
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 12 February 2013
Sec. Pharmacology of Anti-Cancer Drugs
Volume 4 - 2013 | https://doi.org/10.3389/fphar.2013.00013
 Marco Tafani1,2 Bruna Pucci2 Andrea Russo3
Marco Tafani1,2 Bruna Pucci2 Andrea Russo3 Luana Schito4 Laura Pellegrini2,5 Giulietta A. Perrone6
Luana Schito4 Laura Pellegrini2,5 Giulietta A. Perrone6 Lidia Villanova2,7
Lidia Villanova2,7 Luisa Salvatori8 Linda Ravenna8
Luisa Salvatori8 Linda Ravenna8 Elisa Petrangeli1,8
Elisa Petrangeli1,8 Matteo A. Russo1,2*
Matteo A. Russo1,2*HIF1α and NFkB are two transcription factors very frequently activated in tumors and involved in tumor growth, progression, and resistance to chemotherapy. In fact, HIF1α and NFkB together regulate transcription of over a thousand genes that, in turn, control vital cellular processes such as adaptation to the hypoxia, metabolic reprograming, inflammatory reparative response, extracellular matrix digestion, migration and invasion, adhesion, etc. Because of this wide involvement they could control in an integrated manner the origin of the malignant phenotype. Interestingly, hypoxia and inflammation have been sequentially bridged in tumors by the discovery that alarmin receptors genes such as RAGE, P2X7, and some TLRs, are activated by HIF1α; and that, in turn, alarmin receptors strongly activate NFkB and proinflammatory gene expression, evidencing all the hallmarks of the malignant phenotype. Recently, a large number of drugs have been identified that inhibit one or both transcription factors with promising results in terms of controlling tumor progression. In addition, many of these molecules are natural compounds or off-label drugs already used to cure other pathologies. Some of them are undergoing clinical trials and soon they will be used alone or in combination with standard anti-tumoral agents to achieve a better treatment of tumors with reduction of metastasis formation and, more importantly, with a net increase in survival. This review highlights the central role of HIF1α activated in hypoxic regions of the tumor, of NFkB activation and proinflammatory gene expression in transformed cells to understand their progression toward malignancy. Different molecules and strategies to inhibit these transcription factors will be reviewed. Finally, the central role of a new class of deacetylases called Sirtuins in regulating HIF1α and NFkB activity will be outlined.
The currently prevalent theory on cancer pathogenesis still assumes that the basic events for carcinogenesis and metastasis are mutations that are accumulated by a single cell during its life. Although Weinberg demonstrated that a combination of mutated oncogenes and/or suppressor genes transfected in a normal cell can produce a fully transformed cell, there is no demonstration that another set of genes can produce the malignant phenotype, invading and forming metastasis.
Hanahan and Weinberg (2011) proposed that microenvironment could participate to the progression in many ways: providing VEGF, cytokines, and other growth and survival factors, mostly from activated mesenchymal and inflammatory cells; and creating a reactive oxygen species (ROS)-rich microenvironment which could favor new mutations.
Transformation is characterized by loss of control of proliferation and/or of apoptosis and it is due to an accumulation of mutations with gain-of-function (oncogenes) or loss-of-function (oncosuppressor genes) of gene families related to the cell cycle control and apoptosis control (Hanahan and Weinberg, 2011; Larsson, 2011). Mutations of genes belonging to the DNA repair mechanisms may be responsible for the upstream steps in this sequence, increasing the chances of accumulation of the mutations needed to have a transformed phenotype. Experimental models of transformation definitely have established the cause/effect relationship between certain mutations of these genes and the precise generation of a transformed phenotype (Elenbaas et al., 2001; Ince et al., 2007).
Genomic instability, chemical mutagens, and radiations are responsible for random mutations that can involve transformation-related genes. Epigenetic changes (methylation or acetylation status of DNA) and alterations in chromatin structure maintenance mechanisms can stably achieve biological effects (gain-of-function, loss-of-function) on proliferation and apoptosis control, similar to the classical mutations (Huang et al., 2011; Vanden Berghe, 2012). Mutations can be established in somatic (stem) cells in any period of life or can be present in the zygote being inherited from parents. This last type of mutations is responsible for inherited tumor risk and usually regards a loss-of-function of oncosuppressor genes. A gain-of-function of oncogenes (such as ret oncogene) can be observed only exceptionally (Traugott and Moley, 2010). This can be explained by the fact that the presence of oncogene mutations, disrupting normal morphogenesis and development, lead to premature embryonic or fetal death.
Progression is characterized by the acquisition of the malignant phenotype that leads to a clinically significant tumor. Malignancy includes ability to grow above the limited dimensions conditioned by diffusion of oxygen and nutrients in the absence of newly formed vessels (neoangiogenesis), ability to extrude and/or inactivate entire families of molecules (resistance to drugs), invasion of adjacent tissues (degradation of BM and ECM), ability to detach from original tissue (changes in adhesive molecules and properties), to migrate in response to a chemoattractant (receptors for chemokines and other chemoattractants), to homing in a specific site that will harbor the new tumor (expression of new sets of adhesive molecules which will encounter their countereceptors on an otherwise activated distant endothelium; Furuta et al., 2010; Zigler et al., 2010; Noman et al., 2011; Nasr and Pelletier, 2012). Most of these genes have been individually studied and analyzed for their mutation, epigenetic changes, and other abnormalities to determine their contributes to the malignancy.
However, the understanding of the progression and all the properties of a malignant cell in terms of mutations of all the necessary genes, has been disappointing and unrealistic. These genes are so numerous that the stochastical occurrence of their mutations during the entire human life is statistically improbable or impossible. Today it is generally accepted that, although mutations of progression-related genes may contribute to the malignancy, other factors, not necessarily mutations, are responsible for the pathogenetic sequence leading to the malignant phenotype.
In the last decade the tissue environment in which the tumor originate and manifest has been subjected to an intensive study. Results of this analysis show that microenvironment of both host tissue and tumor tissue contributes in many ways to the progression and to the final destination of a tumor. A plethora of papers have shown that the contribution depends on cell involved, on local interaction among cells, on paracrine signals generated, on the level of local hypoxia, on the presence of an active local immune-inflammatory response with activated leukocytes and on many other factors (Zigler et al., 2010; Noman et al., 2011; Coleman et al., 2012; Hanahan and Coussens, 2012; Hao et al., 2012; Mucaj et al., 2012; Muratori and Tamagnone, 2012; Nasr and Pelletier, 2012).
Among these so many different contributions, it is difficult to evaluate the precise role of each factor as well as their position in the pathway originating a malignant tumor. In addition, they are too heterogeneous to be included in a logical and sequential pathway in the attempt to fully explain the various facets of the malignant phenotype. Any available unitary framework, including the one recently proposed by Hanahan and Weinberg (2011), is unable to contain all the heterogeneous observations and experiments.
However, papers from many laboratories converged toward an unitary explanation of the progression of the early transformed cells. From one side it has been demonstrated that a great promotion to the malignancy can come from gene adaptation to the hypoxia (Shay and Celeste Simon, 2012). From the other side it has been shown that many proinflammatory genes are overexpressed by malignant cells (Tafani et al., 2011a,b; Jin et al., 2012; Schito et al., 2012; Yao et al., 2012; De Santis et al., 2013). Bridging hypoxia adaptation and proinflammatory gene expression by cancer cells, we suggested the hypothesis that these two very complex cell responses, when sequentially activated, could be a good candidate framework to explain all the properties of the malignant phenotype. In addition, we suggested that transformed (still non-progressed) tumor stem cells could best adapt to generate this kind of responses.
In the next paragraph we will analyze the molecular and biological effects of the hypoxia on transformed cancer cells.
Early transformed cancer cells are able to proliferate and form small tumors in the absence of neoangiogenesis. Oxygen and nutrients can diffuse from host normal tissue vessels over a radius of no more than 200 μm (Brahimi-Horn et al., 2007). When the small tumor reaches more than 400 μm in diameter, a hypoxic environment is generated, especially in the center of the tumor (Toffoli and Michiels, 2008).
Hypoxia produces two basic consequences: (a) Necrosis of cells that are more distant from vessels of host tissue; (b) Activation of HIF1α in surviving tumor cells closer to the vessels and sublethally damaged; the HIF1α-driven gene expression allows them to survive and grow increasing their commitment to malignancy (Figure 1).
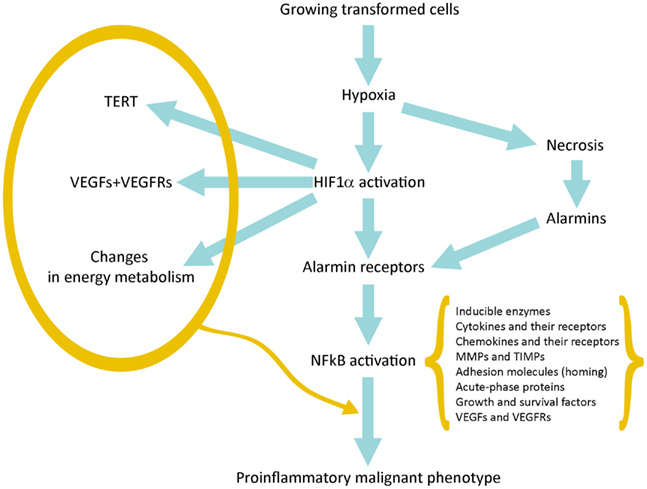
Figure 1. Schematic representation of the pathway linking hypoxia and HIF1α activation with inflammation and tumor progression.
Activation of HIF1α leads to the expression of hundreds genes (Table 1). Many of them provide a first impulse (commitment) toward tumor progression. VEGFs and their receptors are responsible for neoangiogenesis and for the possibility to grow above the limit of 400 μm in diameter; telomerase activation increases the proliferative potential and the number of possible cycles; and changes in intermediate and energy metabolism are the best known metabolic effects of this adaptation (Brahimi-Horn et al., 2007; Mucaj et al., 2012).

Table 1. Adaptation to hypoxia in transformed (stem) cells.
Necrotic damage include plasma membrane fragmentation and release of intracellular molecules, some of which constitute alarmins or Damage-Associated Molecular Patterns (DAMPs). The interaction of released alarmins with their receptors triggers a proinflammatory gene expression in various cell types: resident innate immunity cells or leukocytes, which usually express in their plasma membrane a number of alarmin receptors and tumor cells in which alarmin receptors have been induced by hypoxia (Tafani et al., 2011a). Alarmin receptor signaling leads to the activation of NFkB and then to the proinflammatory gene expression. This proinflammatory microenvironment can contribute to tumor progression (see below).
By examining the functions of the genes activated by HIF1α it is evident that a number of these genes or gene families play a critical role in pushing a transformed cell toward the acquisition of many hallmarks of malignancy. In particular, the overexpression of VEGFs and their receptors (Ahluwalia and Tarnawski, 2012) activate a tumor-specific neoangiogenesis, allowing the early tumor to grow over the dimensions (200–300 μm), imposed by the simple diffusion of oxygen and nutrients. The activation of telomerase (TERT) increases the length of telomeres and the proliferative potential, immortalizing the involved tumor cells (Guan et al., 2012). A further contribute to the proliferative potential is given by the HIF1α-dependent activation of typical proproliferative genes such as c-myc and cyclin D1 (Zhu et al., 2010; Fer and Melillo, 2011). In addition, HIF1α activates OCT4 and Notch facilitating stem cell renewal, contributing to the immortalization (Lee et al., 2012; Qiang et al., 2012). Resistance to chemotherapy is achieved by overexpression of ABC transporters (Maugeri-Saccà et al., 2011). The overexpression of a number of key-molecules such as ALDA, PGK, GLUT-1, beautifully explains the reprograming of the tumor energy metabolism (increased glucose transport and consumption and high glycolysis with lactate production; Semenza et al., 1996; Lam et al., 2009; Mucaj et al., 2012).
Most of the invasion and metastasis genes are co-controlled by HIF1α and by NFkB. Therefore, they will be examined in the next paragraph.
Finally, more importantly, we have observed that in a hypoxic environment a number of cell types, including cancer and normal stem cells, express de novo or overexpress different alarmin receptors (similar to those present in activated leukocytes or CD45+ cells; Tafani et al., 2011a). RAGE, P2X7, TLRs, and others, upon activation by alarmins released by necrotic cells, converge in the activation of NFkB with a robust proinflammatory gene expression (Figure 1). This represents the key event to bridge the adaptation to the hypoxia with the expression of hundreds of genes related to the Inflammatory Reparative Response (IRR) and, very importantly, to the acquisition of classical properties to the malignant phenotype. Table 1 summarizes genes or gene families principally involved in the hypoxia adaptation contributing to the malignant progression. This picture include also the so-called EMT (epithelial-mesenchymal transition), in which all the involved genes can be HIF1α and/or NFkB-dependent (Micalizzi et al., 2010).
Once NFkB has been activated through many different pathways, a complex gene response occurs, with the expression of hundreds of genes belonging to specific gene families including a large number of members functionally related to the inflammatory and reparative response (see Table 2). Individually most of these genes have been implicated in the acquisition of crucial properties of the malignant phenotype, providing a coherent theoretical framework to explain the acquisition of most of the malignant hallmarks as an integrated response and adaptation to the tumor environment.

Table 2. IRR gene expression and malignant phenotype biological properties.
Inducible enzymes produced in activated leukocytes upon activation of NFkB are responsible for mediator molecules production such as prostaglandins, leukotrienes, and NO, leading to the manifestation and amplification of the IRR. Their presence in tumor microenvironment and their expression by tumor cells itself has been one of the earliest observation involving inflammation in the pathogenesis of cancer and its progression (Wang and Dubois, 2006). Molecules produced by these enzymes contribute to the many aspects of tumor progression such as neoangiogenesis, recruitment of leukocyte to the tumor microenvironment, and changes for EMT (Micalizzi et al., 2010). Almost 15 years ago a landmark epidemiological study suggested that the use of low-dose aspirin for cardiovascular prevention drastically reduced the risk for colon cancer (Gustafson-Svärd et al., 1997). These epidemiological observations stimulated a number of other retrospective studies and controlled clinical trials on aspirin and other COX2 inhibitors in preventing tumors and their progression, giving rise to a new era in the understanding the role of inflammation in tumor pathogenesis.
Cytokines characterizes IRR directly influencing target leukocytes, polarizing the response as Th1 or Th2 and stimulating the proliferation of target cells (CD45+) to reinforce and amplify the IRR (DiDonato et al., 2012). Cytokines are present in most human tumor microenvironment, being produced by cancer cells itself and/or by leukocyte infiltrate (DiDonato et al., 2012). Interestingly, tumor cells express also receptors for various cytokines in parallel with their degree of malignancy (DiDonato et al., 2012). Therefore, thanks to the presence of cytokine receptors, tumor cells can be strongly influenced in their biology, such as proliferation rate (IL-2) and in their polarization (Th1 cytokines) and, probably, in the expression of adhesion molecules and their countereceptors, thus influencing the homing for metastasis (DiDonato et al., 2012).
MMPs and TIMPs are NFkB-dependent genes normally expressed in activated leukocytes, but it is well known that disruption of the MMP/TIMP activity ratio with a gain-of-function of proteasic activity over basement membrane and extracellular matrix proteins is present in malignant tumors and parallels the invasive potential (Tobar et al., 2010; Choi et al., 2011b). Then the key event for demolishing the physiological tissue barrier (limits) and for invasion to start is basically controlled by both HIF1α and NFkB through the expression of these genes.
The activation of NFkB in leukocytes finely reprograms the expression of adhesion molecules for migration and for homing at constitutive district tissue or at damaged site. A NFkB-dependent and/or cytokine-dependent new expression of adhesive molecules occurs also in tumor cells, allowing a number of biological changes typically related with malignancy. These changes include the ability to detach from the original tissue (i.e., cadherins), the ability to migrate following a specific chemotactic gradient and a path of ECM molecules (receptors for chemokines and integrins), and, finally, the identification of the homing site represented by activated endothelial cells (ICAM-1, selectins, and their countereceptors; Marcu et al., 2010).
Tumor cells express both chemokines and their receptors in parallel with their degree of malignancy (Lu and Kang, 2010). The production of chemokines give rise to a gradient which is probably the main responsible for the attraction of leukocytes and mononuclear infiltration in advanced tumors (Lu and Kang, 2010). More importantly, the expression of chemokine receptors is a crucial event for the occurrence of metastasis. In fact, metastasis is a complex event which include a number of steps with the participation of hundreds of genes. Detachment from the primary tumor tissue must be followed by a vectorial migration along a chemotactic gradient, which implies the presence of specific receptors for the chemoattractant. CXCR4, a receptor for SDF1α, is the best characterized in tumor cells and has been definitely associated with progression and prediction of metastasis in many human tumors (Lu and Kang, 2010). Both chemokines and their receptors are under the control of NFkB.
The occurrence of a clinically relevant tumor, detectable by the present imaging techniques, needs to grow at the dimension of a few mm in diameter and then, by default, it needs a process of neoangiogenesis, with an adequate expression of VEGFs and VEGFRs in the tumor microenvironment. VGEFs can be produced both by activated leukocytes and mesenchymal cells present in the tumor microenvironment or, more importantly, by tumor cells themselves under the influence of activated HIF1α and NFkB (Ono, 2008). In the last case it has been demonstrated that cancer cells (probably tumor stem cells and progenitors) may express also VEGFRs, suggesting the possibility that tumor cells can contribute to the formation of their new vascular tree (Ono, 2008).
HIF1α and NFkB control a number of growth and survival factors and their receptors. This has been demonstrated in activated leukocytes (involved in tissue repair) and in hypoxia-activated tumor cells. This is an additional advantage for tumor growth and a prerequisite for the establishment of a secondary metastatic tumor. The “Seed and soil” hypothesis predicts that a favorable tissue environment is relevant for the occurrence of a metastasis (Langley and Fidler, 2011). In this case growth and survival factors can be provided both by activated leukocytes or mesenchymal cells of the microenvironment and by tumor cells themselves in which proliferative pathways are already activated (transforming oncogenes) or in which these genes are overexpressed upon NFkB activation (Brahimi-Horn et al., 2007).
Acute-phase proteins have been considered plasma markers useful to evaluate the systemic IRR. They include soluble and cell bound isoforms, such as C reactive protein, pentraxin-3, and other pentraxins; their functions are only partially elucidated. Similarly to the other NFkB-dependent genes, they appear expressed or overexpressed in hypoxia-activated tumor cells and in activated leukocytes. Their functions in tumor progression is still debated. From one side, they appear to inhibit tumor cell proliferation and to decrease with progression (Ronca et al., submitted), from the other hand they can be highly expressed in malignant cells compared to the host normal tissue (Hiratsuka et al., 2008).
NFkB activation include also the expression of a number of negative key-regulator proteins of IRR, such as SOCS-1 (Strebovsky et al., 2012). This latter protein is a member of SOCS family which suppress the cytokine signaling via JAK/STAT, down-regulate TLR expression and signaling and decrease the NFkB activity and duration (Strebovsky et al., 2012). This family and other negative regulators are considered part of the normal feed-back control of the IRR. As predicted by our hypothesis, SOCS-1 decreased in hypoxia-activated cells, as a physiological response of HIF1α-NFkB integrated activation (De Santis et al., 2013).
A number of epidemiological studies and some clinical controlled trials support the idea that the negative modulation of IRR reduces the risk and the incidence of some tumors and, in addition, may slow or inhibit their progression toward malignancy. These observations and the experimental studies linking hypoxia, IRR, and tumor progression have suggested new strategies for preventing tumors, reducing their incidence, slowing their progression, substantially increasing survival, and decreasing death ratio for malignancy (Liu et al., 2011).
Recently and because of its central role in tumor progression, HIF1α has become the target of an increasing number of inhibitors developed with the aim to block or reduce tumor growth and possibly progression (Semenza, 2012; Xia et al., 2012; Figure 2). However, it must be noted that most of these compounds are FDA approved molecules used for treatment of cancer or other pathologies or natural products and that most of the studies and discoveries on inhibitors or activators of HIF1α have been made in cell-based systems or xenografts by research laboratories and not by pharmaceutical companies (Semenza, 2012). Inhibitors identified so far exert their action through a variety of different mechanisms ranging from decreased mRNA and protein levels of HIF1α, to prevention of HIF1α dimerization or DNA binding, to inhibition of HIF1α binding to co-activators. Some representative compounds acting at different steps of HIF1α pathway are reported on Figure 3.
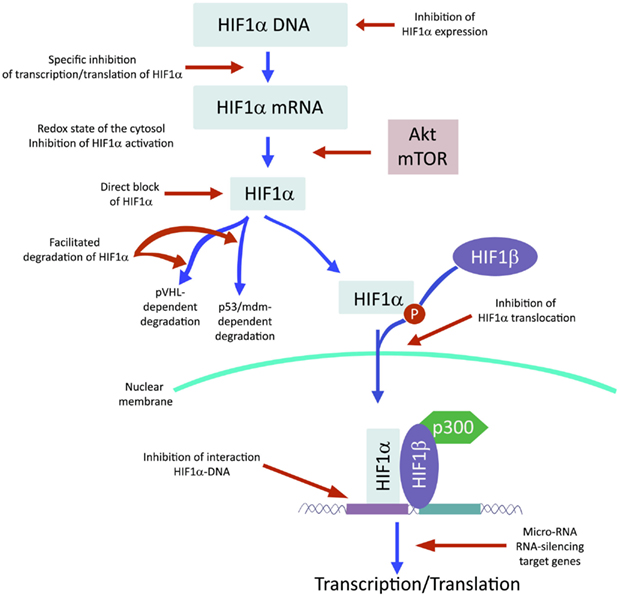
Figure 2. HIF1α activation can be inhibited at different steps along the pathway.
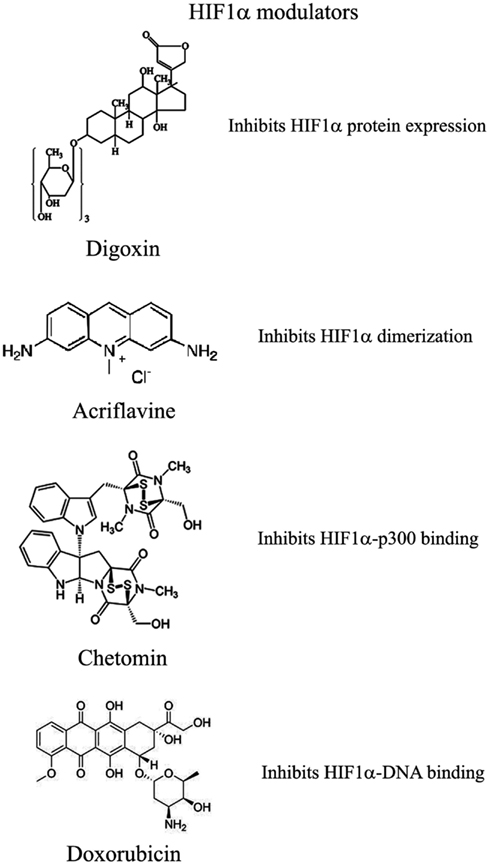
Figure 3. Selection of compounds that can be used to modulate HIF1α pathway.
Among those: (1) PI3Kinase inhibitors wortmannin and LY294002 as well as mTOR inhibitor rapamycin have been shown to reduce HIF1α protein levels in different cell lines (Jiang et al., 2001; Majumder et al., 2004). In fact, these studies demonstrate that the rapamycin-sensitive functions of mTOR are not essential for the accumulation of HIF1α but are important for full expression of this protein as well as for integrating oxygen and nutrient poor conditions (Majumder et al., 2004). (2) Inhibition of HIF1α protein expression and decreased growth of tumor xenografts has been obtained also with cardiac glycosides such as digoxin, ouabain, etc (Semenza, 2012). Digoxin has been used for treatment of heart disease for a long time and it is therefore of particular interest for a combinatorial therapy with other conventional anti-tumor agents (Zhang et al., 2008). (3) Microtubule targeting agents such as 2 methoxyestradiol (2ME2) and its synthetic derivatives prevents HIF1α translation and nuclear accumulation with a corresponding anti-tumor activity (Mabjeesh et al., 2003). However, the exact mechanism of action of such compounds has not been fully elucidated. (4) Class II histone deacetylase (HDAC) inhibitors such as trichostatin and LAQ824 increased HIF1α ubiquitination and degradation of HIF1α by an unknown mechanism (Qian et al., 2006). The emerging role of class III HDAC on HIF1α stabilization will be discussed in detail below. (5) Locked nucleic acid (LNA)-based oligonucleotides are third generation of antisense technology that offer high stability and long lasting target inhibition. EZN-2968 is a LNA directed against HIF1α that has shown great inhibition of HIF1α and HIF1α-dependent genes and that is currently under phase I clinical study because of its ability to reduce tumor growth in xenografts (Greenberger et al., 2008). (6) Ibuprofen and other NSAIDs decreased HIF1α and HIF2α protein levels in prostate cancer cells by a yet to define mechanism that could involve either the PI3K or the proteasome (Palayoor et al., 2003). However, HIF degradation by ibuprofen was not due to its COX2 inhibition activity (Isaacs et al., 2002). (7) Natural antibiotic geldanamycin (GA) and antifungal radicicol prevent binding of Hsp90 to HIF1α thereby decreasing its stability with subsequent proteasomal degradation (Isaacs et al., 2002). In particular, derivates of GA are now in clinical trials. Similarly, Antimycin A, an antibiotic from Streptomyces sp. that induces apoptosis and inhibits the mitochondrial electron transport chain from cytochrome b to cytochrome C1, decreased HIF1α protein level by an unknown mechanism (Maeda et al., 2006). (8) A plethora of natural products possess inhibitory effects of HIF1α. Many of these substances increase HIF1α degradation by activating proteasomal system or by unknown mechanisms. In particular, moracin O and P derived from Morus species (mulberry bark) activates HIF1α degradation (Dat et al., 2009). Other HIF1α inhibitors are manassantin B from the aquatic plant Saururus cernus that probably exerts its effect by degrading HIF1α and inhibiting VEGF secretion (Hossain et al., 2005). Curcumin and berberine, derived respectively from the Indian spice turmeric and from the chinese goldthread increases HIF1α proteasomal degradation (Choi et al., 2006). Similar results were obtained with resveratrol, a compound found in grapes and other plants, and with flavonoids such as methylalpinumisoflavone from the tropical legumaceous Lonchocarpus glabrescens (Park et al., 2007; Liu et al., 2009). Sibiriquinone A from red sage suppressed HIF1α accumulation and VEGF secretion through HIF1α degradation (Dat et al., 2007). This partial list clearly demonstrates that natural products are an important source of HIF1α inhibitors that act through a variety of different mechanisms many of which still unknown.
Acriflavine and Korean red ginseng. Acriflavine, an antibacterial agent, binds to the PAS-B subdomain of HIF1α and HIF2α thereby preventing the binding to HIF1β, an effect that results in reduced VEGF production and tumor growth (Lee et al., 2009). Water extract of red ginseng inhibited HIF1α and 1β dimerization with no toxic effects, however, the mechanism of action and the anti-tumoral effects are not known (Choi et al., 2011a).
Synthetic polyamides binding to and inhibiting HRE elements recognized by HIF1α prevented VEGF synthesis (Semenza, 2012). Doxorubicin and daunorubicin bind to DNA and prevent HIF binding, transcription of target genes, and tumor growth (Tanaka et al., 2012). Finally, echinomycin, an antibiotic isolated from Streptomyces echinatus, binds to DNA and inhibits HIF1α activity (Wang et al., 2011).
Chetomin, a metabolite from the fungus chaetomium, prevents HIF-p300 binding by acting on p300 structure and inhibit transcription of HIF target genes (Kung et al., 2004). In nude mice, Chetomin prevented tumor growth without affecting body weight (Kung et al., 2004). Bortezomib, a proteasome inhibitor, binds to the domain of HIF1α that interacts with p300 thereby preventing a functional interaction between these two factors and blocking transcription of target genes (Befani et al., 2012).
The transcription factor NFkB plays a central role during tumorigenesis because promotes the expression of more than 500 genes involved in crucial cellular pathways such as suppression of apoptosis, increased migration and invasion, increased digestion of extracellular matrix, increased expression of adhesion molecules, etc (Gupta et al., 2010; Figure 4). NFkB is regulated by many post-translational modifications such as methylation, acetylation, phosphorylation, ubiquitination. Moreover, once activated, NFkB translocates and accumulates in the nucleus where binds to the DNA and activates transcription of a plethora of genes (Gupta et al., 2010). Therefore, similarly to HIF1α, also NFkB can be modulated by acting on different steps of the pathways that controls its activity. Again, many natural products mostly with anti-inflammatory properties are NFkB inhibitors (Gupta et al., 2010). More than 700 inhibitors of NFkB have been identified so far and their importance is due to the central role that this transcription factor has for many pathologies, beside inflammation and cancer (Wilczynski et al., 2011). Some representative compounds acting at different steps of NFkB pathway are reported on Figure 5.
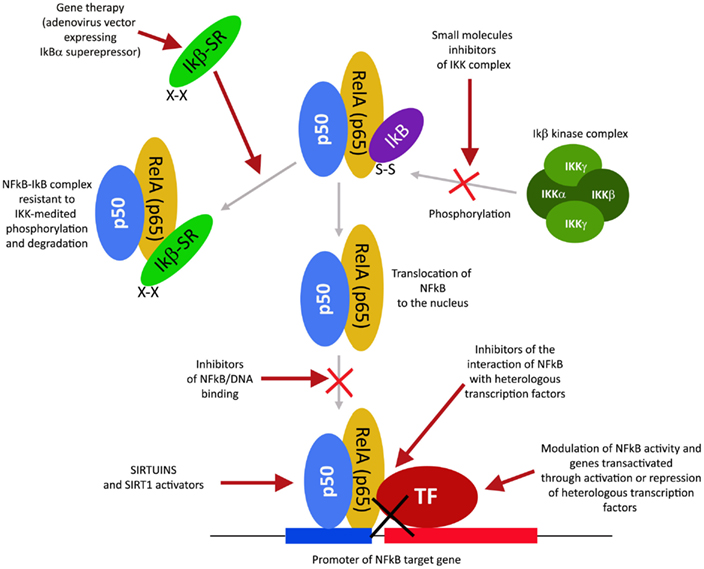
Figure 4. NFkB activation pathway and its different inhibition steps.
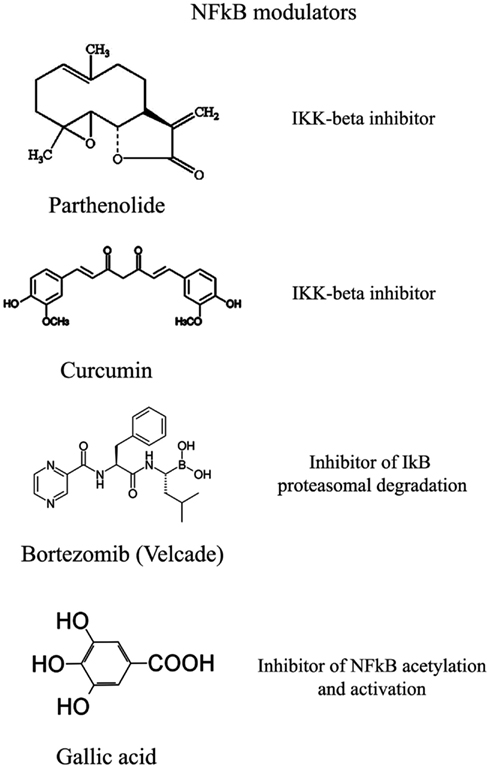
Figure 5. Selection of compounds that can be used to modulate NFkB pathway.
Moreover, it is possible to group the large number of NFkB inhibitors by considering their mechanism of action. In particular:
This class of NFkB inhibitors prevents the activation of IKK kinases. IKK phosphorylates and increases the degradation of the inhibitory subunit IkB, thereby activating NFkB (Gupta et al., 2010). However, only for few IKK inhibitors the mechanism of action is known (Gupta et al., 2010). Among these IKK inhibitors there are natural compounds such as B-carboline (Karin et al., 2004), an indole alkaloid extracted from several plants and that acts as a benzodiazepine agonist, parthenolide, a natural compound isolated from feverfew that acts by interacting with Cys-179 of IKK and, curcumin (Lubbad et al., 2009; Gupta et al., 2010). In particular, parthenolide seems to have an anti-tumoral activity also on cancer stem cells (Gunn et al., 2011). Anti-inflammatory drugs are IKK inhibitors although with an unknown mechanisms (Wilczynski et al., 2011). Among these there are: aspirin, ibuprofen, sulindac, sulfasalazine, and other NSAIDs (Kast, 2006). Other NFkB inhibitors are some kinase inhibitors such as SB203580, PD0980589, tyrosine kinase inhibitors, betaine, etc. (Vanden Berghe et al., 1998). Finally also both IKK dominant negative kinase delivered by adenoviruses and antibodies anti-IKK are alternative strategies that could inhibit NFkB activation (Gupta et al., 2010). By contrast NFkB can be inhibited by activating phosphatases that reduce IkB phosphorylation. For example, cytosine arabinoside dephosphorylates NFKB and induces apoptosis in tumor cells (Sreenivasan et al., 2003).
This strategy exploits the fact that inhibition of IkB degradation by the proteasomal system, results in inhibition of NFkB. A well studied and used proteasome inhibitor is Bortezomib (also mentioned as HIF1α inhibitor) that has been shown to reduce tumor growth in xenograft models and has been successfully used in first-line therapy in combination with other drugs such as cisplatinum (Wilczynski et al., 2011). Inhibition of HIF1α, VEGF, and tumor vascularization by Bortezomib are additional benefits that accompany NFkB inhibition. Other proteasome inhibitors similar to bortezomib include ALLnL, lactacystine, MG132, etc. Finally, second-generation proteasome inhibitors are carfilzomib and salinosporamide that act at nanomolar range, have a lower toxicity and can be delivered orally (Wilczynski et al., 2011; Kale and Moore, 2012).
Similar to phosphorylation by kinases and dephosphorylation by phosphatases, acetylation/deacetylation by acetyl tranferases and deacetylases is considered an equally important post translation modification that control the activity of many proteins (Gray and Teh, 2001). NFkB has been shown to be acetylated on several lysines an event that increase its activation (Kiernan et al., 2003). Therefore, as discussed in major details in the following section, the recent discovery of a new class of deacetylases named Sirtuins has increased the attention toward the possibility to control NFkB activity through these enzymes. In fact, sirtuins activation causes an inhibition of NFkB. Similarly, NFkB can be inhibited by inhibiting acetyl transferases such as p300 and CREB-binding protein (Chen and Greene, 2004). In fact, gallic acid from gallnuts or oak bark and anacardic acid have been shown to inhibit NFkB acetylation and consequent activation (Choi et al., 2009).
This approach is based on the fact that preventing NFkB by accumulating in the nucleus also prevents its DNA association and transcription of target genes. However, such mechanism has been documented only for SN50 a peptide with a hydrophobic membrane-translocating region and the NLS of NFkB. SN50 competes with NFkB for the nuclear translocation machinery thereby preventing NFkB nuclear translocation. Unfortunately, SN50 prevents nuclear accumulation of a large number of transcription factors (Sun et al., 2012). A more promising inhibitor is a compound derived from a fungal antibiotic called dehydroxymethylepoxyquinomicin (DHMEQ) that specifically inhibits NFkB nuclear accumulation with anti-inflammatory and anti-tumoral activity (Kozakai et al., 2012).
Sirtuins owe their name to silent information regulator 2 (Sir2), identified in yeast and linked to lifespan extension (Houtkooper et al., 2012). In mammals there are seven Sir2 homologs (SIRTs 1-7). Sirtuins are either class III nicotinamide adenine dinucleotide-(NAD+)-dependent deacetylase or ADP-ribosyl transferases (Houtkooper et al., 2012). Their dependence from NAD+ directly links sirtuins activity to the metabolic state of the cells. For this reason sirtuins have been implicated in many physiological functions such as gene silencing, cell death, longevity, inflammation, and cancer (Houtkooper et al., 2012).
Sirtuins have also shown to associate, deacetylate, and regulate the activity of both HIF1α and NFkB. However, only for SIRT1, 2, 3, and 6 this regulatory function as been demonstrated.
SIRT1 deacetylates both HIF1α and NFkB. In the case of NFkB all the results so far point toward an inhibition of its signaling following deacetylation by SIRT1 (Morris, 2012). In fact, both in vitro and in vivo experiments have shown that SIRT1 or activation of SIRT1 by resveratrol and other polyphenols decreases inflammatory response by deacetylating and inhibiting NFkB. These results are particularly interesting considering the central role of NFkB in many cellular pathways involved in inflammation, aging, cancer, etc. Controversial results have been reported, instead, for SIRT1/HIF1α signaling. In fact, it is not yet clear if SIRT1 is influenced or not by hypoxia. Some reports indicates that hypoxia increases SIRT1 levels whereas others that hypoxia decreases SIRT1 (Lim et al., 2010; Laemmle et al., 2012). Under hypoxia SIRT1 deacylates HIF1α however, such reaction in some cases decreases HIF1α activity, whereas in other increases HIF1α activity. Obviously, more data must by accumulated on different cell lines, tissue and in vivo models before the real function of SIRT1 on HIF1α can be delineated. Moreover, it is also possible that SIRT1 action of HIF1α differs in different tissues and organs.
SIRT6 is another nuclear sirtuin that controls both HIF1α and NFkB acetylation status and transcriptional activity. In the case of HIF1α, SIRT6 functions as a corepressor of HIF1α transcriptional activity, deacetylating histone 3 lysine 8 (H3K9) at HIF1α target gene promoters. In fact, regulation of glucose flux by SIRT6 appears critical because SIRT6 deficiency causes a lethal hypoglycemia (Zhong et al., 2010). Interestingly, a similar mechanism is used by SIRT6 to inhibit NFkB function. Also in this case SIRT6 deacetylates H3K9 on the promoter of selected NFkB target genes thereby decreasing accessibility to NFkB to these promoters (Kawahara et al., 2009). Importantly, in SIRT1 deficient mice, SIRT6 has shown a compensatory effect by attenuating the increased NFkB activity due to an increased acetylation state (Schug et al., 2010). In conclusion both SIRT1 and SIRT6, although with different mechanisms, represent negative regulators of NFkB activity.
SIRT2 has been shown to deacetylate subunit p65 of NFkB on lysine 310 (K310) in the cytoplasm (Rothgiesser et al., 2010). In this way SIRT2 inhibits NFkB activation and transcription of NFkB target genes following TNF stimulation (Rothgiesser et al., 2010). In fact, SIRT2 silenced cells have an increased activation of NFkB and a lower percentage of cell death following TNF exposure (Rothgiesser et al., 2010). Therefore, NFkB can be deacetylated by SIRT2 in the cytosol and by SIRT1 in the nucleus.
SIRT3 controls HIF1α activation indirectly. In fact, SIRT3 reduces mitochondrial ROS and activates cellular pathways and enzymes scavenging ROS (Finley et al., 2011; Pellegrini et al., 2012). In particular, by decreasing ROS levels, SIRT3 stabilizes HIF degrading enzyme prolyl hydroxylase (PHD) lowering HIF1α levels (Finley et al., 2011). Interestingly, SIRT3 deficiency is associated with tumor growth in xenografts and SIRT3 expression is lowered in several cancers and cancer cell lines (Finley et al., 2011).
Giving the fact that sirtuins regulates both HIF1α and NFkB and the central role that these two transcription factors have during tumor progression, the possibility to act on sirtuins in order to control HIF1α and NFkB has drawn much attention. Therefore, presently, a great deal of efforts have been put in producing Sirtuins modulators. Several natural compounds such as resveratrol, quercetin, piceatannol, and other polyphenols have been shown to modulate sirtuins function and particularly SIRT1 (Chung et al., 2010; Gertz et al., 2012). However, their action is not limited to SIRT1 but influences other enzymes such as phosphodiesterases (PDEs) and AMP kinase (AMPK; Dallas et al., 2008). An up-to-date and accurate review of inhibitors and activators of sirtuins has been recently published (Villalba and Alcaín, 2012).
The recognition of the central role of HIF in tumor growth and progression and the in vitro and in vivo demonstration of tumor growth inhibition by many HIF1α inhibitors, is currently translating in clinical trials for some of them. In particular, 2ME2 is undergoing Phase II clinical trials in patients with breast, prostate, and ovarian cancer (Semenza, 2012; Xia et al., 2012). Similarly also molecules derived from 2ME2 have been selected for evaluation of Phase I clinical trials (Semenza, 2012; Xia et al., 2012). Analogs of geldanamycin (GA) are in Phase II clinical trials in patients with VHL disease, breast cancer, etc (Semenza, 2012; Xia et al., 2012). Bortezomib (velcade) has been approved in the US by FDA for use in multiple myeloma, based on the results from the Phase II trial. Two open-label, Phase III trials established the efficacy of bortezomib 1.3 mg/m2 (with or without dexamethasone) in patients with relapsed/refractory multiple myeloma (Semenza, 2012; Xia et al., 2012). EZN-2968, is currently in Phase I clinical trials (Semenza, 2012; Xia et al., 2012).
Finally, an important consideration is that many of HIF1α interacting drugs are in clinical cancer trials or are already approved for the treatment of cancer or other diseases (Xia et al., 2012).
Several classes of NFkB inhibitors are currently being tested in conjunction with chemotherapy and radiotherapy. In fact, a large number of clinical trials are testing the efficacy and specificity of rationally designed drugs that inhibit NFkB (for a specific trial in the US see the NIH service at: ClincalTrials.gov). In the case of NFkB for example the proteasome inhibitor Bortezomib (also used to inhibit HIF1α) has surprisingly limited side effects and therefore currently appears to be no reason to reject NFκB as drug target on the basis of potential adverse effects which might be induced by inhibition of this transcription factor (Wilczynski et al., 2011). Recently, in a phase II clinical trial, curcumin was found to be beneficial for patient with advanced pancreatic cancer (Wilczynski et al., 2011; Grynkiewicz and Slifirski, 2012). These are only two examples of a large number of NFkB inhibitors that are being currently tested. Hopefully, in the next few years several NFkB inhibitors that can increase the therapeutic efficiency of chemotherapy and radiotherapy will be successfully employed in treatment of cancer patients.
Considering the relatively short time that these enzymes are under investigation, very few sirtuin modulators are under trials. In fact, currently, there are no modulators that can specifically regulate a single sirtuin. However, resveratrol and a number of its derivatives have shown beneficial effects on a randomized double-blind cross-over trials in humans with effects similar to calorie restriction and activation of AMPK, SIRT1, and PGC-1α levels (Timmers et al., 2011). Inhibitors of SIRT1 and SIRT2 have been proposed for treatment of cancer but are far from clinical trials (Morris, 2012).
Considering the central role of HIF1α and NFkB in metabolic reprogramming, inflammation, and cancer, and considering also the fact that both transcription factors are regulated by sirtuins, the possibility to develop more specific modulators acting on different steps of these molecular net, hold promising results toward new therapies with higher success rates.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ahluwalia, A., and Tarnawski, A. S. (2012). Critical role of hypoxia sensor–HIF-1α in VEGF gene activation. Implications for angiogenesis and tissue injury healing. Curr. Med. Chem. 19, 90–97.
Befani, C. D., Vlachostergios, P. J., Hatzidaki, E., Patrikidou, A., Bonanou, S., Simos, G., et al. (2012). Bortezomib represses HIF-1α protein expression and nuclear accumulation by inhibiting both PI3K/Akt/TOR and MAPK pathways in prostate cancer cells. J. Mol. Med. 90, 45–54.
Brahimi-Horn, M. C., Chiche, J., and Pouysségur, J. (2007). Hypoxia and cancer. J. Mol. Med. 85, 1301–1307.
Chen, L. F., and Greene, W. C. (2004). Shaping the nuclear action of NF-kappaB. Nat. Rev. Mol. Cell Biol. 5, 392–401.
Choi, H., Chun, Y. S., Kim, S. W., Kim, M. S., and Park, J. W. (2006). Curcumin inhibits hypoxia inducible factor-1 by degrading aryl hydrocarbon receptor nuclear translocator: a mechanism of tumor growth inhibition. Mol. Pharmacol. 70, 1664–1671.
Choi, K. C., Lee, Y. H., Jung, M. G., Kwon, S. H., Kim, M. J., Jun, W. J., et al. (2009). Gallic acid suppresses lipopolysaccharide-induced nuclear factor-kappaB signaling by preventing RelA acetylation in A549 lung cancer cells. Mol. Cancer Res. 7, 2011–2021.
Choi, Y. J., Choi, H., Cho, C. H., and Park, J. W. (2011a). Red ginseng deregulates hypoxia-induced genes by dissociating the HIF-1 dimer. J. Nat. Med. 65, 344–352.
Choi, J. Y., Jang, Y. S., Min, S. Y., and Song, J. Y. (2011b). Overexpression of MMP-9 and HIF-1α in breast cancer cells under hypoxic conditions. J. Breast Cancer 14, 88–95.
Chung, S., Yao, H., Caito, S., Hwang, J. W., Arunachalam, G., and Rahman, I. (2010). Regulation of SIRT1 in cellular functions: role of polyphenols. Arch. Biochem. Biophys. 501, 79–90.
Coleman, R., Gnant, M., Morgan, G., and Clezardin, P. (2012). Effects of bone-targeted agents on cancer progression and mortality. J. Natl. Cancer Inst. 104, 1059–1067.
Dallas, C., Gerbi, A., Tenca, G., Juchaux, F., and Bernard, F. X. (2008). Lipolytic effect of a polyphenolic citrus dry extract of red orange, grapefruit, orange (SINETROL) in human body fat adipocytes. Mechanism of action by inhibition of cAMP-phosphodiesterase (PDE). Phytomedicine 15, 783–792.
Dat, N. T., Jin, X., Lee, J. H., Lee, D., Hong, Y. S., Lee, K., et al. (2007). Abietane diterpenes from Salvia miltiorrhiza inhibit the activation of hypoxia-inducible factor-1. J. Nat. Prod. 70, 1093–1097.
Dat, N. T., Jin, X., Lee, K., Hong, Y. S., Kim, Y. H., and Lee, J. J. (2009). Hypoxia-inducible factor-1 inhibitory benzofurans and chalcone-derived diels-alder adducts from Morus species. J. Nat. Prod. 72, 39–43.
De Santis, E., Di Vito, M., Perrone, G. A., Mari, E., Osti, M., De Antoni, E., et al. (2013). Overexpression of pro-inflammatory genes and down-regulation of SOCS-1 in human PTC and in hypoxic BCPAP cells. Biomed. Pharmacother. 67, 7–16.
DiDonato, J. A., Mercurio, F., and Karin, M. (2012). NF-κB and the link between inflammation and cancer. Immunol. Rev. 246, 379–400.
Elenbaas, B., Spirio, L., Koerner, F., Fleming, M. D., Zimonjic, D. B., Donaher, J. L., et al. (2001). Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 15, 50–65.
Fer, N., and Melillo, G. (2011). The HIF-1α-c-Myc pathway and tumorigenesis: evading the apoptotic gate-keeper. Cell Cycle 10, 3228.
Finley, L. W., Carracedo, A., Lee, J., Souza, A., Egia, A., Zhang, J., et al. (2011). SIRT3 opposes reprogramming of cancer cell metabolism through HIF1α destabilization. Cancer Cell 19, 416–428.
Furuta, E., Okuda, H., Kobayashi, A., and Watabe, K. (2010). Metabolic genes in cancer: their roles in tumor progression and clinical implications. Biochim. Biophys. Acta 1805, 141–152.
Gertz, M., Nguyen, G. T., Fischer, F., Suenkel, B., Schlicker, C., Fränzel, B., et al. (2012). A molecular mechanism for direct sirtuin activation by resveratrol. PLoS ONE 7:e49761. doi:10.1371/journal.pone.0049761
Gray, S. G., and Teh, B. T. (2001). Histone acetylation/deacetylation and cancer: an “open” and “shut” case? Curr. Mol. Med. 1, 401–429.
Greenberger, L. M., Horak, I. D., Filpula, D., Sapra, P., Westergaard, M., Frydenlund, H. F., et al. (2008). A RNA antagonist of hypoxia-inducible factor-1alpha, EZN-2968, inhibits tumor cell growth. Mol. Cancer Ther. 7, 3598–3608.
Grynkiewicz, G., and Slifirski, P. (2012). Curcumin and curcuminoids in quest for medicinal status. Acta Biochim. Pol. 59, 201–212.
Guan, J. Z., Guan, W. P., Maeda, T., and Makino, N. (2012). Different levels of hypoxia regulate telomere length and telomerase activity. Aging Clin. Exp. Res. 24, 213–217.
Gunn, E. J., Williams, J. T., Huynh, D. T., Iannotti, M. J., Han, C., Barrios, F. J., et al. (2011). The natural products parthenolide and andrographolide exhibit anti-cancer stem cell activity in multiple myeloma. Leuk. Lymphoma 52, 1085–1097.
Gupta, S. C., Sundaram, C., Reuter, S., and Aggarwal, B. B. (2010). Inhibiting NF-κB activation by small molecules as a therapeutic strategy. Biochim. Biophys. Acta 1799, 775–787.
Gustafson-Svärd, C., Lilja, I., Hallböök, O., and Sjödahl, R. (1997). Cyclo-oxygenase and colon cancer: clues to the aspirin effect? Ann. Med. 29, 247–252.
Hanahan, D., and Coussens, L. M. (2012). Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 21, 309–322.
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674.
Hao, N. B., Lü, M. H., Fan, Y. H., Cao, Y. L., Zhang, Z. R., and Yang, S. M. (2012). Macrophages in tumor microenvironments and the progression of tumors. Clin. Dev. Immunol. 2012, 948098.
Hiratsuka, S., Watanabe, A., Sakurai, Y., Akashi-Takamura, S., Ishibashi, S., Miyake, K., et al. (2008). The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat. Cell Biol. 10, 1349–1355.
Hossain, C. F., Kim, Y. P., Baerson, S. R., Zhang, L., Bruick, R. K., Mohammed, K. A., et al. (2005). Saururus cernuus lignans–potent small molecule inhibitors of hypoxia-inducible factor-1. Biochem. Biophys. Res. Commun. 333, 1026–1033.
Houtkooper, R. H., Pirinen, E., and Auwerx, J. (2012). Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 13, 225–238.
Huang, J., Plass, C., and Gerhauser, C. (2011). Cancer chemoprevention by targeting the epigenome. Curr. Drug Targets 12, 1925–1956.
Ince, T. A., Richardson, A. L., Bell, G. W., Saitoh, M., Godar, S., Karnoub, A. E., et al. (2007). Transformation of different human breast epithelial cell types leads to distinct tumor phenotypes. Cancer Cell 12, 160–170.
Isaacs, J. S., Jung, Y. J., Mimnaugh, E. G., Martinez, A., Cuttitta, F., and Neckers, L. M. (2002). Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1 alpha-degradative pathway. J. Biol. Chem. 77, 29936–29944.
Jiang, B. H., Jiang, G., Zheng, J. Z., Lu, Z., Hunter, T., and Vogt, P. K. (2001). Phosphatidylinositol 3-kinase signaling controls levels of hypoxia-inducible factor 1. Cell Growth Differ. 12, 363–369.
Jin, S., Mutvei, A. P., Chivukula, I. V., Andersson, E. R., Ramsköld, D., Sandberg, R., et al. (2012). Non-canonical Notch signaling activates IL-6/JAK/STAT signaling in breast tumor cells and is controlled by p53 and IKKα/IKKβ. Oncogene. doi:10.1038/onc.2012.517
Kale, A. J., and Moore, B. S. (2012). Molecular mechanisms of acquired proteasome inhibitor resistance. J. Med. Chem. 55, 10317–10327.
Karin, M., Yamamoto, Y., and Wang, Q. M. (2004). The IKK NF-kappa B system: a treasure trove for drug development. Nat. Rev. Drug Discov. 3, 17–26.
Kast, R. E. (2006). Aspirin, TNF-alpha, NFkB, and survival in multiple myeloma: the importance of measuring TNF-alpha. Inflammopharmacology 14, 256–259.
Kawahara, T. L., Michishita, E., Adler, A. S., Damian, M., Berber, E., Lin, M., et al. (2009). SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell 136, 62–74.
Kiernan, R., Brès, V., Ng, R. W., Coudart, M. P., El Messaoudi, S., Sardet, C., et al. (2003). Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J. Biol. Chem. 278, 2758–2766.
Kozakai, N., Kikuchi, E., Hasegawa, M., Suzuki, E., Ide, H., Miyajima, A., et al. (2012). Enhancement of radiosensitivity by a unique novel NF-κB inhibitor, DHMEQ, in prostate cancer. Br. J. Cancer 107, 652–657.
Kung, A. L., Zabludoff, S. D., France, D. S., Freedman, S. J., Tanner, E. A., Vieira, A., et al. (2004). Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell 6, 33–43.
Laemmle, A., Lechleiter, A., Roh, V., Schwarz, C., Portmann, S., Furer, C., et al. (2012). Inhibition of SIRT1 impairs the accumulation and transcriptional activity of HIF-1α protein under hypoxic conditions. PLoS ONE 7:e33433. doi:10.1371/journal.pone.0033433
Lam, W., Bussom, S., and Cheng, Y. C. (2009). Effect of hypoxia on the expression of phosphoglycerate kinase and antitumor activity of troxacitabine and gemcitabine in non-small cell lung carcinoma. Mol. Cancer Ther. 8, 415–423.
Langley, R. R., and Fidler, I. J. (2011). The seed and soil hypothesis revisited – the role of tumor-stroma interactions in metastasis to different organs. Int. J. Cancer 128, 2527–2535.
Larsson, L. G. (2011). Oncogene- and tumor suppressor gene-mediated suppression of cellular senescence. Semin. Cancer Biol. 21, 367–376.
Lee, K., Zhang, H., Qian, D. Z., Rey, S., Liu, J. O., and Semenza, G. L. (2009). Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc. Natl. Acad. Sci. U.S.A. 106, 17910–17915.
Lee, S. W., Jeong, H. K., Lee, J. Y., Yang, J., Lee, E. J., Kim, S. Y., et al. (2012). Hypoxic priming of mESCs accelerates vascular-lineage differentiation through HIF1-mediated inverse regulation of Oct4 and VEGF. EMBO Mol. Med. 4, 924–938.
Lim, J. H., Lee, Y. M., Chun, Y. S., Chen, J., Kim, J. E., and Park, J. W. (2010). Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell 38, 864–878.
Liu, J. L., Ma, H. P., Lu, X. L., Sun, S. H., Guo, X., and Li, F. C. (2011). NF-κB induces abnormal centrosome amplification by upregulation of CDK2 in laryngeal squamous cell cancer. Int. J. Oncol. 39, 915–924.
Liu, Y., Veena, C. K., Morgan, J. B., Mohammed, K. A., Jekabsons, M. B., Nagle, D. G., et al. (2009). Methylalpinumisoflavone inhibits hypoxia-inducible factor-1 (HIF-1) activation by simultaneously targeting multiple pathways. J. Biol. Chem. 284, 5859–5868.
Lu, X., and Kang, Y. (2010). Hypoxia and hypoxia-inducible factors: master regulators of metastasis. Clin. Cancer Res. 16, 5928–5935.
Lubbad, A., Oriowo, M. A., and Khan, I. (2009). Curcumin attenuates inflammation through inhibition of TLR-4 receptor in experimental colitis. Mol. Cell. Biochem. 322, 127–135.
Mabjeesh, N. J., Escuin, D., LaVallee, T. M., Pribluda, V. S., Swartz, G. M., Johnson, M. S., et al. (2003). 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell 3, 363–375.
Maeda, M., Hasebe, Y., Egawa, K., Shibanuma, M., and Nose, K. (2006). Inhibition of angiogenesis and HIF-1alpha activity by antimycin A1. Biol. Pharm. Bull. 29, 1344–1348.
Majumder, P. K., Febbo, P. G., Bikoff, R., Berger, R., Xue, Q., McMahon, L. M., et al. (2004). mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat. Med. 10, 594–601.
Marcu, K. B., Otero, M., Olivotto, E., Borzi, R. M., and Goldring, M. B. (2010). NF-kappaB signaling: multiple angles to target OA. Curr. Drug Targets 11, 599–613.
Maugeri-Saccà, M., Vigneri, P., and De Maria, R. (2011). Cancer stem cells and chemosensitivity. Clin. Cancer Res. 17, 4942–4947.
Micalizzi, D. S., Farabaugh, S. M., and Ford, H. L. (2010). Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J. Mammary Gland Biol. Neoplasia 15, 117–134.
Morris, B. J. (2012). Seven sirtuins for seven deadly diseases of aging. Free Radic. Biol. Med. doi:10.1016/j.freeradbiomed.2012.10.525 [pii: S0891-5849(12)01756-X].
Mucaj, V., Shay, J. E., and Simon, M. C. (2012). Effects of hypoxia and HIFs on cancer metabolism. Int. J. Hematol. 95, 464–470.
Muratori, C., and Tamagnone, L. (2012). Semaphorin signals tweaking the tumor microenvironment. Adv. Cancer Res. 114, 59–85.
Nasr, Z., and Pelletier, J. (2012). Tumor progression and metastasis: role of translational deregulation. Anticancer Res. 32, 3077–3084.
Noman, M. Z., Messai, Y., Carré, T., Akalay, I., Méron, M., Janji, B., et al. (2011). Microenvironmental hypoxia orchestrating the cell stroma cross talk, tumor progression and antitumor response. Crit. Rev. Immunol. 31, 357–377.
Ono, M. (2008). Molecular links between tumor angiogenesis and inflammation: inflammatory stimuli of macrophages and cancer cells as targets for therapeutic strategy. Cancer Sci. 99, 1501–1506.
Palayoor, S. T., Tofilon, P. J., and Coleman, C. N. (2003). Ibuprofen-mediated reduction of hypoxia-inducible factors HIF-1alpha and HIF-2alpha in prostate cancer cells. Clin. Cancer Res. 9, 3150–3157.
Park, S. Y., Jeong, K. J., Lee, J., Yoon, D. S., Choi, W. S., Kim, Y. K., et al. (2007). Hypoxia enhances LPA-induced HIF-1alpha and VEGF expression: their inhibition by resveratrol. Cancer Lett. 258, 63–69.
Pellegrini, L., Pucci, B., Villanova, L., Marino, M. L., Marfe, G., Sansone, L., et al. (2012). SIRT3 protects from hypoxia and staurosporine-mediated cell death by maintaining mitochondrial membrane potential and intracellular pH. Cell Death Differ. 19, 1815–1825.
Qian, D. Z., Kachhap, S. K., Collis, S. J., Verheul, H. M., Carducci, M. A., Atadja, P., et al. (2006). Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res. 66, 8814–8821.
Qiang, L., Wu, T., Zhang, H. W., Lu, N., Hu, R., Wang, Y. J., et al. (2012). HIF-1α is critical for hypoxia-mediated maintenance of glioblastoma stem cells by activating Notch signaling pathway. Cell Death Differ. 19, 284–294.
Rothgiesser, K. M., Erener, S., Waibel, S., Lüscher, B., and Hottiger, M. O. (2010). SIRT2 regulates NF-κB dependent gene expression through deacetylation of p65 Lys310. J. Cell. Sci. 123, 4251–4258.
Schito, L., Rey, S., Tafani, M., Zhang, H., Wong, C. C., Russo, A., et al. (2012). Hypoxia-inducible factor 1-dependent expression of platelet-derived growth factor B promotes lymphatic metastasis of hypoxic breast cancer cells. Proc. Natl. Acad. Sci. U.S.A. 109, E2707–E2716.
Schug, T. T., Xu, Q., Gao, H., Peres-da-Silva, A., Draper, D. W., Fessler, M. B., et al. (2010). Myeloid deletion of SIRT1 induces inflammatory signaling in response to environmental stress. Mol. Cell. Biol. 30, 4712–4721.
Semenza, G. L. (2012). Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 33, 207–214.
Semenza, G. L., Jiang, B. H., Leung, S. W., Passantino, R., Concordet, J. P., Maire, P., et al. (1996). Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J. Biol. Chem. 271, 32529–32537.
Shay, J. E., and Celeste Simon, M. (2012). Hypoxia-inducible factors: crosstalk between inflammation and metabolism. Semin. Cell Dev. Biol. 23, 389–394.
Sreenivasan, Y., Sarkar, A., and Manna, S. K. (2003). Mechanism of cytosine arabinoside-mediated apoptosis: role of Rel A (p65) dephosphorylation. Oncogene 22, 4356–4369.
Strebovsky, J., Walker, P., and Dalpke, A. H. (2012). Suppressor of cytokine signaling proteins as regulators of innate immune signaling. Front. Biosci. 17, 1627–1639.
Sun, Y. X., Dai, D. K., Liu, R., Wang, T., Luo, C. L., Bao, H. J., et al. (2012). Therapeutic effect of SN50, an inhibitor of nuclear factor-κB, in treatment of TBI in mice. Neurol. Sci. PMID: 22437493 [Epub ahead of print].
Tafani, M., Schito, L., Pellegrini, L., Villanova, L., Marfe, G., Anwar, T., et al. (2011a). Hypoxia-increased RAGE and P2X7R expression regulates tumor cell invasion through phosphorylation of Erk1/2 and Akt and nuclear translocation of NF-{kappa}B. Carcinogenesis 32, 1167–1175.
Tafani, M., Di Vito, M., Frati, A., Pellegrini, L., De Santis, E., Sette, G., et al. (2011b). Pro-inflammatory gene expression in solid glioblastoma microenvironment and in hypoxic stem cells from human glioblastoma. J. Neuroinflammation 8, 32.
Tanaka, T., Yamaguchi, J., Shoji, K., and Nangaku, M. (2012). Anthracycline inhibits recruitment of hypoxia-inducible transcription factors and suppresses tumor cell migration and cardiac angiogenic response in the host. J. Biol. Chem. 287, 34866–34882.
Timmers, S., Konings, E., Bilet, L., Houtkooper, R. H., van de Weijer, T., Goossens, G. H., et al. (2011). Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 14, 612–622.
Tobar, N., Villar, V., and Santibanez, J. F. (2010). ROS-NFkappaB mediates TGF-beta1-induced expression of urokinase-type plasminogen activator, matrix metalloproteinase-9 and cell invasion. Mol. Cell. Biochem. 340, 195–202.
Toffoli, S., and Michiels, C. (2008). Intermittent hypoxia is a key regulator of cancer cell and endothelial cell interplay in tumors. FEBS J. 275, 2991–3002.
Vanden Berghe, W. (2012). Epigenetic impact of dietary polyphenols in cancer chemoprevention: lifelong remodeling of our epigenomes. Pharmacol. Res. 65, 565–576.
Vanden Berghe, W., Plaisance, S., Boone, E., De Bosscher, K., Schmitz, M. L., Fiers, W., et al. (1998). p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways are required for nuclear factor-kappaB p65 transactivation mediated by tumor necrosis factor. J. Biol. Chem. 273, 3285–3290.
Villalba, J. M., and Alcaín, F. J. (2012). Sirtuin activators, and inhibitors. Biofactors 38, 349–359.
Wang, Y., Liu, Y., Malek, S. N., Zheng, P., and Liu, Y. (2011). Targeting HIF1α eliminates cancer stem cells in hematological malignancies. Cell Stem Cell 8, 399–411.
Wilczynski, J., Duechler, M., and Czyz, M. (2011). Targeting NF-κB and HIF-1 pathways for the treatment of cancer: part I. Arch. Immunol. Ther. Exp. (Warsz.) 59, 289–299.
Xia, Y., Choi, H. K., and Lee, K. (2012). Recent advances in hypoxia-inducible factor (HIF)-1 inhibitors. Eur. J. Med. Chem. 49, 24–40.
Yao, J., Hu, R., Sun, J., Lin, B., Zhao, L., Sha, Y., et al. (2012). Oroxylin A prevents inflammation-related tumor through down-regulation of inflammatory gene expression by inhibiting NF-κB signaling. Mol. Carcinog. doi:10.1002/mc.21958
Zhang, H., Qian, D. Z., Tan, Y. S., Lee, K., Gao, P., Ren, Y. R., et al. (2008). Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc. Natl. Acad. Sci. U.S.A. 105, 19579–19586.
Zhong, L., D’Urso, A., Toiber, D., Sebastian, C., Henry, R. E., Vadysirisack, D. D., et al. (2010). The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell 140, 280–293.
Zhu, P., Ning, Y., Yao, L., Chen, M., and Xu, C. (2010). The proliferation, apoptosis, invasion of endothelial-like epithelial ovarian cancer cells induced by hypoxia. J. Exp. Clin. Cancer Res. 29, 124.
Keywords: hypoxia, inflammation, cancer, HIF1α inhibitors, NFkB inhibitors, sirtuin activators
Citation: Tafani M, Pucci B, Russo A, Schito L, Pellegrini L, Perrone GA, Villanova L, Salvatori L, Ravenna L, Petrangeli E and Russo MA (2013) Modulators of HIF1α and NFkB in cancer treatment: is it a rational approach for controlling malignant progression? Front. Pharmacol. 4:13. doi: 10.3389/fphar.2013.00013
Received: 05 December 2012; Paper pending published: 30 December 2012;
Accepted: 23 January 2013; Published online: 12 February 2013.
Edited by:
Sarah Crawford, Connecticut State University, USAReviewed by:
Jian Hui Wu, McGill University, CanadaCopyright: © 2013 Tafani, Pucci, Russo, Schito, Pellegrini, Perrone, Villanova, Salvatori, Ravenna, Petrangeli and Russo. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Matteo A. Russo, Department of Experimental Medicine, Sapienza University of Rome, Viale Regina Elena, 324 – 00161 Roma, Italy. e-mail:bWF0dGVvYW50b25pby5ydXNzb0B1bmlyb21hMS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.