 Karin Jurkat-Rott1*
Karin Jurkat-Rott1*


95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 11 June 2012
Sec. Pharmacology of Ion Channels and Channelopathies
Volume 3 - 2012 | https://doi.org/10.3389/fphar.2012.00112
This article is part of the Research Topic Mechanisms of ion channels voltage-dependency View all 17 articles
In voltage-gated cation channels, a recurrent pattern for mutations is the neutralization of positively charged residues in the voltage-sensing S4 transmembrane segments. These mutations cause dominant ion channelopathies affecting many tissues such as brain, heart, and skeletal muscle. Recent studies suggest that the pathogenesis of associated phenotypes is not limited to alterations in the gating of the ion-conducting alpha pore. Instead, aberrant so-called omega currents, facilitated by the movement of mutated S4 segments, also appear to contribute to symptoms. Surprisingly, these omega currents conduct cations with varying ion selectivity and are activated in either a hyperpolarized or depolarized voltage range. This review gives an overview of voltage sensor channelopathies in general and focuses on pathogenesis of skeletal muscle S4 disorders for which current knowledge is most advanced.
The basic motif of the alpha subunit of voltage-gated cation channels is a tetrameric association of four domains I–IV, each consisting of six transmembrane helical segments S1–S6, connected by intracellular and extracellular loops. At the resting potential, open probability is low. Activation results from a depolarization-induced conformational change leading to the opening of the alpha pore. When the pore is pharmacologically blocked, charge movements in the electrical membrane field are measurable as so-called gating currents. These gating currents are a result of the outward movement of highly conserved voltage-sensitive S4 segments which display an arginine or lysine residue at every third amino acid position, almost vertically aligned (denoted as R1, R2, R3, etc.). At resting potential, the outermost S4 charge separates the extracellular fluid from the cytoplasm. With membrane depolarization, each S4 moves outwardly shifting deeper situated arginines to the critical position of extra-intracellular separation. During their outward movement, the S4 segments move in a spiral path through canaliculi of the channel protein made up of segments S1–S3 (Figures 1 and 2). Likewise, during recovery from channel inactivation, the S4 segments are thought to move back to their original position. Because of the narrow constriction separating extracellular from intracellular compartments, mutations of the S4 charges may cause either a hyperpolarization-activated or a depolarization-activated non-specific cation leak called the omega current (Figure 2).
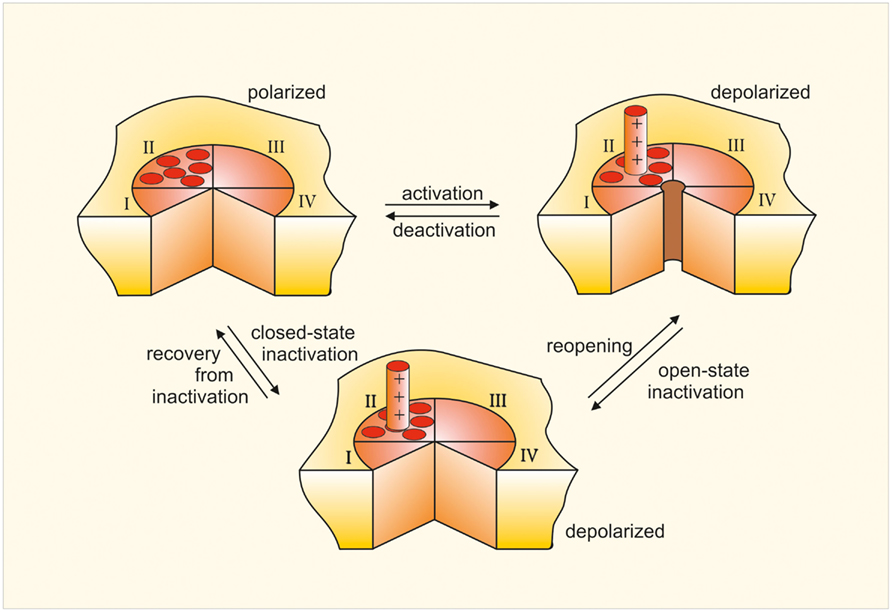
Figure 1. Scheme of a voltage-gated ion channel. Bird’s eye view of the channel consisting of four similar repeats (I to IV). The channel has been cut open between repeats I and IV to show the opening and closing of the central pore. The model also shows one of the four voltage sensors S4, which moves outward when the membrane becomes depolarized and remains in this position until repolarization.
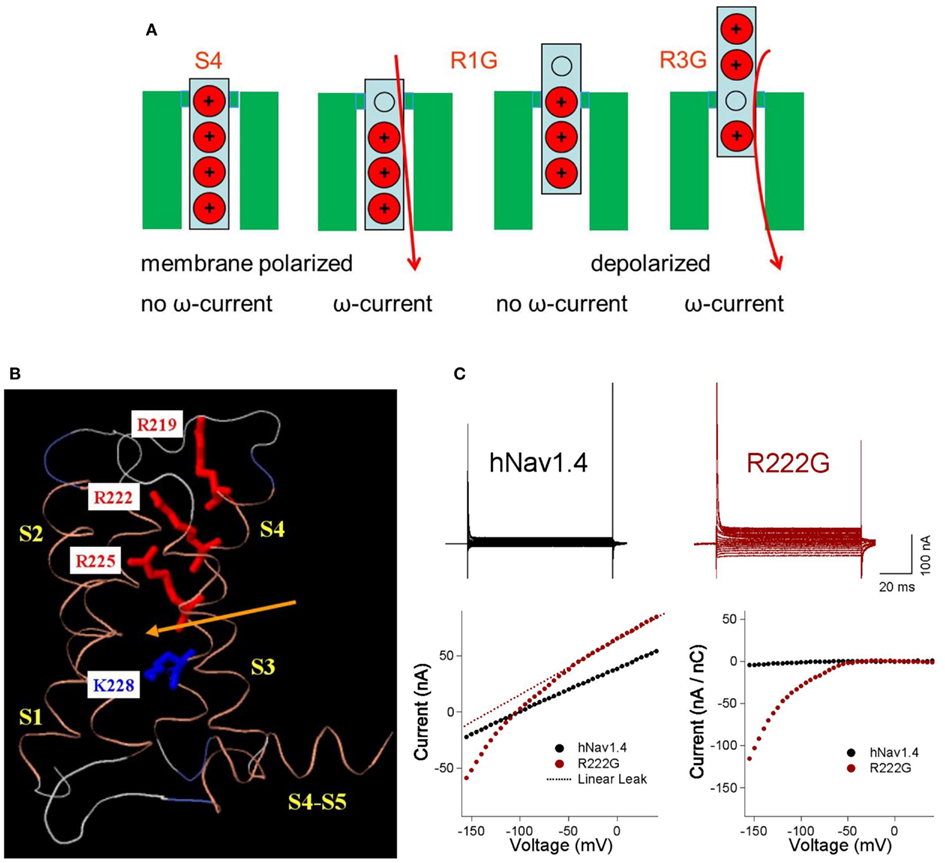
Figure 2. Omega pores and currents dependent on the R position. (A) Replacement of the outermost arginine (red) by a neutral amino acid (gray) such as glycine (R1G) opens a conductive pathway through the polarized membrane, resulting in an omega current (red). At depolarized potentials at which the S4 segment moves outward, the conductive pathway is closed by a deeper arginine and the omega current ceases. In contrast the replacement of a deeper arginine (R3G) only opens the omega pore if the membrane is depolarized. (B) Homology model of domain I in hNav1.4 based on crystal structure of NaVAb (activated-closed; crystal structure at 0 mV), using Modeller. Positions of arginine and lysine residues of DIS4 are shown, relative to the putative gating pore constriction (arrow). Modified from Groome and Winston (2012). (C) Comparison of current-voltage (I/V) traces for wild type hNav1.4 and R222G, with plots of raw I/V, linear leak, and normalized current (linear leak subtracted from IV and normalized to gating current at +40 mV). The mutation R222G causes HypoPP type 2. External solution contained 120 mM K+ and 1 μM TTX. Modified from Holzherr et al. (2010).
An omega current was first described in the voltage-gated potassium channel of drosophila Shaker. In this channel the mutation R1H in S4 generates an inward-directed hyperpolarization-induced proton current that becomes increasingly prominent as the proton reversal potential EH is shifted to more positive values (Starace and Bezanilla, 2004). R1 substitutions to residues other than histidine result in a hyperpolarization-induced inward current carried by alkali metal cations rather than protons (Tombola et al., 2005) indicating that this mutation creates a short-circuit connection of intracellular and extracellular compartments in the canaliculus. This current varies with the identity of the substituted residue: R1S > R1C > R1V ∼ R1A, has the selectivity Cs+ > K+ > Li+, and is not affected by alpha pore blockers. Shaker mutants R2H and R3H conduct protons at potentials corresponding to the voltage-dependent movement of S4 charges, as the current is maximal at potentials close to midpoint of the QON-voltage relation (Starace et al., 1997; Starace and Bezanilla, 2001). Further depolarization blocks the omega pore, consistent with the model that outward S4 movement shifts the short-circuiting mutant residues out of the canaliculus.
Studies on Shaker potassium channel and domain II of rat brain sodium channel Nav1.2 suggest that substitution of a single S4 arginine may be insufficient to produce a non-proton omega current, but that two adjacent mutations (i.e., R2/R3 or II-R1/R2) are required (Sokolov et al., 2005; Gamal El-Din et al., 2010). In this respect, the presence of alanine at the position equivalent to R0 in Shaker may therefore enhance the omega current of R1S. The motif of adjacent arginine replacements recurs in some of the potassium channel disorders and may be explained by the degree of accessibility of arginines to the extracellular or intracellular compartments. However, this does not hold true for all channels suggesting that position and orientation of the S4 segment and its environment play an important role as well. Based on current knowledge of S4 position, in this review R0 designates charge positions of S4 outside of the canaliculus and R1 the first charge inside of it.
For human diseases, the channelopathies result from mutations in voltage-gated ion channels. In several of these disorders, mutations of specific charges of S4 segments are known, such as in epilepsy, hemiplegic migraine, long QT (LQT) syndrome, paramyotonia congenita, and periodic paralyzes (Table 1). The underlying mutations generally modify voltage sensitivity of gating and maximal amplitude of alpha current. Additionally, some of these mutations have been reported to produce an omega current. Most of these are inward currents that result in enhanced membrane depolarization, contributing to the symptoms of the channelopathy. Depending on the position of the S4 mutation and the voltage threshold of activation of the channel, the omega current can appear in very different voltage ranges.
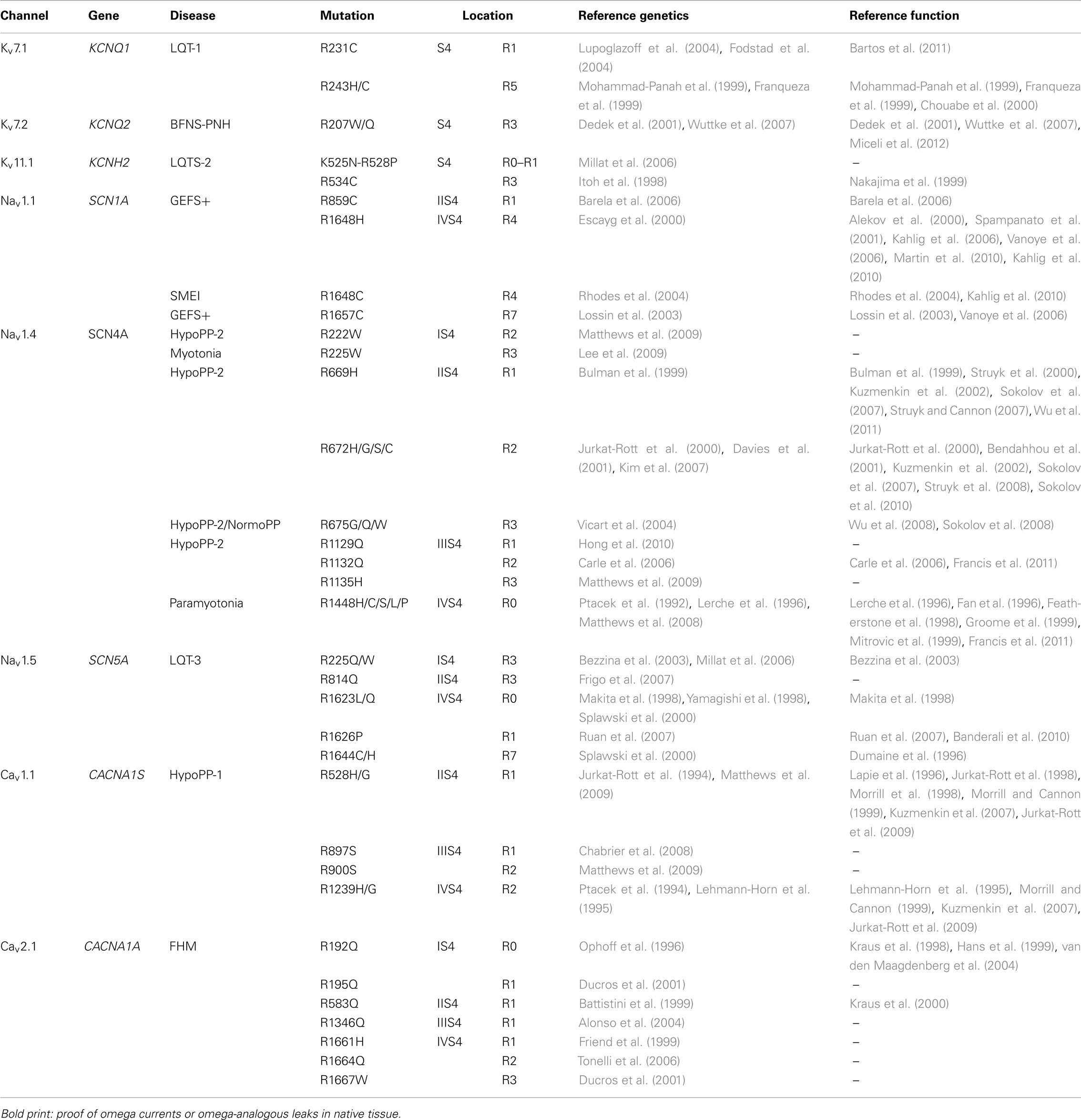
Table 1. Voltage sensor mutations in K+, Na+, and Ca2 + channelopathies.
This review briefly outlines the clinical features and pathogenesis of the S4 channelopathies with a focus on mutation-dependent changes of alpha and omega currents as well as their contributions to the phenotype. It also introduces the concept of membrane bi-stability in general and in computer simulations incorporating omega current. Also discussed are the consequences of intracellular sodium overload and tissue degeneration to explain the non-episodic features of the channelopathies and the ideas behind best practice treatment. For further reading, there are reviews with more emphasis on S4 structure and function (Catterall, 2010), S4 mutations in channels of skeletal muscle (Cannon, 2010; Jurkat-Rott et al., 2010), or drug action in periodic paralyzes and related channelopathies (Matthews and Hanna, 2010; Tricarico and Camerino, 2011).
Epilepsy is characterized by recurring episodes of synchronized electrical discharges of neurons caused by their facilitated depolarization within the central nervous system. The symptoms of a seizure depend on age of the patient, the underlying cause, and the brain region involved.
Generalized epilepsy with febrile seizures plus (GEFS+) is a childhood-onset syndrome featuring febrile seizures (FS) and afebrile epileptic convulsions within the same pedigree. The penetrance is ∼60%. In two-thirds of affected individuals either FS persist after the sixth year of life or afebrile generalized tonic-clonic seizures additionally occur (FS+). GEFS+ type II is caused among others by missense mutations in the alpha subunit of the neuronal sodium channel Nav1.1, encoded by SCN1A. Mutations frequently destabilize the fast-inactivated state, resulting in a persistent inward sodium current that depolarizes the membrane potential to a value closer to the Nav1.1 threshold (facilitated spike generation). Additionally, a few mutations result in loss-of-function by trafficking defects (Catterall et al., 2010; Escayg and Goldin, 2010). S4 mutations II-R1C, IV-R4H, and IV-R7C (Figure 3) result in additional loss-of-function effects such as decreased current amplitude and stabilization of the inactivated state (Alekov et al., 2000; Spampanato et al., 2001; Lossin et al., 2003; Barela et al., 2006; Vanoye et al., 2006). Apparently, loss-of-function in inhibitory neurons produces overexcitability (Martin et al., 2010) similar to gain-of-function effects in excitatory neurons (Kahlig et al., 2006, 2010).
Severe myoclonic epilepsy of infancy (SMEI), or Dravet syndrome, is characterized by clonic or tonic-clonic seizures that occur in the first year of life, are often prolonged, and are typically associated with fever. During the course of the disease, patients develop additional afebrile generalized and partial seizures. Cognitive deterioration appears in early childhood. In contrast to GEFS+, the syndrome is resistant to pharmacotherapy, although stiripentol seems to have a positive effect by enhancing GABAergic neurotransmission. Cranial magnetic resonance (MR) imaging in patients reveals focal and generalized atrophy. Because some patients with SMEI have a family history of febrile or afebrile seizures, and in some families GEFS+ and SMEI overlap, SMEI may therefore be regarded as the most severe phenotype of the GEFS+ spectrum (Mullen and Scheffer, 2009). SMEI is caused by mutations in SCN1A encoding Nav1.1. Most SMEI mutations cause loss-of-function due to nonsense mutations demonstrating that haploinsufficiency of SCN1A is pathogenic (Oakley et al., 2011). The loss-of-function of Nav1.1 channels results in reduced action potential (AP) firing in hippocampal inhibitory neurons, thus generating overexcitability. IV-R4C causes gain-of-function by destabilizing fast inactivation and producing persistent currents (Rhodes et al., 2004) that may be inhibited by the sodium channel blocker ranolazine (Kahlig et al., 2010; Figure 3).
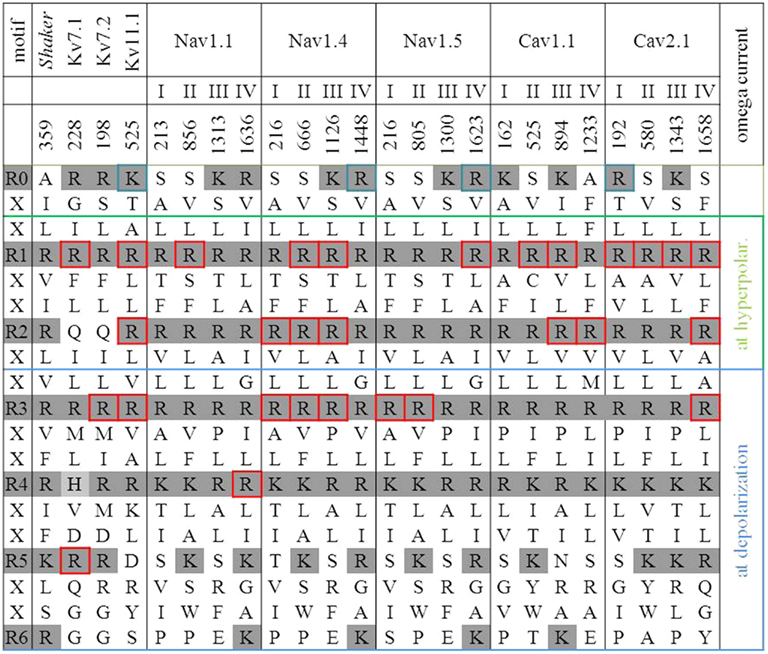
Figure 3. S4 sequences of channels with an R0 to R6 mutation. Positively charged residues in R0 to R6 positions are delineated with a shaded background (gray). The constriction of the omega pore lies between R2 and R3. Therefore an inward movement of S4 due to hyperpolarization opens the omega pore if R1 or R2 is replaced by a neutral amino acid. An outward movement of S4 due to depolarization opens the omega pore if R3 or R4 is replaced by a neutral amino acid. These gray-background arginines are boxed. The Shaker K+ channel serves as reference. Neutral replacements of its arginines R2 and R3 have been described as proton transporters.
Benign familial neonatal seizures (BFNS) are dominantly inherited with a penetrance of 85%. The seizures manifest within the first weeks of life and typically disappear spontaneously after weeks to months. Seizures may have a partial onset or may appear as generalized. Accordingly, ictal EEGs show focal and generalized discharges. Interictal EEGs are mostly normal. The risk of seizures recurring in adulthood is ∼15%, but psychomotor development is usually normal. Causative mutations have been identified in Kv7.2 and Kv7.3 potassium channels encoded by KCNQ2 and KCNQ3 respectively that interact with each other and constitute the so-called M-current, an important regulator of membrane potential near the AP threshold. Co-expression of heteromeric wild type and mutant Kv7.2/Kv7.3 channels usually reveals a potassium current reduction of ∼20–30% in the setting of haploinsufficiency. This causes BFNS by depolarizing the membrane and facilitating AP firing (Maljevic et al., 2008). In contrast, S4 mutations of the pattern R3W/Q (Figure 3) produce a dominant negative effect on co-expressed wild type brought about by a drastic depolarizing shift of the activation curve and slowing of the activation time course (Dedek et al., 2001; Wuttke et al., 2007; Miceli et al., 2012). This suppression of inhibitory neurons explains the hyperexcitability that affects central as well as peripheral neurons. The latter locus of hyperexcitability leads to additional neuromyotonia also termed peripheral nerve hyperexcitability (PNH). This disorder is characterized by short spells of spontaneous skeletal muscle overactivity resulting in twitching, undulation, or rippling of the muscles and painful cramps, so that the phenotype for these S4 mutation carriers is BFNS-PNH. Omega currents have been implicated in BFNS for the analogous Kv7.4 channel. There, corresponding R3W/Q mutations generate non-specific depolarization-induced cation outward currents of ∼1% of the pore current amplitude, at the +40 mV voltage step (Miceli et al., 2012). In Kv7 channels, single mutations of R1 and R3 fulfill the concept of adjacent arginine replacements because there is a glutamine at position R2.
Long QT syndrome is named for an elongated QT interval in the electrocardiogram. It is caused by lengthening of the repolarization phase of the cardiac ventricular AP. Because duration of the cardiac AP is dispersed in different regions of the ventricle depending on regional channel expression, any disproportionate prolongation of the AP increases the probability for re-entry of the depolarization wave, early after-depolarizations and thus, arrhythmia. Associated ECG findings may be ventricular bigemini and torsade de pointes in which the QRS complex twists around the isoelectric axis in the electrocardiogram. By this, syncope and sudden death may result in young and otherwise healthy individuals. LQT syndrome (LQTS) is caused among others by loss-of-function of potassium channels Kv7.1 encoded by KCNQ1 or Kv11.1 encoded by KCNH2 and by gain-of-function of the Nav1.5 sodium channel encoded by SCN5A.
Kv7.1 currents deactivate very slowly allowing adaptive shortening of the AP during tachycardia by incomplete deactivation. Therefore, loss-of-function in Kv7.1 leads to arrhythmia especially at elevated heart rate due to physical or emotional stress. R1C decreases current amplitude which explains the LQT in the electrocardiogram, but also produces a hyperpolarizing shift of the voltage dependence of activation and tail currents, thereby explaining the additional atrial fibrillation in these patients (Bartos et al., 2011). R5H/C produce a slowed rate of activation, a positive voltage shift of activation and/or dominant suppression of co-expressed wild type (Franqueza et al., 1999; Mohammad-Panah et al., 1999; Chouabe et al., 2000). R1, R3, and R5 mutants all fulfill the concept of adjacent arginine replacements because there is a glutamine at R2 and a histidine at R4 (Figure 3).
Kv11.1 activates and deactivates relatively slowly in comparison to a rapid inactivation. LQT mutations suppress repolarization of the myocardial AP and lengthen the QT interval by either loss-of-function or haploinsufficiency. A dominant negative effect may be achieved by current reduction of the tetrameric channel complex. For the LQT double mutation of R0/R1, there is no functional data but there two adjacent arginines are changed (Figure 3). The point mutation R3C reduces current amplitude and accelerates deactivation thereby slowing repolarization of the cardiac AP in its final phase (Nakajima et al., 1999).
Nav1.5 initiates the cardiac AP. LQT mutants frequently conduct a persistent inward current during membrane depolarization and single channel recordings show fluctuation between normal and non-inactivating gating modes. I-R3Q/W shift the steady-state inactivation curve to the right (Bezzina et al., 2003). IV-R0L/Q delay inactivation (Makita et al., 1998), IV-R1P impairs inactivation or shifts the steady-state inactivation curve to the left (Ruan et al., 2007) and decreases inactivation upon stretch modulation (Banderali et al., 2010). Mutations IV-R7C/H show dispersed re-openings (Dumaine et al., 1996; Figure 3). It has yet to be established that omega currents play a role in the pathogenesis of Kv or Nav LQTS channelopathies, but the finding of mutations in these cardiac disorders in R1–R3 residues of S4 raises that possibility (Table 1; Sokolov et al., 2007).
Familial hemiplegic migraine (FHM) presents with characteristic unilateral migraine headaches accompanied by nausea, phono-, and photophobia. Episodes are typically precipitated by an aura with symptoms of both hyper- and hypo-excitability such as aphasia, dysarthria, vertigo, homonymous hemianopsia, cheiro-oral paresthesias, and some degree of (primarily) unilateral paresis. Additionally, ataxia and cerebellar degeneration are frequently part of the FHM phenotype. Up to 50% of cases are caused by mutations in Cav2.1 encoded by CACNA1A. Current pathogenesis models of migraine with aura suggests cortical spreading depression which consists of an initial brief spike of increased neuronal activity followed by long-lasting suppression of excitability spreading across the cortex at 1–3 mm/min. The depression wave is associated with long-lasting depolarization associated with elevation of extracellular potassium and intracellular sodium. The progress of FHM correlates to the succession of symptoms during the aura initiating the migraine attacks. FHM1 includes sporadic hemiplegic migraine with progressive cerebellar ataxia. The aura may be prolonged and confusion and loss of consciousness may occur. In the interval, some families additionally present with epilepsy, retinal degeneration, hypacusis, and persistent cerebellar dysfunction with Purkinje cell atrophy. While migraine is mainly caused by gain-of-function, loss-of-function may lead to additional ataxia. Of the seven known S4 mutations in Cav2.1, there is functional data for only two. I-R0Q increases open probability and channel density in vitro (Kraus et al., 1998; Hans et al., 1999). In a knock-in mouse this mutation generates increased current density in cerebellar neurons, enhanced transmission at the neuromuscular junction, and reduced threshold and increased velocity of cortical spreading depression, effects that are compatible with an omega current (van den Maagdenberg et al., 2004; Figure 3). II-R1Q shifts activation to the left but decreases peak current (Kraus et al., 2000). Omega currents produced by either of these mutations have yet to be demonstrated.
Myotonia is an involuntary slowed relaxation after a forceful voluntary muscle contraction, experienced by the patient as muscle stiffness. After making a forceful fist or eyelid closure, the patient cannot reopen their hand or eye. Repetition decreases the myotonia, a phenomenon called warm-up. Electrical hyperexcitability of the muscle fiber membrane is the basis for myotonia, apparent in the form of repetitive APs in the EMG. Needle insertions into the resting muscle elicit myotonic bursts, i.e., runs of APs with amplitude and frequency modulation that sound like dive bombers. Autosomal dominantly inherited myotonia can be caused among others by mutations in SCN4A, the gene encoding the alpha subunit of the voltage-gated sodium channel of skeletal muscle, Nav1.4 (Heine et al., 1993), essential for generation of the muscle fiber AP. Most mutations destabilize the fast-inactivated state so that the channel inactivates slower and incompletely (Lerche et al., 1993). The resulting subthreshold membrane potential facilitates AP generation. I-R3W causes typical myotonia with warm-up phenomenon and transient weakness (Lee et al., 2009). However, no functional data are available.
Paramyotonia (PC) is also caused by dominant SCN4A missense mutations. The cardinal symptom is cold-induced muscle stiffness that increases with continued activity called paradoxical myotonia, or paramyotonia for short. In most families, on intensive cooling the stiffness gives way to flaccid weakness or even to paralysis which is caused by severe membrane depolarization (Lehmann-Horn et al., 1987; Lerche et al., 1996). Families with IV-R0H/C/S/L/P also have episodes of generalized periodic paralysis (Lehmann-Horn and Jurkat-Rott, 1999). Such attacks occur spontaneously and can be triggered by rest or elevated serum potassium. They are of much shorter duration than the cold-induced weakness that usually lasts for several hours even after the muscles are re-warmed. During cooling, PC muscle fibers slowly depolarize to an extent greater than that observed in normal muscle fibers. The depolarization is associated with long-lasting bursts of APs which cease when the membrane potential reaches −55 mV. At this potential sodium channels inactivate so that muscle becomes inexcitable and paralyzed. Causative mutations generally slow the entry into, and accelerate recovery from, fast inactivation (Yang et al., 1994). IV-R0 mutations slow deactivation, in addition to destabilizing the fast-inactivated state (Fan et al., 1996; Lerche et al., 1996; Featherstone et al., 1998; Groome et al., 1999; Mitrovic et al., 1999). No omega current is observed (Francis et al., 2011), possibly because R0 is located outside of the canaliculus (Figures 2 and 3).
Autosomal dominant hypokalemic periodic paralysis (HypoPP) is characterized by episodes of flaccid generalized muscle weakness accompanied by low serum potassium levels. Triggers for the weakness are carbohydrates and insulin with subsequent hypokalemia, rest after exercise, and cooling. Oral potassium administration accelerates the recovery of muscle strength. Native patient muscle fibers show long-lasting membrane depolarization paradoxically triggered by lowering of extracellular potassium, resulting in an inactivation of sodium channels necessary for AP generation (Jurkat-Rott et al., 2000). This effect is exacerbated in the presence of insulin (Bond and Gordon, 1993; Ruff, 1999). Permanent progressive weakness is additionally found in ∼60% of patients whereby muscle degenerates and is increasingly replaced by fatty tissue. The causative mutations are located in the skeletal muscle calcium channel Cav1.1 encoded by CACNA1S (HypoPP-1) and the skeletal muscle sodium channel Nav1.4 encoded by SCN4A. Given the finding that all but one of these mutations neutralize a positively charged S4 residue, it is not surprising that a role for omega currents as a causal factor in the pathogenesis of the channelopathies has been most clearly demonstrated for the periodic paralyzes.
Functional studies of the alpha currents of either of these channels do not satisfactorily explain the phenotype. In Cav1.1, II-R1H/G and IV-R2H/G (Figure 3) lead to loss-of-function in the sense of current amplitude reduction, slowing of activation, reduced open probability, left shift of steady-state inactivation, reduced amplitude and broadening of APs (Lehmann-Horn et al., 1995; Lapie et al., 1996; Jurkat-Rott et al., 1998; Morrill et al., 1998; Morrill and Cannon, 1999; Kuzmenkin et al., 2007). None of these gating defects are expected to lead to increased depolarization. Similarly, in Nav1.4, II-R1H, II-R2H/G/S/C, and II-R3G/W/Q show reduced current amplitude, left shift of steady-state fast inactivation, enhanced fast and slow inactivation, slowed recovery after long depolarizations, and reduced AP amplitudes (Bulman et al., 1999; Jurkat-Rott et al., 2000; Struyk et al., 2000; Bendahhou et al., 2001; Kuzmenkin et al., 2002; Carle et al., 2006; Wu et al., 2008, 2011). These defects would not contribute to depolarization either.
In Nav1.4, II-R1H, II-R2H/G/C/S, and III-R2Q mutations generate omega currents that are activated by hyperpolarization, are still active at the usual resting potential, but are closed by depolarizations large enough to activate and thus, move S4 (Figure 3). Their amplitude is about 1% of the alpha pore current. Substitution of these arginines by the bulky histidine residue enables proton transport only; other cations do not seem to have enough space to pass (Sokolov et al., 2007; Struyk and Cannon, 2007; Francis et al., 2011). Substitution of these arginines with uncharged resides such as glycine allow an inwardly rectifying flow of small monovalent cations whereby potassium and cesium are preferred over sodium or lithium (Sokolov et al., 2007; Struyk et al., 2008). Figure 2C shows current-voltage relationships for R222G/hNaV1.4 channels from experiments performed in our lab as an example (Holzherr et al., 2010). Omega pores conduct currents that show an above-linear increase in amplitude with hyper- or depolarization although the electrical field is focused to a single amino acid (Ohm resistor) and not constantly increasing within the membrane (constant field theory). The non-linearity of the omega current reflects the stochastic process of a voltage-dependent open probability and follows a Boltzmann distribution.
Guanidinium, a derivative of arginine, has greater than 10 times the conductance through the omega pore than monovalent cations, while divalent and trivalent cations block the omega pore at mM concentrations (Sokolov et al., 2010). In that study, a screen of guanidine derivatives as potential blockers of the pathogenic omega current in HypoPP yielded one compound, 1-(2-4 xylyl) guanine that exhibited similar potency for block compared to the divalent cations tested. In addition, there is an inward cation leak current at hyperpolarized potentials in native muscle of R1H knock-in mice with omega features (Wu et al., 2011). The conclusion from these alternative approaches to study omega currents is that they contribute to the depolarization leading to inexcitability and weakness. Nevertheless, despite similarities of sodium channel omega currents with those in Shaker produced by R1H mutations, their blockade is quite different, suggesting differences in shape, size, and exact cation pathway along the canaliculi of these channels. Perhaps this phenomenon is related to the postulation that two adjacent mutated charges promote omega currents in voltage-gated potassium channels, a requisite that does not hold true for sodium channels.
In contrast to the hyperpolarization-induced omega currents from substitutions of II-R1 and II-R2, II-R3G/Q/W mutations generate Nav1.4 cation currents that are activated by depolarization (Figure 3). This means that the omega currents are conducted when S4 is in the activated or inactivated state, but not in the resting state. This omega current is deactivated at hyperpolarized membrane potentials (Sokolov et al., 2008). The associated phenotype differs slightly in the ictal serum potassium levels which can be low or normal (normokalemic periodic paralysis, NormoPP). Also, the reaction to oral potassium administration may be different than for HypoPP – anything from amelioration to worsening of the weakness.
For II-R1H/Q and IV-R2H/G Cav1.1 channel mutations (Figure 3), no direct omega currents have been shown. However, in native patient muscle there is an inward cation leak current at hyperpolarized potentials with omega features (Jurkat-Rott et al., 2009). Also, based on these measurements, the periodic paralysis episodes can be successfully modeled. The underlying idea is that depolarized muscle fibers will be paralyzed, and that polarized fibers will be fully functional. If transitions between these two situations are reversible, the membrane potential is bi-stable.
P-states are the electrically stable resting membrane potentials of a cell in the nomenclature of Jurkat-Rott et al. (2009). Normal cells reveal a bimodal distribution around the stable membrane potentials P1 and P2. At physiological conditions, most fibers are highly negative polarized and in the excitable P1-state which follows the predictions of the Goldman–Hodgkin–Katz equation. Upon an increased sodium conductance, e.g., by omega pores, the fraction of cells in the (depolarized) P2-state is increased. With slow depolarization, voltage-gated sodium channels might inactivate via closed-state inactivation and render the cells inexcitable. In contrast, cells that remain in the P1-state are only slightly depolarized and thus still excitable. This bi-stability is the result of inwardly rectifying potassium channels (Kir), which open with hyperpolarization and close with depolarization (Figure 4A). The bi-stability is the reason that only a small increase in sodium conductance, e.g., by an omega pore, is required to shift cells from the P1- to the P2-state (Figure 4B). Vice versa, a repolarizing conductance can shift the membrane back to the P1-state (Figure 4). Transitions between the P1- and P2-states can occur when any trigger drives the system closer to a limit point or shifts a limit point. If the limit points of both states (LP1 and LP2) are situated at different K+ values, the system shows hysteresis (Figure 4C). If external K+ is considered as a control parameter, it is shown to play an important role in driving the system to its limits, and set the stage during “physiological” hypokalemia, for the paradoxical depolarization triggered by a slight increase in sodium conductance provided by an omega current. A significant role for the activated omega pore as a causal factor in the pathogenesis of HypoPP is supported by the computer model whose details are given in the Appendix and whose parameters are given in Table 2.
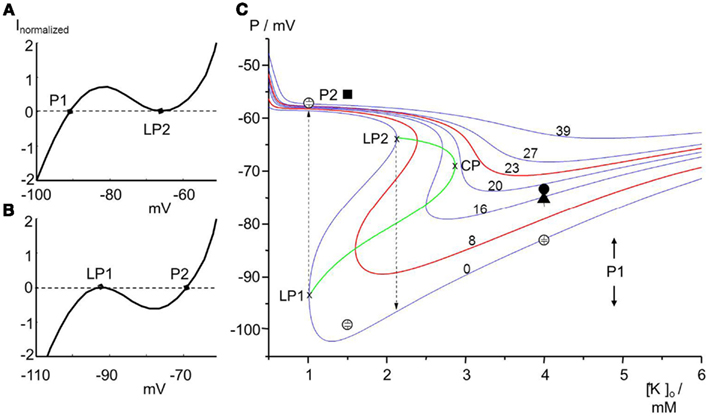
Figure 4. Effects of omega currents schematically (A) and in a computer simulation (B). (A) The current-voltage relationship of Kir potassium channels shows a voltage range characterized by a “negative” resistance. This negativity leads to membrane bi-stability. Whereas small instantaneous changes of P1 will be compensated for (P1 is therefore a stable membrane potential), larger depolarizing artifacts will cause a jump of P1 to the limit point LP2. P1 can be regained by substantial repolarizing influences like the Na/K pump. (B) The curve is downwardly shifted by an omega Na+ current. Limit point LP1 is a very instable membrane potential that will be shifted to P2 by even smallest instantaneous changes. In the P2-state the cell membrane will be electrically stable. (C) Membrane potentials P for various [K+]o values and for various omega pore conductances (in μS/cm2). Reducing [K+]o first leads to hyperpolarization until the limit point (LP1) is reached at which the membrane potential becomes instable and jumps to the depolarized state of about −58 mV. From there, increasing [K+]o takes the potential along the curve until LP2 is reached. LP2 is the starting point for the repolarization. The curves of LP1 and LP2 meet in the cusp point, CP. The region inside the cusp (bounded by LP1, LP2, and CP) is bi-stable. In contrast omega pore conductances larger than 18 μS/cm2 result in gradual depolarization without bi-stability. The model reveals that an omega pore shifts LP1 and LP2 (the cusp) to the right, i.e., less severe hypokalemia is required to shift cells from the P1- to the P2-state. The membrane potentials yielded by the computer simulation were compared with values measured for muscle fibers by use of microelectrodes: open symbols stand for human controls (−83 ± 5 mV in 95% of fibers at 4 mM K+, −99 ± 3 mV for 87% at 1.5 mM K+, and −58 mV for 91% at 1 mM K+); filled symbols for HypoPP patients with either Cav1.1-R1239H (circle: −74 ± 5 mV in 76% of fibers at 4 mM K+) or Cav1.1-R528H (triangle: −75 ± 5 mV in 91% of fibers at 4 mM K+); at 1.5 mM K+, 95% of the patients’ fibers −56 mV (square; n = 127). Modified after Jurkat-Rott et al. (2010).
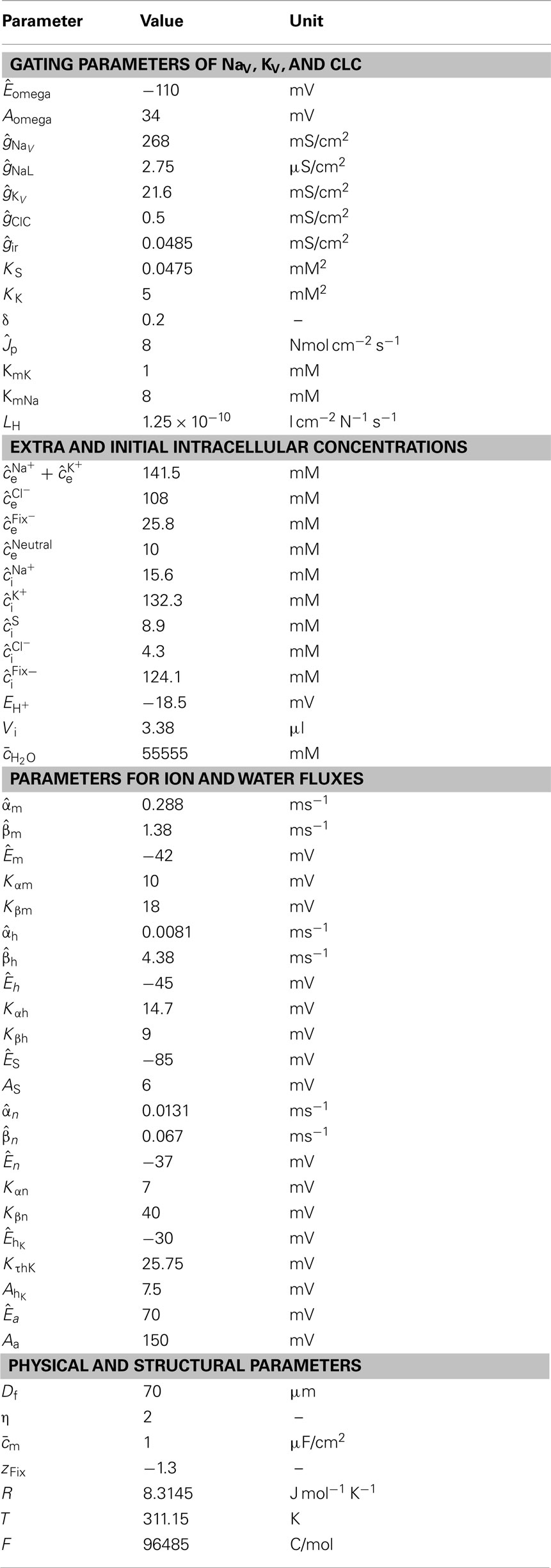
Table 2. Parameters of the catastrophe model.
A drop of serum K+ will decrease Kir conductance. Although the Nernst equation predicts hyperpolarization when external K+ is reduced, the hypokalemia-induced reduction of Kir conductance may shift the cells from the P1- to the P2-state (Figure 4C). Therefore, cells show a bimodal distribution of membrane potentials especially at low K+. This phenomenon is called paradoxical depolarization (Jurkat-Rott and Lehmann-Horn, 2007; Struyk and Cannon, 2008; Jurkat-Rott et al., 2009). Not surprisingly, decreased conductance in mutant Kir channels contributes to the pathogenesis of episodic paralysis associated with thyrotoxicosis (Kir 2.6; Ryan et al., 2010; Cheng et al., 2011), periodic paralysis and cardiac arrhythmia in Tawil–Anderson’s syndrome (Kir 2.1; Plaster et al., 2001) and underlies barium-induced augmentation of paradoxical depolarization of skeletal muscle fibers in low external K+ (Kir 2.1; Struyk and Cannon, 2008). Decreased conductance of the ATP-sensitive K channel in skeletal muscle may also be a causative factor in hypokalemic-induced depolarization, at least in response to insulin (Tricarico et al., 1998). It has been proposed that part of the therapeutic effect of acetazolamide in HypoPP is its action to promote the activation of several different types of skeletal muscle potassium channels, which may offset the decreased conductance of Kir (for review see Matthews and Hanna, 2010).
If the omega pore conductance is >18 μS/cm2 (for the conditions in Figure 4C), the bi-stability is replaced by a single stable resting potential. The value depends on the conductance ratio gK/gω with gK carried by additional (e.g., delayed rectifying) Kv channels which are less dependent on extracellular K+ than are Kir channels. In this monostable system, the membrane potential can vary between −74 and −58 mV (Figure 4C).
Usually the majority of normal cells do not become inexcitable in response to decreased serum K+, because the limit points are located at very low K+ that do not occur physiologically. However in the presence of an omega current, the limit points are shifted to higher K+, and transitions between the states are possible under physiological conditions. HypoPP seems to be caused by such a shift. For instance, computer simulation of the effect of R669H on Vrest in response to low external K+ predicts a decrease in the threshold for paradoxical depolarization of muscle fibers, similar to that observed experimentally with barium poisoning (Struyk and Cannon, 2008). Cation ionophores like amphotericin B and gramicidin can mimic the omega pore current and serve as a successful, pharmacological in vitro model for HypoPP (Jurkat-Rott et al., 2009). Based on the ionophore experiments, the fraction of fibers in the P2-state may correspond to the chronic weakness in HypoPP patients. Typical triggers such as glucose and insulin causing hypokalemia will further increase this fraction and precipitate a paralytic attack. The pharmacological computer model predicts that smaller omega currents require a more severe hypokalemia of the patients to shift cells into the P2-state than larger omega currents.
By tackling the question as to whether sodium influx (e.g., through omega pores) increases the cytoplasmic sodium concentration, a 23Na MR sequence was developed (Nielles-Vallespin et al., 2007) that indeed revealed a muscle sodium overload in HypoPP patients (Figure 5). In addition, 1H-MR images (MRI) with a short-tau inversion recovery (STIR) sequence displayed an edema in the muscles of these HypoPP patients (Figures 5A–F). Therefore the question arose as to whether the edema was caused by a cytoplasmic sodium accumulation and osmotic imbalance, or by an interstitial edema, e.g., by an inflammation. To answer this question the MRI technique was further improved to a 23Na MRI inversion recovery sequence (Na-IR) which partially suppresses the signal raised by free sodium in the extracellular fluid and thus mainly represents cytoplasmic sodium (Nagel et al., 2011). In the HypoPP patients the nature of the edema was cytoplasmic. It seemed to be cytotoxic as all patients who showed the edema presented with a chronic weakness and muscle degeneration with age (Figures 5G–I).
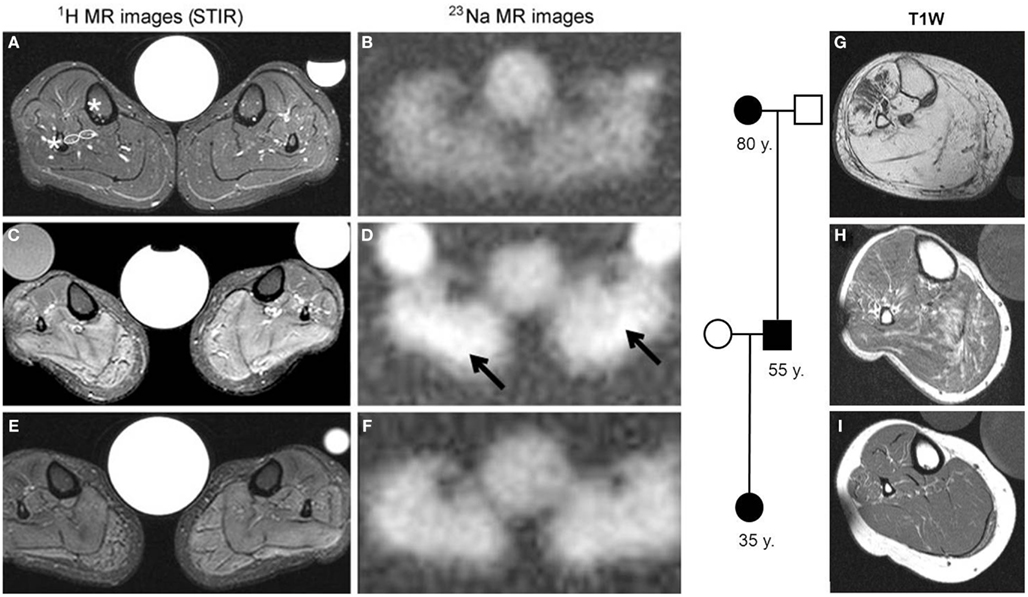
Figure 5. 1H and 23Na measurements in the calf muscles of HypoPP patients. (A–F) T2-weighted STIR 1H (left) and 23Na-MR images (right) from a healthy control (A,B) and the propositus of a HypoPP family, a 37-year-old female harboring the Cav1.1-R1239H mutation (C–F). The images in (C,D) were taken before treatment and the images in (E,F) were taken after treatment with 250 mg/d acetazolamide for 4 weeks. Note the very high proton intensities in STIR (C) and the elevated Na+ concentration before treatment (D), arrows pointing at highest Na signal intensities) and their improvement after treatment. The central reference contains 0.3% NaCl solution; occasional side tubes containing 0.3% NaCl in 1% agarose (left) and 0.6% NaCl in H2O (right) were additional standards. (G–I) Axial T1-weighted MR images from family members patients: the female’s 80-year-old grandmother whose limb muscles were almost completely replaced with fat (G), the female’s 35-year-old sister (I), and the 55-year-old uncle (H,G). Modified after Jurkat-Rott et al. (2009).
In parallel with the concept of degeneration caused by sodium overload, ictal cytotoxic edema during the long-lasting depolarization (spreading depression) has been frequently reported in FHM (for example Chabriat et al., 2000; Butteriss et al., 2003) and has been suggested to sustain the long-lasting aura (Iizuka et al., 2006) and contribute to neurodegeneration (Carreño et al., 2011). Likewise, ictal edema is associated with neuronal degeneration and sclerosis of the hippocampus in FSs (Scott et al., 2003, 2006; Sokol et al., 2003). Therefore, it is possible that intracellular sodium overload may contribute to neuronal as well as muscle channelopathies.
Guided by the experience that acetazolamide has favorable effects on the episodic weakness in the periodic paralyzes (Resnick et al., 1968), this agent was administered to HypoPP patients and reduced the chronic weakness of the patients as well as the sodium overload and edema in the MRI (Figure 5). In the pharmacological in vitro model of HypoPP using an ionophore, acetazolamide shifted many fibers from the P2- to the P1-state. This in vitro effect was considered to be responsible for the positive, in vivo effects (Jurkat-Rott et al., 2009). Thus, drugs that repolarize the fiber membrane may prevent progression of the cell degeneration by reducing the cytotoxic edema. Supporting the idea that FHM and epilepsy share similar pathomechanisms to HypoPP, it is not surprising that acetazolamide is standard treatment option for both epilepsy (Wolf, 2011) and FHM (Russell and Ducros, 2011).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The work was supported by research grants from the Else Kröner-Fresenius-Stiftung and the BMBF (IonoNeurOnet), both to Karin Jurkat-Rott and Frank Lehmann-Horn (2010_A27), and by NIH 1R15NS064556-01 to James Groome. Frank Lehmann-Horn is endowed Senior Research Professor of the non-profit Hertie-Foundation. We thank M. Fauler and V. Winston (ISU) for their contributions to the computer models.
Alekov, A., Rahman, M. M., Mitrovic, N., Lehmann-Horn, F., and Lerche, H. (2000). A sodium channel mutation causing epilepsy in man exhibits subtle defects in fast inactivation and activation in vitro. J. Physiol. (Lond.) 529, 533–539.
Alonso, I., Barros, J., Tuna, A., Seixas, A., Coutinho, P., Sequeiros, J., and Silveira, I. (2004). A novel R1347Q mutation in the predicted voltage sensor segment of the P/Q-type calcium-channel alpha-subunit in a family with progressive cerebellar ataxia and hemiplegic migraine. Clin. Genet. 65, 70–72.
Banderali, U., Juranka, P. F., Clark, R. B., Giles, W. R., and Morris, C. E. (2010). Impaired stretch modulation in potentially lethal cardiac sodium channel mutants. Channels (Austin) 4, 12–21.
Barela, A. J., Waddy, S. P., Lickfett, J. G., Hunter, J., Anido, A., Helmers, S. L., Goldin, A. L., and Escayg, A. (2006). An epilepsy mutation in the sodium channel SCN1A that decreases channel excitability. J. Neurosci. 26, 2714–2723.
Bartos, D. C., Duchatelet, S., Burgess, D. E., Klug, D., Denjoy, I., Peat, R., Lupoglazoff, J. M., Fressart, V., Berthet, M., Ackerman, M. J., January, C. T., Guicheney, P., and Delisle, B. P. (2011). R231C mutation in KCNQ1 causes long QT syndrome type 1 and familial atrial fibrillation. Heart Rhythm 8, 48–55.
Battistini, S., Stenirri, S., Piatti, M., Gelfi, C., Righetti, P. G., Rocchi, R., Giannini, F., Battistinin, N., Guazzi, C. G., Faerrarri, M., and Carrera, P. (1999). A new CACNA1A gene mutation in acetazolamide-responsive familial hemiplegic migraine and ataxia. Neurology 53, 38–43.
Bendahhou, S., Cummins, T. R., Griggs, R. C., Fu, Y. H., and Ptácek, L. J. (2001). Sodium channel inactivation defects are associated with acetazolamide-exacerbated hypokalemic periodic paralysis. Ann. Neurol. 50, 417–420.
Bezzina, C. R., Rook, M. B., Groenewegen, W. A., Herfst, L. J., van der Wal, A. C., Lam, J., Jongsma, H. J., Wilde, A. A., and Mannens, M. M. (2003). Compound heterozygosity for mutations (W156X and R225W) in SCN5A associated with severe cardiac conduction disturbances and degenerative changes in the conduction system. Circ. Res. 92, 159–168.
Bond, E. F., and Gordon, A. M. (1993). Insulin-induced membrane changes in (K+) depleted rat skeletal muscle. Am. J. Physiol. 265, C257–C265.
Bulman, D. E., Scoggan, K. A., van Oene, M. D., Nicolle, M. W., Hahn, A. F., Tollar, L. L., and Ebers, G. C. (1999). A novel sodium channel mutation in a family with hypokalemic periodic paralysis. Neurology 53, 1932–1936.
Butteriss, D. J., Ramesh, V., and Birchall, D. (2003). Serial MRI in a case of familial hemiplegic migraine. Neuroradiology 45, 300–303.
Cannon, S. C. (2010). Voltage-sensor mutations in channelopathies of skeletal muscle. J. Physiol. (Lond.) 588, 1887–1895.
Carle, T., Lhuillier, L., Luce, S., Strenberg, D., Devuyst, O., Fontaine, B., and Tabti, N. (2006). Gating defects of a novel Na+ channel mutant causing hypokalemic peiodic paralysis. Biochem. Biophys. Res. Commun. 348, 653.
Carreño, O., García-Silva, M. T., García-Campos, Ó., Martínez-de Aragón, A., Cormand, B., and Macaya, A. (2011). Acute striatal necrosis in hemiplegic migraine with de novo CACNA1A mutation. Headache 51, 1542–1546.
Catterall, W. A. (2010). Ion channel voltage sensors: structure, function, and pathophysiology. Neuron 67, 915–928.
Catterall, W. A., Kalume, F., and Oakley, J. C. (2010). NaV1.1 channels and epilepsy. J. Physiol. (Lond.) 588, 1849–1859.
Chabriat, H., Vahedi, K., Clark, C. A., Poupon, C., Ducros, A., Denier, C., Le Bihan, D., and Bousser, M. G. (2000). Decreased hemispheric water mobility in hemiplegic migraine related to mutation of CACNA1A gene. Neurology 54, 510–512.
Chabrier, S., Monnier, N., and Lunardi, J. (2008). Early onset of hypokalaemic periodic paralysis caused by a novel mutation of the CACNA1S gene. J. Med. Genet. 45, 686–688.
Cheng, C.-J., Lin, S.-H., Lo, Y.-F., Yang, S.-S., Hsu, Y.-J., Cannon, S. C., and Huang, C.-L. (2011). Identification and functional characterization of Kir2.6 mutations associated with non-familial hypokalemic periodic paralysis. J. Biol. Chem. 286, 27425–27435.
Chouabe, C., Neyroud, N., Richard, P., Denjoy, I., Hainque, B., Romey, G., Drici, M. D., Guicheney, P., and Barhanin, J. (2000). Novel mutations in KvLQT1 that affect Iks activation through interactions with Isk. Cardiovasc. Res. 45, 971–980.
Davies, N. P., Eunson, L. H., Samuel, M., and Hanna, M. G. (2001). Sodium channel gene mutations in hypokalemic periodic paralysis: an uncommon vause in the UK. Neurology 57, 1323–1325.
Dedek, K., Kunath, B., Kananura, C., Reuner, U., Jentsch, T. J., and Steinlein, O. K. (2001). Myokymia and neonatal epiepsy caused by a mutation in the voltage sensor of the KCNQ2 K+ channel. Proc. Natl. Acad. Sci. U.S.A. 98, 12272–12277.
Ducros, A., Denier, C., Joutel, A., Cecillon, M., Lescoat, C., Vahedi, K., Darcel, F., Vicaut, E., Bousser, M. G., and Tournier-Lasserve, E. (2001). The clinical spectrum of familial hemiplegic migraine associated with mutations in a neuronal calcium channel. N. Engl. J. Med. 345, 17–24.
Dumaine, R., Wang, Q., Keating, M. T., Hartmann, H. A., Schwartz, P. J., Brown, A. M., and Kirsch, G. E. (1996). Multiple mechanisms of Na+ channel – linked long-QT syndrome. Circ. Res. 78, 916–924.
Escayg, A., and Goldin, A. L. (2010). Sodium channel SCN1A and epilepsy: mutations and mechanisms. Epilepsia 51, 1650–1168.
Escayg, A., MacDonald, B. T., Meisler, M. H., Baulac, S., Huberfeld, G., An-Gourfinkel, I., Brice, A., LeGuern, E., Moulard, B., Chaigne, D., Buresi, C., and Malafosse, A. (2000). Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat. Genet. 24, 343–345.
Fan, Z., George, A. L. Jr., Kyle, J. W., and Makielski, J. C. (1996). Two human paramyotonia congenital mutations have opposite effects on lidocaine block of Na+ channels expressed in a mammalian cell line. J. Physiol. (Lond.) 496, 275–286.
Featherstone, D. E., Fujimoto, E., and Ruben, P. C. (1998). A defect in skeletal muscle sodium channel deactivation exacerbates hyperexcitability in human paramyotonia congenita. J. Physiol. (Lond.) 506, 627–638.
Fodstad, H., Swan, H., Laitinen, P., Piippo, K., Paavonen, K., Viitasalo, M., Toivonen, L., and Kontula, K. (2004). Four potassium channel mutations account for 73% of the genetic spectrum underlying long-QT syndrome (LQTS) and provide evidence for a strong founder effect in Finland. Ann. Med. 36, 53–63.
Francis, D. G., Rybalchenko, V., Struyk, A., and Cannon, S. C. (2011). Leaky sodium channels from voltage sensor mutations in periodic paralysis, but not paramyotonia. Neurology 76, 1635–1641.
Franqueza, L., Lin, M., Shen, J., Splawski, I., Keating, M. T., and Sanguinetti, M. C. (1999). Long QT syndrome-associated mutations in the S4-S5 linker of KvLQT1 potassium channels modify gating and interaction with minK subunits. J. Biol. Chem. 274, 21063–21070. [Erratum in: J. Biol. Chem. 274, 25188].
Friend, K. L., Crimmins, D., Phan, T. G., Sue, C. M., Colley, A., Fung, V. S., Morris, J. G., Sutherland, G. R., and Richards, R. I. (1999). Detection of a novel missense mutation and second recurrent mutation in the CACNA1A gene in individuals with EA-2 and FHM. Hum. Genet. 105, 261–265.
Frigo, G., Rampazzo, A., Bauce, B., Pilichou, K., Beffagna, G., Danieli, G. A., Nava, A., and Martini, B. (2007). Homozygous SCN5A mutation in Brugada syndrome with monomorphic ventricular tachycardia and structural heart abnormalities. Europace 9, 391–397.
Gamal El-Din, T. M., Heldstab, H., Lehmann, C., and Greeff, N. G. (2010). Double gaps along Shaker S4 demonstrate omega currents at three different closed states. Channels (Austin) 4, 93–100.
Groome, J. R., Fujimoto, E., George, A. L., and Ruben, P. C. (1999). Differential effects of homologous S4 mutations in human skeletal muscle sodium channels on deactivation gating from open and inactivated states. J. Physiol. (Lond.) 516, 687–698.
Groome, J. R., and Winston, V. (2012). Voltage sensor module in NaV1.4: S1-S3 charges regulate fast inactivation. Biophys. Soc. Abstr. 1655, Pos B425.
Hans, M., Luvisetto, S., Williams, M. E., Spagnolo, M., Urrutia, A., Tottene, A., Brust, P. F., Johnson, E. C., Harpold, M. M., Stauderman, K. A., and Pietrobon, D. (1999). Functional consequences of mutations in the human alpha1A calcium channel subunit linked to familial hemiplegic migraine. J. Neurosci. 19, 1610–1619.
Heine, R., Pika, U., and Lehmann-Horn, F. (1993). A novel SCN4A mutation causing myotonia aggravated by cold and potassium. Hum. Mol. Genet. 2, 1349–1353.
Holzherr, B. D., Groome, J. R., Fauler, M., Nied, E., Lehmann-Horn, F., and Jurkat-Rott, K. (2010). Characterization of a novel hNaV1.4 mutation causing hypokalemic periodic paralysis. Biophys. Soc. Abstr. LB201, Pos L201.
Hong, D., Luan, X., Chen, B., Zheng, R., Zhang, W., Wang, Z., and Yuan, Y. (2010). Both hypokalaemic and normokalaemic periodic paralysis in different members of a single family with novel R1129Q mutation in SCN4A gene. J. Neurol. Neurosurg. Psychiatr. 81, 703–704.
Iizuka, T., Sakai, F., Suzuki, K., Igarashi, H., and Suzuki, N. (2006). Implication of augmented vasogenic leakage in the mechanism of persistent aura in sporadic hemiplegic migraine. Cephalgia 26, 332–335.
Itoh, T., Tanaka, T., Nagai, R., Kamiya, T., Sawayama, T., Nakayama, T., Tomoike, H., Sakurada, H., Yazaki, Y., and Nakamura, Y. (1998). Genomic organization and mutational analysis of HERG, a gene responsible for familial long QT syndrome. Hum. Genet. 102, 435–439.
Jurkat-Rott, K., Holzherr, B., Fauler, M., and Lehmann-Horn, F. (2010). Sodium channelopathies of skeletal muscle result from gain or loss of function. Pflugers Arch. 460, 239–248.
Jurkat-Rott, K., and Lehmann-Horn, F. (2007). Do hyperpolarization-induced proton currents contribute to the pathogenesis of hypokalemic periodic paralysis, a voltage sensor channelopathy? J. Gen. Physiol. 130, 1–5.
Jurkat-Rott, K., Lehmann-Horn, F., Elbaz, A., Heine, R., Gregg, R. G., Hogan, K., Powers, P. A., Lapie, P., Vale-Santos, J. E., Weissenbach, J., and Fontaine, B. (1994). A calcium channel mutation causing hypokalemic periodic paralysis. Hum. Mol. Genet. 3, 1415–1419.
Jurkat-Rott, K., Mitrovic, N., and Lehmann-Horn, F. (2000). Voltage-sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current. Proc. Natl. Acad. Sci. U.S.A. 97, 9549–9554.
Jurkat-Rott, K., Uetz, U., Pika-Hartlaub, U., Powell, J., Fontaine, B., Melzer, W., and Lehmann-Horn, F. (1998). Calcium currents and transients of native and heterologously expressed mutant skeletal muscle DHP receptor alpha1 subunits (R528H). FEBS Lett. 423, 198–204.
Jurkat-Rott, K., Weber, M. A., Fauler, M., Guo, X. H., Holzherr, B. D., Paczulla, A., Nordsborg, N., Joechle, W., and Lehmann-Horn, F. (2009). K+-dependent paradoxical membrane depolarization and Na+ overload, major and reversible contributors to weakness by ion channel leaks. Proc. Natl. Acad. Sci. U.S.A. 106, 4036–4041.
Kahlig, K. M., Lepist, I., Leung, K., Rajamani, S., and George, A. L. (2010). Ranolazine selectively blocks persistent current evoked by epilepsy-associated Naν1.1 mutations. Br. J. Pharmacol. 161, 1414–1426.
Kahlig, K. M., Misra, S. N., and George, A. L. Jr. (2006). Impaired inactivation gate stabilization predicts increased persistent current for an epilepsy-associated SCN1A mutation. J. Neurosci. 26, 10958–10966.
Kim, J. B., Kim, M. H., Lee, S. J., Kim, D. J., and Lee, B. C. (2007). The genotype and clinical phenotype of Korean patients with familial hypokalemic periodic paralysis. J. Korean Med. Sci. 22, 946–951.
Kraus, R. L., Sinnegger, M. J., Glossmann, H., Hering, S., and Striessnig, J. (1998). Familial hemiplegic migraine mutations change alpha1A Ca2+ channel kinetics. J. Biol. Chem. 273, 5586–5590.
Kraus, R. L., Sinnegger, M. J., Koschak, A., Glossmann, H., Stenirri, S., Carrera, P., and Striessnig, J. (2000). Three new familial hemiplegic migraine mutants affect P/Q-type Ca(2+) channel kinetics. J. Biol. Chem. 275, 9239–9243.
Kuzmenkin, A., Hang, C., Kuzmenkina, E., and Jurkat-Rott, K. (2007). Gating of the HypoPP-1 mutations: I. Mutant-specific effects and cooperativity. Pflugers Arch. 454, 495–505.
Kuzmenkin, A., Muncan, V., Jurkat-Rott, K., Hang, C., Lerche, H., Lehmann-Horn, F., and Mitrovic, N. (2002). Enhanced inactivation and pH sensitivity of Na+ channel mutations causing hypokalaemic periodic paralysis type II. Brain 125, 835–843.
Lapie, P., Goudet, C., Nargeot, J., Fontaine, B., and Lory, P. (1996). Electrophysiological properties of the hypokalaemic periodic paralysis mutation (R528H) of the skeletal muscle alpha 1s subunit as expressed in mouse L cells. FEBS Lett. 382, 244–248.
Lee, S. C., Kim, H. S., Park, Y. E., Choi, Y. C., Park, K. H., and Kim, D. S. (2009). Clinical diversity of SCN4A-mutation-associated skeletal muscle sodium channelopathy. J. Clin. Neurol. 5, 186–191.
Lehmann-Horn, F., and Jurkat-Rott, K. (1999). Voltage-gated ion channels and hereditary disease. Physiol. Rev. 79, 1317–1371.
Lehmann-Horn, F., Rüdel, R., and Ricker, K. (1987). Membrane defects in paramyotonia congenita (Eulenburg). Muscle Nerve 10, 633–641.
Lehmann-Horn, F., Sipos, I., Jurkat-Rott, K., Heine, R., Brinkmeier, H., Fontaine, B., Kovacs, L., and Melzer, W. (1995). Altered calcium currents in human hypokalemic periodic paralysis myotubes expressing mutant L-type calcium channels. Soc. Gen. Physiol. Ser. 50, 101–113.
Lerche, H., Heine, R., Pika, U., George, A. L., Mitrovic, N., Browatzki, M., Weiss, T., Bastide-Rivet, M., Franke, C., LoMonaco, M., Ricker, K., and Lehmann-Horn, F. (1993). Human sodium channel myotonia: slowed channel inactivation due to substitutions for a glycine within the III/IV linker. J. Physiol. (Lond.) 470, 13–22.
Lerche, H., Mitrovic, N., Dubowitz, V., and Lehmann-Horn, F. (1996). Paramyotonia congenita: the R1448P Na+ channel mutation in adult human skeletal muscle. Ann. Neurol. 39, 599–608.
Lossin, C., Rhodes, T. H., Desai, R. R., Vanoye, C. G., Wang, D., Carniciu, S., Devinsky, O., and George, A. L. Jr. (2003). Epilepsy-associated dysfunction in the voltage-gated neuronal sodium channel SCN1A. J. Neurosci. 23, 11289–11295.
Lupoglazoff, J. M., Denjoy, I., Villain, E., Fressart, V., Simon, F., Bozio, A., Berthet, M., Benammar, N., Hainque, B., and Guicheney, P. (2004). Long QT syndrome in neonates: conduction disorders associated with HERG mutations and sinus bradycardia with KCNQ1 mutations. J. Am. Coll. Cardiol. 43, 826–830.
Makita, N., Shirai, N., Magashima, M., Matsuoka, R., Yamada, Y., Tohse, N., and Kitabatake, A. (1998). A de novo missense mutation of human cardiac Na(+) channel exhibiting novel molecular mechanism of long QT syndrome. FEBS Lett. 423, 5–9.
Maljevic, S., Wuttke, T. V., and Lerche, H. (2008). Nervous system Kv7 disorders: breakdown of a subthreshold brake. J. Physiol. (Lond.) 586, 1791–1801.
Martin, M. S., Dutt, K., Papale, L. A., Dubé, C. M., Dutton, S. B., de Haan, G., Shankar, A., Tufik, S., Meisler, M. H., Baram, T. Z., Goldin, A. L., and Escayg, A. (2010). Altered function of the SCN1A voltage-gated sodium channel leads to gamma-aminobutyric acid-ergic (GABAergic) interneuron abnormalities. J. Biol. Chem. 285, 9823–9834.
Matthews, E., and Hanna, M. G. (2010). Muscle channelopathies: does the predicted gating pore offer new treatment insights for hypokalemic periodic paralysis? J. Physiol. (Lond.) 588, 1879–1886.
Matthews, E., Labrum, R., Sweeney, M. G., Sud, R., Haworth, A., Chinnery, P. F., Meola, G., Schorge, S., Kullmann, D. M., Davis, M. B., and Hanna, M. G. (2009). Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis. Neurology 72, 1544–1547.
Matthews, E., Tan, S. V., Fialho, D., Sweeney, M. G., Sud, R., Haworth, A., Stanley, E., Cea, G., Davis, M. B., and Hanna, M. G. (2008). What causes paramyotonia in the United Kingdom? Common and new SCN4A mutations revealed. Neurology 70, 50–53.
Miceli, F., Vargas, E., Bezanilla, F., and Taglialatela, M. (2012). Gating currents from K(v)7 channels carrying neuronal hyperexcitability mutations in the voltage-sensing domain. Biophys. J. 102, 1372–1382.
Millat, G., Chevalier, P., Restier-Miron, L., Da Costa, A., Bouvagnet, P., Kugener, B., Fayol, L., Gonzàlez Armengod, C., Oddou, B., Chanavat, V., Froidefond, E., Perraudin, R., Rousson, R., and Rodriguez-Lafrasse, C. (2006). Spectrum of pathogenic mutations and associated polymorphisms in a cohort of 44 unrelated patients with long QT syndrome. Clin. Genet. 70, 214–227.
Mitrovic, N., George, A. L. Jr., Rüdel, R., Lehmann-Horn, F., and Lerche, H. (1999). Mutant channels contribute <50% to Na+ current in paramyotonia congenita muscle. Brain 122, 1085–1092.
Mohammad-Panah, R., Demolombe, S., Neyroud, N., Guicheney, P., Kyndt, F., van den Hoff, M., Baró, I., and Escande, D. (1999). Mutations in a dominant-negative isoform correlate with phenotype in inherited cardiac arrhythmias. Am. J. Hum. Genet. 64, 1015–1023.
Morrill, J. A., Brown, R. H. Jr., and Cannon, S. C. (1998). Gating of the L-type Ca channel in human skeletal myotubes: an activation defect caused by the hypokalemic periodic paralysis mutation R528H. J. Neurosci. 18, 10320–10334.
Morrill, J. A., and Cannon, S. C. (1999). Effects of mutations causing hypokalaemic periodic paralysis on the skeletal muscle L-type Ca2+ channel expressed in Xenopus laevis oocytes. J. Physiol. (Lond.) 520, 321–336.
Mullen, S. A., and Scheffer, I. E. (2009). Translational research in epilepsy genetics. Arch. Neurol. 66, 21–26.
Nagel, A. M., Amarteifio, E., Lehmann-Horn, F., Jurkat-Rott, K., Semmler, W., Schad, L. R., and Weber, M. A. (2011). 3 Tesla sodium inversion recovery magnetic resonance imaging allows improved visualization of intracellular sodium content changes in muscular channelopathies. Invest. Radiol. 46, 759–766.
Nakajima, T., Furukawa, T., Hirano, Y., Tanaka, T., Sakurada, H., Takahashi, T., Nagai, R., Itoh, T., Katayama, Y., Nakamura, Y., and Hiraoka, M. (1999). Voltage-shift of the current activation in HERG S4 mutation (R534C) in LQT2. Cardiovasc. Res. 44, 283–293.
Nielles-Vallespin, S., Weber, M. A., Bock, M., Bongers, A., Speier, P., Combs, S. E., Wöhrle, J., Lehmann-Horn, F., Essig, M., and Schad, L. R. (2007). 3D radial projection technique with ultrashort echo times for sodium MRI: clinical applications in human brain and skeletal muscle. Magn. Reson. Med. 57, 74–81.
Oakley, J. C., Kalume, F., and Catterall, W. A. (2011). Insights into pathophysiology and therapy from a mouse model of Dravet syndrome. Epilepsia 52, 59–61.
Ophoff, R. A., Terwindt, G. M., Vergouwe, M. N., van Eijk, R., Oefner, P. J., Hoffman, S. M. G., Lamerdin, J. E., Mohrenweiser, H. W., Bulman, D. E., Ferrari, M., Haan, J., Lindhout, D., van Ommen, G.-J. B., Hofker, M. H., Ferrari, M. D., and Frants, R. R. (1996). Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca(2+) channel gene CACNL1A4. Cell 87, 543–552.
Plaster, N. M., Tawil, R., Tristani-Firouzi, M., Canun, S., Bendahhou, S., Tsunoda, A., Donaldson, M. R., Iannaccone, S. T., Brunt, E., Barohn, R., Clark, J., Deymeer, F., George, A. L. Jr., Fish, F. A., Hahn, A., Nitu, A., Ozdemir, C., Serdaroglu, P., Subramony, S. H., Wolfe, G., Fu, Y.-H., and Ptacek, L. J. (2001). Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Anderson’s syndrome. Cell 105, 511–519.
Ptacek, L. J., George, A. L. Jr., Barchi, R. L., Griggs, R. C., Riggs, J. E., Robertson, M., and Leppert, M. F. (1992). Mutations in an S4 segment of the adult skeletal muscle sodium channel cause paramyotonia congenita. Neuron 8, 891–897.
Ptacek, L. J., Tawil, R., Griggs, R. C., Engel, A. G., Layzer, R. B., Kwiecinski, H., McManis, P. G., Santiago, L., Moore, M., Fouad, G., Bradley, P., and Leppert, M. F. (1994). Dihydropyridine receptor mutations cause hypokalemic periodic paralysis. Cell 77, 863–868.
Resnick, J. S., Engel, W. K., Griggs, R. C., and Stam, A. C. (1968). Acetazolamide prophylaxis in hypokalemic periodic paralysis. N. Engl. J. Med. 278, 582–586.
Rhodes, T. H., Lossin, C., Vanoye, C. G., Wang, D. W., and George, A. L. Jr. (2004). Noninactivating voltage-gated sodium channels in severe myoclonic epilepsy of infancy. Proc. Natl. Acad. Sci. U.S.A. 101, 11147–11152.
Ruan, Y., Liu, N., Bloise, R., Napolitano, C., and Priori, S. G. (2007). Gating properties of SCN5A mutations and the response to mexiletine in long-QT syndrome type 3 patients. Circulation 116, 1137–1144.
Ruff, R. L. (1999). Insulin acts in hypokalemic periodic paralysis by reducing inward rectifier K current. Neurology 53, 1556.
Russell, M. B., and Ducros, A. (2011). Sporadic and familial hemiplegic migraine: pathophysiological mechanisms, clinical characteristics, diagnosis, and management. Lancet Neurol. 10, 457–470.
Ryan, D. P., da Silva, M. R. D., Soong, T. W., Fontaine, B., Donaldson, M. R., Kung, A. W. C., Jongjaroenprasert, W., Liang, M. C., Khoo, D. H. C., Cheah, J. S., Ho, S. C., Bernstein, H. S., Maciel, R. M. B., Brown, R. H. Jr., and Ptacek, L. J. (2010). Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell 140, 88–98.
Scott, R. C., King, M. D., Gadian, D. G., Neville, B. G., and Connelly, A. (2003). Hippocampal abnormalities after prolonged febrile convulsion: a longitudinal MRI study. Brain 126, 2551–2557.
Scott, R. C., King, M. D., Gadian, D. G., Neville, B. G., and Connelly, A. (2006). Prolonged febrile seizures are associated with hippocampal vasogenic edema and developmental changes. Epilepsia 47, 1493–1498.
Sokol, D. K., Demyer, W. E., Edwards-Brown, M., Sanders, S., and Garg, B. (2003). From swelling to sclerosis: acute change in mesial hippocampus after prolonged febrile seizure. Seizure 12, 237–240.
Sokolov, S., Scheuer, T., and Catterall, W. A. (2005). Ion permeation through a voltage- sensitive gating pore in brain sodium channels having voltage sensor mutations. Neuron 47, 183–189.
Sokolov, S., Scheuer, T., and Catterall, W. A. (2007). Gating pore current in an inherited ion channelopathy. Nature 446, 76–78.
Sokolov, S., Scheuer, T., and Catterall, W. A. (2008). Depolarization-activated gating pore current conducted by mutant sodium channels in potassium-sensitive normokalemic periodic paralysis. Proc. Natl. Acad. Sci. U.S.A. 105, 19980–19985.
Sokolov, S., Scheuer, T., and Catterall, W. A. (2010). Ion permeation and block of the gating pore in the voltage sensor of NaV1.4 channels with hypokalemic periodic paralysis mutations. J. Gen. Physiol. 136, 225–236.
Spampanato, J., Escayg, A., Meisler, M. H., and Goldin, A. L. (2001). Functional effects of two voltage-gated sodium channel mutations that cause generalized epilepsy with febrile seizures plus type 2. J. Neurosci. 21, 7481–7490.
Splawski, I., Shen, J., Timothy, K. W., Lehmann, M. H., Priori, S., Robinson, J. L., Moss, A. J., Schwartz, P. J., Towbin, J. A., Vincent, G. M., and Keating, M. T. (2000). Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 102, 1178–1185.
Starace, D. M., and Bezanilla, F. (2001). Histidine scanning mutagenesis of basic residues of the S4 segment of the Shaker K+ channel. J. Gen. Physiol. 117, 469–490.
Starace, D. M., and Bezanilla, F. (2004). A proton pore in a potassium channel voltage sensor reveals a focused electric field. Nature 427, 548–553.
Starace, D. M., Stefani, E., and Bezanilla, F. (1997). Voltage-dependent proton transport by the voltage sensor of the Shaker K+ channel. Neuron 19, 1319–1327.
Struyk, A. F., and Cannon, S. C. (2007). A Na+ channel mutation linked to hypokalemic periodic paralysis exposes a proton-selective gating pore. J. Gen. Physiol. 130, 11–20.
Struyk, A. F., and Cannon, S. C. (2008). Paradoxical depolarization of Ba2+-treated muscle exposed to low extracellular K+: insights into resting potential abnormalities in hypokalemic paralysis. Muscle Nerve 37, 326–337.
Struyk, A. F., Markin, V. S., Francis, D., and Cannon, S. (2008). Gating pore currents in DIIS4 mutations of NaV1.4 associated with periodic paralysis: saturation of ion flux and implications for disease pathogenesis. J. Gen. Physiol. 132, 447–464.
Struyk, A. F., Scoggan, K. A., Bulman, D. E., and Cannon, S. C. (2000). The human skeletal muscle Na channel mutation R669H associated with hypokalemic periodic paralysis enhances slow inactivation. J. Neurosci. 20, 8610–8617.
Tombola, F., Pathak, M. M., and Isacoff, E. Y. (2005). Voltage-sensing arginines in a potassium channel permeate and occlude cation-selective pores. Neuron 45, 379–388.
Tonelli, A., D’Angelo, M. G., Salati, R., Villa, L., Germinasi, C., Frattini, T., Meola, G., Turconi, A. C., Bresolin, N., and Bassi, M. T. (2006). Early onset, non fluctuating spinocerebellar ataxia and a novel missense mutation in CACNA1A gene. J. Neurol. Sci. 241, 13–17.
Tricarico, D., and Camerino, D. C. (2011). Recent advances in the pathogenesis and drug action in periodic paralyses and related channelopathies. Front. Pharmacol. 2:8. doi:10.3389/fphar.2011.00008
Tricarico, D., Pierno, S., Mallamaci, R., Brigiani, G. S., Capriulo, R., Santoro, G., and Camerino, D. C. (1998). The biophysical and pharmacological characteristics of skeletal muscle ATP-sensitive K+ channels are modified in K+-depleted rat, an animal model of hypokalemic periodic paralysis. Mol. Pharmacol. 54, 197–206.
van den Maagdenberg, A. M., Pietrobon, D., Pizzorusso, T., Kaja, S., Broos, L. A., Cesetti, T., van de Ven, R. C., Tottene, A., van der Kaa, J., Plomp, J. J., Frants, R. R., and Ferrari, M. D. (2004). A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron 41, 701–710.
Vanoye, C. G., Lossin, C., Rhodes, T. H., and George, A. L. Jr. (2006). Single-channel properties of human NaV1.1 and mechanism of channel dysfunction in SCN1A-associated epilepsy. J. Gen. Physiol. 127, 1–14.
Vicart, S., Sternberg, D., Fournier, E., Ochsner, F., Laforet, P., Kuntzer, T., Eymard, B., Hainque, B., and Fontaine, B. (2004). New mutations of SCN4A cause a potassium-sensitive normokalemic periodic paralysis. Neurology 63, 2120–2127.
Wallinga, W., Meijer, S. L., Alberink, M. J., Vliek, M., Wienk, E. D., and Ypey, D. L. (1999). Modelling action potentials and membrane currents of mammalian skeletal muscle fibres in coherence with potassium concentration changes in the T-tubular system. Eur. Biophys. J. 28, 317–329.
Wu, F., Mi, W., Burns, D. K., Fu, Y., Gray, H. F., Struyk, A. F., and Cannon, S. C. (2011). A sodium channel knockin mutant (NaV1.4-R669H) mouse model of hypokalemic periodic paralysis. J. Clin. Invest. 121, 4082–4094.
Wu, L., Wu, W., Yan, G., Wang, X., and Liu, J. (2008). The construction and preliminary investigation of the cell model of a novel mutation R675Q in the SCN4A gene identified in a Chinese family with normokalemic periodic paralysis. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 25, 629–632.
Wuttke, T. V., Jurkat-Rott, K., Paulus, W., Garncarek, M., Lehmann-Horn, F., and Lerche, H. (2007). Peripehral nerve hyperexcitability due to dominant-negative KCNQ2 mutations. Neurology 69, 2045–2053.
Yamagishi, H., Furutani, M., Kamisago, M., Morikawa, Y., Kojima, Y., Hino, Y., Furutani, Y., Kimura, M., Imamura, S., Takao, A., Momma, K., and Matsuoka, R. (1998). A de novo missense mutation (R1623Q) of the SCN5A gene in a Japanese girl with sporadic long QT sydrome. Mutations in brief no. 140. Online. Hum. Mutat. 11, 481.
Yang, N., Ji, S., Zhou, M., Ptacek, L. J., Barchi, R. L., Horn, R., and George, A. L. Jr. (1994). Sodium channel mutations in paramyotonia congenita exhibit similar biophysical phenotypes in vitro. Proc. Natl. Acad. Sci. U.S.A. 91, 12785–12789.
Using catastrophe theory, the effects of an omega current were simulated in a one-compartment computer model of skeletal muscle fibers. Both changes of ion concentrations and consecutive water shifts were considered. The size of the T-tubular system was accounted for by a specific factor. It was not considered a separate compartment since the low-resistance T-tubular openings justified the assumption of an equilibrium. Parameters of the catastrophe model are given in Table 2; notations and abbreviations at the end of the Appendix.
The membrane was equipped with the following conductances: a background Na+ leak conductance (gNaL), the omega pore conductance (gleak), a voltage-gated Na+ (gNaV) and K+ (gKv) conductance, an inward-rectifying K+ (gIR) and a voltage-dependent Cl− (gCl) conductance, a hydraulic conductivity (LH), and a Na+/K+ pump flux (Jp). The model was based on the charge-difference approach (3) in which the membrane potential E is calculated from the amount of charges in the intracellular compartment Qi and the membrane area Am:
Vi was the cell volume, Df the fiber diameter and η a factor correcting the size of the T-tubular membrane. The cell was only permeable for Na, K, Cl, and H2O. Fix are intracellular anions (e.g., proteins, phosphates), which – in this model – cannot pass through the membrane. Their overall charge valence zFix is set to −1.3. This was necessary to avoid very negative membrane potentials at low potassium concentrations. S are the positive charged ions, that block inward-rectifying K+ channels from inside upon depolarization. Its valence zS is set to +2, so they represent Mg2+. Multiple positively charged polyamines that increase the strength of rectification were not considered.
The amounts of ions and water in the cell n are state variables in this model. They are integrated over time by numerically solving the differential equations that describe their fluxes J:
The fluxes of ions through channels and water through an aquaporin (modeled by a simple hydraulic conductivity) are computed by the following equations:
The driving forces for the fluxes were the electrochemical gradients (E − Eion) for ions or the osmotic pressure difference ΔΠ for water:
The Na/K pump was modeled by
The parameters KmK and KmNa were taken from Wallinga et al. (1999). The maximum pump flux was chosen to get an intracellular Na+ concentration of 15 mM at control conditions. Intracellular ion concentrations were calculated from the cell volume Vi and the amount of ions:
The voltage-gated Na+ (NaV), K+ (KV), and Cl− (ClC1) conductances were based on the Hodgkin–Huxley type formalism; identifiers had their classical meaning. The equations taken from Wallinga et al. (1999) are given here for completeness:
The gating variables m, h, S (NaV slow inactivation), n, and hK (inactivation of KV) were state variables and defined by the following differential equations
The gating variable of ClC1 was assumed to reach the steady-state instantaneously
For the inward-rectifying potassium channels the same model as in Wallinga et al. (1999) was used, parameters were chosen to get appropriate values for the limit points at control conditions and a resting conductivity at of 41 μS/cm2. The equations were:
The constant background sodium leak conductance was then adapted to reach a K+ to Na+ permeability ratio between 0.01 and 0.02 and a membrane potential of approximately –86 mV (at = 3.5 mM).
For modeling the H+ leak conductance, it was assumed – based on the 31P MR spectroscopy measurements (Jurkat-Rott et al., 2009) – that the H+ equilibrium potential was constant and further, that every H+ was exchanged by one Na+ ion, presumably by a Na+/H+ exchanger. Thus the omega current was treated as a sodium current (indirectly) driven by the electrochemical gradient for protons. Its conductance was modeled as a pore with a voltage-dependent open probability that follows a Boltzmann distribution. The equation
was fitted to the HypoPP muscle fiber results. The Na+ flux (indirectly) mediated by the omega pore was calculated by
The model was implemented in MatLab Version 7.3 (The Mathworks, Inc.). Initial-value problems were solved with ODE15s, a solver for sets of stiff ordinary differential equations. Then bifurcation analysis was done by using the continuation routines of the toolbox CL_MATCONT Version 2.4.


Keywords: epilepsy and neuromyotonia, long QT syndrome, familial hemiplegic migraine, myotonia and paramyotonia, hyperkalemic and hypokalemic periodic paralysis, sodium overload, cytotoxic edema, degeneration
Citation: Jurkat-Rott K, Groome J and Lehmann-Horn F (2012) Pathophysiological role of omega pore current in channelopathies. Front. Pharmacol. 3:112. doi: 10.3389/fphar.2012.00112
Received: 22 April 2012; Accepted: 23 May 2012;
Published online: 11 June 2012.
Edited by:
Gildas Loussouarn, University of Nantes, FranceReviewed by:
Stephen Cannon, University of Texas Southwestern Medical Center, USACopyright: © 2012 Jurkat-Rott, Groome and Lehmann-Horn. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Karin Jurkat-Rott, Department of Neurophysiology, Ulm University, Albert-Einstein-Allee 11, 89069 Ulm, Germany. e-mail:a2FyaW4uanVya2F0LXJvdHRAdW5pLXVsbS5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.