

- Department of Cellular and Molecular Pharmacology, RFUMS/The Chicago Medical School, North Chicago, IL, USA
The cannabinoid receptor 1 (CB1R) is the G-protein coupled receptor responsible for the majority of the endocannabinoid signaling in the human brain. It is widely distributed in the limbic system, basal ganglia, and cerebellum, which are areas responsible for cognition, memory, and motor control. Because of this widespread distribution, it is not surprising that drugs that activate CB1R have expected behavioral outcomes consistent with dysregulated signaling from these areas (e.g., memory loss, cognitive deficits, etc). In the context of this review, we present evidence for the role of CB1R signaling in the prefrontal cortex (PFC), an area involved in executive functions, with emphasis on the developmental regulation of CB1R signaling in the acquisition of mature PFC function. We further hypothesize how alterations in CB1R signaling specifically during adolescent maturation might confer liability to psychiatric disorders.
This article reviews the available evidence of long-lasting cognitive deficits induced by cannabis consumption during adolescence and its relationship to schizophrenia, and presents a working model for dissecting the mechanisms involved in this dysfunction based on the current clinical and preclinical knowledge. Because of the role of the prefrontal cortex (PFC) in higher cognitive functions, we have limited our discussion and conclusions to the role of endogenous and exogenous cannabinoid signaling in prefrontal circuits. Importantly, the machinery involved in cannabinoid signaling is found in other cortical and subcortical regions (e.g., amygdala, striatum), which have also been implicated in other aspects of schizophrenia’s symptomatology and therefore are beyond the scope of this Perspective Review.
Cannabis, Cortical Development, and the Liability for Schizophrenia
Adolescence is the transitional period between childhood and adulthood that is accompanied by refinement of cognitive functions thought to underlie the acquisition of mature behavior (Casey et al., 2000; Spear, 2000). Many of these executive functions, such as problem solving, working memory, abstract thinking, and increased inhibitory control are processed by the PFC. Accordingly, the PFC is one of the last brain regions to develop and undergo changes during late adolescence that include increased myelination and synaptic pruning (Andersen, 2003; Chambers et al., 2003; Crews et al., 2007). This active remodeling of brain areas creates a developmental window where environmental factors can affect the normal trajectories of cortical circuits. Indeed, cannabis use during adolescence has been associated with increased liability of developing neuropsychiatric and abuse disorders later in life (Arseneault et al., 2002, 2004; Zammit et al., 2002; Caspi et al., 2005; Moore et al., 2007). Specifically, there is a concern that cannabis use/abuse during adolescence results in increased risk of developing schizophrenia-spectrum disorders in adulthood, although the mechanisms underlying such an association still remain unclear (Sewell et al., 2009). Given that adolescence is a period of increased liability to drug addiction due to inherent novelty-seeking (Spear, 2000; Chambers et al., 2003), it is of utmost importance to determine the mechanisms of this impairment for sensible public policy implementation.
Administration of cannabinoids to otherwise healthy adult individuals causes psychotic symptoms including paranoia, perceptual alterations, conceptual disorganization, and fragmented thinking, similar to the positive symptoms experienced by patients with schizophrenia (D’Souza et al., 2004). Notably, controlled cannabis administration in patients with schizophrenia exacerbates the positive symptoms and increases the cognitive deficits, suggesting a common underlying mechanism (D’Souza et al., 2005). In addition, adult subjects administered cannabinoids display deficits in memory recall, attention, and inhibitory control (Hart et al., 2001; D’Souza et al., 2004; Henquet et al., 2006). Although the long-term effects of chronic cannabis use are still debated, acute cannabinoid administration shows mostly transient effects in adult individuals, suggesting that only a specific dysregulation by cannabinoids during developmentally sensitive periods like adolescence could be responsible for the long-lasting deficits seen in prefrontal functioning.
In support of this hypothesis, morphological examination of brains from cannabis users has demonstrated significant smaller gray matter volumes in cortical regions when cannabis use starts before the age of 17 (Wilson et al., 2000). Early-onset use of cannabis also results in attentional dysfunctions reflected in reduced phasic alertness and attention (Ehrenreich et al., 1999). Moreover, an early vs. late adolescent exposure to cannabis can give rise to specific impairments in visual scanning, a test that assesses the reaction time to the processing of visual stimuli (Ehrenreich et al., 1999). Pope et al. (2003) have found that early-onset users (before age 17) show deficits in verbal IQ compared to late-onset users, and differ significantly from control subjects in other verbal functions involving memory and recall. More recently, Fontes et al. (2011) have established that individuals with early-adolescence onset of cannabis use (before age 15) score poorer in prefrontal-lobe related tasks, and significantly worse than the late-adolescent-onset group. All in all, the bulk of evidence seems to indicate that cannabis use during or even within specific periods of adolescence can lead to enduring cognitive impairments involving the PFC.
The precise neural substrates underlying cannabinoid-induced cognitive impairments remain elusive; however, evidence suggests that cannabis affects the integration of cortical information. In cortical structures, information processing is thought to be mediated by the synchronization of neuronal networks, such that spatio-temporal integration of networks functionally encodes cortical-related behaviors and cognition. This is typically manifested as changes in cortical oscillations at different frequency bands and synchrony between different regional networks (Buzsaki and Draguhn, 2004). This is especially important in the light that patients with schizophrenia show abnormal oscillations in the theta (4–7 Hz) and gamma range (30–200 Hz), and abnormal synchrony between cortical structures, suggesting aberrant integration of large and local neuronal circuits (Spencer et al., 2003; Uhlhaas and Singer, 2010). Similarly, cannabis smoking decreases theta oscillations, and this correlates with detrimental effects on working memory task performance (Ilan et al., 2004). More recently, D’Souza et al. have determined that cannabis administration dose-dependently reduces the amplitude of event-related potentials (ERPs, a measure of network responses to a stimulus) in an auditory task assessing attention and cognition (D’Souza et al. 2012). These data indicate that cannabis compromises both context updating and allocation of attentional resources. Similar reductions in ERPs have been observed in schizophrenia and are associated with decreased shift in attention (Rissling et al., 2010). Altogether, cannabis disrupts cortical network dynamics similar to those reported in schizophrenia, suggesting that exogenous cannabinoids can alter the physiology of brain circuits involved in higher-order cognitive processing.
Endocannabinoids and CB1 Receptor Signaling in the Central Nervous System
The endocannabinoid system is comprised by the receptors, synthesizing and inactivating enzymes that mediate the actions of natural endogenous cannabinoids (Kreitzer and Regehr, 2002; Lovinger, 2008). The cannabinoid receptor 1 (CB1R) is the G-protein coupled receptor responsible for most endocannabinoid signaling in the brain (Matsuda et al., 1990). It is widely expressed in the vertebrate brain, and at especially high levels in the cortex, hippocampus, cerebellum, and the various nuclei of the basal ganglia where its expression is mostly presynaptic (Herkenham et al., 1991; Pettit et al., 1998; Egertova and Elphick, 2000). Under physiological conditions, CB1R binds the endogenous cannabinoids anandamide (AEA) and 2-arachydonylglycerol (2-AG). These fatty acid-derived ligands are produced in the postsynaptic membrane as a result of neurotransmitter binding to its cognate receptors (Lovinger, 2008). Endocannabinoids then diffuse in a retrograde manner and bind to the presynaptically located CB1R. Here, CB1R signaling is mostly mediated by coupling to pertussis-sensitive Gi/o proteins which effectively inhibit the enzyme adenylate cyclase (Vogel et al., 1993) leading to a local reduction of cAMP levels that decreases PKA activity and leads to activation of A-type K channels. In addition, CB1R signaling inhibits N- and Q-type Ca++ currents which are necessary for neurotransmitter release at the presynaptic site (Lovinger, 2008). Because of this generic mode of action, endocannabinoid-mediated CB1R signaling is capable of modulating a broad range of neurotransmitters including glutamate, GABA, dopamine, serotonin, and acetylcholine (Lovinger, 2008). In this sense, the endocannabinoid system has a homeostatic role in controlling excessive excitation or inhibition and acts continuously through an intrinsic “tone” (Marsicano et al., 2003; Katona and Freund, 2008).
The Role of CB1R Signaling in Cognitive Functions
Interest in CB1R signaling arises from the fact that it is the receptor responsible for binding the psychoactive components of cannabis, most prominently Δ9-THC, leading to the behavioral abnormalities associated with marijuana use, including but not limited to memory retrieval, euphoria, and in rare cases psychosis (Iversen, 2003). Given that cannabis contains more than 70 known cannabinoids (Elsohly and Slade, 2005), synthetic cannabinoids have been utilized over the years to systematically dissect the role of CB1 receptor in the development and function of the PFC.
Similar to the effect of cannabis in humans, administration of CB1R agonists in rodents during the adolescent transition to young adulthood (from PD35 to PD65) can lead to behavioral impairments in adulthood that model those observed in schizophrenia. Accordingly, rats chronically treated with the CB1R agonist WIN55,212-2 (WIN) from PD40 to 65 and tested in adult (PD75-150) exhibit deficits in sensorimotor gating, short-term memory, and invest less effort for reward, a proxy for anhedonia (Schneider and Koch, 2003). WIN-treated rats also displayed reduced object-social discrimination, social interaction, and spontaneous social behavior as adults, indicating permanent deficits in recognition memory and increased anxiety (Schneider et al., 2008). Similarly, rats treated with the synthetic cannabinoid CP55,940 from the juvenile period (PD30) to PD51 also display reduced object recognition and social interactions during adulthood (O’Shea et al., 2004). The residual effects in object recognition and anxiety have also been observed in adult rats treated with Δ9-THC from PD32 to 55 (Quinn et al., 2008). Interestingly, rats treated during adulthood lacked any permanent deficits on these behavioral measures, suggesting that cannabinoids had a significant effect only when treatment spanned adolescence (Schneider and Koch, 2003; O’Shea et al., 2004; Bortolato et al., 2005; Schneider et al., 2008). Notably, a recent study found that rats administered WIN from PD45 to 60 exhibited significant long-lasting effects in a hippocampal-dependent task, while a more PFC-dependent task was impaired only transiently (no impairment after 30 days; Abush and Akirav, 2012). Thus, there seems to be a differential sensitivity to cannabinoids that is dependent on the interaction of the specific developmental window of CB1R activation with the specific ontogeny of brain structures that are rich in CB1 receptors.
CB1 Receptor Expression and Signaling in the Frontal Cortex
In considering the role of CB1R in the development of the central nervous system, it is well known that CB1R expression plays an important role during the prenatal and early postnatal period. During this time, CB1R plays a critical role in neurogenesis, axon migration, and synaptogenesis (for review Harkany et al., 2007). Consistent with these findings, CB1R expression appears as early as gestational day (GD) 14 and by GD16 is clearly expressed across the developing frontal cortex, hippocampus, striatum, and midbrain (Berrendero et al., 1998). Another study determined that CB1R binding can be detected at different levels by PD10 and increases steadily until adolescence in several limbic regions, including the frontal cortex, striatum, and mesencephalon, after which binding starts declining (Rodriguez de Fonseca et al., 1993). In addition, we have recently demonstrated that CB1R mRNA expression drops in all cortical areas during the adolescent transition to adulthood, most prominently in the limbic/associative cortex (prelimbic, infralimbic, and cingulate). More importantly, this decrease of CB1R expression is associated with a functional decline in CB1R-mediated signaling on excitatory synaptic transmission in the PFC (Heng et al., 2011). Altogether, these data suggest that developmental maturation of the PFC is accompanied by a net downregulation of CB1R function.
In cortical areas, the CB1 receptor is known to be expressed in pyramidal neurons and to a much greater extent in GABAergic interneurons (Marsicano and Lutz, 1999; Hill et al., 2007), indicating that CB1R has a greater modulatory role in GABA release. In the neocortex, CB1R is expressed at high levels in a subpopulation of cholecystokinin (CCK)-positive basket interneurons and to a lower extent in calbindin-positive interneurons (Katona et al., 1999; Marsicano and Lutz, 1999; Wedzony and Chocyk, 2009). Thus, these CB1-positive GABAergic interneuron populations are independent of the other well-characterized fast-spiking parvalbumin-positive basket cells (Katona et al., 1999; Bodor et al., 2005). Both types of basket cells mediate the perisomatic inhibition of pyramidal cells (Trettel and Levine, 2003) and are electrically coupled to their own class (Hestrin and Galarreta, 2005), two distinctive properties that makes them efficient at synchronizing the activity of pyramidal cells. However, CCK- and PV-positive interneurons are differentially sensitive to cannabinoid-mediated effects (Freund, 2003). PV-positive interneurons do not express CB1R (Katona et al., 1999; Marsicano and Lutz, 1999; Bodor et al., 2005; Eggan et al., 2010), and their inhibitory synapses with pyramidal cells are exempted from cannabinoid-mediated retrograde signaling (Harkany et al., 2004; Galarreta et al., 2008).
In addition to the effects on GABAergic transmission, Fortin et al. demonstrated in juvenile animals that only glutamatergic inputs to layer 5 pyramidal neurons – the main output layer of the cortex – are subjected to cannabinoid-mediated suppression of excitation and not inhibition, while Layer 2/3 pyramidal neurons undergo CB1-mediated suppression of excitation and inhibition, indicating that cannabinoids only control the glutamatergic input onto L5 pyramidal cells but both the glutamatergic and GABAergic input to L2/3 pyramidal cells (Trettel and Levine, 2002; Fortin et al., 2004; Fortin and Levine, 2007). This is consistent with the preferential laminar distribution of CB1R and CCK-positive neurons/baskets in Layer 2/3 (Marsicano and Lutz, 1999; Egertova and Elphick, 2000), while deeper layers show CB1R positive varicosities suggestive of axons and terminals (Eggan et al., 2010). Therefore, the developmental decrease in CB1R expression observed in our study (Heng et al., 2011) would not only facilitate excitatory output from the PFC during maturation, but would also increase the cortico-cortical transmission of information received through Layer 2/3.
CB1R Signaling, GABAergic Interneurons, and Cortical Network Oscillations: A Working Model for Prefrontal Dysfunction Induced by Cannabinoid Exposure during Adolescence
Initial studies in the hippocampus indicated that acute administration of exogenous cannabinoids reduces cortical network oscillations in the theta and gamma band (Robbe et al., 2006; Hajos et al., 2008). Recently, Kucewicz et al. (2011) have demonstrated that systemic administration of a potent CB1R agonist (CP55,940) reduces gamma oscillations in the PFC as well as hippocampal theta oscillations, which correlates with reduced performance in a spatial working memory task. Notably, the reduced gamma oscillations observed in schizophrenia are thought to be an expression of reduced GABAergic interneuron transmission in cortical circuits (Uhlhaas and Singer, 2006, 2010). Interestingly, at the cellular level this decrease in gamma oscillations has been linked to reduced function of PV-positive interneurons (Lodge et al., 2009; Sohal et al., 2009; Volman et al., 2011) which lack CB1R. Based on the abundant expression of CB1R in CCK-positive GABAergic interneurons, it would suggest that a dysfunction of this population is at least partially responsible for abnormal oscillations caused by acute CB1R agonist administration. In support of this hypothesis, CCK-positive and PV-positive interneurons are anatomically and functionally coupled for sustaining fast oscillations in the hippocampus (Armstrong and Soltesz, 2012). Future studies will need to determine the role of CB1R signaling in the regulation of such interactions for maintaining gamma-range synchronous activity in the frontal cortex.
From the developmental perspective, studies from animal models indicate that a remodeling of local inhibitory circuits within the PFC during adolescence requires proper glutamatergic drive from the ventral hippocampus (see review by Tseng et al., 2009). In this regard, CB1Rs are also well positioned to exert presynaptic inhibition of excitatory synaptic transmission onto local GABAergic circuits in the PFC. Thus, repeated exposure to THC or excessive activation of CB1 receptors during adolescence could significantly reduce the level of glutamatergic drive needed for the functional maturation of prefrontal GABAergic interneurons. This would result in reduced inhibitory tone to cortical pyramidal cells and consequently decreased synchronization of the prefrontal network.
Based on the evidence presented above, cannabinoid signaling acts in at least three distinct synapses within the PFC to regulate both prefrontal input and output (Figure 1): (i) GABAergic synapses of CCK-positive interneurons onto pyramidal neurons, (ii) glutamatergic afferents (e.g., thalamus, hippocampus) onto pyramidal neurons, and (iii) glutamatergic afferents onto PV-positive interneurons. While any of these synapses could be the target of acute cannabinoid intoxication, only those synapses that are developmentally regulated could explain the long-lasting deficits seen with early adolescent-onset cannabis use/abuse. The data presently indicate that only the glutamatergic inputs (ii, iii), especially those contacting GABAergic PV-positive interneurons, are developmentally regulated. Thus, adolescent exposure to cannabis could disrupt the glutamatergic facilitation of PV-positive interneuron function and underlie the cognitive impairments seen in adulthood. It remains to be determined whether CCK-positive interneurons follow dramatic developmental changes similar to their PV-positive counterparts, and if so, how would this play in the generation of prefrontal synchrony.
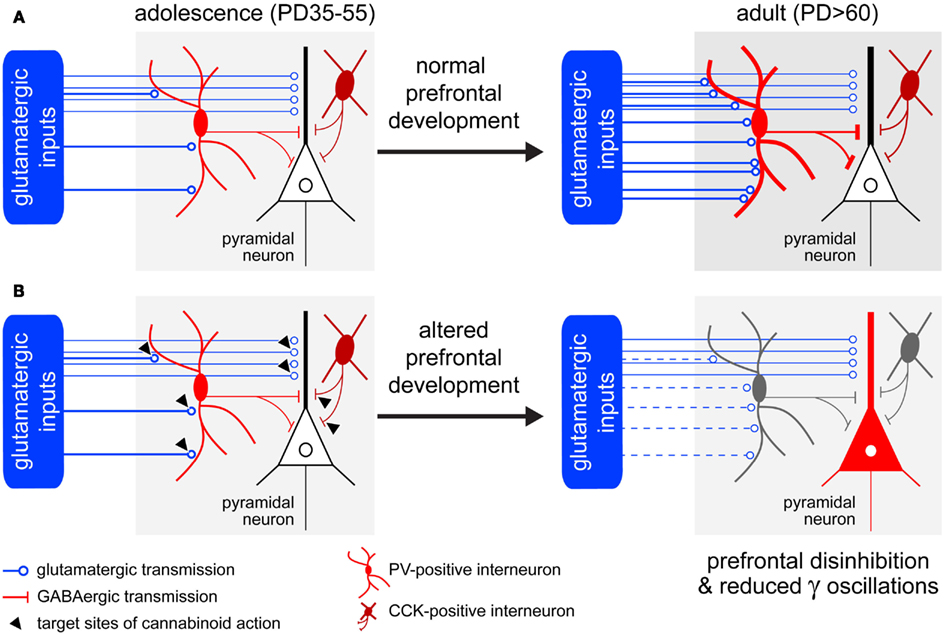
Figure 1. Working model of prefrontal dysfunction induced by cannabinoid exposure during adolescence. (A) During normal development, the adolescent facilitation of glutamatergic transmission onto prefrontal PV-positive interneurons is thought to determine the maturation local GABAergic transmission in the PFC (Tseng et al., 2009). (B) Based on the expression and function of CB1R in the cortex, the acute effect of cannabinoids on prefrontal dysfunction could occur by suppression of synaptic transmission at any of the following synapses (black triangles): (i) CCK-positive interneuron → pyramidal neurons, (ii) glutamatergic afferents → pyramidal neurons, and (iii) glutamatergic afferents → PV-positive interneurons. Regarding the long-lasting effects of adolescent exposure to cannabinoids, we hypothesize that cannabinoid-induced (e.g., Δ9-THC) suppression of glutamatergic drive onto PV-positive cells will prevent the normal maturation of these interneurons. If such interneuronal activity does not become enhanced during this transitional period, the control of PFC inhibition will be profoundly reduced, resulting in a lack of synchrony in the PFC and consequent impairment of cognitive functions.
All in all, these findings suggest that a downregulation of cortical GABAergic function underlies the effects of CB1R stimulation on cortical functioning. In turn, pyramidal neuron excitability will be increased, resulting in an overall disinhibition of the PFC and dysregulation of prefrontal-dependent processes. Overstimulation of the CB1R over sensitive developmental periods could alter the trajectory of any of the above-mentioned circuits leading to the deficits associated with schizoaffective disorders.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by Rosalind Franklin University, National Institutes of Health Grant R01-MH086507, and the Brain Research Foundation.
References
Abush, H., and Akirav, I. (2012). Short- and long-term cognitive effects of chronic cannabinoids administration in late-adolescence rats. PLoS ONE 7, e31731. doi:10.1371/journal.pone.0031731
Andersen, S. L. (2003). Trajectories of brain development: point of vulnerability or window of opportunity? Neurosci. Biobehav. Rev. 27, 3–18.
Armstrong, C., and Soltesz, I. (2012). Basket cell dichotomy in microcircuit function. J. Physiol. (Lond.) 590, 683–694.
Arseneault, L., Cannon, M., Poulton, R., Murray, R., Caspi, A., and Moffitt, T. E. (2002). Cannabis use in adolescence and risk for adult psychosis: longitudinal prospective study. BMJ 325, 1212–1213.
Arseneault, L., Cannon, M., Witton, J., and Murray, R. M. (2004). Causal association between cannabis and psychosis: examination of the evidence. Br. J. Psychiatry 184, 110–117.
Berrendero, F., Garcia-Gil, L., Hernandez, M. L., Romero, J., Cebeira, M., De Miguel, R., Ramos, J. A., and Fernandez-Ruiz, J. J. (1998). Localization of mRNA expression and activation of signal transduction mechanisms for cannabinoid receptor in rat brain during fetal development. Development 125, 3179–3188.
Bodor, A. L., Katona, I., Nyiri, G., Mackie, K., Ledent, C., Hajos, N., and Freund, T. F. (2005). Endocannabinoid signaling in rat somatosensory cortex: laminar differences and involvement of specific interneuron types. J. Neurosci. 25, 6845–6856.
Bortolato, M., Aru, G. N., Frau, R., Orru, M., Luckey, G. C., Boi, G., and Gessa, G. L. (2005). The CB receptor agonist WIN 55,212-2 fails to elicit disruption of prepulse inhibition of the startle in Sprague-Dawley rats. Psychopharmacology (Berl.) 177, 264–271.
Buzsaki, G., and Draguhn, A. (2004). Neuronal oscillations in cortical networks. Science 304, 1926–1929.
Casey, B. J., Giedd, J. N., and Thomas, K. M. (2000). Structural and functional brain development and its relation to cognitive development. Biol. Psychol. 54, 241–257.
Caspi, A., Moffitt, T. E., Cannon, M., Mcclay, J., Murray, R., Harrington, H., Taylor, A., Arseneault, L., Williams, B., Braithwaite, A., Poulton, R., and Craig, I. W. (2005). Moderation of the effect of adolescent-onset cannabis use on adult psychosis by a functional polymorphism in the catechol-O-methyltransferase gene: longitudinal evidence of a gene X environment interaction. Biol. Psychiatry 57, 1117–1127.
Chambers, R. A., Taylor, J. R., and Potenza, M. N. (2003). Developmental neurocircuitry of motivation in adolescence: a critical period of addiction vulnerability. Am. J. Psychiatry 160, 1041–1052.
Crews, F., He, J., and Hodge, C. (2007). Adolescent cortical development: a critical period of vulnerability for addiction. Pharmacol. Biochem. Behav. 86, 189–199.
D’Souza, D. C., Abi-Saab, W. M., Madonick, S., Forselius-Bielen, K., Doersch, A., Braley, G., Gueorguieva, R., Cooper, T. B., and Krystal, J. H. (2005). Delta-9-tetrahydrocannabinol effects in schizophrenia: implications for cognition, psychosis, and addiction. Biol. Psychiatry 57, 594–608.
D’Souza, D. C., Fridberg, D. J., Skosnik, P. D., Williams, A., Roach, B., Singh, N., Carbuto, M., Elander, J., Schnakenberg, A., Pittman, B., Sewell, R. A., Ranganathan, M., and Mathalon, D. (2012). Dose-related modulation of event-related potentials to novel and target stimuli by intravenous delta(9)-THC in humans. Neuropsychopharmacology. 37, doi:10.1038/npp.2012.8
D’Souza, D. C., Perry, E., Macdougall, L., Ammerman, Y., Cooper, T., Wu, Y. T., Braley, G., Gueorguieva, R., and Krystal, J. H. (2004). The psychotomimetic effects of intravenous delta-9-tetrahydrocannabinol in healthy individuals: implications for psychosis. Neuropsychopharmacology 29, 1558–1572.
Egertova, M., and Elphick, M. R. (2000). Localisation of cannabinoid receptors in the rat brain using antibodies to the intracellular C-terminal tail of CB. J. Comp. Neurol. 422, 159–171.
Eggan, S. M., Melchitzky, D. S., Sesack, S. R., Fish, K. N., and Lewis, D. A. (2010). Relationship of cannabinoid CB1 receptor and cholecystokinin immunoreactivity in monkey dorsolateral prefrontal cortex. Neuroscience 169, 1651–1661.
Ehrenreich, H., Rinn, T., Kunert, H. J., Moeller, M. R., Poser, W., Schilling, L., Gigerenzer, G., and Hoehe, M. R. (1999). Specific attentional dysfunction in adults following early start of cannabis use. Psychopharmacology (Berl.) 142, 295–301.
Elsohly, M. A., and Slade, D. (2005). Chemical constituents of marijuana: the complex mixture of natural cannabinoids. Life Sci. 78, 539–548.
Fontes, M. A., Bolla, K. I., Cunha, P. J., Almeida, P. P., Jungerman, F., Laranjeira, R. R., Bressan, R. A., and Lacerda, A. L. (2011). Cannabis use before age 15 and subsequent executive functioning. Br. J. Psychiatry 198, 442–447.
Fortin, D. A., and Levine, E. S. (2007). Differential effects of endocannabinoids on glutamatergic and GABAergic inputs to layer 5 pyramidal neurons. Cereb. Cortex 17, 163–174.
Fortin, D. A., Trettel, J., and Levine, E. S. (2004). Brief trains of action potentials enhance pyramidal neuron excitability via endocannabinoid-mediated suppression of inhibition. J. Neurophysiol. 92, 2105–2112.
Freund, T. F. (2003). Interneuron diversity series: rhythm and mood in perisomatic inhibition. Trends Neurosci. 26, 489–495.
Galarreta, M., Erdelyi, F., Szabo, G., and Hestrin, S. (2008). Cannabinoid sensitivity and synaptic properties of 2 GABAergic networks in the neocortex. Cereb. Cortex 18, 2296–2305.
Hajos, M., Hoffmann, W. E., and Kocsis, B. (2008). Activation of cannabinoid-1 receptors disrupts sensory gating and neuronal oscillation: relevance to schizophrenia. Biol. Psychiatry 63, 1075–1083.
Harkany, T., Guzman, M., Galve-Roperh, I., Berghuis, P., Devi, L. A., and Mackie, K. (2007). The emerging functions of endocannabinoid signaling during CNS development. Trends Pharmacol. Sci. 28, 83–92.
Harkany, T., Holmgren, C., Hartig, W., Qureshi, T., Chaudhry, F. A., Storm-Mathisen, J., Dobszay, M. B., Berghuis, P., Schulte, G., Sousa, K. M., Fremeau, R. T. Jr., Edwards, R. H., Mackie, K., Ernfors, P., and Zilberter, Y. (2004). Endocannabinoid-independent retrograde signaling at inhibitory synapses in layer 2/3 of neocortex: involvement of vesicular glutamate transporter 3. J. Neurosci. 24, 4978–4988.
Hart, C. L., Van Gorp, W., Haney, M., Foltin, R. W., and Fischman, M. W. (2001). Effects of acute smoked marijuana on complex cognitive performance. Neuropsychopharmacology 25, 757–765.
Heng, L., Beverley, J. A., Steiner, H., and Tseng, K. Y. (2011). Differential developmental trajectories for CB1 cannabinoid receptor expression in limbic/associative and sensorimotor cortical areas. Synapse 65, 278–286.
Henquet, C., Rosa, A., Krabbendam, L., Papiol, S., Fananas, L., Drukker, M., Ramaekers, J. G., and Van Os, J. (2006). An experimental study of catechol-o-methyltransferase Val158Met moderation of delta-9-tetrahydrocannabinol-induced effects on psychosis and cognition. Neuropsychopharmacology 31, 2748–2757.
Herkenham, M., Lynn, A. B., Johnson, M. R., Melvin, L. S., De Costa, B. R., and Rice, K. C. (1991). Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J. Neurosci. 11, 563–583.
Hestrin, S., and Galarreta, M. (2005). Electrical synapses define networks of neocortical GABAergic neurons. Trends Neurosci. 28, 304–309.
Hill, E. L., Gallopin, T., Ferezou, I., Cauli, B., Rossier, J., Schweitzer, P., and Lambolez, B. (2007). Functional CB1 receptors are broadly expressed in neocortical GABAergic and glutamatergic neurons. J. Neurophysiol. 97, 2580–2589.
Ilan, A. B., Smith, M. E., and Gevins, A. (2004). Effects of marijuana on neurophysiological signals of working and episodic memory. Psychopharmacology (Berl.) 176, 214–222.
Katona, I., and Freund, T. F. (2008). Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nat. Med. 14, 923–930.
Katona, I., Sperlagh, B., Sik, A., Kafalvi, A., Vizi, E. S., Mackie, K., and Freund, T. F. (1999). Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J. Neurosci. 19, 4544–4558.
Kreitzer, A. C., and Regehr, W. G. (2002). Retrograde signaling by endocannabinoids. Curr. Opin. Neurobiol. 12, 324–330.
Kucewicz, M. T., Tricklebank, M. D., Bogacz, R., and Jones, M. W. (2011). Dysfunctional prefrontal cortical network activity and interactions following cannabinoid receptor activation. J. Neurosci. 31, 15560–15568.
Lodge, D. J., Behrens, M. M., and Grace, A. A. (2009). A loss of parvalbumin-containing interneurons is associated with diminished oscillatory activity in an animal model of schizophrenia. J. Neurosci. 29, 2344–2354.
Marsicano, G., Goodenough, S., Monory, K., Hermann, H., Eder, M., Cannich, A., Azad, S. C., Cascio, M. G., Gutierrez, S. O., Van Der Stelt, M., Lopez-Rodriguez, M. L., Casanova, E., Schutz, G., Zieglgansberger, W., Di Marzo, V., Behl, C., and Lutz, B. (2003). CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 302, 84–88.
Marsicano, G., and Lutz, B. (1999). Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur. J. Neurosci. 11, 4213–4225.
Matsuda, L. A., Lolait, S. J., Brownstein, M. J., Young, A. C., and Bonner, T. I. (1990). Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346, 561–564.
Moore, T. H., Zammit, S., Lingford-Hughes, A., Barnes, T. R., Jones, P. B., Burke, M., and Lewis, G. (2007). Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet 370, 319–328.
O’Shea, M., Singh, M. E., Mcgregor, I. S., and Mallet, P. E. (2004). Chronic cannabinoid exposure produces lasting memory impairment and increased anxiety in adolescent but not adult rats. J. Psychopharmacol. (Oxford) 18, 502–508.
Pettit, D. A., Harrison, M. P., Olson, J. M., Spencer, R. F., and Cabral, G. A. (1998). Immunohistochemical localization of the neural cannabinoid receptor in rat brain. J. Neurosci. Res. 51, 391–402.
Pope, H. G. Jr., Gruber, A. J., Hudson, J. I., Cohane, G., Huestis, M. A., and Yurgelun-Todd, D. (2003). Early-onset cannabis use and cognitive deficits: what is the nature of the association? Drug Alcohol Depend. 69, 303–310.
Quinn, H. R., Matsumoto, I., Callaghan, P. D., Long, L. E., Arnold, J. C., Gunasekaran, N., Thompson, M. R., Dawson, B., Mallet, P. E., Kashem, M. A., Matsuda-Matsumoto, H., Iwazaki, T., and Mcgregor, I. S. (2008). Adolescent rats find repeated Delta(9)-THC less aversive than adult rats but display greater residual cognitive deficits and changes in hippocampal protein expression following exposure. Neuropsychopharmacology 33, 1113–1126.
Rissling, A. J., Makeig, S., Braff, D. L., and Light, G. A. (2010). Neurophysiologic markers of abnormal brain activity in schizophrenia. Curr. Psychiatry Rep. 12, 572–578.
Robbe, D., Montgomery, S. M., Thome, A., Rueda-Orozco, P. E., Mcnaughton, B. L., and Buzsaki, G. (2006). Cannabinoids reveal importance of spike timing coordination in hippocampal function. Nat. Neurosci. 9, 1526–1533.
Rodriguez de Fonseca, F., Ramos, J. A., Bonnin, A., and Fernandez-Ruiz, J. J. (1993). Presence of cannabinoid binding sites in the brain from early postnatal ages. Neuroreport 4, 135–138.
Schneider, M., and Koch, M. (2003). Chronic pubertal, but not adult chronic cannabinoid treatment impairs sensorimotor gating, recognition memory, and the performance in a progressive ratio task in adult rats. Neuropsychopharmacology 28, 1760–1769.
Schneider, M., Schomig, E., and Leweke, F. M. (2008). Acute and chronic cannabinoid treatment differentially affects recognition memory and social behavior in pubertal and adult rats. Addict. Biol. 13, 345–357.
Sewell, R. A., Ranganathan, M., and D’Souza, D. C. (2009). Cannabinoids and psychosis. Int. Rev. Psychiatry 21, 152–162.
Sohal, V. S., Zhang, F., Yizhar, O., and Deisseroth, K. (2009). Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 459, 698–702.
Spear, L. P. (2000). The adolescent brain and age-related behavioral manifestations. Neurosci. Biobehav. Rev. 24, 417–463.
Spencer, K. M., Nestor, P. G., Niznikiewicz, M. A., Salisbury, D. F., Shenton, M. E., and Mccarley, R. W. (2003). Abnormal neural synchrony in schizophrenia. J. Neurosci. 23, 7407–7411.
Trettel, J., and Levine, E. S. (2002). Cannabinoids depress inhibitory synaptic inputs received by layer 2/3 pyramidal neurons of the neocortex. J. Neurophysiol. 88, 534–539.
Trettel, J., and Levine, E. S. (2003). Endocannabinoids mediate rapid retrograde signaling at interneuron right-arrow pyramidal neuron synapses of the neocortex. J. Neurophysiol. 89, 2334–2338.
Tseng, K. Y., Chambers, R. A., and Lipska, B. K. (2009). The neonatal ventral hippocampal lesion as a heuristic neurodevelopmental model of schizophrenia. Behav. Brain Res. 204, 295–305.
Uhlhaas, P. J., and Singer, W. (2006). Neural synchrony in brain disorders: relevance for cognitive dysfunctions and pathophysiology. Neuron 52, 155–168.
Uhlhaas, P. J., and Singer, W. (2010). Abnormal neural oscillations and synchrony in schizophrenia. Nat. Rev. Neurosci. 11, 100–113.
Vogel, Z., Barg, J., Levy, R., Saya, D., Heldman, E., and Mechoulam, R. (1993). Anandamide, a brain endogenous compound, interacts specifically with cannabinoid receptors and inhibits adenylate cyclase. J. Neurochem. 61, 352–355.
Volman, V., Behrens, M. M., and Sejnowski, T. J. (2011). Downregulation of parvalbumin at cortical GABA synapses reduces network gamma oscillatory activity. J. Neurosci. 31, 18137–18148.
Wedzony, K., and Chocyk, A. (2009). Cannabinoid CB1 receptors in rat medial prefrontal cortex are colocalized with calbindin- but not parvalbumin- and calretinin-positive GABA-ergic neurons. Pharmacol. Rep. 61, 1000–1007.
Wilson, W., Mathew, R., Turkington, T., Hawk, T., Coleman, R. E., and Provenzale, J. (2000). Brain morphological changes and early marijuana use: a magnetic resonance and positron emission tomography study. J. Addict. Dis. 19, 1–22.
Keywords: prefrontal cortex, adolescence, cannabinoids, schizophrenia
Citation: Caballero A and Tseng KY (2012) Association of cannabis use during adolescence, prefrontal CB1 receptor signaling, and schizophrenia. Front. Pharmacol. 3:101. doi: 10.3389/fphar.2012.00101
Received: 09 April 2012; Accepted: 05 May 2012;
Published online: 28 May 2012.
Edited by:
Joseph F. Cheer, University of Maryland School of Medicine, USAReviewed by:
Steven R. Laviolette, University of Western Ontario, CanadaTommy Pattij, VU University Medical Center, Netherlands
Copyright: © 2012 Caballero and Tseng. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Kuei Y. Tseng, Department of Cellular and Molecular Pharmacology, RFUMS/The Chicago Medical School, 3333 Green Bay Road, North Chicago, IL 60064, USA. e-mail:a3VlaS15dWFuLnRzZW5nQHJvc2FsaW5kZnJhbmtsaW4uZWR1