
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 06 June 2012
Sec. Pharmacology of Ion Channels and Channelopathies
Volume 3 - 2012 | https://doi.org/10.3389/fphar.2012.00106
 Silke B. Bodendiek1 Clio Rubinos2 Maria Pilar Trelles2 Nichole Coleman1
Silke B. Bodendiek1 Clio Rubinos2 Maria Pilar Trelles2 Nichole Coleman1 David Paul Jenkins1
David Paul Jenkins1 Heike Wulff1†
Heike Wulff1† Miduturu Srinivas2*†
Miduturu Srinivas2*†The paucity of specific pharmacological agents has been a major impediment for delineating the roles of gap junction (GJ) channels formed by connexin proteins in physiology and pathophysiology. Here, we used the selective optimization of side activities (SOSA) approach, which has led to the design of high affinity inhibitors of other ion channels, to identify a specific inhibitor for channels formed by Cx50, a connexin subtype that is primarily expressed in the lens. We initially screened a library of common ion channel modulating pharmacophores for their inhibitory effects on Cx50 GJ channels, and identified four new classes of compounds. The triarlymethane (TRAM) clotrimazole was the most potent Cx50 inhibitor and we therefore used it as a template to explore the structure activity relationship (SAR) of the TRAMs for Cx50 inhibition. We describe the design of T122 (N-[(2-methoxyphenyl)diphenylmethyl]-1,3-thiazol-2-amine) and T136 (N-[(2-iodophenyl)diphenylmethyl]-1,3-thiazol-2-amine), which inhibit Cx50 with IC50s of 1.2 and 2.4 μM. Both compounds exhibit at least 10-fold selectivity over other connexins as well as major neuronal and cardiac voltage-gated K+ and Na+ channels. The SAR studies also indicated that the TRAM pharmacophore required for connexin inhibition is significantly different from the pharmacophore required for blocking the calcium-activated KCa3.1 channel. Both T122 and T136 selectively inhibited Cx50 GJ channels in lens epithelial cells, suggesting that they could be used to further explore the role of Cx50 in the lens. In addition, our results indicate that a similar approach may be used to find specific inhibitors of other connexin subtypes.
Connexins (Cx) are a family of proteins with four transmembrane regions, which are encoded by 21 genes in humans and which form hexameric connexons (= hemichannels). These connexons can either function as transmembrane ion channels or assemble into gap junctions (GJ) by the docking of two hemichannels from adjacent cells and directly mediate signaling between cells by passing ions, metabolites and signaling molecules up to 1 kDa in mass. Both hemichannels and GJ channels formed by different connexins play important roles in tissue homeostasis and have therefore been proposed as potential new targets for the treatment of epilepsy, cardiac arrhythmia, cancer, stroke, essential tremor, and corneal wound healing (Nemani and Binder, 2005; Salameh and Dhein, 2005; Bodendiek and Raman, 2010; Kandouz and Batist, 2010).
Since the 1980s several endogenous as well as exogenous molecules that modulate GJ channels have been discovered. The peptidic GJ channel activator rotigaptide, which mainly activates Cx43, advanced into clinical trials for the treatment of atrial fibrillation (phase II, terminated in 2007) and endothelial dysfunction (Lang et al., 2008), while GAP-134, an orally available di-peptidic analog of rotigaptide, recently completed phase I clinical trials for the treatment of atrial fibrillation (ClinicalTrials.Gov). However, apart from these exceptions, the development of GJ channel modulators as pharmacological tools and potential therapeutics is still in its infancy. Most existing modulators are either of low potency and exhibit little selectivity either for connexin channels or among individual connexin subtypes. For example, one of the most commonly used GJ channel blockers, carbenoxolone, a more water soluble derivative of the pentacyclic triterpenoid glycyrrhetinic acid, reversibly inhibits GJ currents in human fibroblasts with an IC50 of 3 μM and reduces Cx26 and Cx38 hemichannel currents in Xenopus oocytes with IC50s of 21 and 34 μM, respectively. However, at similar concentrations carbenoxolone also inhibits several other targets such as the enzyme 11β-hydroxysteroid dehydrogenase (IC50 ∼5 μM; Monder et al., 1989), voltage-gated Ca2+ currents (IC50 48 μM; Vessey et al., 2004), and the structurally similar pannexin channels (IC50 2–5 μM; Locovei et al., 2007). At even lower concentrations carbenoxolone inhibits P2x7 receptors (IC50 175 nM; Suadicani et al., 2006). Other commonly used connexin blockers like the long-chain alcohols heptanol and octanol, the diphenylborate 2-APB or flufenamic acid are similarly either of low potency or lack selectivity for connexin channels (for a recent review see: Bodendiek and Raman, 2010).
Potent connexin subtype selective modulators are urgently needed to further elucidate the physiological and pathophysiological roles of the different connexins and to perform proof-of-concept studies validating connexins as potential drug targets for various diseases for which they have been proposed as novel targets. We therefore screened a small library of compounds containing ion channel modulating pharmacophores for their effects on Cx50 GJ channels. Cx50 was used as an exemplary connexin because it is expressed robustly in expression systems. Cx50 is mainly expressed in the crystalline lens. In lens epithelial cells, it is co-expressed with Cx43 and plays an important role in postnatal lens growth (White et al., 1998; Rong et al., 2002). In fiber cells, where it is co-expressed with Cx46, it has been shown to be an important component of the lens microcirculation, essential for maintenance of lens transparency (Mathias et al., 1997, 2010). Genetic deletion of Cx50 causes mild cataracts and significantly decreases lens growth (White et al., 1998; Rong et al., 2002), while missense and frame shift mutations have been found in families with inherited cataracts (Berthoud and Beyer, 2009; Mathias et al., 2010). To further study the role of Cx50 channels in the lens, a potent and selective blocker would be of great interest. Such an inhibitor is likely to be useful to dissect the contribution of the coupling provided by Cx50 to lens development and transparency. In this study, we describe the design of two Cx50 inhibitors with IC50s of 1.2 and 2.4 μM. Both compounds exhibit excellent selectivity for Cx50 over Cx43, and Cx46, which are also expressed in the lens (<18% inhibition at 10 μM), and strongly reduced junctional currents in primary lens epithelial cells isolated on postnatal day 6, a developmental time-point where Cx50 provides the majority of the coupling in the epithelium. These new pharmacological tool compounds will be useful to further explore the role of Cx50 in lens physiology and pathophysiology and for structure function studies of connexins.
Clotrimazole (CAS No. 23593-75-1), triphenylmethane (CAS No. 519-73-3), triphenylmethyl chloride (CAS No. 76-83-5), triphenylmethanol (CAS No. 76-84-6), 3,3,3-triphenylpropionic acid (T51, CAS No. 900-91-4), (R)-(+)-α,α-diphenyl-2-pyrrolidinemethanol (T52, CAS No. 22348-32-9), (S)-(−)-α,α-diphenyl-2-pyrrolidinemethanol (T53, CAS No. 112068-01-6), diphenyl-4-pyridylmethane (T160, CAS No. 3678-72-6), and triphenylmethylamine (T162, CAS No. 5824-40-8) were purchased from Sigma-Aldrich (St. Louis, MO). Tetraphenylmethane (T161, CAS No. 630-76-2) was purchased from Alfa Aesar (Ward Hill, MA). 2-Chlorotrityl chloride (T3-Cl, CAS No. 42074-68-0) and diphenyl-4-pyridylmethanol (T50, CAS No. 1620-30-0) were purchased from TCI America (Portland, OR). T1-T4, T9, T11, T13, T20, T34, T35, T39, T41, T43, T44, T54, T57, T61, T64, T66-T75, T78-T80, T85, and T97 were available in the Wulff laboratory compound library and had been previously synthesized and characterized (Wulff et al., 2000). The remaining compounds were synthesized using Grignard, chlorination and alkylation reactions described as general methods A, B, and C (see also Figure 3). New chemical entities (NCEs) were characterized by melting point (Melting Point B-540, Büchi), 1H-NMR (Avance 500, Bruker), mass spectrometry (MS: LCQ, Thermo Scientific; HRMS: LTQ-Orbitrap XL Thermo Scientific), and/or combustion analysis (2400 Series II combustion analyzer, Perkin Elmer). All MS and HRMS spectra were recorded with ESI as ionization mode if not stated otherwise. In cases where no sufficient analytical data for previously reported compounds (T109, T117, T129, T141, T143, T144, T154-OH, and T165) were available 1H-NMR, MS and/or combustion data in addition to melting points are also provided.
Triaryl methanols were synthesized through a Grignard reaction by stirring 25 mmol of magnesium turnings and 10–15 mmol of the required arylbromide in 50 mL of anhydrous diethyl ether. To initiate the reaction, catalytic amounts of iodine were added. The remaining 15–20 mmol of the required arylbromide were diluted with or dissolved in anhydrous diethyl ether (50 mL) and added slowly allowing gentle reflux. The reaction mixture was refluxed until all magnesium was consumed. Then, a solution of the required benzophenone (25 mmol) in anhydrous diethyl ether (50 mL) was added drop wise and the resulting mixture was heated under reflux for 12–24 h. After completion of the reaction the mixture was cooled to 0°C, poured into 100 mL of cold water and acidified with concentrated HCl. The organic phase was separated, and the aqueous phase was extracted three times with diethyl ether. The organic phases were combined, washed with NaHCO3 (10%) and dried over sodium sulfate. After evaporation of the solvent the crude triaryl methanols were obtained either as solid or as oily residues, which were recrystallized from petroleum ether (40–60°C) several times, if necessary.
The triaryl methyl chlorides were obtained according to McNaughton-Smith et al. (2008) by adding a five-fold excess of acetyl chloride to a stirred solution of the respective triaryl methanol in dichloromethane. After stirring the reaction mixture at room temperature for 12–24 h the solvent was evaporated and toluene (2 × 50 mL) was added and again removed under vacuum to afford the crude triaryl chlorides.
To a solution of the respective triaryl chloride (5 mmol) in anhydrous acetonitrile (100 mL) an excess of the respective amine (10–20 mmol) as hydrogen acceptor was added and the resulting mixture was refluxed for several hours. The progress of the reaction was monitored by TLC. Work up I: The mixture was poured into cold water (400 mL) and kept at 4°C for a few hours. The precipitate was filtered off, thoroughly washed with water to remove any remaining amine, and recrystallized from ethanol. Work up II: the solvent was evaporated and the crude residue was purified by column chromatography and/or recrystallization.
1-[(2-Chlorophenyl)(diphenyl)methyl]-4-methyl-2-phenyl-1H-imidazole (T89) was synthesized from T3-Cl (2.5 g, 7.98 mmol) and 4-methyl-2-phenylimidazole (1.26 g, 7.98 mmol), and triethylamine (1.11 ml, 7.98 mmol) as hydrogen acceptor in anhydrous acetonitrile (100 mL). After 24 h of refluxing the solvent was evaporated to afford a creamy residue, which was dissolved in dichloromethane (200 mL), washed with water (2 × 50 mL), and dried over Na2SO4. Evaporation of the solvent gave the crude product which was recrystallized from petroleum ether (40–60°C)/dichloromethane. T89 was obtained as a white powder (650 mg, 18.7%): Mp 226°C; 1H NMR (DMSO-d6) δ: 1.41 (s, 3H, −CH3), 6.92 (d, 1H, 3J = 6.9 Hz), 7.17–7.27 (m, 13H), 7.32 (dt, 1H, 3J = 7.3 Hz, 4J = 1.3 Hz), 7.39 (t, 2H, 3J = 7.7 Hz), 7.42 (d, 1H, 3J = 7.8 Hz), 7.83 (d, 2H, 3J = 7.5 Hz); MS (ESI) m/z calcd 435.2 [M + H]+; found 435.5 [M + H]+; Anal. calcd for C29H23ClN2: C, 80.08; H, 5.33; N, 6.44; found: C, 79.70; H, 5.09; N, 6.59.
1-[(2-Chlorophenyl)diphenylmethyl]-2,5-dihydro-1H-pyrrole-2,5-dione (T91) was synthesized from T3-Cl (1.6 g, 5 mmol) and maleimide (1.94 g, 20 mmol) according to general method C as a slightly yellowish powder (320 mg, 17.12%): Mp 93°C; 1H NMR (DMSO-d6) δ: 6.43 (d, 1H, 3J = 1 Hz), 7.09 (dd, 1H, 3J = 7.8 Hz, 4J = 1.6 Hz), 7.22–7.27 (m, 8H), 7.29–7.32 (m, 5H), 7.39 (dd, 1H, 3J = 7.8 Hz, 4J = 1.3 Hz), MS (ESI) m/z calcd 338.1 [C23H16NO2]+; found 338.2 [C23H16NO2]+.
N-[(2-Chlorophenyl)diphenylmethyl]-N-(3-aminopropyl)imidazoleamine (T94) was synthesized from T3-Cl (1.6 g, 5 mmol) and N-(3-aminopropyl)imidazole (1.88 g, 15 mmol) according to general method C as white powder (510 mg, 25.4%): Mp 89°C; 1H NMR (DMSO-d6) δ: 1.93–1.96 (m, 4H, 2x -CH2), 2.84 (t, 1H, 3J = 7.6 Hz, N-H), 4.03 (t, 2H, 3J = 6.8 Hz, CH2), 6.83 (s, 1H), 7.10 (sbroad, 1H), 7.14 (t, 2H, 3J = 7.3 Hz), 7.25 (t, 4H, 3J = 7.7 Hz), 7.31–7.35 (m, 2H), 7.37–7.39 (m, 5H), 7.53 (dd, 1H, 3J = 7.5 Hz, 4J = 2.0 Hz), 7.56 (sbroad, 1H); MS (ESI) m/z calcd 402.2 [M + H]+; found 402.3 [M + H]+; Anal. calcd for C25H24ClN3: C, 74.71; H, 6.02; N, 10.45; found: C, 74.06; H, 6.44; N, 10.32.
N-[(2-Chlorophenyl)diphenylmethyl]-N-(3-aminopropionitrile)amine (T95) was synthesized from T3-Cl (1.6 g, 5 mmol) and 3-aminopropionitrile (1.12 mL, 1.05 g, 15 mmol) according to general method C as a white yellowish powder (610 mg, 35.2%): Mp 140°C; 1H NMR (DMSO-d6) δ: 2.13 (q, 2H, 3J = 7.1 Hz, NH-CH2), 2.70 (t, 2H, 3J = 6.4 Hz, CH2-CN), 3.19 (t, 1H, 3J = 8.4 Hz, N-H), 7.18 (t, 2H, 3J = 7.3 Hz), 7.29 (t, 4H, 3J = 7.7 Hz), 7.33-7.40 (m, 3H), 7.42–7.45 (m, 4H), 7.69 (dbroad, 1H, 3J = 7.7 Hz); HRMS (ESI) m/z calcd 347.1315 [M + H]+; found 347.1290 [M + H]+; Anal. calcd for C22H19ClN2: C, 76.18; H, 5.52; N, 8.08; found: C, 75.53; H, 5.34; N, 8.06.
N-[(2-Chlorophenyl)diphenylmethyl]-3-(trifluoromethoxy)aniline (T102) was synthesized from T3-Cl (1.6 g, 5 mmol) and 3-(trifluoromethoxy)aniline (2.0 mL, 2.66 g, 15 mmol) according to general method C and recrystallized from methanol as white powder (1.34 g, 59%): Mp 144.2°C; 1H NMR (CDCl3) δ: 6.46 (d, 1H, 3J = 8 Hz), 6.49 (s, 1H, N-H), 6.67 (d, 1H, 3J = 8 Hz), 7.02 (t, 1H, 3J = 8.3 Hz), 7.11 (s, 1H), 7.32 (t, 2H, 3J = 7 Hz, phenyl-Cl-H4 and -H5), 7.36-7.46 (m, 11 H), 7.66 (d, 1H, 3J = 7 Hz); HRMS (ESI) m/z calcd 277.0784 [C19H14Cl]+, 178.0480 [C7H7NOF3]+; found 277.0764 [C19H14Cl]+, 178.0461 [C7H7NOF3]+; Anal. calcd for C26H19ClF3NO: C, 68.80; H, 4.22; N, 3.09; found: C, 68.86; H, 4.02; N, 3.13.
N-[(2-Chlorophenyl)diphenylmethyl]-1,3-benzothiazol-2-amine (T103) was synthesized from T3-Cl (1.6 g, 5 mmol) and 2-aminobenzothiazole (2.25 g, 15 mmol) according to general method C and recrystallized from methanol as an off-white powder (1.12 g, 52%): Mp 156.3°C; 1H NMR (DMSO-d6) δ: 6.95 (t, 1H, 3J = 7.5 Hz), 7.00 (d, 1H, 3J = 8.5 Hz), 7.06 (t, 1H, 3J = 7.3 Hz), 7.20–7.23 (m, 2H), 7.28–7.31 (m, 11H), 7.49 (m, 1H), 7.60 (d, 1H, 3J = 7.5 Hz), 8.96 (s, 1H, N-H); HRMS (ESI) m/z calcd 427.1036 [M + H]+; found 427.1060 [M + H]+; Anal. calcd for C26H19ClN2S: C, 73.14; H, 4.49; N, 6.56; S, 7.51; found: C, 73.05; H, 4.39; N, 6.63; S, 8.07.
N-[(2-Chlorophenyl)diphenylmethyl]-2-(trifluoromethoxy)aniline (T104) was synthesized from T3-Cl (1.6 g, 5 mmol) and 2-trifluoromethoxyaniline (2.04 mL, 2.7 g, 15 mmol) according to general method C as a slightly yellowish powder (1.4 g, 62%): Mp 123.5°C; 1H NMR (DMSO-d6) δ: 5.91 (s, 1H, N-H), 6.12 (d, 1H, 3J = 7.8 Hz), 6.60 (dt, 1H, 3J = 8.2, Hz, 4J = 1.3 Hz), 6.57 (dt, 1H, 3J = 7.9 Hz, 4J = 1.3 Hz), 7.18–7.38 (m, 14H), 7.58 (d, 1H, 3J = 7.4 Hz); HRMS (ESI) m/z calcd 277.0784 [C19H14Cl]+, 178.0480 [C7H7NOF3]+; found 277.0773 [C19H14Cl]+, 178.0465 [C7H7NOF3]+; Anal. calcd for C26H19ClF3NO: C, 68.8; H, 4.22; N, 3.09; found: C, 69.19; H, 4.26; N, 3.10.
1-(4-{[(2-Chlorophenyl)diphenylmethyl]amino}phenyl)ethan-1-one (T105) was synthesized from T3-Cl (1.6 g, 5 mmol) and 4-aminoacetophenone (2.0 g, 15 mmol) according to general method C as a white powder (400 mg, 19.4%): Mp 166.7°C; 1H NMR (DMSO-d6) δ: 2.30 (s, 3H, -OCH3), 6.55 (swide, 2H), 7.20–7.25 (m, 6H), 7.29–7.35 (m, 8H), 7.46 (d, 2H, 3J = 8.6 Hz), 7.56 (d, 1H, 3J = 7.4 Hz); HRMS (ESI) m/z calcd 412.1468 [M + H]+; found 412.1454 [M + H]+; Anal. calcd for C27H22ClNO: C, 78.73; H, 5.38; N, 3.40; found: C, 78.42; H, 5.48; N, 3.38.
N-[(2-Chlorophenyl)diphenylmethyl]-4-methoxyaniline (T106) was synthesized from T3-Cl (1.6 g, 5 mmol) and p-anisidine (1.85 g, 15 mmol) according to general method C and recrystallized from methanol as a slightly redish-beige powder (1.24 g, 62%): Mp 142.5°C; 1H NMR (DMSO-d6) δ: 3.52 (s, 3H, -OCH3), 6.10 (s, 1H), 6.43 (s, 4H), 7.18 (t, 2H, 3J = 6 Hz), 7.26–7.30 (m, 11H), 7.59 (d, 1H, 3J = 7.5 Hz); HRMS (ESI) m/z calcd: 399.13899 [M]+; found: 399.1381 [M]+; Anal. calcd for C26H22ClNO: C, 78.09; H, 5.54; N, 3.50; found: C, 77.85; H, 5.62; N, 3.53.
1-Chloro-2-(phenoxydiphenylmethyl)benzene (T107): To a solution of T3-Cl (0.95 g, 3 mmol) in anhydrous acetone (50 mL) phenol (282 mg, 3 mmol), K2CO3 (1.93 g, 14 mmol) and catalytic amounts of KI were added. Afterward the resulting mixture was refluxed for several hours. The progress of the reaction was monitored by TLC. After completion of the reaction K2CO3 was filtered off and the solvent was evaporated. The solid residue was dissolved in CH2Cl2 and the solution was extracted three times with NaOH (0.5 M). The pooled organic phases were dried over Na2SO4 and concentrated in vacuo. The residue was recrystallized from ethanol to give an off-white, slightly yellowish powder (80 mg, 7%): Mp 125°C; 1H NMR (DMSO-d) δ: 7.07 (dd, 2H, 3J = 8.1 Hz, 4J = 1.6 Hz), 7.12 (dt, 2H, 3J = 7.7 Hz, 4J = 1.4 Hz), 7.15–7.17 (m, 5H), 7.26 (dt, 2H, 3J = 7.6 Hz, 4J = 1.7 Hz), 7.29–7.31 (m, 8H); MS (FAB-NBA) m/z calcd 293 [C19H14ClO]+, 277 [C19H14Cl]+; found 293 [C19H14ClO]+, 277 [C19H14Cl]+.
N-[(2-Chlorophenyl)diphenylmethyl]aniline (T109) was synthesized from T3-Cl (0.95 g, 3 mmol) and aniline (279 mg, 3 mmol) as described for T107 and recrystallized from ethanol as a yellowish-beige powder (750 mg, 2 mmol, 67.6%): Mp 131.4°C (ethanol), Lit. 121°C (benzene; Gomberg and Van Slyke, 1911); 1H NMR (DMSO-d6) δ: 6.41–6.44 (m, 2H, N-H), 6.49 (d, 2H, 3J = 8.1 Hz), 6.80 (t, 2H, 3J = 7.8 Hz), 7.17–7.20 (m, 2H), 7.25–7.33 (m, 11H), 7.59 (d, 1H, 3J = 7.8 Hz); HRMS (ESI) m/z calcd 369.1284 [M]+; found 369.1277 [M]+.
2-{[(2-Chlorophenyl)diphenylmethyl]sulfanyl}pyrimidine (T112) was synthesized from T3-Cl (1.6 g, 5 mmol) and 2-mercaptopyrimidine (1.68 g, 15 mmol) according to general method C and recrystallized from methanol as an off-white powder (1.16 g, 60%): Mp 151.6°C; 1H NMR (DMSO-d6) δ: 7.00 (t, 1H, 3J = 4.8 Hz), 7.20 (t, 2H, 3J = 7.2 Hz), 7.26 (t, 4H, 3J = 7.6 Hz), 7.33–7.37 (m, 7H), 7.81–7.82 (m, 1H), 8.28 (d, 2H, 3J = 4.9 Hz); HRMS (ESI) m/z calcd 389.0879 [M + H]+; found 389.0869 [M + H]+; Anal. calcd for C23H17ClN2S: C, 71.03; H, 4.41; N, 7.2; S, 8.24; found: C, 71.04; H, 4.26; N, 7.13; S, 8.56.
(2-Methoxyphenyl)diphenylmethanol (T117) was synthesized from 2-bromoanisole (3.09 mL, 4.68 g, 25 mmol) according to general method A as an off-white powder (4.9 g, 17 mmol, 67.6%): Mp 128.7°C; Lit. 128–129°C (Baeyer, 1907); 1H NMR (DMSO-d6) δ: 3.67 (s, 3H, -OCH3), 5.28 (s, 1H, -OH), 6.54 (d, 1H, 3J = 7.7 Hz), 6.84 (t, 1H, 3J = 7.6 Hz), 6.97 (d, 1H, 3J = 8.2 Hz), 7.25–7.32 (m, 11H); HRMS (ESI) m/z calcd 273.12795 [M-OH]+; found 273.1270 [M-OH]+; Anal. calcd for C20H18O2: C, 82.73; H, 6.25; found: C, 82.45; H, 6.20.
N-[(2-Methoxyphenyl)diphenylmethyl]-1,3-thiazol-2-amine (T122): In a first-step compound T117 was chlorinated with acetyl chloride according to method B to afford 1-(chlorodiphenylmethyl)-2-methoxybenzene (T117-Cl). Without further purification and characterization T117-Cl (3.7 g, 12 mmol) was reacted with 2-aminothiazole (2.9 g, 30 mmol) according to general method C to afford T122 as an off-white powder (1.67 g, 37%): Mp 162.3°C; 1H NMR (DMSO-d6) δ: 3.40 (s, 3H, -OCH3), 6.49 (d, 1H, 3J = 3.5 Hz, thiazole-H4), 6.87 (d, 1H, 3J = 3.3, thiazole-H3), 6.89 (d, 1H, 3J = 7.5 Hz), 6.97 (d, 1H, 3J = 8.1 Hz), 7.12 (d, 1H, 3J = 7.7 Hz), 7.19–7.29 (m, 11H), 8.00 (s, 1H, N-H); HRMS (ESI) m/z calcd 373.1375 [M + H]+; found 373.1371 [M + H]+; Anal. calcd for C23H20N2OS: C, 74.16; H, 5.41; N, 7.52; S, 8.61; found: C, 74.26; H, 5.31; N, 7.29; S, 8.87.
N-[(2-Methoxyphenyl)diphenylmethyl]pyrimidin-2-amine (T123): T117 was chlorinated to afford T117-Cl as described for T122. In a next step T117-Cl (6.48 g, 21 mmol) and 2-aminopyrimidine (5.02 g, 52.75 mmol) were reacted according to general method C to give T123 as an off-white powder (2.15 g, 27.9%): Mp 166.5°C; 1H NMR (CD3COCD3) δ: 3.44 (s, 3H, -OCH3), 6.48 (t, 1H, 3J = 4.8 Hz), 6.83–6.86 (m, 2H, N-H), 6.92 (d, 1H, 3J = 7.8 Hz), 7.14–7.16 (m, 2H), 7.20–7.26 (m, 6H), 7.29–7.31 (m, 4H) 8.02 (s, 2H); HRMS (ESI) m/z calcd 368.1763 [M + H]+; found 368.1757 [M + H]+; Anal. calcd for C24H21N3O: C, 78.45; H, 5.76; N, 11.44; found: C, 78.37; H, 5.71; N, 11.18.
N-[(2-Methylphenyl)diphenylmethyl]-1,3-thiazol-2-amine (T124): (2-Methylphenyl)diphenylmethanol (T118) was synthesized from 1-bromo-2-methylbenzene (3.01 mL, 4.28 g, 25 mmol) according to general method A and then chlorinated according to method B to afford 1-(chlorodiphenylmethyl)-2-methylbenzene (T118-Cl). Without further purification and characterization T118-Cl (2.9 g, 9.9 mmol) was immediately reacted with 2-aminothiazole (2.48 g, 24.8 mmol) according to general method C. T124 was obtained as a beige powder after recrystallization from ethanol (2.69 g, 76%): Mp 158.3°C; 1H NMR (CDCl3) δ: 1.97 (s, 3H, phenyl-CH3), 6.26 (d, 1H, 3J = 4 Hz, thiazole-H4), 6.86 (s, 1H, N-H), 7.04 (d, 1H, 3J = 4 Hz, thiazole-H3), 7.12 (d, 2H, 3J = 8 Hz), 7.20-7.27 (m, 12H); HRMS (ESI) m/z calcd 357.1426 [M + H]+, 373.1375 [M + H2O]+; found 357.1420 [M + 1]+, 373.1357 [M + H2O]+; Anal. calcd for C23H20N2S: C, 77.49; H, 5.65; N, 7.86; S, 8.99; found: C, 76.87; H, 5.67; N, 7.7; S, 9.48.
N-[(2-Methylphenyl)diphenylmethyl]-pyrimidin-2-amine (T125): (2-Methylphenyl)diphenylmethanol (T118) was synthesized and afterward chlorinated (T118-Cl) as described for T124. Without further purification and characterization T118-Cl (2.9 g, 9.9 mmol) was immediately reacted with 2-aminopyrimidine (2.36 g, 24.8 mmol) according to general method C. T125 was obtained as a white powder after recrystallization from ethanol (1.46 g, 42%): Mp 183.1°C; 1H NMR (CDCl3) δ: 1.99 (s, 3H, phenyl-CH3), 6.43 (t, 1H, 3J = 4.8 Hz, pyrimidine-H5), 7.05 (d, 1H, 3J = 7.1 Hz), 7.08 (d, 1H, 3J = 7.7 Hz), 7.14-7.29 (m, 14H), 8.05 (sbroad, 2H); HRMS (ESI) m/z calcd 352.1814 [M + H]+; found 352.1808 [M + H]+; Anal. calcd for C24H21N3: C, 82.02; H, 6.02; N, 11.96; found: C, 81.79; H, 5.93; N, 11.86.
N-{Diphenyl[2-(trifluoromethoxy)phenyl]methyl}-1,3-thiazol-2-amine (T126): Diphenyl[2-(trifluoromethoxy)phenyl]methanol (T119) was synthesized from 1-bromo-2-(trifluoromethoxy)benzene (3.69 mL, 6.03 g, 25 mmol) according to general method A. Without further purification and characterization T119 was chlorinated according to method B to afford 1-(chlorodiphenylmethyl)-2-(trifluoromethoxy)benzene (T119-Cl). Without further purification and characterization T119-Cl (g, 14.38 mmol) was immediately reacted with 2-aminothiazole (3.6 g, 35.94 mmol) according to general method C. T126 was obtained as an off-white powder after recrystallization from ethanol (2.25 g, 36.7%): Mp 178.8°C; 1H NMR (CDCl3) δ: 6.24 (d, 1H, 3J = 3.6 Hz, thiazole-H4), 6.82 (s, 1H, N-H), 6.98 (d, 1H, 3J = 3.6 Hz, thiazole-H3), 7.10–7.29 (m, 13H), 7.41 (dd, 1H, 3J = 8.1 Hz, 4J = 1.4 Hz); HRMS (ESI) m/z calcd 427.1092 [M + H]+; found 427.1087 [M + H]+; Anal. calcd for C23H17F3N2OS: C, 64.78; H, 4.02; N, 6.57; S, 7.52; found: C, 64.64; H, 3.89; N, 6.56; S, 8.1.
N-{Diphenyl[2-(trifluoromethoxy)phenyl]methyl}pyrimidin-2-amine (T127): Diphenyl[2-(trifluoromethoxy)phenyl]methanol (T119) was synthesized and afterward chlorinated (T119-Cl) as described for T126. Without further purification and characterization T119-Cl (5.2 g, 14.38 mmol) and 2-aminopyrimidine (3.42 g, 35.94 mmol) were reacted according to general method C. T127 was obtained as a white powder after recrystallization from ethanol (1.2 g, 19.8%): Mp 135°C; 1H NMR (DMSO-d6) δ: 6.54 (t, 1H, 3J = 4.8 Hz, pyrimidine-H4), 7.00 (s, 1H, N-H), 7.15-7.20 (m, 3H), 7.24–7.29 (m, 9H), 7.39 (ddd, 1H, 3J = 8.2 Hz, 3J = 7.4 Hz, 4J = 1.7 Hz), 7.51 (dd, 1H, 3J = 8 Hz, 4J = 1.7 Hz), 8.07 (sbroad, 2H, pyrimidine-H3 and -H5); HRMS (ESI) m/z calcd 422.1480 [M + H]+; found 422.1471 [M + H]+; Anal. calcd for C24H18F3N3O: C, 68.4; H, 4.31; N, 9.97; found: C, 68.3; H, 4.58; N, 9.99.
N-[(2-Fluorophenyl)diphenylmethyl]pyrimidine-2-amine (T128): (2-Fluorophenyl)diphenylmethanol (T36) was synthesized from bromobenzene (2.63 mL, 3.93 g, 25 mmol) and 2-fluorobenzophenone (4.22 mL, 5.0 g, 25 mmol) according to general method A (yellowish solid, 6.5 g, 93.41%). Spectroscopic data were in accordance with literature (Wulff et al., 2000). In a next step T36 was chlorinated according to method B to afford 1-(chlorodiphenylmethyl)-2-fluorobenzene (T36-Cl). Without further purification and characterization T36-Cl (3 g, 10.28 mmol) was immediately reacted with 2-aminopyrimidine (2.44 g, 25.7 mmol) according to general method C. T128 was obtained as slightly yellowish powder after recrystallization from ethanol (1.73 g, 47.3%): Mp 161.1°C; 1H NMR (DMSO-d6) δ: 6.53 (t, 1H, 3J = 5 Hz, pyrimidine-H4), 7.01 (dd, 1H, 3J = 12 Hz, 3J = 8 Hz), 7.11 (t, 1H, 3J = 7.9 Hz), 7.18 (t, 2H, 3J = 6.4 Hz), 7.23–7.30 (m, 9H), 7.34 (s, 1H, N-H), 7.40 (dt, 1H, 3J = 8 Hz, 4J = 1.3 Hz), 8.07 (sbroad, 2H, pyrimidine-H3 and -H5); HRMS (ESI) m/z calcd: 356.1563 [M + H]+; found: 356.1563 [M + H]+; Anal. calcd for C23H18FN3: C, 77.73; H, 5.10; N, 11.82; found: C, 77.55; H, 4.95; N, 11.42.
N-[(3-Chlorophenyl)diphenylmethyl]-1,3-thiazol-2-amine (T129): (3-Chlorophenyl)diphenylmethanol (T2) was synthesized from 1-bromo-3-chlorobenzene (2.94 mL, 4.79 g, 25 mmol) according to general method A (5.0 g, 67.8%). Spectroscopic data were in accordance with literature (Wulff et al., 2000). T2 was then chlorinated according to general method B to afford 1-chloro-3-(chlorodiphenylmethyl)benzene (T2-Cl). Without further purification and characterization T2-Cl (2.75 g, 8.78 mmol) was reacted with 2-aminothiazole (2.2 g, 21.95 mmol) according to general method C. T129 was obtained as a beige powder after recrystallization from ethanol (3.0 g, 91%): Mp 164.5°C; 1H NMR (DMSO-d6) δ: 6.56 (d, 1H, 3J = 3.6 Hz, thiazole-H4), 6.81 (d, 1H, 3J = 3.7 Hz, thiazole-H3), 7.18–7.31 (m, 13H), 7.35 (t, 1H, 3J = 1.8 Hz), 8.57 (s, 1H, N-H); HRMS (ESI) m/z calcd 377.0879 [M + H]+; found 377.0861 [M + H]+; Anal. calcd for C22H17ClN2S: C, 70.11; H, 4.55; N, 7.43; S, 8.51; found: C, 69.94; H, 4.46; N, 7.48; S, 9.09.
N-[(3-Chlorophenyl)diphenylmethyl]pyrimidin-2-amine (T130): (3-Chlorophenyl)diphenylmethanol (T2) was synthesized and afterward chlorinated (T2-Cl) as described for T129. Without further purification and characterization T2-Cl (2.75 g, 8.78 mmol) and 2-aminopyrimidine (2.1 g, 21.95 mmol) were reacted according to general method C. T130 was obtained as a white powder after recrystallization from ethanol (1.65 g, 51%): Mp 124.4°C; 1H NMR (DMSO-d6) δ: 6.53 (t, 1H, 3J = 4.8 Hz, pyrimidine-H4), 7.16 (t, 2H, 3J = 7.2 Hz), 7.22–7.28 (m, 7H), 7.30-7.32 (m, 4H), 7.35 (sbroad, 1H), 7.87 (s, 1H, N-H), 8.10 (d, 2H, 3J = 4.3 Hz, pyrimidine-H3 and -H5); HRMS (ESI) m/z calcd 372.1262 [M + H]+; found 372.1253 [M + H]+; Anal. calcd for C23H18ClN3: C, 74.29; H, 4.88; N, 11.3; found: C, 74.14; H, 4.76; N, 11.21.
N-[(2-Bromophenyl)diphenylmethyl]-1,3-thiazol-2-amine (T131): (2-Bromophenyl)diphenylmethanol (T116) was synthesized according to method A (bromobenzene (2.84 mL, 4.24 g, 27 mmol) and 2-bromobenzophenone (6.52 g, 25 mmol). The crude oily T116 was chlorinated in a next step according to method B to afford 1-bromo-2-(chlorodiphenylmethyl)benzene (T116-Cl). Without further purification and characterization T116-Cl was immediately reacted with 2-aminothiazole (6.3 g, 62.5 mmol) according to general method C. T131 was obtained as white powder [silica column: cyclohexane/ethylacetate (8/2); 1.05 g, 10%]: Mp 142°C; 1H NMR (DMSO-d6) δ: 6.52 (d, 1H, 3J = 3.7 Hz, thiazole-H4), 6.74 (d, 1H, 3J = 3.6 Hz, thiazole-H3), 7.17–7.20 (m, 3H), 7.28 (m, 7H), 7.32–7.29 (m, 2H), 7.41 (dd, 1H, 3J = 8 Hz, 4J = 1.4 Hz), 7.52 (d, 1H, 3J = 7.8 Hz) 8.36 (s, 1H, N-H); HRMS (ESI) m/z calcd 421.0374 [M + H]+, 423.0353, found 421.0366 [M + H]+, 423.0335.
N-[(2-Bromophenyl)diphenylmethyl]-pyrimidin-2-amine (T132): (2-Bromophenyl)diphenylmethanol (T116) was synthesized and afterward chlorinated (T116-Cl) as described for T131. T116-Cl (4.5 g, 12.5 mmol) was immediately without further purification and characterization reacted with 2-aminopyrimidine (1.2 g, 12.6 mmol) according to general method C. T132 was obtained as an off-white to yellowish powder after recrystallization from ethanol (850 mg, 16%): Mp 158.7°C; 1H NMR (DMSO-d6) δ: 6.57 (t, 1H, 3J = 4.5 Hz, pyrimidine-H4), 7.10 (s, 1H), 7.14–7.32 (m, 13H), 7.49 (d, 2H, 3J = 8.5 Hz, pyrimidine-H3 and -H5), 8.10 (s, 1H, N-H); HRMS (ESI) m/z calcd 416.0762 [M + H]+, 418.0742, 419.0776; found 416.0758 [M + H]+, 418.0728, 419.0752; Anal. calcd for C23H18BrN3: C, 66.36; H, 4.36; N, 10.09; found: C, 66.16; H, 4.23; N, 9.74.
N-{diphenyl[3-(trifluoromethyl)phenyl]methyl}-1,3-thiazol-2-amine (T133): Diphenyl[3-(trifluoromethoxy)phenyl]methanol (T121) was synthesized from 1-bromo-3-(trifluoromethoxy)benzene (3.49 mL, 5.63 g, 25 mmol) and benzophenone (4.56 g, 25 mmol) according to general method A. T121 was then chlorinated according to method B to afford 1-(chlorodiphenylmethyl)-3-(trifluoromethoxy)benzene (T121-Cl). Without further purification and characterization T121-Cl (4.5 g, 12.98 mmol) was immediately reacted with 2-aminothiazole (3.25 g, 32 mmol) according to general method C. T133 was obtained as a white powder after recrystallization from ethanol (3.64 g, 68%): Mp 120.5°C; 1H NMR (DMSO-d6) δ: 6.56 (d, 1H, 3J = 3.7 Hz, thiazole-H4), 6.79 (d, 1H, 3J = 3.7 Hz, thiazole-H3), 7.19–7.22 (m, 2H), 7.27–7.32 (m, 8H), 7.50 (t, 1H, 3J = 8.1 Hz), 7.57 (t, 2H, 3J = 9.1 Hz), 7.67 (s, 1H), 8.64 (s, 1H, N-H); HRMS (ESI) m/z calcd 411.1137 [M + H]+; found 411.1137 [M + H]+; Anal. calcd for C23H17F3N2S: C, 67.3; H, 4.17; N, 6.82; S, 7.81; found: C, 67.39; H, 3.98; N, 6.85; S, 7.82.
N-{Diphenyl[3-trifluoromethyl)phenyl]methyl}pyrimidin-2-amine (T134): Diphenyl[3-(trifluoromethoxy)phenyl]methanol (T121) was synthesized and afterward chlorinated (T121-Cl) as described for T133. In a next step T121-Cl (4.5 g, 12.98 mmol) was immediately reacted with 2-aminopyrimidine (3.1 g, 32.4 mmol) according to general method C. T134 was obtained as an off-white powder after recrystallization from ethanol (1.95 g, 37%): Mp 161.4°C; 1H NMR (DMSO-d6) δ: 6.53 (t, 1H, 3J = 4.8 Hz, pyrimidine-H4), 7.17 (t, 2H, 3J = 7.18 Hz, 2× phenyl-H4), 7.25 (t, 4H, 3J = 7.7 Hz, 2× phenyl-H3 and -H5), 7.31 (d, 4H, 3J = 7.7 Hz, 2x phenyl-H2 and -H6), 7.48 (t, 1H, 3J = 7.8 Hz, phenyl-CF3-H5), 7.53 (d, 1H, 3J = 7.8 Hz, phenyl-CF3-H6), 7.62 (d, 1H, 3J = 7.9 Hz, phenyl-CF3-H4), 7.67 (swide, 1H, phenyl-CF3-H2), 8.00 (s, 1H, N-H), 8.09 (d, 2H, 3J = 4 Hz, pyrimidine-H3 and -H5); HRMS (ESI) m/z calcd 406.1526 [M + H]+, found 406.1521 [M + H]+, Anal. calcd for C24H18F3N3: C, 71.1; H, 4.48; N, 10.36; found: C, 71.03; H, 4.29; N, 10.26.
N-[(2-Iodophenyl)diphenylmethyl]-1,3-thiazol-2-amine (T136): (2-Iodophenyl)diphenylmethanol (T135) was synthesized according to general method A from bromobenzene (2.1 mL, 3.13 g, 20 mmol) and 2-iodobenzophenone (6 g, 19.5 mmol). The crude oily T135 was then chlorinated according to method B to afford 1-(chlorodiphenylmethyl)-2-iodobenzene (T135-Cl). Without further purification and characterization T135-Cl was immediately reacted with 2-aminothiazole (1.5 g, 15 mmol) according to general method C. T136 was obtained as a slightly yellowish powder (silica column cyclohexane/ethylacetate (8/2); 80 mg, 2.8%): Mp 81°C; 1H NMR (DMSO-d6) δ: 6.52 (d, 1H, 3J = 2.6 Hz, thiazole-H4), 6.78 (d, 1H, 3J = 2.6 Hz, thiazole-H3), 6.96–6.98 (m, 1H), 7.20–7.32 (m, 12H), 7.88 (d, 1H, 3J = 7.6 Hz, phenyl-I-H3), 8.25 (s, 1H, N-H); HRMS (ESI) m/z calcd: 469.0236 [M + H]+; found: 469.0213 [M + H]+.
N-[(2-Iodophenyl)diphenylmethyl]-pyrimidin-2-amine (T137): (2-Iodophenyl)diphenyl methanol (T135) was synthesized and afterward chlorinated (T135-Cl) as described for T136. In a next step T135-Cl (2.4 g, 6 mmol) was immediately without further purification and characterization reacted with 2-aminopyrimidine (1.43 g, 15 mmol) according to general method C. T137 was obtained as an off-white to yellowish powder [silica column cyclohexane/ethylacetate (8/2); 570 mg, 20.5%]: Mp 160.7°C; 1H NMR (DMSO-d6) δ: 6.58 (dt, 1H, 3J = 4.7 Hz, 4J = 1.2 Hz), 6.93 (t, 1H, 3J = 7.5 Hz), 7.05 (sbroad, 1H), 7.19–7.32 (m, 11H), 7.44 (d, 1H, 3J = 8 Hz), 7.82 (d, 1H, 3J = 7.8 Hz), 8.1 (sbroad, 2H, pyrimidine-H3 and -H5); MS (ESI) m/z calcd 469.4 [M + H]+; found 496.3 [M + H]+; Anal. calcd for C23H18IN3: C, 59.62; H, 3.92; N, 9.07; found: C, 60.38; H, 3.74; N, 8.84.
N-(Triphenylmethyl)pyrimidin-2-amine (T141) was synthesized from (chlorophenylmethyl)benzene (1.34 g, 4.82 mmol) and 2-aminopyrimidine (1.48 g, 15.56 mmol) according to general method C as a white powder (1.18 g, 72.4%): Mp 174.2°C; Lit. 174–175°C (Dahlbom and Ekstrand, 1944). Spectroscopic data were in accordance with literature (Zunszain et al., 2002).
1-[(3-Chlorophenyl)diphenylmethyl]-1H-pyrazole (T142): (3-Chlorophenyl)diphenylmethanol (T2) was synthesized and afterward chlorinated (T2-Cl) as described for T129. Without further purification and characterization T2-Cl (3.3 g, 10.54 mmol) and pyrazole (1.77 g, 26 mmol) were reacted according to general method C. T142 was obtained as a white powder after recrystallization from ethanol (3.1 g, 85.3%): Mp 127.3°C; 1H NMR (DMSO-d6) δ: 6.33 (t, 1H, 3J = 2.1 Hz), 6.99–7.04 (m, 5H), 7.10 (t, 1H, 3J = 1.9 Hz), 7.34–7.43 (m, 9H), 7.66 (d, 1H, 3J = 1.6 Hz); HRMS (ESI) m/z calcd 69.04528 [C3H5N2]+, 277.0784 [C19H14Cl]+, found 69.0432 [C3H5N2]+, 277.0757 [C19H14Cl]+; Anal. calcd for C22H17ClN2: C, 76.63; H, 4.97; N, 8.12; found: C, 76.13; H, 4.92; N, 8.13.
1-[(3-Chlorophenyl)diphenylmethyl]-1H-imidazole (T143): (3-Chlorophenyl)diphenylmethanol (T2) was synthesized and afterward chlorinated (T2-Cl) as described for T129. Without further purification and characterization T2-Cl (3.3 g, 10.54 mmol) and imidazole (1.77 g, 26 mmol) were reacted according to general method C. T143 was obtained as a white powder after recrystallization from ethanol (1.3 g, 35.77%): Mp 121.3°C, Lit. 122–124°C (Bartroli et al., 1992); 1H NMR (DMSO-d6) δ: 6.94 (sbroad, 1H, imidazole-H5), 7.01 (sbroad, 1H, imidazole-H4), 7.03–7.04 (m, 2H), 7.06 (td, 1H, 3J = 6.8 Hz, 4J = 2.1 Hz), 7.09–7.1 (m, 3H), 7.36–7.47 (m, 9H), 8.46 (s, 1H, imidazole-H2); HRMS (ESI) m/z calcd for C22H17ClN2 345.1159 [M + H]+, found 345.1148 [M + H]+.
1-[(4-Chlorophenyl)diphenylmethyl]-1H-imidazole (T144): (4-Chlorophenyl)diphenylmethanol (T1) was synthesized from magnesium turnings (1.2 g, 50 mmol), 1-bromo-4-chlorobenzene (11.5 g, 60 mmol) and benzophenone (9.12 g, 50 mmol) according to general method A. Spectroscopic data were in accordance with literature (Wulff et al., 2000). The crude T1 was chlorinated according to method B to afford 1-chloro-4-(chlorodiphenylmethyl)benzene (T1-Cl). Without further purification and characterization T1-Cl (∼8 g, 25.5 mmol) was immediately reacted with imidazole (4.34 g, 63.75 mmol) according to general method C. T144 was obtained as a white powder (0.6 g, 3.5%): Mp 141.2°C, Lit. 140–142°C (Bartroli et al., 1992); 1H NMR (DMSO-d6) δ: 6.90 (sbroad, 1H), 6.99 (sbroad, 1H), 7.07–7.09 (m, 6H), 7.36 (m, 7H), 7.47 (d, 2H, 3J = 8.7 Hz); HRMS (ESI) m/z calcd for C22H17ClN2 345.1159 [M + H]+, found 345.1126 [M + H]+.
N-[(4-Chlorophenyl)diphenylmethyl]pyrimidin-2-amine (T145): (4-Chlorophenyl)diphenylmethanol (T1) was synthesized and afterward chlorinated (T1-Cl) as described for T144. Without further purification and characterization T1-Cl (15.7 g, 50 mmol) was reacted with 2-aminopyrimidine (11.88 g, 128 mmol) according to general method C. T145 was obtained as a white powder after recrystallization from ethanol (2.3 g, 12%): Mp 117.8°C; 1H NMR (DMSO-d6) δ: 6.51-6.54 (m, 3H, pyrimidne-H4), 7.15 (t, 2H, 3J = 7.2 Hz), 7.24 (t, 3H, 3J = 7.7 Hz), 7.28–7.34 (m, 6H), 7.80 (s, 1H, N-H), 8.09 (d, 2H, 3J = 3.5 Hz, pyrimidne-H3 and -H5), 8.19 (d, 2H, 3J = 4.8 Hz); HRMS (ESI) m/z calcd 372.1268 [M + H]+, found 372.1261 [M + H]+.
N-[(2-Chlorophenyl)diphenylmethyl]-4-(trifluoromethoxy)aniline (T150) was synthesized from T3-Cl (1.6 g, 5 mmol) and 4-(trifluoromethoxy)aniline (2.01 mL, 2.66 g, 15 mmol) according to general method C and recrystallized from ethanol as an off-white powder (1.5 g, 66%): Mp 99°C; 1H NMR (DMSO-d6) δ: 6.54 (d, 2H, 3J = 8.9 Hz), 6.82–6.80 (m, 3H, N-H), 7.18-7.22 (m, 2H), 7.26–7.36 (m, 11H), 7.58 (d, 1H, 3J = 7.9 Hz, phenyl-Cl-H3); HRMS (ESI) m/z calcd 454.1180 [M + H]+, 178.0474 [C7H6F3NO + H]+, found 454.1156 [M + H]+, 178.0461 [C7H6F3NO + H]+; Anal. calcd for C26H19ClF3NO: C, 68.8; H, 4.22; N, 3.09; found: C, 68.52; H, 4.09; N, 3.05.
(2-Aminophenyl)diphenylmethanol (T154-OH) was synthesized from phenylmagnesium bromide (1.89 g, 15.3 mmol) and 2-aminobenzophenone (2 g, 10.2 mmol) according to general method A (125 mg, 4.5%) as a red crystalline solid: Mp 117.4–118.9°C; Lit. 116–117°C (Misra et al., 1983); 1H NMR (DMSO-d6) δ: 5.07 (s, 2H, -NH2) 6.23 (d, 1H, 3J = 7.8 Hz), 6.34 (t, 1H, 3J = 7.4 Hz), 6.60 (d, 1H, 3J = 8.0 Hz), 6.73 (s, 1H, -OH), 7.31–7.17 (m, 10H); MS (ESI) m/z calcd 258.13 [C19H16N]+, 180.1 [C13H10N]+, found 258.3 [C19H16N]+, 180.1 [C13H10N]+; Anal. calcd for C19H17NO: C, 82.88; H, 6.22; N, 5.09; found: C, 82.42; H, 6.09; N, 5.17.
Bis-(2-methoxyphenyl)(phenyl)methanol (T165) was synthesized from magnesium turnings (0.6 g, 25 mmol), 1-bromo-2-methoxybenzene (4.0 mL, 5.99 g, 32 mmol) and 2-methoxybenzophenone (5.0 g, 23.5 mmol) according to general method A and recrystallized from ethanol as a white powder (1.2 g, 15.8%): Mp 115.8°C; Lit. 115°C (Baeyer, 1907); 1H NMR (DMSO-d6) δ: 3.39 (s, 6H, -OCH3), 5.37 (s, 1H, -OH), 6.87 (dt, 2H, 3J = 7.3 Hz, 4J = 0.7 Hz), 6.98 (d, 2H, 3J = 7.9 Hz), 7.03 (dd, 2H, 3J = 7.8 Hz, 3J = 1.5 Hz), 7.15-7.17 (m, 3H), 7.20–7.21 (m, 2H) 7.23–7.27 (m, 2H); MS (ESI) m/z calcd 343.38 [M + Na+], 303.14 [M-OH]+, found 343.1 [M + Na+], 303.1 [M-OH]+; Anal. calcd for C21H20O3: C, 78.73; H, 6.29; found: C, 78.14; H, 6.97.
N-[bis(2-methoxyphenyl)(phenyl)methyl]-1,3-thiazol-2-amine (T166): In a first-step T165 was chlorinated according to general method B (T165-Cl). The crude T165-Cl (530 mg, 1.56 mmol) was immediately reacted with 2-aminothiazole (392 mg, 3.9 mmol) according to general method C. After recrystallization from ethanol T166 was obtained as a white powder (250 mg, 39.8%): Mp 150°C; 1H NMR (DMSO-d6) δ: 3.39 (s, 6H, -OCH3), 6.44 (d, 1H, 3J = 3.6 Hz), 6.88 (dt, 2H, 3J = 7.8 Hz, 4J = 1.0 Hz), 6.90 (d, 1H, 3J = 3.6 Hz), 6.95 (d, 2H, 3J = 8.2 Hz), 7.08 (d, 2H, 3J = 7.6 Hz), 7.16 (d, 2H, 3J = 7.1 Hz), 7.21–7.29 (m, 5H), 7.81 (s, 1H); HRMS (ESI) m/z calcd for C24H22N2O2S 403.1480 [M + H]+, found 403.1465 [M + H]+.
All electrophysiological experiments described here were performed on N2A neuroblastoma cells that were transiently co-transfected with connexin and enhanced green fluorescent protein cDNAs as described previously (Srinivas et al., 2001). The connexins used in the study were rCx26, rCx32, rCx43, rCx46, and mCx50 (where r and m refer to rat, and mouse cDNAs, respectively). Transiently transfected cells were dissociated at 8–12 h after transfection, plated at low density on 1 cm round glass coverslips, and used within 48 h thereafter.
HEK-293 cells stably expressing hKCa3.1 were obtained from Khaled Houamed, University of Chicago, IL. The cloning of hKCa2.3 (19 CAG repeats) and hKCa3.1 has been previously described (Wulff et al., 2001). The hKCa2.3 clone was later stably expressed in COS-7 cells at Aurora Biosciences Corp., San Diego, CA. Cell lines stably expressing other mammalian ion channels were gifts from several sources: hKCa1.1 in HEK-293 cells (Andrew Tinker, University College London); rKv4.2 in LTK cells (Michael Tamkun, University of Colorado, Boulder); Kv11.1 (HERG) in HEK-293 cells (Craig January, University of Wisconsin, Madison); hNav1.4 in HEK-293 cells (Frank Lehmann-Horn, University of Ulm, Germany) and Cav1.2 in HEK-293 cells (Franz Hofmann, Munich, Germany). Cells stably expressing mKv1.1, mKv1.3, hKv1.5, and mKv3.1 have been previously described (Grissmer et al., 1994); N1E-115 neuroblastoma cells (expressing Nav1.2) were obtained from ATCC. Rat Nav1.5 in pSP64T was provided by Roland G. Kallen (University of Pennsylvania), inserted into pcDNA-3.1(+) as described (Sankaranarayanan et al., 2009), and transiently transfected into COS-7 cells together with eGFP-C1 with Fugene-6 (Roche) according to the manufacturer’s protocol. Human Kv7.2 and Kv7.3 in PTLN or pcDNA-3 was provided by Bernhard Attali (Weizmann Institute of Science, Rehovot, Israel).
Junctional current measurements were performed on N2A neuroblastoma cells transiently transfected with cDNAs corresponding to individual connexins or on mouse primary lens epithelial cells isolated from postnatal day 6. Dissociation of lens epithelial was performed as described previously (White et al., 2007). Junctional conductance was measured between cell pairs using the dual whole-cell voltage-clamp technique with Axopatch 1D patch-clamp amplifiers (Molecular Devices, CA) at room temperature. Each cell of a pair was initially held at a common holding potential of 0 mV. To evaluate junctional coupling, 200 msec hyperpolarizing pulses from the holding potential of 0 mV to −20 mV were applied to one cell to establish a transjunctional voltage gradient (Vj), and junctional current was measured in the second cell (held at 0 mV). The solution bathing the cells contained 140 mM NaCl, 5 mM KCl, 2 mM CsCl, 2 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, 5 mM dextrose, 2 mM pyruvate, and 1 mM BaCl2, pH 7.4. Patch electrodes had resistances of 3–5 MΩ when filled with internal solution containing 130 mM CsCl, 10 mM EGTA, 0.5 mM CaCl2, 3 mM MgATP, 2 mM Na2ATP, and 10 mM HEPES, pH 7.2. Macroscopic recordings were filtered at 0.2–0.5 kHz and sampled at 1–2 kHz. Data were acquired using pClamp software (Axon Instruments) and plotted using Origin 6.0 software (OriginLab Corp, Northampton, MA). Drugs were applied with a gravity-fed perfusion system. Solution exchanges were complete within 10–20 s. All compounds were applied to Cx50 expressing N2A cells at an initial concentration of 10 μM. Compounds that reduced Cx50 junctional currents by >80% at 10 μM were then applied at lower concentrations ranging from 0.5 to 5 μM. Concentration-response curves for drug-induced uncoupling were typically determined by exposure of each cell pair to 0.5 or 1 μM, 5 and 10 μM of each drug. Concentrations of drugs ([D]) that caused a half-maximal inhibition (IC50) and the Hill coefficients (nh) of concentration-response relationships were estimated by fitting the data to the equation: gj,% control = 1/[1 + ([D]/EC50)nh ] where gj (% control) is fraction of the conductance (gj) in the absence and presence of the drug. Data are presented as means ± S.E.M.
Experiments on K+ and Na+ channels were performed with an EPC-10 amplifier (HEKA, Lambrecht/Pfalz, Germany) in the whole-cell configuration of the patch-clamp technique with a holding potential of –80 mV. Pipette resistances averaged 2.0 MΩ. Solutions of triarylmethanes in Ringer were freshly prepared directly before the experiments from 10 mM stock solutions in DMSO. The final DMSO concentration never exceeded 1%. For measurements of KCa2 and KCa3.1 currents we used an internal pipette solution containing (in mM): 145 K+ aspartate, 2 MgCl2, 10 HEPES, 10 K2EGTA, and 5.96 (250 nM free Ca2+) or 8.55 CaCl2 (1 μM free Ca2+), pH 7.2, 290–310 mOsm. Free Ca2+ concentrations were calculated with MaxChelator assuming a temperature of 25°C, a pH of 7.2 and an ionic strength of 160 mM. To reduce currents from native chloride channels in COS-7 and HEK-293 cells, Na+ aspartate Ringer was used as an external solution (in mM): 160 Na+ aspartate, 4.5 KCl, 2 CaCl2, 1 MgCl2, 5 HEPES, pH 7.4, 290–310 mOsm. KCa2 and KCa3.1 currents were elicited by 200-ms voltage ramps from −120 to 40 mV applied every 10 s and the fold-increase of slope conductance at −80 mV by drug taken as a measure of channel activation.
KCa1.1 currents were elicited by 200-ms voltage steps from −80 to 60 mV applied every 10 s (1 μM free Ca2+), and channel modulation measured as a change in mean current amplitude. Kv1.1, Kv1.3, Kv1.4, Kv1.5, Kv3.1, Kv3.2, and Kv4.2 currents were recorded in normal Ringer solution with a Ca2+-free pipette solution containing (in mM): 145 KF, 10 HEPES, 10 EGTA, 2 MgCl2, pH 7.2, 300 mOsm. Currents were elicited by 200-ms depolarizing pulses to 40 mV applied every 10 s. HERG (Kv11.1) currents were recorded with a KCl-based pipette solution (4 mM ATP) and with a two-step pulse from –80 mV first to 20 mV for 2 s and then to –50 mV for 2 s. The reduction of both peak and tail current by the drug was determined. Current from co-expressed Kv7.2/7.3 channels was elicited by depolarizing pulses from the holding potential (−80 mV) to +40 mV for 500 ms followed by hyperpolarization to −120 mV for 200 ms. Nav1.2 currents from N1E-115 cells and Nav1.4 currents from stably transfected HEK cells were recorded with 20 ms pulses from −80 to −10 mV every 10 s with a KCl-based pipette solution and normal Ringer as an external solution. Blockade of Na+ current was determined by measuring the reduction of peak maximum conductance.
To identify Cx50 inhibitors we first screened a small library of compounds containing known ion channel pharmacophores including the antihistamine astemizole, several psoralens and related heterocycles, benzothiazoles, triterpenes, and flavanoid glycosides as well as the antifungal agent clotrimazole. From this library we identified four novel low micromolar inhibitors of Cx50: Astemizole, rutin (a flavonoid glycoside), PAA-10 (an alkyl substituted dibenzazocinone), and clotrimazole (see Figure 1 for structures). All compounds produced significant inhibition of the Cx50 junctional current at a concentration of 10 μM (Figure 1). The inhibition of junctional currents caused by clotrimazole, astemizole and PAA-10 was completely reversible upon washout. In contrast, the effects of rutin were only partially reversible. Of these four hits, the triarylmethane clotrimazole seemed the most drug-like and attractive compound to us. Astemizole is known to affect many other ion channels including the cardiac K+ channel HERG (Kv11.1; Suessbrich et al., 1996), a liability not generally encountered with triarylmethanes (Toyama et al., 2008). We also discarded rutin as a template since preliminary experiments showed that the rutin aglycon, quercetin, had no effect on Cx50 at 10 μM (data not shown) demonstrating that the sugar moiety is essential for connexin inhibition. The dibenzazocinone PAA-10 would of course also have been a possible lead but we preferred to perform structure activity relationship (SAR) studies around clotrimazole since our laboratory had a library of 80 triarylmethanes that were immediately available for an SAR analysis on Cx50. These compounds had been previously synthesized for an SAR study to determine the structural requirements for inhibition of the intermediate-conductance calcium-activated potassium channel KCa3.1 (a.k.a. IKCa1, SK4). By using the so-called selective optimization of side activities (SOSA) approach, which allows for the selective optimization of the side activity of an old drug (Wermuth, 2004), our group successfully designed a triarylmethane, TRAM-34 (T34), that selectively blocked KCa3.1 channels without affecting cytochrome P450-dependent enzymes, the main target of clotrimazole (Wulff et al., 2000). This previous work had demonstrated that it is possible to achieve selectivity for different targets by appropriately modifying the triarylmethane (TRAM) pharmacophore, which was another reason for us to choose clotrimazole as a template for our current study on Cx50.
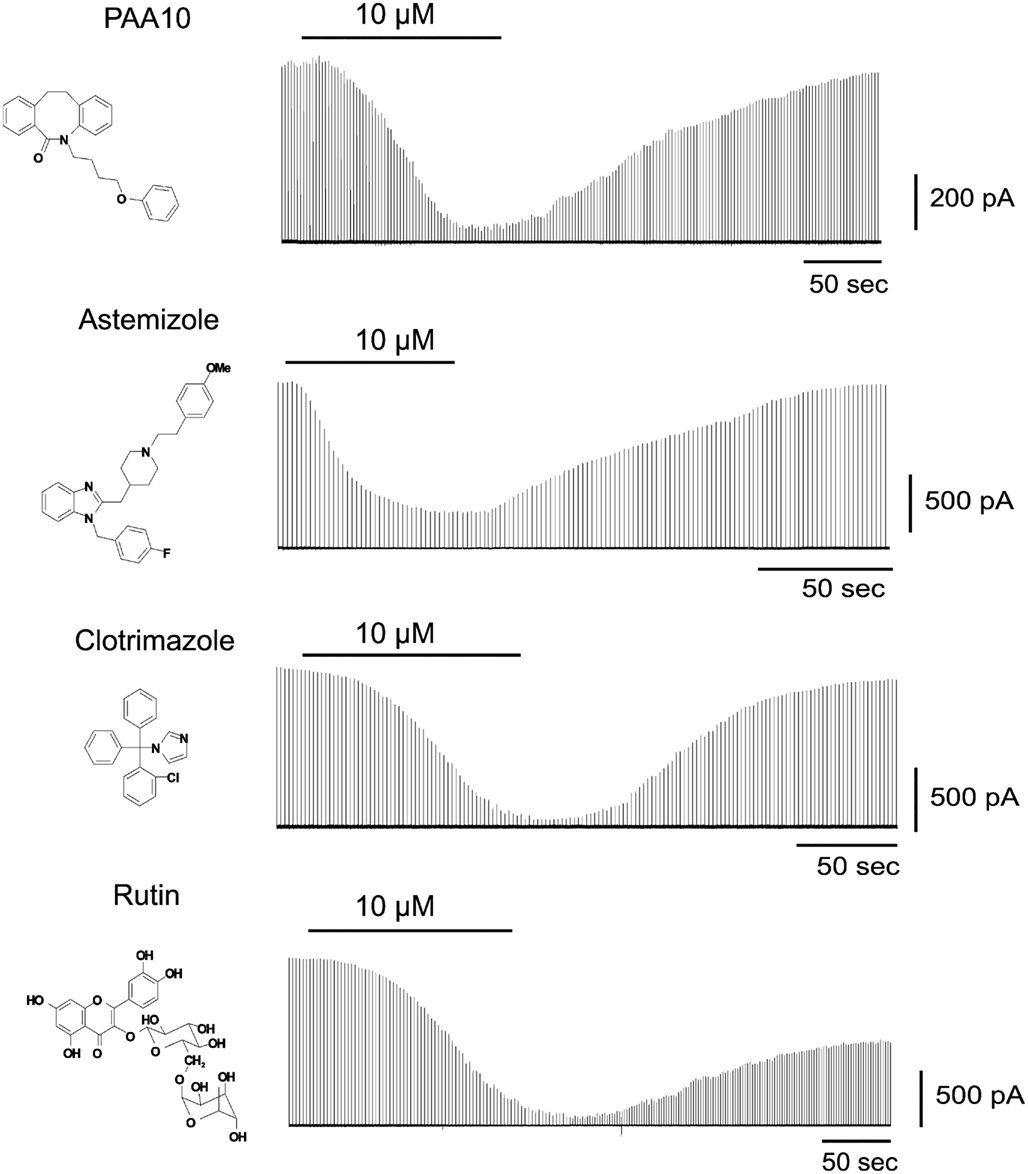
Figure 1. Time course of the effect of PAA-10, astemizole, clotrimazole, and rutin on the junctional currents of Cx50 channels expressed in N2A cell pairs measured by dual whole-cell patch-clamp.
Clotrimazole reversibly inhibited Cx50 expressed in N2A cells with an IC50 of 5 μM and a Hill slope of ∼ 2.1 (Figure 2). At concentrations of 10 μM clotrimazole had no effect on channels built out of Cx32, Cx36, and Cx46 (Figure 2). GJ channel conductance in all cases was measured by using the dual whole-cell patch-clamp technique as described in the Section “Materials and Methods” (Srinivas et al., 2001; Srinivas and Spray, 2003; Cruikshank et al., 2004).
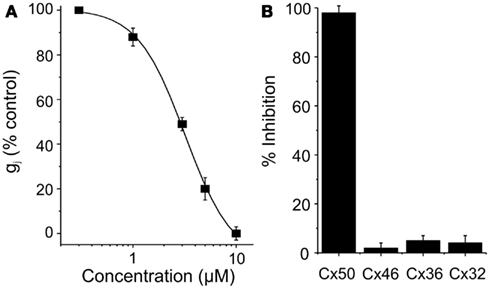
Figure 2. (A) Dose-concentration curve for Cx50 inhibition by clotrimazole. (B) Connexin-selectivity profile of clotrimazole (10 μM). Shown is percentage of current inhibition (mean ± SEM).
Using clotrimazole as a template we explored the SAR of the triphenylmethane scaffold according to the synthetic strategy shown in Figure 3. In a Grignard reaction mono-substituted benzophenones and bromobenzenes were reacted in anhydrous diethyl ether to yield the corresponding triphenylmethanols. These alcohols were then either ammonolyzed, cyanated with copper cyanide or chlorinated using acetyl chloride. The triphenylmethane chlorides were further reacted in a nucleophilic substitution to give the respective triphenylmethane derivatives (further details on exact conditions and quantities are given in the see Materials and Methods). We first substituted the imidazole ring of clotrimazole with several other heterocycles, differently substituted carbocycles or aliphatic functional groups while keeping the 2-chlorophenyldiphenyl methane basic structure (Figure 3). Except for T44 (pyrrol), T69 (2-aminopyridine), T89 (4-methyl-2-phenylimidazole) and the bicyclic T71 (phthalimide) and T103 (2-aminobenzothiazole), most of the heterocyclic derivatives blocked Cx50 in the low micromolar range. Spacer linked carbocycles (T102, T104, T106, T107, T109, T150) in contrast showed no effect on Cx50 at concentrations of 10 μM with the exception of T106, which was found to be a weak blocker with an IC50 10 μM (Figure 4). We next tested the heterocyclic substituted triarylmethanes for selectivity over KCa3.1 (Figure 4). As previously reported (Wulff et al., 2000), clotrimazole and T34 (= TRAM-34) are nanomolar KCa3.1 blockers, that exhibit IC50 values of 70 and 20 nM, respectively, and are therefore not useful as Cx50 inhibitors. In contrast, the aminothiazole and aminopyrimidine substituted T66 and T68 were found to be 15- to 200-fold less potent on KCa3.1 and T66 even exhibited a moderate three-fold selectivity for Cx50 over KCa3.1.
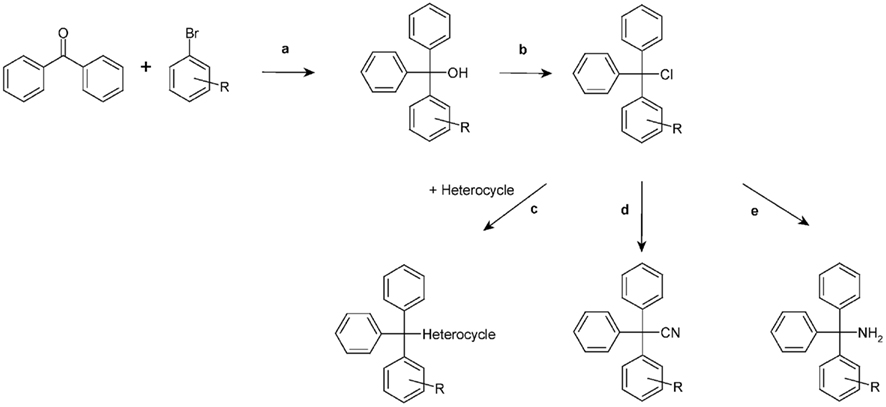
Figure 3. Chemical synthesis scheme. a, Grignard reaction; b, CH3COCl; c, Excess of amine; d, CuCN; e, NH3.
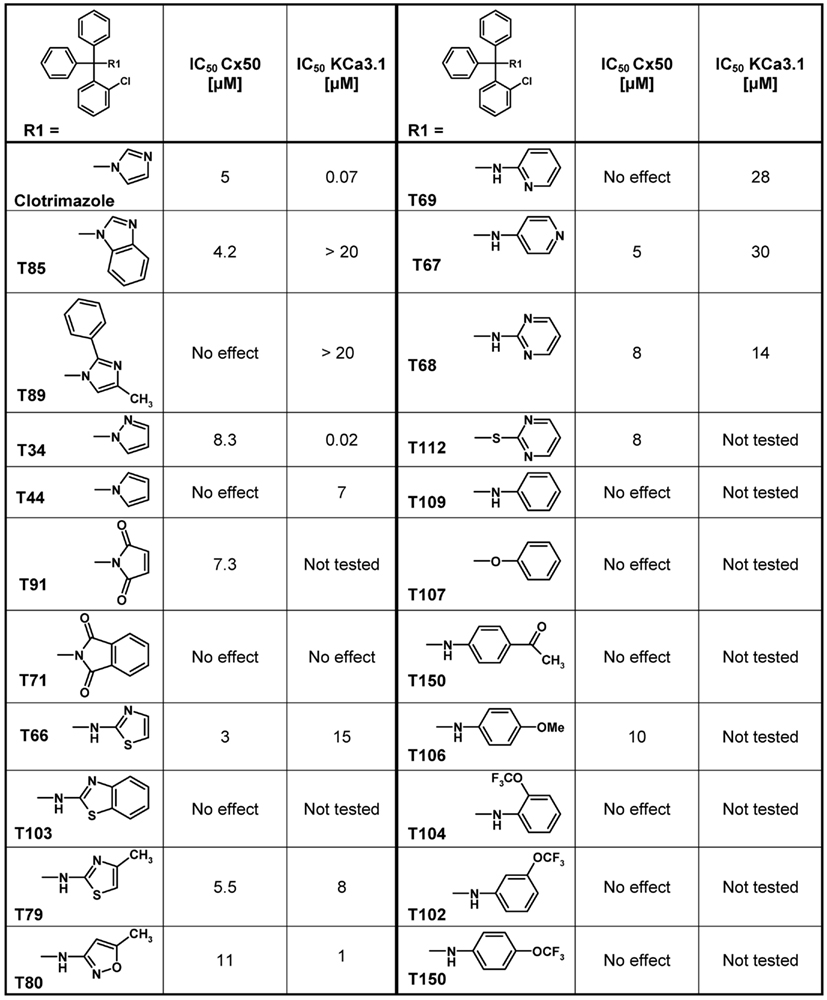
Figure 4. Table showing the structures and IC50 values for Cx50 and KCa3.1 inhibition of heterocyclic substituted triarylmethanes. Concentrations of triarylmethanes that caused a half-maximal inhibition (IC50) values were obtained by fitting the data to the Hill equation, as described in the methods. Means of current inhibition and SD were determined by application of two or three concentrations of each triarylmethane to multiple cells (n ranging from 3 to 8 per concentration). The SD values are not shown for clarity; SD values typically ranged between 5 and 15%.
Since the alcohol T3, which is the first-step intermediate for the heterocyclic substituted triarylmethanes, was also found to reduce Cx50 currents with an IC50 of 2 μM we further synthesized and tested several triarylmethane alcohols, amines, nitriles, and ureas on Cx50 (Table A1 in Appendix). While several of the alcohols including the p-chloro substituted T1, the m-chlorosubstituted T2 as well as the non-substituted triphenylmethanol and triphenylamine (T162) exhibited IC50 values for Cx50 in the 1–2 μM range (Table A1 in Appendix), all these compounds lacked selectivity over KCa3.1 and were further found to inhibit other connexins like Cx43, Cx46 at similar concentrations as Cx50 (data not shown). In addition, the inhibition produced by these compounds was often enhanced by a prior application of the compounds. We therefore did not study these compounds further and instead concentrated our synthetic efforts on the heterocyclic substituted triarylmethanes and explored the substitution position of the chlorine atom on one of the phenyl rings by moving it from the ortho- to the meta- or para-position or completely removing it (Figure 5). All four imidazole ring containing compounds (T97, clotrimazole, T143, T144) but only the o-chloro and m-chloro substituted pyrazole derivatives (T34 and T142) inhibited Cx50 channels with IC50s of 5–8 μM. In the 2-aminothiazole and 2-aminopyrimidine series only the o-chloro substituted T66 and T68 were active, while the other regio-isomers or the unsubstituted analogs showed no effect at 10 μM. Because the imidazole and pyrazole-substituted triarylmethanes were poorly selective for Cx50 over KCa3.1, we studied the effect of modifications of the o-chloro substituent in the 2-aminothiazole and 2-aminopyrimdine series. Specifically, we replaced the o-chloro substituent with other halogens (F, Br, I), the more lipophilic CF3 or OCF3 groups or electron-donating methyl or methoxy groups. All 14 compounds exhibited IC50 values in the low micromolar range (Figure 6) with the methoxy-substituted T122 (IC50 1.2 μM) and the iodo-substituted T136 (IC50 2.4 μM) being the most potent (Figure 6).
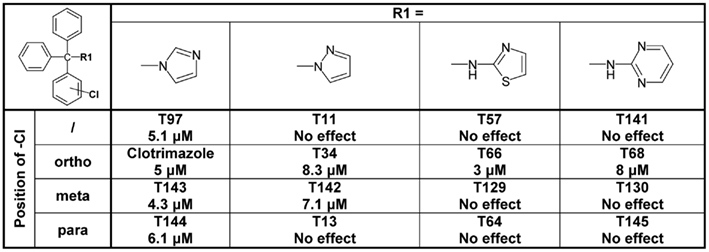
Figure 5. Table showing the structures and IC50 values for Cx50 inhibition for imidazole, pyrazole, aminothiazole, and aminopyrimidine substituted triarylmethanes bearing a chlorine substituent in various positions on one of the phenyl rings. Means of current inhibition and SD were determined by application of two or three concentrations of each triarylmethane to multiple cells (n ranging from 3 to 8 per concentration). The SD values are not shown for clarity; SD values typically ranged between 5 and 15%.
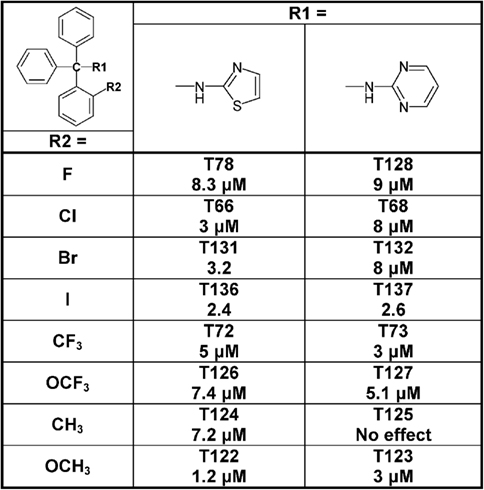
Figure 6. Table showing the structures and IC50 values for Cx50 inhibition for aminothiazole and aminopyrimdine substituted triarylmethanes with various functional groups in ortho-position on one of the phenyl rings. Means of current inhibition and SD were determined by application of two or three concentrations of each triarylmethane to multiple cells (n ranging from 3 to 8 per concentration). The SD values are not shown for clarity; SD values typically ranged between 5 and 15%.
The effects of T122 and T136 on Cx50 junctional channels were further characterized using a five-point dose response curve (Figure 7A). Non-linear least-squares fit of the individual data points to the Hill equation (see Materials and Methods) yielded IC50 values of 1.2 and 2.4 μM for the inhibition of Cx50 GJ channels by T122 and T136, respectively. In both cases, the Hill coefficients were ≈ 2 (1.6 for T122, 1.7 for T136), indicating that binding of two TRAM molecules was required to inhibit Cx50 GJ channels. Both compounds exhibited high selectivity for Cx50 channels over other connexin subtypes. The effects of T122 and T136 on GJ channels formed by several other connexins, including Cx26, Cx32, Cx40, Cx43, and Cx46 are illustrated in Figure 7B. At a concentration of 10 μM, sufficient to cause near-maximal decreases in Cx50 junctional current, T122 and T136 did not significantly inhibit Cx26, Cx32, Cx46, or Cx43 GJ channel currents. The reduction of junction conductance was less than 20% in each of these cases. These results demonstrate that inhibition of Cx50 GJ channels by T122 and T136 is highly connexin-selective.
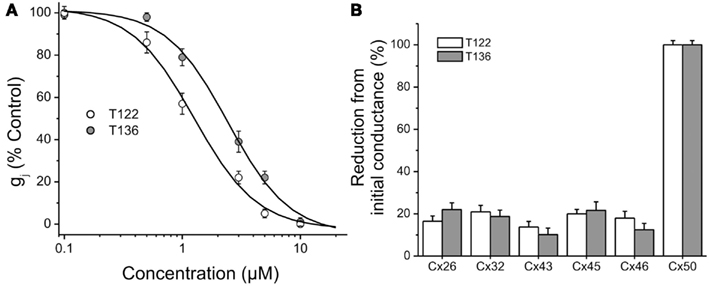
Figure 7. Connexin-selectivity of T122 and T136. (A) Concentration dependence of T122 and T136 on Cx50 gap junction channels. Each point represents the mean ± SEM of gj (% of the initial conductance) values obtained from 4 to 10 cell pairs. The solid line is a fit of the data points to the Hill equation (see Materials and Methods). The EC50 and Hill slope values are indicated in the text. Each cell pair was exposed to only a single concentration. (B) Bar graph illustrating that T122 (10 μM) and T136 (10 μM) has no significant effect on Cx26, Cx32, Cx43, Cx45, and Cx46 gap junction channels. Each bar represents the mean ± SEM of four to six cell pairs.
In order to more broadly evaluate the selectivity of T122 and T136 we further determined their effect on a panel of 12 potassium and sodium channels from various gene families (Table 1). While both compounds exerted practically no effect on the neuronal Nav1.2 and the skeletal muscle Nav1.4 channel or Kv channels from the Kv4, Kv7, Kv11, or KCa2 (SK) family, they exhibited only moderate selectivity over Kv1, Kv2, Kv3-family channels, Kv11.1 (hERG), and the calcium-activated K+ channels KCa1.1 and KCa3.1. T122 in particular reduced Kv1.1 currents by 65% at 10 μM, while T136 was found to have an IC50 of 1.3 μM for KCa3.1.
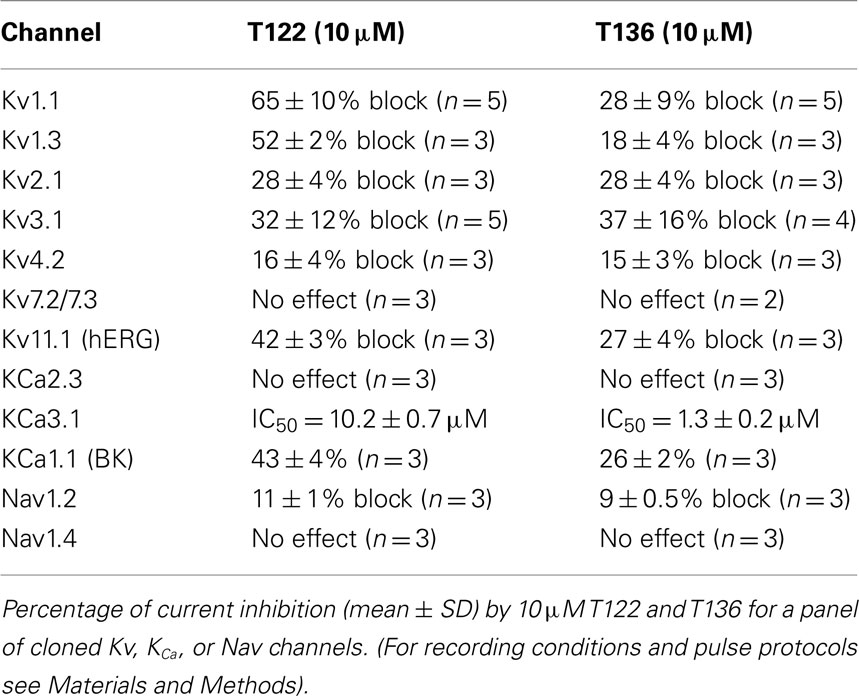
Table 1. Selectivity over other ion channels.
Cx50 is strongly expressed in the lens in both the epithelium and in fibers. In the epithelium, the functional contribution of Cx50 to epithelial cell coupling is highest during the first postnatal week (∼70–75% of total coupling on average) with the remainder being contributed by Cx43. Therefore, we determined whether T122 and T136 (10 μM) also inhibited coupling provided by Cx50 in epithelial cells (Figure 8). The effect of T122 and T136 on junctional currents between epithelial cells isolated from mouse lenses on postnatal day 6 is shown in Figure 8A. Both compounds strongly reduced junctional currents, an effect that was reversible on washout of the drug. The reduction of junctional currents caused by T122 and T136 ranged from 65 to 87% of the initial conductance (means ± SEM are 67 ± 9%, n = 7 for T122; and 64 ± 8%, n = 6 for T136). These values were similar to the reduction produced by quinine, which also selectively inhibits Cx50, but not Cx43 GJ channels (Figure 8B).
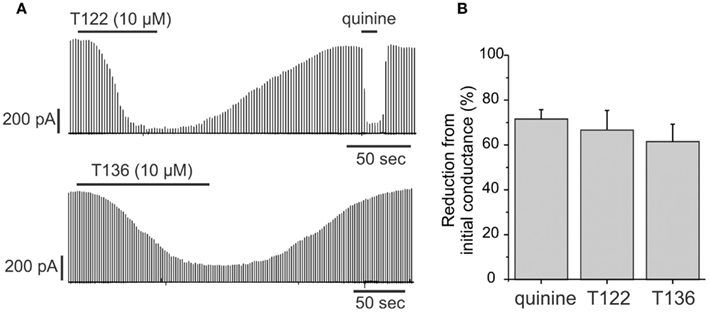
Figure 8. T122 and T136 reduce junctional conductance in lens epithelial cells. (A) Effect of 10 μM T122 (top) and 10 μM T136 (bottom) on junctional currents in epithelial cells isolated from P6 lenses. The magnitude of inhibition is similar to that produced by quinine. The recordings in T122 and T136 are from two different cell pairs. (B) Bar graph summarizing the effect of T122 (10 μM) and T136 (10 μM), compared to that of quinine on coupling in epithelial cells. Each bar represents the mean ± SEM of four to six cell pairs.
A major reason for the poorly developed pharmacology of GJ channel modulators is the intercellular location of these channels, which makes it extremely difficult to design high-throughput screening assays. In our search for Cx50 inhibitors we therefore decided to screen a small library containing known ion channel modulators using conventional dual whole-cell voltage-clamp. Our library was enriched in so-called “privileged” structures which are small molecule pharmacophores that are able to bind to multiple targets and which are therefore highly likely to exert biological effects (Evans et al., 1988; Horton et al., 2003). By appropriately decorating such “privileged” scaffolds their potency and selectivity can often be directed toward a single target with relatively high affinity. If the template for such an SAR study is an “old” drug, the approach is also called SOSA approach (selective optimization of the side activity of an old drug) as suggested by Wermuth (2004). Using this approach we found four structurally very different compounds able to inhibit Cx50 channels in the low micromolar range including the triarylmethane (TRAM) clotrimazole (IC50 5 μM; Figure 1). Using clotrimazole as a template, we have tested a series of previously known or newly synthesized differently substituted TRAMs for their effects on Cx50 channels and identified several compounds inhibiting Cx50 in the low micromolar range including T122, which exhibits an IC50 of 1.2 μM for Cx50 and excellent selectivity over other connexins. Analyzing at the structural requirements for selective Cx50 inhibition we find that the TRAM pharmacophore should contain a heteroaromatic ring system in R1 position. Smaller, less bulky substitutions with functional groups such as OH, NH, or H also result in potent Cx50 blockers (Table A1 in Appendix), however, these types of compounds lack selectivity over other connexins and are therefore not useful as pharmacological tools. In addition to a heteroaromatic substituent in R1 position, the second requirement is that one of the phenyl rings of the triphenylmethane should be substituted preferably in ortho-position (Figure 5), whereby it seems to be of little consequence whether the substituent is electron withdrawing (CF3 in T72) or donating (OCH3 in T122), as long as it is lipophilic. Lastly, the heteroaromatic ring system, which can be directly attached or linked by a one-atom spacer (nitrogen or sulfur), should contain at least one hydrogen-bond accepting heteroatom, which might be directly interacting with a hydrogen-bond donor in the connexin protein (e.g., see clotrimazole with its imidazole ring versus the inactive T44 and T109). It also seems that steric bulk is a limiting factor for potency, because the bulkier bicyclic derivatives T103 and T71 are ineffective while the smaller T66 and T91 inhibit Cx50.
Since our compounds were derived from clotrimazole, which inhibits KCa3.1 channels with an IC50 of 70 nM, we also tested the more active compounds for selectivity over KCa3.1. For nanomolar KCa3.1 inhibition Wulff et al. (2000) previously proposed a propeller-shaped pharmacophore consisting of the triphenyl moiety with an o-halogen on one of the phenyl rings and an unsubstituted, polar π-electron-rich heterocycle of limited size such as pyrazole, tetrazole, or an even smaller nitrile group in R1 position. A similar KCa3.1-inhibiting TRAM pharmacophore was described by McNaughton-Smith et al. (2008) as exemplified by ICA-17043 (Senicapoc®), which contains a carboxamide moiety in R1 position and entered clinical trials for sickle cell anemia. Considering this knowledge about the TRAM pharmacophore for KCa3.1 inhibition in our current Cx50-focused SAR study, we could achieve a drop in potency on KCa3.1 of more than 1000-fold by inserting a one-atom linker between the triphenyl moiety and the heterocycle in R1 position. This insertion, which results in a “kink” in the perfect propeller shape of clotrimazole or TRAM-34 shifted selectivity toward Cx50 [clotrimazole (IC50 of 70 nM for KCa3.1 and 5 μM Cx50) versus T122 (IC50 of 10 μM for KCa3.1 and 1.2 μM for Cx50)]. At concentrations of 10 μM the two most potent compounds, T122 and T136, showed excellent selectivity over other tested connexin channels including channels built out of Cx43 and Cx46, which are also expressed in the lens (inhibition < 18%). In addition, T122 and T136 showed very good selectivity over sodium channels (<11%) and moderate selectivity over most of the tested potassium channels (18–43%), except for T122, which blocked Kv1.1 by 65%. Both compounds also inhibited Cx50 mediated coupling in primary lens epithelial cells. Thus, compared to the existing pharmacological agents such as quinine and 2-APB, which exhibit poor selectivity for Cx50 channels, T122 and T136 are likely better agents for studying the role of Cx50 in lens development and the maintenance of lens transparency.
Taken together, we here identified the triphenylmethane scaffold as a new pharmacophore for Cx50 inhibition and synthesized two new compounds that inhibit cloned and native Cx50 channels in the low micromolar range. Even more importantly, we were able to develop a connexin subtype specific inhibitor starting with a lead compound that exhibited several fold higher selectivity for K+ channels over Cx50, indicating that application of a similar approach, i.e., identification of new lead compound(s) from existing ion channel libraries followed by selective optimization of their side activities, may lead to the development of specific blockers for other connexin channels such as Cx43 or Cx26.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by grants from National Institute of Neurological Disorders And Stroke [R21NS064417] and the National Eye Institute [RO1EY13869]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute Of Neurological Disorders And Stroke or the National Eye Institute.
Baeyer, A. (1907). The investigation of derivatives of triphenylcarbinol. First paper. Justus Liebigs Ann. Chem. 354, 152–204.
Bartroli, J., Alguero, M., Boncompte, E., and Forn, J. (1992). Synthesis and antifungal activity of a series of difluorotritylimidazoles. Arzneimittelforschung 42, 832–835.
Berthoud, V. M., and Beyer, E. C. (2009). Oxidative stress, lens gap junctions, and cataracts. Antioxid. Redox Signal. 11, 339–353.
Bodendiek, S. B., and Raman, G. (2010). Connexin modulators and their potential targets under the magnifying glass. Cur. Med. Chem. 17, 4191–4230.
Cruikshank, S. J., Hopperstad, M., Younger, M., Connors, B. W., Spray, D. C., and Srinivas, M. (2004). Potent block of Cx36 and Cx50 gap junction channels by mefloquine. Proc. Natl. Acad. Sci. U.S.A. 101, 12364–12369.
Dahlbom, R., and Ekstrand, T. (1944). Trityl derivatives of sulfanilamides. Sven Kem. Tidskr. 56, 304–314.
Evans, B. E., Rittle, K. E., Bock, M. G., DiPardo, R. M., Freidinger, R. M., Whitter, W. L., Lundell, G. F., Veber, D. F., Anderson, P. S., and Chang, R. S. (1988). Methods for drug discovery: development of potent, selective, orally effective cholecystokinin antagonists. J. Med. Chem. 31, 2235–2246.
Grissmer, S., Nguyen, A. N., Aiyar, J., Hanson, D. C., Mather, R. J., Gutman, G. A., Karmilowicz, M. J., Auperin, D. D., and Chandy, K. G. (1994). Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol. Pharmacol. 45, 1227–1234.
Horton, D. A., Bourne, G. T., and Smythe, M. L. (2003). The combinatorial synthesis of bicyclic privileged structures or privileged substructures. Chem. Rev. 103, 893–930.
Kandouz, M., and Batist, G. (2010). Gap junctions and connexins as therapeutic targets in cancer. Expert Opin. Ther. Targets 14, 681–692.
Lang, N. N., Myles, R. C., Burton, F. L., Hall, D. P., Chin, Y. Z., Boon, N. A., and Newby, D. E. (2008). The vascular effects of rotigaptide in vivo in man. Biochem. Pharmacol. 76, 1194–1200.
Locovei, S., Scemes, E., Qiu, F., Spray, D. C., and Dahl, G. (2007). Pannexin1 is part of the pore forming unit of the P2X(7) receptor death complex. FEBS Lett. 581, 483–488.
Mathias, R. T., Rae, J. L., and Baldo, G. J. (1997). Physiological properties of the normal lens. Physiol. Rev. 77, 21–50.
Mathias, R. T., White, T. W., and Gong, X. (2010). Lens gap junctions in growth, differentiation, and homeostasis. Physiol. Rev. 90, 179–206.
McNaughton-Smith, G. A., Burns, J. F., Stocker, J. W., Rigdon, G. C., Creech, C., Arrington, S., Shelton, T., and de Franceschi, L. (2008). Novel inhibitors of the Gardos channel for the treatment of sickle cell disease. J. Med. Chem. 51, 976–982.
Misra, B. K., Rao, Y. R., and Mahapatra, S. N. (1983). Friedel-Crafts acylation of some aromatic compounds with 2-isocyanatobenzoyl chloride. Indian J. Chem. Sect. B 22B, 1132–1138.
Monder, C., Stewart, P. M., Lakshmi, V., Valentino, R., Burt, D., and Edwards, C. R. (1989). Licorice inhibits corticosteroid 11 beta-dehydrogenase of rat kidney and liver: in vivo and in vitro studies. Endocrinology 125, 1046–1053.
Nemani, V. M., and Binder, D. K. (2005). Emerging role of gap junctions in epilepsy. Histol. Histopathol. 20, 253–259.
Rong, P., Wang, X., Niesman, I., Wu, Y., Benedetti, L. E., Dunia, I., Levy, E., and Gong, X. (2002). Disruption of Gja8 (alpha 8 connexin) in mice leads to microphthalmia associated with retardation of lens growth and lens fiber maturation. Development 129, 167–174.
Salameh, A., and Dhein, S. (2005). Pharmacology of gap junctions. New pharmacological targets for treatment of arrhythmia, seizure and cancer? Biochim. Biophys. Acta 1719, 36–58.
Sankaranarayanan, A., Raman, G., Busch, C., Schultz, T., Zimin, P. I., Hoyer, J., Kohler, R., and Wulff, H. (2009). Naphtho[1,2-d]thiazol-2-ylamine (SKA-31), a new activator of KCa2 and KCa3.1 potassium channels, potentiates the endothelium-derived hyperpolarizing factor response and lowers blood pressure. Mol. Pharmacol. 75, 281–295.
Srinivas, M., Hopperstad, M. G., and Spray, D. C. (2001). Quinine blocks specific gap junction channel subtypes. Proc. Natl. Acad. Sci. U.S.A. 98, 10942–10947.
Srinivas, M., and Spray, D. C. (2003). Closure of gap junction channels by arylaminobenzoates. Mol. Pharmacol. 63, 1389–1397.
Suadicani, S. O., Brosnan, C. F., and Scemes, E. (2006). P2X7 receptors mediate ATP release and amplification of astrocytic intercellular Ca2+ signaling. J. Neurosci. 26, 1378–1385.
Suessbrich, H., Waldegger, S., Lang, F., and Busch, A. E. (1996). Blockade of HERG channels expressed in Xenopus oocytes by the histamine receptor antagonists terfenadine and astemizole. FEBS Lett. 385, 77–80.
Toyama, K., Wulff, H., Chandy, K. G., Azam, P., Raman, G., Saito, T., Fujiwara, Y., Mattson, D. L., Das, S., Melvin, J. E., Pratt, P. F., Hatoum, O. A., Gutterman, D. D., Harder, D. R., and Miura, H. (2008). The intermediate-conductance calcium-activated potassium channel KCa3.1 contributes to atherogenesis in mice and humans. J. Clin. Invest. 118, 3025–3037.
Vessey, J. P., Lalonde, M. R., Mizan, H. A., Welch, N. C., Kelly, M. E., and Barnes, S. (2004). Carbenoxolone inhibition of voltage-gated Ca channels and synaptic transmission in the retina. J. Neurophysiol. 92, 1252–1256.
Wermuth, C. G. (2004). Selective optimization of side activities: another way for drug discovery. J. Med. Chem. 47, 1303–1314.
White, T. W., Gao, Y., Li, L., Sellitto, C., and Srinivas, M. (2007). Optimal lens epithelial cell proliferation is dependent on the connexin isoform providing gap junctional coupling. Invest. Ophthalmol. Vis. Sci. 48, 5630–5637.
White, T. W., Goodenough, D. A., and Paul, D. L. (1998). Targeted ablation of connexin50 in mice results in microphthalmia and zonular pulverulent cataracts. J. Cell. Biol. 143, 815–825.
Wulff, H., Gutman, G. A., Cahalan, M. D., and Chandy, K. G. (2001). Delination of the clotrimazole/TRAM-34 binding site on the intermediate conductance calcium-activated potassium channel IKCa1. J. Biol. Chem. 276, 32040–32045.
Wulff, H., Miller, M. J., Haensel, W., Grissmer, S., Cahalan, M. D., and Chandy, K. G. (2000). Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: a potential immunosuppressant. Proc. Natl. Acad. Sci. U.S.A. 97, 8151–8156.
Zunszain, P. A., Shah, M. M., Miscony, Z., Javadzadeh-Tabatabaie, M., Haylett, D. G., and Ganellin, C. R. (2002). Tritylamino aromatic heterocycles and related carbinols as blockers of Ca2+-activated potassium ion channels underlying neuronal hyperpolarization. Arch. Pharm. 335, 159–166.
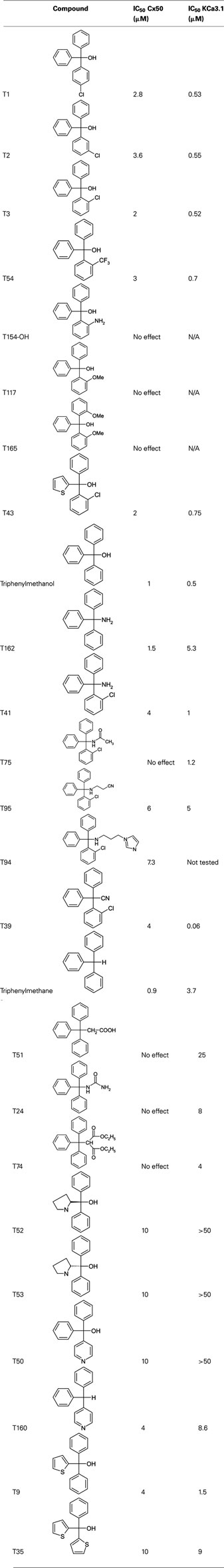
Table A1. IC50 values for Cx50 inhibition for triarymethane alcohols, amines, nitriles and ureas.
Keywords: connexin 50, gap junctions, triarylmethane, inhibitors, pharmacophore, channel, SAR, lens
Citation: Bodendiek SB, Rubinos C, Trelles MP, Coleman N, Jenkins DP, Wulff H and Srinivas M (2012) Triarylmethanes, a new class of Cx50 inhibitors. Front. Pharmacol. 3:106. doi: 10.3389/fphar.2012.00106
Received: 20 March 2012; Accepted: 18 May 2012;
Published online: 06 June 2012.
Edited by:
Michael Pusch, Istituto di Biofisica, Consiglio Nazionale delle Ricerche, ItalyReviewed by:
Alonso P. Moreno, Cardiovascular and Training Institute at the University of Utah, USACopyright: © 2012 Bodendiek, Rubinos, Trelles, Coleman, Jenkins, Wulff and Srinivas. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Miduturu Srinivas, Department of Biological Sciences, State University of New York – State College of Optometry, 33 W 42nd Street, New York, NY 10036, USA. e-mail:bXNyaW5pdmFzQHN1bnlvcHQuZWR1
†Heike Wulff and Miduturu Srinivas have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.