

- Section on Synaptic Pharmacology, Laboratory for Integrative Neuroscience, National Institute on Alcohol Abuse and Alcoholism, U.S. National Institutes of Health, Rockville, MD, USA
The nigrostriatal dopaminergic system is implicated in action control and learning. A large body of work has focused on the contribution of this system to modulation of the corticostriatal synapse, the predominant synapse type in the striatum. Signaling through the D2 dopamine receptor is necessary for endocannabinoid-mediated depression of corticostriatal glutamate release. Here we review the known details of this mechanism and discuss newly discovered signaling pathways interacting with this system that ultimately exert dynamic control of cortical input to the striatum and striatal output. This topic is timely with respect to Parkinson’s disease given recent data indicating changes in the striatal endocannabinoid system in patients with this disorder.
Endocannabinoids and Synaptic Plasticity
Discovery of the cannabinoid signaling system grew out of studies of the actions of cannabis-derived drugs that contain Δ9-Tetrahyrocannabinol (Δ 9-THC). The discovery of a Δ9-THC-activated G protein-coupled receptor (GPCR), termed the cannabinoid 1 or CB1 receptor (Matsuda et al., 1990), stimulated the search for endogenous ligands (endocannabinoids, eCBs) that could activate the receptor. Eventually, two lipid metabolites were found to have CB1 agonist action. Arachidonoyl ethanolamide (AEA or anandamide) is derived from phosphatidylethanolamide, with several potential synthesis pathways described in the literature (Devane et al., 1992; Di Marzo et al., 1994; Liu et al., 2008). The other major endocannabinoid, 2-arachidonoyl-glycerol (2-AG) is produced from membrane lipids via a two-stage reaction catalyzed by phospholipase C and diacylglycerol lipase (DAGL; Mechoulam et al., 1995). Enzymes that catalyze the breakdown of AEA (fatty acid amide hydrolase, FAAH) and 2-AG (monoacylglycerol lipase) have also been characterized. Both eCBs are known to activate CB1 receptors, and the other known cannabinoid receptor termed CB2, and act throughout the body and brain. It is worth noting, however, that AEA has been reported to have many additional actions, most notably acting as an agonist for the vanilloid-activated transient receptor potential vanilloid 1 (TRPV1) channel (Zygmunt et al., 1999; Smart et al., 2000).
The CB1 receptor couples to the Gi/o subtypes of G-proteins. Like the other Gi/o-activating GPCRs, CB1 receptors inhibit adenylyl cyclase, activate G-protein-activated potassium channels (GIRKs), inhibit voltage-gated calcium channels (VGCCs), and inhibit neurotransmitter release (Howlett and Fleming, 1984; Howlett et al., 1986; Ishac et al., 1996; Jin et al., 1999). Within the central nervous system, including the striatum, CB1 receptors are located almost exclusively on presynaptic axon terminals (Katona et al., 1999; Rodriguez et al., 2001; Mátyás et al., 2006). It is thus not surprising that the most appreciated function of these receptors is to inhibit neurotransmitter release. Within the striatum, presynaptic CB1 receptors are found on GABAergic and glutamatergic axon terminals. Activation of CB1 with synthetic agonists has been shown to inhibit release of GABA, glutamate, and dopamine (DA) within the striatum (Cadogan et al., 1997; Szabo et al., 1998; Kathmann et al., 1999; Gerdeman and Lovinger, 2001; Sidló et al., 2008), although the effects on DA release appear to involve indirect effects of other neurotransmitters (Sidló et al., 2008).
eCBs and CB1 receptors are also implicated in several forms of synaptic plasticity involving presynaptic expression mechanisms. There are three common forms: depolarization-induced suppression of excitatory or inhibitory transmission (DSE/DSI), short-term depression (STD) activated by Gq-coupled postsynaptic metabotropic receptors, and long-term depression (LTD; Lovinger, 2008; Heifets and Castillo, 2009). The time courses of these synaptic depressant phenomena differ, with DSE/DSI and STD persisting only as long as the eCB signal acts on CB1 receptors, while LTD continues indefinitely even after CB1 activation is terminated (Lovinger, 2008; Heifets and Castillo, 2009). It should be noted that the mechanisms involved in DSE/DSI and STD can interact, such that activation of metabotropic receptors can increase the magnitude and extend the time course of depolarization-induced synaptic suppression (Varma et al., 2001; Brenowitz and Regehr, 2005). One common feature of all of these forms of eCB-mediated synaptic depression is the need for a postsynaptic stimulus, followed by a presynaptic CB1-mediated synaptic depression. Thus, eCB-mediated DSE/DSI, STD, and LTD have been categorized as involving “retrograde signaling,” a type of synaptic plasticity that has come to be appreciated as widespread in the nervous system (Wilson and Nicoll, 2001; Kano et al., 2009).
DA Roles in Corticostriatal Synaptic Plasticity
Corticostriatal Short-Term Depression
The dorsal striatum is the entry nucleus for basal ganglia processing of cortical information. Cortical fibers innervate the striatum and synapse on striatal medium spiny neurons (MSNs), release glutamate and, therefore, drive the activity of these GABAergic projection cells. As such, the corticostriatal synapse represents a crucial, initial step in the complex series of mechanisms underlying basal ganglia control of actions. Indeed, aberrant corticostriatal function is implicated in a variety of basal ganglia disorders, including Parkinson’s disease (PD), Huntington’s disease, obsessive-compulsive disorder, and addiction (Ingham et al., 1989; Calabresi et al., 1992; Berke and Hyman, 2000; Klapstein et al., 2001; Zaja-Milatovic et al., 2005; Welch et al., 2007). Investigation over the past quarter century has revealed that glutamatergic signaling at the corticostriatal synapse is tightly modulated by a complex array of neurotransmitters and cognate receptors located on either presynaptic or postsynaptic elements. The eCBs have emerged as important modulators of these inputs. The finding that CB1 activation depresses corticostriatal glutamatergic synaptic transmission (Gerdeman and Lovinger, 2001; Huang et al., 2001), provides one mechanism contributing to intrastriatal CB1 agonist injection-induced hypomotility and catalepsy (Gough and Olley, 1978).
Within the striatum there is evidence for expression of several types of eCB-dependent synaptic depression. Studies to date have focused on characterizing plasticity at synapses onto MSNs. Although eCB-mediated DSE/DSI have proven rather difficult to measure (Kreitzer and Malenka, 2005; Narushima et al., 2006a), direct activation of metabotropic receptors, such as neurotensin I, appear to be sufficient to induce STD at glutamatergic synapses onto MSNs (Yin et al., 2008).
Several studies have examined actions of D2 receptors on striatal glutamatergic transmission with mixed results. The majority of reports indicate that D2 activation has little effect when afferents are activated at low frequencies, while synaptic depression can be induced when D2 agonists are applied in conjunction with more frequent afferent stimulation or during generalized depolarization (Brown and Arbuthnott, 1983; Hsu et al., 1995; Flores-Hernández et al., 1997; Calabresi et al., 1999; Bamford et al., 2004a; Yin and Lovinger, 2006). One interpretation of these findings is that presynaptic D2 heteroceptors located on glutamatergic terminals selectively suppress inputs from highly active afferents (Bamford et al., 2004b). An alternative idea is that the combination of D2 activation and elevated afferent activity stimulates production of eCBs that then inhibit synaptic transmission (Kreitzer and Malenka, 2005; Yin and Lovinger, 2006). Manipulations of postsynaptic calcium levels and signaling mechanisms prevent D2-induced synaptic depression, while expression of this depression likely involves a presynaptic decrease in glutamate release probability (Yin and Lovinger, 2006).
Thus, a retrograde signaling role for eCBs has been postulated to underlie this synaptic depression. Recent studies from the Calabresi laboratory suggest that adenosine activation of adenosine A2A receptors is one factor that suppresses D2 receptor-stimulated eCB production (Tozzi et al., 2007). Blockade of these receptors, which occurs after consumption of caffeinated beverages, relieves this suppression. This allows for eCB-mediated synaptic depression even when glutamatergic afferents are activated only at low frequencies. This interesting finding suggests one way in which striatal A2A receptors may participate in striatal-based learning and memory (Yu et al., 2009). Synaptic depression induced by activation of neurotensin I receptors also involves conjoint activity of D2 receptors and glutamatergic synaptic transmission leading to eCB actions (Yin et al., 2008). Thus, several neurotransmitters and neuromodulators appear to play key roles in DA stimulation of eCB-mediated retrograde signaling. The synaptic depression involving D2 activation in the presence of relatively moderate synaptic activation is fully reversible when agonist is removed from the slice, and can thus be characterized as STD (Yin and Lovinger, 2006).
Corticostriatal Long-Term Depression
The bulk of work on eCB-mediated synaptic plasticity in striatum has focused on LTD at glutamatergic synapses. Analysis of CB1 mRNA expression patterns indicates that CB1 receptors are present in deep-layer cortical projection neurons (Marsicano and Lutz, 1999). In contrast, extra-cortical areas that also supply glutamatergic input to the dorsal striatum, the thalamic parafascicular nucleus and central medial, paracentral, and centrolateral thalamic complex, are devoid of any detectable CB1 mRNA signal (Marsicano and Lutz, 1999) and their striatal afferents do not immunostain for CB1 protein (Uchigashima et al., 2007). Thus, eCB/CB1-mediated plasticity at glutamatergic striatal synapses is most likely restricted to corticostriatal inputs.
Studies beginning in the early 1990s showed that high frequency afferent stimulation in striatal slices induces a long-lasting decrease in the amplitude of single stimulus-evoked glutamatergic excitatory postsynaptic potentials or currents measured in MSNs (Calabresi et al., 1992; Lovinger et al., 1993; Walsh, 1993). This is a Hebbian form of lasting synaptic plasticity, with induction requiring high frequency afferent stimulation coupled with postsynaptic depolarization (HFS; Calabresi et al., 1992; Choi and Lovinger, 1997a; Figure 1). eCB-LTD at glutamatergic synapses involves the postsynaptic activation of (group 1) metabotropic glutamate receptors (mGluRs), likely combined with concurrent activation of L-type VGCCs secondary to postsynaptic depolarization (Choi and Lovinger, 1997a; Gubellini et al., 2001; Wang et al., 2006). These postsynaptic events stimulate retrograde synaptic signaling (Choi and Lovinger, 1997a,b), involving the production and release of eCBs that signal to presynaptically localized CB1 receptors (Gerdeman et al., 2002; Robbe et al., 2002; Kreitzer and Malenka, 2005; Figure 1). CB1 receptors couple to Gi/o G-proteins which, upon receptor activation, likely inhibit VGCCs and adenylyl cyclase activity in the presynaptic terminal. Maintained expression of eCB-LTD may require adenylyl cyclase inhibition and decreased production of cAMP, resulting in a decrease in the probability of glutamate release (Heifets et al., 2008; Mato et al., 2008). Corticostriatal eCB-LTD is also pharmacologically inducible with agonists of mGluRs or L-type VGCC activators (Kreitzer and Malenka, 2005; Adermark and Lovinger, 2007).
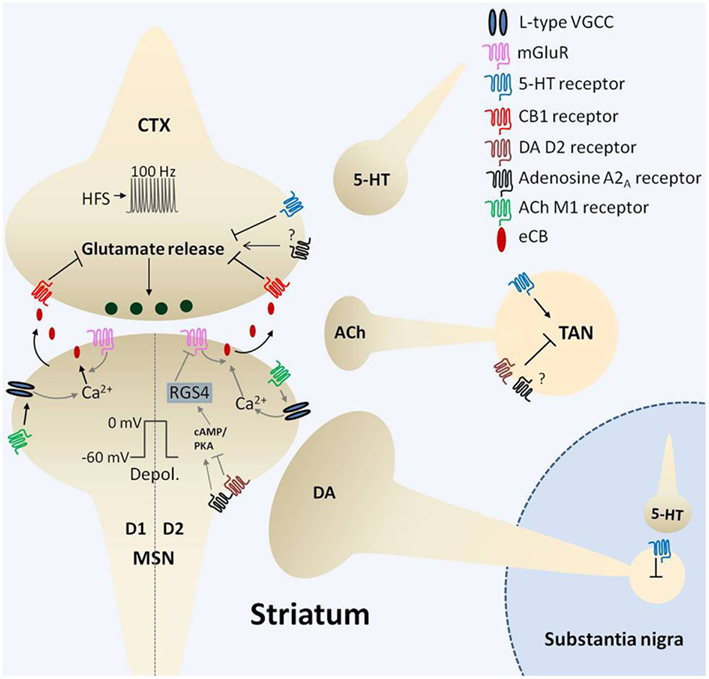
Figure 1. Mechanisms of corticostriatal long-term depression (LTD). Four, one second bouts of afferent (corticostriatal, CTX) stimulation at 100 Hz (high frequency stimulation, HFS) synchronized with postsynaptic medium spiny neuron (MSN) depolarization (depol.) to 0 mV every ten seconds induces endocannabinoid (eCB)-mediated LTD (eCB-LTD). Induction of eCB-LTD involves metabotropic glutamate receptor (mGluR) activation as well as suppression of cholinergic tone that requires dopamine (DA) D2 receptor activation on tonically active cholinergic interneurons (TANs). Relief of cholinergic tone on M1 acetylcholine (ACh) receptors, coupled with postsynaptic depolarization, enhances activation of L-type voltage-gated calcium channels (VGCCs) resulting in calcium influx (Ca2+). This triggers eCB production and subsequent liberation of eCBs that signal retrogradely to activate presynaptic cannabinoid type 1 receptors (CB1), resulting in a persistent suppression of glutamate release. Adenosine A2A receptors also play a role in eCB-LTD, with current evidence indicating the location of action on both TANs and D2 receptor expressing (indirect pathway) MSNs. A balance between A2A and D2 receptor activation may govern downstream effectors modulating eCB production, including PKA/cAMP and RGS4, which in turn regulate mGluR5-induced signaling. The role of A2A receptors on presynaptic terminals is presently unclear. Serotonin (5-HT) modulation of substantia nigra pars compacta DA cell firing and excitation of TANs likely influences this form of plasticity. Moreover, serotonergic activation of corticostriatal 5-HT1b receptors induces LTD that is mutually occlusive with eCB-LTD. Thus, a complex interplay of several neurotransmitter systems orchestrates the fine tuning of the corticostriatal synapse.
D2 DA Receptor Dependence
The mechanisms underlying HFS-induced corticostriatal eCB-LTD are similar to LTD observed at GABAergic and glutamatergic synapses in many brain regions (Lovinger, 2008; Heifets and Castillo, 2009). However, the situation in striatum is further complicated by the involvement of DA and D2 receptors in eCB-LTD induction. Calabresi et al. (1992, 1997) initially showed that striatal glutamatergic LTD is lost following DA depletion, or after blockade or knockout of D2Rs, and this finding has been widely repeated (Tang et al., 2001; Kreitzer and Malenka, 2005, 2007; Wang et al., 2006). But where in the striatal microcircuitry is the site of D2 activation that is crucial for eCB-LTD? A recent study suggested that one possible site may be the tonically active cholinergic interneurons (TANS; Wang et al., 2006). These investigators discovered that the ability of D2 receptors on TANS to “pause” the activity of, and thereby reduce ACh release from the cholinergic neuron, relieves an inhibitory cholinergic tone on M1 receptor-expressing MSNs. This tone appears to involve M1 suppression of L-type VGCC function in MSNs (Figure 1). Relieving this tonic inhibition with M1 blockers promotes eCB production and subsequent release. Consistent with this idea, both Calabresi and Surmeier laboratories showed that bath application of pirenzepine depresses baseline corticostriatal glutamatergic transmission (Wang et al., 2006; Tozzi et al., 2011), a finding that is refuted by Kreitzer and Malenka (2007). This M1 activation induced effect is blocked by a CB1 antagonist (Wang et al., 2006). One of the attractive aspects of a role for the cholinergic system in modulating corticostriatal eCB-LTD is that it indicates a potential consequence of the burst TAN activity followed by a pause in firing during incentivized behaviors (Shimo and Hikosaka, 2001). HFS then theoretically represents the “burst” phase and, through subsequent D2 receptor-mediated inhibition of TAN activity, also induces the pause epoch. Thus, eCB-LTD in this context may be implicated as a form of plasticity underlying incentivized learning.
The other possible site of D2 activation is the D2-expressing indirect pathway MSN. Supporting this idea, evidence emerged indicating that eCB-LTD is restricted to glutamatergic synapses onto indirect pathway MSNs, in contrast to direct pathway MSNs that express D1 in preference to D2 receptors, when assessed using the classic HFS protocol and microstimulation (Kreitzer and Malenka, 2007). Subsequently, Surmeier’s group provided further evidence, using a negative spike-timing-dependent protocol, that it is possible to observe eCB-LTD only in D2 MSNs (Shen et al., 2008). This further implicated the D2 receptor on indirect pathway MSNs in eCB-LTD as activation of a TAN-localized D2 receptor should theoretically induce eCB-LTD equally in both D1 and D2 MSNs (see Wang et al., 2006). Despite these interesting findings, no direct evidence yet exists supporting or refuting the role of D2 receptor activation on indirect pathway MSNs in eCB-LTD.
Whether activation of D2 receptors located on TANs or indirect pathway MSNs, or both, contribute to eCB-LTD, it is important to remember that D2 activation plays a modulatory, not a necessary role per se. This is illustrated by the finding that activation of mGluRs (Kreitzer and Malenka, 2005) or L-type VGCCs alone induce eCB-LTD that occludes HFS-induced LTD and occurs in the presence of a D2 antagonist (Adermark and Lovinger, 2007). It was recently shown in indirect pathway MSNs that (microstimulator-delivered) HFS-induced calcium entry through L-type VGCCs induces an intracellular calcium release that ultimately results in the production of AEA as the eCB signaling molecule mediating this form of eCB-LTD (Ade and Lovinger, 2007; Lerner and Kreitzer, 2012). Complicating the picture, moderate frequency (10–20 Hz) stimulation-induced eCB-LTD, which is known to be calcium independent (Ronesi and Lovinger, 2005), was shown to be 2-AG-mediated (Lerner and Kreitzer, 2012). In light of the calcium-independence of this form of eCB-LTD, the degree of its demonstrated D2 receptor-dependence begs further inquiry (Lerner and Kreitzer, 2012).
If indirect pathway MSN D2 receptors are involved in eCB-LTD induction, what signaling pathways mediated by these receptors link them to eCB production? One possibility is activation of phospholipase C via Gβ/γ signaling (Lee et al., 2004; Pollack, 2004). However, the PLC signaling pathway is thought to involve cooperative D1 and D2 receptor actions, contrasting with the usual segregation of these receptors onto different MSN subpopulations as well as the separate roles of these receptors in LTP and LTD. Furthermore, there is little or no direct evidence that D2 activation leading to phospholipase stimulation actually occurs in MSNs. Inhibition of adenylyl cyclase by D2 receptors could also contribute to eCB production (Kheirbek et al., 2009; Lerner and Kreitzer, 2012). Indeed, Kheirbek et al. (2009) have suggested that this involves inhibition of the adenylyl cyclase 5 isoform implicated in LTD. Interestingly, Lerner and Kreitzer (2012) suggest a key role in LTD induction for the regulator of G protein signaling 4 (RGS4) protein that is a potential downstream target of cAMP/PKA signaling. The roles of these signaling pathways remain to be determined when LTD is induced with different stimulus paradigms and in direct pathway MSNs.
Adenosine A2A Receptor Modulation
Data now exists indicating a role for the adenosine A2A receptor in eCB-LTD modulation. The Surmeier group showed that a competitive interaction exists between the A2A and the D2 DA receptor in the induction of spike-timing-dependent eCB-LTD (Shen et al., 2008). They showed that the negative timing protocol-induced LTD in D2 MSNs is governed by a balance between D2 and A2A receptor activation, such that D2 blockade abolishes LTD, but D2 blockade with combined A2A activation allows for LTP induction (Shen et al., 2008). This opposing interaction between D2 and A2A was recently supported by findings showing that (microstimulator-delivered) HFS- and moderate frequency-induced eCB-LTD in D2 MSNs is blocked by D2 blockade or A2A activation (Lerner and Kreitzer, 2012). Thus, D2 receptor activation promotes LTD, while A2A activation promotes LTP.
Further supporting the interaction between DA and adenosine systems in eCB-LTD, the Calabresi group demonstrated that D2 DA receptor activation alone does not induce striatal glutamatergic synaptic depression, but does induce plasticity when an A2A receptor antagonist is co-applied to striatal slices (Tozzi et al., 2011). This synaptic depression is disrupted by CB1 receptor blockade. While D2 and A2A receptors are expressed by indirect pathway MSNs, qPCR data definitively shows co-expression of these receptors on TANs (Song et al., 2000), and this is supported by other findings (Brown et al., 1990; James and Richardson, 1993; Tozzi et al., 2011). This again begs the question of whether the site of action of D2 activation/A2A inhibition-induced glutamatergic synaptic depression is on TANs or indirect pathway MSNs (Figure 1). Supporting a role for the cholinergic interneuron, only the specific combination of D2 activation/A2A inhibition suppresses TAN firing activity. Furthermore, this drug-induced depression of glutamatergic synapses is blocked (or occluded) in the presence of an M1 muscarinic antagonist (Tozzi et al., 2011), and this receptor is reportedly expressed on both direct and indirect pathway MSNs. As such, this eCB-dependent form of synaptic depression is likely expressed at corticostriatal synapses onto both direct and indirect pathway MSNs (Tozzi et al., 2011). It should be noted, however, that inducing synaptic depression by drug application may not be recruiting the same mechanisms recruited by the HFS protocol classically used to induce eCB-LTD.
Further complicating the role of the A2A receptor in modulating eCB-LTD, anatomical evidence now exists demonstrating the presence of this receptor on glutamatergic terminals contacting direct pathway MSN dendritic spines (Quiroz et al., 2009). Activation of A2A receptors alone with CGS21680 increases glutamatergic transmission onto direct pathway MSNs, while blockade of these receptors decreases release (Quiroz et al., 2009). Interestingly, applying a significantly lower concentration of CGS21680 by itself has no effect, but in the presence of a CB1 agonist, partially inhibits the CB1-mediated suppression of glutamatergic transmission (Martire et al., 2011). Taken together, these findings suggest a possible role for direct A2A modulation of the corticostriatal synapse alone or in combination with CB1 receptors.
Nitric Oxide
Studies have also implicated the gaseous neuromodulator nitric oxide (NO) in striatal synaptic plasticity. Roles for NO in synaptic plasticity in hippocampus, cerebellum, and other brain regions had been postulated since the early 1990s (Schuman and Madison, 1991), but the subject has not been without controversy (see Zorumski and Izumi, 1993; Malenka and Bear, 2004). Experiments in the Calabresi laboratory have indicated that NOS inhibitors prevent LTD induction (Calabresi et al., 1999; Bagetta et al., 2011), while data from the Haas laboratory provide a more confusing picture of the NOS role in LTD. These investigators originally reported that the occurrence of HFS-induced LTD was enhanced by NOS inhibition and in eNOS knockout mice (Doreulee et al., 2003). A subsequent report from the same group showed that LTD induced by group I mGluR-activation was converted to STD in the presence of a NOS inhibitor or in eNOS knockout mice (Sergeeva et al., 2007), while CB1 antagonists blocked LTD altogether. These investigators also showed that an LTD-like decrease in evoked synaptic responses could be induced by application of NO donors. One prominent molecular target of NO is the striatally enriched soluble guanylate cyclase (GC), suggesting the possibility that activation of this enzyme and subsequent cGMP formation might be sufficient to induce LTD. Indeed, Calabresi et al. (1999) have demonstrated that the cGMP phosphodiesterase inhibitor zaprinast, and intracellular application of cGMP itself, can induce long-lasting synaptic depression during low-frequency synaptic activation. Intracellular application of a soluble GC inhibitor prevents LTD induced by HFS or NO donor application. Protein kinase G activation would be expected to result from activation of this signaling cascade, and a PKG inhibitor was shown to prevent LTD (Calabresi et al., 1999). This pharmacologically induced depression occludes HFS-induced LTD, consistent with overlapping mechanisms of induction/expression.
Several questions remain about how NO signaling participates in striatal LTD and the nature of the relationship between NO, DA, and eCBs in LTD. Calabresi et al. (1992, 1999) have reported that antagonists of D1-like DA receptors prevent LTD induction by HFS. Further investigation revealed that LTD was intact in gene-targeted knockout mice lacking the D1 receptor (Centonze et al., 2003). However, in mice lacking the D1 receptor, a D1-like antagonist still prevented HFS-induced LTD, suggesting involvement of D5 receptors (Centonze et al., 2003). Experiments demonstrating that D5 receptors are expressed by striatal interneurons that express NO synthase (NOS) led these investigators to postulate that the role of D5 receptors in LTD might involve NO production by these neurons. Additional direct evidence for a D5 role in eCB-LTD would provide stronger support for this idea. What accounts for the apparent variability in NO involvement when LTD is induced by different means in the Haas laboratory papers? It should be noted that these investigators examined LTD solely in field potential recordings where the synaptically driven firing of striatal neurons is the only measurement. This is at best an indirect measure of synaptic efficacy. Thus, it is possible that mechanisms other than changes in glutamatergic synaptic efficacy account for their findings. In the context of this review, it is not clear if NO and eCBs act separately or in concert to induce or facilitate LTD. The fact that NOS inhibitors and CB1 antagonists have been reported to completely eliminate LTD would suggest that both are necessary and likely act via part of the same mechanism. However, the fact that STD induced by the mGluR agonist DHPG remains in the presence of the NOS inhibitor provides a potential clue to the sequence of mechanisms involving these neuromodulators. Perhaps cCBs stimulate NO production that is then required for maintained LTD, which is suggested as one possibility by Sergeeva et al. (2007). However, NOS inhibitor exposure prevents induction of HFS-induced LTD (Calabresi et al., 1999), making this scenario seem less plausible. It must be noted that some investigators have reported evidence consistent with the idea that NO increases cortical activation of MSN firing (West and Grace, 2004; Ondracek et al., 2008), and of course there is the work implicating NO in LTP in several brain regions including hippocampus (Schuman and Madison, 1991) and even striatum (Doreulee et al., 2003). Thus, it is unclear why this effect would not counteract LTD upon indiscriminate MSN sampling.
Direct vs. indirect pathway
As the field marches forward, it is important to note that the pathway specificity data gathered by Kreitzer and Malenka (2007) runs counter to the findings of other laboratories (Wang et al., 2006; Bagetta et al., 2011). Differences in stimulation electrode size (micro vs. macro) have been hypothesized as the reason for this discrepancy. Kreitzer and Malenka (2007) used a microstimulator, arguing that macrostimulation results in eCB overflow from neighboring cells, therefore resulting in direct pathway MSN eCB-LTD (Kreitzer and Malenka, 2008). Supporting this, a negative timing protocol, where repeated theta bursts of postsynaptic cell spiking precede afferent theta burst activation induced by a microstimulator, induces eCB-LTD at indirect, but not direct pathway MSNs (Shen et al., 2008).
Using macrostimulation, evidence continues to mount supporting the occurrence of eCB-LTD at corticostriatal synapses onto both direct and indirect pathway MSNs. A recent study from the Calabresi laboratory observed eCB-LTD in both direct and indirect pathway cell types using intracellular biocytin and immunolabeling for either substance P (direct pathway) or A2A receptor (indirect pathway; Bagetta et al., 2011). Intriguingly, they did not observe eCB-LTD at direct pathway MSNs upon recording from GFP-positive cells in slices taken from D1-eGFP BAC transgenic mice. However, they observed eCB-LTD in GFP-negative MSNs in slices taken from D2-eGFP BAC transgenic mice (Bagetta et al., 2011). The authors conclude that an alteration of D1 DA receptor signaling, secondary to eGFP expression, may be blocking the expression of eCB-LTD. Direct evidence of such an assertion is still needed, however. In light of the requirement for D1 DA receptor antagonism to achieve negative timing-induced eCB-LTD at direct pathway MSNs (using a microstimulator; Shen et al., 2008), alteration of D1 DA receptor signaling in the D1-eGFP BAC transgenic mouse may explain the loss of eCB-LTD in eGFP-positive MSNs of these animals (Bagetta et al., 2011). Though the recordings showing a lack of eCB-LTD in eGFP-positive MSNs in D1-eGFP BAC mice calls into question the pathway specificity claims of eCB-LTD, the substantial differences in techniques used by the different investigators leave open the possibility that specificity occurs under certain experimental conditions.
Rather than a clear preference for one pathway, a possible bias may exist for plasticity to occur at indirect over direct pathway MSNs. Ultimately, it will be useful to know if eCB-LTD occurs at corticostriatal synapses in vivo, and if so, is there pathway specificity related to behavior.
Striatal GABAergic Synaptic Plasticity
Examination of GABAergic synapses onto striatal MSNs has also revealed eCB-mediated synaptic depression, including LTD (Szabo et al., 1998; Narushima et al., 2006b; Adermark et al., 2009). Kano and colleagues have found small-magnitude DSI following MSN depolarization (Narushima et al., 2006b, 2007). This group has also observed significant short-lasting synaptic depression following combined postsynaptic depolarization and activation of muscarinic acetylcholine receptors (Narushima et al., 2007). In this context, it is also worth noting the work of Maccarrone and coworkers that has implicated eCBs in synaptic depression at GABAergic inputs to MSNs triggered by D2 receptor activation (Centonze et al., 2004). These investigators observed that GABAergic synaptic transmission is depressed by application of cocaine or a D2 receptor agonist to striatal slices. Synaptic depression is accompanied by a decrease in the frequency of spontaneous IPSCs, suggesting a presynaptic mechanism of the cocaine and D2 agonist actions. The cocaine-induced synaptic depression is prevented by D2 antagonists, and reduced by CB1 antagonists, and CB1 agonist application produces synaptic depression (Szabo et al., 1998; Narushima et al., 2006a; Adermark et al., 2009). Application of cocaine or D2 agonist to slices also increases tissue AEA levels (Centonze et al., 2004). Thus, eCBs and CB1 receptors have crucial roles in short-lasting synaptic depression produced by various stimuli.
In contrast to the high frequencies of synaptic activation used to induce glutamatergic eCB-LTD, LTD at GABAergic synapses is activated at low stimulus frequencies (1–5 Hz). Moreover, depolarization of the postsynaptic cell is not necessary to induce eCB-LTD at GABAergic synapses, as it is at glutamatergic synapses. The lower threshold for eCB-LTD induction at inhibitory synapses is likely a reflection of the significantly higher expression of CB1 on striatal GABAergic terminals relative to their glutamatergic counterparts (Uchigashima et al., 2007). However, similar to the glutamatergic synapses, activation of mGluRs and VGCC currents are implicated in GABAergic eCB-LTD (Adermark et al., 2009). It is not yet clear if DA plays a role in this form of plasticity. In addition, it is not known whether this GABAergic eCB-LTD occurs at MSN–MSN or fast-spiking interneuron-MSN synapses, both of which are inhibitory and contain presynaptic CB1 (Freiman et al., 2006; Uchigashima et al., 2007). What we do know is that the net effect of eCB-LTD at GABAergic and glutamatergic synapses appears to be disinhibition and inhibition of MSN activation, respectively. These opposing effects create a situation in which striatal output can be modulated as a consequence of glutamatergic input in a frequency-dependent manner (Adermark and Lovinger, 2009). It will be interesting to determine if dopaminergic modulation adds another level of complexity to this dual-plasticity scenario.
Serotonin and DA/eCB Striatal Synaptic Plasticity
In addition to the dense striatal dopaminergic innervation, serotonergic fibers arising from the dorsal raphe nucleus also project to the dorsal striatum. Existing data on serotonin (5-HT) effects on striatal function suggest interplay between 5-HT and DA systems. 5-HT acts at multiple points in the striatal machinery critical for DA-dependent eCB-LTD, having effects on dopaminergic nigrostriatal cells, striatal TANS, and corticostriatal terminals.
Such a tight interaction between both monoaminergic systems would suggest a level of functional competition. This idea is supported by the observation that neonatal DA depletion in rats results in serotonergic hyperinnervation selectively in the striatum (Breese et al., 1984; Stachowiak et al., 1984). Surprisingly, these animals are not akinetic in adulthood, but are hyperactive (Shaywitz et al., 1976; Erinoff et al., 1979; Luthman et al., 1989). This observation would suggest that intact striatal function is dependent upon a dynamic interplay between the dopaminergic and serotonergic signaling systems. Supporting this notion, Mathur et al. (2011) showed that application of 5-HT or a 5-HT1b receptor agonist induces LTD of glutamate release. Evidence supports a presynaptic locus of expression of this form of LTD and a presynaptic localization of 5-HT1b (Mathur et al., 2011). This study showed that blockade of 5-HT reuptake with a selective 5-HT reuptake inhibitor coupled with a burst stimulation protocol induces a 5-HT1b dependent depression of corticostriatal glutamate release. This endogenous 5-HT-LTD is mutually occlusive of eCB-LTD, demonstrating a direct interaction of 5-HT and DA-dependent, CB1-mediated control of corticostriatal glutamate release (Mathur et al., 2011).
The serotonergic innervation of the human striatum appears heterogeneous (Wallman et al., 2011), a finding that may carry important implications for serotonergic control of selected actions. These 5-HT innervation poor zones are also weak in tyrosine hydroxylase immunostaining (Wallman et al., 2011). Thus, one conclusion might be that such zones may correspond to striosomes (Wallman et al., 2011), in which case 5-HT signaling may have a greater affect on matrix/motor-associated processing (Crittenden and Graybiel, 2011). It should be noted, however, that the study by Wallman et al. (2011) did not control for cell population density or white matter composition, nor did they co-immunostain for a striosomal marker. As such, substantive links between serotonergic innervation heterogeneity and action control are lacking. However, the co-innervation of striatum by dopaminergic and serotonergic fibers underscores the potential interplay between these systems and carries implications for striatal disorders involving DA dysfunction.
Other targets of 5-HT may also directly influence eCB-LTD. Exogenous or endogenous 5-HT increases the spontaneous firing rate of rat striatal TANs (Blomeley and Bracci, 2005; Bonsi et al., 2007). Considering the data supporting a role for relief of M1 receptor activation in eCB-LTD induction, 5-HT-mediated activation of TANS could increase the threshold for eCB-LTD induction (Wang et al., 2006; Figure 1). 5-HT may also be modulating striatal DA release, representing another level of DA and 5-HT interaction influencing eCB-LTD. Genetic deletion of the 5-HT2c receptor increases burst firing of substantia nigra pars compacta dopaminergic neurons and increases the frequency of spike trains (Abdallah et al., 2009). Matching this enhancement, dorsal striatal DA concentrations in 5-HT2c knockouts are significantly elevated. Given the critical role of DA D2 receptor activation in eCB-LTD, serotonergic modulation of nigrostriatal DA release may exquisitely influence this form of plasticity. Thus, multiple levels of interaction between 5-HT and DA systems exist that may be necessary for the proper tuning of corticostriatal synaptic transmission.
Pathophysiology
Parkinson’s disease is a progressive neurodegenerative disorder that is characterized by a loss of neurons in the substantia nigra that supply the dopaminergic innervation to the dorsal striatum. PD is effectively treated with levodopa (or L-DOPA), the biosynthetic pathway precursor molecule to DA. However, prolonged L-DOPA treatment results in side effects involving abnormal involuntary movements, or dyskinesia. Since the dorsal striatum is richly endowed with a dopaminergic innervation, and neuronal degeneration preferentially occurs in substantia nigra as opposed to the ventral tegmental area in PD, cell loss dramatically affects the dorsal striatum. Given that corticostriatal eCB-LTD is D2 receptor dependent, the profound implication is that this form of synaptic plasticity is lost as striatal DA content diminishes, possibly contributing to the cardinal PD clinical features of tremor, rigidity, and bradykinesia. eCB-LTD is indeed lost upon striatal DA depletion (Calabresi et al., 1992; Kreitzer and Malenka, 2007). Furthermore, corticostriatal eCB-LTD is implicated in the development of L-DOPA-induced dyskinesia (LID). Picconi et al. (2011) showed that non-dyskinetic parkinsonian rats (treated with L-DOPA) expressed eCB-LTD, while dyskinetic rats did not. Phosphodiesterase inhibition restores eCB-LTD in these dyskinetic rats and alleviates the occurrence of dyskinetic behaviors (Picconi et al., 2011). Thus, eCB-LTD is positioned as a key target for therapeutic intervention in PD.
Unlike the progressive degeneration of striatal dopaminergic innervation seen in PD, striatal serotonergic innervation is relatively spared and, upon treatment with levodopa (or L-DOPA), this innervation is enriched (Rylander et al., 2010). Striatal serotonergic fibers synthesize and release L-DOPA-derived DA; this ectopic DA release is responsible for the development of LID (Carta et al., 2007). Interestingly, serotonergic neuron autoreceptor (5-HT1a/b) agonists possess efficacy in alleviating LID (Carta et al., 2007). In light of the host of studies now supporting a role for the dysregulation of corticostriatal glutamatergic signaling in the occurrence of LID (Huot and Brotchie, 2011), it is tempting to speculate that the mechanism of action underlying 5-HT1b agonist efficacy for the treatment of LID is the dampening of corticostriatal glutamate release. This idea is consistent with the findings by Picconi et al. (2011) showing a loss of eCB-LTD only in L-DOPA-treated parkinsonian rodents that express LID.
The striatal DA depletion seen in PD also induces a dramatic MSN dendritic dysmorphology (Zaja-Milatovic et al., 2005; Deutch et al., 2007). Specifically, a significant pruning of dendritic spines occurs, culminating in the loss of corticostriatal synapses. The Surmeier laboratory elegantly showed that this dendritic pruning is restricted to indirect MSNs (Day et al., 2006). It is interesting to speculate that if eCB-LTD is restricted to the indirect pathway (Kreitzer and Malenka, 2007) that the loss of corticostriatal synapses onto these cells contributes to an inability to detect eCB-LTD in the parkinsonian state. However, this would seem unlikely given that both the Malenka and Surmeier laboratories showed that DA-denervation-induced impairment of eCB-LTD in D2 MSNs is restored simply by bath applying a D2 agonist (Kreitzer and Malenka, 2007; Shen et al., 2008). This finding would imply that the most parsimonious reason for the loss of eCB-LTD, at least in D2 MSNs, in the DA denervated state is loss of D2 receptor activation. Interestingly, timing-dependent induction of LTD is observed in D1 MSNs only in the presence of a D1 receptor antagonist or in the DA denervated state (Shen et al., 2008). This finding raises the interesting question of why eCB-LTD induced with HFS in D1 MSNs or MSNs sampled indiscriminately is blocked in the presence of a D2 antagonist or in the DA denervated state (Calabresi et al., 1992; Wang et al., 2006). This issue again highlights the caveat that different plasticity induction protocols are employed across different studies and laboratories and that both D1 and D2 MSNs are not always discriminated during sampling. For instance, it would be interesting to see if the induction protocol used in the Kreitzer and Malenka (2007) study evokes eCB-LTD in direct pathway MSNs in the DA denervated state.
Clearly, more work is needed to determine the contribution of eCB-LTD onto D1 and D2 MSNs to parkinsonism. Nevertheless, advances in targeting eCB-LTD of corticostriatal synapses may have already been realized. For instance, muscarinic antagonists have been utilized for years as treatment for PD (Chen and Swope, 2007), which would support the idea that cholinergic tone is an important modulator of eCB-LTD. Specifically regarding the M1 receptor, antagonists of this target yield a mild improvement in motor symptoms in PD animal models, but fail to achieve the efficacy seen with broad muscarinic antagonist administration (Xiang et al., 2012). This finding would suggest that muscarinic activation at other basal ganglia synapse types are importantly modulated by this receptor class. Regarding the role of adenosine, administration of an A2A antagonist to reserpine-treated mice prevents reserpine-induced catalepsy (Peterson et al., 2012). Considering the eCB system, but perhaps not the corticostriatal synapse in particular, findings in DA-depleted animals indicate that motor dysfunction is alleviated by inhibiting eCB metabolism with systemic injection of a FAAH inhibitor in mice (Kreitzer and Malenka, 2007). Finally, data now implicate a role for the RGS4 protein in modulating eCB production in indirect pathway MSNs: inhibiting RGS4 restores eCB-LTD in the presence of a D2 antagonist and DA denervated RGS4 null mice are resistant to some features of motor dysfunction typical of parkinsonism (Lerner and Kreitzer, 2012).
While the striatal hypodopaminergic state results in a paucity of action initiation, it is suggested that aberrant, and perhaps enhanced, mesostriatal DA signaling is an important component in the formation of repeated, habitual actions seen in addiction to drugs that act on DA signaling, such as cocaine or amphetamine (Lovinger et al., 2003). A role for DA signaling in the learning of motor sequences is now established. Upon learning a fixed-ratio of eight lever presses to earn a food reward, substantia nigra dopaminergic cells fire most vigorously during the initiation and termination of the entire lever-pressing sequence (Jin and Costa, 2010). This finding may explain the action initiation and termination deficiency seen in PD, and reinforces the concept of dopaminergic control of striatal-mediated action learning and control. Implicating striatal DA-dependent eCB-LTD in action learning, genetic deletion of CB1 or systemic pharmacological blockade of this receptor disrupts habit formation, a behavior dependent upon the dorsolateral striatum (Hilário et al., 2007).
Conclusion
Though the last two decades have seen considerable advances in our knowledge of DA modulation of eCB-LTD at the corticostriatal synapse, more work is needed to fully characterize this plasticity system. Currently, the core issues requiring consensus are the site of action of the D2 DA receptor and the pathway specificity of this form of synaptic plasticity. As detailed above, data generated from various laboratories offer discrepant conclusions regarding these issues. It is important to remember, however, that the methods used to generate these divergent data vary from laboratory to laboratory. Thus, the literature may accurately reflect the capability of the system, but determining when and under what conditions these varying outcomes are expressed in vivo represents the major challenge for future investigation of corticostriatal eCB-LTD.
As the molecular and circuit mechanisms of eCB-LTD are further elucidated, another major challenge will be to determine the contribution of eCB-LTD to action learning. We really have only scratched the surface of this issue (Hilário et al., 2007). In the future, applying knowledge derived from in vitro analyses to in vivo manipulation of this system using conditional knockout and optogenetic approaches, for example, combined with sophisticated behavioral measures will be necessary to fully describe the roles of this plasticity in physiological and pathophysiological states.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abdallah, L., Bonasera, S. J., Hopf, F. W., O’Dell, L., Giorgetti, M., Jongsma, M., Carra, S., Pierucci, M., Di Giovanni, G., Esposito, E., Parsons, L. H., Bonci, A., and Tecott, L. H. (2009). Impact of serotonin 2C receptor null mutation on physiology and behavior associated with nigrostriatal dopamine pathway function. J. Neurosci. 29, 8156–8165.
Ade, K. K., and Lovinger, D. M. (2007). Anandamide regulates postnatal development of long-term synaptic plasticity in the rat dorsolateral striatum. J. Neurosci. 27, 2403–2409.
Adermark, L., and Lovinger, D. M. (2007). Combined activation of L-type Ca2+ channels and synaptic transmission is sufficient to induce striatal long-term depression. J. Neurosci. 27, 6781–6787.
Adermark, L., and Lovinger, D. M. (2009). Frequency-dependent inversion of net striatal output by endocannabinoid-dependent plasticity at different synaptic inputs. J. Neurosci. 29, 1375–1380.
Adermark, L., Talani, G., and Lovinger, D. M. (2009). Endocannabinoid-dependent plasticity at GABAergic and glutamatergic synapses in the striatum is regulated by synaptic activity. Eur. J. Neurosci. 29, 32–41.
Bagetta, V., Picconi, B., Marinucci, S., Sgobio, C., Pendolino, V., Ghiglieri, V., Fusco, F. R., Giampà, C., and Calabresi, P. (2011). Dopamine-dependent long-term depression is expressed in striatal spiny neurons of both direct and indirect pathways: implications for Parkinson’s disease. J. Neurosci. 31, 12513–12522.
Bamford, N. S., Robinson, S., Palmiter, R. D., Joyce, J. A., Moore, C., and Meshul, C. K. (2004a). Dopamine modulates release from corticostriatal terminals. J. Neurosci. 24, 9541–9552.
Bamford, N. S., Zhang, H., Schmitz, Y., Wu, N. P., Cepeda, C., Levine, M. S., Schmauss, C., Zakharenko, S. S., Zablow, L., and Sulzer, D. (2004b). Heterosynaptic dopamine neurotransmission selects sets of corticostriatal terminals. Neuron 42, 653–663.
Berke, J. D., and Hyman, S. E. (2000). Addiction, dopamine, and the molecular mechanisms of memory. Neuron 25, 515–532.
Blomeley, C., and Bracci, E. (2005). Excitatory effects of serotonin on rat striatal cholinergic interneurones. J. Physiol. (Lond.) 569, 715–721.
Bonsi, P., Cuomo, D., Ding, J., Sciamanna, G., Ulrich, S., Tscherter, A., Bernardi, G., Surmeier, D. J., and Pisani, A. (2007). Endogenous serotonin excites striatal cholinergic interneurons via the activation of 5-HT 2C, 5-HT6, and 5-HT7 serotonin receptors: implications for extrapyramidal side effects of serotonin reuptake inhibitors. Neuropsychopharmacology 32, 1840–1854.
Breese, G. R., Baumeister, A. A., McCown, T. J., Emerick, S. G., Frye, G. D., Crotty, K., and Mueller, R. A. (1984). Behavioral differences between neonatal and adult 6-hydroxydopamine-treated rats to dopamine agonists: relevance to neurological symptoms in clinical syndromes with reduced brain dopamine. J. Pharmacol. Exp. Ther. 231, 343–354.
Brenowitz, S. D., and Regehr, W. G. (2005). Associative short-term synaptic plasticity mediated by endocannabinoids. Neuron 45, 419–431.
Brown, J. R., and Arbuthnott, G. W. (1983). The electrophysiology of dopamine (D2) receptors: a study of the actions of dopamine on corticostriatal transmission. Neuroscience 10, 349–355.
Brown, S. J., James, S., Reddington, M., and Richardson, P. J. (1990). Both A1 and A2a purine receptors regulate striatal acetylcholine release. J. Neurochem. 55, 31–38.
Cadogan, A. K., Alexander, S. P., Boyd, E. A., and Kendall, D. A. (1997). Influence of cannabinoids on electrically evoked dopamine release and cyclic AMP generation in the rat striatum. J. Neurochem. 69, 1131–1137.
Calabresi, P., Centonze, D., Gubellini, P., Marfia, G. A., and Bernardi, G. (1999). Glutamate-triggered events inducing corticostriatal long-term depression. J. Neurosci. 19, 6102–6110.
Calabresi, P., Maj, R., Pisani, A., Mercuri, N. B., and Bernardi, G. (1992). Long-term synaptic depression in the striatum: physiological and pharmacological characterization. J. Neurosci. 12, 4224–4233.
Calabresi, P., Saiardi, A., Pisani, A., Baik, J. H., Centonze, D., Mercuri, N. B., Bernardi, G., and Borrelli, E. (1997). Abnormal synaptic plasticity in the striatum of mice lacking dopamine D2 receptors. J. Neurosci. 17, 4536–4544.
Carta, M., Carlsson, T., Kirik, D., and Björklund, A. (2007). Dopamine released from 5-HT terminals is the cause of L-DOPA-induced dyskinesia in parkinsonian rats. Brain 130, 1819–1833.
Centonze, D., Battista, N., Rossi, S., Mercuri, N. B., Finazzi-Agrò, A., Bernardi, G., Calabresi, P., and Maccarrone, M. A. (2004) A critical interaction between dopamine D2 receptors and endocannabinoids mediates the effects of cocaine on striatal gabaergic transmission. Neuropsychopharmacology 29, 1488–1497.
Centonze, D., Grande, C., Saulle, E., Martin, A. B., Gubellini, P., Pavón, N., Pisani, A., Bernardi, G., Moratalla, R., and Calabresi, P. (2003). Distinct roles of D1 and D5 dopamine receptors in motor activity and striatal synaptic plasticity. J. Neurosci. 23, 8506–8512.
Chen, J. J., and Swope, D. M. (2007). Pharmacotherapy for Parkinson’s disease. Pharmacotherapy 27, 161S–173S.
Choi, S., and Lovinger, D. M. (1997a). Decreased probability of neurotransmitter release underlies striatal long-term depression and postnatal development of corticostriatal synapses. Proc. Natl. Acad. Sci. U.S.A. 94, 2665–2670.
Choi, S., and Lovinger, D. M. (1997b). Decreased frequency but not amplitude of quantal synaptic responses associated with expression of corticostriatal long-term depression. J. Neurosci. 17, 8613–8620.
Crittenden, J. R., and Graybiel, A. M. (2011). Basal ganglia disorders associated with imbalances in the striatal striosome and matrix compartments. Front. Neuroanat. 5:59. doi:10.3389/fnana.2011.00059
Day, M., Wang, Z., Ding, J., An, X., Ingham, C. A., Shering, A. F., Wokosin, D., Ilijic, E., Sun, Z., Sampson, A. R., Mugnaini, E., Deutch, A. Y., Sesack, S. R., Arbuthnott, G. W., and Surmeier, D. J. (2006). Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat. Neurosci. 9, 251–259.
Deutch, A. Y., Colbran, R. J., and Winder, D. J. (2007). Striatal plasticity and medium spiny neuron dendritic remodeling in parkinsonism. Parkinsonism Relat. Disord. 13, S251–S258.
Devane, W. A., Hanus, L., Breuer, A., Pertwee, R. G., Stevenson, L. A., Griffin, G., Gibson, D., Mandelbaum, A., Etinger, A., and Mechoulam, R. (1992). Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258, 1946–1949.
Di Marzo, V., Fontana, A., Cadas, H., Schinelli, S., Cimino, G., Schwartz, J. C., and Piomelli, D. (1994). Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 372, 686–691.
Doreulee, N., Sergeeva, O. A., Yanovsky, Y., Chepkova, A. N., Selbach, O., Gödecke, A., Schrader, J., and Haas, H. L. (2003). Cortico-striatal synaptic plasticity in endothelial nitric oxide synthase deficient mice. Brain Res. 964, 159–163.
Erinoff, L., MacPhail, R. C., Heller, A., and Seiden, L. S. (1979). Age-dependent effects of 6-hydroxydopamine on locomotor activity in the rat. Brain Res. 164, 195–205.
Flores-Hernández, J., Galarraga, E., and Bargas, J. (1997). Dopamine selects glutamatergic inputs to neostriatal neurons. Synapse 25, 185–195.
Freiman, I., Anton, A., Monyer, H., Urbanski, M. J., and Szabo, B. (2006). Analysis of the effects of cannabinoids on identified synaptic connections in the caudate-putamen by paired recordings in transgenic mice. J. Physiol. (Lond.) 575, 789–806.
Gerdeman, G., and Lovinger, D. M. (2001). CB1cannabinoid receptor inhibits synaptic release of glutamate in rat dorsolateral striatum. J. Neurophysiol. 85, 468–471.
Gerdeman, G. L., Ronesi, J., and Lovinger, D. M. (2002). Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat. Neurosci. 5, 446–451.
Gough, A. L., and Olley, J. E. (1978). Catalepsy induced by intrastriatal injections of delta9-THC and 11-OH-delta9-THC in the rat. Neuropharmacology 17, 137–144.
Gubellini, P., Saulle, E., Centonze, D., Bonsi, P., Pisani, A., Bernardi, G., Conquet, F., and Calabresi, P. (2001). Selective involvement of mGlu1 receptors in corticostriatal LTD. Neuropharmacology 40, 839–846.
Heifets, B. D., and Castillo, P. E. (2009). Endocannabinoid signaling and long-term synaptic plasticity. Annu. Rev. Physiol. 71, 283–306.
Heifets, B. D., Chevaleyre, V., and Castillo, P. E. (2008). Interneuron activity controls endocannabinoid-mediated presynaptic plasticity through calcineurin. Proc. Natl. Acad. Sci. U.S.A. 105, 10250–10255.
Hilário, M. R., Clouse, E., Yin, H. H., and Costa, R. M. (2007). Endocannabinoid signaling is critical for habit formation. Front. Integr. Neurosci. 1:6. doi:10.3389/neuro.07/006.2007
Howlett, A. C., and Fleming, R. M. (1984). Cannabinoid inhibition of adenylate cyclase. Pharmacology of the response in neuroblastoma cell membranes. Mol. Pharmacol. 26, 532–538.
Howlett, A. C., Qualy, J. M., and Khachatrian, L. L. (1986). Involvement of Gi in the inhibition of adenylate cyclase by cannabimimetic drugs. Mol. Pharmacol. 29, 307–313.
Hsu, K. S., Huang, C. C., Yang, C. H., and Gean, P. W. (1995). Presynaptic D2 dopaminergic receptors mediate inhibition of excitatory synaptic transmission in rat neostriatum. Brain Res. 690, 264–268.
Huang, C. C., Lo, S. W., and Hsu, K. S. (2001). Presynaptic mechanisms underlying cannabinoid inhibition of excitatory synaptic transmission in rat striatal neurons. J. Physiol. (Lond.) 532, 731–748.
Huot, P., and Brotchie, J. M. (2011). 5-HT(1A) receptor stimulation and L-DOPA-induced dyskinesia in Parkinson’s disease: bridging the gap between serotonergic and glutamatergic mechanisms. Exp. Neurol. 231, 195–198.
Ingham, C. A., Hood, S. H., and Arbuthnott, G. W. (1989). Spine density on neostriatal neurones changes with 6-hydroxydopamine lesions and with age. Brain Res. 503, 334–338.
Ishac, E. J., Jiang, L., Lake, K. D., Varga, K., Abood, M. E., and Kunos, G. (1996). Inhibition of exocytotic noradrenaline release by presynaptic cannabinoid CB1 receptors on peripheral sympathetic nerves. Br. J. Pharmacol. 118, 2023–2028.
James, S., and Richardson, P. J. (1993). The subcellular distribution of [3H]-CGS 21680 binding sites in the rat striatum: copurification with cholinergic nerve terminals. Neurochem. Int. 23, 115–122.
Jin, W., Brown, S., Roche, J. P., Hsieh, C., Celver, J. P., Kovoor, A., Chavkin, C., and Mackie, K. (1999). Distinct domains of the CB1 cannabinoid receptor mediate desensitization and internalization. J. Neurosci. 19, 3773–3780.
Jin, X., and Costa, R. M. (2010). Start/stop signals emerge in nigrostriatal circuits during sequence learning. Nature 466, 457–462.
Kano, M., Ohno-Shosaku, T., Hashimotodani, Y., Uchigashima, M., and Watanabe, M. (2009). Endocannabinoid-mediated control of synaptic transmission. Physiol. Rev. 89, 309–380.
Kathmann, M., Bauer, U., Schlicker, E., and Göthert, M. (1999). Cannabinoid CB1 receptor-mediated inhibition of NMDA- and kainate-stimulated noradrenaline and dopamine release in the brain. Naunyn Schmiedebergs Arch. Pharmacol. 359, 466–470.
Katona, I., Sperlágh, B., Sík, A., Käfalvi, A., Vizi, E. S., Mackie, K., and Freund, T. F. (1999). Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J. Neurosci. 19, 4544–4558.
Kheirbek, M. A., Britt, J. P., Beeler, J. A., Ishikawa, Y., McGehee, D. S., and Zhuang, X. (2009). Adenylyl cyclase type 5 contributes to corticostriatal plasticity and striatum-dependent learning. J. Neurosci. 29, 12115–12124.
Klapstein, G. J., Fisher, R. S., Zanjani, H., Cepeda, C., Jokel, E. S., Chesselet, M. F., and Levine, M. S. (2001). Electrophysiological and morphological changes in striatal spiny neurons in R6/2 Huntington’s disease transgenic mice. J. Neurophysiol. 86, 2667–2677.
Kreitzer, A. C., and Malenka, R. C. (2005). Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J. Neurosci. 25, 10537–10545.
Kreitzer, A. C., and Malenka, R. C. (2007). Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature 445, 643–647.
Kreitzer, A. C., and Malenka, R. C. (2008). Striatal plasticity and basal ganglia circuit function. Neuron 60, 543–554.
Lee, S. P., So, C. H., Rashid, A. J., Varghese, G., Cheng, R., Lança, A. J., O’Dowd, B. F., and George, S. R. (2004). Dopamine D1 and D2 receptor Co-activation generates a novel phospholipase C-mediated calcium signal. J. Biol. Chem. 279, 35671–35678.
Lerner, T. N., and Kreitzer, A. C. (2012). RGS4 is required for dopaminergic control of striatal LTD and susceptibility to Parkinsonian motor deficits. Neuron 73, 347–359.
Liu, J., Wang, L., Harvey-White, J., Huang, B. X., Kim, H. Y., Luquet, S., Palmiter, R. D., Krystal, G., Rai, R., Mahadevan, A., Razdan, R. K., and Kunos, G. (2008). Multiple pathways involved in the biosynthesis of anandamide. Neuropharmacology 54, 1–7.
Lovinger, D. M. (2008). Presynaptic modulation by endocannabinoids. Handb. Exp. Pharmacol. 184, 435–477.
Lovinger, D. M., Partridge, J. G., and Tang, K. C. (2003). Plastic control of striatal glutamatergic transmission by ensemble actions of several neurotransmitters and targets for drugs of abuse. Ann. N. Y. Acad. Sci. 1003, 226–240.
Lovinger, D. M., Tyler, E. C., and Merritt, A. (1993). Short- and long-term synaptic depression in rat neostriatum. J. Neurophysiol. 70, 1937–1949.
Luthman, J., Fredriksson, A., Sundström, E., Jonsson, G., and Archer, T. (1989). Selective lesion of central dopamine or noradrenaline neuron systems in the neonatal rat: motor behavior and monoamine alterations at adult stage. Behav. Brain Res. 33, 267–277.
Marsicano, G., and Lutz, B. (1999). Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur. J. Neurosci. 11, 4213–4225.
Martire, A., Tebano, M. T., Chiodi, V., Ferreira, S. G., Cunha, R. A., Köfalvi, A., and Popoli, P. (2011). Pre-synaptic adenosine A2A receptors control cannabinoid CB1 receptor-mediated inhibition of striatal glutamatergic neurotransmission. J. Neurochem. 116, 273–280.
Mathur, B. N., Capik, N. A., Alvarez, V. A., and Lovinger, D. M. (2011). Serotonin induces long-term depression at corticostriatal synapses. J. Neurosci. 31, 7402–7411.
Mato, S., Lafourcade, M., Robbe, D., Bakiri, Y., and Manzoni, O. J. (2008). Role of the cyclic-AMP/PKA cascade and of P/Q-type Ca++ channels in endocannabinoid-mediated long-term depression in the nucleus accumbens. Neuropharmacology 54, 87–94.
Matsuda, L. A., Lolait, S. J., Brownstein, M. J., Young, A. C., and Bonner, T. I. (1990). Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346, 561–564.
Mátyás, F., Yanovsky, Y., Mackie, K., Kelsch, W., Misgeld, U., and Freund, T. F. (2006). Subcellular localization of type 1 cannabinoid receptors in the rat basal ganglia. Neuroscience 137, 337–361.
Mechoulam, R., Ben-Shabat, S., Hanus, L., Ligumsky, M., Kaminski, N. E., Schatz, A. R., Gopher, A., Almog, S., Martin, B. R., Compton, D. R., Pertwee, R. G., Griffin, G., Bayewitch, M., Barg, J., and Vogel, Z. (1995). Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 50, 83–90.
Narushima, M., Hashimoto, K., and Kano, M. (2006a). Endocannabinoid-mediated short-term suppression of excitatory synaptic transmission to medium spiny neurons in the striatum. Neurosci. Res. 54, 159–164.
Narushima, M., Uchigashima, M., Hashimoto, K., Watanabe, M., and Kano, M. (2006b). Depolarization-induced suppression of inhibition mediated by endocannabinoids at synapses from fast-spiking interneurons to medium spiny neurons in the striatum. Eur. J. Neurosci. 24, 2246–2252.
Narushima, M., Uchigashima, M., Fukaya, M., Matsui, M., Manabe, T., Hashimoto, K., Watanabe, M., and Kano, M. (2007). Tonic enhancement of endocannabinoid-mediated retrograde suppression of inhibition by cholinergic interneuron activity in the striatum. J. Neurosci. 27, 496–506.
Ondracek, J. M., Dec, A., Hoque, K. E., Lim, S. A., Rasouli, G., Indorkar, R. P., Linardakis, J., Klika, B., Mukherji, S. J., Burnazi, M., Threlfell, S., Sammut, S., and West, A. R. (2008). Feed-forward excitation of striatal neuron activity by frontal cortical activation of nitric oxide signaling in vivo. Eur. J. Neurosci. 27, 1739–1754.
Peterson, J. D., Goldberg, J. A., and Surmeier, D. J. (2012). Adenosine A2a receptor antagonists attenuate striatal adaptations following dopamine depletion. Neurobiol. Dis. 45, 409–416.
Picconi, B., Bagetta, V., Ghiglieri, V., Paillè, V., Di Filippo, M., Pendolino, V., Tozzi, A., Giampà, C., Fusco, F. R., Sgobio, C., and Calabresi, P. (2011). Inhibition of phosphodiesterases rescues striatal long-term depression and reduces levodopa-induced dyskinesia. Brain 134, 375–387.
Pollack, A. (2004). Coactivation of D1 and D2 dopamine receptors: in marriage, a case of his, hers, and theirs. Sci. STKE 255, 50.
Quiroz, C., Luján, R., Uchigashima, M., Simoes, A. P., Lerner, T. N., Borycz, J., Kachroo, A., Canas, P. M., Orru, M., Schwarzschild, M. A., Rosin, D. L., Kreitzer, A. C., Cunha, R. A., Watanabe, M., and Ferré, S. (2009). Key modulatory role of presynaptic adenosine A2A receptors in cortical neurotransmission to the striatal direct pathway. ScientificWorldJournal 9, 1321–1344.
Robbe, D., Kopf, M., Remaury, A., Bockaert, J., and Manzoni, O. J. (2002). Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc. Natl. Acad. Sci. U.S.A. 99, 8384–8388.
Rodriguez, J. J., Mackie, K., and Pickel, V. M. (2001). Ultrastructural localization of the CB1 cannabinoid receptor in mu-opioid receptor patches of the rat caudate putamen nucleus. J. Neurosci. 21, 823–833.
Ronesi, J., and Lovinger, D. M. (2005). Induction of striatal long-term synaptic depression by moderate frequency activation of cortical afferents in rat. J. Physiol. 562, 245–256.
Rylander, D., Parent, M., O’Sullivan, S. S., Dovero, S., Lees, A. J., Bezard, E., Descarries, L., and Cenci, M. A. (2010). Maladaptive plasticity of serotonin axon terminals in levodopa-induced dyskinesia. Ann. Neurol. 68, 619–628.
Schuman, E. M., and Madison, D. V. (1991). A requirement for the intercellular messenger nitric oxide in long-term potentiation. Science 254, 1503–1506.
Sergeeva, O. A., Doreulee, N., Chepkova, A. N., Kazmierczak, T., and Haas, H. L. (2007). Long-term depression of cortico-striatal synaptic transmission by DHPG depends on endocannabinoid release and nitric oxide synthesis. Eur. J. Neurosci. 26, 1889–1894.
Shaywitz, B. A., Yager, R. D., and Klopper, J. H. (1976). Selective brain dopamine depletion in developing rats: an experimental model of minimal brain dysfunction. Science 191, 305–308.
Shen, W., Flajolet, M., Greengard, P., and Surmeier, D. J. (2008). Dichotomous dopaminergic control of striatal synaptic plasticity. Science 321, 848–851.
Shimo, Y., and Hikosaka, O. (2001). Role of tonically active neurons in primate caudate in reward-oriented saccadic eye movement. J. Neurosci. 21, 7804–7814.
Sidló, Z., Reggio, P. H., and Rice, M. E. (2008). Inhibition of striatal dopamine release by CB1 receptor activation requires nonsynaptic communication involving GABA, H2O2, and KATP channels. Neurochem. Int. 52, 80–88.
Smart, D., Gunthorpe, M. J., Jerman, J. C., Nasir, S., Gray, J., Muir, A. I., Chambers, J. K., Randall, A. D., and Davis, J. B. (2000). The endogenous lipid anandamide is a full agonist at the human vanilloid receptor (hVR1). Br. J. Pharmacol. 129, 227–230.
Song, W. J., Tkatch, T., and Surmeier, D. J. (2000). Adenosine receptor expression and modulation of Ca(2+) channels in rat striatal cholinergic interneurons. J. Neurophysiol. 83, 322–332.
Stachowiak, M. K., Bruno, J. P., Snyder, A. M., Stricker, E. M., and Zigmond, M. J. (1984). Apparent sprouting of striatal serotonergic terminals after dopamine-depleting brain lesions in neonatal rats. Brain Res. 291, 164–167.
Szabo, B., Dörner, L., Pfreundtner, C., Nörenberg, W., and Starke, K. (1998). Inhibition of GABAergic inhibitory postsynaptic currents by cannabinoids in rat corpus striatum. Neuroscience 85, 395–403.
Tang, K., Low, M. J., Grandy, D. K., and Lovinger, D. M. (2001). Dopamine-dependent synaptic plasticity in striatum during in vivo development. Proc. Natl. Acad. Sci. U.S.A. 98, 1255–1260.
Tozzi, A., de Iure, A., Di Filippo, M., Tantucci, M., Costa, C., Borsini, F., Ghiglieri, V., Giampà, C., Fusco, F. R., Picconi, B., and Calabresi, P. (2011). The distinct role of medium spiny neurons and cholinergic interneurons in the D/AA receptor interaction in the striatum: implications for Parkinson’s disease. J. Neurosci. 31, 1850–1862.
Tozzi, A., Tscherter, A., Belcastro, V., Tantucci, M., Costa, C., Picconi, B., Centonze, D., Calabresi, P., and Borsini, F. (2007). Interaction of A2A adenosine and D2 dopamine receptors modulates corticostriatal glutamatergic transmission. Neuropharmacology 53, 783–789.
Uchigashima, M., Narushima, M., Fukaya, M., Katona, I., Kano, M., and Watanabe, M. (2007). Subcellular arrangement of molecules for 2-arachidonoyl-glycerol-mediated retrograde signaling and its physiological contribution to synaptic modulation in the striatum. J. Neurosci. 27, 3663–3676.
Varma, N., Carlson, G. C., Ledent, C., and Alger, B. E. (2001). Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. J. Neurosci. 21, 188.
Wallman, M. J., Gagnon, D., and Parent, M. (2011). Serotonin innervation of human basal ganglia. Eur. J. Neurosci. 33, 1519–1532.
Walsh, J. P. (1993). Depression of excitatory synaptic input in rat striatal neurons. Brain Res. 608, 123–128.
Wang, Z., Kai, L., Day, M., Ronesi, J., Yin, H. H., Ding, J., Tkatch, T., Lovinger, D. M., and Surmeier, D. J. (2006). Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron 50, 443–452.
Welch, J. M., Lu, J., Rodriguiz, R. M., Trotta, N. C., Peca, J., Ding, J. D., Feliciano, C., Chen, M., Adams, J. P., Luo, J., Dudek, S. M., Weinberg, R. J., Calakos, N., Wetsel, W. C., and Feng, G. (2007). Cortico-striatal synaptic defects and OCD-like behaviours in Sapap3-mutant mice. Nature 448, 894–900.
West, A. R., and Grace, A. A. (2004). The nitric oxide-guanylyl cyclase signaling pathway modulates membrane activity States and electrophysiological properties of striatal medium spiny neurons recorded in vivo. J. Neurosci. 24, 1924–1935.
Wilson, R. I., and Nicoll, R. A. (2001). Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 410, 588–592.
Xiang, Z., Thompson, A. D., Jones, C. K., Lindsley, C. W., and Conn, P. J. (2012). Roles of the m1 muscarinic acetylcholine receptor subtype in the regulation of basal ganglia function and implications for the treatment of Parkinson’s disease. J. Pharmacol. Exp. Ther. 340, 595–603.
Yin, H. H., Adermark, L., and Lovinger, D. M. (2008). Neurotensin reduces glutamatergic transmission in the dorsolateral striatum via retrograde endocannabinoid signaling. Neuropharmacology 54, 79–86.
Yin, H. H., and Lovinger, D. M. (2006). Frequency-specific and D2 receptor-mediated inhibition of glutamate release by retrograde endocannabinoid signaling. Proc. Natl. Acad. Sci. U.S.A. 103, 8251–8256.
Yu, C., Gupta, J., Chen, J. F., and Yin, H. H. (2009). Genetic deletion of A2A adenosine receptors in the striatum selectively impairs habit formation. J. Neurosci. 29, 15100–151003.
Zaja-Milatovic, S., Milatovic, D., Schantz, A. M., Zhang, J., Montine, K. S., Samii, A., Deutch, A. Y., and Montine, T. J. (2005). Dendritic degeneration in neostriatal medium spiny neurons in Parkinson disease. Neurology 64, 545–547.
Zorumski, C. F., and Izumi, Y. (1993). Nitric oxide and hippocampal synaptic plasticity. Biochem. Pharmacol. 46, 777–785.
Keywords: long-term depression, CB1, serotonin, 5-HT1b, adenosine, Parkinson’s disease, medium spiny neuron
Citation: Mathur BN and Lovinger DM (2012) Endocannabinoid–dopamine interactions in striatal synaptic plasticity. Front. Pharmacol. 3:66. doi: 10.3389/fphar.2012.00066
Received: 07 December 2011; Accepted: 02 April 2012;
Published online: 19 April 2012.
Edited by:
Joseph F. Cheer, University of Maryland School of Medicine, USAReviewed by:
Dalton J. Surmeier, Northwestern University, USAPaolo Calabresi, Santa Maria della Misericordia Hospital, Italy
Copyright: © 2012 Mathur and Lovinger. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: David M. Lovinger, Section on Synaptic Pharmacology, Laboratory for Integrative Neuroscience, National Institute on Alcohol Abuse and Alcoholism, U.S. National Institutes of Health, Rockville, TS-13, 5625 Fishers Lane, Bethesda, MD 20892, USA. e-mail:bG92aW5kYXZAbWFpbC5uaWguZ292