Juknevicius, I., Segal, Y., Kren, S., Lee, R., and Hostetter, T. H. (2004). Effect of aldosterone on renal transforming growth factor-beta. Am. J. Physiol. Renal Physiol. 286, F1059–F1062. doi: 10.1152/ajprenal.00202.2003
Kagawa, C. M. (1960). Blocking the renal electrolyte effects of mineralocorticoids with an orally active steroidal spirolactone. Endocrinology 67, 125–132. doi: 10.1210/endo-67-1-125
Kim, Y., Kleppel, M. M., Butkowski, R., Mauer, S. M., Wieslander, J., and Michael, A. F. (1991). Differential expression of basement membrane collagen chains in diabetic nephropathy. Am. J. Pathol. 138, 413–420.
Lang, F., Bohmer, C., Palmada, M., Seebohm, G., Strutz-Seebohm, N., and Vallon, V. (2006). (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol. Rev. 86, 1151–1178. doi: 10.1152/physrev.00050.2005
Lee, G. S., Nast, C. C., Peng, S. C., Artishevsky, A., Ihm, C. G., Guillermo, R., et al. (1998). Differential response of glomerular epithelial and mesangial cells after subtotal nephrectomy. Kidney Int. 53, 1389–1398. doi: 10.1046/j.1523-1755.1998.00871.x
Levy, D. G., Rocha, R., and Funder, J. W. (2004). Distinguishing the antihypertensive and electrolyte effects of eplerenone. J. Clin. Endocrinol. Metab. 89, 2736–2740. doi: 10.1210/jc.2003-032149
Lewis, E. J., Hunsicker, L. G., Clarke, W. R., Berl, T., Pohl, M. A., Lewis, J. B., et al. (2001). Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N. Engl. J. Med. 345, 851–860. doi: 10.1056/NEJMoa011303
Lijnen, P., Staessen, J., Fagard, R., and Amery, A. (1982). Increase in plasma aldosterone during prolonged captopril treatment. Am. J. Cardiol. 49, 1561–1563. doi: 10.1016/0002-9149(82)90390-3
McInnes, G. T., Perkins, R. M., Shelton, J. R., and Harrison, I. R. (1982). Spironolactone dose-response relationships in healthy subjects. Br. J. Clin. Pharmacol. 13, 513–518.
McMahon, E. G. (2001). Recent studies with eplerenone, a novel selective aldosterone receptor antagonist. Curr. Opin. Pharmacol. 1, 190–196. doi: 10.1016/S1471-4892(01)00022-4
Muldowney, J. A. 3rd., Schoenhard, J. A., and Benge, C. D. (2009). The clinical pharmacology of eplerenone. Expert Opin. Drug Metab. Toxicol. 5, 425–432. doi: 10.1517/17425250902837973
Nagai, Y., Miyata, K., Sun, G. P., Rahman, M., Kimura, S., Miyatake, A., et al. (2005). Aldosterone stimulates collagen gene expression and synthesis via activation of ERK1/2 in rat renal fibroblasts. Hypertension 46, 1039–1045. doi: 10.1161/01.HYP.0000174593.88899.68
Naray-Fejes-Toth, A., Canessa, C., Cleaveland, E. S., Aldrich, G., and Fejes-Toth, G. (1999). sgk is an aldosterone-induced kinase in the renal collecting duct. Effects on epithelial na+ channels. J. Biol. Chem. 274, 16973–16978. doi: 10.1074/jbc.274.24.16973
Palmer, B. F. (2004). Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N. Engl. J. Med. 351, 585–592. doi: 10.1056/NEJMra035279
Parving, H. H., Lehnert, H., Brochner-Mortensen, J., Gomis, R., Andersen, S., and Arner, P. (2001). Effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. Ugeskr. Laeger 163, 5519–5524. doi: 10.1056/NEJMoa011489
Pfeiffer, A., Middelberg-Bisping, K., Drewes, C., and Schatz, H. (1996). Elevated plasma levels of transforming growth factor-beta 1 in NIDDM. Diabetes Care 19, 1113–1117. doi: 10.2337/diacare.19.10.1113
Pitt, B. (1995). “Escape” of aldosterone production in patients with left ventricular dysfunction treated with an angiotensin converting enzyme inhibitor: implications for therapy. Cardiovasc. Drugs Ther. 9, 145–149. doi: 10.1007/BF00877755
Quinkler, M., Zehnder, D., Eardley, K. S., Lepenies, J., Howie, A. J., Hughes, S. V., et al. (2005). Increased expression of mineralocorticoid effector mechanisms in kidney biopsies of patients with heavy proteinuria. Circulation 112, 1435–1443. doi: 10.1161/CIRCULATIONAHA.105.539122
Remuzzi, G., Ruggenenti, P., and Perico, N. (2002). Chronic renal diseases: renoprotective benefits of renin-angiotensin system inhibition. Ann. Intern. Med. 136, 604–615. doi: 10.7326/0003-4819-136-8-200204160-00010
Riser, B. L., Varani, J., Cortes, P., Yee, J., Dame, M., and Sharba, A. K. (2001). Cyclic stretching of mesangial cells up-regulates intercellular adhesion molecule-1 and leukocyte adherence: a possible new mechanism for glomerulosclerosis. Am. J. Pathol. 158, 11–17. doi: 10.1016/S0002-9440(10)63938-7
Ruggenenti, P., Fassi, A., Ilieva, A. P., Bruno, S., Iliev, I. P., Brusegan, V., et al. (2004). Preventing microalbuminuria in type 2 diabetes. N. Engl. J. Med. 351, 1941–1951. doi: 10.1056/NEJMoa042167
Sato, A., Hayashi, K., Naruse, M., and Saruta, T. (2003). Effectiveness of aldosterone blockade in patients with diabetic nephropathy. Hypertension 41, 64–68. doi: 10.1161/01.HYP.0000044937.95080.E9
Sica, D. A. (2002). Eplerenone: a new aldosterone receptor antagonist–are the FDAs restrictions appropriate? J. Clin. Hypertens. (Greenwich) 4, 441–445. doi: 10.1111/j.1524-6175.2002.01510.x
Takebayashi, K., Matsumoto, S., Aso, Y., and Inukai, T. (2006). Aldosterone blockade attenuates urinary monocyte chemoattractant protein-1 and oxidative stress in patients with type 2 diabetes complicated by diabetic nephropathy. J. Clin. Endocrinol. Metab. 91, 2214–2217. doi: 10.1210/jc.2005-1718
Tamsma, J. T., Van Den Born, J., Bruijn, J. A., Assmann, K. J., Weening, J. J., Berden, J. H., et al. (1994). Expression of glomerular extracellular matrix components in human diabetic nephropathy: decrease of heparan sulphate in the glomerular basement membrane. Diabetologia 37, 313–320. doi: 10.1007/BF00398060
Terada, Y., Kuwana, H., Kobayashi, T., Okado, T., Suzuki, N., Yoshimoto, T., et al. (2008). Aldosterone-stimulated SGK1 activity mediates profibrotic signaling in the mesangium. J. Am. Soc. Nephrol. 19, 298–309. doi: 10.1681/ASN.2007050531
Vallon, V., Schroth, J., Lang, F., Kuhl, D., and Uchida, S. (2009). Expression and phosphorylation of the Na+-Cl- cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am. J. Physiol. Renal Physiol. 297, F704–F712. doi: 10.1152/ajprenal.00030.2009
Wada, T., Furuichi, K., Sakai, N., Iwata, Y., Yoshimoto, K., Shimizu, M., et al. (2000). Up-regulation of monocyte chemoattractant protein-1 in tubulointerstitial lesions of human diabetic nephropathy. Kidney Int. 58, 1492–1499. doi: 10.1046/j.1523-1755.2000.00311.x
Weir, M. R., and Rolfe, M. (2010). Potassium homeostasis and renin-angiotensin-aldosterone system inhibitors. Clin. J. Am. Soc. Nephrol. 5, 531–548. doi: 10.2215/CJN.07821109
Welling, P. A., and Ho, K. (2009). A comprehensive guide to the ROMK potassium channel: form and function in health and disease. Am. J. Physiol. Renal Physiol. 297, F849–F863. doi: 10.1152/ajprenal.00181.2009
Wolf, G. (2006). Renal injury due to renin-angiotensin-aldosterone system activation of the transforming growth factor-beta pathway. Kidney Int. 70, 1914–1919.
Yamaguchi, H., Igarashi, M., Hirata, A., Tsuchiya, H., Sugiyama, K., Morita, Y., et al. (2004). Progression of diabetic nephropathy enhances the plasma osteopontin level in type 2 diabetic patients. Endocr. J. 51, 499–504. doi: 10.1507/endocrj.51.499
Yokoi, H., Sugawara, A., Mukoyama, M., Mori, K., Makino, H., Suganami, T., et al. (2001). Role of connective tissue growth factor in profibrotic action of transforming growth factor-beta: a potential target for preventing renal fibrosis. Am. J. Kidney Dis. 38, S134–S138. doi: 10.1053/ajkd.2001.27422
Keywords: urinary albumin to creatinine ratio, UACR, nephropathy, albuminuria, hyperkalemia, mineralocorticoid receptor
Citation: Orena S, Maurer TS, She L, Eudy R, Bernardo V, Dash D, Loria P, Banker ME, Tugnait M, Okerberg CV, Qian J and Boustany-Kari CM (2013) PF-03882845, a non-steroidal mineralocorticoid receptor antagonist, prevents renal injury with reduced risk of hyperkalemia in an animal model of nephropathy. Front. Pharmacol. 4:115. doi: 10.3389/fphar.2013.00115
Received: 03 July 2013; Paper pending published: 02 August 2013;
Accepted: 27 August 2013; Published online: 14 October 2013.
Edited by:
Ryan M. Fryer, Boehringer-Ingelheim Pharmaceuticals, Inc., USAReviewed by:
Rink-Jan Lohman, The University of Queensland, AustraliaHemal H. Patel, University of California, San Diego, USA
Copyright © 2013 Orena, Maurer, She, Eudy, Bernardo, Dash, Loria, Banker, Tugnait, Okerberg, Qian and Boustany-Kari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carine M. Boustany-Kari, CardioMetabolic Diseases Research, Boehringer Ingelheim, Ridgefield, CT 06844, USA e-mail:Y2FyaW5lLmJvdXN0YW55QGJvZWhyaW5nZXItaW5nZWxoZWltLmNvbQ==
†Present address: Stephen Orena, Nutrition Research Institute, Kannapolis, USA
Rena Eudy, Metrum Research Group, Tariffville, USA
Carine M. Boustany-Kari, CardioMetabolic Diseases Research, Boehringer Ingelheim, Ridgefield, USA