

94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 21 May 2012
Sec. Pharmacogenetics and Pharmacogenomics
Volume 3 - 2012 | https://doi.org/10.3389/fphar.2012.00092
Tamoxifen is a first-line endocrine agent in the mechanism-based treatment of estrogen receptor positive (ER+) mammary carcinoma and applied to breast cancer patients all over the world. Endoxifen is a secondary and highly active metabolite of tamoxifen that is formed among others by the polymorphic cytochrome P450 2D6 (CYP2D6). It is widely accepted that CYP2D6 poor metabolizers exert a pronounced decrease in endoxifen steady-state plasma concentrations compared to CYP2D6 extensive metabolizers. Nevertheless, an in-depth understanding of the chain of cause and effect between CYP2D6 genotype, endoxifen steady-state plasma concentration, and subsequent tamoxifen treatment benefit still remains to be evolved. In this study, physiologically based pharmacokinetic (PBPK)-modeling was applied to mechanistically investigate the impact of CYP2D6 phenotype on endoxifen formation in female breast cancer patients undergoing tamoxifen therapy. A PBPK-model of tamoxifen and its pharmacologically important metabolites N-desmethyltamoxifen (NDM-TAM), 4-hydroxytamoxifen (4-OH-TAM), and endoxifen was developed and validated. This model is able to simulate the pharmacokinetics (PK) after single and repeated oral tamoxifen doses in female breast cancer patients in dependence of the CYP2D6 phenotype. A detailed model-based analysis of the mass balance offered support for a recent hypothesis stating a more prominent role for endoxifen formation from 4-OH-TAM. In the future this model provides a good basis to further investigate the linkage of PK, mode of action, and treatment outcome in dependence of factors such as phenotype, ethnicity, or co-treatment with CYP2D6 inhibitors.
Tamoxifen is a first-line endocrine agent in the adjuvant treatment of estrogen receptor positive (ER+)-mammary carcinoma and applied within a regular 5 year therapy regimen to patients all over the world (Fisher et al., 2001; Goetz et al., 2008). Although it represents a milestone in the mechanism-based treatment of ER+-breast cancer, there is evidence that not all patients benefit from therapy (Jordan, 2006, 2007). In this context, the question of whether or not cytochrome P450 2D6 (CYP2D6) polymorphisms significantly affect treatment outcome still remains controversially discussed (Hoskins et al., 2009; Seruga and Amir, 2010; Brauch et al., 2011; Rae, 2011).
An extensive hepatic metabolism of tamoxifen via CYP-catalyzed phase I reactions leads to the formation of primary and secondary metabolites (Coller et al., 2002; Desta et al., 2004). These are further glucuronidated or sulfated in phase II reactions catalyzed by uridine-5′-diphospho-glucuronosyltransferases (UGT) and sulfonyl-transferases (SULT; Nishiyama et al., 2002; Kaku et al., 2004; Sun et al., 2007; Figure 1). The involvement of the polymorphic CYP2D6 in the formation of the active secondary metabolite endoxifen leads to inter-individual variability in its plasma concentration due to for example single nucleotide polymorphisms affecting CYP2D6 enzyme activity (Zanger et al., 2001; Coller et al., 2002; Stearns et al., 2003; Johnson et al., 2004; Lim et al., 2006). It has been reported in the literature that CYP2D6 poor metabolizers (PM) show a pronounced decrease in endoxifen steady-state plasma concentrations in comparison to CYP2D6 extensive metabolizers (EM; Stearns et al., 2003; Jin et al., 2005; Gjerde et al., 2008; Irvin et al., 2011; Madlensky et al., 2011; Murdter et al., 2011). Most clinical trials reported up to now, either assessed the correlation between CYP2D6 genotype and endoxifen steady-state plasma concentration, or between CYP2D6 genotype and therapy outcome (Goetz et al., 2005, 2007; Borges et al., 2006; Bijl et al., 2009; Lammers et al., 2010). However, the linkage between CYP2D6 genotype, endoxifen steady-state plasma concentration, and subsequent treatment benefit needs further investigation to achieve an in-depth understanding of the chain of cause and effect (Lash et al., 2009; Fleeman et al., 2011).
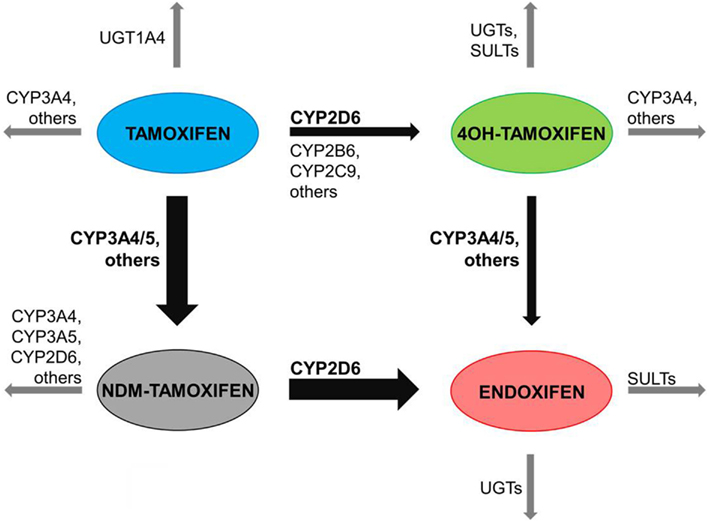
Figure 1. Main human biotransformation routes of tamoxifen. Tamoxifen is mainly metabolized via the cytochrome P450 isoenzyme (CYP) 3A4/5 (CYP3A4/5) – catalyzed N-demethylation to N-desmethyltamoxifen (NDM-tamoxifen) and subsequently 4-hydroxylated by the polymorphic CYP2D6 to endoxifen. In a minor route, tamoxifen is 4-hydroxylated by CYP2D6 and other CYP iso-forms to 4-hydroxytamoxifen (4-OH-tamoxifen) and consequently N-demethylated to endoxifen. Sequential phase I or II reactions of tamoxifen and metabolites occur via flavin-containing monooxygenases (FMO), CYPs, uridine-5′-diphospho-glucuronosyltransferases (UGT), or sulfonyl-transferases (SULT).
To better understand the mass balance of tamoxifen and the rate and extent of formation of its active metabolites, a physiologically based pharmacokinetic (PBPK)-model is desirable. PBPK-modeling comprises substance-specific as well as anatomical and physiological species-specific parameters within the mechanistic framework of a generic model structure (Willmann et al., 2003a). In short, every compartment represents a single organ which is defined by its physiological volume and is further divided into sub-compartments representing the intracellular, vascular (plasma and red blood cells), and interstitial space (Figure 2; sub-compartments not shown). In the context of such a PBPK-model, the influence of CYP2D6 enzyme activity on endoxifen pharmacokinetics (PK) can be studied (Zanger et al., 2001; Coller et al., 2002). The PBPK-model described in this study will further provide a starting point to investigate the pharmacogenomics of tamoxifen treatment response after integration of a mechanistic description of the mode of action of tamoxifen and its active metabolites.
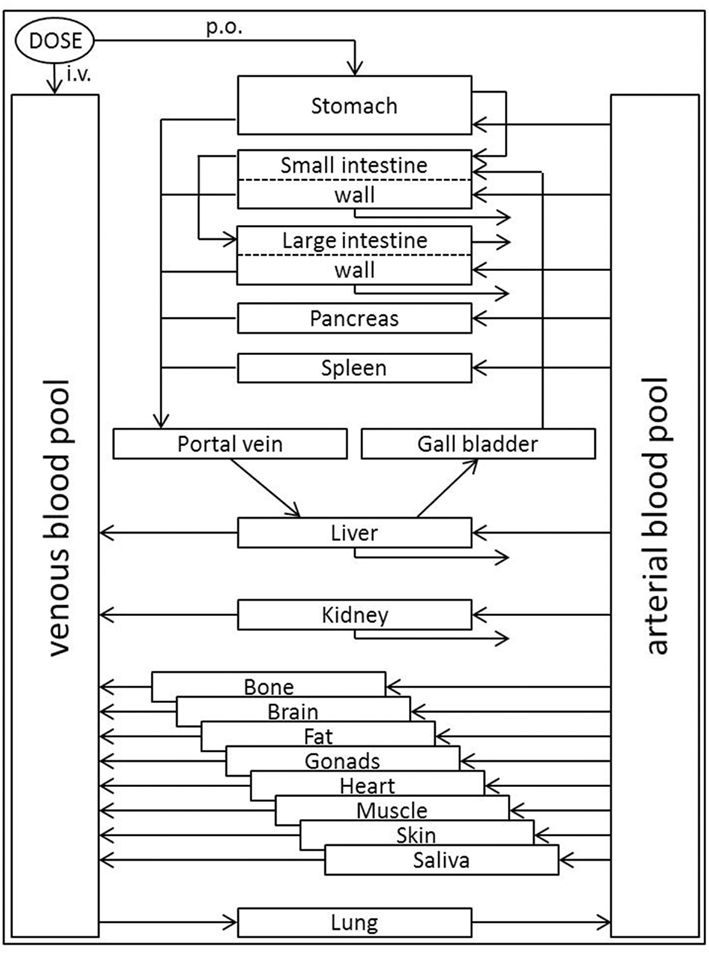
Figure 2. Generic structure of the whole-body PBPK-model. The model organism is built by compartments, each typically representing a single organ defined by its physiological volume. Organs are interconnected via respective blood flows which occur, except for pulmonary circulation, from the arterial blood pool to the venous blood pool thus accounting for inter-compartmental mass-transfer. Application of substances can be defined as intravenous (i.v.), per oral (p.o.), or into any desired compartment. In addition to clearance events in intestinal wall, liver, and kidney, metabolism processes can be implemented into any compartment. Transport processes that significantly influence a compound’s PK may be inserted between compartments.
To establish a PBPK-model for tamoxifen, plasma concentration–time data after intravenous (i.v.) administration of tamoxifen was highly desirable. Because – to the best of our knowledge – no human data following i.v. dosing of tamoxifen is publically available, a PBPK-model for the rat, for which i.v. data is available, was first developed in order to describe the disposition kinetics of tamoxifen. The rat PBPK-model was subsequently scaled to an average European female individual of CYP2D6 EM phenotype receiving oral tamoxifen. This female subject PBPK-model was in the next step extended by PBPK-models for N-desmethyltamoxifen (NDM-TAM), 4-hydroxytamoxifen (4-OH-TAM), and endoxifen. Finally, the CYP2D6 EM-specific PBPK-model was scaled to CYP2D6 PM using prior knowledge about phenotype-specific CYP2D6 enzyme activity.
The modus operandi during each PBPK-model development step was kept consistent and constant. A PBPK-model was built with reference data in terms of species- and substance-specific parameters (i.e., other generic parameters were kept on default values) and model predictions were compared to corresponding experimental data taken from the literature. If necessary, fine-tuning of parameters in a reasonable order of magnitude was performed to improve model adequacy as detailed in the results.
Physiologically based pharmacokinetic-models were developed and coupled by means of the computational systems biology software platform including PK-Sim® 4.2.4 and MoBi® 2.3.5 (Bayer Technology Services GmbH, Leverkusen, Germany)1. Subsequent population simulations were conducted using the MoBi® Toolbox for MATLAB® 2.2 (Bayer Technology Services GmbH, Leverkusen, Germany; see text footnote 1 with MATLAB® from The MathWorks, Inc., Natick, USA)2.
A detailed description of the development of a coupled parent-metabolite PBPK-model and population simulations comprising explicitly modeled metabolites formed via polymorphic enzymes was recently published (Eissing et al., 2011).
The physico-chemical parameters of tamoxifen that served as input parameters for PK-Sim® are summarized in Table 1. As can be seen in Figure 3, however, the volume of distribution of tamoxifen in rats was not adequately described by the set of physico-chemical parameters. After re-adjustment of the lipophilicity input parameter in PK-Sim® (clog MA = 4.23 instead of 3.43; Table 1), the plasma kinetics of tamoxifen in rat could be very well described. The available experimental data demonstrated an influence of gender on tamoxifen PK that could solely be attributed to a gender-specific clearance in the model (Figure 3; Table 4).
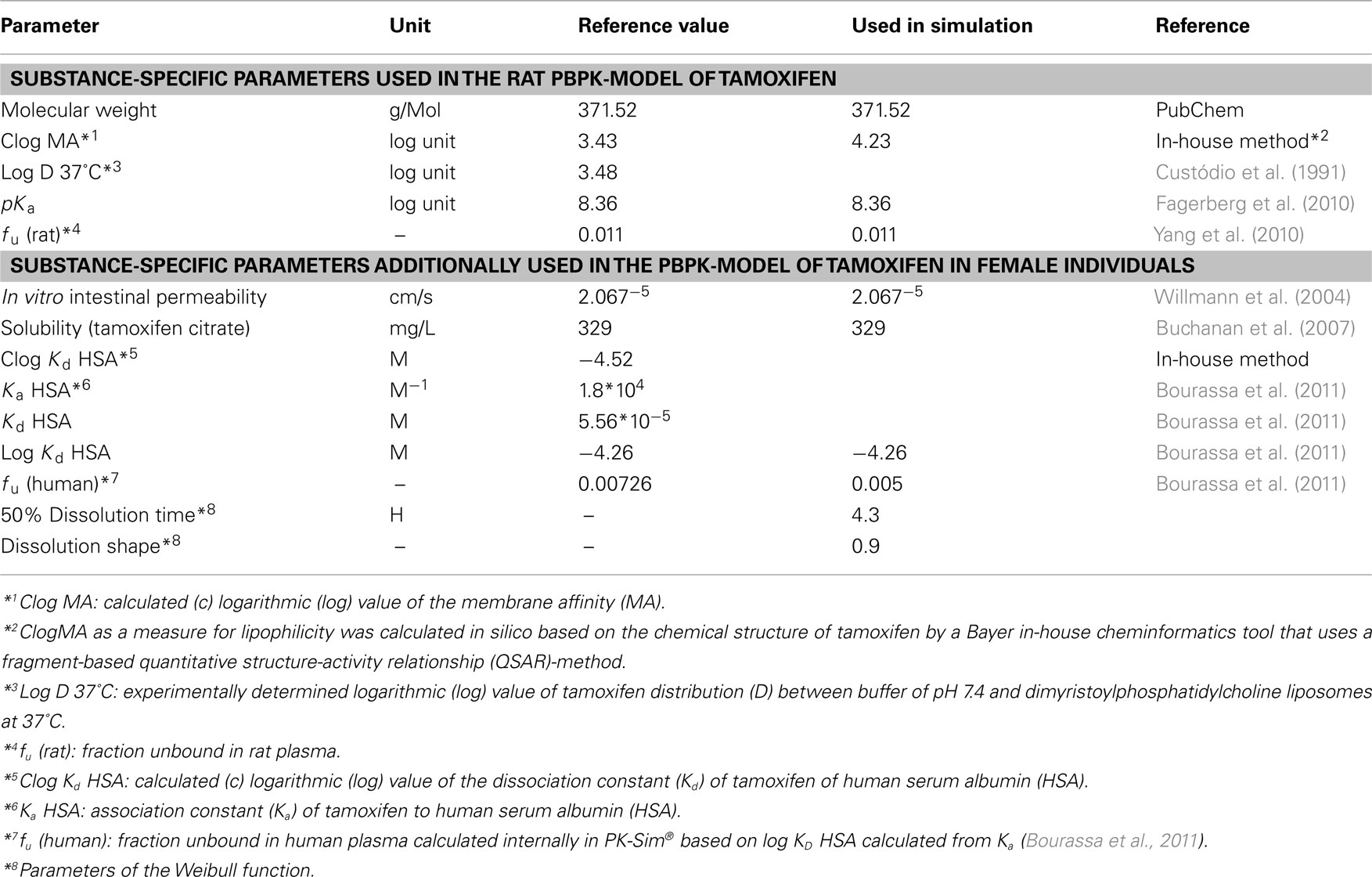
Table 1. Substance-specific physico-chemical parameters of tamoxifen used in the PBPK-model development.
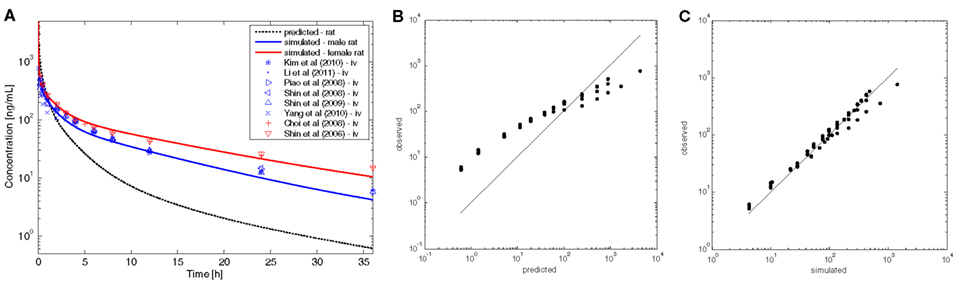
Figure 3. Results of the rat PBPK-model for intravenous tamoxifen. (A) Plasma concentration–time profiles following application of i.v. single dose (SD) of 2 mg/kg tamoxifen to rat. The gray dashed line represents the predicted profile, colored lines represent simulated profiles after refinement of lipophilicity. Blue and red symbols show experimental data taken from the literature. PBPK-models for male (blue) and female rat (red) differ exclusively in terms of the input parameter clearance. (B) Predicted versus observed data to show model predictivity. (C) Simulated versus observed data to show model improvement after refinement of lipophilicity.
In summary, the established PBPK-model is able to correctly describe tamoxifen disposition kinetics in rats on the basis of the adjusted lipophilicity parameter.
The rat PBPK-model was subsequently scaled to human physiology (i.e., to an average European female breast cancer patient; Table 2), while the substance-related physico-chemical input parameters remained unchanged. The fraction unbound of tamoxifen in plasma (fu) differs between rat and human and was, therefore, also adapted (Yang et al., 2010; Bourassa et al., 2011; Table 1).
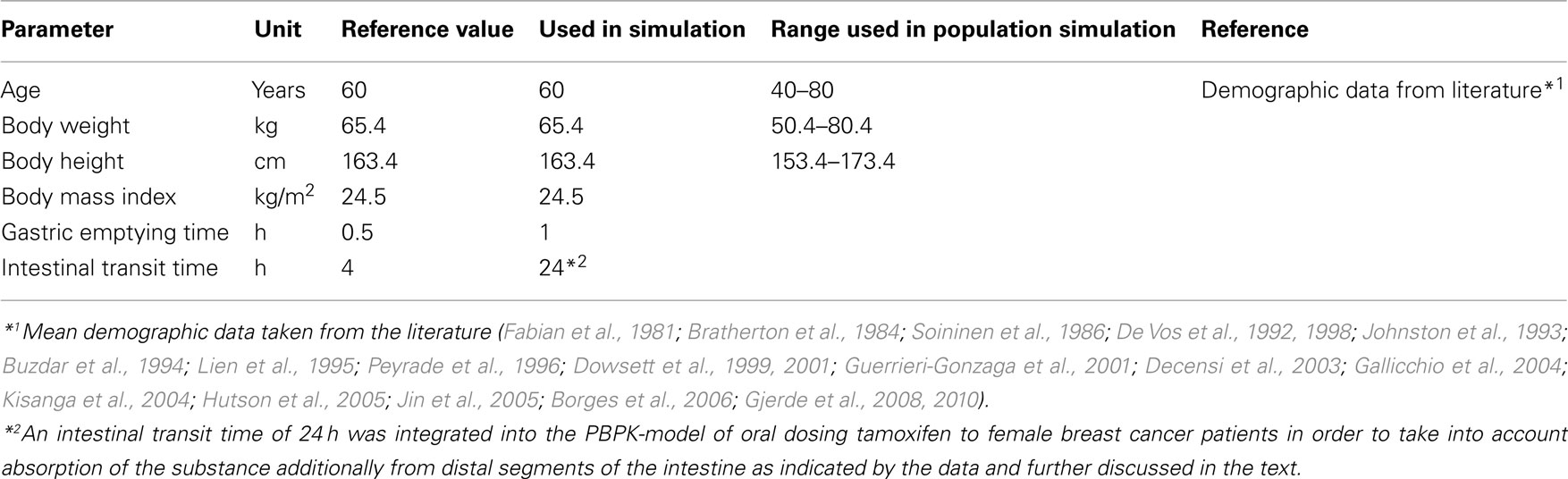
Table 2. Species-specific model parameters for the European female individuals.
According to the current dose regimen of tamoxifen for breast cancer patients, simulations were conducted for 20 mg tamoxifen per os (p.o.) and compared to literature data. These data were either taken from investigations following administration of 20 mg tamoxifen p.o. or data were normalized to a dose of 20 mg tamoxifen p.o. assuming linear PK in the applied dose range (Etienne et al., 1989; Fuchs et al., 1996; Stearns et al., 2003; Kisanga et al., 2004; Jin et al., 2005; Furlanut et al., 2007; Gjerde et al., 2008, 2010; Jaremko et al., 2010; Madlensky et al., 2011; Murdter et al., 2011).
As tamoxifen is a Biopharmaceutics Classification System (BCS) Class II compound and hence poorly soluble, an empirical dissolution function was integrated into the PBPK-model (Table 1) in order to account for solubility limitation of oral absorption (Tukker et al., 1986; Bergstrom et al., 2003; Kasim et al., 2003; Choi and Kang, 2008; Fagerberg et al., 2010). Furthermore, tamoxifen and its metabolites have been reported to undergo entero-hepatic circulation and subsequent re-absorption (Fromson et al., 1973a,b; Lien et al., 1988; Buckley and Goa, 1989; Kisanga et al., 2005). This phenomenon was described in the model assuming prolonged intestine transit time (ITT; Table 2). As the model was built with the continuous tube model of the gastrointestinal tract (GI-tract), which does not include the large intestine, this prolonged ITT additionally takes into account the probability that absorption processes may not only occur in proximal segments of the intestine but also in distal parts of the GI-tract (Willmann et al., 2003b, 2004).
In a first simulation, the elimination of tamoxifen in humans was predicted from the specific hepatic clearance (CLspec) of a female rat in combination with a scaling factor of 0.434 that was taken from reported differences of initial formation rates of tamoxifen metabolites between female rat liver microsomes and female human liver microsomes (Lim et al., 1994; Table 4). This approach turned out to slightly over-predict the clearance observed in humans (Figure 4). Therefore, the input parameter for the total hepatic clearance was re-adjusted in a second step in order to match the experimental data (Table 4; Etienne et al., 1989; Fuchs et al., 1996; Furlanut et al., 2007). In addition, a slight decrease of fu (Table 1) improved the agreement between the simulated and experimentally observed data (Figure 4). This tamoxifen PBPK-model for an average female breast cancer patient formed the basis for the coupled model that also includes the three metabolites.
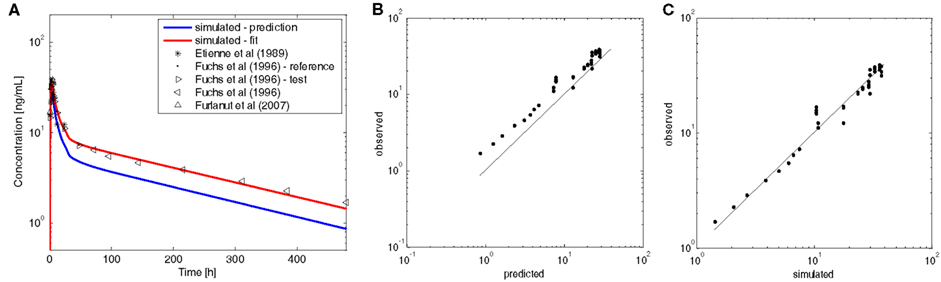
Figure 4. European female individual PBPK-model for oral tamoxifen versus experimental data. (A) Plasma concentration–time profiles of tamoxifen following administration of a single oral dose of 20 mg tamoxifen to an average European female individual. The blue line represents the predicted profile; the red line shows simulation results after refinement of the specific hepatic clearance (CLspec) and fu. Black symbols indicate experimental data taken from the literature. (B) Predicted versus observed data to show model predictivity. (C) Simulated versus observed data to show model improvement after adjustment of CLspec and fu.
At first, PBPK-models for the three metabolites (NDM-TAM, 4-OH-TAM, and endoxifen) in the average European female individual (Table 2) had to be established according to the PBPK-model for tamoxifen. As – to the best of our knowledge, no plasma concentration–time data following i.v. dosing of the three metabolites are available, neither in rats nor in humans, the input parameters were taken from the literature and integrated into the respective PBPK-models. Lipophilicity values predicted with the in-house method were scaled by the same difference of reference versus simulated as described for tamoxifen. Likewise, fu values were scaled by the same factor of reference versus simulated as for tamoxifen (Tables 1 and 3). For endoxifen, plasma concentration–time data following single oral administration of endoxifen were recently determined in healthy male volunteers. In addition, the total body clearance of endoxifen was reported by the authors, which was taken as reference input parameter to describe endoxifen clearance (Table 5; Ahmad et al., 2010). These data were used to optimize the lipophilicity value of endoxifen in the same way as described above for tamoxifen (data not shown). It was found that a lipophilicity value of 3.8 yields the best agreement with the reported data (Table 3). Hence, the value was used for all further simulations of endoxifen.
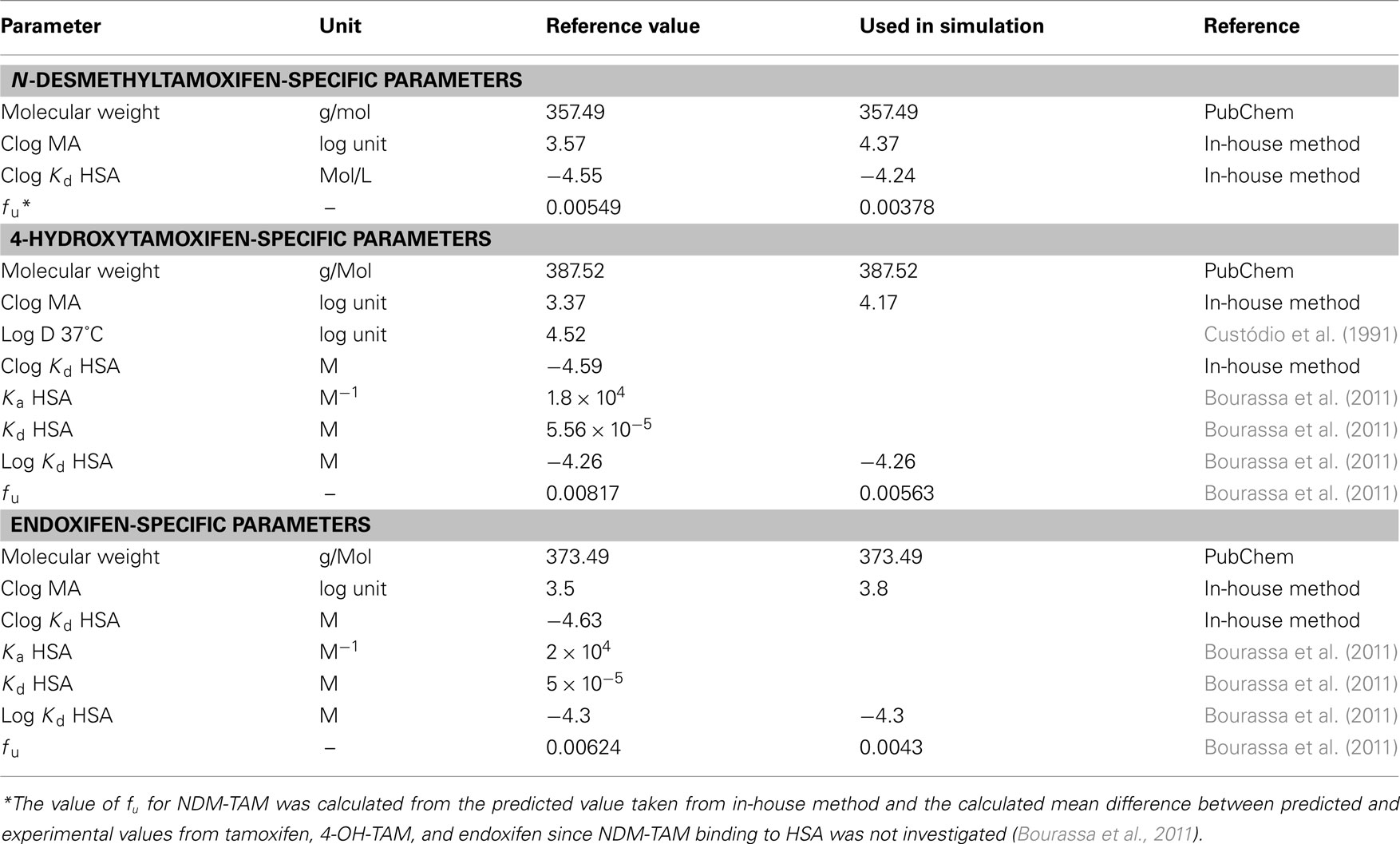
Table 3. Substance-specific parameters for PBPK-model development of NDM-TAM, 4-OH-TAM, and endoxifen in European female individuals.
To couple the tamoxifen model with the models for NDM-TAM, 4-OH-TAM, and endoxifen, first the total hepatic clearance of tamoxifen was split into several processes. Each process serves as a source for the metabolites, which are consequently formed in the intracellular space of the liver. All hepatic clearance processes were implemented as Michaelis–Menten kinetic processes (Table 4). Vmax-values derived from Desta et al. (2004) for implemented biotransformation routes of tamoxifen via CYPs and Vmax-values derived from Kaku et al. (2004) for glucuronidation of tamoxifen were scaled by two specific factors (10 and 2, respectively; Table 4) to be consistent with the previously determined total hepatic CLspec of tamoxifen in human female liver and observed data. The biotransformation steps thus provide the link between the two PBPK-models of NDM-TAM and 4-OH-TAM to the parent drug PBPK-model (Tables 4 and 5). In terms of NDM-TAM and 4-OH-TAM clearance processes, Michaelis–Menten kinetics from human liver microsome assays were integrated following the same logic (Nishiyama et al., 2002; Desta et al., 2004), thereby establishing a link to the endoxifen PBPK-model. All implemented clearance processes are deemed to best represent a CYP2D6 EM phenotype (Figure 1, Tables 4 and 5).
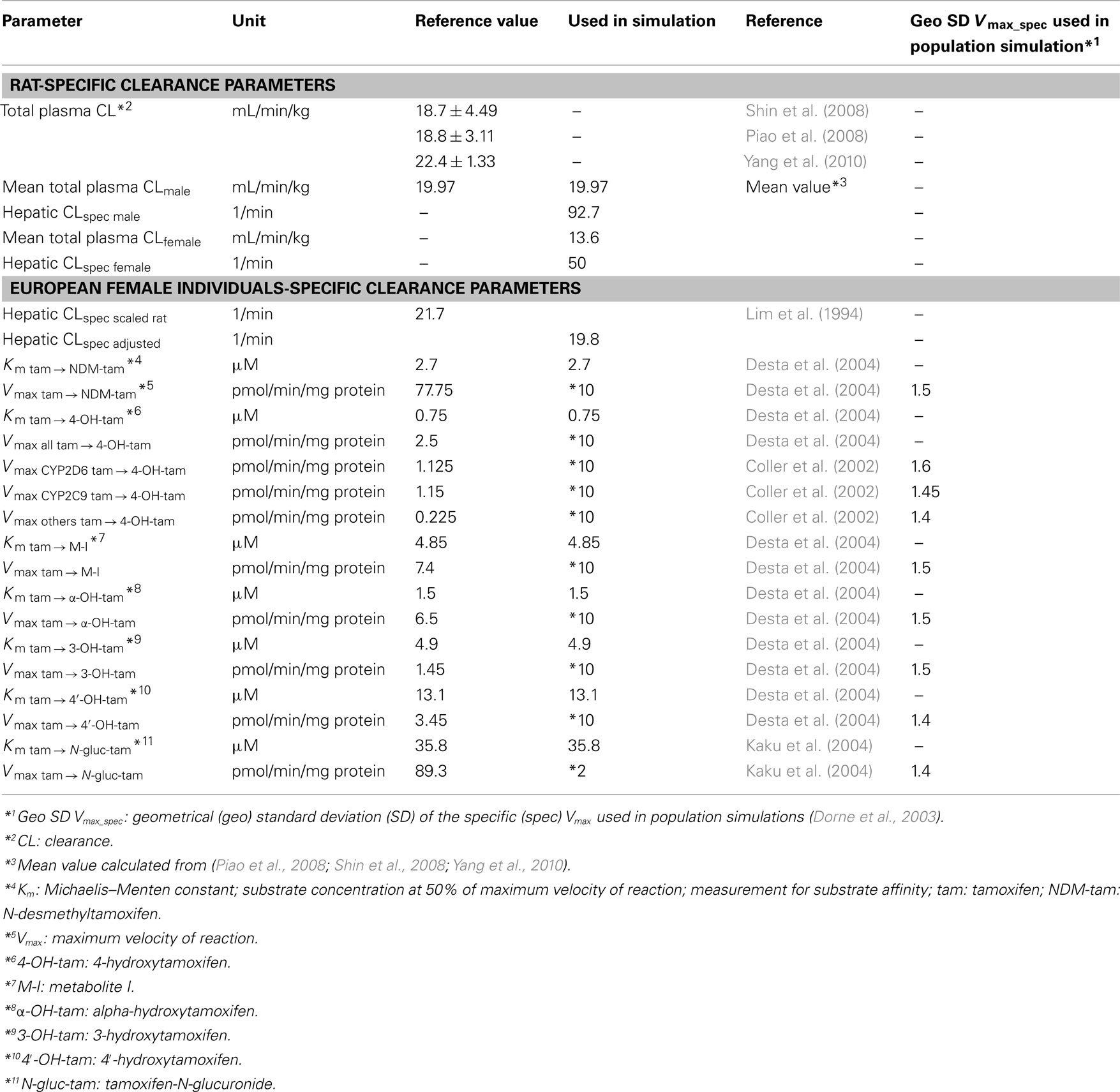
Table 4. Parameters describing tamoxifen clearance in rat and European female individuals.
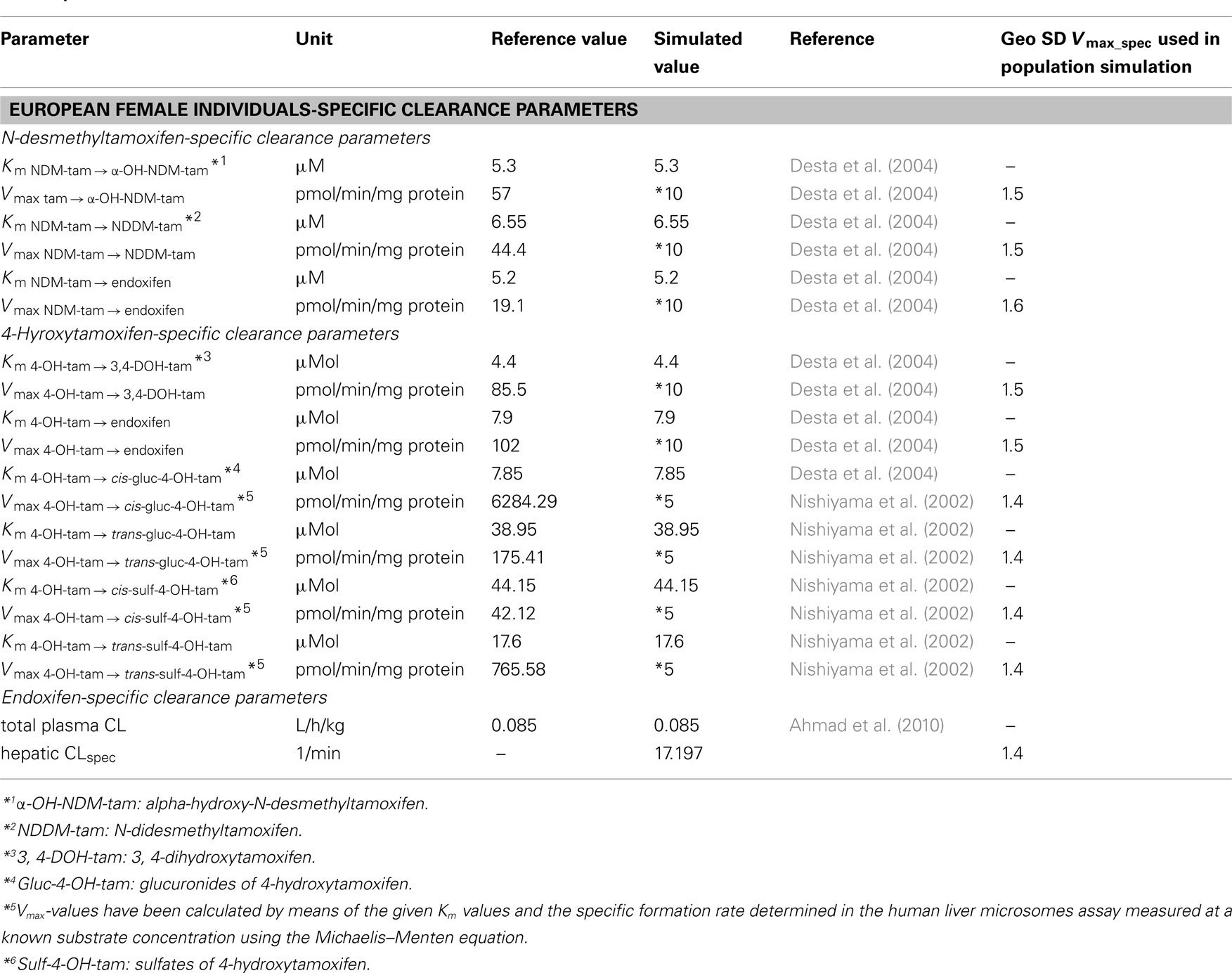
Table 5. Parameters describing the clearance of NDM-TAM, 4-OH-TAM, and endoxifen in European female individuals.
In vitro data indicate that about 45% of total 4-OH-TAM are formed via CYP2D6 out of tamoxifen and that CYP2C9 catalyzes the formation of 46% of total 4-OH-TAM, whereas the remaining 9% of total 4-OH-TAM is formed out of tamoxifen via other CYP iso-enzymes (Coller et al., 2002). This mass balance of 4-OH-TAM-formation out of tamoxifen was integrated into the coupled PBPK-model (Table 4).
The coupled PBPK-model was subsequently used to simulate the plasma concentration–time profiles of tamoxifen and the metabolites included over a period of 12 months with a once daily administration of 20 mg tamoxifen. The simulation results were compared to observed data (Figure 5; Etienne et al., 1989; Fuchs et al., 1996; Stearns et al., 2003; Jin et al., 2005; Furlanut et al., 2007; Gjerde et al., 2008, 2010; Jaremko et al., 2010; Irvin et al., 2011; Madlensky et al., 2011; Murdter et al., 2011). Data collected later than 12 months were used as if they were collected after 12 months assuming that steady-state kinetics are reached by 1 year (Buzdar et al., 1994). The simulated tamoxifen plasma concentration was well in line with the observed data (Figure 5; solid blue line). However, the profiles of the metabolites (Figure 5; dashed lines) were still over-predicted due to the not yet scaled metabolites’ clearance processes (Table 5).
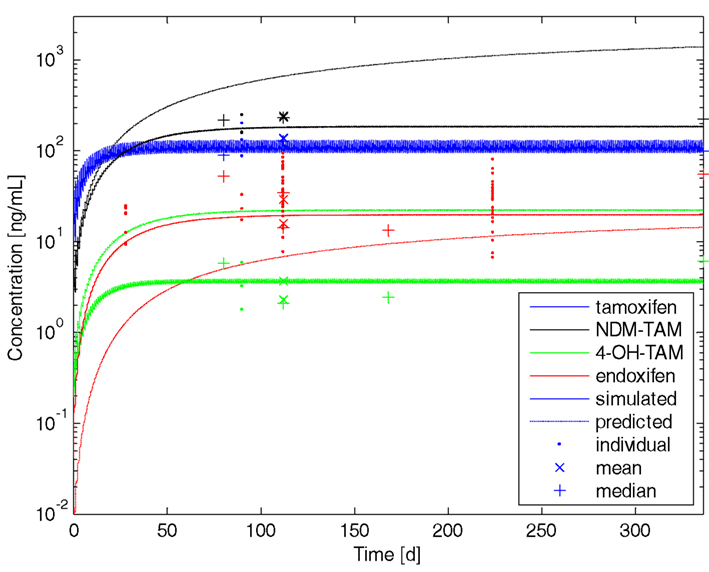
Figure 5. Tamoxifen and metabolites steady-state kinetics in an average European female individual of CYP2D6 EM phenotype. Resulting plasma concentration–time profiles of tamoxifen (blue), N-desmethyltamoxifen (NDM-TAM; black), 4-hydroxytamoxifen (4-OH-TAM; green), and endoxifen (red) following once daily administration of 20 mg tamoxifen p.o. to an average European female individual of CYP2D6 extensive metabolizer phenotype over a period of 12 months. Dashed lines represent predicted profiles of metabolites, solid lines show simulated profiles after adjustment of Vmax-values. Dots represent individual data, stars represent mean data, and plus’ represent median data taken from the literature.
Consistent with the PBPK-model development of the parent drug, clearance data taken from Desta et al. (2004) were subsequently scaled by a factor of 10 (Tables 4 and 5). Data taken from Nishiyama et al. (2002) was adjusted in terms of Vmax by a factor of 5 (Table 5). No further refinement of endoxifen clearance proved necessary (Table 5).
The final comparison of simulated and experimental data reveals that the PBPK-model describes the formation and maintenance of tamoxifen and metabolites steady-state kinetics (Figure 5; solid lines) very well. It is noticeable from the observed data that tamoxifen and its metabolites show a high inter-individual variability in plasma concentrations (Langan-Fahey et al., 1990; Peyrade et al., 1996). To address the inter-individual variability, PBPK-population simulations were set up for both, the CYP2D6 extensive metabolizer and the poor metabolizer.
The coupled PBPK-model of tamoxifen, NDM-TAM, 4-OH-TAM, and endoxifen representing an average European female subject of CYP2D6 EM phenotype was adapted to a CYP2D6 PM phenotype by integrating known CYP2D6 PM phenotype-specific enzyme activities (Coller et al., 2002).
In the CYP2D6 EM phenotype-specific coupled PBPK-model, it is assumed that CYP2D6 catalyzes the formation of 4-OH-TAM out of tamoxifen to an extent of 45% and is exclusively responsible for endoxifen formation out of NDM-TAM (Coller et al., 2002; Desta et al., 2004; Murdter et al., 2011; Figure 1, Tables 4 and 5). The two CYP2D6 EM phenotype-specific CYP2D6 Vmax-values were thus scaled by a factor of 0.015 in order to represent the negligible enzyme activity of CYP2D6 in female individuals of PM phenotype (Coller et al., 2002). All other parameters of the CYP2D6 EM phenotype-specific PBPK-model parameterization were kept constant. Simulations of 20 mg tamoxifen daily for a period of 12 months were performed as described above.
The implemented mass balance of metabolite formation resulting from the enzyme kinetics reported by several groups assumes that approximately 10% of endoxifen is formed via 4-OH-TAM (Coller et al., 2002; Nishiyama et al., 2002; Desta et al., 2004; Kaku et al., 2004). Recently, one group postulated that 20–30% of total endoxifen is formed via 4-OH-TAM (Murdter et al., 2011). In order to evaluate this hypothesis, the coupled CYP2D6 EM phenotype-specific PBPK-model was modified to alternatively reflect the formation of 20, 30, and 40%, respectively, of total endoxifen from 4-OH-TAM. Each model variant was scaled to the CYP2D6 PM phenotype by the procedure described above (see also Table 6).
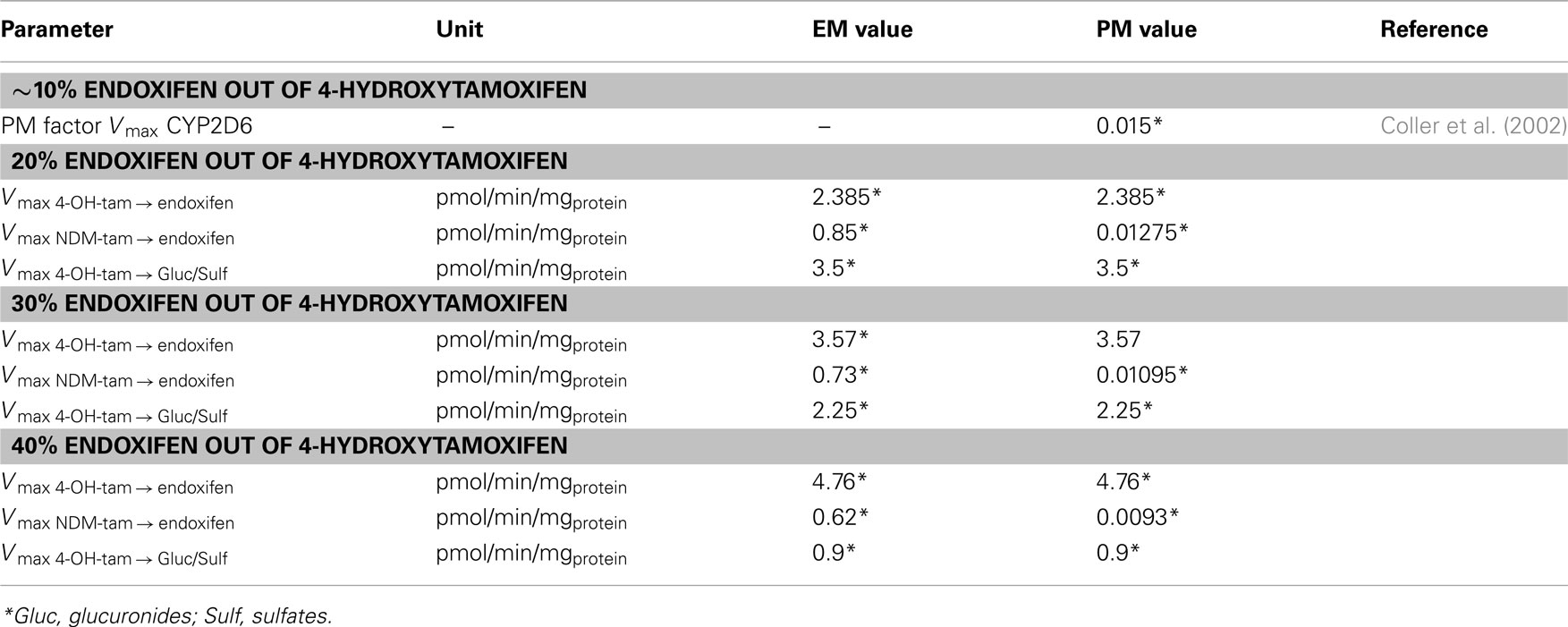
Table 6. Modification of the coupled CYP2D6 EM phenotype-specific PBPK-model parameterization to alternatively reflect the formation of 20–40% of total endoxifen formation out of 4-OH-TAM and subsequent scale factors for the CYP2D6 PM phenotype-specific PBPK-model parameterization.
Based on a few simple input parameters (ethnicity, gender, age, weight, and height; Table 2), generic populations of individuals were created taking into account inter-individual variability in physiological parameters such as organ volumes and blood flow rates (Willmann et al., 2007). In addition, variability can be superimposed on non-generic parameters. Tables 1, 4, and 5 present the chosen settings for generic and non-generic parameters that were applied to create 1000 female individuals. These can be used to re-parameterize each of the different coupled PBPK-models of tamoxifen, NDM-TAM, 4-OH-TAM, and endoxifen for an average European female subject of CYP2D6 EM phenotype accounting for ∼10–40% of endoxifen formation out of 4-OH-TAM, as well as the PM variants thereof (Table 6). Daily dosing of 20 mg tamoxifen for a period of 12 months was simulated in each individual.
Population simulation results of the four CYP2D6 EM European female individual populations and the four scaled CYP2D6 PM European female individual populations were compared to reported data from female patients of known CYP2D6 phenotype (Figure 6; Gjerde et al., 2008, 2010; Irvin et al., 2011; Madlensky et al., 2011; Murdter et al., 2011).
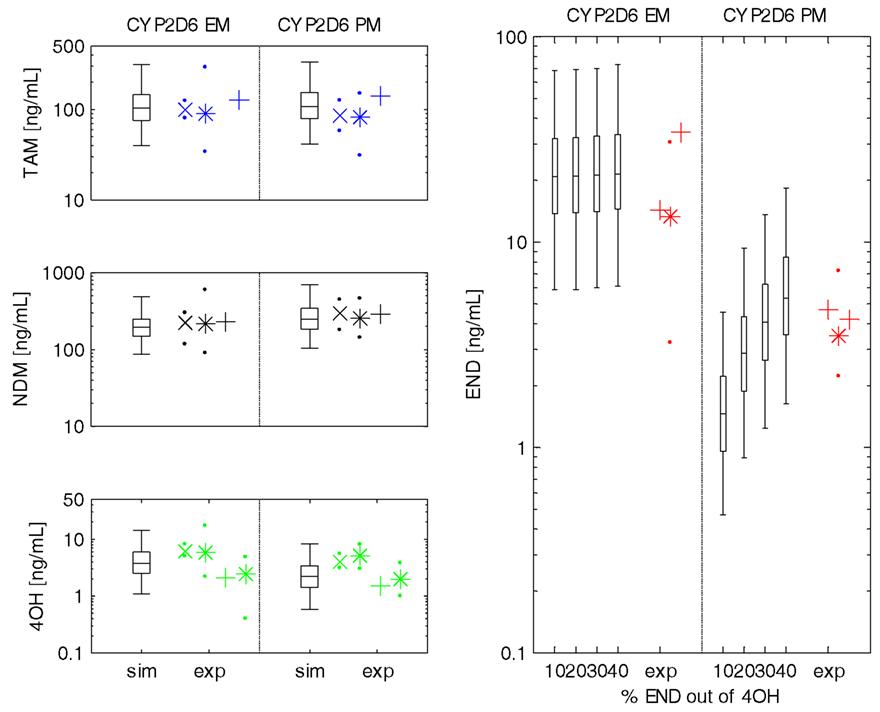
Figure 6. Steady-state plasma concentrations of tamoxifen, NDM-TAM, 4-OH-TAM, and endoxifen in populations of CYP2D6 EM and PM phenotype. Steady-state plasma concentrations for European female subject populations (N = 1000) of CYP2D6 extensive metabolizer phenotype (left) and CYP2D6 poor metabolizer phenotype (right). Box–Whisker-plots indicate the percentiles 5, 25, 50, 75, and 95 of the population simulations considering ∼10–40% of total endoxifen being formed out of 4-hydroxytamoxifen (x-axis). Symbols represent experimental data used for model validation (from left to right: Gjerde et al., 2010, median + 25th/75th percentile (x-symbol + dots); Gjerde et al., 2008, median + range (*-symbol + dots); Madlensky et al., 2011, median (+-symbol); Murdter et al., 2011, median + range (*-symbol + dots); Irvin et al., 2011; median (+-symbol)). Abbreviations in y-axis-label: TAM, tamoxifen; NDM, N-desmethyltamoxifen; 4OH, 4-hydroxytamoxifen; END, endoxifen. Abbreviations in x-axis-label: sim, simulated results; exp, experimental data taken from literature.
Simulation results of the four CYP2D6 EM individual populations show that the model parameterizations are able to account for the high inter-individual variability in PK of tamoxifen and match all reported median plasma concentration data of NDM-TAM, 4-OH-TAM, and endoxifen. There is no obvious difference in simulation results between the four assumed mass balances of endoxifen formation out of 4-OH-TAM (Figure 6; not all data shown).
Predictions following the four population simulations of tamoxifen and metabolites PK in 1000 European female subjects of CYP2D6 PM phenotype sufficiently describe the corresponding plasma concentration data of tamoxifen, NDM-TAM, and 4-OH-TAM in all four assumed mass balances (Figure 6; left column, not all data shown). Endoxifen plasma concentration data is best reflected in the scaled PBPK-model of CYP2D6 PM phenotype from the CYP2D6 EM phenotype-specific PBPK-model assuming 30% endoxifen formation out of 4-OH-TAM (Figure 6; right column) thereby supporting the hypothesis postulated by Murdter et al. (2011).
Tamoxifen was approved by the U.S. food and drug administration (FDA) for the treatment of metastatic breast cancer in 1977 and its indication has been expanded gradually during the following years (Desta et al., 2004). Although millions of women worldwide have been treated with the drug, there is still a lot to be learnt about tamoxifen.
First, tamoxifen has neither been investigated in terms of optimal biological dose-finding nor has its PK been determined following an i.v. administration to humans, leaving basic PK parameters such as volume of distribution, total plasma clearance, and absolute bioavailability after oral administration undetermined (Decensi et al., 2003). Second, biotransformation routes of tamoxifen are complex and lead to the formation of a number of active metabolites that significantly contribute to its anti-tumoral activity (Murdter et al., 2011).
Several polymorphic enzymes are involved in tamoxifen biotransformation, most importantly CYP2D6 that determines endoxifen exposure in tamoxifen-treated women. Nevertheless, after stratifying for CYP2D6 polymorphisms, endoxifen steady-state plasma concentration remains variable. Murdter et al. demonstrated that CYP2D6 genotype explains about 38% of endoxifen variability, which is well in line with results published by another group (Madlensky et al., 2011; Murdter et al., 2011). This leads to the assumption that there could be further influencing factors such as CYP2C9/2C19 or UGT polymorphisms that may also have a significant impact on tamoxifen PK and biotransformation to endoxifen (Blevins-Primeau et al., 2009; Ruiter et al., 2010; Murdter et al., 2011; Van Schaik et al., 2011).
Besides the long use of tamoxifen, the consequences of the high inter-individual variability in the metabolite pattern on treatment outcome, in particular in CYP2D6 PM and IM individuals, is still a subject of vivid discussions.
The presented coupled PBPK-model of tamoxifen and its most important metabolites provides a useful starting point to investigate the influence of certain covariates on tamoxifen PK and biotransformation to endoxifen such as CYP2D6 phenotype, ethnicity, or co-administration of other drugs.
In general, it is preferred to build at first a PBPK-model for i.v. administration in order to simulate the drug’s distribution kinetic in the absence of absorption. This was not possible for tamoxifen, because i.v. PK data of this drug has – to the best of our knowledge – never been measured in humans. Therefore, a rat i.v. model has been developed based on preclinical PK data (Shin et al., 2006, 2008; Choi and Kang, 2008; Piao et al., 2008; Shin and Choi, 2009; Kim et al., 2010; Yang et al., 2010; Li et al., 2011). After determination of an appropriate lipophilicity input parameter, this model was scaled to human individuals by exchanging the species-specific physiological parameters while the species-independent physico-chemical parameters of tamoxifen remained unchanged. Based on this i.v. scaling approach, the volume of distribution of tamoxifen in an average adult female individual weighing 65 kg was calculated to be 34 L/kg which is largely in accordance with the apparent terminal distribution volume estimated by Lien et al. (1989; 52–61 L/kg) that was obtained after repeated oral tamoxifen dosing.
In the next modeling step, the route of drug delivery was changed to p.o. administration. After adjustment of model parameters that affect the dissolution rate, the absorption kinetics could be very well described. The necessity for these adjustments arose from the fact that tamoxifen is a poorly soluble (BCS Class II) drug and thus, dissolution is very likely to determine the rate of absorption. Moreover, this adjustment has only a minor influence on the peak-trough ratios after repeated dosing, and does not drastically affect the average steady-state tamoxifen levels, because the bioavailability was not affected by the changes. Based on the presented model, oral absorption of tamoxifen is almost complete (97%). Together with a small first pass effect in the liver (3%), the simulated oral bioavailability of tamoxifen amounts to 94% (data not shown).
Overall, the resulting PBPK-model of tamoxifen SD in an average European female patient was able to describe the observed plasma concentration–time profiles (Figure 4). The slight adaptation of CLspec (Table 4) is supported by the fact that the resulting total plasma clearance calculated with the model confers to a reported total plasma clearance reported in the literature as the model assumes a fraction absorbed of 0.97 (Fuchs et al., 1996). So far, the model has only demonstrated its applicability in European breast cancer patients and is – strictly speaking – limited to this ethnicity. A translation to other ethnicities, however, should be easily achievable on the basis of prior knowledge about anthropometric, physiological, and biochemical differences that are included in the databases of PK-Sim®.
Unfortunately, there were only few published plasma concentration–time profiles of SD tamoxifen in female breast cancer patients available. Data is most often presented in the form of steady-state plasma concentration in patients receiving tamoxifen. Consequently, the dosing schedule in the PBPK-model was adapted to a repeated daily dosing of 20 mg tamoxifen in an average European female patient. Using prior in vitro information about tamoxifen’s biokinetics in the form of Michaelis–Menten constants, the total clearance was split into individual processes that formed the sources of metabolites. It proved to be necessary to scale reported Vmax-values of integrated CYP and UGT metabolism of tamoxifen by scaling factors, respectively, in order to reach the dimension of the previously determined hepatic CLspec and match observed plasma concentration–time profiles (Tables 4 and 5; Figures 4 and 5). Besides the fact that the scaled Vmax-values led to a hepatic CLspec that corresponds to the determined hepatic CLspec of the parent drug, adjusting of absolute Vmax-levels may be justified in terms of varying enzyme expression and activity. Human liver microsome assays performed to investigate the formation of tamoxifen’s primary metabolites by Desta et al. (2004) already exerted a difference in CYP3A enzyme activity by a factor of approximately 10. Urinary excretion of tamoxifen was reported to be very low and hence was not included into the PBPK-model (Soininen et al., 1986).
After implementation of the individual clearance processes, the parent drug PBPK-model was extended by PBPK-models for its two most important primary metabolites and the sequential PBPK-model of endoxifen, which was performed via linkage of the PBPK-models through specific enzyme activities of hepatic intracellular metabolism (Figure 2; Coller et al., 2002; Nishiyama et al., 2002; Desta et al., 2004; Kaku et al., 2004). Simulation of 12 months daily dosing tamoxifen with the coupled PBPK-model showed a good match of resulting metabolites plasma concentrations with experimental data after scaling Vmax-values in a similar approach as in the tamoxifen PBPK-model (Tables 4 and 5; Figure 5).
Thus, we have established a coupled PBPK-model that is able to reflect the mass balance based on reported enzyme kinetics and that links formation of endoxifen to preceding 4-hydroxylation or N-demethylation of tamoxifen (Desta et al., 2004). According to this model, the formation of 45% of total 4-OH-TAM out of tamoxifen proceeds via the polymorphic CYP2D6. In case of endoxifen formation from NDM-TAM, CYP2D6 is exclusively responsible for biotransformation. Performed simulations of tamoxifen, NDM-TAM, 4-OH-TAM, and endoxifen steady-state plasma concentration development during a period of 12 months of daily dosing 20 mg tamoxifen led to a good description of tamoxifen, NDM-TAM, and 4-OH-TAM in each the two phenotype populations. Endoxifen steady-state plasma concentration was slightly underestimated only in the CYP2D6 PM phenotype population (Figure 6).
The quantitative contribution of the two primary tamoxifen metabolites to the formation of endoxifen has not been investigated in vivo so far. The mass balance integrated into the EM phenotype-specific PBPK-model of tamoxifen metabolism accounts for ∼10% of total endoxifen formation out of 4-OH-TAM and is based on several published data (Coller et al., 2002; Nishiyama et al., 2002; Desta et al., 2004; Kaku et al., 2004). However, Murdter et al. (2011) postulated in their very recently published investigation of tamoxifen metabolism in breast cancer patients that approximately 20–30% of total endoxifen may be formed out of 4-OH-TAM.
Hence, the baseline PBPK-model of tamoxifen metabolism in a CYP2D6 EM phenotype was adjusted to reflect 20, 30, and 40% of total endoxifen formation out of 4-OH-TAM. All four EM phenotype-specific PBPK-model parameterizations were subsequently scaled to PM phenotype by the factor derived from Coller et al. (2002) and compared to observed data. The resulting model parameterizations revealed that an assumption of 30% of total endoxifen formed out of 4-OH-TAM provides best prediction of endoxifen steady-state plasma concentration in the CYP2D6 PM phenotype, thus supporting the hypothesis (Murdter et al., 2011). Further confirmation in vitro and in vivo may be promising.
The role of CYP2C9 in 4-hydroxylation of tamoxifen – a matter of fact that has been already assessed in experimental settings in vitro – is further supported in vivo by Murdter et al. and could in addition be well described by the established CYP2D6 phenotype-specific PBPK-models leading to a less pronounced decrease of 4-OH-TAM in CYP2D6 PMs compared to endoxifen as also reported in the literature (Coller et al., 2002; Stearns et al., 2003; Murdter et al., 2011).
The results of population simulations that consider physiological variability in European female patient populations representing each of the two phenotypes are able to account to a large extend for the reported high inter-individual variability in plasma concentrations of tamoxifen and its metabolites.
A PBPK-model of tamoxifen and its pharmacologically important metabolites NDM-TAM, 4-OH-TAM, and endoxifen was developed. This model is able to simulate the PK after single and repeated oral tamoxifen doses in female breast cancer patients in dependence of the CYP2D6 phenotype. The analysis regarding the mass balance thereby supports a recent hypothesis stating a more prominent role for endoxifen formation from 4-OH-TAM. The established models can explain PK variability within a population and provide a basis for further investigations concerning tamoxifen PK under varying scenarios such as in populations of differing geographical origin that exert characteristic distributions of CYP2D6 phenotypes (Sistonen et al., 2007). Furthermore, the impact of concomitant treatment with compounds inhibiting CYP2D6 can in the future be elucidated with this model. Ultimately, by bridging the gap between CYP2D6 phenotype, PK, and resulting impact on tumor cell growth, a future PBPK/PD-model of tamoxifen and its metabolites may provide a useful tool to investigate the role of CYP2D6 phenotype on the outcome of breast cancer therapy using tamoxifen.
Kristin Dickschen is funded by and Thomas Eissing, Jörg Lippert, Kirstin Thelen, and Stefan Willmann are employes of Bayer Technology Services GmbH, the company that owns and commercializes the software platform used for the simulations described in the manuscript (PK-Sim® and MoBi®), as well as parent company stock owners.
The authors acknowledge financial support by the German Federal Ministry of Education and Research for the grants [0315280F] (FORSYS-Partner project “A Systems Biology Approach toward Predictive Cancer Therapy”), and [0315747] (Virtual Liver).
Ahmad, A., Shahabuddin, S., Sheikh, S., Kale, P., Krishnappa, M., Rane, R. C., and Ahmad, I. (2010). Endoxifen, a new cornerstone of breast cancer therapy: demonstration of safety, tolerability, and systemic bioavailability in healthy human subjects. Clin. Pharmacol. Ther. 88, 814–817.
Bergstrom, C. A., Strafford, M., Lazorova, L., Avdeef, A., Luthman, K., and Artursson, P. (2003). Absorption classification of oral drugs based on molecular surface properties. J. Med. Chem. 46, 558–570.
Bijl, M., Van Schaik, R., Lammers, L., Hofman, A., Vulto, A., Van Gelder, T., Stricker, B., and Visser, L. (2009). The CYP2D6*4 polymorphism affects breast cancer survival in tamoxifen users. Breast Cancer Res. Treat. 118, 125–130.
Blevins-Primeau, A. S., Sun, D., Chen, G., Sharma, A. K., Gallagher, C. J., Amin, S., and Lazarus, P. (2009). Functional significance of UDP-glucuronosyltransferase variants in the metabolism of active tamoxifen metabolites. Cancer Res. 69, 1892–1900.
Borges, S., Desta, Z., Li, L., Skaar, T. C., Ward, B. A., Nguyen, A., Jin, Y., Storniolo, A. M., Nikoloff, D. M., Wu, L., Hillman, G., Hayes, D. F., Stearns, V., and Flockhart, D. A. (2006). Quantitative effect of CYP2D6 genotype and inhibitors on tamoxifen metabolism: implication for optimization of breast cancer treatment. Clin. Pharmacol. Ther. 80, 61–74.
Bourassa, P., Dubeau, S., Maharvi, G. M., Fauq, A. H., Thomas, T. J., and Tajmir-Riahi, H. A. (2011). Binding of antitumor tamoxifen and its metabolites 4-hydroxytamoxifen and endoxifen to human serum albumin. Biochimie 93, 1089–1101.
Bratherton, D. G., Brown, C. H., Buchanan, R., Hall, V., Kingsley Pillers, E. M., Wheeler, T. K., and Williams, C. J. (1984). A comparison of two doses of tamoxifen (Nolvadex) in postmenopausal women with advanced breast cancer: 10 mg bd versus 20 mg bd. Br. J. Cancer 50, 199–205.
Brauch, H. B., Schroth, W., Ingle, J. N., and Goetz, M. P. (2011). CYP2D6 and tamoxifen: awaiting the denouement. J. Clin. Oncol. 29, 4589–4590.
Buchanan, C. M., Buchanan, N. L., Edgar, K. J., Little, J. L., Malcolm, M. O., Ruble, K. M., Wacher, V. J., and Wempe, M. F. (2007). Pharmacokinetics of tamoxifen after intravenous and oral dosing of tamoxifen-hydroxybutenyl-beta-cyclodextrin formulations. J. Pharm. Sci. 96, 644–660.
Buckley, M. M., and Goa, K. L. (1989). Tamoxifen. A reappraisal of its pharmacodynamic and pharmacokinetic properties, and therapeutic use. Drugs 37, 451–490.
Buzdar, A. U., Hortobagyi, G. N., Frye, D., Ho, D., Booser, D. J., Valero, V., Holmes, F. A., Birmingham, B. K., Bui, K., and Yeh, C. (1994). Bioequivalence of 20-mg once-daily tamoxifen relative to 10-mg twice-daily tamoxifen regimens for breast cancer. J. Clin. Oncol. 12, 50–54.
Choi, J. S., and Kang, K. W. (2008). Enhanced tamoxifen bioavailability after oral administration of tamoxifen in rats pretreated with naringin. Arch. Pharm. Res. 31, 1631–1636.
Coller, J. K., Krebsfaenger, N., Klein, K., Endrizzi, K., Wolbold, R., Lang, T., Nussler, A., Neuhaus, P., Zanger, U. M., Eichelbaum, M., and Murdter, T. E. (2002). The influence of CYP2B6, CYP2C9 and CYP2D6 genotypes on the formation of the potent antioestrogen Z-4-hydroxy-tamoxifen in human liver. Br. J. Clin. Pharmacol. 54, 157–167.
Custódio, J. A., Almeida, L. M., and Madeira, V. M. C. (1991). A reliable and rapid procedure to estimate drug partitioning in biomembranes. Biochem. Biophys. Res. Commun. 176, 1079–1085.
De Vos, D., Slee, P. H., Briggs, R. J., and Stevenson, D. (1998). Serum and urine levels of tamoxifen and its metabolites in patients with advanced breast cancer after a loading dose and at steady-state levels. Cancer Chemother. Pharmacol. 42, 512–514.
De Vos, D., Slee, P. H., Stevenson, D., and Briggs, R. J. (1992). Serum elimination half-life of tamoxifen and its metabolites in patients with advanced breast cancer. Cancer Chemother. Pharmacol. 31, 76–78.
Decensi, A., Robertson, C., Viale, G., Pigatto, F., Johansson, H., Kisanga, E. R., Veronesi, P., Torrisi, R., Cazzaniga, M., Mora, S., Sandri, M. T., Pelosi, G., Luini, A., Goldhirsch, A., Lien, E. A., and Veronesi, U. (2003). A randomized trial of low-dose tamoxifen on breast cancer proliferation and blood estrogenic biomarkers. J. Natl. Cancer Inst. 95, 779–790.
Desta, Z., Ward, B. A., Soukhova, N. V., and Flockhart, D. A. (2004). Comprehensive evaluation of tamoxifen sequential biotransformation by the human cytochrome P450 system in vitro: prominent roles for CYP3A and CYP2D6. J. Pharmacol. Exp. Ther. 310, 1062–1075.
Dorne, J. L., Walton, K., and Renwick, A. G. (2003). Human variability in CYP3A4 metabolism and CYP3A4-related uncertainty factors for risk assessment. Food Chem. Toxicol. 41, 201–224.
Dowsett, M., Cuzick, J., Howell, A., and Jackson, I. (2001). Pharmacokinetics of anastrozole and tamoxifen alone, and in combination, during adjuvant endocrine therapy for early breast cancer in postmenopausal women: a sub-protocol of the ‘Arimidex and tamoxifen alone or in combination’ (ATAC) trial. Br. J. Cancer 85, 317–324.
Dowsett, M., Tobias, J. S., Howell, A., Blackman, G. M., Welch, H., King, N., Ponzone, R., Von Euler, M., and Baum, M. (1999). The effect of anastrozole on the pharmacokinetics of tamoxifen in post-menopausal women with early breast cancer. Br. J. Cancer 79, 311–315.
Eissing, T., Kuepfer, L., Becker, C., Block, M., Coboeken, K., Gaub, T., Goerlitz, L., Jaeger, J., Loosen, R., Ludewig, B., Meyer, M., Niederalt, C., Sevestre, M., Siegmund, H. U., Solodenko, J., Thelen, K., Telle, U., Weiss, W., Wendl, T., Willmann, S., and Lippert, J. (2011). A computational systems biology software platform for multiscale modeling and simulation: integrating whole-body physiology, disease biology, and molecular reaction networks. Front. Physiol. 2:4. doi:10.3389/fphys.2011.00004
Etienne, M. C., Milano, G., Fischel, J. L., Frenay, M., Francois, E., Formento, J. L., Gioanni, J., and Namer, M. (1989). Tamoxifen metabolism: pharmacokinetic and in vitro study. Br. J. Cancer 60, 30–35.
Fabian, C., Sternson, L., El-Serafi, M., Cain, L., and Hearne, E. (1981). Clinical pharmacology of tamoxifen in patients with breast cancer: correlation with clinical data. Cancer 48, 876–882.
Fagerberg, J. H., Tsinman, O., Sun, N., Tsinman, K., Avdeef, A., and Bergstrom, C. A. (2010). Dissolution rate and apparent solubility of poorly soluble drugs in biorelevant dissolution media. Mol. Pharm. 7, 1419–1430.
Fisher, B., Dignam, J., Bryant, J., and Wolmark, N. (2001). Five versus more than five years of tamoxifen for lymph node-negative breast cancer: updated findings from the National Surgical Adjuvant Breast and Bowel Project B-14 randomized trial. J. Natl. Cancer Inst. 93, 684–690.
Fleeman, N., Martin Saborido, C., Payne, K., Boland, A., Dickson, R., Dundar, Y., Fernandez Santander, A., Howell, S., Newman, W., Oyee, J., and Walley, T. (2011). The clinical effectiveness and cost-effectiveness of genotyping for CYP2D6 for the management of women with breast cancer treated with tamoxifen: a systematic review. Health Technol. Assess. 15, 1–102.
Fromson, J. M., Pearson, S., and Bramah, S. (1973a). The metabolism of tamoxifen (I.C.I. 46,474). I. In laboratory animals. Xenobiotica 3, 693–709.
Fromson, J. M., Pearson, S., and Bramah, S. (1973b). The metabolism of tamoxifen (I.C.I. 46,474). II. In female patients. Xenobiotica 3, 711–714.
Fuchs, W. S., Leary, W. P., Van Der Meer, M. J., Gay, S., Witschital, K., and Von Nieciecki, A. (1996). Pharmacokinetics and bioavailability of tamoxifen in postmenopausal healthy women. Arzneimittelforschung 46, 418–422.
Furlanut, M., Franceschi, L., Pasqual, E., Bacchetti, S., Poz, D., Giorda, G., and Cagol, P. (2007). Tamoxifen and its main metabolites serum and tissue concentrations in breast cancer women. Ther. Drug. Monit. 29, 349–352.
Gallicchio, L., Tkaczuk, K., Lord, G., Danton, M., Lewis, L. M., Lim, C. K., and Flaws, J. A. (2004). Medication use, tamoxifen (TAM), and TAM metabolite concentrations in women with breast cancer. Cancer Lett. 211, 57–67.
Gjerde, J., Geisler, J., Lundgren, S., Ekse, D., Varhaug, J. E., Mellgren, G., Steen, V. M., and Lien, E. A. (2010). Associations between tamoxifen, estrogens, and FSH serum levels during steady state tamoxifen treatment of postmenopausal women with breast cancer. BMC Cancer 10, 313. doi:10.1186/1471-2407-10-313
Gjerde, J., Hauglid, M., Breilid, H., Lundgren, S., Varhaug, J. E., Kisanga, E. R., Mellgren, G., Steen, V. M., and Lien, E. A. (2008). Effects of CYP2D6 and SULT1A1 genotypes including SULT1A1 gene copy number on tamoxifen metabolism. Ann. Oncol. 19, 56–61.
Goetz, M. P., Kamal, A., and Ames, M. M. (2008). Tamoxifen pharmacogenomics: the role of CYP2D6 as a predictor of drug response. Clin. Pharmacol. Ther. 83, 160–166.
Goetz, M. P., Knox, S. K., Suman, V. J., Rae, J. M., Safgren, S. L., Ames, M. M., Visscher, D. W., Reynolds, C., Couch, F. J., Lingle, W. L., Weinshilboum, R. M., Fritcher, E. G., Nibbe, A. M., Desta, Z., Nguyen, A., Flockhart, D. A., Perez, E. A., and Ingle, J. N. (2007). The impact of cytochrome P450 2D6 metabolism in women receiving adjuvant tamoxifen. Breast Cancer Res. Treat. 101, 113–121.
Goetz, M. P., Rae, J. M., Suman, V. J., Safgren, S. L., Ames, M. M., Visscher, D. W., Reynolds, C., Couch, F. J., Lingle, W. L., Flockhart, D. A., Desta, Z., Perez, E. A., and Ingle, J. N. (2005). Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes. J. Clin. Oncol. 23, 9312–9318.
Guerrieri-Gonzaga, A., Baglietto, L., Johansson, H., Bonanni, B., Robertson, C., Sandri, M. T., Canigiula, L., Lampreda, C., Diani, S., Lien, E. A., and Decensi, A. (2001). Correlation between tamoxifen elimination and biomarker recovery in a primary prevention trial. Cancer Epidemiol. Biomarkers Prev. 10, 967–970.
Hoskins, J. M., Carey, L. A., and McLeod, H. L. (2009). CYP2D6 and tamoxifen: DNA matters in breast cancer. Nat. Rev. Cancer 9, 576–586.
Hutson, P. R., Love, R. R., Havighurst, T. C., Rogers, E., and Cleary, J. F. (2005). Effect of exemestane on tamoxifen pharmacokinetics in postmenopausal women treated for breast cancer. Clin. Cancer Res. 11, 8722–8727.
Irvin, W. J. Jr., Walko, C. M., Weck, K. E., Ibrahim, J. G., Chiu, W. K., Dees, E. C., Moore, S. G., Olajide, O. A., Graham, M. L., Canale, S. T., Raab, R. E., Corso, S. W., Peppercorn, J. M., Anderson, S. M., Friedman, K. J., Ogburn, E. T., Desta, Z., Flockhart, D. A., Mcleod, H. L., Evans, J. P., and Carey, L. A. (2011). Genotype-guided tamoxifen dosing increases active metabolite exposure in women with reduced CYP2D6 metabolism: a multicenter study. J. Clin. Oncol. 29, 3232–3239.
Jaremko, M., Kasai, Y., Barginear, M. F., Raptis, G., Desnick, R. J., and Yu, C. (2010). Tamoxifen metabolite isomer separation and quantification by liquid chromatography-tandem mass spectrometry. Anal. Chem. 82, 10186–10193.
Jin, Y., Desta, Z., Stearns, V., Ward, B., Ho, H., Lee, K. H., Skaar, T., Storniolo, A. M., Li, L., Araba, A., Blanchard, R., Nguyen, A., Ullmer, L., Hayden, J., Lemler, S., Weinshilboum, R. M., Rae, J. M., Hayes, D. F., and Flockhart, D. A. (2005). CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. J. Natl. Cancer Inst. 97, 30–39.
Johnson, M. D., Zuo, H., Lee, K. H., Trebley, J. P., Rae, J. M., Weatherman, R. V., Desta, Z., Flockhart, D. A., and Skaar, T. C. (2004). Pharmacological characterization of 4-hydroxy-N-desmethyl tamoxifen, a novel active metabolite of tamoxifen. Breast Cancer Res. Treat. 85, 151–159.
Johnston, S. R. D., Haynes, B. P., Sacks, N. P. M., Mckinna, J. A., Griggs, L. J., Jarman, M., Baum, M., Smith, I. E., and Dowsett, M. (1993). Effect of oestrogen receptor status and time on the intra-tumoural accumulation of tamoxifen and N-desmethyltamoxifen following short-term therapy in human primary breast cancer. Breast Cancer Res. Treat. 28, 241–250.
Jordan, V. C. (2006). Tamoxifen (ICI46,474) as a targeted therapy to treat and prevent breast cancer. Br. J. Pharmacol. 147(Suppl. 1), S269–S276.
Jordan, V. C. (2007). New insights into the metabolism of tamoxifen and its role in the treatment and prevention of breast cancer. Steroids 72, 829–842.
Kaku, T., Ogura, K., Nishiyama, T., Ohnuma, T., Muro, K., and Hiratsuka, A. (2004). Quaternary ammonium-linked glucuronidation of tamoxifen by human liver microsomes and UDP-glucuronosyltransferase 1A4. Biochem. Pharmacol. 67, 2093–2102.
Kasim, N. A., Whitehouse, M., Ramachandran, C., Bermejo, M., Lennernäs, H., Hussain, A. S., Junginger, H. E., Stavchansky, S. A., Midha, K. K., Shah, V. P., and Amidon, G. L. (2003). Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol. Pharm. 1, 85–96.
Kim, C. S., Choi, S. J., Park, C. Y., Li, C., and Choi, J. S. (2010). Effects of silybinin on the pharmacokinetics of tamoxifen and its active metabolite, 4-hydroxytamoxifen in rats. Anticancer Res. 30, 79–85.
Kisanga, E. R., Gjerde, J., Guerrieri-Gonzaga, A., Pigatto, F., Pesci-Feltri, A., Robertson, C., Serrano, D., Pelosi, G., Decensi, A., and Lien, E. A. (2004). Tamoxifen and metabolite concentrations in serum and breast cancer tissue during three dose regimens in a randomized preoperative trial. Clin. Cancer Res. 10, 2336–2343.
Kisanga, E. R., Mellgren, G., and Lien, E. A. (2005). Excretion of hydroxylated metabolites of tamoxifen in human bile and urine. Anticancer Res. 25, 4487–4492.
Lammers, L. A., Mathijssen, R. H., Van Gelder, T., Bijl, M. J., De Graan, A. J., Seynaeve, C., Van Fessem, M. A., Berns, E. M., Vulto, A. G., and Van Schaik, R. H. (2010). The impact of CYP2D6-predicted phenotype on tamoxifen treatment outcome in patients with metastatic breast cancer. Br. J. Cancer 103, 765–771.
Langan-Fahey, S. M., Tormey, D. C., and Jordan, V. C. (1990). Tamoxifen metabolites in patients on long-term adjuvant therapy for breast cancer. Eur. J. Cancer 26, 883–888.
Lash, T. L., Lien, E. A., Sorensen, H. T., and Hamilton-Dutoit, S. (2009). Genotype-guided tamoxifen therapy: time to pause for reflection? Lancet Oncol. 10, 825–833.
Li, C., Lim, S. C., Kim, J., and Choi, J. S. (2011). Effects of myricetin, an anticancer compound, on the bioavailability and pharmacokinetics of tamoxifen and its main metabolite, 4-hydroxytamoxifen, in rats. Eur. J. Drug Metab. Pharmacokinet. 36, 175–182.
Lien, E. A., Anker, G., and Ueland, P. M. (1995). Pharmacokinetics of tamoxifen in premenopausal and postmenopausal women with breast cancer. J. Steroid Biochem. Mol. Biol. 55, 229–231.
Lien, E. A., Solheim, E., Kvinnsland, S., and Ueland, P. M. (1988). Identification of 4-hydroxy-N-desmethyltamoxifen as a metabolite of tamoxifen in human bile. Cancer Res. 48, 2304–2308.
Lien, E. A., Solheim, E., Lea, O. A., Lundgren, S., Kvinnsland, S., and Ueland, P. M. (1989). Distribution of 4-hydroxy-N-desmethyltamoxifen and other tamoxifen metabolites in human biological fluids during tamoxifen treatment. Cancer Res. 49, 2175–2183.
Lim, C. K., Yuan, Z. X., Lamb, J. H., White, I. N., De Matteis, F., and Smith, L. L. (1994). A comparative study of tamoxifen metabolism in female rat, mouse and human liver microsomes. Carcinogenesis 15, 589–593.
Lim, Y. C., Li, L., Desta, Z., Zhao, Q., Rae, J. M., Flockhart, D. A., and Skaar, T. C. (2006). Endoxifen, a secondary metabolite of tamoxifen, and 4-OH-tamoxifen induce similar changes in global gene expression patterns in MCF-7 breast cancer cells. J. Pharmacol. Exp. Ther. 318, 503–512.
Madlensky, L., Natarajan, L., Tchu, S., Pu, M., Mortimer, J., Flatt, S. W., Nikoloff, D. M., Hillman, G., Fontecha, M. R., Lawrence, H. J., Parker, B. A., Wu, A. H., and Pierce, J. P. (2011). Tamoxifen metabolite concentrations, CYP2D6 genotype, and breast cancer outcomes. Clin. Pharmacol. Ther. 89, 718–725.
Murdter, T. E., Schroth, W., Bacchus-Gerybadze, L., Winter, S., Heinkele, G., Simon, W., Fasching, P. A., Fehm, T., Eichelbaum, M., Schwab, M., and Brauch, H. (2011). Activity levels of tamoxifen metabolites at the estrogen receptor and the impact of genetic polymorphisms of phase I and II enzymes on their concentration levels in plasma. Clin. Pharmacol. Ther. 89, 708–717.
Nishiyama, T., Ogura, K., Nakano, H., Ohnuma, T., Kaku, T., Hiratsuka, A., Muro, K., and Watabe, T. (2002). Reverse geometrical selectivity in glucuronidation and sulfation of cis- and trans-4-hydroxytamoxifens by human liver UDP-glucuronosyltransferases and sulfotransferases. Biochem. Pharmacol. 63, 1817–1830.
Peyrade, F., Frenay, M., Etienne, M. C., Ruch, F., Guillemare, C., Francois, E., Namer, M., Ferrero, J. M., and Milano, G. (1996). Age-related difference in tamoxifen disposition. Clin. Pharmacol. Ther. 59, 401–410.
Piao, Y., Shin, S. C., and Choi, J. S. (2008). Effects of oral kaempferol on the pharmacokinetics of tamoxifen and one of its metabolites, 4-hydroxytamoxifen, after oral administration of tamoxifen to rats. Biopharm. Drug Dispos. 29, 245–249.
Rae, J. M. (2011). Personalized tamoxifen: what is the best way forward? J. Clin. Oncol. 29, 3206–3208.
Ruiter, R., Bijl, M. J., Van Schaik, R. H., Berns, E. M., Hofman, A., Coebergh, J. W., Van Noord, C., Visser, L. E., and Stricker, B. H. (2010). CYP2C19*2 polymorphism is associated with increased survival in breast cancer patients using tamoxifen. Pharmacogenomics 11, 1367–1375.
Seruga, B., and Amir, E. (2010). Cytochrome P450 2D6 and outcomes of adjuvant tamoxifen therapy: results of a meta-analysis. Breast Cancer Res. Treat. 122, 609–617.
Shin, S. C., and Choi, J. S. (2009). Effects of epigallocatechin gallate on the oral bioavailability and pharmacokinetics of tamoxifen and its main metabolite, 4-hydroxytamoxifen, in rats. Anticancer Drugs 20, 584–588.
Shin, S. C., Choi, J. S., and Li, X. (2006). Enhanced bioavailability of tamoxifen after oral administration of tamoxifen with quercetin in rats. Int. J. Pharm. 313, 144–149.
Shin, S. C., Piao, Y. J., and Choi, J. S. (2008). Effects of morin on the bioavailability of tamoxifen and its main metabolite, 4-hydroxytamoxifen, in rats. In vivo 22, 391–395.
Sistonen, J., Sajantila, A., Lao, O., Corander, J., Barbujani, G., and Fuselli, S. (2007). CYP2D6 worldwide genetic variation shows high frequency of altered activity variants and no continental structure. Pharmacogenet. Genomics 17, 93–101.
Soininen, K., Kleimola, T., Elomaa, I., Salmo, M., and Rissanen, P. (1986). The steady-state pharmacokinetics of tamoxifen and its metabolites in breast cancer patients. J. Int. Med. Res. 14, 162–165.
Stearns, V., Johnson, M. D., Rae, J. M., Morocho, A., Novielli, A., Bhargava, P., Hayes, D. F., Desta, Z., and Flockhart, D. A. (2003). Active tamoxifen metabolite plasma concentrations after coadministration of tamoxifen and the selective serotonin reuptake inhibitor paroxetine. J. Natl. Cancer Inst. 95, 1758–1764.
Sun, D., Sharma, A. K., Dellinger, R. W., Blevins-Primeau, A. S., Balliet, R. M., Chen, G., Boyiri, T., Amin, S., and Lazarus, P. (2007). Glucuronidation of active tamoxifen metabolites by the human UDP glucuronosyltransferases. Drug Metab. Dispos. 35, 2006–2014.
Tukker, J. J., Blankenstein, M. A., and Nortier, J. W. (1986). Comparison of bioavailability in man of tamoxifen after oral and rectal administration. J. Pharm. Pharmacol. 38, 888–892.
Van Schaik, R. H., Kok, M., Sweep, F. C., Van Vliet, M., Van Fessem, M., Meijer-Van Gelder, M. E., Seynaeve, C., Lindemans, J., Wesseling, J., Van ’t Veer, L. J., Span, P. N., Van Laarhoven, H., Sleijfer, S., Foekens, J. A., Linn, S. C., and Berns, E. M. (2011). The CYP2C19*2 genotype predicts tamoxifen treatment outcome in advanced breast cancer patients. Pharmacogenomics 12, 1137–1146.
Willmann, S., Hohn, K., Edginton, A., Sevestre, M., Solodenko, J., Weiss, W., Lippert, J., and Schmitt, W. (2007). Development of a physiology-based whole-body population model for assessing the influence of individual variability on the pharmacokinetics of drugs. J. Pharmacokinet. Pharmacodyn. 34, 401–431.
Willmann, S., Lippert, J., Sevestre, M., Solodenko, J., Fois, F., and Schmitt, W. (2003a). PK-Sim®: a physiologically based pharmacokinetic ‘whole-body’ model. Drug Discov. Today Biosilico 1, 121–124.
Willmann, S., Schmitt, W., Keldenich, J., and Dressman, J. B. (2003b). A physiologic model for simulating gastrointestinal flow and drug absorption in rats. Pharm. Res. 20, 1766–1771.
Willmann, S., Schmitt, W., Keldenich, J., Lippert, J., and Dressman, J. B. (2004). A physiological model for the estimation of the fraction dose absorbed in humans. J. Med. Chem. 47, 4022–4031.
Yang, S. H., Suh, J. H., and Lee, M. G. (2010). Pharmacokinetic interaction between tamoxifen and ondansetron in rats: non-competitive (hepatic) and competitive (intestinal) inhibition of tamoxifen metabolism by ondansetron via CYP2D subfamily and 3A1/2. Cancer Chemother. Pharmacol. 65, 407–418.
Keywords: tamoxifen, endoxifen, CYP2D6, pharmacokinetics, PBPK, population simulation
Citation: Dickschen K, Willmann S, Thelen K, Lippert J, Hempel G and Eissing T (2012) Physiologically based pharmacokinetic modeling of tamoxifen and its metabolites in women of different CYP2D6 phenotypes provides new insight into the tamoxifen mass balance. Front. Pharmacol. 3:92. doi: 10.3389/fphar.2012.00092
Received: 03 April 2012; Paper pending published: 19 April 2012;
Accepted: 27 April 2012; Published online: 21 May 2012.
Edited by:
José A. G. Agúndez, University of Extremadura, SpainReviewed by:
Luis Abel Quiñones, University of Chile, ChileCopyright: © 2012 Dickschen, Willmann, Thelen, Lippert, Hempel and Eissing. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Kristin Dickschen, Computational Systems Biology, Bayer Technology GmbH, Building 9115, 51368 Leverkusen, Germany. e-mail:a3Jpc3Rpbi5kaWNrc2NoZW5AYmF5ZXIuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.