
 Kathleen A. Martin
Kathleen A. Martin
- Section of Cardiovascular Medicine, Department of Internal Medicine, Yale School of Medicine, Yale University, New Haven, CT, USA
Diabetes mellitus (DM) is a complex metabolic disorder arising from lack of insulin production or insulin resistance (Diagnosis and classification of diabetes mellitus, 2007). DM is a leading cause of morbidity and mortality in the developed world, particularly from vascular complications such as atherothrombosis in the coronary vessels. Aldose reductase (AR; ALR2; EC 1.1.1.21), a key enzyme in the polyol pathway, catalyzes nicotinamide adenosine dinucleotide phosphate-dependent reduction of glucose to sorbitol, leading to excessive accumulation of intracellular reactive oxygen species (ROS) in various tissues of DM including the heart, vasculature, neurons, eyes, and kidneys. As an example, hyperglycemia through such polyol pathway induced oxidative stress, may have dual heart actions, on coronary blood vessel (atherothrombosis) and myocardium (heart failure) leading to severe morbidity and mortality (reviewed in Heather and Clarke, 2011). In cells cultured under high glucose conditions, many studies have demonstrated similar AR-dependent increases in ROS production, confirming AR as an important factor for the pathogenesis of many diabetic complications. Moreover, recent studies have shown that AR inhibitors may be able to prevent or delay the onset of cardiovascular complications such as ischemia/reperfusion injury, atherosclerosis, and atherothrombosis. In this review, we will focus on describing pivotal roles of AR in the pathogenesis of cardiovascular diseases as well as other diabetic complications, and the potential use of AR inhibitors as an emerging therapeutic strategy in preventing DM complications.
Introduction
In mammalian cells, under normoglycemia (3.8–6.1 mmol/L), cellular glucose is predominantly phosphorylated into glucose 6-phosphate by hexokinase, and enters the glycolytic pathway. Only trace amounts of non-phosphorylated glucose (about 3%) enter the polyol pathway (Morrison et al., 1970). However, under hyperglycemic condition (>7 mmol/L), there is increased flux through the polyol pathway, accounting for greater than 30% of glucose metabolism (Gonzalez et al., 1984; Yabe-Nishimura, 1998). The rate limiting step of the polyol pathway is the reduction of glucose to sorbitol catalyzed by aldose reductase (AR), at the expense of reduced nicotinamide adenosine dinucleotide phosphate (NADPH). Sorbitol is, in turn, converted to fructose by sorbitol dehydrogenase (SDH) with the oxidized form of nicotinamide adenine dinucleotide (NAD+) as a co-factor (Yabe-Nishimura, 1998; El-Kabbani et al., 2004; Figure 1). The polyol pathway was first identified in the seminal vesicle by Hers (1956) who demonstrated the conversion of blood glucose into fructose, an energy source for sperm cells. AR has since been isolated and purified from a number of human and animal tissues including various regions of the eyes (Srivastava et al., 1984), testis (Kawasaki et al., 1989), liver (Petrash and Srivastava, 1982), placenta (Das and Srivastava, 1985a; Vander Jagt et al., 1990a), ovary (Iwata et al., 1990), kidney (Ansari et al., 1991; Ohta et al., 1991), erythrocyte (Das and Srivastava, 1985b), cardiac (Vander Jagt et al., 1990b) and skeletal muscle (Cromlish and Flynn, 1983; Morjana and Flynn, 1989; Vander Jagt et al., 1990b), and the brain (Wermuth et al., 1982; Cromlish et al., 1985). AR is located in the cytoplasm of most cells (Flynn, 1982) but is not uniformly distributed in all cell types of an organ. For example, in the kidney the enzyme is present in the Henle’s loop, collecting tubules, outer and inner medulla, but not in the cortex (Terubayashi et al., 1989; Ohta et al., 1991).
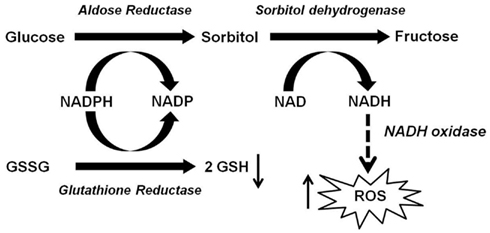
Figure 1. Role of aldose reductase (AR) in hyperglycemia-induced oxidative stress. Excessive amount of glucose is shunted to the polyol pathway, where AR reduces glucose into sorbitol at the expense of NADPH. Since NADPH is essential for generation of GSH (intracellular antioxidant) from GSSG, the depletion of NADPH by the AR pathway may impair intracellular antioxidant defense. Sorbitol is then converted to fructose by SDH with the production of NADH, potentially leading to increased ROS via NADH oxidase.
Contribution of Aldose Reductase to Diabetes-Induced Oxidative Stress
Diabetes mellitus (DM) is characterized by chronic hyperglycemia and disturbances of carbohydrate, fat, and protein metabolism resulting from an absolute or relative deficiency of insulin (Diagnosis and classification of diabetes mellitus, 2007). Increased oxidative stress is thought to play an important role in the pathogenesis of diabetic complications, as supported by increased levels of oxidized DNA, proteins, and lipids (Wiernsperger, 2003). The induction of oxidative stress in DM can result from multiple mechanisms. Excessive levels of glucose can disrupt the electron transport chain in the mitochondria, leading to overproduction of superoxide anions (Nishikawa et al., 2000). High glucose can also stimulate oxidative stress via the auto-oxidation of glucose (Wolff and Dean, 1987) and through non-enzymatic glycation (Mullarkey et al., 1990). Reactive oxygen species (ROS) is generated in the process of advanced glycation endproducts (AGEs) formation (Kennedy and Lyons, 1997; Yim et al., 2001) and interaction between AGEs and their receptors RAGE can also lead to ROS production (Schmidt et al., 1994). Moreover, glycation can inactivate antioxidant enzymes, impairing antioxidant defense, as observed with glycation of superoxide dismutase (Kawamura et al., 1992; Morgan et al., 2002).
Another important mechanism whereby high glucose can induce oxidative stress is the polyol pathway. Previous studies using AR deficient mice have shown that polyol pathway is an important source of diabetes-induced oxidative stress (Lee and Chung, 1999; Obrosova et al., 2003, 2005; Drel et al., 2006, 2008; Ho et al., 2006). There are three potential mechanisms by which the polyol pathway contributes to oxidative stress. First, under hyperglycemic condition, 30% of the glucose is channeled into AR-dependent polyol pathway, which depetes NADPH and consequently reduces GSH level (Cheng and Gonzalez, 1986). Second, oxidative stress is generated during the conversion of sorbitol into fructose by SDH (i.e., the second step of polyol pathway). In this step, the co-factor NAD+ is converted to NADH by SDH. NADH is a substrate for NADH oxidase leading to production of superoxide anions (Morre et al., 2000). Third, the polyol pathway converts glucose to fructose, and fructose can be further metabolized into fructose-3-phosphate and 3-deoxyglucosone, which are more potent non-enzymatic glycation agent than glucose (Hamada et al., 1996a,b). Thus, the flux of glucose through the polyol pathway would increase AGEs formation, ultimately leading to ROS generation. Thus there is crosstalk between AR-dependent and AR independent sources of oxidative stress making it difficult to establish the relative contributions of each. Additionally, the pathways leading to production of oxidative stress is both tissue and cell dependent. Relative contributions of oxidative stress remains an outstanding question.
Aldose Reductase and Atherothrombotic Cardiovascular Disease in Diabetes
Latest estimates predict that the global prevalence of DM will increase by 165% from 11 million in 2000 (prevalence of 4.0%) to 29 million in 2050 (prevalence of 7.2%; Boyle et al., 2001). Atherothrombotic cardiovascular events account for up to 80% of all deaths among DM patients (Haffner et al., 1998). While standard preventative treatments to combat atherothrombosis include glycemic control and low dose aspirin, challenges remain. The effectiveness of tight glycemic control has recently been the subject of considerable debate, with some studies suggesting that it is of marginal benefit in preventing cardiovascular events relative to less stringent glycemic control (American Diabetes Association, 2000, 2003; Wilson and Perry, 2009). While low dose aspirin is protective in many patients, some individuals exhibit aspirin-resistance (Grotemeyer, 1991; Grotemeyer et al., 1993; Helgason et al., 1994; Pappas et al., 1994; Buchanan and Brister, 1995; Marshall et al., 1997; Andersen et al., 2002; Macchi et al., 2002; Grundmann et al., 2003; Zimmermann et al., 2003). Therefore there is an urgent need for novel pharmacological agents to reduce the increasing burden of atherothrombotic cardiovascular disease.
The pathogenesis of the diabetic complications is complex with multiple mechanisms proposed, including (1) non-enzymatic glycation, (2) protein kinase C (PKC) activation, (3) hexosamine pathway activation, and (4) mitochondrial respiratory chain disruption. However, one of the major proposed mechanisms for the development of the diabetic complications involves the polyol pathway, which mediates the metabolic and osmotic alterations in many tissues (e.g., neurons, platelets). Increased glucose flux through the polyol pathway has been associated with the pathogenesis of diabetic complications via several potential mechanisms, including sorbitol-osmotic effects, depletion of myoinositol (Kinoshita et al., 1962) and subsequent perturbations in Na+/K+ ATPase activity (Greene et al., 1987; Steele et al., 1993), disturbances in cellular redox and free radical defense, increased oxidative, and glycation stress, activation of PKC (Steele et al., 1993; Hamada et al., 2000; Hamada and Nakamura, 2004), nitric oxide (NO)-mediated vascular tone (Tesfamariam et al., 1993), and induction of hyperglycemic pseudohypoxia (Van den Enden et al., 1995; Figure 2). Moreover, polymorphic markers of the human AR gene demonstrate a strong association with a susceptibility to develop diabetic complications. This suggests that the polyol pathway plays an important role in the pathogenesis of DM in human patients. Indeed a number of AR inhibitors are currently being investigated to prevent diabetic complications such as cardiomyopathy, neuropathy, nephropathy, and retinopathy (Johnson et al., 2004; Giannoukakis, 2006; Ramirez and Borja, 2008).
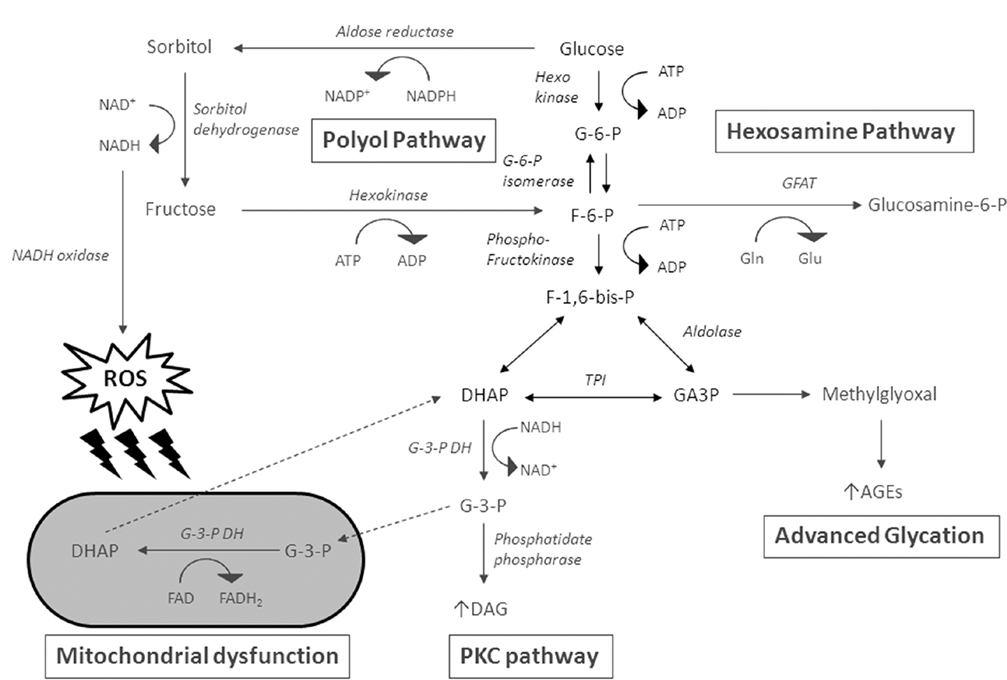
Figure 2. Glucose flux through the polyol pathway has been associated with the pathogenesis of diabetic complications via several potential mechanisms. Intracellular accumulation of sorbitol causes osmotic stress. The end production of the polyol pathway, fructose, is converted to fructose-6-phosphate (F-6-P) by hexokinase, and is further converted to glucosamine-6-phosphate by glutamine: fructose-6-phosphate amidotransferase (GFAT). Fructose-6-phosphate may also form fructose-1,6-bisphosphate (F-1,6-P), which is converted to dihydroxyacetone phosphate (DHAP). DHAP and glyceraldehdye-3-phosphate (GA3P) are interconvertible by triosephosphate isomerase. They can lead to the formation of methylglyoxal, resulting in advanced glycation end-product. DHAP can further be converted to diacylglycerol (DAG), leading to PKC activation. The continuous conversion of glycerol-3-phosphate (G-3-P) to DHAP results in concomitant transfer of electrons from reduced cytosolic NADH to mitochondrial oxidized FAD, which can generate high mitochondrial membrane potentials and inhibition of the electron transport chain at complex III. The oxidation of NADH by NADH oxidase produces reactive oxygen species (ROS), which can attack the mitochondrial membrane.
Diabetes has been viewed as a coronary heart disease and myocardial infarction risk equivalent, in part due to its association with a hypercoagulable state and elevated concentration of procoagulant factors, including fibrinogen and von Willebrand factor (Kessler et al., 1998; Boden and Rao, 2007). Even acute increases in blood glucose concentration cause spontaneous platelet aggregation, while AR inhibition significantly inhibits platelet aggregation (May et al., 1990), and has anti-platelet activity both in vitro and in vivo (Tawata et al., 1992), indicating a direct contribution to platelet aggregation. During chronic hyperglycemia, platelets from diabetic patients have increased responsiveness to collagen and adenosine diphosphate (ADP), which can be normalized by treatment with the AR inhibitor, sorbinil (Jennings et al., 1990). Previous animal studies also demonstrated that AR inhibition improved platelet hyperaggregation in streptozotocin-induced diabetic rats (Hara et al., 1995; Hotta et al., 1995). A recent proteomic study has shown that AR is abundantly expressed in human platelets, and its inhibitor, epalrestat, reduces platelet aggregation (Schulz et al., 2010), supporting a crucial role of AR in platelet aggregation. Consistent with these findings, inhibition of AR has also been demonstrated to attenuate the hyperglycemia-induced platelet hyperaggregation in human platelet by reducing oxidative stress (Tang et al., 2011). All these findings suggest that AR plays a central role in platelet aggregation, particularly during hyperglycemic conditions. Oxidative stress generated by the AR-dependent polyol pathway likely plays a major role in diabetic platelet hyperaggregation.
Interestingly, generalized overexpression of human AR in diabetic mice demonstrated increased expression of inflammatory markers and uptake of modified lipoprotein in macrophages. This AR overexpression increases atherosclerosis on a low-density lipoprotein receptor knockout background; a relatively low endogenous AR expression is found in wild-type mice (Vikramadithyan et al., 2005). Another study in ApoE−/− mice also demonstrated that human AR expression is proatherogenic and that expression, specifically in endothelial cells, leads to more severe disease (Vedantham et al., 2011). AR also contributes to diabetes abnormalities in vascular smooth muscle cell growth by increasing the intracellular oxidative stress, translocation, and phosphorylation of signaling targets (e.g., PKC) as well as release of TNF-α and related cytokines (Ramana et al., 2005; Srivastava et al., 2006; Reddy et al., 2009). Hyperglycemia-stimulated release of TNF-α and related cytokines from VSMCs might potentially mediate diabetes-induced acceleration of atherogenesis and endothelial dysfunction in humans. These data suggest that AR plays a critical role in atherothrombotic cardiovascular disease, and hyperglycemia in diabetic patients provides sufficient substrate for the vasculotoxic effects of this enzyme.
Besides diabetic vasculopathy, AR has also been found to play an important role in diabetic cardiomyopathy, characterized by myocardial contractile dysfunction independent of coronary artery disease (Rubler et al., 1972). A study using mouse hearts demonstrated that the activity of AR was increased (but its gene expression was suppressed) during the early stage of diabetes (Iwata et al., 2007). Despite low abundance of AR in mouse hearts, it is believed that the increased AR activity (as with hyperglycemia) may exacerbate myocardial dysfunction, leading to diabetic cardiomyopathy. AR may lead to hyperosmotic stress and may induce cardiac myocyte apoptosis (Galvez et al., 2003). Recently, the activity of AR was found to increase NADH/NAD+ ratio in diabetic rat heart, and inhibition of AR in diabetic hearts lowered the NADH/NAD+ ratio, normalizing the response to glucose metabolism and improving cardiac function (Ramasamy et al., 1997). Furthermore, the AR inhibitor, fidarestat, has been shown to improve contractile dysfunction and normalize Ca2+ signaling in the hearts of diabetic db/db obese mice. The intracellular superoxide induced by diabetes was also attenuated by treatment with fidarestat, suggesting that the polyol pathway activity contributes to contractile dysfunction by increasing superoxide formation in cardiac myocytes under hyperglycemic condition (Dong and Ren, 2007).
Aldose Reductase and Myocardial Ischemia/Reperfusion Injury
Myocardial ischemia/reperfusion (I/R) injury is one of the major causes of morbidity and mortality in patients with DM. Previous studies have indicated that ROS formed in the ischemic heart activate AR by modifying its cysteine residues to sulfenic acids (Kaiserova et al., 2008). Increased activity of AR in I/R rat hearts depletes intracellular NADPH, thereby reducing cellular GSH levels, increasing oxidative stress, as NADPH is also needed for the activity of glutathione reductase. AR was also reported to act as a mediator of late phase ischemic preconditioning. The increased AR activity at 24 h after ischemic preconditioning reduced the formation of HNE and the accumulation of HNE-modified proteins during myocardial I/R (Shinmura et al., 2002). Thus, a complete picture concerning the role of AR during myocardial ischemia remains elusive.
In recent years, it has been shown that AR is a key component of I/R injury in diabetic as well as non-diabetic heart (Ramasamy et al., 1997; Hwang et al., 2004). The protective mechanism contributed by AR inhibition is thought to be due to the preservation of high-energy phosphates and maintenance for a lower cytosolic NADH/NAD+ ratio, which can prevent the depletion of ATP and redox imbalance during myocardial I/R. Further studies showed that AR mediated the myocardial I/R injury in mice by depleting the ATP level thus increasing ROS generation (Iwata et al., 2006). Oxidative stress generated by AR is believed to be in part contributed to by enhanced mitochondrial permeability transition pore openings (Ananthakrishnan et al., 2009). Moreover, the AR-dependent polyol pathway was also found to contribute to myocardial contractile dysfunction and tissue damage by increasing oxidative stress in I/R rat hearts (Tang et al., 2008, 2010). Therefore, it is believed that the pharmacological inhibition of AR presents a novel adjunctive approach for protecting ischemic hearts in both diabetic and non-diabetic patients.
Apart from AR, SDH (converting sorbitol to fructose) has also been found to be another novel target for adjunctive protection of the ischemic myocardium. Studies indicate that inhibition of SDH attenuated the increased cytosolic NADH/NAD+ ratio and increased glycolysis as well as glucose oxidation (Hwang et al., 2003). This further supported the role of the polyol pathway in myocardial I/R injury, and suggests a mechanism for SDH competing with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) for NAD+. Thus, AR and SDH are both potential targets for pharmacological intervention for myocardial I/R injury.
Aldose Reductase and Other Complications in Diabetes
The pathogenic role of AR in diabetes is not limited to cardiovascular complications, and similar mechanisms are also involved in other complications, such as retinopathy, nephropathy, and neuropathy. The AR-dependent polyol pathway plays a major role in diabetic cataractogenesis. Previous studies showed that structurally diverse AR inhibitors prevented cataract formation in streptozotocin-induced diabetic rats (Sun et al., 2006; Drel et al., 2008). The key role for AR in diabetic cataractogenesis is further supported by studies in AR-overexpressing mice. Sugar cataracts form in transgenic diabetic mice expressing human AR in the lens, but not in wild-type streptozotocin-induced diabetic mice which have very low expression of AR (Varma and Kinoshita, 1974; Lee et al., 1995). AR siRNA transfection and inhibition suppressed high glucose-induced ROS formation, NF-kappaB activation, and apoptosis in rat lens epithelial cells (Nambu et al., 2008). Studies on slow cataract formation showed that metabolic imbalance caused by increased AR activity plays a major role in slow cataract development in mature diabetic animals (Sun et al., 2006; Drel et al., 2008), which is more relevant to diabetic patients. Two further studies suggested an important role for AR in high glucose- and diabetes-induced impairment of lenticular signaling (Ramana et al., 2003; Zatechka et al., 2003). Therefore, increased AR activity is likely to contribute to diabetic cataract formation through oxidative signaling mechanisms.
The AR-dependent polyol pathway is one of the more promising targets for diabetic neuropathy. Increased AR activity leads to more severe diabetic neuropathy (Yagihashi et al., 2001; Song et al., 2003) and decreased levels of GSH (Song et al., 2003). Previous studies demonstrated that hyperglycemia-induced oxidative stress led to the activation of mitogen-activated protein kinase (MAPK), which may have contributed to neuronal pathogenesis (Wang et al., 1998; Purves et al., 2001). Fidarestat, an AR inhibitor, was shown to prevent activation of MAPK and nerve conduction velocity deficits in diabetes (Price et al., 2004), indicating that AR inhibitors could reduce the diabetes-induced oxidative stress. Other studies using AR knockout mice (Ho et al., 2000) also demonstrated that AR deficiency could prevent diabetes-induced oxidative stress in nerve cells in the retina (Cheung et al., 2005). Moreover, both AR deficiency and AR inhibition reduced oxidative stress in the peripheral nerves and markedly protected mice from diabetes-induced functional deficits (Ho et al., 2006). All these findings suggest that AR contributes to the pathogenesis of diabetic neuropathy via oxidative stress.
AR is differentially expressed in mammalian kidney, where AR expression is low under physiological condition in the glomerulus but significantly increased in diabetic human patients (Corder et al., 1979; Kasajima et al., 2001). In diabetic rats, it was found that hyperglycemia-induced increase in glomerular sorbitol levels was attenuated by treatment with an AR inhibitor, sorbinil (Beyer-Mears et al., 1984). Hyperactivation of AR in renal cells have been linked with aberrant activation of PKC (Ishii et al., 1998; Kapor-Drezgic et al., 1999; Noh and King, 2007), generation of advanced glycation products, increased expression of TGF-β and generation of ROS (Oates and Mylari, 1999). A recent study using mice with AR deficiency in all tissues except in the renal medulla, showed that genetic ablation of AR significantly ameliorates the development of diabetic nephropathy in streptozotocin-induced diabetic mice (Liu et al., 2011). Together these data suggest that activation of AR by hyperglycemia in the renal glomeruli contributes to the onset and progression of diabetic nephropathy via oxidative stress.
Summary
Accumulating evidence in experimental studies has demonstrated the mechanistic role of AR in various metabolic diseases associated with diabetes and its complications. Although a number of AR inhibitors have been tested or are currently undergoing testing in clinical trials (reviewed in Giannoukakis, 2008), the clinical efficacy is uncertain and there are concerns with associated adverse effects such as hepatic damage. One of the possible reasons for the discrepancy between experimental animal studies and human clinical studies (besides species differences) is the length of time between the onset of diabetes and start of the AR inhibitor treatment. In many experimental studies, treatment with AR inhibitors are often commenced before the onset of diabetic complications and induction of AR. In contrast, treatment with AR inhibitors are usually administrated to patients with longstanding DM where the affected tissues (e.g., nerves and retina) have already undergone extensive damage. Thus it is not surprising that the clinical efficacy of AR inhibitors is relatively low. However, human platelets (a critical contributor to atherothrombosis in DM) has a short life span with a high turnover rate and thus may respond to AR inhibitor therapy in conjunction with low dose aspirin. As discussed in this review, under hyperglycemic conditions, activation of the AR pathway upregulates many other glucose toxicity pathways (e.g., non-enzymatic glycation, PKC pathway, hexosamine pathway, and disruption of mitochondrial respiratory chain), so treatment with AR inhibitors alone may not be as effective. AR inhibitors may serve as an effective adjunct therapy for prevention of diabetic complications.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the National Institutes of Health (NIH), the National Heart, Lung, and Blood Institute (NHLBI), and the American Heart Association (AHA).
References
American Diabetes Association. (2000). Implications of the United Kingdom Prospective Diabetes Study. Diabetes Care 23(Suppl. 1), S27–S31.
American Diabetes Association. (2003). Implications of the diabetes control and complications trial. Diabetes Care 26(Suppl. 1), S25–S27.
Ananthakrishnan, R., Kaneko, M., Hwang, Y. C., Quadri, N., Gomez, T., Li, Q., Caspersen, C., and Ramasamy, R. (2009). Aldose reductase mediates myocardial ischemia-reperfusion injury in part by opening mitochondrial permeability transition pore. Am. J. Physiol. Heart Circ. Physiol. 296, H333–H341.
Andersen, K., Hurlen, M., Arnesen, H., and Seljeflot, I. (2002). Aspirin non-responsiveness as measured by PFA-100 in patients with coronary artery disease. Thromb. Res. 108, 37–42.
Ansari, N. H., Bhatnagar, A., Liu, S. Q., and Srivastava, S. K. (1991). Purification and characterization of aldose reductase and aldehyde reductase from human kidney. Biochem. Int. 25, 755–765.
Beyer-Mears, A., Ku, L., and Cohen, M. P. (1984). Glomerular polyol accumulation in diabetes and its prevention by oral sorbinil. Diabetes 33, 604–607.
Boden, G., and Rao, A. K. (2007). Effects of hyperglycemia and hyperinsulinemia on the tissue factor pathway of blood coagulation. Curr. Diab. Rep. 7, 223–227.
Boyle, J. P., Honeycutt, A. A., Narayan, K. M., Hoerger, T. J., Geiss, L. S., Chen, H., and Thompson, T. J. (2001). Projection of diabetes burden through 2050: impact of changing demography and disease prevalence in the U.S. Diabetes Care 24, 1936–1940.
Buchanan, M. R., and Brister, S. J. (1995). Individual variation in the effects of ASA on platelet function: implications for the use of ASA clinically. Can. J. Cardiol. 11, 221–227.
Cheng, H. M., and Gonzalez, R. G. (1986). The effect of high glucose and oxidative stress on lens metabolism, aldose reductase, and senile cataractogenesis. Metab. Clin. Exp. 35, 10–14.
Cheung, A. K., Fung, M. K., Lo, A. C., Lam, T. T., So, K. F., Chung, S. S., and Chung, S. K. (2005). Aldose reductase deficiency prevents diabetes-induced blood-retinal barrier breakdown, apoptosis, and glial reactivation in the retina of db/db mice. Diabetes 54, 3119–3125.
Corder, C. N., Braughler, J. M., and Culp, P. A. (1979). Quantitative histochemistry of the sorbitol pathway in glomeruli and small arteries of human diabetic kidney. Folia Histochem. Cytochem. (Krakow) 17, 137–145.
Cromlish, J. A., and Flynn, T. G. (1983). Purification and characterization of two aldose reductase isoenzymes from rabbit muscle. J. Biol. Chem. 258, 3416–3424.
Cromlish, J. A., Yoshimoto, C. K., and Flynn, T. G. (1985). Purification and characterization of four NADPH-dependent aldehyde reductases from pig brain. J. Neurochem. 44, 1477–1484.
Das, B., and Srivastava, S. K. (1985a). Purification and properties of aldehyde reductases from human placenta. Biochim. Biophys. Acta 840, 324–333.
Das, B., and Srivastava, S. K. (1985b). Purification and properties of aldose reductase and aldehyde reductase II from human erythrocyte. Arch. Biochem. Biophys. 238, 670–679.
Dong, F., and Ren, J. (2007). Fidarestat improves cardiomyocyte contractile function in db/db diabetic obese mice through a histone deacetylase Sir2-dependent mechanism. J. Hypertens. 25, 2138–2147.
Drel, V. R., Pacher, P., Ali, T. K., Shin, J., Julius, U., El-Remessy, A. B., and Obrosova, I. G. (2008). Aldose reductase inhibitor fidarestat counteracts diabetes-associated cataract formation, retinal oxidative-nitrosative stress, glial activation, and apoptosis. Int. J. Mol. Med. 21, 667–676.
Drel, V. R., Pacher, P., Stevens, M. J., and Obrosova, I. G. (2006). Aldose reductase inhibition counteracts nitrosative stress and poly(ADP-ribose) polymerase activation in diabetic rat kidney and high-glucose-exposed human mesangial cells. Free Radic. Biol. Med. 40, 1454–1465.
El-Kabbani, O., Ruiz, F., Darmanin, C., and Chung, R. P. (2004). Aldose reductase structures: implications for mechanism and inhibition. Cell. Mol. Life Sci. 61, 750–762.
Flynn, T. G. (1982). Aldehyde reductases: monomeric NADPH-dependent oxidoreductases with multifunctional potential. Biochem. Pharmacol. 31, 2705–2712.
Galvez, A. S., Ulloa, J. A., Chiong, M., Criollo, A., Eisner, V., Barros, L. F., and Lavandero, S. (2003). Aldose reductase induced by hyperosmotic stress mediates cardiomyocyte apoptosis: differential effects of sorbitol and mannitol. J. Biol. Chem. 278, 38484–38494.
Giannoukakis, N. (2006). Drug evaluation: ranirestat – an aldose reductase inhibitor for the potential treatment of diabetic complications. Curr. Opin. Investig. Drugs 7, 916–923.
Giannoukakis, N. (2008). Ranirestat as a therapeutic aldose reductase inhibitor for diabetic complications. Expert Opin. Investig. Drugs 17, 575–581.
Gonzalez, R. G., Barnett, P., Aguayo, J., Cheng, H. M., and Chylack, L. T. Jr. (1984). Direct measurement of polyol pathway activity in the ocular lens. Diabetes 33, 196–199.
Greene, D. A., Lattimer, S. A., and Sima, A. A. (1987). Sorbitol, phosphoinositides, and sodium-potassium-ATPase in the pathogenesis of diabetic complications. N. Engl. J. Med. 316, 599–606.
Grotemeyer, K. H. (1991). Effects of acetylsalicylic acid in stroke patients. Evidence of nonresponders in a subpopulation of treated patients. Thromb. Res. 63, 587–593.
Grotemeyer, K. H., Scharafinski, H. W., and Husstedt, I. W. (1993). Two-year follow-up of aspirin responder and aspirin non responder. A pilot-study including 180 post-stroke patients. Thromb. Res. 71, 397–403.
Grundmann, K., Jaschonek, K., Kleine, B., Dichgans, J., and Topka, H. (2003). Aspirin non-responder status in patients with recurrent cerebral ischemic attacks. J. Neurol. 250, 63–66.
Haffner, S. M., Lehto, S., Ronnemaa, T., Pyorala, K., and Laakso, M. (1998). Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N. Engl. J. Med. 339, 229–234.
Hamada, Y., Araki, N., Koh, N., Nakamura, J., Horiuchi, S., and Hotta, N. (1996a). Rapid formation of advanced glycation end products by intermediate metabolites of glycolytic pathway and polyol pathway. Biochem. Biophys. Res. Commun. 228, 539–543.
Hamada, Y., Araki, N., Horiuchi, S., and Hotta, N. (1996b). Role of polyol pathway in nonenzymatic glycation. Nephrol. Dial. Transplant. 11(Suppl. 5), 95–98.
Hamada, Y., and Nakamura, J. (2004). Clinical potential of aldose reductase inhibitors in diabetic neuropathy. Treat. Endocrinol. 3, 245–255.
Hamada, Y., Nakamura, J., Naruse, K., Komori, T., Kato, K., Kasuya, Y., Nagai, R., Horiuchi, S., and Hotta, N. (2000). Epalrestat, an aldose reductase ihibitor, reduces the levels of Nepsilon-(carboxymethyl)lysine protein adducts and their precursors in erythrocytes from diabetic patients. Diabetes Care 23, 1539–1544.
Hara, T., Nakamura, J., Koh, N., Sakakibara, F., Hamada, Y., Sasaki, H., Naruse, K., Nakashima, E., Takeuchi, N., and Inukai, S. (1995). An aldose reductase inhibitor, TAT, reduces ADP-induced platelet hyperaggregation in streptozotocin-induced diabetic rats with neuropathy. J. Lab. Clin. Med. 126, 541–547.
Heather, L. C., and Clarke, K. (2011). Metabolism, hypoxia and the diabetic heart. J. Mol. Cell. Cardiol. 50, 598–605.
Helgason, C. M., Bolin, K. M., Hoff, J. A., Winkler, S. R., Mangat, A., Tortorice, K. L., and Brace, L. D. (1994). Development of aspirin resistance in persons with previous ischemic stroke. Stroke 25, 2331–2336.
Hers, H. G. (1956). The mechanism of the transformation of glucose in fructose in the seminal vesicles. Biochim. Biophys. Acta 22, 202–203.
Ho, E. C., Lam, K. S., Chen, Y. S., Yip, J. C., Arvindakshan, M., Yamagishi, S., Yagihashi, S., Oates, P. J., Ellery, C. A., Chung, S. S., and Chung, S. K. (2006). Aldose reductase-deficient mice are protected from delayed motor nerve conduction velocity, increased c-Jun NH2-terminal kinase activation, depletion of reduced glutathione, increased superoxide accumulation, and DNA damage. Diabetes 55, 1946–1953.
Ho, H. T., Chung, S. K., Law, J. W., Ko, B. C., Tam, S. C., Brooks, H. L., Knepper, M., and Chung, S. S. (2000). Aldose reductase-deficient mice develop nephrogenic diabetes insipidus. Mol. Cell. Biol. 20, 5840–5846.
Hotta, N., Koh, N., Sakakibara, F., Nakamura, J., Hamada, Y., Hara, T., Takeuchi, N., Inukai, S., Kasama, N., Fukasawa, H., and Kakuta, H. (1995). An aldose reductase inhibitor, TAT, prevents electroretinographic abnormalities and ADP-induced hyperaggregability in streptozotocin-induced diabetic rats. Eur. J. Clin. Invest. 25, 948–954.
Hwang, Y. C., Bakr, S., Ellery, C. A., Oates, P. J., and Ramasamy, R. (2003). Sorbitol dehydrogenase: a novel target for adjunctive protection of ischemic myocardium. FASEB J. 17, 2331–2333.
Hwang, Y. C., Kaneko, M., Bakr, S., Liao, H., Lu, Y., Lewis, E. R., Yan, S., Ii, S., Itakura, M., Rui, L., Skopicki, H., Homma, S., Schmidt, A. M., Oates, P. J., Szabolcs, M., and Ramasamy, R. (2004). Central role for aldose reductase pathway in myocardial ischemic injury. FASEB J. 18, 1192–1199.
Ishii, H., Tada, H., and Isogai, S. (1998). An aldose reductase inhibitor prevents glucose-induced increase in transforming growth factor-beta and protein kinase C activity in cultured mesangial cells. Diabetologia 41, 362–364.
Iwata, K., Matsuno, K., Nishinaka, T., Persson, C., and Yabe-Nishimura, C. (2006). Aldose reductase inhibitors improve myocardial reperfusion injury in mice by a dual mechanism. J. Pharmacol. Sci. 102, 37–46.
Iwata, K., Nishinaka, T., Matsuno, K., Kakehi, T., Katsuyama, M., Ibi, M., and Yabe-Nishimura, C. (2007). The activity of aldose reductase is elevated in diabetic mouse heart. J. Pharmacol. Sci. 103, 408–416.
Iwata, N., Inazu, N., and Satoh, T. (1990). The purification and properties of aldose reductase from rat ovary. Arch. Biochem. Biophys. 282, 70–77.
Jennings, P. E., Nightingale, S., Le Guen, C., Lawson, N., Williamson, J. R., Hoffman, P., and Barnett, A. H. (1990). Prolonged aldose reductase inhibition in chronic peripheral diabetic neuropathy: effects on microangiopathy. Diabet. Med. 7, 63–68.
Johnson, B. F., Nesto, R. W., Pfeifer, M. A., Slater, W. R., Vinik, A. I., Chyun, D. A., Law, G., Wackers, F. J., and Young, L. H. (2004). Cardiac abnormalities in diabetic patients with neuropathy: effects of aldose reductase inhibitor administration. Diabetes Care 27, 448–454.
Kaiserova, K., Tang, X. L., Srivastava, S., and Bhatnagar, A. (2008). Role of nitric oxide in regulating aldose reductase activation in the ischemic heart. J. Biol. Chem. 283, 9101–9112.
Kapor-Drezgic, J., Zhou, X., Babazono, T., Dlugosz, J. A., Hohman, T., and Whiteside, C. (1999). Effect of high glucose on mesangial cell protein kinase C-delta and -epsilon is polyol pathway-dependent. J. Am. Soc. Nephrol. 10, 1193–1203.
Kasajima, H., Yamagishi, S., Sugai, S., Yagihashi, N., and Yagihashi, S. (2001). Enhanced in situ expression of aldose reductase in peripheral nerve and renal glomeruli in diabetic patients. Virchows Arch. 439, 46–54.
Kawamura, N., Ookawara, T., Suzuki, K., Konishi, K., Mino, M., and Taniguchi, N. (1992). Increased glycated Cu,Zn-superoxide dismutase levels in erythrocytes of patients with insulin-dependent diabetis mellitus. J. Clin. Endocrinol. Metab. 74, 1352–1354.
Kawasaki, N., Tanimoto, T., and Tanaka, A. (1989). Characterization of aldose reductase and aldehyde reductase from rat testis. Biochim. Biophys. Acta 996, 30–36.
Kennedy, A. L., and Lyons, T. J. (1997). Glycation, oxidation, and lipoxidation in the development of diabetic complications. Metab. Clin. Exp. 46, 14–21.
Kessler, L., Wiesel, M. L., Attali, P., Mossard, J. M., Cazenave, J. P., and Pinget, M. (1998). Von Willebrand factor in diabetic angiopathy. Diabetes Metab. 24, 327–336.
Kinoshita, J. H., Merola, L. O., Satoh, K., and Dikmak, E. (1962). Osmotic changes caused by the accumulation of dulcitol in the lenses of rats fed with galactose. Nature 194, 1085–1087.
Lee, A. Y., Chung, S. K., and Chung, S. S. (1995). Demonstration that polyol accumulation is responsible for diabetic cataract by the use of transgenic mice expressing the aldose reductase gene in the lens. Proc. Natl. Acad. Sci. U.S.A. 92, 2780–2784.
Lee, A. Y., and Chung, S. S. (1999). Contributions of polyol pathway to oxidative stress in diabetic cataract. FASEB J. 13, 23–30.
Liu, H., Luo, Y., Zhang, T., Wu, Q., Yuan, L., Chung, S. S., Oates, P. J., and Yang, J. Y. (2011). Genetic deficiency of aldose reductase counteracts the development of diabetic nephropathy in C57BL/6 mice. Diabetologia 54, 1242–1251.
Macchi, L., Christiaens, L., Brabant, S., Sorel, N., Allal, J., Mauco, G., and Brizard, A. (2002). Resistance to aspirin in vitro is associated with increased platelet sensitivity to adenosine diphosphate. Thromb. Res. 107, 45–49.
Marshall, P. W., Williams, A. J., Dixon, R. M., Growcott, J. W., Warburton, S., Armstrong, J., and Moores, J. (1997). A comparison of the effects of aspirin on bleeding time measured using the Simplate method and closure time measured using the PFA-100, in healthy volunteers. Br. J. Clin. Pharmacol. 44, 151–155.
May, J., Loesche, W., and Heptinstall, S. (1990). Glucose increases spontaneous platelet aggregation in whole blood. Thromb. Res. 59, 489–495.
Morgan, P. E., Dean, R. T., and Davies, M. J. (2002). Inactivation of cellular enzymes by carbonyls and protein-bound glycation/glycoxidation products. Arch. Biochem. Biophys. 403, 259–269.
Morjana, N. A., and Flynn, T. G. (1989). Aldose reductase from human psoas muscle. Purification, substrate specificity, immunological characterization, and effect of drugs and inhibitors. J. Biol. Chem. 264, 2906–2911.
Morre, D. M., Lenaz, G., and Morre, D. J. (2000). Surface oxidase and oxidative stress propagation in aging. J. Exp. Biol. 203, 1513–1521.
Morrison, A. D., Clements, R. S. Jr., Travis, S. B., Oski, F., and Winegrad, A. I. (1970). Glucose utilization by the polyol pathway in human erythrocytes. Biochem. Biophys. Res. Commun. 40, 199–205.
Mullarkey, C. J., Edelstein, D., and Brownlee, M. (1990). Free radical generation by early glycation products: a mechanism for accelerated atherogenesis in diabetes. Biochem. Biophys. Res. Commun. 173, 932–939.
Nambu, H., Kubo, E., Takamura, Y., Tsuzuki, S., Tamura, M., and Akagi, Y. (2008). Attenuation of aldose reductase gene suppresses high-glucose-induced apoptosis and oxidative stress in rat lens epithelial cells. Diabetes Res. Clin. Pract. 82, 18–24.
Nishikawa, T., Edelstein, D., Du, X. L., Yamagishi, S., Matsumura, T., Kaneda, Y., Yorek, M. A., Beebe, D., Oates, P. J., Hammes, H. P., Giardino, I., and Brownlee, M. (2000). Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404, 787–790.
Noh, H., and King, G. L. (2007). The role of protein kinase C activation in diabetic nephropathy. Kidney Int. Suppl. 106, S49–S53.
Oates, P. J., and Mylari, B. L. (1999). Aldose reductase inhibitors: therapeutic implications for diabetic complications. Expert Opin. Investig. Drugs 8, 2095–2119.
Obrosova, I. G., Minchenko, A. G., Vasupuram, R., White, L., Abatan, O. I., Kumagai, A. K., Frank, R. N., and Stevens, M. J. (2003). Aldose reductase inhibitor fidarestat prevents retinal oxidative stress and vascular endothelial growth factor overexpression in streptozotocin-diabetic rats. Diabetes 52, 864–871.
Obrosova, I. G., Pacher, P., Szabo, C., Zsengeller, Z., Hirooka, H., Stevens, M. J., and Yorek, M. A. (2005). Aldose reductase inhibition counteracts oxidative-nitrosative stress and poly(ADP-ribose) polymerase activation in tissue sites for diabetes complications. Diabetes 54, 234–242.
Ohta, M., Tanimoto, T., and Tanaka, A. (1991). Localization, isolation and properties of three NADPH-dependent aldehyde reducing enzymes from dog kidney. Biochim. Biophys. Acta 1078, 395–403.
Pappas, J. M., Westengard, J. C., and Bull, B. S. (1994). Population variability in the effect of aspirin on platelet function. Implications for clinical trials and therapy. Arch. Pathol. Lab. Med. 118, 801–804.
Petrash, J. M., and Srivastava, S. K. (1982). Purification and properties of human liver aldehyde reductases. Biochim. Biophys. Acta 707, 105–114.
Price, S. A., Agthong, S., Middlemas, A. B., and Tomlinson, D. R. (2004). Mitogen-activated protein kinase p38 mediates reduced nerve conduction velocity in experimental diabetic neuropathy: interactions with aldose reductase. Diabetes 53, 1851–1856.
Purves, T., Middlemas, A., Agthong, S., Jude, E. B., Boulton, A. J., Fernyhough, P., and Tomlinson, D. R. (2001). A role for mitogen-activated protein kinases in the etiology of diabetic neuropathy. FASEB J. 15, 2508–2514.
Ramana, K. V., Friedrich, B., Bhatnagar, A., and Srivastava, S. K. (2003). Aldose reductase mediates cytotoxic signals of hyperglycemia and TNF-alpha in human lens epithelial cells. FASEB J. 17, 315–317.
Ramana, K. V., Friedrich, B., Tammali, R., West, M. B., Bhatnagar, A., and Srivastava, S. K. (2005). Requirement of aldose reductase for the hyperglycemic activation of protein kinase C and formation of diacylglycerol in vascular smooth muscle cells. Diabetes 54, 818–829.
Ramasamy, R., Oates, P. J., and Schaefer, S. (1997). Aldose reductase inhibition protects diabetic and nondiabetic rat hearts from ischemic injury. Diabetes 46, 292–300.
Ramirez, M. A., and Borja, N. L. (2008). Epalrestat: an aldose reductase inhibitor for the treatment of diabetic neuropathy. Pharmacotherapy 28, 646–655.
Reddy, A. B., Ramana, K. V., Srivastava, S., Bhatnagar, A., and Srivastava, S. K. (2009). Aldose reductase regulates high glucose-induced ectodomain shedding of tumor necrosis factor (TNF)-alpha via protein kinase C-delta and TNF-alpha converting enzyme in vascular smooth muscle cells. Endocrinology 150, 63–74.
Rubler, S., Dlugash, J., Yuceoglu, Y. Z., Kumral, T., Branwood, A. W., and Grishman, A. (1972). New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am. J. Cardiol. 30, 595–602.
Schmidt, A. M., Hori, O., Brett, J., Yan, S. D., Wautier, J. L., and Stern, D. (1994). Cellular receptors for advanced glycation end products. Implications for induction of oxidant stress and cellular dysfunction in the pathogenesis of vascular lesions. Arterioscler. Thromb. 14, 1521–1528.
Schulz, C., Leuschen, N. V., Frohlich, T., Lorenz, M., Pfeiler, S., Gleissner, C. A., Kremmer, E., Kessler, M., Khandoga, A. G., Engelmann, B., Ley, K., Massberg, S., and Arnold, G. J. (2010). Identification of novel downstream targets of platelet glycoprotein VI activation by differential proteome analysis: implications for thrombus formation. Blood 115, 4102–4110.
Shinmura, K., Bolli, R., Liu, S. Q., Tang, X. L., Kodani, E., Xuan, Y. T., Srivastava, S., and Bhatnagar, A. (2002). Aldose reductase is an obligatory mediator of the late phase of ischemic preconditioning. Circ. Res. 91, 240–246.
Song, Z., Fu, D. T., Chan, Y. S., Leung, S., Chung, S. S., and Chung, S. K. (2003). Transgenic mice overexpressing aldose reductase in Schwann cells show more severe nerve conduction velocity deficit and oxidative stress under hyperglycemic stress. Mol. Cell. Neurosci. 23, 638–647.
Srivastava, S., Ramana, K. V., Tammali, R., Srivastava, S. K., and Bhatnagar, A. (2006). Contribution of aldose reductase to diabetic hyperproliferation of vascular smooth muscle cells. Diabetes 55, 901–910.
Srivastava, S. K., Ansari, N. H., Hair, G. A., and Das, B. (1984). Aldose and aldehyde reductases in human tissues. Biochim. Biophys. Acta 800, 220–227.
Steele, J. W., Faulds, D., and Goa, K. L. (1993). Epalrestat. A review of its pharmacology, and therapeutic potential in late-onset complications of diabetes mellitus. Drugs Aging 3, 532–555.
Sun, W., Oates, P. J., Coutcher, J. B., Gerhardinger, C., and Lorenzi, M. (2006). A selective aldose reductase inhibitor of a new structural class prevents or reverses early retinal abnormalities in experimental diabetic retinopathy. Diabetes 55, 2757–2762.
Tang, W. H., Kravtsov, G. M., Sauert, M., Tong, X. Y., Hou, X. Y., Wong, T. M., Chung, S. K., and Man Chung, S. S. (2010). Polyol pathway impairs the function of SERCA and RyR in ischemic-reperfused rat hearts by increasing oxidative modifications of these proteins. J. Mol. Cell. Cardiol. 49, 58–69.
Tang, W. H., Stitham, J., Gleim, S., Di Febbo, C., Porreca, E., Fava, C., Tacconelli, S., Capone, M., Evangelista, V., Levantesi, G., Wen, L., Martin, K., Minuz, P., Rade, J., Patrignani, P., and Hwa, J. (2011). Glucose and collagen regulate human platelet activity through aldose reductase induction of thromboxane. J. Clin. Invest. 121, 4462–4476.
Tang, W. H., Wu, S., Wong, T. M., Chung, S. K., and Chung, S. S. (2008). Polyol pathway mediates iron-induced oxidative injury in ischemic-reperfused rat heart. Free Radic. Biol. Med. 45, 602–610.
Tawata, M., Aida, K., Noguchi, T., Ozaki, Y., Kume, S., Sasaki, H., Chin, M., and Onaya, T. (1992). Anti-platelet action of isoliquiritigenin, an aldose reductase inhibitor in licorice. Eur. J. Pharmacol. 212, 87–92.
Terubayashi, H., Sato, S., Nishimura, C., Kador, P. F., and Kinoshita, J. H. (1989). Localization of aldose and aldehyde reductase in the kidney. Kidney Int. 36, 843–851.
Tesfamariam, B., Palacino, J. J., Weisbrod, R. M., and Cohen, R. A. (1993). Aldose reductase inhibition restores endothelial cell function in diabetic rabbit aorta. J. Cardiovasc. Pharmacol. 21, 205–211.
Van den Enden, M. K., Nyengaard, J. R., Ostrow, E., Burgan, J. H., and Williamson, J. R. (1995). Elevated glucose levels increase retinal glycolysis and sorbitol pathway metabolism. Implications for diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 36, 1675–1685.
Vander Jagt, D. L., Hunsaker, L. A., Robinson, B., Stangebye, L. A., and Deck, L. M. (1990a). Aldehyde and aldose reductases from human placenta. Heterogeneous expression of multiple enzyme forms. J. Biol. Chem. 265, 10912–10918.
Vander Jagt, D. L., Robinson, B., Taylor, K. K., and Hunsaker, L. A. (1990b). Aldose reductase from human skeletal and heart muscle. Interconvertible forms related by thiol-disulfide exchange. J. Biol. Chem. 265, 20982–20987.
Varma, S. D., and Kinoshita, J. H. (1974). The absence of cataracts in mice with congenital hyperglycemia. Exp. Eye Res. 19, 577–582.
Vedantham, S., Noh, H., Ananthakrishnan, R., Son, N., Hallam, K., Hu, Y., Yu, S., Shen, X., Rosario, R., Lu, Y., Ravindranath, T., Drosatos, K., Huggins, L. A., Schmidt, A. M., Goldberg, I. J., and Ramasamy, R. (2011). Human aldose reductase expression accelerates atherosclerosis in diabetic apolipoprotein E-/- mice. Arterioscler. Thromb. Vasc. Biol. 31, 1805–1813.
Vikramadithyan, R. K., Hu, Y., Noh, H. L., Liang, C. P., Hallam, K., Tall, A. R., Ramasamy, R., and Goldberg, I. J. (2005). Human aldose reductase expression accelerates diabetic atherosclerosis in transgenic mice. J. Clin. Invest. 115, 2434–2443.
Wang, X., Martindale, J. L., Liu, Y., and Holbrook, N. J. (1998). The cellular response to oxidative stress: influences of mitogen-activated protein kinase signalling pathways on cell survival. Biochem. J. 333(Pt 2), 291–300.
Wermuth, B., Burgisser, H., Bohren, K., and von Wartburg, J. P. (1982). Purification and characterization of human-brain aldose reductase. Eur. J. Biochem. 127, 279–284.
Wiernsperger, N. F. (2003). Oxidative stress as a therapeutic target in diabetes: revisiting the controversy. Diabetes Metab. 29, 579–585.
Wilson, G., and Perry, T. (2009). Is tight glycemic control in type 2 diabetes really worthwhile? No. Can. Fam. Physician. 55, 581–588.
Wolff, S. P., and Dean, R. T. (1987). Glucose autoxidation and protein modification. The potential role of “autoxidative glycosylation” in diabetes. Biochem. J. 245, 243–250.
Yabe-Nishimura, C. (1998). Aldose reductase in glucose toxicity: a potential target for the prevention of diabetic complications. Pharmacol. Rev. 50, 21–33.
Yagihashi, S., Yamagishi, S. I., Wada Ri, R., Baba, M., Hohman, T. C., Yabe-Nishimura, C., and Kokai, Y. (2001). Neuropathy in diabetic mice overexpressing human aldose reductase and effects of aldose reductase inhibitor. Brain 124, 2448–2458.
Yim, M. B., Yim, H. S., Lee, C., Kang, S. O., and Chock, P. B. (2001). Protein glycation: creation of catalytic sites for free radical generation. Ann. N. Y. Acad. Sci. 928, 48–53.
Zatechka, D. S. Jr., Kador, P. F., Garcia-Castineiras, S., and Lou, M. F. (2003). Diabetes can alter the signal transduction pathways in the lens of rats. Diabetes 52, 1014–1022.
Keywords: aldose reductase, oxidative stress, diabetes mellitus, atherosclerosis, thrombosis
Citation: Tang WH, Martin KA and Hwa J (2012) Aldose reductase, oxidative stress, and diabetic mellitus. Front. Pharmacol. 3:87. doi: 10.3389/fphar.2012.00087
Received: 06 March 2012; Paper pending published: 26 March 2012;
Accepted: 19 April 2012; Published online: 09 May 2012.
Edited by:
Yi Jin, University of Pennsylvania, USACopyright: © 2012 Tang, Martin and Hwa. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: John Hwa, Section of Cardiovascular Medicine, Department of Internal Medicine, Yale School of Medicine, Cardiovascular Research Center, Yale University, 300 George Street, Room 759H, New Haven, CT 06511, USA. e-mail:am9obi5od2FAeWFsZS5lZHU=.